

Summary
Fecal microbiota transplantation (FMT) from healthy donor to patient is a treatment for microbiome-associated diseases. Although the success of FMT requires donor bacteria to engraft in the patient’s gut, the forces governing engraftment in humans are unknown. Here, we use an ongoing clinical experiment, the treatment of recurrent Clostridium difficile infection, to uncover the rules of engraftment in humans. We built a statistical model that predicts which bacterial species will engraft in a given host, and developed Strain Finder, a method to infer strain genotypes and track them over time. We find that engraftment can be predicted largely from the abundance and phylogeny of bacteria in the donor and the pre-FMT patient. Further, donor strains within a species engraft in an all-or-nothing manner and previously undetected strains frequently colonize patients receiving FMT. We validated these findings for metabolic syndrome, suggesting that the same principles of engraftment extend to other indications.
Graphical Abstract
eTOC
Smillie et al. profile the gut microbiota of recurrent Clostridium difficile patients during fecal microbiota transplantation (FMT) and uncover the principles of microbiota engraftment in humans. They validate their findings across several FMT datasets and in another disease context, metabolic syndrome.
Introduction
Fecal microbiota transplantation (FMT) is an emerging treatment for infectious and autoimmune diseases, in which donor feces are implanted in a patient’s intestinal tract. This treatment cures recurrent Clostridium difficile infection (rCDI) in eighty-five percent of cases (van Nood et al., 2013) and there is some evidence it may be effective for other diseases, including inflammatory bowel disease (Moayyedi et al., 2015; Suskind et al., 2015), metabolic syndrome (Ridaura et al., 2013; Vrieze et al., 2012), and autism (Hsiao et al., 2013). Putative mechanisms of FMT efficacy focus on the trillions of bacteria that inhabit the gastrointestinal tract, the gut microbiota. FMT is thought to restore these bacteria (Shahinas et al., 2012; Youngster et al., 2014), which may then alter host metabolism (Floch, 2015; Trompette et al., 2014), inhibit pathogens (Britton and Young, 2014) and effect changes in host immunity (Furusawa et al., 2013; Ivanov et al., 2009; Round and Mazmanian, 2010).
Precision engineering of the gut microbiota with bacterial isolates in pure culture offers the therapeutic potential of FMT without the risks associated with the use of raw fecal matter (Petrof and Khoruts, 2014). Whether this next generation of microbiome-based therapeutics will effectively replace FMT will depend on whether: (i) the “active ingredients” of FMT that carry out a desired mechanism can be identified, (ii) these strains engraft in a patient’s gut, and (iii) they are sufficiently abundant to produce a clinical response.
While mechanism may be studied using in vitro or animal models of disease (as for traditional small molecule drugs), engraftment and abundance in humans are less well understood. If engraftment is governed by simple rules, such as the law of mass action, then it may be highly deterministic and easy to predict. Alternatively, if engraftment is governed by contextual factors, such as genetics, diet, antibiotics, and the immune system, then it may vary considerably among patients and be difficult to predict. A quantitative model of bacterial engraftment would accelerate drug discovery efforts by pinpointing the bacteria that engraft at high abundance in a given host. However, no such model exists, and despite significant advances in our understanding of FMT (Li et al., 2016), surprisingly few principles of bacterial engraftment in a human host are known.
Direct tests of bacterial engraftment are difficult, as animal models do not capture important aspects of human biology, and experiments in humans present regulatory and ethical challenges. However, there is already an ongoing large-scale experiment of engraftment in humans: the use of FMT to treat recurrent C. difficile infection.
C. difficile is a gram-positive pathogen that causes severe diarrhea and is responsible for 500,000 infections, resulting in 30,000 deaths, per year (Lessa et al., 2015). It is often carried asymptomatically in the gut, where it is normally inhibited by gut commensals. Disruptions to this inhibition, often via broad-spectrum antibiotics, allow C. difficile to proliferate. First-line treatment with antibiotics can cure this infection, but in twenty percent of cases, C. difficile spores persist and reinitiate the cycle of infection. FMT is thought to break this cycle by restoring the protective gut microbiota that inhibit the growth of C. difficile and prevent recurrence of the infection (Aroniadis and Brandt, 2013).
Bacterial engraftment is therefore thought to be responsible for the efficacy of FMT, yet few studies have examined the factors that promote the engraftment of individual strains. Here, we develop a method for strain inference, Strain Finder, and combine it with techniques from machine learning to quantitatively model bacterial engraftment in diverse human hosts. We uncover the principal factors that govern bacterial engraftment after FMT and show that these rules appear to generalize to the treatment of another disease, metabolic syndrome.
Results
Profiling fecal microbiota transplantation with shotgun metagenomics
Prior studies of recurrent C. difficile (Chang et al., 2008; Seekatz et al., 2014; Weingarden et al., 2015; Youngster et al., 2014) have used 16S rRNA sequencing to reveal species-level changes in the gut microbiota after FMT. However, these studies lack sufficient phylogenetic resolution to measure the engraftment of single strains (Thompson et al., 2005). Thus, to study engraftment in this human experiment, we used higher resolution deep shotgun metagenomics sequencing to follow nineteen recurrent C. difficile patients after FMT (Fig. 1). Feces from one of four donors were administered to each patient. Stool samples and clinical metadata were collected from the donor and the patient before FMT, and in follow-up visits ranging from one day to four months after FMT. In total, we sequenced 79 stool samples at a mean depth of 1.3 × 1010 bp, yielding high resolution snapshots of each patient’s gut microbiota before and after FMT.
Figure 1: Study design for the use of fecal microbiota transplantation to treat recurrent Clostridium difficile infection.
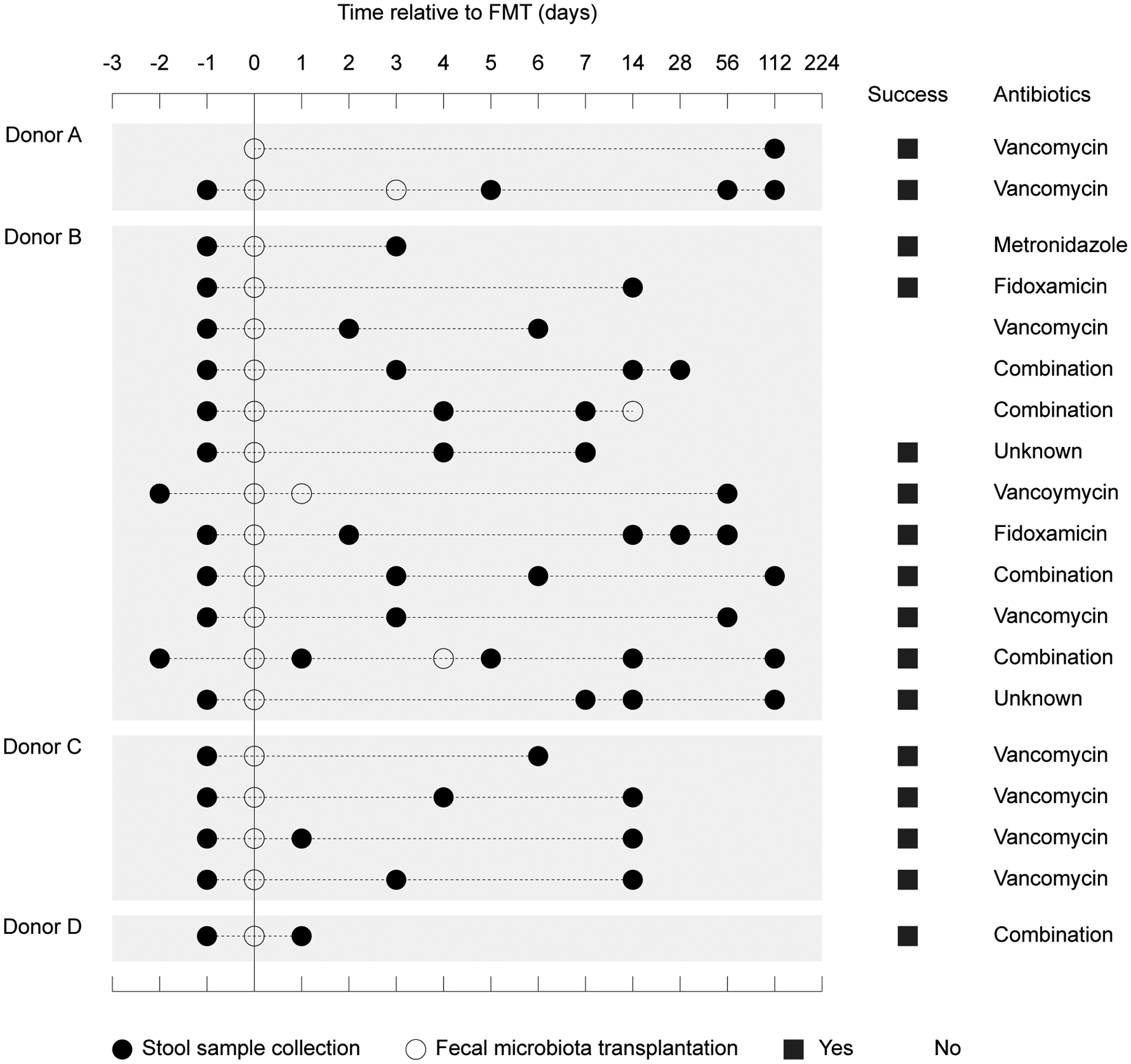
Nineteen patients with recurrent Clostridium difficile infection were treated with feces from one of four donors. Stool samples were collected and FMTs were performed at the indicated time points. For each patient, we show the class of antibiotics they were most recently treated with and the success of the overall treatment.
To determine the abundances of bacterial taxa in these samples, we mapped each metagenome to a set of 649 non-redundant reference genomes from the Human Microbiome Project (Consortium, 2012). Because bacterial genes vary in copy number and horizontal gene transfer may obscure evolutionary relationships, we focused on 31 single-copy phylogenetic markers from the AMPHORA database (Wu and Eisen, 2008). We estimated the abundances of bacterial taxa as their mean depth of coverage, normalized to the total sequencing depth of each sample. For simplicity, we refer to these bacterial taxa as mg-OTUs (“metagenomic OTUs”), following the convention used in 16S rRNA studies. These mg-OTUs roughly correspond to bacterial species (see Methods).
We identified 306 mg-OTUs in this dataset (Fig. S1A and Table S1). The number of mg-OTUs per sample was weakly, but significantly associated with sequencing depth (Kendall’s tau = 0.19, p-value = 0.02). The mg-OTUs comprise all major taxonomic groups in the human gut, including the Bacteroidetes, Firmicutes, Actinobacteria, and Proteobacteria, along with less prevalent phyla such as the Fusobacteria and Verrucomicrobia. C. difficile was included in our set of reference genomes, but was only detected at low abundances: 5 of 79 samples had reads covering at least 20% of the C. difficile reference, and no samples had reads covering more than 50% of the reference. One explanation for the low abundance of C. difficile in these samples is that patients had received extensive antibiotics leading up to sample collection.
Prior to FMT, the patient’s gut was marked by reduced species diversity and imbalances in many gut taxa (Fig. S1B and Fig. S1G). Whereas pre-FMT patients clustered into two community types, dominated by Enterobacteriales and Lactobacillales, samples from donors and post-FMT patients were dominated by Bacteroidales and Clostridiales (Fig. S1C). To visualize the effects of FMT, we applied principal coordinates analysis to the weighted UniFrac distances among all samples (Fig. S1E). Principal Coordinate 1 (PC1) was significantly associated with dysbiosis, as pre-FMT patients and donors formed distinct clusters along this axis (Wilcoxon test, p-value = 0.002). FMT led to increased PC1 usage in nearly all patients (Wilcoxon signed rank test, p-value = 0.001), which became more similar to the donor (Fig. S1F, Wilcoxon test, p-value = 0.01). However, because patients received antibiotics before FMT, this analysis is unable to separate the effects of FMT from bacterial growth following the cessation of antibiotics.
A machine learning model predicts bacterial engraftment
After FMT, the gut microbiota of the patient is distinct from that of the donor and of itself before FMT. On average, only 35% of the mg-OTUs in the donor (N= 436, SD = 27%) and 42% of the mg-OTUs in the pre-FMT patient (N = 287, SD = 31%) were detected on the first follow-up visit after FMT. The post-FMT patient also acquires new bacteria, as 39% of the mg-OTUs in these samples (N = 295, SD = 28%) were undetected prior to FMT. These bacteria may be acquired from the environment or resurrected from low abundances (below the detection limit) in the donor or the patient. To examine the relationships among samples, we clustered them according to their mg-OTU abundances (Fig. 2). Post-FMT patient samples do not cluster with the correct pre-FMT patient or donor samples (Figs. 2A and 2B), nor with weighted averages of these samples (Fig. S2). The gut microbiota of the patient is therefore not a simple combination of the donor and the pre-FMT communities, but instead, is a complex mixture of bacteria from the donor, the patient, and the environment.
Figure 2: A machine learning model predicts the gut microbiota of the post-FMT patient.
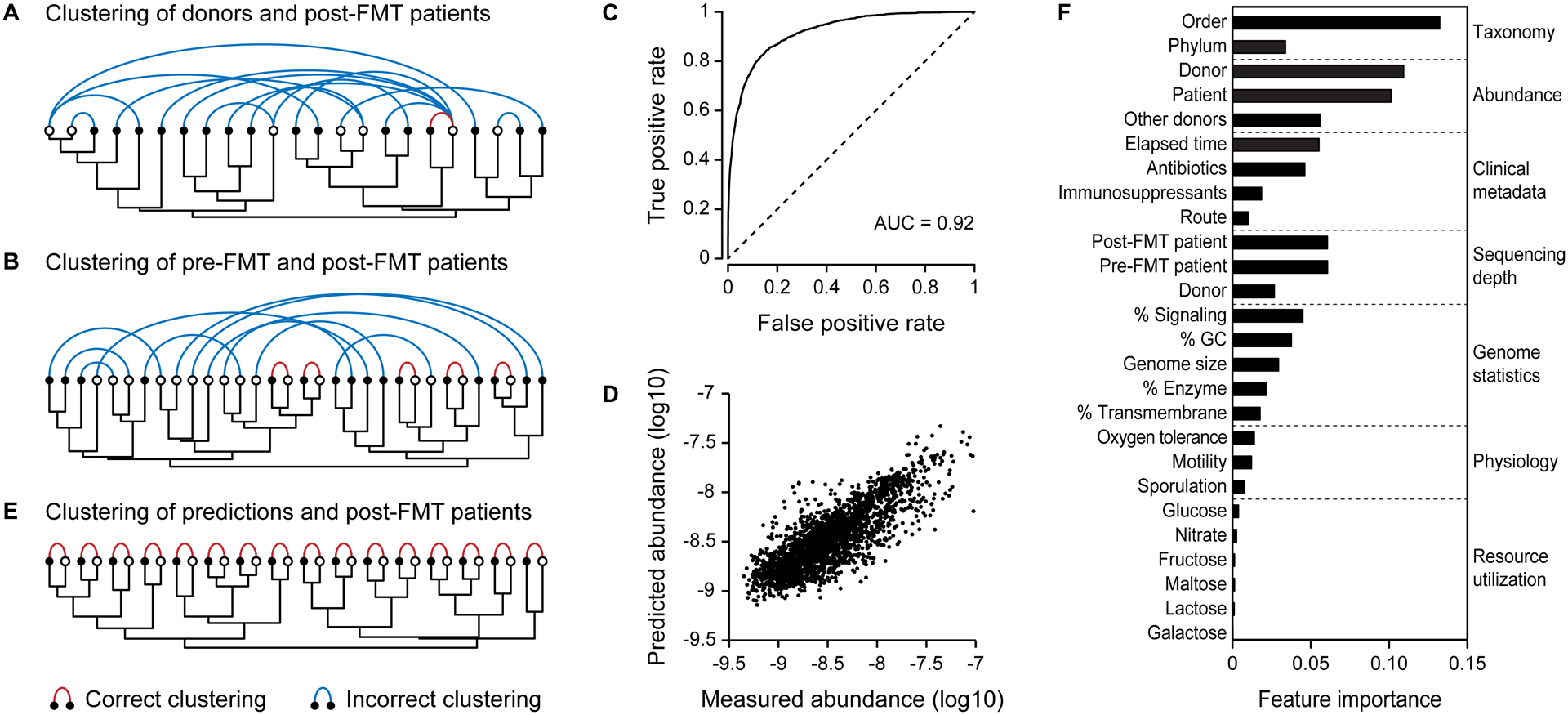
Samples were clustered according to their species compositions (dendrograms). Samples from the same patient are connected with colored arcs. (A) Donor samples (white dots) do not cluster with the corresponding post-FMT patient samples (black dots). (B) Pre-FMT patient samples (white dots) do not cluster with the corresponding post-FMT samples (black dots). (C) The receiver operating characteristic (ROC) curve for the model of mg-OTU presence. (D) Predicted abundances were correlated to the measured abundances in the post-FMT samples. (E) Samples from post-FMT patients (black dots) cluster perfectly with their predicted values (white dots). (F) The mean relative importance of each feature across both models. Because sequencing depth was only used in the model of OTU presence, only this feature importance is reported.
Although FMT outcomes cannot be inferred from simple combinations of the donor and patient gut microbiota, it is possible that after accounting for all clinical and metagenomics data, these post-FMT communities may be predictable. To test this hypothesis, we used data from the donor and the pre-FMT patient to build a machine learning model of the presence and the abundance of mg-OTUs in the patient after FMT. The inputs to this model were the clinical metadata, along with the abundance, phylogeny, and several genomic features of each mg-OTU (Table S2). The model consists of two steps: first, it uses Random Forest classification to predict which mg-OTUs are present in all samples, then it uses Random Forest regression to predict their abundances. In the second step, we remove the sequencing depth of the post-FMT sample from the model because this feature was used to normalize mg-OTU abundances.
We used Random Forest because it is nonlinear, it accepts categorical and continuous predictors, and it is robust to overfitting. Random Forest internally estimates the test error using “out of bag” samples that are hidden from the training algorithm, eliminating the need for cross-validation or a separate test set. The statistics reported in this work therefore reflect the model’s performance on test data (i.e. test accuracy), rather than training data (i.e. training accuracy).
Both the incidence and abundance of mg-OTUs in the post-FMT patient were predictable (Figs. 2C and 2D, AUC = 0.92 and r2 = 0.40 for all time points). These predictions were not only accurate, but also specific, as they clustered perfectly with the gut microbiota of the post-FMT patients (Fig. 2E). The model’s accuracy, despite not including major determinants of the gut microbiota, such as diet, bacterial species interactions, host genetics, and the immune system, suggests that knowledge of these factors is not strictly necessary to predict the post-FMT gut community. However, it is possible that the addition of these features would yield even more accurate predictions of bacterial engraftment after FMT.
We next assessed the model’s accuracy when the mg-OTU is missing from the donor and the pre-FMT patient. As expected, in 91% of such cases the mg-OTU is also absent from the post-FMT community, and the model predicts this result 88% of the time. However, when we focus on the 810 mg-OTUs that were unseen before the FMT and later found in the post-FMT patient, the model predicts that the mg-OTU is present in the post-FMT community in 38% of such cases, representing a 3-fold increase in the probability of engraftment. Therefore, even when there is no direct evidence that an mg-OTU is present in the donor or the patient, the model can successfully predict its colonization of the post-FMT patient.
In addition to measuring the model’s performance on test data, we used permutation tests and extensive subsampling to further confirm that our model is not overfitting. As expected, the model is no longer accurate when the abundances of mg-OTUs are shuffled among the post-FMT patients (Fig. S3A). We confirmed that the model can predict long-term outcomes by removing all samples collected within fourteen days of FMT (Fig. S3B). The model is also accurate when we subsample one mg-OTU from each genus and average the results across all such models, indicating that redundancy in our set of reference genomes does not significantly affect our results (Fig. S3C). To minimize the potential for overfitting, we built a reduced model using only mg-OTU abundances, sequencing depth, the mg-OTU phylum, and the amount of elapsed time since the FMT. Despite its simplicity, this reduced model accurately predicts the presence and abundance of mg-OTUs in the post-FMT patient (Fig. S3D).
Resolving strain-level dynamics with Strain Finder
Until now, we have used reference genome alignments to study engraftment. However, these species-level data cannot determine whether a single strain is shared by the donor and the patient, preventing direct measurement of engraftment. Shotgun metagenomic sequencing has the potential to generate higher resolution, strain-level data because it captures the full bacterial genome. However, this approach yields short reads that are difficult to assemble into strain genotypes with existing methods because two strains may differ by relatively few SNPs.
Prior attempts to circumvent this problem have used copy number variation (Greenblum et al., 2015) and single nucleotide variants (Li et al., 2016; Schloissnig et al., 2013) as proxies of strains. However, these methods cannot recover the strain genotypes that are present in a sample because they do not solve the problem of metagenome assembly, which requires linking SNPs into larger strain haplotypes. One solution to this problem is to assemble strain genotypes using the SNP frequencies themselves. Here, the intuition is that SNPs derived from the same strain will be present at roughly equivalent frequencies across samples. We use this intuition to develop a maximum-likelihood method, Strain Finder, which infers the genotypes and frequencies of strains in complex metagenomics samples (see Methods).
We validated this method in silico by simulating 6,000 alignments with varying numbers of strains (N = 2 – 32), samples (N = 2 – 64), SNPs (N = 4 – 1024), and sequencing depths (25 – 1000X). In total, these alignments encompassed 74,400 unique strains distributed across 126,000 samples. To assess the accuracy of strain inference, we searched for a distance metric that could account for the similarity between the estimated strains and the true strains, both in terms of their estimated genotypes and frequencies. The weighted UniFrac distance (Lozupone et al., 2011) measures the number of SNPs shared by our predictions and the true strains, weighted by their frequencies along every branch of the strain phylogeny. Because this metric accounts for the phylogenetic relationships among strains, it elegantly handles situations in which two closely related strains are merged into a single strain profile, penalizing only the algorithm’s failure to identify the SNPs that discriminate between those two strains.
With sufficient data, Strain Finder can accurately predict over 32 strains in as few as 16 samples (Fig. S4). The weighted UniFrac distances between our predictions and the true strains were less than 0.10 for many parameters, indicating that Strain Finder captures 90% of the frequency-weighted SNPs, and that the total deviance of the predicted frequencies from the true frequencies is less than 10%. We compared these predictions to two null models, in which the strain frequencies are sampled from a Dirichlet multinomial distribution and the strain genotypes are sampled from either a discrete uniform distribution (Null Model 1) or from the alignment data itself (Null Model 2). Strain Finder outperforms both null model (Fig. S4).
We also tested Strain Finder against another strain inference method, ConStrains, which is not based on a complete statistical model. Following the methods in (Luo et al., 2015), we used 16 Escherichia coli genomes to simulate 16 alignments of 8 samples each (see Methods). Across these alignments, we varied the number of strains (N = 4 – 16) and the depth of coverage (25 – 1,000X). We ran Strain Finder with default parameters and ConStrains in two modes: separately for each sample (Constrains 1) and on all samples combined (Constrains 2). While Strain Finder and ConStrains were both more accurate than Null Model 1, only Strain Finder outperformed Null Model 2 (Fig. 4, p-value < 1e-10 for all comparisons, Wilcoxon test on weighted UniFrac distances to true strains). Strain Finder was more accurate than ConStrains across all simulated datasets (Fig. 4, p-value < 1e-10 for all comparisons, Wilcoxon test on weighted UniFrac distances to true strains).
Figure 4: Strain Finder outperforms ConStrains on simulated metagenomic alignments generated from a set of Escherichia coli genomes.
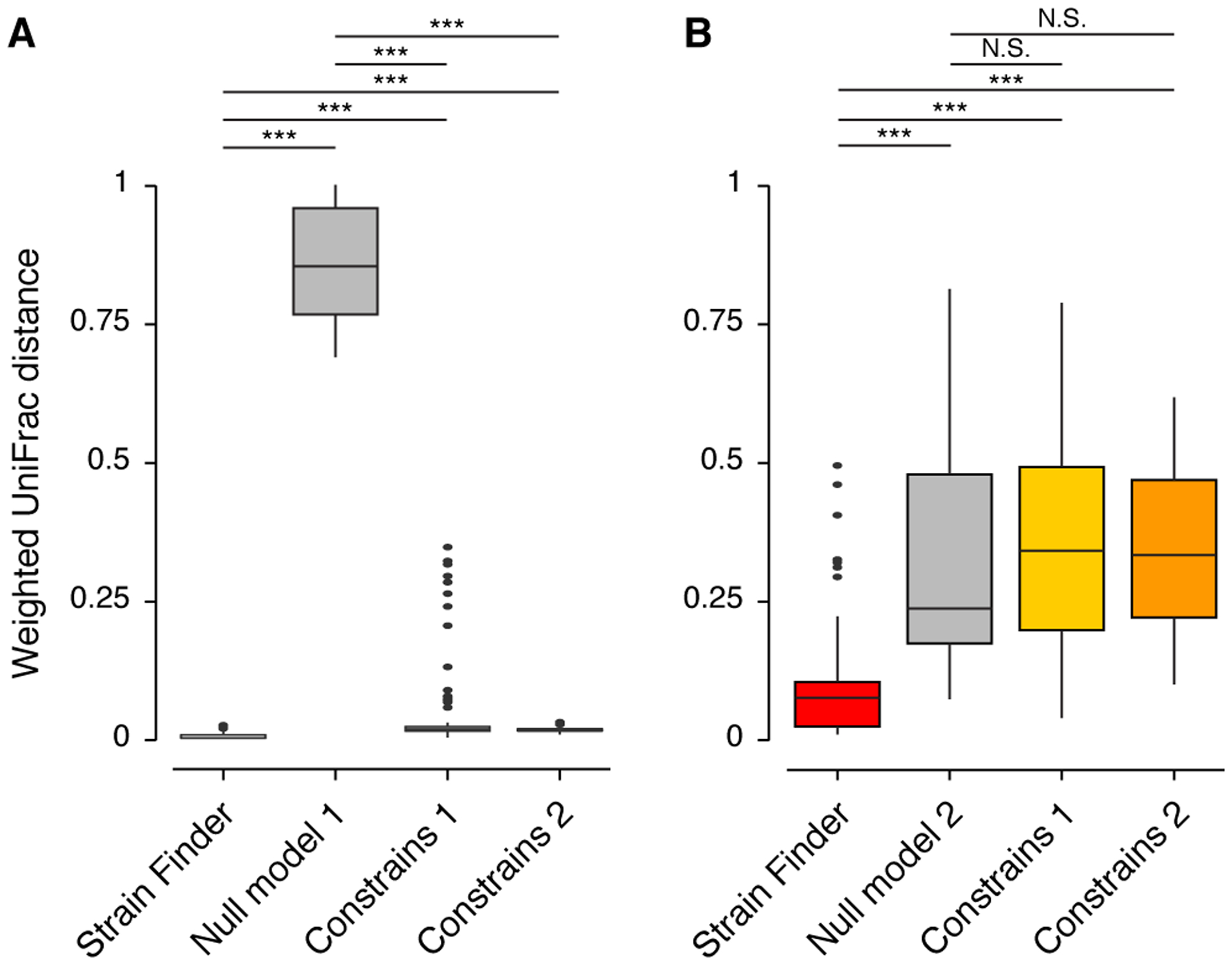
We simulated 16 metagenomic alignments of 8 samples each, with varying numbers of strains (N = 4, 8, 12, and 16) and depths of coverage (N = 25, 100, 500, and 1,000X). ConStrains was run on each sample separately (CS Model 1) and on all samples combined (CS Model 2). (A) The weighted UniFrac distances from the true strain profiles to the predictions of Strain Finder, ConStrains, and Null Model 1. (B) The weighted UniFrac distances from the true strain profiles to the predictions of Strain Finder, both ConStrains models, and Null Model 2. Asterisks denote significant comparisons (*** = p-value < 1e-10) as determined by a Wilcox test. N.S. denotes non-significant comparisons.
We also find empirical support for Strain Finder’s accuracy on real-world metagenomics data: with no a priori knowledge of the underlying structure of the data, Strain Finder recovers the correct donor-patient pairs using only the similarity of their strain profiles (see below).
To systematically determine the impacts of FMT on bacterial engraftment in the human gut, we used Strain Finder to infer the strain genotypes and frequencies of 306 bacterial species across the 79 metagenomics samples in our dataset. In total, we identified 1,091 bacterial strains, with each sample containing an average of 130 strains with a relative abundance of at least one percent of their respective species (Fig. S1D). Within a species, strains differed by a median of 182 SNPs across the thirty-one AMPHORA genes (IQR = 9 – 321), corresponding to a sequence identity of roughly 99.5% at these loci. For comparison, a sequence identity of 97% at the more highly conserved 16S rRNA gene is widely used to delineate bacterial species. Analysis of this vast dataset allows us to track the transfer of hundreds of bacterial strains from the donor to the patient during FMT, and to follow the persistence of these strains for months after treatment.
The engraftment and persistence of bacterial strains
At the strain level, we observe several patterns of bacterial dynamics after FMT (Fig. 5A). When an mg-OTU is present in the patient, but absent from the donor, the composition of strains tends to remain stable after FMT (Fig. 5A, Klebsiella pneumoniae). Conversely, when an mg-OTU is present in the donor, but absent from the patient, the patient tends to adopt the strain composition of the donor (Fig. 5A, Faecalibacterium prausnitzii). Competition between strains in the donor and the patient can lead to a range of outcomes: the patient strains may resist invasion by donor strains, the donor strains may outcompete the patient strains, the donor strains and patient strains may coexist, or the patient may acquire new strains (Fig. 5A, E. coli and Dorea longicatena).
Figure 5: Complete sets of donor strains and previously undetected strains engraft in the patient after FMT.
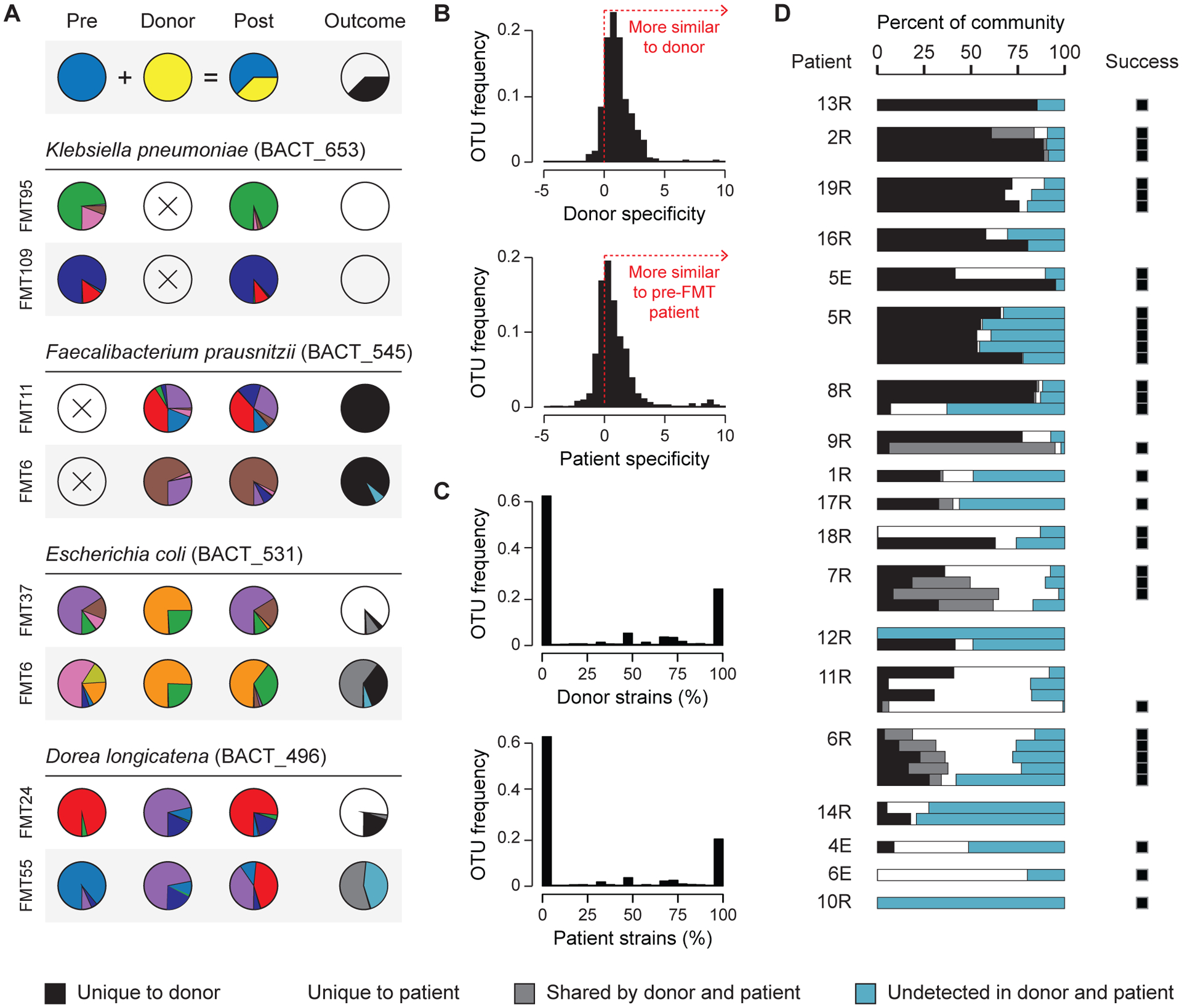
(A) The strain compositions of mg-OTUs in the pre-FMT patient, the donor, and the post-FMT patient. The frequencies of strains that are unique to the donor, unique to the patient, shared by the donor and the patient, and undetected are shown on the right. (B) Strain specificity of mg-OTUs in the donor (N = 3,090) and the pre-FMT patient (N = 2,024). Strain specificity is measured as the log-ratio of (i) the distance from the donor (or pre-FMT patient) to the post-FMT patient, and (ii) the distance from an unrelated donor (or unrelated pre-FMT patient) to the post-FMT patient. (C) Across mg-OTUs, the percentages of strains from the donor (N = 3,090) and the pre-FMT patient (N = 2,024) that were transferred to the post-FMT patient. (D) The fraction of each community that is unique to the donor, unique to the patient, present in the donor and the patient, and previously undetected. Treatment success is provided on the right.
Bacterial strains from the donor were directly transmitted to the patient’s gut during FMT. In total, we identified 439 unique strains that were found in the donor and the post-FMT patient. The presence of strains in the donor was predictive of their presence in the post-FMT patient (Fisher test, p-value < 1e-10). While these findings are consistent with direct transfer, an alternative hypothesis is that unrelated subjects harbor similar strains, which may reflect a healthy or unperturbed state. We therefore examined strain specificity, finding that in terms of strain composition, the post-FMT patient more closely resembles its donor than other donors (Fig. 5B, Wilcoxon test on 455 mg-OTUs, p-value < 1e-10). We also confirmed that the post-FMT patient is more similar to its pre-FMT self than to other pre-FMT patients (Fig. 5B, Wilcoxon test on 232 mg-OTUs, p-value < 1e-10). Shared strains in the donor and the patient are therefore best explained by direct transmission, rather than incidental similarity.
After FMT, strains from the donor colonize the patient’s gut for months or even longer. To study persistence, we focused on donor-derived strains, which we define as being unique to the donor before FMT and present in the patient after FMT. We expected the number of donor-derived strains to decline over time, as they are replaced by strains from the patient and the environment. In agreement with this hypothesis, when we focused on patients with multiple follow-up visits, we identified 125 donor-derived strains on the first visit after FMT (mean = 10, SD = 15). This number declined to 82 strains by the final follow-up visit (mean = 7, SD = 6), including 58 strains that were detected more than one month after FMT. Donor-derived strains may therefore persist in the patient’s gut for months after FMT, although studies with larger sample sizes are required to more rigorously assess their long-term dynamics.
We searched for strains that have high engraftment rates relative to their sister taxa, as such strains may represent promising candidates for microbial therapeutics. However, in contrast to bacterial species, closely related strains did not vary significantly in their overall levels of engraftment (Kruskal-Wallis test, adjusted p-value > 0.05). While this negative result may be due to low statistical power, simulations indicate that our test is powered to detect mean differences in engraftment rates exceeding 15%. Therefore, differences in the engraftment of individual strains are expected to be small, relative to the vast interspecific differences in engraftment that were previously observed.
Although only a fraction of bacterial species engrafts in the patient’s gut, bacterial strains engraft in an all-or-nothing manner, in which no strains or complete sets of strains colonize the patient (Fig. 5C). The engraftment of partial sets of strains is infrequent, comprising 15% of all transferred donor mg-OTUs. This surprising observation reveals a strong coupling among closely related strains: rather than transferring independently of one another, strains within a species transfer together as a cohesive unit. This observation suggests that species traits are the major determinants of bacterial transmission, while functional differences between stains are relatively less important for patient colonization after FMT.
Once donor strains have been transferred to the patient, they adopt the same composition in the patient that they held in the donor. The strain compositions of mg-OTUs in the donor were nearly perfectly correlated to those in the post-FMT patient (median cosine similarity of 455 mg-OTUs = 0.90). This result is significant when compared to a control group of unrelated donors (median cosine similarity of 472 mg-OTUs = 0.14; Wilcoxon test, p-value < 1e-10). The strain compositions of mg-OTUs in the patients were also strongly correlated before and after FMT (median cosine similarity of 232 mg-OTUs = 0.93). Therefore, while the abundances of bacterial species in the patient are shaped by the host, the abundances of bacterial strains are controlled by their input levels (i.e. dose dependence).
Strain signatures reveal engraftment rates following FMT
Because the donor and the pre-FMT patient have distinct strain signatures that are stable through time, we were able to use these signatures to infer the origins of the post-FMT gut microbiota with high confidence. We used these estimates to calculate the probability of engraftment for each bacterial species in the donor’s gut microbiota (Table S3). Consistent with our previous findings, the probability of engraftment of a bacterial species was strongly correlated to its mean abundance in the donor gut microbiota (Kendall’s tau = 0.50, p-value < 1e-10). While Prevotella copri and B. vulgatus had some of the highest engraftment rates, the closely related P. tannerae and B. pectinophilus had some of the lowest engraftment rates, underscoring the importance of understanding bacterial engraftment for engineering the human gut microbiota.
We used the inferred origin of each bacterial species to determine the contributions of the donor, the patient, and the environment, to the recovery of the patient’s gut microbiota after FMT (Fig. 5D). These contributions vary substantially among patients, with donor-derived strains comprising as little as one percent, or as much as eighty percent, of the total community. Strains that were previously undetected in the donor or the patient, which were either below the detection limit or derived from the environment, contribute substantially to the gut microbiota after FMT. FMT may therefore facilitate the expansion of lowly abundant strains, or the colonization of new strains from environmental reservoirs (i.e. ingested food). However, further work with appropriate controls is required to rigorously determine the mechanisms underlying the expansion of these previously undetected strains in the post-FMT patient.
FMT success, defined as the clinical resolution of diarrhea without relapse after eight weeks, was not associated with the fraction of cells in the community that engrafted (Mann-Whitney test, p-value > 0.05). This raises the possibility that the engraftment of individual strains, rather than the total community, may underlie the efficacy of FMT in treating recurrent C. difficile infection. Consistent with this hypothesis, a mixture of six bacterial strains can treat recurrent C. difficile infection in mice (Lawley et al., 2012). There is significant interest in identifying an analogous strain mixture for humans, but the discovery of strain mixtures is time-consuming and manufacturing a diverse strain mixture, in order to maximize the probability of engraftment, may be prohibitively expensive. An understanding of engraftment may accelerate drug discovery by pinpointing which bacteria will engraft in a given host.
Discussion
Determinants of bacterial engraftment in a human host
The most important factors in our model were the bacterial abundances, the bacterial taxonomy and the amount of elapsed time since the FMT (Fig. 2F). The importance of each feature was estimated by removing it from the model and measuring the resultant increase in error. Sequencing depth, while biologically uninformative, was important because it determines the bacterial detection limit, which is leveraged by our model. No measured clinical factors, such as the type of antibiotics used, the route of FMT administration, or whether the patient had taken immunosuppressants had a significant impact on our predictions.
We found evidence for the direct transmission of bacterial species from the donor to the patient: the presence of mg-OTUs in the donor was highly predictive of their presence in the post-FMT patient (Fisher test, p-value < 1e-10). In addition to acquiring bacteria from the donor, the patient also retains species from before FMT. The presence of mg-OTUs in the patient before FMT was highly predictive of their presence after FMT (Fisher test, p-value < 1e-10). Efforts to engineer the gut microbiota with FMT should therefore account for the effects of bacteria from both the donor and the patient before FMT.
We explored two mechanisms that may shape bacterial abundances after FMT: dose dependence and host control. Under a model of dose dependence, the abundances in the patient after FMT are determined by their input levels in the donor and the patient. In contrast, under a model of host control, these abundances are determined by selective forces in the patient’s gut. mg-OTU abundances in the donor were strongly correlated to mg-OTU abundances in the patient after FMT (Fig. 3A, N = 346, Kendall’s tau = 0.28, p-value < 1e-10) and we observed an even stronger correlation between mg-OTU abundances in the patient before and after FMT (Fig. 3B, N = 175, Kendall’s tau = 0.40, p-value < 1e-10). While these findings are consistent with a model of dose dependence, an alternative hypothesis is that unrelated hosts select for similar levels of each mg-OTU and that these results are therefore explained by host control.
Figure 3: The abundance and phylogeny of bacterial species are the strongest determinants of bacterial engraftment.
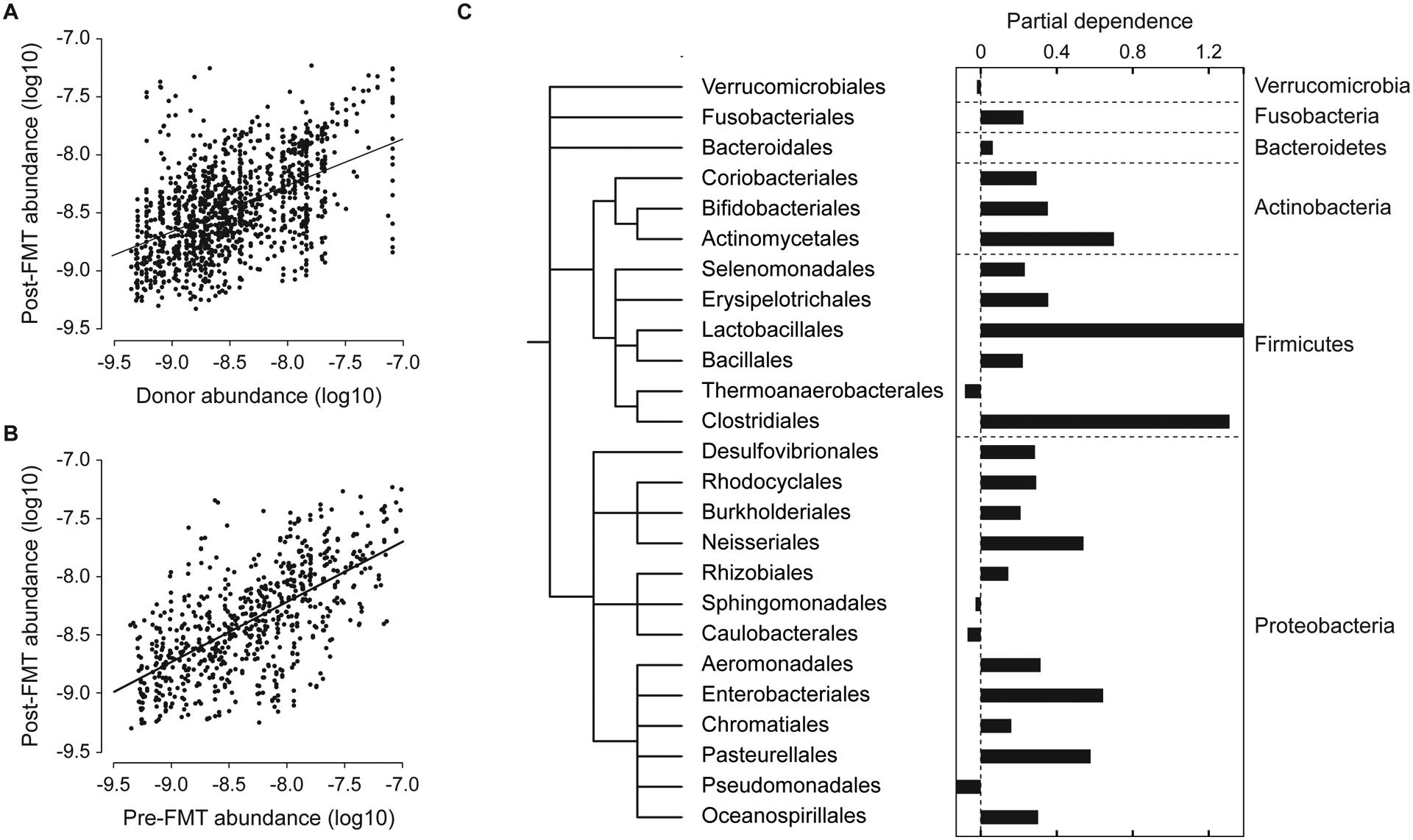
(A) The abundances of mg-OTUs in the donor are strongly correlated to their abundances in the post-FMT patient. (B) The abundances of mg-OTUs in the patient are strongly correlated before and after FMT. (C) The partial dependence of engraftment on each taxonomic order, reflecting the orders’ marginal effects on the probability of engraftment in the reduced model. Orders are arranged on the bacterial taxonomy, with phylum labels on the right.
We hypothesized that by focusing on mg-OTUs that were present in both the donor and the patient before FMT, it would be possible to measure each sample’s impacts on mg-OTU abundances in the post-FMT patient. Using partial regression, we controlled for the correlation in abundances between the pre-FMT and post-FMT patient, and found that donor abundances were no longer significant in the model (F-test, N = 76, p-value > 0.05). When we performed the reciprocal test, controlling for the correlation between the donor and the post-FMT patient, the abundances in the patient remained significant (F-test, N = 76, p-value = 1e-4), suggesting that host control, rather than dose dependence, determines bacterial abundances after FMT.
The next most important factor shaping engraftment was the bacterial phylogeny. We used partial dependence plots to measure the marginal impact of each bacterial order on the probability of engraftment, while controlling for abundance, elapsed time, and sequencing depth in our reduced model (Fig. 3C). Clades with high marginal effects on engraftment may represent bacteria that survive the FMT and engraft with high efficiency in the patient’s gut. Conversely, clades with low marginal effects on engraftment may represent bacteria that have low rates of engraftment in the patient’s gut.
We attempted to identify the traits that underlie this phylogenetic signal using predictions of bacterial physiology from genome sequences (Markowitz et al., 2014). Although the FMTs were performed aerobically, neither oxygen tolerance nor the ability to sporulate significantly impacted abundances after FMT. In addition to these traits, other factors may contribute to this phylogenetic signal, including the ability to degrade host mucins and dietary carbohydrates (Koropatkin et al., 2012), the ability to associate with colonic crypts (Lee et al., 2013), and the ability to evade the immune system (Cullen et al., 2015). However, these traits are difficult to predict from genome sequences or are present in too few organisms to leave statistical signals.
Validating our model of bacterial engraftment across multiple FMT trials
While metagenomic sequencing provides high resolution for the detection of individual strains, its high cost makes it difficult to study FMT trials with large numbers of donors, patients, and mg-OTUs. To confirm that our model of engraftment generalizes to larger datasets, we therefore performed 16S rRNA sequencing on the 79 samples in this study (Youngster et al., 2014), along with 83 samples from another study of FMT for the treatment of recurrent C. difficile infection. We combined these data with the sequence data from three prior studies of FMT (Seekatz et al., 2014; Song et al., 2013; Staley et al., 2016), yielding a final dataset comprising 375 samples from 93 patients and 69 donors (Table S4). We mapped the 16S rRNA sequences from these studies onto the Greengenes OTUs clustered at 97% sequence identity, yielding OTUs that roughly correspond to bacterial species.
For each of the five FMT trials, we separately trained a model of bacterial engraftment, predicting the post-FMT gut microbiota using only the OTU abundances in the donor and the pre-FMT patient, the phylum and order of each OTU, the amount of elapsed time since the FMT, and the sequencing depth of each sample (Table S2). No clinical metadata were used in these models because they were not consistently available across studies. Because these datasets differ with respect to several factors that are known to induce strong batch effects (e.g. DNA extraction and amplification, the targeted region of the 16S rRNA, and DNA sequencing technology), we were unable to train a model on one dataset and test it against the remaining datasets. However, when we tested these models on out-of-bag test data from the same FMT trial, they accurately predicted the composition of the post-FMT gut microbiota (Figs. 6A and 6B, AUC = 0.84, r2 = 0.36), confirming that our modeling approach can be extended to larger FMT trials. Although they were independently trained on separate datasets, these models discovered similar rules of engraftment: the relative effects of bacterial orders on the predicted levels of engraftment were correlated across all models (Fig. 6C, mean Kendall’s tau = 0.62, p-value < 1e-2 for all tests).
Figure 6: Engraftment modeling is accurate in a meta-analysis of five FMT trials for the treatment of recurrent C. difficile.
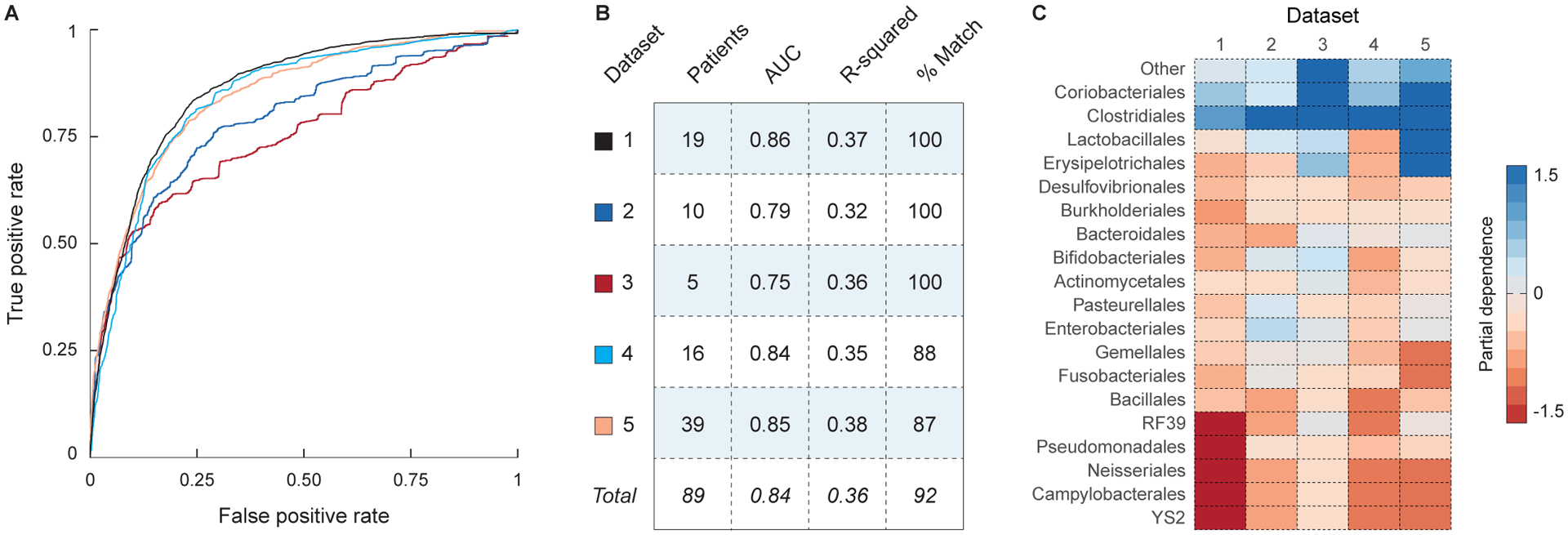
Models of bacterial engraftment were trained on the 16S rRNA sequence data from five FMT trials for the treatment of recurrent C. difficile infection. (A) ROC curves for the predictions of OTU presence in each of the five FMT datasets (see legend in panel B). (B) Statistics describing the performance of each model, including the number of patients in the dataset, the AUC (for predictions of OTU presence), the r-squared (for predictions of OTU abundance), and the percent of predictions that correctly cluster with their target samples (see Methods). (C) Heatmap showing the partial dependence of the models of OTU presence on each bacterial order. High values indicate that the taxon has a favorable impact on engraftment.
Extending our model of engraftment to other diseases
To assess whether our findings generalize beyond recurrent C. difficile infection, we analyzed a separate metagenomics dataset from a study of FMT for the treatment of metabolic syndrome (Li et al., 2016). This study, comprising five patients treated with allogeneic stool samples and five controls treated with autologous stool samples, differs from our study of recurrent C. difficile infection in several ways. While recurrent C. difficile is an infectious disease that causes severe gut dysbiosis and can be effectively treated with FMT, metabolic syndrome is a multifactorial disorder that is weakly associated with the gut microbiota for which the efficacy of FMT is unknown. Moreover, the treatment of recurrent C. difficile patients with antibiotics may free up niche space in the gut, facilitating the engraftment of donor bacteria. In contrast, the patients with metabolic syndrome did not receive antibiotics prior to FMT, potentially making them less amenable to colonization by exogenous bacteria.
Despite the considerable differences between these studies, the gut microbiota of patients with metabolic syndrome were predictable after FMT, both in terms of mg-OTU presence (Fig. S5, AUC = 0.82) and mg-OTU abundance (Fig. S5, Kendall’s tau = 0.39, p-value < 1e-10). Here, we conservatively used our reduced model of bacterial engraftment, which only includes the pre-FMT abundances, the mg-OTU phylum, sequencing depth, and elapsed time. The post-FMT samples did not cluster with their corresponding samples from the donor or the pre-FMT patient, but clustered perfectly with our predictions (Fig. S5). Our approach therefore extends beyond the treatment of recurrent C. difficile infection to the treatment of metabolic syndrome.
To determine whether the same underlying principles drive both models of engraftment, we estimated the importance of each feature in our model. Consistent with our previous findings for recurrent C. difficile infection, the abundances and phylum of the mg-OTU were among the most important features for predicting bacterial engraftment (Fig. S5). To elucidate the effects of the bacterial phylogeny on engraftment for metabolic syndrome, we augmented our reduced model with the taxonomic order of each mg-OTU and calculated the marginal effects of each order on our predictions. The marginal effects were highly correlated to those estimated for recurrent C. difficile infection (Fig. 7, Kendall’s tau = 0.50, p-value < 1e-10). Together, these findings suggest that the principles of engraftment we discovered for recurrent C. difficile infection may generalize to other disease indications, including metabolic syndrome.
Figure 7: Shared phylogenetic principles drive models of engraftment for recurrent C. difficile infection and metabolic syndrome.
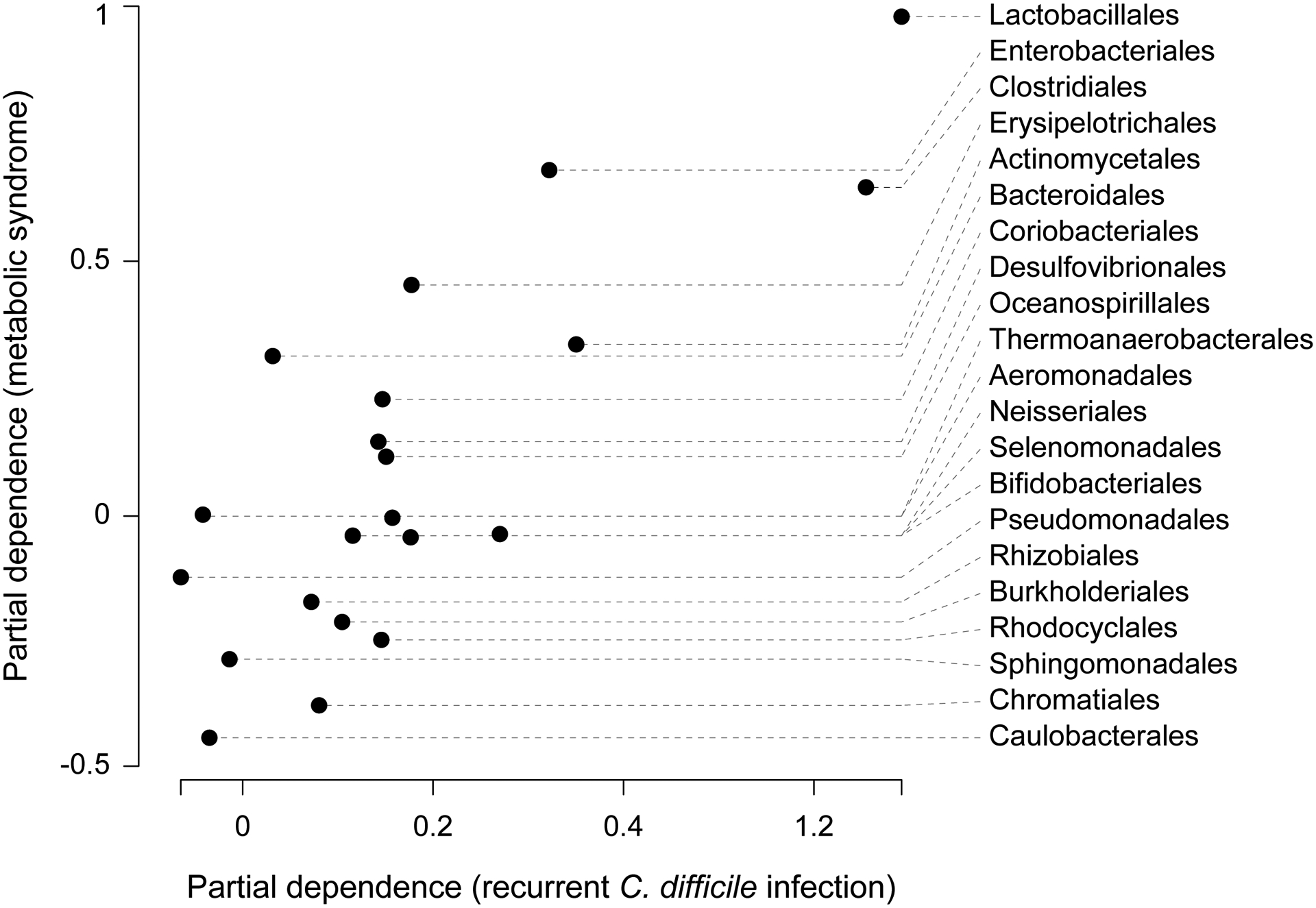
Partial dependence reflects the impact of each bacterial order on the predicted frequency of bacterial engraftment, with high values indicating that a taxon has a favorable impact on engraftment. Partial dependence values estimated for recurrent C. difficile infection and metabolic syndrome are strongly correlated (Kendall’s tau = 0.50, p-value < 1e-10). Prevalent gut commensals, such as Lactobacillales and Clostridiales, had consistently positive impacts on predicted levels of engraftment, while bacteria that are rarely found in the gut, such as Caulobacterales and Chromatiales, had consistently negative effects.
Conclusions
In addition to recurrent C. difficile infection (Khanna et al., 2016), significant efforts are underway to develop bacterial therapeutics for several other diseases. Recent work shows that species belonging to Bacteroides and Bifidobacterium are important to eliciting T-cell responses in mice with checkpoint inhibitors (Sivan et al., 2015; Vétizou et al., 2015). However, these experiments use germ-free mice in controlled environments, where the gut microbiota is relatively easy to manipulate. The success of this treatment in humans will depend on whether these bacteria engraft in hosts with previously established gut microbiota. In another study, butyrate-producing bacteria were found to promote the differentiation of colonic Tregs, leading to the attenuation of colitis in mice (Atarashi et al., 2013; Furusawa et al., 2013). In humans, it will be necessary to identify butyrate producers that not only engraft in the gut, but also reach sufficient abundances to impact levels of colonic butyrate. In both examples, the engraftment of bacteria in a human host is needed to design therapies to engineer the human gut microbiota.
Because our model of engraftment was developed for recurrent C. difficile infection, additional work is needed to develop and test models for other diseases. Recurrent C. difficile may be particularly amenable to modeling for several reasons: it is treated with antibiotics, the disease mechanism is associated with the depletion of gut bacteria, and its resolution involves a transition from a dysbiotic state to a healthy state. Nevertheless, the modeling framework we provide may be extended to other studies, including the 145 registered clinical trials investigating the use of FMT to treat indications including inflammatory bowel disease, metabolic syndrome, and cancer (U.S. National Institutes of Health). Our model of engraftment for the treatment of metabolic syndrome suggests that this is not only possible, but that the same principles of engraftment may generalize to these other diseases. By elucidating the principles of bacterial engraftment in the human gut, such work will advance our understanding of FMT and accelerate the development of bacterial therapeutics that target these diseases.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eric Alm (ejalm@mit.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study cohort:
This study was reviewed and approved by the Partners Human Research Committee (IRB) as well as by the Food and Drug Administration (FDA) (Investigational New Drug application number 15199), and all donors and recipients provided written informed consent to participate. We have previously published descriptions of the trial design, patient selection, donor screening, sample collection and sample processing (Youngster et al., 2014). A repository of pre-screened frozen donor stool from healthy, non-pregnant adults 18–50 years of age on no medications with normal BMI (18.5 – 25mg/m2) was established. Extensive donor screening was performed and donor inocula were processed and prepared as previously described (Youngster et al., 2014). Recipients received pre-screened, frozen stool from an unrelated donor via colonoscopic or nasogastric tube administration for the treatment of recurrent or relapsing CDI between 2012 and 2013 following inclusion/exclusion criteria as previously published. (Youngster et al., 2014).
Validation cohort:
The study was conducted at Brigham and Women’s Hospital and included subjects with recurrent CDI, defined by 3 or more a confirmed episodes and failure with standard anti-CDI antibiotics. Main exclusion criteria included neutropenia or evidence of bowel perforation. Stool samples were collected pre and post FMT under a protocol reviewed and approved by the Partners Human Research Committee (IRB) at Brigham and Women’s Hospital. All recipients provided written informed consent to participate. FMT was performed either via colonoscopy or via capsule, pending patient preference, between March, 2015 and March, 2016. Donor material, regardless of delivery modality, was obtained from OpenBiome, the largest stool bank in the United States. Screening, sample collection, and sample processing were performed according to established protocols at OpenBiome previously described (Fischer et al., 2016).
METHOD DETAILS
Metagenomic data collection (study cohort):
A total of 7 stool samples from 4 donors and 67 stool samples from 19 patients were analyzed before and after FMT for the treatment of recurrent or relapsing CDI. Stool samples were stored at −80°C until DNA extractions from stool were carried out using the QIAamp DNA Stool Mini Kit (Qiagen, Inc., Valencia, CA, USA). Shotgun sequencing for metagenomics using the Illumina GAIIx platform was performed as previously described (Consortium, 2012). Clinical metadata collected included sample collection date, most recent antibiotic administered prior to FMT, route of administration, use of immunosuppression, general health scale as previously published (Youngster et al., 2014), overall stool frequency, and presence of CDI recurrence at 8 weeks post FMT. Immunosuppression was defined as use of prednisone 40 mg or less or steroid-equivalent dose. Patients on major immunosuppressive agents were excluded: high-dose corticosteroids (greater than 40 mg oral prednisone or steroid-equivalent dose), calcineurin inhibitors, mTOR inhibitors, lymphocyte-depleting biologic agents, anti-TNFα agents, and chemotherapeutic anti-neoplastic agents.
16S rRNA data collection (validation cohort):
A total of 52 samples from 10 donors and 18 patients were analyzed before and after FMT for the treatment of recurrent or relapsing CDI. Stool samples were shipped in RNAlater to the University of Michigan Microbial Systems Laboratory for 16S rRNA sequencing. DNA extractions were performed with the PowerSoil-htp 96 Well Soil DNA isolation kit (MO BIO Laboratories). The V4 region of the bacterial 16S rRNA gene was amplified using custom barcoded primers and sequenced as previously described using an Illumina MiSeq sequencer (Kozich et al., 2013).
QUANTIFICATION AND STATISTICAL ANALYSIS
Reference alignments:
We used AMPHORA (Wu and Eisen, 2008) to identify thirty-one single-copy phylogenetic markers in a set of 649 non-redundant reference genomes from the Human Microbiome Project (Consortium, 2012). We mapped the metagenomics reads from each sample to these genes using BWA-mem (Li and Durbin, 2009) with the “-a” flag and a cutoff of ninety percent sequence identity. In order to ensure that these alignments represented strains, rather than species, this cutoff was conservatively selected to be below the species boundary (Fig. S6). These alignments were filtered as follows. First, we removed all monomorphic sites, retaining only positions with SNPs. Next, we removed all samples with zero coverage at greater than 25% of the alignment sites, as such samples may reflect cases where the reference genome is not representative of bacteria in the sample. Finally, we removed alignment sites with atypical coverage, defined as being greater than 1.5 standard deviations away from the mean coverage.
mg-OTU abundances:
To estimate the abundances of mg-OTUs from metagenomic alignments, we calculated the mean depth of coverage of each alignment, normalized to the total sequencing depth. We externally validated this approach using the 16S rRNA sequences that were generated from the same samples. At the genus level, the abundances inferred from the 16S rRNA and the metagenomic sequence data were in significant agreement (Fig. S7, median Spearman’s rho across samples = 0.60). For comparison, we also estimated the abundances in the metagenomic data using the mOTUs pipeline (Sunagawa et al., 2013), which was significantly less accurate for this particular dataset (one-sided Wilcoxon signed rank test on Spearman’s correlation coefficients, p-value = 5e-5).
16S rRNA abundances:
To estimate the abundances of OTUs in the 16S rRNA sequence data, we used the UPARSE pipeline (Edgar, 2013). Primers and barcodes were trimmed from the de-multiplexed reads. USEARCH (Edgar, 2010) was used to map these reads onto the Greengenes reference OTUs (DeSantis et al., 2006) clustered at 97% sequence identity (version gg_12_8). The sequence identity of the mapping was 0.97. Singleton OTUs were discarded, but no other quality filtering or length trimming was performed because low-quality reads would not map to the reference database. OTU abundances were estimated as the total number of mapped reads for each OTU, normalized to the total sequencing depth.
Principal coordinates analysis:
We used the phyloseq package in R (McMurdie and Holmes, 2013) to calculate the weighted UniFrac distances between all samples (i.e. beta-diversity). The NCBI taxonomy was used to estimate the phylogenetic relationships among all organisms. Principal coordinates analysis on this pairwise dissimilarity matrix was performed using the ‘pcoa’ function in the ape package in (Paradis et al., 2004).
Model of engraftment:
We used the Random Forest package in R (Liaw and Wiener, 2002) to predict the presence (using Random Forest classification) and the abundance (using Random Forest regression) of each mg-OTU in every post-FMT patient sample. For a dataset comprising M samples and N mg-OTUs, these models are trained on (M × N) total instances. The inputs to these predictions are listed in Table S2. We used an iterative subsampling scheme to ensure that all examples and features in the training set were independent. At each iteration, we randomly selected one sample from each patient, and one mg-OTU from each bacterial species, and trained a model on this reduced dataset. After 100 iterations of subsampling, we averaged the predictions across all such models. All results reflect the model’s accuracy on test data (i.e. the test error) calculated from out-of-bag samples. The predictions of mg-OTU presence used Random Forest classification with default parameters. We accounted for class imbalances, which may bias the predictions in favor of the majority class, by setting the “sampsize” argument to the minimum class size. For mg-OTUs that were predicted to be present in a sample, we next predicted their log-transformed abundances using Random Forest regression with default parameters. Both predictions used the same input features, with the exception of the sequencing depth of the post-FMT sample, which was only used to predict mg-OTU presence, and not mg-OTU abundances.
The use of multiple time points in the model:
Samples collected from the same patient at different time points are not statistically independent. Therefore, in all statistical tests, we conservatively used only the final sample collected from each patient (unless otherwise indicated). In order to display all of the data, plots show data from all time points.
The use of sequencing depth in the model:
Sequencing depth was used in the model of OTU (or mg-OTU) presence. However, because the OTU (or mg-OTU) abundances are normalized to the sequencing depth, we removed this feature from the data before predicting OTU abundances.
Clustering:
We clustered the gut microbiota of the donor, the pre-FMT and post-FMT patient, and our predictions according to their OTU (or mg-OTU) abundances. When samples from multiple time points were available, we used the mean OTU (or mg-OTU) abundances across these time points. For each subject, we constructed a vector of log-transformed abundances and standardized this vector by subtracting the mean and dividing by the standard deviation. We calculated pairwise distances among samples using the cosine dissimilarity function. For the metagenomic engraftment models, subjects were clustered based on this dissimilarity matrix, using complete-linkage clustering with the ‘hclust’ function in R. For the 16S rRNA models of engraftment, we used the Hungarian algorithm to calculate the optimal assignments between the post-FMT samples and their predicted values, minimizing the total dissimilarity among samples.
Feature importance:
Feature importance was calculated in the Random Forest by removing each feature from the model and measuring the decrease in accuracy (for presence) or the increase in the mean-squared error (for abundance). To combine these importance scores into a single metric in Figure 2, we rescaled each set of scores to the (0, 1) interval and calculated the average of these rescaled scores across both models.
Phylogenetic effects:
To estimate the marginal impact of each bacterial order on the probability of engraftment, we augmented our reduced model of OTU (or mg-OTU) presence with the order of each OTU and trained this model on 1,000 subsampled datasets (as previously described). We used the partialPlot function in the Random Forest package to estimate each bacterial order’s marginal effect on the model’s predictions, then averaged these estimates across all such models. The Interactive Tree of Life (Letunic and Bork, 2007) was used to visualize all phylogenetic trees.
Strain inference:
Our strain inference method first aligns all of the metagenomic reads against a reference sequence and tabulates the SNPs at every position of the alignment. We assume that these SNPs are conditionally independent given the underlying strain frequencies (z), the strain genotypes (π), and the sequencing error rate (ε). The probability of generating an alignment of M samples and L positions is then , where xij is a 4-vector of nucleotide counts in position j of sample i. Strain Finder assumes that within each sample, the alignment data at any given position can be modeled as a multinomial distribution, where the probability of observing SNP k at position j in sample i is equal to the frequency of strains with nucleotide k at position j in sample i. To account for sequencing error, we assume that with some probability ε, a random nucleotide is observed instead. The probability of any alignment can then be calculated from three latent parameters: the (M × N) strain frequencies, the (N × L × 4) strain genotypes, and the error rate: , where is the depth of sample i at position j, is the multinomial coefficient, zil is the frequency of strain l in sample i, and πljk is a dummy variable that is equal to 1 when strain l has nucleotide k at position j and 0 otherwise. The log-likelihood function is then: . Strain Finder finds maximum likelihood estimates of the strain frequencies (z) and strain genotypes (π) by iteratively updating them using the expectation-maximization (EM) algorithm. To estimate strain frequencies, Strain Finder uses the OpenOpt NLP solver with SLSQP to maximize the log-likelihood function while constraining the strain frequencies within a sample to sum to one. To estimate strain genotypes, Strain Finder exhaustively searches genotype space, or in cases with large numbers of strains, solves a continuous optimization problem as follows. Strain Finder allows strains to have ‘fuzzy’ genotypes, where the nucleotide probabilities vary continuously from 0 to 1 at every site. It then uses the OpenOpt NLP solver with SLSQP to maximize the L2-penalized log-likelihood, then discretizes the final genotypes by selecting the dominant nucleotide at every site. The L2 penalty biases the estimated genotypes toward discrete values. Because it is more accurate than the optimization, the exhaustive search was used for all strain estimates, except for the simulations of 16 and 32 strains. As the EM algorithm only converges to a local optimum, we repeat the strain inference for thousands of initial conditions to approximate a global solution. For each mg-OTU, Bayes information criterion was used to select an optimal number of strains. Strain Finder is available at http://www.github.com/cssmillie/StrainFinder.git.
Simulations:
To validate our strain inference method, we simulated alignments while varying the numbers of samples (2, 4, 8, 16, 32, and 64), numbers of strains (2, 4, 8, 16, and 32), sequencing depths (25, 100, 500, and 1000 nucleotides per position) and alignment lengths (4, 16, 64, 256, and 1024 SNP positions). For each simulation, we select random strain frequencies and random strain genotypes, which are then used to generate an alignment. Strain frequencies are sampled from a Dirichlet distribution with uniform concentration parameters. To select random strain genotypes, we use Dendropy (Sukumaran and Holder, 2010) to generate a random phylogenetic tree using an unconstrained Kingman coalescent process. We simulate the evolution of the strain genotypes on this tree, producing strain genotypes with realistic evolutionary relationships. This process is conservative, as it generates alignments with mostly dimorphic sites, which are most challenging for our model to predict. To generate a random alignment, we sample nucleotides from a multinomial distribution, where the event probabilities are determined by the strain frequencies and genotypes. More explicitly, the probability of sampling nucleotide k at position j in sample i is equal to the frequency of strains with nucleotide k at position j in sample i. We performed 10 simulations for each parameter combination and averaged the results, for a total of 6,000 simulations. To evaluate the accuracy of this method, we used the Hungarian algorithm to find optimal assignments between the inferred strains and the true strains, minimizing the total edit distance among their genotypes. We then calculated the similarity of the inferred strains and the true strains in terms of their genotypes and frequencies across samples.
ConStrains comparison:
We obtained the full genome sequences of 16 Escherichia coli strains from NCBI: O157 H7, 536, APEC 01, CFT073, IAI1, SE11, ETEC H10407, K12 MG1655, HS, DH1, 2011C-3493, NRG 857C, BLK9, SLK172, CFSAN051542, and SF-468. We simulated 128 synthetic metagenomes following the methods of (Luo et al., 2015). For a given number of strains (N = 4, 8, 12, or 16), we sampled 1,000 strain compositions from a Dirichlet multinomial with uniform concentration parameters, then calculated the entropy of each strain composition. We selected 8 strain compositions corresponding to the 1st through 9th deciles of the entropy distribution, for a total of 32 strain compositions. We used NeSSM (Jia et al., 2013) to simulate 80 bp metagenomic reads for each strain composition at four different sequencing depths (25X, 100X, 500X, and 1000X). Reads were aligned to the MetaPhlAn Escherichia coli marker genes (Segata et al., 2012) using Bowtie 2 with default parameters (Langmead and Salzberg, 2012). Strain Finder and ConStrains were run on the resulting alignments, resulting in strain estimates for each sample. RAxML (Stamatakis, 2014) was used to estimate the phylogenetic relationships among the true strains and the strains inferred by Strain Finder, ConStrains, and each null model. These midpoint-rooted phylogenetic trees were used to calculate the weighted UniFrac distances between the true and inferred strain profiles, which were used to assess each method’s accuracy.
Specificity:
Donor specificity was calculated as the ratio of the distance from the donor to the post-FMT patient, relative to the mean distance from other donors to the post-FMT patient. Patient specificity was calculated as the ratio of the distance from the pre-FMT patient to the post-FMT patient, relative to the mean distance from other pre-FMT patients to the post-FMT patient. Distances were calculated using the Jensen-Shannon divergence and these ratios were log-transformed to make them symmetric about zero.
Probability of engraftment:
We used our strain estimates to determine whether each bacterial species from the donor engrafted in each patient. For a given bacterial species, we assume that the number of successful engraftment events is a binomial random variable with a fixed probability of success. Because we have a limited number of observations for each mg-OTU, we estimated this probability using a Bayesian estimate of the mean: , where x is the number of successes, N is the number of trials, and reflects the prior probability of engraftment, which we estimated from all bacterial species across all FMTs. We provide a 95% confidence interval for the probability of engraftment, estimated using the ‘binom.test’ function in R.
DATA AND SOFTWARE AVAILABILITY
Strain Finder is available at http://www.github.com/cssmillie/StrainFinder.git. The C. difficile 16S and shotgun metagenomic data are available from the European Nucleotide Archive under accessions PRJEB23489 and PRJEB23524.
Supplementary Material
Table S1, Related to Figure 2: List of bacterial reference genomes used in this study.
Table S2, Related to Figure 2: List of features used in the model of bacterial engraftment.
Table S3, Related to Figure 5: Engraftment probabilities of donor OTUs.
Table S4, Related to Figure 6: Summary of the 16S rRNA datasets used in this study.
Table S5, Related to Figure 5: Estimated strain frequencies and genotypes for all mg-OTUs.
Highlights.
Gut microbiota of 18 C difficile patients profiled during fecal microbiota transplantation
Developed Strain Finder, a method to infer strain genotypes and track them over time
Bacterial abundance and phylogeny are the strongest determinants of microbiota engraftment
Unlike bacterial species, closely related strains engraft in an all-or-nothing pattern
Declaration of Interests
This work was supported by funding from the Crohn’s & Colitis Foundation, the Center for Microbiome Informatics and Therapeutics, and National Institutes of Health grant DK043351. D.G. is an employee of Janssen Pharmaceuticals. M.B.S. and E.J.A. are founders and employees of Finch Therapeutics. E.J.A. receives research funding from Assembly Biosciences. A.K. has received support from Crestovo. M.J.S. has received support and has consulted for Crestovo. The University of Minnesota Conflicts of Interest Program is managing conflicts for A.K. and M.J.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aroniadis OC, and Brandt LJ (2013). Fecal microbiota transplantation: past, present and future. Curr. Opin. Gastroenterol 29, 79–84. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. [DOI] [PubMed] [Google Scholar]
- Britton RA, and Young VB (2014). Role of the Intestinal Microbiota in Resistance to Colonization by Clostridium difficile. Gastroenterology 146, 1547–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, and Young VB (2008). Decreased Diversity of the Fecal Microbiome in Recurrent Clostridium difficile—Associated Diarrhea. J. Infect. Dis 197, 435–438. [DOI] [PubMed] [Google Scholar]
- Consortium, T.H.M.P. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen TW, Schofield WB, Barry NA, Putnam EE, Rundell EA, Trent MS, Degnan PH, Booth CJ, Yu H, and Goodman AL (2015). Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science 347, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, and Andersen GL (2006). Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol 72, 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Edgar RC (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, nmeth.2604. [DOI] [PubMed] [Google Scholar]
- Floch MH (2015). Intestinal Microbiota Metabolism of a Prebiotic to Treat Hepatic Encephalopathy. Clin. Gastroenterol. Hepatol 13, 209. [DOI] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. [DOI] [PubMed] [Google Scholar]
- Greenblum S, Carr R, and Borenstein E (2015). Extensive Strain-Level Copy-Number Variation across Human Gut Microbiome Species. Cell 160, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al. (2013). Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Consortium (2012). Structure, Function and Diversity of Human Microbiome in an Adult Reference Population. Nat. E-Pub Ahead Print Doi 10. [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. (2009). Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia B, Xuan L, Cai K, Hu Z, Ma L, and Wei C (2013). NeSSM: A Next-Generation Sequencing Simulator for Metagenomics. PLOS ONE 8, e75448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Pardi DS, Kelly CR, Kraft CS, Dhere T, Henn MR, Lombardo M-J, Vulic M, Ohsumi T, Winkler J, et al. (2016). A Novel Microbiome Therapeutic Increases Gut Microbial Diversity and Prevents Recurrent Clostridium difficile Infection. J. Infect. Dis jiv766. [DOI] [PubMed] [Google Scholar]
- Koropatkin NM, Cameron EA, and Martens EC (2012). How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol 10, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, and Schloss PD (2013). Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol 79, 5112–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C, et al. (2012). Targeted Restoration of the Intestinal Microbiota with a Simple, Defined Bacteriotherapy Resolves Relapsing Clostridium difficile Disease in Mice. PLoS Pathog 8, e1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Donaldson GP, Mikulski Z, Boyajian S, Ley K, and Mazmanian SK (2013). Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 501, 426–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, et al. (2015). Burden of Clostridium difficile infection in the United States. N. Engl. J. Med 372, 825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, and Bork P (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SS, Zhu A, Benes V, Costea PI, Hercog R, Hildebrand F, Huerta-Cepas J, Nieuwdorp M, Salojärvi J, Voigt AY, et al. (2016). Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science 352, 586–589. [DOI] [PubMed] [Google Scholar]
- Liaw A, and Wiener M (2002). Classification and Regression by randomForest. R News 2, 18–22. [Google Scholar]
- Lozupone C, Lladser ME, Knights D, Stombaugh J, and Knight R (2011). UniFrac: an effective distance metric for microbial community comparison. ISME J. 5, 169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Knight R, Siljander H, Knip M, Xavier RJ, and Gevers D (2015). ConStrains identifies microbial strains in metagenomic datasets. Nat. Biotechnol 33, 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, Chen I-MA, Palaniappan K, Chu K, Szeto E, Pillay M, Ratner A, Huang J, Woyke T, Huntemann M, et al. (2014). IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 42, D560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2013). phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLOS ONE 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moayyedi P, Surette MG, Kim PT, Libertucci J, Wolfe M, Onischi C, Armstrong D, Marshall JK, Kassam Z, Reinisch W, et al. (2015). Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 149, 102–109.e6. [DOI] [PubMed] [Google Scholar]
- van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JFWM, Tijssen JGP, et al. (2013). Duodenal Infusion of Donor Feces for Recurrent Clostridium difficile. N. Engl. J. Med 368, 407–415. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, and Strimmer K (2004). APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20, 289–290. [DOI] [PubMed] [Google Scholar]
- Petrof EO, and Khoruts A (2014). From Stool Transplants to Next-Generation Microbiota Therapeutics. Gastroenterology 146, 1573–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. (2013). Gut Microbiota from Twins Discordant for Obesity Modulate Metabolism in Mice. Science 341, 1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, and Mazmanian SK (2010). Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci 107, 12204–12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A, Waller A, Mende DR, Kultima JR, Martin J, et al. (2013). Genomic variation landscape of the human gut microbiome. Nature 493, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seekatz AM, Aas J, Gessert CE, Rubin TA, Saman DM, Bakken JS, and Young VB (2014). Recovery of the Gut Microbiome following Fecal Microbiota Transplantation. mBio 5, e00893–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, and Huttenhower C (2012). Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 9, 811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahinas D, Silverman M, Sittler T, Chiu C, Kim P, Allen-Vercoe E, Weese S, Wong A, Low DE, and Pillai DR (2012). Toward an Understanding of Changes in Diversity Associated with Fecal Microbiome Transplantation Based on 16S rRNA Gene Deep Sequencing. mBio 3, e00338–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre M-L, et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 350, 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Garg S, Girotra M, Maddox C, von Rosenvinge EC, Dutta A, Dutta S, and Fricke WF (2013). Microbiota Dynamics in Patients Treated with Fecal Microbiota Transplantation for Recurrent Clostridium difficile Infection. PLoS ONE 8, e81330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley C, Kelly CR, Brandt LJ, Khoruts A, and Sadowsky MJ (2016). Complete Microbiota Engraftment Is Not Essential for Recovery from Recurrent Clostridium difficile Infection following Fecal Microbiota Transplantation. mBio 7, e01965–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran J, and Holder MT (2010). DendroPy: a Python library for phylogenetic computing. Bioinformatics 26, 1569–1571. [DOI] [PubMed] [Google Scholar]
- Sunagawa S, Mende DR, Zeller G, Izquierdo-Carrasco F, Berger SA, Kultima JR, Coelho LP, Arumugam M, Tap J, Nielsen HB, et al. (2013). Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods 10, nmeth.2693. [DOI] [PubMed] [Google Scholar]
- Suskind DL, Brittnacher MJ, Wahbeh G, Shaffer ML, Hayden HS, Qin X, Singh N, Damman CJ, Hager KR, Nielson H, et al. (2015). Fecal microbial transplant effect on clinical outcomes and fecal microbiome in active Crohn’s disease. Inflamm. Bowel Dis 21, 556–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JR, Pacocha S, Pharino C, Klepac-Ceraj V, Hunt DE, Benoit J, Sarma-Rupavtarm R, Distel DL, and Polz MF (2005). Genotypic Diversity Within a Natural Coastal Bacterioplankton Population. Science 307, 1311–1313. [DOI] [PubMed] [Google Scholar]
- Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod LP, Harris NL, et al. (2014). Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med 20, 159–166. [DOI] [PubMed] [Google Scholar]
- U.S. National Institutes of Health ClincalTrials.gov. [PubMed]
- Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CPM, et al. (2015). Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze A, Nood EV, Holleman F, Salojärvi J, Kootte RS, Bartelsman JFWM, Dallinga–Thie GM, Ackermans MT, Serlie MJ, Oozeer R, et al. (2012). Transfer of Intestinal Microbiota From Lean Donors Increases Insulin Sensitivity in Individuals With Metabolic Syndrome. Gastroenterology 143, 913–916.e7. [DOI] [PubMed] [Google Scholar]
- Weingarden A, González A, Vázquez-Baeza Y, Weiss S, Humphry G, Berg-Lyons D, Knights D, Unno T, Bobr A, Kang J, et al. (2015). Dynamic changes in short- and long-term bacterial composition following fecal microbiota transplantation for recurrent Clostridium difficile infection. Microbiome 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, and Eisen JA (2008). A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 9, R151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngster I, Sauk J, Pindar C, Wilson RG, Kaplan JL, Smith MB, Alm EJ, Gevers D, Russell GH, and Hohmann EL (2014). Fecal Microbiota Transplant for Relapsing Clostridium difficile Infection Using a Frozen Inoculum From Unrelated Donors: A Randomized, Open-Label, Controlled Pilot Study. Clin. Infect. Dis 58, 1515–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, Related to Figure 2: List of bacterial reference genomes used in this study.
Table S2, Related to Figure 2: List of features used in the model of bacterial engraftment.
Table S3, Related to Figure 5: Engraftment probabilities of donor OTUs.
Table S4, Related to Figure 6: Summary of the 16S rRNA datasets used in this study.
Table S5, Related to Figure 5: Estimated strain frequencies and genotypes for all mg-OTUs.
Data Availability Statement
Strain Finder is available at http://www.github.com/cssmillie/StrainFinder.git. The C. difficile 16S and shotgun metagenomic data are available from the European Nucleotide Archive under accessions PRJEB23489 and PRJEB23524.