

Abstract
Lynch Syndrome (LS) is the most common cause of hereditary colorectal cancer (CRC) and is secondary to germline alterations in one of four DNA mismatch repair (MMR) genes. Here we aimed to provide novel insights into the initiation of MMR deficient (MMRd) colorectal carcinogenesis by characterizing the expression profile of MMRd intestinal stem cells (ISC). A tissue-specific MMRd mouse model (Villin-Cre;Msh2LoxP/LoxP) was crossed with a reporter mouse (Lgr5-EGFP-IRES-creERT2) to trace and isolate ISCs (Lgr5+) using flow cytometry. Three different ISC genotypes (Msh2-KO, Msh2-HET, and Msh2-WT) were isolated and processed for mRNAseq and mass spectrometry followed by bioinformatic analyses to identify expression signatures of complete MMRd and haplo-insufficiency. These findings were validated using qRT-PCR, immunohistochemistry, and whole transcriptomic sequencing in mouse tissues, organoids, and a cohort of human samples, including normal colorectal mucosa, pre-malignant lesions, and early-stage CRC from LS patients and Familial Adenomatous Polyposis (FAP) patients as controls. Msh2-KO ISCs clustered together with differentiated intestinal epithelial cells from all genotypes. Gene set enrichment analysis indicated inhibition of replication, cell cycle progression, and the Wnt pathway and activation of epithelial signaling and immune reaction. An expression signature derived from MMRd ISCs successfully distinguished MMRd neoplastic lesions of LS patients from FAP controls. SPP1 was specifically upregulated in MMRd ISCs and colocalized with LGR5 in LS colorectal pre-malignant lesions and tumors. These results show that expression signatures of MMRd ISC recapitulate the initial steps of LS carcinogenesis and have the potential to unveil novel biomarkers of early cancer initiation.
Keywords: Lynch syndrome, Mismatch repair deficiency, Intestinal stem cells, Gene expression, Proteomics, Biomarkers
Introduction
Lynch Syndrome (OMIM# 120435, LS) is a hereditary cancer syndrome predisposing patients to develop colorectal cancers (CRC) as well as tumors of the endometrium, ovary, stomach, and small intestine1. LS has an estimated prevalence of 1 in 279, thus affecting over 1 million individuals in the US2. LS is secondary to germline mutations in one of the DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2) that control post-replicative DNA proofreading, thus ensuring genomic integrity3. MMR deficiency (MMRd) accelerates the acquisition of secondary somatic mutations in oncogenes and tumor suppressor genes that regulate different pathways, including cell fate, transcription, growth factors, and other DNA repair mechanisms, thus promoting carcinogenesis4. LS has an autosomal dominant inheritance causing an estimated lifetime risk of CRC development of 20–50% depending on the germline MMR gene that carries the mutation, and also a young age of onset, typically in the fourth decade of life5–7. Despite the recommendation of annual or biannual endoscopic surveillance starting at age 20–25, LS patients continue developing interval cancers and are counseled to consider risk-reducing surgeries8, 9.
The epithelium of the small and large intestine contains niches of stem cells located at the bottom of specialized finger-like invaginations that are arranged in functional units called crypts, which are surrounded by connective tissue, and the underlying lamina propria. These fast-cycling stem cells, refered to as crypt base columnar (CBC) cells, generate daughter cells that exit the stem cell niche to integrate into the transit-amplifying compartment by migrating upwards to the lumen of the gut. This process takes 4–5 days and gives rise to several differentiated and specialized cell subtypes, including enterocytes (nutrient uptake), goblet (mucus production), enteroendocrine (hormone production), and paneth cells (growth factor production exclusively in the small intestine)10–12.
Studies in animal models have demonstrated that pluripotent stem cells acquiring an initiating mutational event become the ‘cell of origin’ in several malignancies, including intestinal cancers13, 14. In fact, the transcriptomic and proteomic profiles of intestinal stem cells (ISC) under physiologic and APC-inactivating conditions have been successfully characterized in genetically engineered animal models that allow for lineage tracing of cells expressing Lgr5, a Wnt target gene and stem cell marker15, 16. Furthermore, inactivation of MMR function via Msh2 deletion in mouse embryonic stem cells generates a mutator phenotype causing genomic instability and accumulation of subsequent somatic mutations leading to cancer development17, thus demonstrating characteristics of a cancer stem cell18. Therefore, we hypothesized that MMRd ISCs display a unique transcriptomic and proteomic profile that is different from their daughter cells. It is essential to understand the molecular and cellular landscape of MMRd tissue-specific stem cells in order to unravel the mechanisms behind the earliest stages of cancer initiation before macroscopic lesions are detectable, thus identifying specific targets for the development of novel cancer interception strategies and biomarkers for early detection of cancer in LS19.
Here, we present, for the first time, the whole transcriptomic and proteomic landscape of the intestinal epithelium with haploinsufficiency and complete deficiency of MMR functioning, which mimics the biology of normal colorectal and neoplastic epithelium in LS patients, respectively. This gene expression signature of MMRd ISCs derived from a genetically-engineered mouse model uncovers activated molecular pathways involved in the initiation and early steps of MMRd colorectal carcinogenesis.
Materials and Methods
Mice.
C57BL/6J strain of conditional knockout mice for MMR gene Msh2 (Msh2LoxP/LoxP) were crossed with Villin-Cre (VC) transgene expressing mice to obtain VC-Msh2LoxP/LoxP mice20. We further crossed VC-Msh2LoxP/LoxP mice with a reporter Lgr5EGFP-IRES-creERT2 mice to track and isolate Lgr5EGFP+ stem cells by flow cytometry15. We generated Lgr5EGFP-IRES-creERT2;VC-Msh2LoxP/LoxP as Msh2 null mice in the intestine (herein referred as Msh2-KO), Lgr5EGFP-IRES-creERT2;VC-Msh2LoxP/+ as Msh2 haplo-insufficient (Msh2-HET), and Lgr5EGFP-IRES-creERT2;VC-Msh2+/+ as mice with wild-type Msh2 function (Msh2-WT). Also, we generated intestinal organoids from Msh2-KO and Msh2-WT for validation of the expression of key genes in ISC. All animal experiments were approved by the institutional animal care and use committee (IACUC) of The University of Texas MD Anderson Cancer Center and the care of the animals was in accordance with institutional guidelines (IACUC protocol # 00000469-RN02).
Crypt isolation from small intestine and Fluorescence-activated cell sorting (FACS).
Crypts from the small intestine of 8-week-old Msh2-WT, Msh2-HET, and Msh2-KO mice were harvested, and single cells from each genotype were subjected to FACS to isolate Lgr5EGFP+ stem cells and Lgr5EGFP- daughter cells.
Transcriptome and mass spectrometry analysis of mouse specimens.
RNA and total cellular extracts (TCE) were isolated from ISCs (Lgr5EGFP+) and daughter cells (Lgr5EGFP-) from all three mouse genotypes. RNA was extracted using TRIzol (Invitrogen) and RNA isolation kit (Ambion) and then subjected to library preparation and sequenced with an Illumina HiSeq4000 instrument. TCE were analyzed using tandem mass spectrometry. Detailed steps in the analysis of RNA-seq and proteomics along with in-depth bioinformatics analysis for differential gene expression and gene set enrichment analysis (GSEA) followed standard protocols and pipelines previously described21.
Transcriptomic analysis of human samples and validation of MMR-deficient stem cell expression signatures.
Tissue samples were acquired through endoscopic biopsies during routine screening colonoscopies from a total of 17 Familial Adenomatous Polyposis (FAP) patients (17 paired adenoma and normal mucosa) and 27 LS patients (11 matched tumor/adenomas and normal mucosa, 3 unmatched tumor/adenomas, and 18 with unmatched normal mucosa, Table S1). All patients were followed at The University of Texas MD Anderson Cancer Center (UTMDACC) for routine surveillance care. Written informed consent was obtained from all study participants, and the UTMDACC Institutional Review Board (IRB) approved this study (IRB #PA12–0327). Total RNA was isolated from tissues that have been flash-frozen or preserved in RNALater and subjected to mRNA sequencing (mRNAseq). This human mRNAseq data set from colorectal normal mucosa, polyp, and tumor samples were used for validation of the gene signatures obtained in mice. FAP normal mucosa and polyps were selected as the human counterparts of Msh2-WT mice (MMR-proficient), LS normal mucosa as Msh2-HET (MMR-haploinsufficient), while LS polyp and tumor samples that were hypermutant (mutation rate≥10/Mb estimated by whole-exome sequencing) or displayed MMRd by microsatellite instability (MSI) via PCR or immunohistochemistry of MMR protein as ‘Msh2-KO’ (MMRd). In addition, transcriptomic data from organoids of normal mucosa from LS22 and sporadic CRC patients23 were used to further validate the MMR-haploinsufficient expression signature.
Gene expression analysis and Chromatin Immunoprecipitation (ChIP) assays.
Total RNA from ISCs and organoids isolated from the different genotypes as well as both Lgr5EGFP+ and Lgr5EGFP- fractions were analyzed by qRT-PCR for validation of the expression of critical genes following previously described methods22. To assess if SPP1 gene expression was epigenetically regulated by histone H3 lysine 27 methylation (H3K27me3), ChIP assays were performed in chromatin extracts of the mismatch repair proficient (MMRp) CRC cell line SW620 and the MMRd endometrial cancer cell line HEC59, which harbors bi-allelic inactivating MSH2 mutations with details provided in Supplementary Material and Methods. Primer sequences are included in Table S2.
Immunofluorescence and imaging of mouse tissues.
Freshly extracted small intestine from 8-week-old mice from Msh2-WT, Msh2-HET, or Msh2-KO were used to stain Lgr5 (GFP+) cells as a marker to visualize ISC and the expression of SPP1 (Table S3).
Fluorescent multiplex immunohistochemistry (IHC) staining of human specimens and automated quantitative imaging.
Formalin-Fixed Paraffin-Embedded (FFPE) tissue specimens of uninvolved normal colorectal mucosa (N=6), tubular/tubulovillius adenoma (N=6), and invasive adenocarcinoma (N=3) from a total of 8 LS patients were used (Table S4). Unstained slides were processed as described above and stained with antibodies against human LGR5 and SPP1. Manual fluorescent multiplex IHC staining was performed following a validated protocol using antibodies and reagents listed in Table S3.
Statistical analysis.
Comparisons between two experimental groups were performed with GraphPad Prism using Student’s unpaired t-test, and among more than two experimental groups were analyzed using one-way ANOVA with Tukey’s post-hoc analysis for multiple comparisons. The data are expressed as means ±SD from three technical replicates and three independent experiments.
Experimental details and computational methods can be found in Supplementary Material and Methods.
Results
Isolation of MMRd mouse ISC.
To understand the biology of MMRd ISCs at the earliest stage of carcinogenesis, we crossed a mouse model of intestinal tissue-specific (Villin-Cre, VC) inactivation of the MMR function via deletion of the essential ATPase domain of Msh2 in exon 12 (Msh2LoxP/LoxP, thus resulting in VC-Msh2LoxP/LoxP) with another mouse line (Lgr5EGFP-IRES-CreERT2 ) that allowed the isolation and tracing of ISCs expressing the validated stem cell marker Lgr5. Then, we isolated ISCs from the entire small intestine of the following mouse genotypes to model different clinical scenarios: Lgr5EGFP-IRES-creERT2+;VC-Msh2+/+ (Msh2-WT) as a counterpart of ISC from the sporadic normal colorectal mucosa; Lgr5EGFP-IRES-creERT2+;VC-Msh2LoxP/+ (Msh2-HET) from normal colorectal mucosa of LS patients; and Lgr5EGFP-IRES-creERT2+;VC-Msh2LoxP/LoxP (Msh2-KO) from early premalignant lesions of LS patients, as well as their corresponding daughter cells that were Lgr5EGFP-IRES-creERT2- (Figure 1). FACS was optimized using crypt preparations from GFP negative mice to specifically isolate epithelial cells expressing high levels of GFP (GFPhi) that were considered as Lgr5EGFP+ ISCs and GFP-/EpCAM+ cells (Lgr5EGFP-) as daughter (non-stem) cells. Cell fractions excluded lymphocytes by labeling total crypt cells with CD45 antibody (Figure S1A). A cohort of 119 mice was used to isolate a sufficient number of stem cells that afforded extraction of RNA and protein for transcriptomics and proteomics analyses, respectively. From a total of 27 Msh2-WT mice, we obtained a mean of 224,388 Lgr5EGFP+ cells per mice; 35 Msh2-HET rendered a mean of 47,873 Lgr5EGFP+ cells; and 57 Msh2-KO a mean of 10,392 Lgr5EGFP+ cells. We observed that the number of stem cells recovered for each genotype decreased exponentially with the deletion of each Msh2 allele (Figure S1B-D and Figure S2), which could possibly be due to premature differentiation of ISC upon deletion of Msh2 allele.
Figure 1.

Schematic outline of the experimental design. Lgr5EGFP-IRES-creERT2 mice were crossed with Villin-Cre;Msh2LoxP/LoxP mice (VC-Msh2LoxP/LoxP). After crypt isolation from Msh2-WT, Msh2-HET, or Msh2-KO mice, FACS was performed to isolate GFP labeled Lgr5EGFP+ stem cells or Lgr5EGFP- daughter cells. Sorted cell populations were used to extract RNA and protein for transcriptomics and proteomics profiling by mRNAseq and tandem Mass Spectrometry, respectively. Bioinformatic analyses were used to identify differentially expressed genes and proteins in MMRd and haploinsufficient ISC. Validation of gene expression signatures was performed using both mouse tissue specimens and organoids as well as human cell lines and tissues from LS patients.
Transcriptomic profile of MMR haploinsufficient and MMRd stem and non-stem cells.
We identified the transcriptomes of Lgr5+ ISCs (Lgr5EGFP+) and their daughter cells (Lgr5EGFP-) in all three mouse genotypes using next-generation whole transcriptomics followed by principal component analysis (PCA). The transcriptome of Lgr5EGFP+ cells of Msh2-KO clustered with daughter cells (Lgr5EGFP- fractions) of all three genotypes (Figure 2A). However, each genotype showed a profile that was distinct from the others when samples were separately analyzed based on stem and daughter cell phenotypes (Figure S3A). Then, we examined the transcriptional differences of MMR haploinsufficient and MMRd ISCs (Msh2-HET and Msh2-KO mice, respectively) to MMRp ISCs (which were represented by Msh2-WT mice and were the comparator for these analyses), and their corresponding daughter cells (differentiated non-stem cells). The comparison of the transcriptomes of Lgr5EGFP+ Msh2-KO and Msh2-WT stem cells identified a total of 340 significantly dysregulated genes (284 upregulated and 56 downregulated, Figure 2B), thus defining an expression profile of MMRd ISC. In contrast, we observed only 39 genes significantly dysregulated in Lgr5EGFP- non-stem cells (Figure S3B). Then, a comparison of Lgr5EGFP+ Msh2-HET and Msh2-WT stem cells rendered a gene profile for MMR haploinsufficiency with a total of 60 genes differentially dysregulated (50 upregulated and 10 downregulated, Figure 2B) and the same comparison of Lgr5EGFP- daughter cells observed 13 genes (Figure S3B). When we changed the reference and compared the transcriptomes of Msh2-KO and Msh2-HET, we observed a total of 182 genes differentially expressed (131 upregulated and 51 downregulated) in the stem cells (Figure 2B) and 13 genes in Lgr5EGFP- non-stem cell fractions (Figure S3C). We observed that 20 genes were dysregulated in common between Lgr5EGFP+ stem cells of Msh2-HET and Msh2-KO (Figure 2C and Table 1), whereas only one gene was commonly dysregulated within Lgr5EGFP- daughter cells of Msh2-HET and Msh2-KO when compared to Msh2-WT. In order to summarize the number of genes expressed in stem and daughter cells as well as the intersection among the different genotypes (Msh2 status) and cell types (Lgr5EGFP+ or Lgr5EGFP-), we generated an UpSet plot matrix that provides a visualization at a glance of these numbers (Supplementary Figure S3C). Overall, the gene profile showing the largest differences was between Msh2-KO and -WT stem cells, and the one with the closest expression was between stem and differentiated cells of the Msh2-KO genotype (Figure S3C). These results indicate that the loss of one or both alleles of Msh2 induces a unique transcriptional profile that perturbs the biology of MMRd ISC. In addition, the transcriptome of Lgr5EGFP+ stem cells of Msh2-KO clustered with daughter cells (Lgr5EGFP- fractions) of all three genotypes in the PCA plot and also displayed the lowest amount of differentially expressed genes compared to their daughter cells, thus indicating that a complete loss of Msh2 may lead to premature differentiation of ISC.
Figure 2. Bioinformatics analysis of gene expression from Lgr5EGFP+ stem and Lgr5EGFP- daughter cells.

(A) PCA plot of expression profiles from Lgr5EGFP+ intestinal stem cells (solid colors) isolated from Msh2-WT (dark yellow), Msh2-HET (dark blue) and Msh2-KO (dark red) and from daughter cells (Lgr5EGFP- and EpCAM+, light shades of respective colors). The first and second principal components are plotted in the X and Y-axis, respectively. Individual samples within each group are connected by a centroid. A total of 21 mice for Msh2-WT, 25 for Msh2-HET and 40 for Msh2-KO were equally distributed among three biological replicates to obtain ~10,000 ISC for each replicate per genotype; (B) Volcano plots illustrate genes expressed in ISC of Msh2-KO and Msh2-HET compared to Msh2-WT and in ISC of Msh2-KO as compared to Msh2-HET. X-axis presents Log2Fold change and Y-axis presents log10 of adjusted P-value for multiple comparisons from DESeq2 differential analysis. The horizontal dashed line represents FDR=0.05, while left and right vertical dashed lines represent Log2FC of ±1, respectively. Significantly down-regulated genes are displayed in green, upregulated in red, and non-significant in black; (C) Venn diagrams showing numbers of significantly expressed genes in Lgr5EGFP+ stem cells and Lgr5EGFP- non-stem cells for each genotype compared with Msh2-WT; (D) Validation of the expression of key signature genes from the MMRd and MMR-haploinsufficient signatures as well as stem and differentiation markers analyzed using qRT-PCR in FACS sorted Lgr5EGFP+ (stem, left panels) and Lgr5EGFP- (non-stem, right panels) cells from Msh2-WT, Msh2-HET and Msh2-KO. Data is presented as fold changes and depicted as relative gene expression levels compared to expression levels in Lgr5EGFP+ cells of Msh2-WT as reference. Expression levels of Gapdh were used as an internal housekeeping gene for normalization. Error bars display ±SD. One-way ANOVA with Tukey’s multiple comparison post-hoc test, *P -value< 0.05, **P-value < 0.01, ***P-value< 0.001, ****P-value<0.0001.
Table 1.
Common significant genes in stem (upper section) and daughter cells (lower section) isolated from Msh2-HET and Msh2-KO mice. Genes selected had FDR≤0.05 and |Log2FC|≥1.
| Gene Symbol | Fold Change Lgr5EGFP+ Het vs WT | Fold Change Lgr5EGFP+ KO vs WT |
|---|---|---|
| Spp1 | 6.019 | 19.335 |
| Nr1h5 | 4.862 | 8.549 |
| Rian | 3.727 | 4.717 |
| Jchain | 3.682 | 3.763 |
| Meg3 | 3.520 | 4.305 |
| Gm28230 | 3.515 | 5.396 |
| Dgkh | 3.045 | 3.907 |
| Arhgap45 | 2.943 | 3.587 |
| Trpv3 | 2.938 | 2.771 |
| P2ry4 | 2.612 | 3.041 |
| Ccn3 | 2.564 | 2.613 |
| B230206H07Rik | 2.518 | 4.196 |
| Tchh | 2.437 | 3.435 |
| Lifr | 2.350 | 2.069 |
| Ahnak | 2.290 | 3.747 |
| Sardh | 2.236 | 3.526 |
| Cacna1h | 2.204 | 2.373 |
| Sned1 | 2.195 | 2.307 |
| Nlrp9b | 2.040 | 4.672 |
| Gm23547 | 0.361 | 0.361 |
| Fold Change Lgr5EGFP- Het vs WT | Fold Change Lgr5EGFP- KO vs WT | |
| Muc5ac | 6.638 | 3.905 |
Validation of stem and non-stem cell specific genes in MMRd ISCs and daughter cells.
We assessed dysregulated genes that overlapped between Msh2-HET and Msh2-KO in Lgr5EGFP+ stem cells and Lgr5EGFP- non-stem cells using qRT-PCR (Table 1). These markers were selected based on their potential roles in tumorigenesis and the correlation between their level of dysregulation and Msh2 allele dosage. We evaluated a total of four markers of stem cells (Spp1, Nr1h5, Ahnak, and Nlrp9b) and one of daughter cells (Muc5ac). Overall, all of them were confirmed to be expressed in both Lgr5EGFP+ and Lgr5EGFP- cells of both Msh2-HET or Msh2-KO mice. Moreover, they were significantly upregulated in stem cells and their expression correlated with the Msh2 allele dosage (Figure 2D, upper left panel). Of note, we observed a high level of upregulation of the marker Spp1 in both Msh2-HET (10-fold) and Msh2-KO (15-fold) stem cells when compared to Msh2-WT. In addition, the relative expression of Spp1 was significantly lower in Lgr5EGFP- daughter cells than in Lgr5EGFP+ stem cells within each respective genotype. In contrast, the expression of Nr1h5 showed significant differences in daughter cells across genotypes, while Ahnak and Nlrp9b did not show differences across genotypes (Figure 2D, upper right panel). In regards to Muc5ac, there were significant differences among daughter cells across different genotypes that were also seen among stem cells, thus making this marker relatively non-specific of cell-of-origin. We attempted to validate the gene expression of the novel stem cell markers observed in the Lgr5EGFP+ stem cells using Msh2-KO and Msh2-WT mouse organoids as an ex vivo model of the stem cell compartment of the intestinal crypt. Initially, we did not confirm the same trends observed in sorted cells (Figure S3D, left panel). Therefore, based on the function of these genes, we reasoned that their expression could be influenced by the immune environment and surrounding stem cell niche. We repeated the expression assessment after stimulating the organoids for 24 hours with colony-stimulating factor 1 (M-CSF1) to recreate cues received by stem cells. Under these conditions, we observed a significant upregulation of Spp1 and Nlrp9b expression in Msh2-KO organoids and no significant changes in the Msh2-WT counterparts (Figure S3D, center panel). These results are consistent with the expression data acquired in vivo and strongly suggest that Spp1 and Nlrp9b expression levels were significantly and specifically enhanced in MMRd ISCs upon interaction with the stem cell niche, including immune cells, thus highlighting its role as a potential biomarker in MMRd carcinogenesis.
Since the transcriptomes derived from Msh2-KO Lgr5EGFP+ ISCs clustered together with daughter cells of all genotypes, we hypothesized that Msh2-KO ISCs lost their stem-ness and underwent premature differentiation. Based on this observation, we performed qRT-PCR analysis of differentiation-specific genes including Krt20 and Alpi (markers for enterocytes), and Muc2 (marker for Goblet cells) and stem cell markers, Ascl2 and Olfm4 in intestinal samples from mice of the three genotypes. We confirmed a significant decrease in expression of stem cell markers, Lgr5, Ascl2, and Olfm4 in stem cells from Msh2-KO mice compared to those in Msh2-WT mice. Subsequently, we observed a strong stimulation in the expression levels of enterocyte markers, Krt20 and Alpi, in the stem cells of Msh2-KO mice (Figure 2D lower left panel). These results were confirmed ex vivo in mouse organoids that showed downregulation of surface stem cell markers and upregulation of differentiation signals (Figure S3D, right panel). In line with these results, no significant differences were observed in the expression of these markers in Lgr5EGFP- cells across all genotypes (Figure 2D lower right panel). Therefore, these results indicate that loss of MMR function influences ISC homeostasis and promotes premature differentiation of ISCs.
Proteomic profile of MMR haploinsufficient and MMR deficient stem and non-stem cells.
To assess the proteomic profile, we isolated total cellular proteins from stem and non-stem fractions of Msh2-WT, Msh2-HET, and Msh2-KO mice and performed tandem mass spectrometry (MS/MS). We identified an average of 1238 gene counts (proteins) from total cell extracts of stem cells from Msh2-WT (Table S5). A total of 797 and 830 proteins were observed from equal amounts of total protein of stem cells from Msh2-HET and Msh2-KO mice, respectively. The number of proteins identified from non-stem cells was higher than stem cell fractions observed in each genotype. Individual analysis of fold change expression obtained directly from mean spectral counts of Lgr5EGFP+ ISCs revealed high levels of differentiation markers such as Krt20 (5.5-fold) and Fabp1 (5.8-fold) in Msh2-HET Lgr5EGFP+ cells (Table S6) and even higher in Msh2-KO Lgr5EGFP+ (Krt20, 6.5-fold; Fabp1, 7.3-fold; Fabp2, 2.0-fold, Table S7) compared to Lgr5EGFP+ Msh2-WT cells. We also observed enrichment for proteins that are involved in cancer progression and migration, such as carbonic anhydrase 1 (Car1, 10.89-fold in Msh2-HET, and 21-fold in Msh2-KO) and actin-binding protein Gelsolin (Gsn, 2.5-fold in Msh2-HET and 9.8-fold in Msh2-KO)24, 25. Thus, the proteomic expression of MMRd stem cells showed enrichment for cancer-associated markers that are involved in their malignant transformation.
Generation of a molecular profile to define a gene signature of MMRd.
Using the transcriptomic and proteomic profiles of Lgr5EGFP+ and Lgr5EGFP- cells for each genotype, we generated a molecular profile that defines a gene signature for MMR haploinsufficiency and MMRd. First, we compared the expression data of Lgr5EGFP+ ISCs obtained from Msh2-KO to Msh2-WT mice and excluded those genes expressed commonly in Lgr5EGFP- fractions in order to generate a list of unique genes that specifically represent LS colorectal neoplasia (pre-cancers and tumors), which is characterized by a complete MMRd. We observed a total of 48 differentially expressed genes (Table 2). The same approach was applied to the analysis of the comparison of the transcriptome of Msh2-HET to Msh2-WT to generate a list of genes exclusively and differentially expressed in MMR haploinsufficient ISCs. This list reflected the expression patterns of LS normal colorectal mucosa and a total of 5 differentially expressed genes were detected (Table 2). GSEA highlighted several relevant pathways in MMRd ISC biology. Among Lgr5EGFP+ cells from Msh2-KO, the top observed pathways were related to Integrin Signaling, Focal Adhesion, and Inflammatory Response. Interestingly, we also observed downregulation of the WNT pathway (Figure 3A). The top enriched pathways in the Msh2-HET ISCs were TGFβ signaling, mitogen-activated protein kinase (MAPK) signaling, and Endochondral Ossification (Figure 3B). We found that the pro-inflammatory gene sets, Prostaglandin-Leukotriene Metabolism and Eicosanoids Synthesis, were depleted in Msh2-HET. In addition, we observed that gene sets for Cytoplasmic Ribosomal Proteins were downregulated in both Msh2-HET and Msh2-KO ISCs (Figure 3C). Finally, we compared our gene signatures reflecting different stages of MMR carcinogenesis with the previously published profile of mouse APC-driven stem cells16. We observed a minimal degree of overlap with the Msh2-KO signature (only 6 genes) and increased numbers of genes shared with Msh2-HET and Msh2-WT, thus consistent with the expectation that APC-driven carcinogenesis overlaps minimally with MMRd during the earliest stages (Figure S3E).
Table 2.
List of genes defining a signature of MMR haploinsufficiency (Msh2-HET, upper section) and MMRd (Msh2-KO, lower section). Genes selected had FDR≤0.05 and |Log2FC|≥1.
| Gene Symbol | Fold Change Lgr5EGFP+ Msh2-Het vs Msh2-WT | Fold Change Msh2-Het Lgr5EGFP+ vs Lgr5EGFP- |
|---|---|---|
| Spp1 | 6.019 | 8.984 |
| Meg3 | 3.520 | 0.413 |
| P2ry4 | 2.612 | 0.442 |
| Defa5 | 0.486 | 0.318 |
| Gm49320 | 0.297 | 0.410 |
| Fold Change Lgr5EGFP+ Msh2-KO vs Msh2-WT | FoldChange Msh2-KO Lgr5EGFP+ vs Lgr5EGFP- | |
| Mup22 | 27.554 | 12.855 |
| Spp1 | 19.335 | 19.562 |
| Slc26a9 | 11.831 | 6.639 |
| Muc6 | 10.515 | 11.907 |
| Ugt8a | 10.462 | 2.432 |
| Cubn | 8.143 | 2.473 |
| Pgc | 7.723 | 32.672 |
| Aqp5 | 7.414 | 14.058 |
| Cyp2c55 | 5.917 | 3.439 |
| Abca12 | 5.825 | 3.805 |
| Slc5a4a | 5.580 | 3.023 |
| Gm28230 | 5.396 | 2.352 |
| Mal | 5.256 | 3.366 |
| Car1 | 4.989 | 2.706 |
| G6pc | 4.820 | 2.433 |
| Sprr1a | 4.709 | 2.328 |
| Slc5a12 | 4.637 | 2.482 |
| Scara3 | 4.513 | 4.933 |
| Gif | 4.484 | 13.338 |
| Slc30a10 | 4.259 | 2.472 |
| Lct | 4.215 | 2.923 |
| Sptssb | 4.087 | 2.970 |
| Sgk2 | 4.058 | 2.795 |
| Clca4a | 4.052 | 2.490 |
| Cyp3a25 | 3.804 | 2.669 |
| Gdpd2 | 3.690 | 2.173 |
| Gkn1 | 3.677 | 3.141 |
| Tmigd1 | 3.602 | 2.732 |
| Slc2a2 | 3.575 | 3.053 |
| Aldh1a1 | 3.476 | 2.309 |
| Fa2h | 3.466 | 2.378 |
| Bst1 | 3.453 | 2.303 |
| 1810065E05Rik | 3.436 | 2.567 |
| Anxa10 | 3.435 | 2.881 |
| Slc10a2 | 3.298 | 2.114 |
| Clu | 3.193 | 4.456 |
| 2010109I03Rik | 3.181 | 2.060 |
| Pdzk1 | 3.000 | 3.215 |
| Cyp2b10 | 2.804 | 3.551 |
| Mafb | 2.766 | 2.497 |
| Slc5a4b | 2.594 | 2.618 |
| Slc16a9 | 2.581 | 2.080 |
| Ifit1 | 2.473 | 2.235 |
| Oas3 | 2.346 | 2.001 |
| Ak4 | 2.264 | 2.020 |
| Sema6a | 2.225 | 2.220 |
| Slc28a1 | 2.168 | 2.053 |
| Itga7 | 2.060 | 0.000 |
Figure 3. Pathways modulated in MMRd and MMR haploinsufficient ISCs.

Bubble chart plots display statistically significant pathways enriched in Msh2-KO ISCs (A), Msh2-HET ISCs (B), and both Msh2-HET and Msh2-KO ISCs (C), using BH-adjusted P-value=0.05 as a cutoff. Pathways bolded were relevant in terms of function to the molecular biology of MMRd ISC. The size of circles represents adjusted P-value (larger circles represent smaller P-value). The colors of bubbles were determined by the sign and amplitude of normalized enrichment score (NES) with positively enriched pathways in red and negatively enriched pathways in green.
Then, we generated a unique list of 78 signature proteins for Msh2-KO (MMRd) as ratio of protein expression from 130 proteins found between Lgr5EGFP+ and Lgr5EGFP- cells of Msh2-KO mice and also from the ratio of protein expression from 117 proteins found between Lgr5EGFP+ cells of Msh2-KO and Msh2-WT (Table S8). From the proteomic analysis, 27 proteins were found to overlap with 197 signature genes from mRNAseq analysis for Msh2-KO (FDR≤0.05, Figure S4A). Of note, our mass spectrometry results indicated that Spp1 protein was found to be expressed only in Lgr5EGFP+ cell fractions of Msh2-KO animals. In addition, we analyzed pathway enrichment using Ingenuity Pathway Analysis (IPA) observing a representative network of proteins related to cellular development, cellular growth, and proliferation as top affected pathways with expression of KRAS as a pivotal network (Figure S4B). Similarly, a unique list of 52 signature proteins for Msh2-HET was derived from comparing the ratio of protein expression from 235 proteins found between Lgr5EGFP+ and Lgr5EGFP- cells of Msh2-HET mice and also from the ratio of protein expression from 187 proteins found between only Lgr5EGFP+ cells of Msh2-HET and Msh2-WT (Table S9). DNA replication, recombination and repair, as well as RNA post-transcriptional modification pathways stood out from the Msh2-HET protein profile, which was PARP1 centered (Figure S4C). Overall, the Msh2-HET and Msh2-KO protein signatures revealed potentially interesting candidates that merits consideration for its influence on carcinogenesis in MMR deficiency.
Validation of the MMRd stem cell signature in LS human specimens.
To assess the biological significance of the MMRd and MMR-haploinsufficient gene signatures derived from ISC in mice, we applied both signatures to whole transcriptomics of normal colorectal mucosa and neoplastic lesions (both adenomas and tumors) from a cohort of LS and FAP patients (as MMRp controls; Table S1). For the Msh2-KO signature, we observed that 41 genes out of 197 signature genes were still significantly dysregulated (FDR≤0.05) and had the same fold change direction in LS hyper-mutant adenomas and tumors. This confirmed that these genes represent and recapitulate the biology of cells derived from an MMRd progenitor. Together, these 41 dysregulated genes were able to clearly separate pre-cancers and early-stage tumors in LS and FAP patients into two distinct clusters, with only two LS samples misclassified (Figure 4A). For the MMR-haploinsufficient signature (Msh2-HET signature), we had to relax the criteria because only one gene out of 27 (AHNAK) was significantly dysregulated and had the same fold change direction. Therefore, we examined 14 genes that had the same fold change direction in mouse and human data sets. Unsupervised hierarchical clustering observed two groups of samples with one group integrated by mostly LS samples with the exception of 4 lesions (Figure S5A). Of note, SPP1 showed a trend towards being upregulated in LS normal mucosa. Overall, the MMRd gene signature (Msh2-KO) was able to correctly classify neoplasms that are MMRd and therefore contain biomarker information that recapitulated early stages of MMRd carcinogenesis in humans. Then, we investigated if the signature comparing Msh2-KO and Msh2-HET was able to distinguish LS pre-cancers and early-stage tumors from normal mucosa. The original signature from mouse ISC contained a total of 182 genes (Figure S3C) and 56 genes retained statistical significance in human and shared the same fold change direction. The resulting gene profile was able to separate the samples into two groups with only three neoplastic lesions misclassified, thus showing a strong performance (Figure 4B). Finally, we combined together with the MMRd and haploinsufficient signatures (Table 2) to validate its performance to differentiate the expression patterns of human organoids derived from the normal mucosa of both LS carriers22 and patients diagnosed with sporadic CRC (as normal mucosa controls, Supplementary Material and Methods)23. We observed that an 11-gene set of significantly expressed genes with corresponding human orthologs were able to precisely differentiate and segregate colorectal normal mucosa organoids of LS from sporadic individuals (Figure S5B).
Figure 4. Expression of Msh2-HET and Msh2-KO signatures in FAP and LS patient samples.

(A) Unsupervised hierarchical clustering heatmaps showing the expression pattern of selected Msh2-KO versus Msh2-WT signature genes in FAP polyps and LS hypermutant/MSI pre-cancer/tumor samples (FDR<0.05 in human and same fold change direction in both mouse and human); (B) Expression patterns of selected Msh2-KO versus Msh2-HET signature genes in normal mucosa from LS patients using row-centered and batch corrected expression data (FDR<0.05 in human and same fold change direction in mouse and human). Dendrograms indicate sample-sample Pearson correlation distances. The significance of genes in human comparison are indicated in a row covariate bar. Log2FC in human and mice for each gene are shown as scatter plots. Sample type is color-coded as follows: blue represents normal tissue from LS patients; red represents hypermutant/MSI adenomas/tumor tissue from LS patients, respectively; FC, fold change; LS, Lynch Syndrome.
Expression of Spp1 in MMR-deficient mouse ISCs and LS patient specimens.
Our combined transcriptomics and proteomics analysis indicated that Spp1 expression is upregulated in ISCs of both MMRd mice and LS patients. To gain mechanistic insights in epigenetic regulation of SPP1, ChIP assays were performed in MMRp and MMRd cell line models. Our results indicated that levels of H3K27me3 epigenetically regulate the expression of SPP1 as a function of the MMR status (Figure S5C). Then, to further confirm the expression of SPP1 in stem cells (Lgr5EGFP+ cells), we performed IHC in intestinal tissue sections of Msh2-WT, Msh2-HET, Msh2-KO mice using antibodies against Spp1 and GFP. Since GFP expression is under control of the Lgr5 promoter in mice, the level of GFP staining correlates to the level of Lgr5 cells. We observed enhanced staining of Spp1 in crypts of Msh2-HET and Msh2-KO mice that co-localized with Lgr5 cells (GFP+) located at the base of the crypts, thus confirming the upregulation of Spp1 in ISCs of MMR haplo-insufficient and MMRd tissues (Figure 5A). Finally, we examined the levels of SPP1 and LGR5 in a series of LS samples (Table S4) representing sequential steps of colorectal carcinogenesis: normal mucosa (n=6), pre-cancers (n=4, tubular adenomas), and tumors (n=3, adenocarcinomas). Quantitative imaging and analysis of single- and double-positive cells for SPP1 and LGR5 (Figure S6) showed the highest proportion of co-expressing stem cells (double-positive cells) in cancers compared to adjacent normal colorectal mucosa and pre-cancers (non-statistically significant trend, P-value=0.15 cancer vs. normal, Figure 5B and 5C). Despite the limited number of samples available for analysis, these results are consistent with the data from the LS mouse model, thus suggesting a potential role of SPP1 in the progression of colorectal MMRd carcinogenesis.
Figure 5. Expression and localization of Spp1 within crypts of MMR mouse models and LS patient specimens.
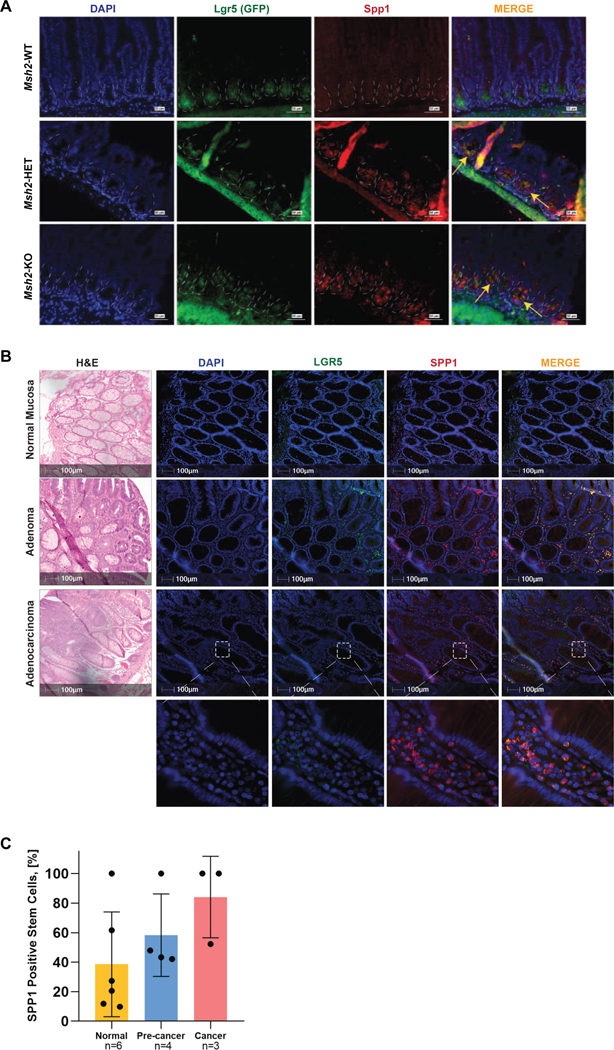
(A) Small intestine from 8 week-old Msh2-WT, Msh2-HET, and Msh2-KO mice was stained with antibodies against GFP to detect Lgr5+ cells (green) and Spp1 (red) by immunofluorescence. Panels show representative images for Lgr5 and Spp1 expression and location within crypts. Yellow arrows in merged images demarcate the co-localization of Lgr5 and Spp1 in crypts of tissues from Msh2-HET and Msh2-KO. Nuclei were counterstained with DAPI (blue). Scale bar is equivalent to 50 μm; (B) Representative images from FFPE tissue sections of H&E, nuclear counterstaining with DAPI (blue), immunostaining with anti-human LGR5 (TSA with Opal-520, green), and anti-SPP1 (TSA with Opal-570, red) antibodies, and composite images acquired using fluorescent multiplex immunohistochemistry. Regions of interest for digital image analysis include normal colon epithelium (top panel), adenomas (middle panel), and adenocarcinoma (lower panel) displaying single positive cells for LGR5 and SPP1, and double-positive cells (MERGE); Scale bar represents 100 μm and scale bar in insets are equivalent to 10 μm. (C) The number of positive cells for each marker and double positives reported was quantified as cell density and expressed by the number of cells per mm2 using inForm advance image analysis software (considering that the total number of nucleated cells is 100%). The percentage of stem cells co-expressing SPP1 and LGR5 (double-positive cells) was higher in pre-cancers and cancers compared to normal adjacent tissue showing a non-statistically significant trend. Graph displays mean±SEM.
Discussion
In this study, we have used a tissue-specific mouse model of LS to identify, for the first time, the transcriptomic and proteomic profiles of ISCs displaying MMRd. We observed a significant loss of Lgr5+ stem cells upon deletion of each Msh2 allele, which posed additional technical challenges as we required a large number of animals to obtain sufficient numbers of cells to perform mRNAseq and mass spec analyses. We validated this observation in MMRd ISCs and intestinal organoids, which revealed a loss of stemness reflected by downregulation of Lgr5, Ascl2, and Olfm4, and upregulation of the differentiation-specific markers Krt20, Alpi, and Muc2. Therefore, our results suggest that stem cells from Msh2-KO prematurely exhibit a differentiated phenotype, as evident by the fact that MMRd stem cells clustered together with differentiated cells of all genotypes regardless of their MMR status. It is plausible that stem cells trigger a natural epigenetic response towards differentiation to avoid malignant transformation. In fact, this mechanism has been therapeutically exploited in the treatment of leukemia, where ‘differentiation therapies’ are used to treat Acute Promyelocytic Leukemia26. More relevant for our disease context was the observation that inhibition of R-Spondin in CRC, a ligand for Lgr5 receptor, led to in-vivo differentiation and loss of stem-cell function27, 28.
Enrichment analysis has pointed towards the dysregulation of other cellular pathways associated with the loss of stemness in MMRd ISC that could drive their transformation into a cancer stem cell phenotype18. We observed the downregulation of ribosomal proteins such as RPS7, which has been reported to act as a tumor suppressor gene inhibiting proliferation by decreasing hypoxia-inducible factor-mediated glycolysis in CRC29, and RPS14, which has been reported to play a significant role in cell proliferation by negatively regulating the transcriptional activity of c-Myc, a key oncogene involved in colorectal carcinogenesis30. Thus, we posit that the MMR system may have a direct role on stem cell maintenance and renewal. As MMRd prompts premature differentiation of stem cells in LS, a relatively small percentage of cells persist that sustain and acquire a cancer stem cell phenotype via dysregulation of key genes from cancer promoting pathways such as those that we have observed in our pathway enrichment analysis. The prematurely differentiated MMRd stem cells that have lost their stem-ness will become de-differentiated under specific conditions, which may yield pluripotency that subsequently drives the onset of carcinogenesis31. Therefore, induction of stem cell differentiation in LS could become a potential avenue for cancer interception that warrants further investigation. Another notable finding is the downregulation of Wnt signaling in MMRd ISC. This observation confirms that the key initiating step in LS carcinogenesis is inactivation of the MMR system within aberrant crypt foci, then leading to flat pre-malignant lesions upon acquisition of additional hits in other key oncogenic drivers, with activation of Wnt signaling at later stages and only in a fraction of the pre-malignant lesions. This agrees with previous models that were based on anecdotal observations and that now can be better substantiated in our results32.
Our gene signature of MMR deficiency in stem cells is a frontier discovery that includes a unique set of genes with the potential of being a biomarker of early cancer progression. In fact, this signature is able to differentiate between MMRd and MMR-proficient neoplasia as well as organoids derived from normal tissues in the same contexts. Several individual genes integrated within this profile have been previously shown to be involved in different aspects of colorectal cancer progression. For example, Aldh1a1 expression, which is normally found in the bottom of the crypt, was shown to be enhanced during progression from normal epithelium to adenoma with increasing expression levels as cells move upwards in the colonic crypt33. Another gene, Spp1, which encodes for the bone sialoprotein Osteopontin was found to be significantly and exclusively expressed in MMR haplo-insufficient and -deficient stem cells. Furthermore, our proteomic profiling recorded spectral counts only in Lgr5EGFP+ stem cells in Msh2-KO mice. These observations were validated both in mouse and human samples where we observed enhanced staining of SPP1 in small intestine crypts of Msh2-HET and Msh2-KO mice co-localizing with Lgr5, and in LS adenoma and tumor samples, respectively. SPP1 has been reported to have a role in invasion and metastasis of several cancer types, including colon, ovarian, and breast, thus acting both in an autocrine and paracrine manner34–36, and specifically promoting stem-cell-like properties in CRC37, 38. In addition, overexpression of SPP1 in tumor cells has been associated with infiltration by tumor-associated macrophages39, thus fueling tumor growth and angiogenesis40. This fact was also thought to weaken CD8+ T cell-mediated immune response leading to immune tolerance by dampening host tumor immune surveillance41, 42. In fact, we were only able to confirm the upregulation of Spp1 in mouse organoids when we culture them in supplemented media with M-CSF1, which has been shown to be involved in the maintenance of the intestinal stem cell niche through different cellular and molecular mechanisms involving Paneth cells43 and VEGF44, as well as mediating the effects of the immune-environment surrounding the stem cell niche, in particular tumor-associated macrophages. Thus, SPP1 functions via adhesive interactions at the tumor/host interface in several malignancies and can be a potential biomarker for MMR deficiency, hence warranting further investigation in LS carcinogenesis.
Our work has several limitations. First, our proteomics data was only able to partially validate the gene expression profiling of stem cells. We believe that this weak correlation between mRNA and protein profiles was due to the limited protein yielded from FACS-sorted stem cells in Msh2-KO mice (only ~100k cells per replicate). Therefore, the lack of comprehensive proteomics coverage due to low-abundance of proteins limits the detection of most proteins with required quantitative precision. These challenges were observed previously in other biological systems, especially in embryonic stem cells where a large number of animals have been required to gather a sufficient number of cells for the appropriate biological replicates to provide adequate statistical power in the analysis45, 46. Second, it would have been ideal to utilize single-cell genomics to investigate and validate MMRd specific gene expression patterns in mouse and human ISC. However, our experiments were designed prior to recent developments that would have helped improve the feasibility and cost-efficiency of single-cell transcriptomics. Third, we have attempted to perform a validation of our newly-discerned MMRd stem cell signature in an internal and limited LS cohort. Itdeally, the validation would have been performed using an independent set of samples with a larger number of tissue specimens. The ultimate value of the gene signature as a whole or individual markers such as SPP1 remains to be established in future studies.
In conclusion, we have identified a gene signature of MMRd ISCs using the transcriptomic and proteomic profiles of the stem and non-stem cells from a MMRd mouse model. We have observed that the MMRd stem cell signature is able to correctly distinguish early MMRd from MMRp early neoplasia from samples of LS and FAP patients. Using systems biology approaches, molecular, and cellular studies in both mouse and human samples, we identified SPP1, which qualifies as a bona fide marker of MMR deficiency in LS patients. In summary, data presented in this study advance our understanding of ISC biology in LS patients that serves as the starting point to develop novel markers of early detection of progression of LS carcinogenesis and potential targets for cancer interception strategies in this patient population.
Supplementary Material
Significance statement:
The transcriptomic and proteomic profile of MMR-deficient intestinal stem cells display a unique set of genes with potential roles as biomarkers of cancer initiation and early progression.
Acknowledgments:
We thank the patients and their families for their participation. We thank the staff of the Advanced Technology Genomics, and the Flow Cytometry and Cellular Imaging Core Facilities at UTMDACC for their assistance with RNA sequencing, FACS sorting of samples, and imaging acquisition of immunofluorescence, respectively.
Funding/Support: This work was supported by grant R01 CA219463 (US National Institutes of Health/National Cancer Institute) to E.V., and a gift from the Feinberg Family Foundation to W.E. and E.V.; by the Multi-Investigator Research Award grant RP160668 (Cancer Prevention and Research Institute of Texas) to I.I.W., and the MD Anderson Lung Cancer Moon Shot Program to I.W.W. and the Translational Molecular Pathology Immune-profiling Laboratory; by the NCI Research Specialist R50 CA243707-01A1 to J.K.B.; and P30 CA13330 and CA016672 (US National Institutes of Health/National Cancer Institute) to the Albert Einstein Cancer Center and The University of Texas MD Anderson Cancer Center Core Support Grant, respectively.
Dr. Vilar has a consulting or advisory role with Janssen Research and Development and Recursion Pharma; has received research support from Janssen Research and Development; Drs. Vilar, Hanash, Bommi, Wu and Katayama are contributors to US provisional patent application based on the work presented in this manuscript. Dr. Wistuba has an advisory board role with Genentech/Roche, Bayer, Bristol-Myers Squibb, Astra Zeneca/Medimmune, Pfizer, HTG Molecular, Asuragen, Merck, GlaxoSmithKline, Oncocyte, Guardant Health and MSD; has received speaker fees from Medscape, MSD, Genentech/Roche, Pfizer, AstraZeneca, Merck; and research support from Genentech, Oncoplex, HTG Molecular, DepArray, Merck, Bristol-Myers Squibb, Medimmune, Adaptive, Adaptimmune, EMD Serono, Pfizer, Takeda, Amgen, Karus, Johnson & Johnson, Bayer, Iovance, 4D, Novartis, and Akoya.
Abbreviations:
- APC
adenomatous polyposis coli
- Ascl2
achaete-scute family bHLH transcription factor 2
- CBC
crypt base columnar
- CMMRD
constitutional mismatch repair deficiency
- CRC
colorectal cancer
- DEGs
differentially expressed genes
- EGFP
enhanced green fluorescent protein
- FACS
florescence-activated cell sorting
- FAP
familial adenomatous polyposis
- GSEA
gene set enrichment analysis
- H&E
hematoxylin and eosin
- Het
heterozygous
- IHC
immunohistochemistry
- IRB
Institutional Review Board
- ISCs
intestinal stem cells
- KO
Knockout
- Krt20
keratin 20
- Lgr5
leucine rich repeat containing G protein-coupled receptor 5
- LS
Lynch Syndrome
- MMRd
mismatch repair-deficient
- MMRp
MMR-proficient
- MS
mass spectrometry
- Msh2
mutS homolog 2
- NES
normalized enrichment score
- qRT-PCR
quantitative reverse transcriptase-polymerase chain reaction
- mRNAseq
RNA sequencing; Spp1, secreted phosphoprotein 1
- UTMDACC
The University of Texas MD Anderson Cancer Center
- WT
wildtype
Footnotes
Declarations
Ethics approval and consent to participate: All patient samples and information were collected with written informed consent. The study conformed to the guidelines from the IRB-approved protocol (MDACC IRB# PA12-0327). Tissue specimens were obtained during routine screening and diagnostic procedures from patients followed at The University of Texas MD Anderson Cancer Center.
Consent for publication: Contained in study consent for MDACC IRB# PA12-0327
Availability of data and materials: mRNA sequencing data have been deposited in GEO. The data sets generated and analyzed during the current study can be accessed for re-analysis in the following links: GSE153385 (18 mouse samples; 7 paired adenoma/mucosa pairs and 10 unmatched normal mucosa specimens from FAP patients; 7 matched tumor/adenomas and normal mucosa pairs and 23 unmatched normal mucosa specimens from LS patients) https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE153385; GSE106500 (13 FAP polyps and 11 LS polyps) which is already accessible to the public at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE106500
Conflict of Interest Disclosures:
No other disclosures are reported.
References
- 1.Lynch HT, Snyder CL, Shaw TG, et al. Milestones of Lynch syndrome: 1895–2015. Nat Rev Cancer 2015;15:181–94. [DOI] [PubMed] [Google Scholar]
- 2.Win AK, Jenkins MA, Dowty JG, et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiol Biomarkers Prev 2017;26:404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol 2010;7:153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol 2011;6:479–507. [DOI] [PubMed] [Google Scholar]
- 5.Bonadona V, Bonaiti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011;305:2304–10. [DOI] [PubMed] [Google Scholar]
- 6.Stoffel E, Mukherjee B, Raymond VM, et al. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology 2009;137:1621–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moller P, Seppala T, Bernstein I, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut 2017;66:464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmeler KM, Lynch HT, Chen LM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med 2006;354:261–9. [DOI] [PubMed] [Google Scholar]
- 9.van Leerdam ME, Roos VH, van Hooft JE, et al. Endoscopic management of Lynch syndrome and of familial risk of colorectal cancer: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy 2019;51:1082–1093. [DOI] [PubMed] [Google Scholar]
- 10.Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat Rev Mol Cell Biol 2014;15:19–33. [DOI] [PubMed] [Google Scholar]
- 11.Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science 2013;340:1190–4. [DOI] [PubMed] [Google Scholar]
- 12.Vermeulen L, Snippert HJ. Stem cell dynamics in homeostasis and cancer of the intestine. Nat Rev Cancer 2014;14:468–80. [DOI] [PubMed] [Google Scholar]
- 13.Visvader JE. Cells of origin in cancer. Nature 2011;469:314–22. [DOI] [PubMed] [Google Scholar]
- 14.Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457:608–11. [DOI] [PubMed] [Google Scholar]
- 15.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449:1003–7. [DOI] [PubMed] [Google Scholar]
- 16.Munoz J, Stange DE, Schepers AG, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J 2012;31:3079–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Wind N, Dekker M, Berns A, et al. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 1995;82:321–30. [DOI] [PubMed] [Google Scholar]
- 18.Vaish M. Mismatch repair deficiencies transforming stem cells into cancer stem cells and therapeutic implications. Mol Cancer 2007;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spira A, Yurgelun MB, Alexandrov L, et al. Precancer Atlas to Drive Precision Prevention Trials. Cancer Res 2017;77:1510–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kucherlapati MH, Lee K, Nguyen AA, et al. An Msh2 conditional knockout mouse for studying intestinal cancer and testing anticancer agents. Gastroenterology 2010;138:993–1002 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang K, Taggart MW, Reyes-Uribe L, et al. Immune Profiling of Premalignant Lesions in Patients With Lynch Syndrome. JAMA Oncol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reyes-Uribe L, Wu W, Gelincik O, et al. Naproxen chemoprevention promotes immune activation in Lynch syndrome colorectal mucosa. Gut 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Costales-Carrera A, Fernandez-Barral A, Bustamante-Madrid P, et al. Comparative Study of Organoids from Patient-Derived Normal and Tumor Colon and Rectal Tissue. Cancers (Basel) 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tetteh PW, Kretzschmar K, Begthel H, et al. Generation of an inducible colon-specific Cre enzyme mouse line for colon cancer research. Proc Natl Acad Sci U S A 2016;113:11859–11864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar R, Raman R, Kotapalli V, et al. Ca(2+)/nuclear factor of activated T cells signaling is enriched in early-onset rectal tumors devoid of canonical Wnt activation. J Mol Med (Berl) 2018;96:135–146. [DOI] [PubMed] [Google Scholar]
- 26.de The H. Differentiation therapy revisited. Nat Rev Cancer 2018;18:117–127. [DOI] [PubMed] [Google Scholar]
- 27.Fischer MM, Yeung VP, Cattaruzza F, et al. RSPO3 antagonism inhibits growth and tumorigenicity in colorectal tumors harboring common Wnt pathway mutations. Sci Rep 2017;7:15270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Storm EE, Durinck S, de Sousa e Melo F, et al. Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature 2016;529:97–100. [DOI] [PubMed] [Google Scholar]
- 29.Zhang W, Tong D, Liu F, et al. RPS7 inhibits colorectal cancer growth via decreasing HIF-1alpha-mediated glycolysis. Oncotarget 2016;7:5800–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou X, Hao Q, Liao JM, et al. Ribosomal protein S14 negatively regulates c-Myc activity. J Biol Chem 2013;288:21793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedmann-Morvinski D, Verma IM. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep 2014;15:244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cerretelli G, Ager A, Arends MJ, et al. Molecular pathology of Lynch syndrome. J Pathol 2020;250:518–531. [DOI] [PubMed] [Google Scholar]
- 33.Huang EH, Hynes MJ, Zhang T, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res 2009;69:3382–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim JH, Skates SJ, Uede T, et al. Osteopontin as a potential diagnostic biomarker for ovarian cancer. JAMA 2002;287:1671–9. [DOI] [PubMed] [Google Scholar]
- 35.Rodrigues LR, Teixeira JA, Schmitt FL, et al. The role of osteopontin in tumor progression and metastasis in breast cancer. Cancer Epidemiol Biomarkers Prev 2007;16:1087–97. [DOI] [PubMed] [Google Scholar]
- 36.Rangaswami H, Bulbule A, Kundu GC. Osteopontin: role in cell signaling and cancer progression. Trends Cell Biol 2006;16:79–87. [DOI] [PubMed] [Google Scholar]
- 37.Cheng Y, Wen G, Sun Y, et al. Osteopontin Promotes Colorectal Cancer Cell Invasion and the Stem Cell-Like Properties through the PI3K-AKT-GSK/3beta-beta/Catenin Pathway. Med Sci Monit 2019;25:3014–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ng L, Wan T, Chow A, et al. Osteopontin Overexpression Induced Tumor Progression and Chemoresistance to Oxaliplatin through Induction of Stem-Like Properties in Human Colorectal Cancer. Stem Cells Int 2015;2015:247892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu Y, Yang J, Xu D, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut 2019;68:1653–1666. [DOI] [PubMed] [Google Scholar]
- 40.Chen P, Zhao D, Li J, et al. Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 2019;35:868–884 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klement JD, Paschall AV, Redd PS, et al. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J Clin Invest 2018;128:5549–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shurin MR. Osteopontin controls immunosuppression in the tumor microenvironment. J Clin Invest 2018;128:5209–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akcora D, Huynh D, Lightowler S, et al. The CSF-1 receptor fashions the intestinal stem cell niche. Stem Cell Res 2013;10:203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Han G, Wang K, et al. Tumor-derived GM-CSF promotes inflammatory colon carcinogenesis via stimulating epithelial release of VEGF. Cancer Res 2014;74:716–26. [DOI] [PubMed] [Google Scholar]
- 45.Lu R, Markowetz F, Unwin RD, et al. Systems-level dynamic analyses of fate change in murine embryonic stem cells. Nature 2009;462:358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwanhausser B, Busse D, Li N, et al. Global quantification of mammalian gene expression control. Nature 2011;473:337–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.