

Abstract
Commonly prescribed selective serotonin reuptake inhibitors (SSRIs) inhibit the serotonin transporter to correct a presumed deficit in extracellular serotonin signaling during depression. These agents bring clinical relief to many who take them; however, a significant and growing number of individuals are resistant to SSRIs. There is emerging evidence that inflammation plays a significant role in the clinical variability of SSRIs, though how SSRIs and inflammation intersect with synaptic serotonin modulation remains unknown. In this work, we use fast in vivo serotonin measurement tools to investigate the nexus between serotonin, inflammation, and SSRIs. Upon acute systemic lipopolysaccharide (LPS) administration in male and female mice, we find robust decreases in extracellular serotonin in the mouse hippocampus. We show that these decreased serotonin levels are supported by increased histamine activity (because of inflammation), acting on inhibitory histamine H3 heteroreceptors on serotonin terminals. Importantly, under LPS-induced histamine increase, the ability of escitalopram to augment extracellular serotonin is impaired because of an off-target action of escitalopram to inhibit histamine reuptake. Finally, we show that a functional decrease in histamine synthesis boosts the ability of escitalopram to increase extracellular serotonin levels following LPS. This work reveals a profound effect of inflammation on brain chemistry, specifically the rapidity of inflammation-induced decreased extracellular serotonin, and points the spotlight at a potentially critical player in the pathology of depression, histamine. The serotonin/histamine homeostasis thus, may be a crucial new avenue in improving serotonin-based treatments for depression.
SIGNIFICANCE STATEMENT Acute LPS-induced inflammation (1) increases CNS histamine, (2) decreases CNS serotonin (via inhibitory histamine receptors), and (3) prevents a selective serotonin reuptake inhibitor (SSRI) from effectively increasing extracellular serotonin. A targeted depletion of histamine recovers SSRI-induced increases in extracellular hippocampal serotonin.
Keywords: carbon fiber microelectrodes, depression, FSCV, H3 receptor, neurochemistry, SSRI
Introduction
Selective serotonin reuptake inhibitors (SSRIs) are some of the most widely prescribed pharmaceutical agents (Pratt et al., 2017; also see https://www.mayoclinic.org/diseases-conditions/depression/in-depth/ssris/art-20044825). The underlying premise supporting SSRI pharmacotherapy is the serotonin hypothesis of depression (Belmaker and Agam, 2008; Krishnan and Nestler, 2008). The postulate here is that extracellular serotonin levels are reduced during depression, especially in the hippocampus (Sheline et al., 1996; Nestler et al., 2002; Stockmeier et al., 2004), and by inhibiting the serotonin transporter (SERT), the extracellular levels of this monoamine can be restored. Because of the difficulties in measuring serotonin, the community has not reached accord on the serotonergic basis for depression and SSRIs exhibit variable clinical efficacy (Artigas et al., 2018). Thus, SSRIs have lost popularity in the research community, and recent work has targeted other means to treat depression including the use of ketamine, psychedelics, electroconvulsive therapy, vagus nerve stimulation, and deep brain stimulation (Mayberg et al., 2005; Lisanby, 2007; Shelton et al., 2010; Andrade, 2015; Carhart-Harris et al., 2016).
There are, however, interesting associations between depression and inflammation that may reinvigorate interests in SSRIs. The comorbidity between inflammation and depression is now well accepted (Dantzer et al., 2011; Miller and Raison, 2016). The critical questions here are how does inflammation affect serotonin neurochemistry and how does this effect intersect with SSRI therapy?
In this study, we use unique electrochemical tools, developed in our laboratory, for studying in vivo serotonin neurochemistry during acute inflammation [lipopolysaccharide (LPS) injection]. We focus the serotonin work on the hippocampus, a dynamic region known to mediate mood and antidepressant response (Malberg et al., 2000; Campbell and MacQueen, 2004; Stockmeier et al., 2004). We found rapid and robust decreases in extracellular hippocampal serotonin (using fast-scan controlled adsorption voltammetry (FSCAV) for basal serotonin levels) in response to intraperitoneal LPS injection. There were no apparent rapid changes in the functionality of the serotonin release/reuptake system via measurements of evoked serotonin with fast-scan cyclic voltammetry (FSCV). In attempting to reverse diminished extracellular serotonin levels via treatment with the SSRI escitalopram (ESCIT), we found that the ability of ESCIT to increase serotonin levels was blunted in LPS-injected animals relative to saline-treated controls, an effect that we found to be SERT independent. The decreased extracellular serotonin in the inflammatory state was supported by increased brain histamine acting via presynaptic histamine type 3 heteroceptors (H3Rs) on serotonergic terminals. We show that the reduced capability of ESCIT to increase serotonin after LPS likely lies in its capacity for also inhibiting histamine reuptake. As such, we show that by dual pharmacological targeting of the serotonergic and histaminergic systems, extracellular serotonin levels can be restored to pre-LPS levels. Our work implies, on a fundamental chemical level, that it is not yet time to give up on SSRI therapy, particularly in the context of elevated inflammation, but rather to consider this treatment in tandem with agents that block the ability of histamine to diminish serotonin release.
Materials and Methods
Chemicals and reagents
Electrodes were calibrated in serotonin solutions of 10, 25, 50, 100 nm made from serotonin hydrochloride (Sigma-Aldrich) dissolved in Tris buffer [15 nm H2NC(CH2OH)2 HCl, 140 mm NaCl, 3.25 mm KCl, 1.2 mm CaCl2, 1.25 mm NaH2PO4·H2O, 1.2 mm MgCl2, and 2.0 mm Na2SO4; Sigma-Aldrich]. Escitalopram oxalate (≥98 mg/kg HPLC; Sigma-Aldrich) at 10 mg/kg, lipopolysaccharide (catalog #0111:B4; Sigma-Aldrich) at 0.2 or 1 mg/kg, α-fluoromethylhistidine dihydrochloride (Toronto Research Chemicals) at 20 mg/kg, GBR 12909 (Sigma-Aldrich) at 15 mg/kg, desipramine hydrochloride (Sigma-Aldrich) at 15 mg/kg, citalopram hydrobromide (Sigma-Aldrich) at 5 mg/kg, sertraline hydrochloride (Sigma-Aldrich) at 10 mg/kg, and decynium-22 (D-22; Sigma-Aldrich) at 0.1 mg/kg were individually dissolved in sterile saline (0.9% NaCl solution, Hospira) and administered via intraperitoneal injections at a volume of 5 ml/kg per animal.
Electrode fabrication
Serotonin and histamine were analyzed using carbon fiber microelectrodes (CFMs) fabricated as previously described (Hashemi et al., 2009; Wood and Hashemi, 2013; Wood et al., 2014). Carbon fibers (7 µm; Goodfellow) were aspirated into glass capillaries (internal diameter, 0.4 mm; outer diameter, 0.6 mm; A-M Systems). A carbon-glass seal was created using a vertical pipette puller (Narishige). The exposed carbon fiber was then trimmed to 150 µm and silver paint was used to adhere the electrode to a connection pin. Last, Nafion was electroplated onto the electrode as previously described (Hashemi et al., 2009).
Animal and surgical procedures
Animal procedures and protocols were in accordance with regulations of the Institutional Animal Care and Use Committee at the University of South Carolina, operating with accreditation from the Association for Assessment and Accreditation of Laboratory Animal Care. Male and female C57BL/6J mice (age, 6–12 weeks; weight, 18–30 g; The Jackson Laboratory) were group housed, with ad libitum access to food and water, and were kept on a 12 h light/dark cycle (lights off at 7:00 A.M., lights on at 7:00 P.M.). Additionally, male and female SERT Met 172 knock-in mice on a 129S6/S4 background were backcrossed with C57BL/6J mice and wild-type (WT) mice [C57 mice with 129 WT SERT gene (Ile172)], 6–20 weeks of age, were also used for these studies (Nackenoff et al., 2016).
Stereotaxic surgery in the CA2 region of the hippocampus and the posterior hypothalamus was performed as previously described (Samaranayake et al., 2015, 2016; Saylor et al., 2019; Abdalla et al., 2020).
Surgery was performed following an intraperitoneal injection of 25% w/v urethane (7 μl/g body weight; Sigma-Aldrich), dissolved in 0.9% NaCl solution (Hospira) to maintain and induce anesthesia. Mouse body temperature was maintained using a heating pad (Braintree Scientific). Stereotaxic surgery (David Kopf Instruments) was performed, and all coordinates were measured in reference to bregma.
Hippocampus.
A Nafion CFM was lowered into the CA2 region [serotonin: anteroposterior (AP), −2.91 mm; mediolateral (ML), +3.35 mm; dorsoventral (DV), −2.5 to −3.0 mm; Paxinos and Franklin, 2013)] and stimulating electrode (insulated stainless steel: diameter: 0.2 mm, untwisted; Plastics One) was placed into the medial forebrain bundle (serotonin: AP, −1.58 mm; ML, +1.00 mm; DV, −4.8 mm; Paxinos and Franklin, 2013).
Posterior hypothalamus.
A Nafion CFM was lowered into the posterior hypothalamus (histamine: AP, −2.45 mm; ML, +0.50 mm; DV, −5.45 to −5.55 mm) and a stimulating electrode was placed into the medial forebrain bundle (histamine: AP, −1.07 mm; ML, +1.10 mm; DV, −5.00 mm; Paxinos and Franklin, 2013).
An Ag/AgCl reference electrode, created by electroplating chloride (30 s in 0.1 m HCl at 5 V) onto a silver wire, was placed into the contralateral hemisphere.
Voltammetric methods and analysis
FSCV and FSCAV were performed while mice were anesthetized using a potentiostat (Dagan), WCCV 3.06 software (Knowmad Technologies), and headstage (either Dagan or Pine Research Instruments). Serotonin and histamine were analyzed using CFMs, as previously described (Hashemi et al., 2009; Wood and Hashemi, 2013; Wood et al., 2014; Samaranayake et al., 2015, 2016). For FSCV, the “Jackson” waveform (Jackson et al., 1995) was used in the hippocampus (scanning from 0.2–1.0 to −0.1 to 0.2 V at 1000 V/s, and the dual serotonin/histamine waveform was used in the posterior hypothalamus (scanning from −0.5 to −0.7 to 1.1 to −0.5 V at 600 V/s; Samaranayake et al., 2015; 2016). Serotonin or histamine release was evoked using a biphasic stimulation applied through a linear constant current stimulus isolator (model NL800A Neurolog, Medical Systems) with 60 Hz, 360 µA each, 2 ms in width and 2 s in length. After FSCV in vivo experiments, ∼5 V was applied to the CFM to lesion tissue for histology. FSCAV experiments followed the collection of control FSCV files, as in prior work (Abdalla et al., 2017). Post-experiment calibrations, in vitro, were completed for each electrode. WCCV software was used for postanalyses. Previously established calibration factors were used for serotonin FSCV (49.5 ± 10.2 nA/μm) and histamine FSCV (histamine: 2.825 nA/µM; serotonin: 11 nA/nM). For FSCAV analysis following reapplication of the serotonin waveform, the third cyclic voltammogram (CV) was selected for quantification and the peak, at between ∼0.4 and 0.85 V, was integrated to determine the charge (in picocoulombs). Each CV was integrated manually and baseline shifts and drifts were manually corrected.
Mathematical modeling was via a previous model for histamine cells in MATLAB (Best et al., 2017).
Experimental design and statistics analyses
For FSCV data, four current–time curves were collected and averaged for each animal to establish a control response. The averages for each individual animal were then combined with those of the other animals in the group to determine an overall group average. We previously reported that a power analysis recommended a minimum n size of 3.5 (rounded to 4) and maintained the animal exclusion criteria [i.e., outliers (via Grubbs' test) and animals that did not survive the experimental paradigm; Saylor et al., 2019]. The SEM was calculated using the average response of each animal (n = # animals). For FSCV, significance was determined between two points with a paired two-tailed Student's t test as (p < 0.05).
For FSCAV data, both parametric and nonparametric approaches were taken to compare the extracellular hippocampal serotonin concentrations for the different treatment conditions.
Bland–Altman plots for the first 30 data points (0–30 min) were used to evaluate the reproducibility between control measurements and treatment measurements. No proportional or systematic errors are detected between control and treatment measurements (see Figs. 5A, 7, 10A,D). A shift of the serotonin baseline is perceived between the control (blue trace) and LPS treatment (red trace) in Figures 5A and 10D. These concentrations are within physiological levels of serotonin concentration in the hippocampus (Abdalla et al., 2020). A representation of the Bland–Altman plot for the baseline (0–30 min) data points between control and thioperamide (H3R antagonist) administration measurements (see Fig. 7) can be seen in Figure 1.
Figure 5.
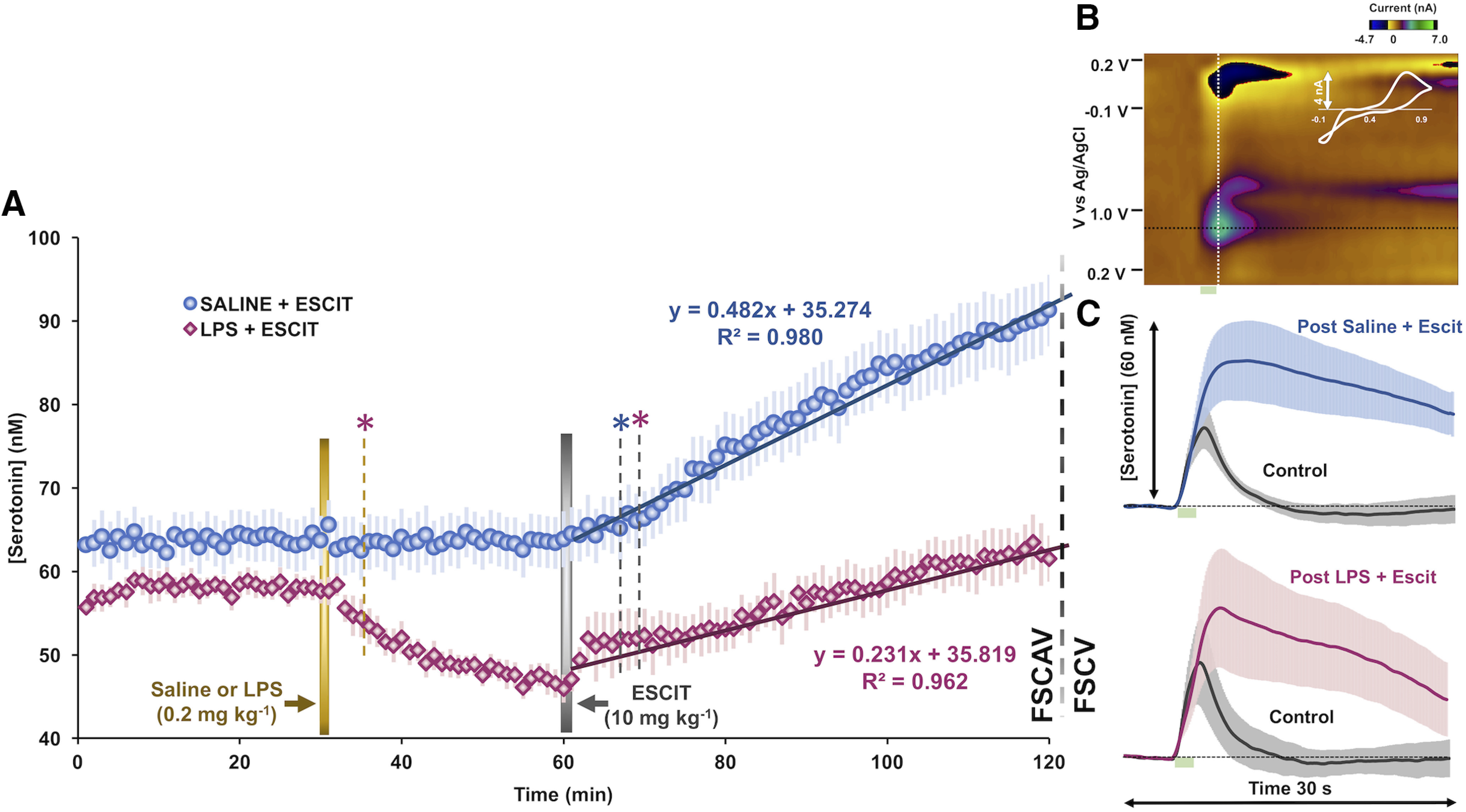
Evoked and extracellular hippocampal serotonin after LPS and SSRI administration. A, Extracellular hippocampal serotonin FSCAV measurements after administration of LPS (0.2 mg/kg, i.p.; n = 10; shown in pink) and vehicle control (intraperitoneal saline; n = 10; shown in blue). ESCIT was administered to each treatment group (10 mg/kg, i.p., n = 10 both). A Student's t test was used to determine the first time a drug treatment induced statistically significant changes in serotonin (p < 0.05). A gold dotted line denotes the first time point at which LPS produced decreased serotonin compared with averaged control (serotonin; pink *). A gray dotted line denotes the first time point that saline- and LPS-treated groups showed significant effects of ESCIT on serotonin compared with the last time point (59 min) before ESCIT was administered (blue and pink *, respectively). Regression analysis of FSCAV data are shown in Figure 3. B, An example serotonin FSCV color plot with a green serotonin event shown at 0.7 V and confirmed with the extracted cyclic voltammogram (shown in white). C, Evoked FSCV serotonin release before (control, shown in gray) and after saline + ESCIT or LPS + ESCIT administration and 120 min of FSCAV data collection (n = 8 saline, shown in blue; n = 6 LPS, shown in pink). Stimulation is marked by a green box at 5–7 s. Error (±SEM) is shown in lighter colored error bars around the traces.
Figure 7.
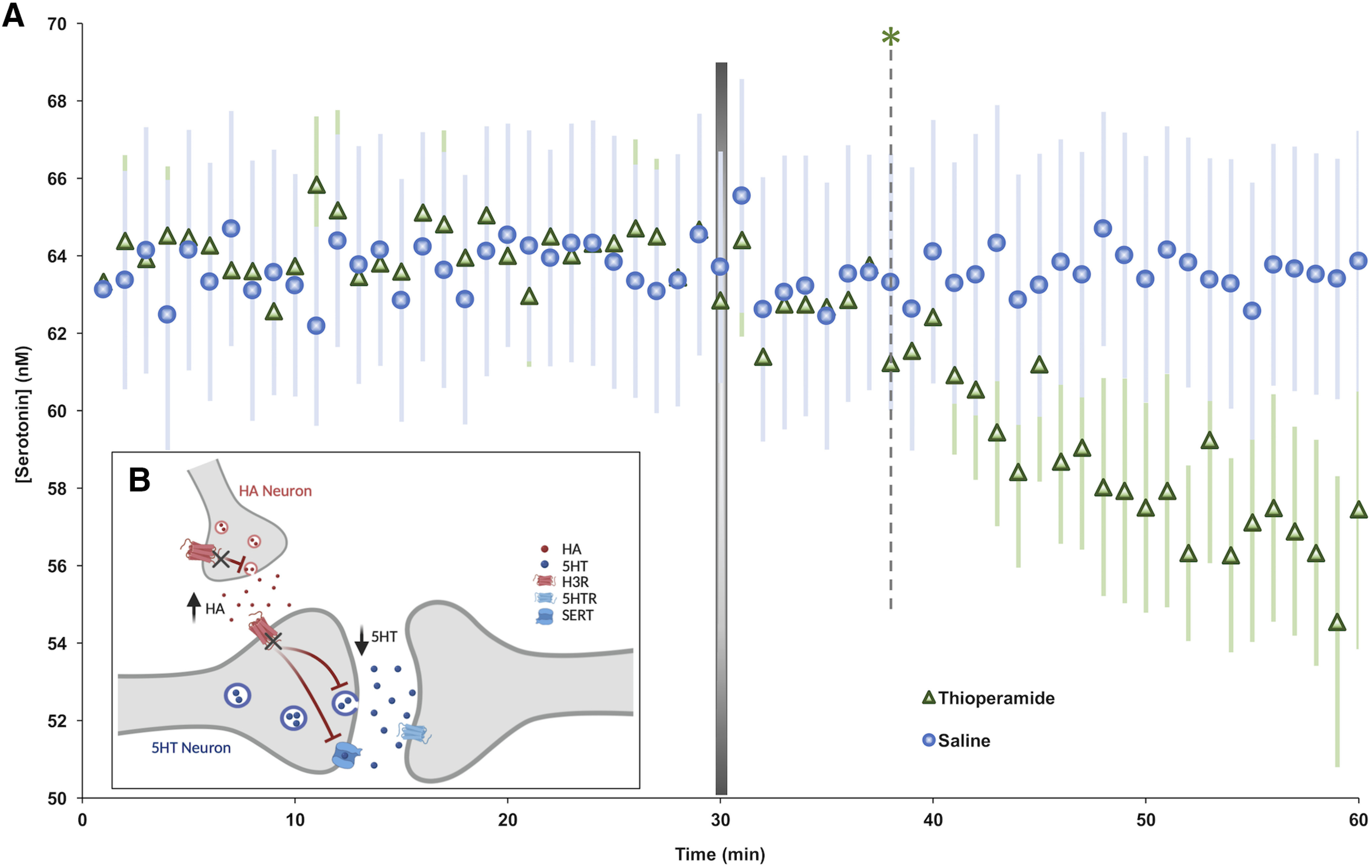
Serotonin decrease in the hippocampus is mediated by histamine. A, Following 30 min of baseline serotonin collection, thioperamide was administered (50 mg/kg, i.p.; green, n = 5). Control animals were given saline (i.p.; blue, n = 10). Error (±SEM) is shown in a lighter color. A Student's t test was used to determine the first time a drug treatment induced statistically significant changes in serotonin from baseline measures. A gray dotted line denotes the first time point at which thioperamide produced decreased serotonin compared with averaged control serotonin (green, *; 8 min). A representative Bland–Altman plot to analyze FSCAV data can be seen in Figure 1. B, Schematic showing how histamine (HA) release acts on H3Rs located on serotonin (i.e., 5-HT) neurons to inhibit serotonin release. The actions of thioperamide on H3Rs are marked.
Figure 10.
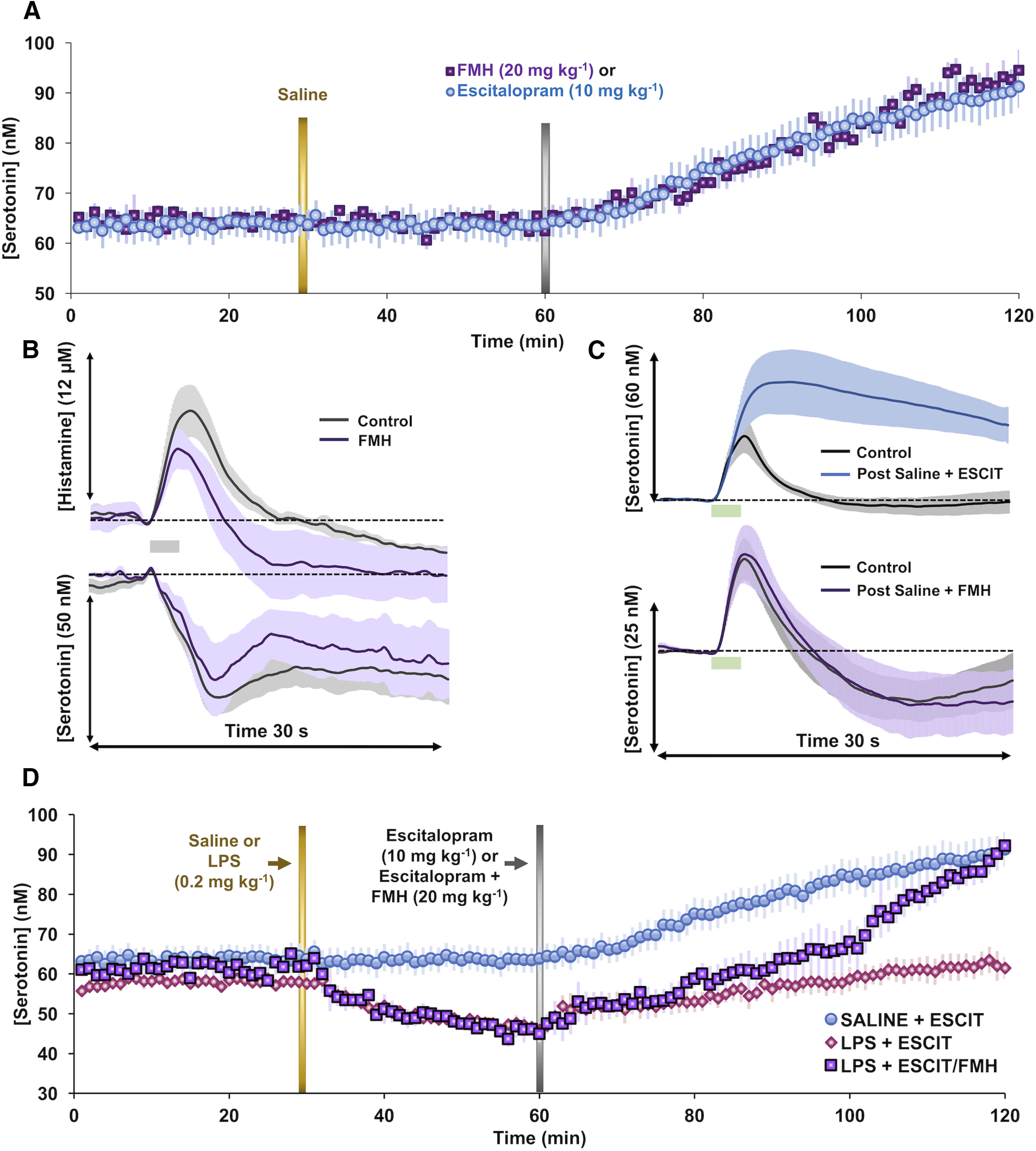
Dual targeting of histamine and serotonin on hippocampal serotonin. A, Control basal hippocampal serotonin is shown for 30 min before saline treatment and 30 min after saline treatment. Mice were then administered either ESCIT (10 mg/kg, i.p.; blue, n = 10) or FMH (20 mg/kg, i.p.; purple, n = 4). B, Evoked hypothalamic histamine and serotonin (black) and 50 min postadministration of FMH (20 mg/kg, i.p.; purple, n = 4) are shown. C, Evoked hippocampal serotonin pre- and post-saline + ESCIT (10 mg/kg, i.p.; blue) and post-saline + FMH (20 mg/kg, i.p.; purple, n = 4) stimulated releases are shown for both treatment groups with control-evoked release shown in gray. D, Basal extracellular hippocampal serotonin after administration of saline and ESCIT (10 mg/kg, i.p.; blue, n = 10); LPS and ESCIT (0.2 and 10 mg/kg, i.p., respectively; pink, n = 10); and LPS, ESCIT, and FMH (0.2, 10, and 20 mg/kg, i.p., respectively; purple, n = 5 each). Additional statistical analyses are found in Figure 2 and Table 1.
Figure 1.

Bland–Altman plot. Representation of the differences between pairs of samples and their means for baseline serotonin FSCAV measurements in control and thioperamide-administered animals (Fig. 7). The blue represents the mean value of the differences. The red dashed lines represent the 95% confidence limits of the limits of agreement.
Significance among stationary differences in serotonin concentration were determined with a two-tailed Student's t test at 95% confidence. The t tests were performed between two points of the time series to confirm the first point at which a serotonin concentration change is statistically significant (see Fig. 5A). The t tests between stationary groups of points were also used to certify a change in concentration after treatment or saline injection (see Figs. 5A, 7, 10D).
The Pearson product-moment correlation coefficient was also used to measure the linear correlation between serotonin concentration increase following ESCIT and α-fluoromethylhistidine dihydrochloride (FMH; a suicide inhibitor of histidine decarboxylase) administration (see Fig. 10A). Before to its use, the data were visually analyzed to check for outliers that could give a false sense of relationship between the time series. Any possible outlier was discarded (via Grubbs' test). A high positive linear correlation was found between the increase of serotonin following ESCIT and FMH administration. A Student's t test was used to certify that the linear correlation is different from zero (r = 0.97, p < 0.001). This means that both serotonin time series increase in tandem.
The distribution of the differences between time points following ESCIT and FMH administration (see Fig. 10A) was also visualized to have a mean of ∼0 and Gaussian shape. An example of the histogram visualization can be seen in Figure 2. The differences between time points were tested not to be significantly different from zero (W statistic = 764.0, p = 0.27, Wilcoxon signed-rank test). In this case, nonparametric tests were preferred to avoid assuming a normal distribution of samples.
Figure 2.

Histogram of ESCIT and FMH differences in the time series. Representation of the frequency of occurrence of differences between serotonin concentrations followed by FMH or ESCIT administration. The blue shaded bars represent the number of time series encountered within a concentration difference interval. The blue vertical line represents the mean of differences, and the red dashed lines, the 95% confidence interval. We can see that the mean of the differences is close to 0.
Regression analysis was used to model the rate of change of extracellular hippocampal serotonin after drug treatment and saline control injection. Significance of a change in the slope of the fitted linear regression was determined with a two-tailed Student's t test to determine whether the slope differs remarkably from zero. All control saline injections fail to provide enough evidence of a change in serotonin concentration over time compared with baseline (see Figs. 5A, 7, 10A,D). Following LPS and thioperamide administration (see Figs. 5, 7, respectively), the slope of the fitted linear regression was tested to be significantly lower than zero. Following ESCIT administration (see Figs. 5, 10D), the slope of the fitted linear regression was tested to be significantly higher than zero. The slopes of the regression lines for ESCIT administration (60–120 min) were also tested to be significantly different between control and LPS injection. An example of this regression analysis can be seen in Figure 3. See Figure 10D for analysis using a one-way ANOVA with Tukey's post hoc test to compare the slopes of each period (all values available in Table 1).
Figure 3.

Regression analysis plots. A–C, Each panel shows the experimental FSCAV time series for LPS treatment between 30 and 60 min (A), LPS + ESCIT treatment between 60 and 120 min (B), and saline plus ESCIT treatment between 60 and 120 min (C). A linear regression is fitted to each time series. The 95% prediction limits and confidence limits of the linear regression are also represented. The coefficient of determination () and the slope SEM are also calculated. The t statistic for the slope of the regression fit is calculated and proved to be significantly different from a slope of zero for all drug treatments.
Table 1.
Analysis of slope across treatment groups
| Group | LSMEAN slope | LSMEAN number | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline1 | 0.01752433 | 1 | |||||||
| Baseline2 | 0.02800934 | 2 | |||||||
| Baseline3 | 0.03261000 | 3 | |||||||
| ESCIT | 0.48186300 | 4 | |||||||
| ESCIT + FMH | 0.70005750 | 5 | |||||||
| LPS+ESCIT | 0.22998300 | 6 | |||||||
| LPS | −0.35329600 | 7 | |||||||
| LPS + FMH | −0.45018750 | 8 | |||||||
| Saline | 0.00547758 | 9 | |||||||
| LSMEAN for effect group [Pr > |t| for H0: LSMean(i) = LSMean(j) (dependent variable: slope)] | |||||||||
| i/j | |||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| 1 | 1.0000 | 1.0000 | <0.0001 | <0.0001 | 0.0286 | <0.0001 | <0.0001 | 1.0000 | |
| 2 | 1.0000 | 1.0000 | <0.0001 | <0.0001 | 0.0453 | <0.0001 | <0.0001 | 1.0000 | |
| 3 | 1.0000 | 1.0000 | <0.0001 | <0.0001 | 0.3019 | 0.0005 | 0.0002 | 1.0000 | |
| 4 | <0.0001 | <0.0001 | <0.0001 | 0.1861 | 0.0042 | <0.0001 | <0.0001 | <0.0001 | |
| 5 | <0.0001 | <0.0001 | <0.0001 | 0.1861 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | |
| 6 | 0.0286 | 0.0453 | 0.3019 | 0.0042 | <0.0001 | <0.0001 | <0.0001 | 0.0164 | |
| 7 | <0.0001 | <0.0001 | 0.0005 | <0.0001 | <0.0001 | <0.0001 | 0.9582 | <0.0001 | |
| 8 | <0.0001 | <0.0001 | 0.0002 | <0.0001 | <0.0001 | <0.0001 | 0.9582 | <0.0001 | |
| 9 | 1.0000 | 1.0000 | 1.0000 | <0.0001 | <0.0001 | 0.0164 | <0.0001 | <0.0001 |
A one-way ANOVA with Tukey's post hoc analysis to compare the slopes of each period for the data presented in Figure 10D. LSMEAN, Least-squares mean.
Results
Systemic LPS decreases extracellular hippocampal serotonin levels
To test whether an acute, systemic injection of LPS affects extracellular serotonin, we applied the following two techniques developed in our laboratory: FSCV for subsecond measurements of evoked release; and FSCAV for minute to minute measurements of extracellular levels, as determined by a preconcentration step that allows several seconds for adsorption of serotonin in the extracellular fluid (ECF) to come into equilibrium with the surface of a CFM (Abdalla et al., 2020). FSCV was used to isolate an evoked serotonin release event in the CA2 region of an anesthetized mouse hippocampus. We focused our studies on this domain because of the implied involvement of this brain region in depression and antidepressant action (Sheline et al., 1996; Nestler et al., 2002; Stockmeier et al., 2004), and we used male and female mice because we had previously shown no significant sex differences in control or ESCIT responses between the sexes (Saylor et al., 2019). LPS (1 mg/kg) was administered intraperitoneal (i.p.), and the serotonin signal was re-evaluated 5 min thereafter and again at 10 min time intervals up to 60 min. There is near perfect agreement in evoked serotonin between the pre-LPS and post-LPS treatment (these data are not normalized and are shown in Fig. 4).
Figure 4.

Evoked hippocampal serotonin after LPS. Evoked serotonin pre-LPS administration (gray) and post-LPS administration (1.0 mg/kg, i.p.; shown in garnet; n = 5). Stimulation is marked by a green box at 5–7 s. The error (±SEM) is shown as shadows around the traces.
In two different cohorts of mice, FSCAV was used to measure extracellular serotonin levels in the same region (Fig. 5). The basal values between cohorts were not significantly different. In the first cohort (Fig. 5, blue trace), which is an average of 10 mice, the first 30 data points were used to generate a baseline value. At 30 min, saline was given intraperitoneally, and the values at 55–60 min (26–30 min post-saline administration) were not significantly different from the baseline values (63.68 ± 3.00 vs 63.64 ± 3.16 nm; t(9) = 2.26, p = 0.92, paired t test). At 60 min, these mice received ESCIT (10 mg/kg, i.p.), and shortly thereafter the serotonin levels increased; the first point at which this increase was significant was at 7 min (26–30 min post-saline administration: 63.64 ± 3.16 vs 65.11 ± 3.51 nm; t(9) = 2.26, p = 0.013, paired t test). In the second cohort (Fig. 5, red trace), we generated a baseline in the same way and administered LPS (0.2 mg/kg, i.p.) instead of saline at 30 min. LPS administration rapidly induced a decrease in serotonin. We fit each trace with linear regressions between 30 and 60 min, and found that only the LPS treatment induced significant changes from baseline serotonin levels (Fig. 5: saline/blue: slope = 0.005 ± 0.01 nm/min; t(28) = 0.39, p = 0.69; LPS/pink: slope = −0.35 ± 0.03 nm/min; t(28) = −13.39, p < 0.0001, slope t test). The first point at which this LPS-induced decrease was significant was at 5 min (57.93 ± 1.37 to vs 54.43 ± 2.17 nm; t(9) = 2.26, p = 0.043, paired t test) with progressively larger reductions ensuing. At 60 min, these mice were given ESCIT (10 mg/kg, i.p.). ESCIT produced an increase in serotonin that was first significant at 9 min (26–30 min post-LPS administration: 46.91 ± 1.48 vs 51.94 ± 3.17 nm; t(9) = 2.26, p = 0.040, paired t test). At 55–60 min post-ESCIT administration, the extracellular serotonin levels in LPS mice were 62.27 ± 3.01 nm; this level is significantly lower than post-ESCIT administration in the saline-treated mice (90.10 ± 4.43 nm; t(9) = 2.26, p = 0.0009, two-sample t test). Further, the rate of increase following ESCIT was significantly lower in LPS-treated animals (control, slope = 0.48 ± 0.01 nm/min; LPS, slope = 0.23 ± 0.05 nm/min; t(58) = −4.90, p < 0.0001, two-sample t test). At 120 min, we saw no difference in electrically evoked serotonin release between the two cohorts (Ampmax: 50.66 ± 12.97 vs 39.47 ± 12.49 nm; t(12) = 2.18, p = 0.55, two-sample t test), nor did we detect a change in the serotonin clearance rate [half-life (t1/2) for descending phase after peak; t1/2: 10.70 ± 2.36 s, 11.30 ± 2.19 s; p = 0.86, t(12) = 2.18, two-sample t test; Fig. 5C].
Brain histamine rapidly increases on systemic LPS injection
In asking which mechanisms might mediate LPS-induced reduction in serotonin, we focused in on histamine. While much is known about histamine as a mediator of inflammation in the periphery, there is almost no information about histamine in the brain during inflammation. The reason that we hypothesized histamine to play an important role here is because of a known histamine/serotonin homeostasis. For example, histamine elevates cell surface SERT expression and diminishes evoked 5-hydroxytryptamine (5-HT) release via H3Rs (Schlicker et al., 1988; Threlfell et al., 2004; Samaranayake et al., 2016; Annamalai et al., 2020).
We have a well established model for evoking serotonin in the CA2 region of the hippocampus, used above (Saylor et al., 2019; Abdalla et al., 2020). Our model for histamine/serotonin modulation is in the posterior hypothalamus (Samaranayake et al., 2015, 2016) because this experiment allows us to evoke histamine and not serotonin, such that the modulatory effects of histamine on serotonin can be observed in the absence of stimulated serotonin. This effect is not accessible in the hippocampus since the stimulation evokes serotonin release. It is not possible to directly compare these two brain regions; thus, we use the hypothalamus data to create and test hypotheses in the hippocampus.
We tested whether the decrease in extracellular serotonin post-LPS administration was because of increased extracellular histamine (shown previously to be a function of H3R activation; Schlicker et al., 1988; Threlfell et al., 2004; Samaranayake et al., 2016). Simultaneous FSCV measurements of hypothalamic histamine and serotonin were performed (Fig. 6A). The interpretation of the dual histamine/serotonin color plot can be found in detail in a previous study (Samaranayake et al., 2016); in brief, the horizontal lines on the color plot denote the peak histamine and serotonin oxidation peaks, and the amplitude of this peak (interchangeable with concentration) with time is then shown in Figure 6, B and C. Histamine was modestly but not significantly increased within 5 min following LPS injection (1.0 mg/kg, i.p.; Fig. 6B; Ampmax: 5.80 ± 1.67 to 8.77 ± 3.27 μm; t(4) = 2.77, p = 0.14, paired t test). Serotonin levels were modestly but not significantly inhibited (Ampmax: 42.00 ± 13.33 to 51.38 ± 18.66 nm; t(4) = 2.77, p = 0.21, paired t test). A lower dose showed similar, but insignificant, trends (Fig. 6C).
Figure 6.
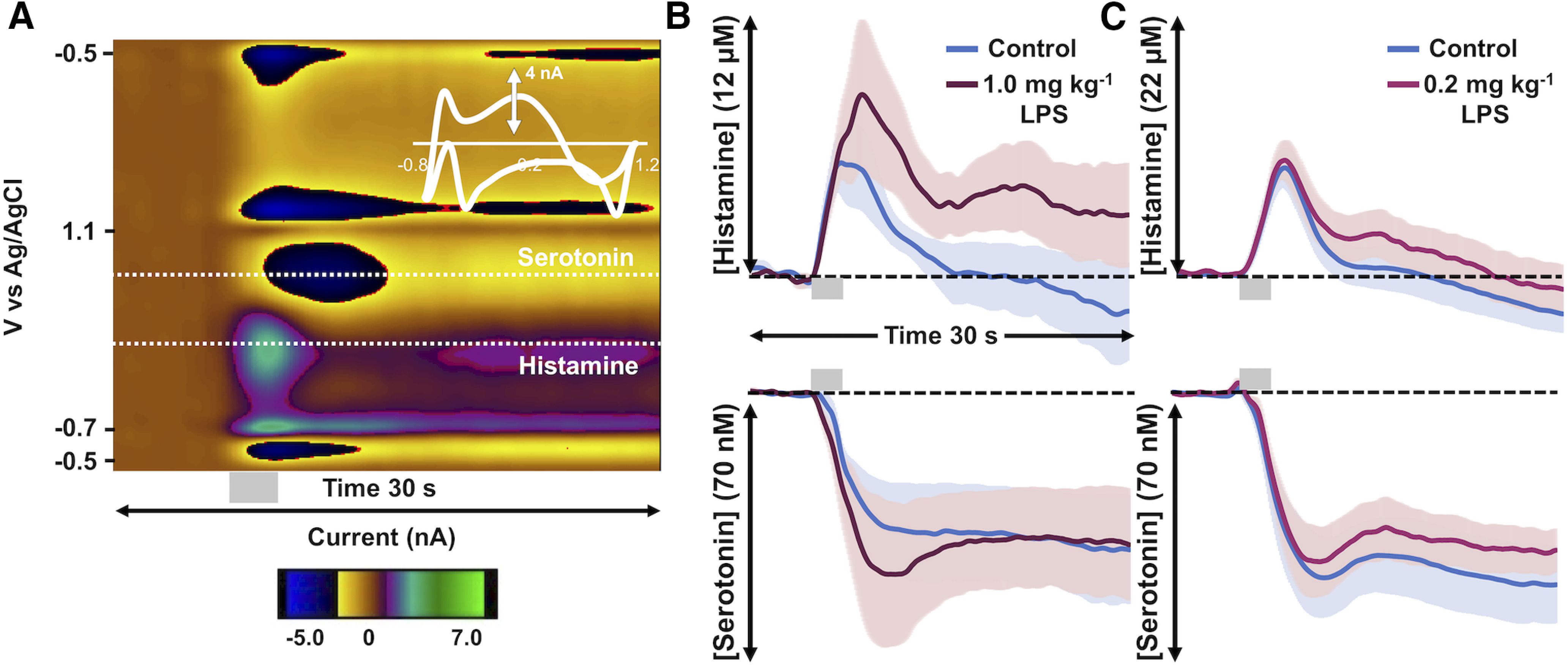
Evoked hypothalamic histamine increases with LPS administration. A, A representative FSCV color plot with a histamine event at 0.3 V and a serotonin event at 0.7 V. The gray bar under the color plot denotes the stimulation from 5 to 7 s. B, Control-evoked hypothalamic histamine (top) and control inhibition of hypothalamic serotonin (bottom) are shown in blue, and 5 min after the administration of LPS at a high dose (1.0 mg/kg, i.p.; n = 5) are shown in garnet. C, Control-evoked hypothalamic histamine (above) and control inhibition of evoked hypothalamic serotonin (below) are shown in blue and following a lower dose of LPS (pink; 0.2 mg/kg, i.p.; n = 10). Stimulation is marked by a gray box at 5–7 s. Error (±SEM) is shown as shadows around the traces.
We used an alternative method, outside of LPS, to pharmacologically alter histamine and assess the resultant effects on serotonin. We previously showed using FSCV that thioperamide, an H3R antagonist, increased evoked extracellular histamine levels that paralleled a larger decrease in extracellular serotonin in the hypothalamus. Here we used FSCAV to monitor extracellular serotonin levels after thioperamide. Baseline serotonin was measured for 30 min before the administration of either thioperamide (green; Fig. 7A) or saline (blue; Fig. 7A). Following thioperamide administration, we observed that extracellular serotonin levels were significantly reduced compared with saline controls (control regression: slope = 0.005 ± 0.014 nm/min; t(28) = 0.39, p = 0.69; post-thioperamide administration regression: slope = −0.27 ± 0.022 nm/min, t(28) = −12.48, p < 0.0001, slope t test). This decrease in serotonin was first significantly different from control values at 8 min post-thioperamide administration (control, 64.11 ± 1.80 nm; 8 min post-thioperamide administration, 61.23 ± 1.95 nm; t(4) 2.78, p = 0.038, paired t test).
SSRIs inhibit reuptake of hypothalamic histamine
To investigate the nexus of histamine, serotonin, and SSRI treatment, we tested the effects of monoamine transport inhibitors on histamine reuptake (under control conditions). In Figure 8, we measured hypothalamic histamine (control, blue) and then pharmacologically inhibited the following transporter proteins (post-drug administration, green): dopamine transporters (DATs), SERTs, and norepinephrine transporters (NETs) with the following agents (intraperitoneal; n = 5 each): GBR 12909 (DAT inhibitor, 15 mg/kg); desipramine (NET inhibitor, 15 mg/kg); escitalopram (SERT inhibitor, 10 mg/kg); citalopram (SERT inhibitor, 5 mg/kg); and sertraline (SERT inhibitor, 10 mg/kg). We found that, in all cases except DAT inhibition, there was slowing of the rate of histamine reuptake that peaked at 60 min.
Figure 8.
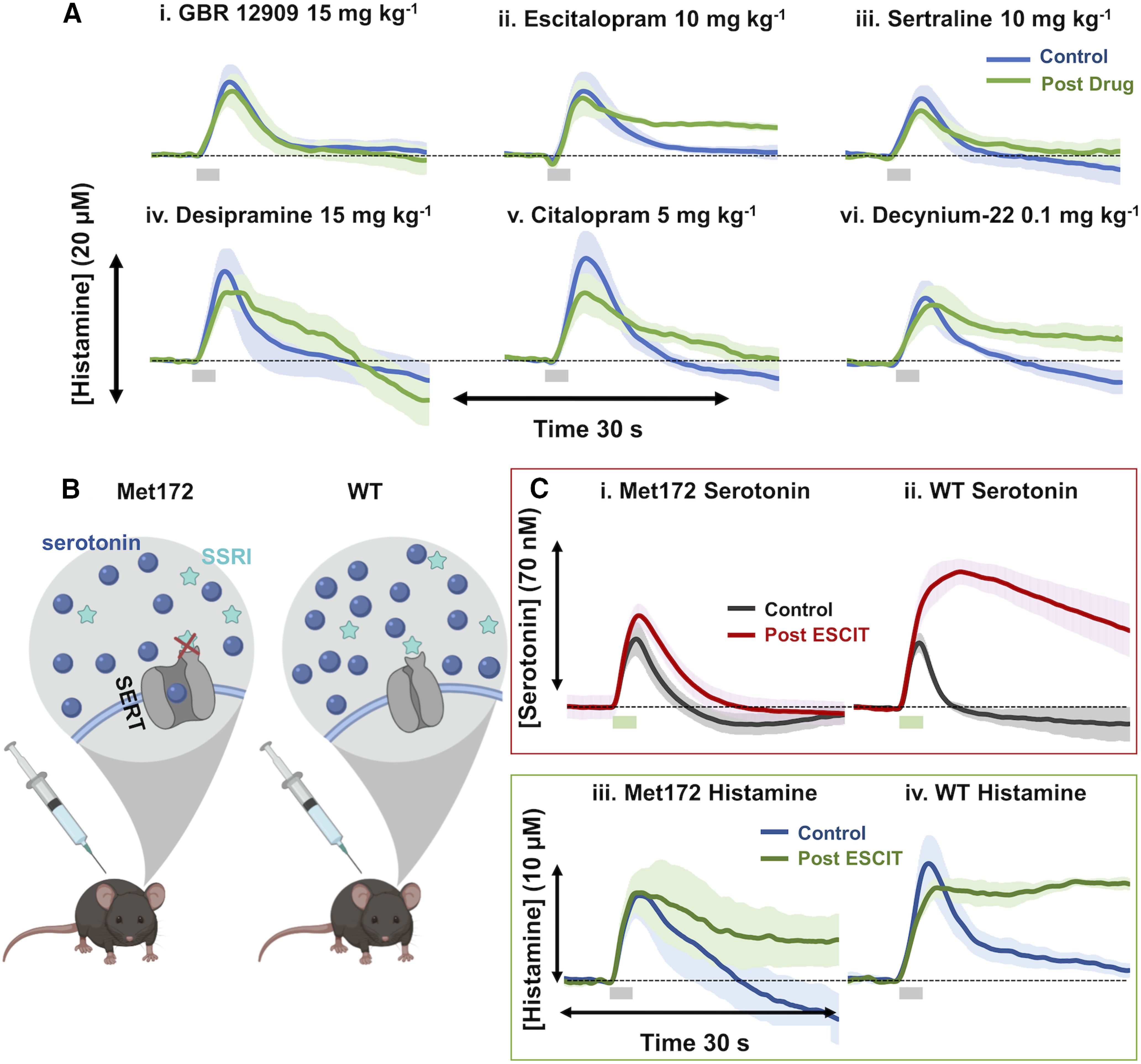
Histamine reuptake is inhibited by monoamine transporter inhibitors. A, Control hypothalamic histamine (blue) and 60 min post-GBR 12909 administration (15 mg/kg; i.p.; n = 5; i), ESCIT (10 mg/kg; n = 5; ii), sertraline (10 mg/kg; n = 5; iii), desipramine (15 mg/kg; n = 5; iv), citalopram (5 mg/kg; n = 5; v), and decynium-22 (0.1 mg/kg; n = 5; vi; green). Stimulation marked by gray box at 5–7 s. Error (±SEM) is around traces. Doses were reported in prior work to create behavioral shifts or neurochemical changes (Hashemi et al., 2009; Park et al., 2009; Zomkowski et al., 2010; Kulikov et al., 2011; Mikail et al., 2012; Horton et al., 2013). B, Schematic showing the inability of SSRIs to bind to SERTs and to prevent serotonin reuptake in SERT Met172 mice. C, Evoked hippocampal serotonin control (gray) and 70+ min post-ESCIT administration (red; 10 mg/kg) in Met172 mice (n = 5; i) and WT (n = 4; ii) with a stimulation marked by a green box below at 5–7 s. Control evoked hypothalamic histamine (blue), and 60 min post-ESCIT administration (green; 10 mg/kg) in Met172 mice (iii) and WT mice (n = 4 each; iv).
We next turned toward organic cation transporters (OCTs) and plasma membrane monoamine transporters (PMATs), which were inhibited with decynium-22 (OCT and PMAT inhibitor, 0.1 mg/kg), providing evidence that this agent also increased the clearance of histamine.
Instead of looking at simple t1/2 here, we took a more sophisticated kinetic approach. We used a previously developed mathematical model for histamine dynamics (Best et al., 2017) to investigate how physiological parameters could be adjusted to best capture curves for ESCIT, citalopram, sertraline, and decynium-22. In all four cases, we found that the major parameter change was a 50% reduction in the transport of extracellular histamine back into the cell consistent with the inhibition of histamine reuptake. Model curves (dashed lines) are compared with experimental curves (solid lines) in Figure 9. DAT inhibition had no effect on the histamine signal and the curve post-NET inhibition trends toward a slower reuptake, but this effect was not significant.
Figure 9.

Modeled transporter data. Each panel shows the experimental curves (solid lines) and model predictions (dashed lines) both pre-drug and post-drug administration for three different SSRIs and decynium-22. The main difference between the pre-drug and post-drug administration model curves was a 50% decrease in the reuptake of histamine from the extracellular space into the histamine varicosity. In the cases of escitalopram and decynium-22, the uptake into glial cells was also partially blocked, which is consistent with the fact that the post-drug administration experimental curves are higher and flatter in those two cases.
To further narrow down which transporters are the largest contributor to histamine reuptake, we used the SERT Met172 mouse. This mouse bears a single amino acid substitution (Ile172 is encoded in humans and mice and here is converted to Met172) that impairs the binding of high-affinity antagonists, such as the SSRIs, without impacting serotonin uptake activity or in vivo serotonin clearance (Fig. 8B; Henry et al., 2006; Nackenoff, 2016; Nackenoff et al., 2016; Simmler and Blakely, 2019). In Figure 8C, we show the effects of ESCIT on changes in extracellular serotonin and histamine using this mouse. In Figure 8C, i and ii, we monitored the effects of ESCIT administration on extracellular hippocampal serotonin levels and confirmed previous findings that ESCIT is ineffective at blocking serotonin clearance in Met172 mice (Nackenoff et al., 2016). In wild-type mice, ESCIT induced an increase in amplitude of hippocampal serotonin release (Ampmax: 29.19 ± 4.25 to 63.30 ± 5.33 nm; t(3) = 3.18, p = 0.008, paired t test) and a slowing of clearance (t1/2: 1.50 ± 0.07 to 7.98 ± 1.67 s; t(3) = 3.18, p = 0.03, paired t test) after 50 min. Conversely, in Figure 8C, iii and iv, ESCIT administration still inhibited histamine clearance in Met172 mice in a manner comparable to that of wild-type mice. Neither curve has a trajectory of return to baseline within 30 seconds. These data effectively rule out a role for SERT in histamine clearance or in the actions of SSRIs to potentiate extracellular histamine levels but nonetheless show that SSRIs inhibit histamine reuptake.
Inhibition of histamine synthesis increases the ability of ESCIT to increase serotonin during LPS treatment
Given that SSRIs inhibit the clearance of histamine (Fig. 8) and that histamine inhibits serotonin release (Figs. 6, 7), we sought to decrease extracellular histamine and observe the resulting effects on serotonin. We achieved this reduction via FMH (20 mg/kg, i.p.), a suicide inhibitor of histidine decarboxylase. In Figure 10B, we show, 50 min post-FMH administration, a decrease in evoked hypothalamic histamine release (from 9.36 ± 2.19 to 6.4 ± 1.40 μm; t(3) = 3.18, p = 0.08, paired t test). Although FMH diminished the reduction observed for serotonin levels, this change was not significant (from 42.79 ± 6.25 to 38.6 ± 12.00 nm; t(3) = 3.18, p = 0.59, paired t test). FSCAV was used to observe the effect of FMH on extracellular hippocampal serotonin levels (Fig. 10A). We observed that FMH induced an increase in basal serotonin level that was indistinguishable from ESCIT administration (10 mg/kg, i.p.) despite this agent not affecting stimulated serotonin release/reuptake as does ESCIT (Pearson's correlation = 0.97; t(58) = 30.38, p < 0.0001, one-sample t test; Fig. 10C). In Figure 10D, we show the data from Figure 5 (ESCIT injection to saline- or LPS-treated animals), here compared with another cohort (purple), where combined ESCIT and FMH treatment was given to LPS-treated animals. Basal serotonin levels following LPS can be restored to control levels post-ESCIT administration with this dual treatment (ESCIT and FMH; from 61.44 ± 2.06 to 87.91 ± 2.72 nm; t(3) = 3.18, p = 0.008, paired t test; control average vs average of 55–60 min post ESCIT and FMH). An ANOVA (F(8,63) = 42.01, mean squared error = 0.81, p < 0.0001) with Tukey's post hoc analysis of the slopes through each treatment period revealed that LPS-treated mice given ESCIT and FMH showed extracellular serotonin increases statistically different from those of LPS-treated mice given ESCIT alone (ESCIT + FMH: slope = 0.70 ± 0.03 nm/min; ESCIT: slope = 0.23 ± 0.05 nm/min; p < 0.0001, Tukey's post hoc test) and similar to those of saline-treated mice given ESCIT (ESCIT + FMH: slope = 0.70 ± 0.03 nm/min; ESCIT: slope = 0.48 ± 0.01 nm/min; p = 0.1861, Tukey's post hoc test).
Discussion
Extracellular serotonin is lower after systemic LPS
Inflammation (chronic and acute) and depression are comorbid (Konsman et al., 2002; Schiepers et al., 2005; Steptoe, 2006; Dantzer et al., 2008, 2011; Kim et al., 2016; Miller and Raison, 2016; Köhler et al., 2017). In this work, we asked how inflammation mediates the fundamental brain chemistry underlying depression phenotypes. As a starting point, we used an acute inflammatory challenge (LPS; O'Connor et al., 2009; Zhu et al., 2010). LPS increases proinflammatory cytokines, inducing sickness-like behavior akin to depressive-like phenotypes (O'Connor et al., 2009; Zhu et al., 2010). LPS does not cross the blood–brain barrier (BBB), but immune reactions to this molecule may impair BBB integrity (Varatharaj and Galea, 2017), activate the vagus nerve (Borovikova et al., 2000; Meneses et al., 2016; Somann et al., 2019), and drive inflammatory responses on the vascular endothelium (Murray et al., 2011).
Because of the implication of the involvement of serotonin in depression, we focused our work on this modulator (Asberg et al., 1976a,b; Owens and Nemeroff, 1994). FSCV-evoked measurements test the functionality of the system via synthesis, packaging, autoreceptor control of release, and reuptake (Wood et al., 2014). LPS did not alter the FSCV signal showing that none of these parameters was altered (Fig. 4). Our findings contrast with work indicating that the inflammatory cytokines IL-1β and TNF-α elevate SERT activity and that LPS increases SERT-mediated serotonin clearance (Zhu et al., 2006, 2010). The electrical stimulations used in FSCV release much less serotonin (10 nm) than the clearance measurements in chronoamperometry experiments (600 nm). Thus, the two findings are difficult to compare since different reuptake mechanisms may be recruited in a concentration-dependent manner.
Given no change in evoked serotonin, we turned to FSCAV to investigate LPS regulation of ambient extracellular serotonin and found a rapid decrease in serotonin after LPS (Fig. 5). Importantly, the time frame of our FSCAV study (serotonin falls significantly within 5 min) agrees with previous work showing that SERT activity increases rapidly in response to LPS in vivo (Zhu et al., 2010). Thirty minutes after LPS or saline, ESCIT was administered. As previously seen in the saline group, ESCIT rapidly increased serotonin levels. Interestingly, in LPS-treated mice, the increase in serotonin post-ESCIT administration was neither as fast nor as large. This finding implies LPS-induced inflammation impairs the capacity of ESCIT to increase extracellular serotonin.
During acute inflammation, extracellular serotonin levels are decreased as a function of increased brain histamine
Histamine is a well known chemical mediator of inflammation (Rocklin, 1976; Schwartz et al., 1980) and in the brain is found in microglia, mast cells (Esposito et al., 2001; Rocha et al., 2014), and neurons where it is thought to be a neuromodulator (Watanabe et al., 1983; Panula et al., 1984). Significant to this work is that histamine inversely modulates serotonin via inhibitory H3 heteroreceptors on serotonin terminals (Schlicker et al., 1988; Threlfell et al., 2004; Samaranayake et al., 2016). Thus, we asked here “is the LPS-induced decrease in extracellular serotonin because of histamine immunomodulation?”
We previously developed an FSCV method to simultaneously measure serotonin and histamine in the posterior hypothalamus (histamine cell body region) and found that evoked histamine resulted in H3R-mediated inhibition of serotonin (Samaranayake et al., 2016). The significance of this model is that histamine is evoked but serotonin is not, so that the effects of histamine on resting serotonin can be evaluated. Here we used this model to investigate the effects of LPS on this histamine/serotonin regulation. We found that the amplitude of evoked histamine tended to increase following higher-dose LPS (1 mg/kg) but not lower-dose LPS (0.2 mg/kg; Fig. 6). This result is difficult to reconcile because a low LPS dose significantly decreases extracellular serotonin levels (Fig. 5). However, FSCV only measures evoked histamine and, since we do not yet have the capacity to measure basal histamine, we do not know about the effect of LPS on glial and mast cell histamine. It is possible that low-dose LPS also increases basal histamine, and this effect accounts for decreased extracellular levels of serotonin at low LPS dose.
We previously showed that histamine-induced serotonin inhibition was attenuated by the H3R antagonist thioperamide (Samaranayake et al., 2016). We have found no evidence that thioperamide acts as an agonist on serotonin autoreceptors; thus, we are confident to attribute the effects of thioperamide on serotonin to histamine. This challenge is a complicated one to interpret since H3 autoreceptors on histamine neurons inhibit histamine (increasing serotonin) and H3 heteroreceptors on serotonin terminals inhibit serotonin (Fig. 7B). Therefore, H3R inhibition has contradictory effects on serotonin. This competition was dose dependent, and at high doses (50 mg/kg) inhibitory effects of histamine on H3 heteroreceptors were dominant (Samaranayake et al., 2016). Here we extend this notion to extracellular serotonin where histamine release is not evoked (Fig. 7). We used high-dose (50 mg/kg) thioperamide and observed that extracellular serotonin decreased after intraperitoneal LPS injection, showing that the decrease in extracellular serotonin is conserved in the absence of histamine stimulation, possibly a reflection of inflammatory cytokine-mediated stimulation of SERT activity (Zhu et al., 2006, 2010). Recently, Annamalai et al. (2020) presented evidence that histamine, acting on H3Rs increases SERT phosphorylation and trafficking, which may synergize with inflammatory cytokine-mediated actions to elevate SERT activity. A further point is that thioperamide has affinity for inhibitory H4Rs on glia and mast cells (Nakamura et al., 2000), which could also contribute to decreased serotonin as a function of increased basal histamine (Fig. 5).
Thus, we have strong evidence pointing toward a role for histamine in decreasing extracellular serotonin after LPS. This finding does not explain the diminished effectiveness of ESCIT in raising extracellular serotonin levels in LPS-treated animals, opening an investigative window for SSRIs to change histamine dynamics.
SSRIs inhibit histamine reuptake, blunting their ability to increase serotonin levels
While there is evidence for histamine uptake into synaptosomes (Barnes and Hough, 2002; Sakurai et al., 2006) and astrocytes (Huszti, 1998, 2003), a histamine-specific transporter has not been identified (Hough, 1988; Brown et al., 2001). Histamine inactivation is thought to be via histamine N-methyltransferase (HNMT; White, 1960; Schayer and Reily, 1973; Pollard et al., 1974; Takemura et al., 1994). Because HNMT is exclusively expressed intracellularly (Nishibori et al., 2000), so there must be a membrane transport mechanism for histamine. Indeed, in our 2016 article (Samaranayake et al., 2016), where we first described the dual histamine/serotonin measurement, when mathematically describing the responses, our models necessitated an active reuptake term to fit the histamine curves.
Monoamine transporters have been described as promiscuous because they are not fully selective for their intended monoamine (Daws, 2009). We pharmacologically tested whether the inhibition of other monoamine transporters affected histamine reuptake. We inhibited SERTs (with three different agents), DATs, and NETs, and found that SERT and NET inhibition slowed the clearance of histamine. Next, we turned toward OCTs and PMATs. OCTs and PMATs are low-affinity transporters with the notable ability to nonselectively transport biogenic amines (Engel et al., 2004; Koepsell et al., 2007), which are expressed at high levels in the CA2 region of the hippocampus (Amphoux et al., 2006; Dahlin et al., 2007; Gasser et al., 2009). Work, including that by Gasser et al. (2006), has shown that the OCTs and PMATs reuptake histamine (and other monoamines) with varying affinities (Amphoux et al., 2006; Baganz et al., 2008; Horton et al., 2013). Our results concur in showing that OCT/PMAT inhibition with D-22 also slowed the clearance of histamine in vivo. These results do not definitively identify which transporter is responsible for histamine reuptake because the different agents have affinity for the different monoamine transporters (Daws, 2009), but of these agents, all that have potential antidepressant activity (sertraline, ESCIT, citalopram, desipramine, and D-22) inhibit histamine reuptake (Hyttel, 1994; Fraser-Spears et al., 2019).
To narrow down a histamine reuptake transporter, we used a transgenic model, the SERT Met172. SERT Met172 mice are a genetic knock-in mice with a single amino acid substitution making them insensitive to a number of SSRIs including ESCIT (Henry et al., 2006; Nackenoff et al., 2016; Simmler and Blakely, 2019). A model that impedes the binding of antagonists of SERT transporter without impairing SERT function is arguably more physiologically relevant than a SERT knock-out mouse (Henry et al., 2006; Thompson et al., 2011). Using this mouse, we observed that ESCIT no longer antagonized serotonin reuptake (Nackenoff et al., 2016); however, ESCIT slowed histamine clearance even more strongly than in wild types, suggesting that a transporter other than SERT, which now the more concentrated ESCIT has affinity for (Owens et al., 2001), dictates ESCIT-sensitive histamine clearance. Moreover, a NET-specific tricyclic antidepressant (desipramine) with little SERT activity also delayed histamine clearance. Given the ability of the OCT/PMAT inhibitor decynium-22 to diminish evoked histamine clearance, it seems likely that SERT-independent effects of SSRIs and the tricyclic antidepressant may be mediated by one of these transporters and sheds light on another.
Acute SSRI injections (used here) do not reflect the clinical SSRI regime where the agents are released steadily over a longer, chronic time period. We address here that human serum levels of SSRIs, administered under clinical regime, were found to be lower than the IC50 values of SSRIs toward PMATs and OCTs (Zhou et al., 2007; Haenisch et al., 2012). SSRIs were thus hypothesized not to act on PMATs at clinical doses (Zhou et al., 2007). However, serum drug levels may not mirror ECF drug levels because of the complex diffusion profile of agents across the blood–brain barrier (O'Brien et al., 2012; Hladky and Barrand, 2014; Vendel et al., 2019). In the brain, much of the SSRI is bound to (and thus concentrated around) SERTs; thus, a steep concentration gradient likely exists between the ECF and the serum. Therefore, it is difficult to ascertain whether SSRIs are active at PMATs from human serum SSRI concentrations. Nonetheless, during inflammation, where increased histamine levels are already contributing to decreased serotonin levels, drugs that inhibit histamine clearance will counteract the intended action of the antidepressant (to elevate extracellular serotonin by reducing serotonin inactivation) and exacerbate the inflammatory process, which may contribute to depression. We next tested this hypothesis by reducing histamine synthesis during LPS treatment.
Dual pharmacological targeting of serotonin and histamine restores SSRI chemical efficacy during acute inflammation
We chose to globally lower histamine levels by inhibiting histamine synthesis at the molecular level via FMH, a suicide inhibitor of histidine decarboxylase. Many studies have shown that this agent can dramatically decrease both peripheral and CNS histamine rapidly following peripheral administration (Garbarg et al., 1980; Maeyama et al., 1982; Watanabe et al., 1990). We confirmed that evoked hypothalamic histamine release is blunted following the administration of FMH (Fig. 10B). Thus, to test our hypothesis that during LPS treatment SSRIs induced increases in histamine counteract SSRI-induced increases in serotonin, we co-administered ESCIT with FMH with or without LPS administration (Fig. 10D). We found, strikingly, that FMH fully reversed the LPS-induced deficits in the ability of ESCIT to increase serotonin levels. Importantly, FMH administration under noninflammatory conditions serves to increase serotonin levels at the same rate as ESCIT despite its completely different mode of action (Fig. 10A,C).
This neurochemical finding must be put into a behavioral context to draw physiologically relevant conclusions. However, we suggest that histamine may be a key player in SSRI action that has thus far not been adequately appreciated. Some studies have suggested a role for histamine in antidepressant treatment (Kanof and Greengard, 1978; Munari et al., 2015), and, in this work, we further probed this connection and solidified a link between histamine and serotonin during acute LPS-induced inflammation (Knigge et al., 1994; Munari et al., 2015). Our work supports the ability of histamine to modulate extracellular serotonin levels in the hippocampus in vivo (note that the effects may be region dependent) and in addition to the target of ESCIT action, is an important factor to consider both in relation to the understanding of serotonergic changes in depression, and for improving the clinical efficacy of existing SSRIs.
Footnotes
This work was supported by National Institutes of Health Grants R01-MH-106563 and R21-MH-109959 (to P.H.). We thank all members of the Hashemi laboratory, Lee Gilman, and Ana Pocivavsek for helpful discussions; and Ian Bain for electrode fabrication. Figures were created using BioRender.com.
The authors declare no competing financial interests.
References
- Abdalla A, Atcherley CW, Pathirathna P, Samaranayake S, Qiang B, Peña E, Morgan SL, Heien ML, Hashemi P (2017) In vivo ambient serotonin measurements at carbon-fiber microelectrodes. Anal Chem 89:9703–9711. 10.1021/acs.analchem.7b01257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdalla A, West A, Jin Y, Saylor RA, Qiang B, Peña E, Linden DJ, Nijhout HF, Reed MC, Best J, Hashemi P (2020) Fast serotonin voltammetry as a versatile tool for mapping dynamic tissue architecture: I. Responses at carbon fibers describe local tissue physiology. J Neurochem 153:33–50. 10.1111/jnc.14854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amphoux A, Vialou V, Drescher E, Brüss M, La Cour CM, Rochat C, Millan MJ, Giros B, Bönisch H, Gautron S (2006) Differential pharmacological in vitro properties of organic cation transporters and regional distribution in rat brain. Neuropharmacology 50:941–952. 10.1016/j.neuropharm.2006.01.005 [DOI] [PubMed] [Google Scholar]
- Andrade C (2015) Intranasal drug delivery in neuropsychiatry: focus on intranasal ketamine for refractory depression. J Clin Psychiatry 76:e628–e631. 10.4088/JCP.15f10026 [DOI] [PubMed] [Google Scholar]
- Annamalai B, Ragu Varman D, Horton RE, Daws LC, Jayanthi LD, Ramamoorthy S (2020) Histamine receptors regulate the activity, surface expression and phosphorylation of serotonin transporters. ACS Chem Neurosci 11:466–476. 10.1021/acschemneuro.9b00664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artigas F, Bortolozzi A, Celada P (2018) Can we increase speed and efficacy of antidepressant treatments? Part I: general aspects and monoamine-based strategies. Eur Neuropsychopharmacol 28:445–456. 10.1016/j.euroneuro.2017.10.032 [DOI] [PubMed] [Google Scholar]
- Asberg M, Traskman L, Thoren P (1976a) 5-HIAA in the cerebrospinal fluid. A biochemical suicide predictor? Arch Gen Psychiatry 33:1193–1197. [DOI] [PubMed] [Google Scholar]
- Asberg M, Thorén P, Träskman L, Bertilsson L, Ringberger V (1976b) "Serotonin depression"—a biochemical subgroup within the affective disorders? Science 191:478–480. 10.1126/science.1246632 [DOI] [PubMed] [Google Scholar]
- Baganz NL, Horton RE, Calderon AS, Owens WA, Munn JL, Watts LT, Koldzic-Zivanovic N, Jeske NA, Koek W, Toney GM (2008) Organic cation transporter 3: keeping the brake on extracellular serotonin in serotonin-transporter-deficient mice. Proc Natl Acad Sci U S A 105:18976–18981. 10.1073/pnas.0800466105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes WG, Hough LB (2002) Membrane-bound histamine N-methyltransferase in mouse brain: possible role in the synaptic inactivation of neuronal histamine. J Neurochem 82:1262–1271. 10.1046/j.1471-4159.2002.01063.x [DOI] [PubMed] [Google Scholar]
- Belmaker RH, Agam G (2008) Major depressive disorder. N Engl J Med 358:55–68. 10.1056/NEJMra073096 [DOI] [PubMed] [Google Scholar]
- Best J, Nijhout HF, Samaranayake S, Hashemi P, Reed M (2017) A mathematical model for histamine synthesis, release, and control in varicosities. Theor Biol Med Model 14:24. 10.1186/s12976-017-0070-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405:458. 10.1038/35013070 [DOI] [PubMed] [Google Scholar]
- Brown RE, Stevens DR, Haas HL (2001) The physiology of brain histamine. Prog Neurobiol 63:637–672. 10.1016/s0301-0082(00)00039-3 [DOI] [PubMed] [Google Scholar]
- Campbell S, MacQueen G (2004) The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci 29:417–426. [PMC free article] [PubMed] [Google Scholar]
- Carhart-Harris RL, Bolstridge M, Rucker J, Day CM, Erritzoe D, Kaelen M, Bloomfield M, Rickard JA, Forbes B, Feilding A (2016) Psilocybin with psychological support for treatment-resistant depression: an open-label feasibility study. Lancet Psychiatry 3:619–627. 10.1016/S2215-0366(16)30065-7 [DOI] [PubMed] [Google Scholar]
- Dahlin A, Xia L, Kong W, Hevner R, Wang J (2007) Expression and immunolocalization of the plasma membrane monoamine transporter in the brain. Neuroscience 146:1193–1211. 10.1016/j.neuroscience.2007.01.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56. 10.1038/nrn2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Lawson MA, Kelley KW (2011) Inflammation-associated depression: from serotonin to kynurenine. Psychoneuroendocrinology 36:426–436. 10.1016/j.psyneuen.2010.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daws LC (2009) Unfaithful neurotransmitter transporters: focus on serotonin uptake and implications for antidepressant efficacy. Pharmacol Ther 121:89–99. 10.1016/j.pharmthera.2008.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel K, Zhou M, Wang J (2004) Identification and characterization of a novel monoamine transporter in the human brain. J Biol Chem 279:50042–50049. 10.1074/jbc.M407913200 [DOI] [PubMed] [Google Scholar]
- Esposito P, Gheorghe D, Kandere K, Pang X, Connolly R, Jacobson S, Theoharides TC (2001) Acute stress increases permeability of the blood–brain-barrier through activation of brain mast cells. Brain Res 888:117–127. 10.1016/s0006-8993(00)03026-2 [DOI] [PubMed] [Google Scholar]
- Fraser-Spears R, Krause-Heuer AM, Basiouny M, Mayer FP, Manishimwe R, Wyatt NA, Dobrowolski JC, Roberts MP, Greguric I, Kumar N (2019) Comparative analysis of novel decynium-22 analogs to inhibit transport by the low-affinity, high-capacity monoamine transporters, organic cation transporters 2 and 3, and plasma membrane monoamine transporter. Eur J Pharmacol 842:351–364. 10.1016/j.ejphar.2018.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbarg M, Barbin G, Rodergas E, Schwartz JC (1980) Inhibition of histamine synthesis in brain by alpha-fluoromethylhistidine, a new irreversible inhibitor: in vitro and in vivo studies. J Neurochem 35:1045–1052. 10.1111/j.1471-4159.1980.tb07858.x [DOI] [PubMed] [Google Scholar]
- Gasser PJ, Lowry CA, Orchinik M (2006) Corticosterone-sensitive monoamine transport in the rat dorsomedial hypothalamus: potential role for organic cation transporter 3 in stress-induced modulation of monoaminergic neurotransmission. J Neurosci 26:8758–8766. 10.1523/JNEUROSCI.0570-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser PJ, Orchinik M, Raju I, Lowry CA (2009) Distribution of organic cation transporter 3, a corticosterone-sensitive monoamine transporter, in the rat brain. J Comp Neurol 512:529–555. 10.1002/cne.21921 [DOI] [PubMed] [Google Scholar]
- Haenisch B, Drescher E, Thiemer L, Xin H, Giros B, Gautron S, Bönisch H (2012) Interaction of antidepressant and antipsychotic drugs with the human organic cation transporters hOCT1, hOCT2 and hOCT3. Naunyn Schmiedebergs Arch Pharmacol 385:1017–1023. 10.1007/s00210-012-0781-8 [DOI] [PubMed] [Google Scholar]
- Hashemi P, Dankoski EC, Petrovic J, Keithley RB, Wightman RM (2009) Voltammetric detection of 5-hydroxytryptamine release in the rat brain. Anal Chem 81:9462–9471. 10.1021/ac9018846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry LK, Field JR, Adkins EM, Parnas ML, Vaughan RA, Zou M-F, Newman AH, Blakely RD (2006) Tyr-95 and Ile-172 in transmembrane segments 1 and 3 of human serotonin transporters interact to establish high affinity recognition of antidepressants. J Biol Chem 281:2012–2023. 10.1074/jbc.M505055200 [DOI] [PubMed] [Google Scholar]
- Hladky SB, Barrand MA (2014) Mechanisms of fluid movement into, through and out of the brain: evaluation of the evidence. Fluids Barriers CNS 11:26. 10.1186/2045-8118-11-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RE, Apple DM, Owens WA, Baganz NL, Cano S, Mitchell NC, Vitela M, Gould GG, Koek W, Daws LC (2013) Decynium-22 enhances SSRI-induced antidepressant-like effects in mice: uncovering novel targets to treat depression. J Neurosci 33:10534–10543. 10.1523/JNEUROSCI.5687-11.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough LB (1988) Cellular localization and possible functions for brain histamine: recent progress. Prog Neurobiol 30:469–505. 10.1016/0301-0082(88)90032-9 [DOI] [PubMed] [Google Scholar]
- Huszti Z (1998) Carrier-mediated high affinity uptake system for histamine in astroglial and cerebral endothelial cells. J Neurosci Res 51:551–558. 10.1002/(SICI)1097-4547(19980301)51:5<551::AID-JNR1>3.0.CO;2-E [DOI] [PubMed] [Google Scholar]
- Huszti Z (2003) Histamine uptake into non-neuronal brain cells. Inflamm Res 52:s3–s6. 10.1007/s000110300028 [DOI] [PubMed] [Google Scholar]
- Hyttel J (1994) Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs). Int Clin Psychopharmacol 9 [Suppl 1]:19–26. [DOI] [PubMed] [Google Scholar]
- Jackson BP, Dietz SM, Wightman RM (1995) Fast-scan cyclic voltammetry of 5-hydroxytryptamine. Anal Chem 67:1115–1120. 10.1021/ac00102a015 [DOI] [PubMed] [Google Scholar]
- Kanof PD, Greengard P (1978) Brain histamine receptors as targets for antidepressant drugs. Nature 272:329–333. 10.1038/272329a0 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2013) The mouse brain in stereotaxic coordinates. Amsterdam: Academic. [Google Scholar]
- Kim Y-K, Na K-S, Myint A-M, Leonard BE (2016) The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog Neuropsychopharmacol Biol Psychiatry 64:277–284. 10.1016/j.pnpbp.2015.06.008 [DOI] [PubMed] [Google Scholar]
- Knigge U, Kjær A, Jørgensen H, Garbarg M, Ross C, Rouleau A, Warberg J (1994) Role of hypothalamic histaminergic neurons in mediation of ACTH and beta-endorphin responses to LPS endotoxin in vivo. Neuroendocrinology 60:243–251. 10.1159/000126757 [DOI] [PubMed] [Google Scholar]
- Koepsell H, Lips K, Volk C (2007) Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res 24:1227–1251. 10.1007/s11095-007-9254-z [DOI] [PubMed] [Google Scholar]
- Köhler C, Freitas T, Maes M, De Andrade N, Liu C, Fernandes B, Stubbs B, Solmi M, Veronese N, Herrmann N (2017) Peripheral cytokine and chemokine alterations in depression: a meta-analysis of 82 studies. Acta Psychiatr Scand 135:373–387. 10.1111/acps.12698 [DOI] [PubMed] [Google Scholar]
- Konsman JP, Parnet P, Dantzer R (2002) Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci 25:154–159. 10.1016/S0166-2236(00)02088-9 [DOI] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ (2008) The molecular neurobiology of depression. Nature 455:894–902. 10.1038/nature07455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulikov AV, Tikhonova MA, Osipova DV, Kulikov VA, Popova NK (2011) Association between tryptophan hydroxylase-2 genotype and the antidepressant effect of citalopram and paroxetine on immobility time in the forced swim test in mice. Pharmacol Biochemi Behav 99:683–687. 10.1016/j.pbb.2011.06.020 [DOI] [PubMed] [Google Scholar]
- Lisanby SH (2007) Electroconvulsive therapy for depression. N Engl J Med 357:1939–1945. 10.1056/NEJMct075234 [DOI] [PubMed] [Google Scholar]
- Maeyama K, Watanabe T, Taguchi Y, Yamatodani A, Wada H (1982) Effect of alpha-fluoromethylhistidine, a suicide inhibitor of histidine decarboxylase, on histamine levels in mouse tissues. Biochem Pharmacol 31:2367–2370. 10.1016/0006-2952(82)90531-7 [DOI] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000) Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20:9104–9110. 10.1523/JNEUROSCI.20-24-09104.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, Schwalb JM, Kennedy SH (2005) Deep brain stimulation for treatment-resistant depression. Neuron 45:651–660. 10.1016/j.neuron.2005.02.014 [DOI] [PubMed] [Google Scholar]
- Meneses G, Bautista M, Florentino A, Díaz G, Acero G, Besedovsky H, Meneses D, Fleury A, Del Rey A, Gevorkian G (2016) Electric stimulation of the vagus nerve reduced mouse neuroinflammation induced by lipopolysaccharide. J Inflamm (Lond) 13:33. 10.1186/s12950-016-0140-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikail HG, Dalla C, Kokras N, Kafetzopoulos V, Papadopoulou-Daifoti Z (2012) Sertraline behavioral response associates closer and dose-dependently with cortical rather than hippocampal serotonergic activity in the rat forced swim stress. Physiol Behav 107:201–206. 10.1016/j.physbeh.2012.06.016 [DOI] [PubMed] [Google Scholar]
- Miller AH, Raison CL (2016) The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 16:22–34. 10.1038/nri.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munari L, Provensi G, Passani MB, Galeotti N, Cassano T, Benetti F, Corradetti R, Blandina P (2015) Brain Histamine is crucial for selective serotonin reuptake inhibitors' behavioral and neurochemical effects. IJNPPY 18:pyv045. 10.1093/ijnp/pyv045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CL, Skelly DT, Cunningham C (2011) Exacerbation of CNS inflammation and neurodegeneration by systemic LPS treatment is independent of circulating IL-1β and IL-6. J Neuroinflammation 8:50. 10.1186/1742-2094-8-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nackenoff AG (2016) Determining the role of serotonin in antidepressant action, as revealed utilizing the SERT Met172 mouse model. Nashville, TN: Vanderbilt University. [Google Scholar]
- Nackenoff AG, Moussa-Tooks AB, McMeekin AM, Veenstra-VanderWeele J, Blakely RD (2016) Essential contributions of serotonin transporter inhibition to the acute and chronic actions of fluoxetine and citalopram in the SERT Met172 mouse. Neuropsychopharmacology 41:1733. 10.1038/npp.2015.335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Itadani H, Hidaka Y, Ohta M, Tanaka K (2000) Molecular cloning and characterization of a new human histamine receptor, HH4R. Biochem Biophys Res Commun 279:615–620. 10.1006/bbrc.2000.4008 [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002) Neurobiology of depression. Neuron 34:13–25. 10.1016/S0896-6273(02)00653-0 [DOI] [PubMed] [Google Scholar]
- Nishibori M, Tahara A, Sawada K, Sakiyama J, Nakaya N, Saeki K (2000) Neuronal and vascular localization of histamine N-methyltransferase in the bovine central nervous system. Eur J Neurosci 12:415–424. 10.1046/j.1460-9568.2000.00914.x [DOI] [PubMed] [Google Scholar]
- O'Brien FE, Dinan TG, Griffin BT, Cryan JF (2012) Interactions between antidepressants and P-glycoprotein at the blood–brain barrier: clinical significance of in vitro and in vivo findings. Br J Pharmacol 165:289–312. 10.1111/j.1476-5381.2011.01557.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor JC, Lawson MA, André C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R (2009) Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry 14:511–522. 10.1038/sj.mp.4002148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens MJ, Nemeroff CB (1994) Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin Chem 40:288–295. 10.1093/clinchem/40.2.288 [DOI] [PubMed] [Google Scholar]
- Owens MJ, Knight DL, Nemeroff CB (2001) Second-generation SSRIs: human monoamine transporter binding profile of escitalopram and R-fluoxetine. Biol Psychiatry 50:345–350. 10.1016/S0006-3223(01)01145-3 [DOI] [PubMed] [Google Scholar]
- Panula P, Yang H, Costa E (1984) Histamine-containing neurons in the rat hypothalamus. Proc Natl Acad Sci U S A 81:2572–2576. 10.1073/pnas.81.8.2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Kile BM, Mark Wightman R (2009) In vivo voltammetric monitoring of norepinephrine release in the rat ventral bed nucleus of the stria terminalis and anteroventral thalamic nucleus. Eur J Neurosci 30:2121–2133. 10.1111/j.1460-9568.2009.07005.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard H, Bischoff S, Schwartz J-C (1974) Turnover of histamine in rat brain and its decrease under barbiturate anesthesia. J Pharmacol Exp Ther 190:88–99. [PubMed] [Google Scholar]
- Pratt LA, Brody DJ, Gu Q (2017) Antidepressant use among persons aged 12 and over: United States, 2011–2014. NCHS Data Brief (283):1–8. [PubMed] [Google Scholar]
- Rocha SM, Pires J, Esteves M, Graça B, Bernardino L (2014) Histamine: a new immunomodulatory player in the neuron-glia crosstalk. Front Cell Neurosci 8:120. 10.3389/fncel.2014.00120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocklin RE (1976) Modulation of cellular-immune responses in vivo and in vitro by histamine receptor-bearing lymphocytes. J Clin Invest 57:1051–1058. 10.1172/JCI108347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai E, Sakurai E, Oreland L, Nishiyama S, Kato M, Watanabe T, Yanai K (2006) Evidence for the presence of histamine uptake into the synaptosomes of rat brain. Pharmacology 78:72–80. 10.1159/000095637 [DOI] [PubMed] [Google Scholar]
- Samaranayake S, Abdalla A, Robke R, Wood KM, Zeqja A, Hashemi P (2015) In vivo histamine voltammetry in the mouse premammillary nucleus. Analyst 140:3759–3765. 10.1039/c5an00313j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaranayake S, Abdalla A, Robke R, Nijhout HF, Reed MC, Best J, Hashemi P (2016) A voltammetric and mathematical analysis of histaminergic modulation of serotonin in the mouse hypothalamus. J Neurochem 138:374–383. 10.1111/jnc.13659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saylor RA, Hersey M, West A, Buchanan AM, Berger SN, Nijhout HF, Reed MC, Best J, Hashemi P (2019) In vivo hippocampal serotonin dynamics in male and female mice: determining effects of acute escitalopram using fast scan cyclic voltammetry. Front Neurosci 13:362. 10.3389/fnins.2019.00362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schayer RW, Reily MA (1973) Formation and fate of histamine in rat and mouse brain. J Pharmacol Exp Ther 184:33–40. [PubMed] [Google Scholar]
- Schiepers OJ, Wichers MC, Maes M (2005) Cytokines and major depression. Prog Neuropsychopharmacol Biol Psychiatry 29:201–217. 10.1016/j.pnpbp.2004.11.003 [DOI] [PubMed] [Google Scholar]
- Schlicker E, Betz R, Göthert M (1988) Histamine H3 receptor-mediated inhibition of serotonin release in the rat brain cortex. Naunyn Schmiedebergs Arch Pharmacol 337:588–590. 10.1007/BF00182737 [DOI] [PubMed] [Google Scholar]
- Schwartz A, Askenase PW, Gershon RK (1980) Histamine inhibition of the in vitro induction of cytotoxic T-cell responses. Immunopharmacology 2:179–190. 10.1016/0162-3109(80)90048-x [DOI] [PubMed] [Google Scholar]
- Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW (1996) Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci U S A 93:3908–3913. 10.1073/pnas.93.9.3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton RC, Osuntokun O, Heinloth AN, Corya SA (2010) Therapeutic options for treatment-resistant depression. CNS Drugs 24:131–161. 10.2165/11530280-000000000-00000 [DOI] [PubMed] [Google Scholar]
- Simmler LD, Blakely RD (2019) The SERT Met172 mouse: an engineered model to elucidate the contributions of serotonin signaling to cocaine action. ACS Chem Neurosci 10:3053–3060. 10.1021/acschemneuro.9b00005 [DOI] [PubMed] [Google Scholar]
- Somann JP, Wasilczuk KM, Neihouser KV, Sturgis J, Albors GO, Robinson JP, Powley TL, Irazoqui PP (2019) Characterization of plasma cytokine response to intraperitoneally administered LPS and subdiaphragmatic branch vagus nerve stimulation in rat model. PLoS One 14:e0214317. 10.1371/journal.pone.0214317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steptoe A (2006) Depression and physical illness. Cambridge, UK: Cambridge UP. [Google Scholar]
- Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY, Uylings HB, Friedman L, Rajkowska G (2004) Cellular changes in the postmortem hippocampus in major depression. Biol Psychiatry 56:640–650. 10.1016/j.biopsych.2004.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura M, Imamura I, Mizuguchi H, Fukui H, Yamatodani A (1994) Tissue distribution of histamine N-methyl transferase-like immunoreactivity in rodents. Life Sci 54:1059–1071. 10.1016/0024-3205(94)00416-1 [DOI] [PubMed] [Google Scholar]
- Thompson BJ, Jessen T, Henry LK, Field JR, Gamble KL, Gresch PJ, Carneiro AM, Horton RE, Chisnell PJ, Belova Y, McMahon DG, Daws LC, Blakely RD (2011) Transgenic elimination of high-affinity antidepressant and cocaine sensitivity in the presynaptic serotonin transporter. Proc Natl Acad Sci U S A 108:3785–3790. 10.1073/pnas.1011920108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Cragg SJ, Kalló I, Turi GF, Coen CW, Greenfield SA (2004) Histamine H3 receptors inhibit serotonin release in substantia nigra pars reticulata. J Neurosci 24:8704–8710. 10.1523/JNEUROSCI.2690-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varatharaj A, Galea I (2017) The blood–brain barrier in systemic inflammation. Brain Behavr Immun 60:1–12. 10.1016/j.bbi.2016.03.010 [DOI] [PubMed] [Google Scholar]
- Vendel E, Rottschäfer V, de Lange EC (2019) Improving the prediction of local drug distribution profiles in the brain with a new 2D mathematical model. Bull Math Biol 81:3477–3507. 10.1007/s11538-018-0469-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Taguchi Y, Hayashi H, Tanaka J, Shiosaka S, Tohyama M, Kubota H, Terano Y, Wada H (1983) Evidence for the presence of a histaminergic neuron system in the rat brain: an immunohistochemical analysis. Neurosci Lett 39:249–254. 10.1016/0304-3940(83)90308-7 [DOI] [PubMed] [Google Scholar]
- Watanabe T, Yamatodani A, Maeyama K, Wada H (1990) Pharmacology of alpha-fluoromethylhistidine, a specific inhibitor of histidine decarboxylase. Trends Pharmacol Sci 11:363–367. [DOI] [PubMed] [Google Scholar]
- White T (1960) Formation and catabolism of histamine in cat brain in vivo. J Physiol 152:299–308. 10.1113/jphysiol.1960.sp006488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KM, Hashemi P (2013) Fast-scan cyclic voltammetry analysis of dynamic serotonin reponses to acute escitalopram. ACS Chem Neurosci 4:715–720. 10.1021/cn4000378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KM, Zeqja A, Nijhout HF, Reed MC, Best J, Hashemi P (2014) Voltammetric and mathematical evidence for dual transport mediation of serotonin clearance in vivo. J Neurochem 130:351–359. 10.1111/jnc.12733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Engel K, Wang J (2007) Evidence for significant contribution of a newly identified monoamine transporter (PMAT) to serotonin uptake in the human brain. Biochem Pharmacol 73:147–154. 10.1016/j.bcp.2006.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C-B, Blakely RD, Hewlett WA (2006) The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 31:2121. 10.1038/sj.npp.1301029 [DOI] [PubMed] [Google Scholar]
- Zhu CB, Lindler KM, Owens AW, Daws LC, Blakely RD, Hewlett WA (2010) Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology 35:2510–2520. 10.1038/npp.2010.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomkowski AD, Engel D, Gabilan NH, Rodrigues ALS (2010) Involvement of NMDA receptors and L-arginine-nitric oxide-cyclic guanosine monophosphate pathway in the antidepressant-like effects of escitalopram in the forced swimming test. Eur Neuropsychopharmacol 20:793–801. 10.1016/j.euroneuro.2010.07.011 [DOI] [PubMed] [Google Scholar]