

Single-cell RNA analysis has been integrated with spatial protein profiling to create a single–cell type map of human tissues.
Abstract
Advances in molecular profiling have opened up the possibility to map the expression of genes in cells, tissues, and organs in the human body. Here, we combined single-cell transcriptomics analysis with spatial antibody-based protein profiling to create a high-resolution single–cell type map of human tissues. An open access atlas has been launched to allow researchers to explore the expression of human protein-coding genes in 192 individual cell type clusters. An expression specificity classification was performed to determine the number of genes elevated in each cell type, allowing comparisons with bulk transcriptomics data. The analysis highlights distinct expression clusters corresponding to cell types sharing similar functions, both within the same organs and between organs.
INTRODUCTION
The marked improvements in massive parallel sequencing coupled with single-cell sample preparations and data deconvolution have allowed single-cell RNA sequencing (scRNA-seq) to become a powerful approach to characterize the gene expression profile in single cells (1, 2). The objective of the international collaborative effort Human Cell Atlas (www.humancellatlas.org) takes advantage of this new technology platform to study the distinctive gene expression profiles on RNA level across diverse cell and tissue types and connect this information with classical cellular descriptions, such as location and morphology (3). In parallel, the development of many millions of publicly available antibodies toward human proteins has enabled single-cell analysis of the corresponding proteins in tissues and organs using immunohistochemistry (4) and fluorescent-based bioimaging (1, 5–7), allowing single-cell spatial mapping in the context of neighboring cells. The objective of the Human Protein Atlas (HPA) (www.proteinatlas.org) effort is to take advantage of these bioimaging approaches to map the expression of all human protein-coding genes across all major human cells, tissues, and organs. More than 10 million bioimages from 37 tissues showing the native protein location in intact tissue samples are publicly available in the HPA, each annotated by a certified pathologist (4). Together, these two platforms thus have the potential to create comprehensive body-wide maps of gene expression at RNA and protein level with the ultimate goal to provide publicly available genome-wide knowledge of protein-coding genes in single cell types across tissues and organs in the human body.
Here, we describe an effort to combine the information from these two efforts to create a publicly available HPA Single Cell Type Atlas with genome-wide expression data from scRNA-seq experiments integrated with the spatial antibody-based bioimaging data. We use an approach outlined in Fig. 1A in which the single–cell type transcriptomics from the scRNA-seq data from a particular cluster of cells is pooled and the average normalized protein-coding transcripts per million (pTPM) as well as a normalized expression are calculated across protein-coding genes. In this manner, the problem with technical noise involving genes having zero counts (so-called dropouts) can be minimized and even genes with very low expression levels can be detected (8). This approach allows the expression profiles for each gene in each cluster to be visualized on a genome-wide and single–cell type level taking advantage of the added information by cumulative counts from hundreds or thousands of cells.
Fig. 1. Annotating 51 cell types from 13 tissues using single-cell transcriptomics data.
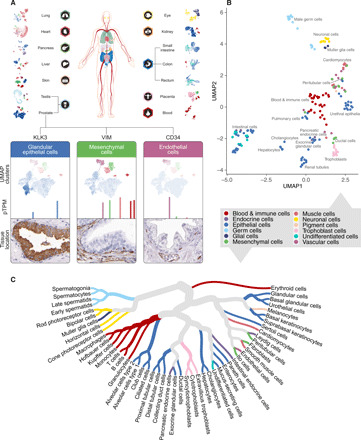
(A) scRNA-seq data from 13 tissues and blood [peripheral blood mononuclear cells (PBMCs)] were processed through a clustering algorithm, and each cluster was annotated using known markers. All cells from a cluster were pooled, and the average transcript per million was calculated for all protein-coding genes. (B) UMAP plot showing the relationship of all cell types from all analyzed tissues. The color-coding corresponds to 12 main cell type groups. (C) Cell type dendrogram showing the relationship between all 51 identified main single cell types based on genome-wide expression.
RESULTS
The tissues included in the study
To make this possible, a survey of scRNA-seq data from nondiseased human tissues and organs was performed. We used three main criteria to include data into the pipeline: (i) publicly available raw data from human tissues with good technical quality with at least 4000 cells analyzed and at least 20 million read counts by the sequencing for each tissue; (ii) high correlation between pseudo-bulk transcriptomics profile from the scRNA-seq data and bulk RNA-seq generated as part of the HPA Tissue Atlas; and (iii) high correlation between the cluster-specific expression and the expected expression pattern of an extensive selection of marker genes representing well-known tissue- and cell type–specific markers, including both markers from the original publications and additional markers used in pathology diagnostics (data S2). Here, we present a dataset containing 13 different human tissues covering most major organs in the human body including ileum (9), colon (10), rectum (9), kidney (11), liver (12), pancreas (13), heart (14), lung (15), prostate (16), testis (17), placenta (18), skin (19), and eye (20), as well as an analysis of human blood (21) (see data S1). No brain samples were included because only single-nuclei data were available, which showed lower correlation to bulk data in comparison to single-cell data (see fig. S1) (22, 23). All raw datasets were gathered into a common cluster analysis, resulting in a total of 192 single–cell type clusters across the datasets (see data S1 for all cluster annotations). In total, the data correspond to 1.47 billion read counts and the average read count per single–cell type cluster was approximately 7.7 million.
Correlation of expression profiles across the 192 cell types
The correlations between bulk RNA-seq and pseudo-bulk single-cell transcriptomics profiles were high for all tissues, ranging from 0.76 to 0.88 (fig. S2). All clusters were manually annotated on the basis of known tissue- and cell type–specific markers and their expected expression in the corresponding clusters (data S2 and fig. S3). As examples of the results, three genes are exemplified in Fig. 1A with cluster expression profiles in prostate. Kallikrein-related peptidase 3 (KLK3), also known as prostate-specific antigen (PSA), was shown to be expressed in two neighboring clusters in prostate, both annotated as glandular epithelial cells. Vimentin (VIM), a well-known marker for mesenchymal cells, was instead expressed in five different clusters, all annotated as mesenchymal-related cell types, including smooth muscle cells and immune cells. CD34, a well-known marker for endothelial cells, was localized to one of these clusters that has been annotated as endothelial cells. A UMAP (uniform manifold approximation and projection) of all clusters (Fig. 1B) revealed, as expected, that profiles of cell types responsible for unique tissue-specific functions have a close association, here shown as distinct tissue-specific groups, e.g., intestinal, hepatic, renal, placental, pulmonary, and neuronal cells. Some cell populations carry out similar cell type–specific functions, and as expected, these clusters from different tissues show high similarity in gene expression, e.g., immune cells (nine tissues), endothelial cells (nine tissues), and fibroblasts (five tissues). Altogether, the 192 single–cell type clusters could be summarized into 51 main cell types belonging to 12 different functional groups of cells (Fig. 1, B and C).
Creation of a Single Cell Type Atlas
On the basis of these new data, a Single Cell Type Atlas has been launched (www.proteinatlas.org/celltype) with data for all protein-coding genes. More than 250,000 interactive UMAP plots are presented in this open access resource showing the primary data for every analyzed cell for all protein-coding genes and all annotated cell types (defined as annotated clusters). Similarly, by pooling the data for every cell in a cluster, we have been able to generate more than 250,000 bar plots showing the calculated transcripts per million (TPM) for each gene and cell type across the entire protein-coding genome. The integration with tissue imaging (Fig. 2A) allows validation of the cell type–specific expression on the protein level by the in situ antibody-based profiling, as exemplified by the immunohistochemical staining of phosphodiesterase 6A (PDE6A) shown by the scRNA-seq analysis to be localized to rod photoreceptor cells in eye (clusters 0, 2, 3, and 4). Similarly, the protein insulin (INS) was shown to be localized to endocrine cells in pancreas (cluster 6), surfactant protein C (SFTPC) was localized to alveolar cells type 2 (AT2) in lung (clusters 1 and 6), and uromodulin (UMOD) was localized to distal tubular cells in kidney (clusters 11 and 12).
Fig. 2. An open access single–cell type transcriptomics atlas of human tissues.
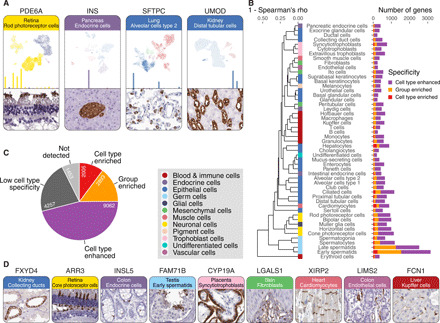
(A) Some examples of data visualization in the HPA Single Cell Type Atlas with UMAP plots from the single-cell analysis in each cluster, the resulting bar plot showing the RNA level gene expression in each cell type, and the corresponding tissue images based on immunohistochemistry. (B) Number of cell type–enriched, group-enriched, and cell type–enhanced genes for each of the 51 cell types based on the single–cell type data. (C) Number of genes classified for single–cell type specificity. (D) Some examples of immunohistochemical tissue images for genes identified as elevated in single cell types: FXYD4, an ion transport regulator localized to renal collecting ducts; ARR3, a protein suggested to play a role in retina-specific signal transduction, localized to the cone photoreceptor cells in eye; INSL5, a protein with essentially unknown function but suggested to play a role as a gut hormone (27), localized to intestinal neuroendocrine cells; FAM71B, an uncharacterized protein, here specifically localized to early spermatids; CYP19A, a member of the cytochrome P450 superfamily of enzymes, here localized to placental syncytiotrophoblasts; LGALS1, a lectin acting as an autocrine negative growth factor regulating cell proliferation, here present in fibroblasts; XIRP2, an actin-binding protein localized to the intercalated discs in cardiomyocytes; LIMS2, a focal adhesion protein modulating cell spreading and migration, here localized to endothelial cells; and finally FCN1, an extracellular lectin involved in innate immunity, localized to hepatic Kupffer cells/macrophages.
Classification of protein-coding genes based on expression profiles
A classification to map the gene expression profile of all protein-coding genes across the different cell types was performed as described earlier (4) to determine the number of genes elevated in particular single cell types and thus showing high or low cell type specificity (table S1). In total, across all cell types, 2005 genes are cell type–enriched, meaning that the expression of a particular gene defined as adjusted TPM (see Materials and Methods) is at least fourfold higher in one cell type as compared to all other cell types analyzed here (Fig. 2C). Similarly, 2893 genes are defined as group-enriched, thus enriched in a group of up to 10 cell types, and 9062 genes are defined as cell type–enhanced, where the expression is at least fourfold higher in one cell type as compared to the mean of all other cell types. A group of genes are also classified as having low cell type specificity (n = 4257), suggesting that they are present at roughly similar levels across all the cell types. Only 11% of the genes were detected in all analyzed cell types, supporting previous estimation of the number of “housekeeping” genes needed in all cells (24, 25). In Fig. 2B, the number of elevated genes (cell type–enriched, group-enriched, or cell type–enhanced) is visualized for all the 51 different cell types. In agreement with previous observations based on bulk transcriptomics (4), testis constitutes the tissue with the highest number of cell type elevated genes, but many elevated genes were also found in the eye (photoreceptor cells, bipolar cells, and horizontal cells) as well as in ciliated cells in lung (see data S3 for a complete list of the classification results). As mentioned above, the integration of multiple analysis platforms allows the validation of the single-cell data with antibody-based image profiling in tissue. Immunohistochemistry shows not only the localization at a single-cell level but also the exact spatial pattern, cell-to-cell variation, and subcellular localization. In Fig. 2D, some examples of this validation are shown, including proteins specifically expressed in rare structures, such as, e.g., renal collecting ducts, retinal photoreceptor cells, early spermatids, intercalated discs in cardiomyocytes, and hepatic Kupffer cells.
The cell type expression landscape
The cell type–specific expression landscape was summarized in a network plot (Fig. 3A), illustrating the number of cell type–enriched and group-enriched genes and their relationships. The analysis highlights distinct expression clusters corresponding to cell types sharing similar functions, both within the same organs and between organs. As expected, many genes are simultaneously enriched in various immune cell linages and the different stages of germ cells in testis. It is also evident that despite that many organs contain epithelial cells, these still have large numbers of genes enriched in only one particular tissue. Cell types of the same origin residing in different tissues, e.g., macrophages, defined as Kupffer cells in liver and Hofbauer cells in placenta, also have genes enriched in only one cell type. Figure 3B shows immunohistochemical examples of proteins that shared elevated expression in cell types present in two different organs but with similar function, such as motility or immune-related functions.
Fig. 3. The number of cell type– and group-enriched genes for each cell type.
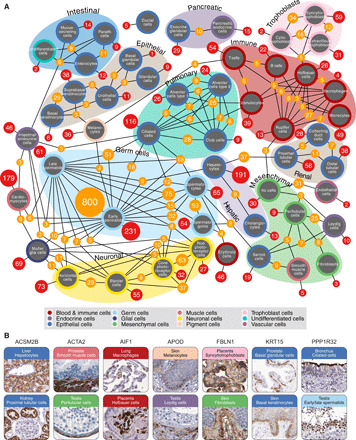
(A) Network plot showing the numbers of cell type–enriched genes (red nodes) and group-enriched genes (orange nodes). Cell type nodes are colored by the 12 different cell type groups as shown in the color legend. Clusters of cell types of related function or origin are visualized with annotations with different background colors. (B) Some examples of group-enriched genes shared by cell types of different tissue origin. The color codes correspond to the 12 different cell type groups as indicated in the color legend: ACSM2B, involved in fatty acid metabolism, localized to liver hepatocytes and renal proximal tubules; ACTA2, a cytoskeleton protein that produces microfilaments allowing the contractile movement of smooth muscle cells, distinctly expressed in smooth muscle cells as well as testicular peritubular cells; AIF1, an actin-binding protein involved in phagocytosis and macrophage activation, expressed in macrophages residing in different organs such as lung and placenta; APOD, a serum glycoprotein involved in the transport of hydrophobic ligands, present in skin melanocytes and testicular Leydig cells that are both highly associated with production of hydrophobic compounds; FBLN1, a glycoprotein incorporated into fibronectin-containing extracellular matrix (ECM), present in skin fibroblasts and trophoblastic cells, both involved in ECM remodeling; KRT15, a basal cell marker, localized to basal cells of all types of epithelia and here present in both prostate and skin epithelia; and PPP1R32, an uncharacterized phosphatase, here localized to both fallopian tube cilia and testis spermatids, cell types that share functions related to motility.
Comparison of bulk and single-cell transcriptomics
The new single–cell type classification of protein-coding genes allowed us to perform a genome-wide comparison between this classification and the previous classification based on bulk transcriptomics. In Fig. 4A, the relationship of classification of all 19,670 protein-coding genes is shown, with most genes overlapping in classification, including a vast majority of the 2005 cell type–enriched genes that are found to be either enriched, group-enriched, or enhanced based on the tissue classification. Relatively few genes have low cell type specificity (n = 4257), but almost all of these have low tissue specificity based on the bulk transcriptomics data. It is noteworthy that the number of elevated genes is higher for our single–cell type classification as compared to the tissue-level classification, supporting the view that many genes that are found across all tissues still have cell type–specific expression profiles. Note that relatively many genes are not detected in the cell type analysis, which is not unexpected, because many tissues were not analyzed due to lack of data for many important tissues, such as the brain.
Fig. 4. A comparison of gene specificity between single cell type and tissue.
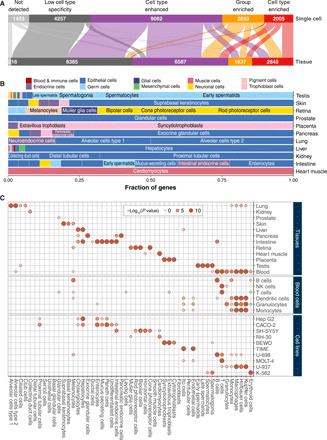
(A) Alluvial diagram showing the number of genes of respective specificity category for single cell (top) and tissue (bottom). (B) Bar plot showing the fraction of single–cell type–enriched genes among the tissue-enriched genes. The color code indicates the cell type groups. The cells with most shared enriched genes with tissues are labeled. (C) Bubble heatmap showing the significance (indicated by dot size and color) of shared enriched genes between single cell types (x axis) and tissues (top), blood lineage (middle), and human cell lines (bottom). Notably, if the overlap of enriched genes is not statistically significant (hypergeometric test, P > 0.05), the corresponding dot is removed.
An investigation of the overlap between the tissue “bulk” expression and the single–cell type expression is shown in Fig. 4B. The analysis showed that most genes with enriched expression in a certain tissue were enriched also based on the single-cell analysis. Tissue-specific expression can thus be attributed to individual cell types present in a particular tissue, exemplified by the many liver-enriched genes that were found to be hepatocyte-enriched. Likewise, all genes enriched in heart muscle by bulk transcriptomics analysis are enriched in cardiomyocytes in the single-cell analysis. The overlap of genes that are enriched at both single–cell type and tissue level is visualized in a network in fig. S4. This highlights the usefulness of scRNA-seq to disentangle the cell type variance across the different tissues in the human body.
Correlation to tissues and blood cells
A hypergeometric test was conducted to show the statistical significance of the overlap between genes that are enriched in the single cell types and genes enriched in tissues, flow-sorted blood cells, and cell lines (Fig. 4C). As noted above, it is reassuring that the cell type–enriched genes generally show a high degree of overlap with the enriched genes defined by bulk transcriptomics from their corresponding tissues. For example, the enriched genes from the liver bulk transcriptomics show overlap with genes elevated in hepatocytes and cholangiocytes based on scRNA-seq. Similarly, the alveolar cells, ciliated cells, and club cells from the single-cell analysis share enriched genes with lung tissue. The scRNA-seq data for the immune cell clusters were also compared with transcriptomics data of flow-sorted single blood cells (Fig. 4C) (26). The macrophages, not present in the HPA Blood Atlas data (26), show as expected overlap with the flow-sorted monocytes. It is also reassuring that the scRNA-seq identified T cells show overlap with the flow-sorted T cells and natural killer (NK) cells published in the HPA Blood Atlas (25), and similarly, enriched genes in scRNA-seq B lymphocytes show overlap with the flow-sorted B cells.
Correlation with human cell lines
Last, we analyzed the overlap of the scRNA-seq analysis with transcriptomics data of in vitro cultivated human cell lines. In Fig. 4C, some examples are shown, with additional 60 cell lines visualized in fig. S5. Overall, there is a high degree of overlap of cell line–enriched genes with the corresponding cell type of origin from the scRNA-seq analysis. For example, the cell line HepG2 shows, as expected, highest degree of overlap with hepatocyte-enriched genes. Similarly, the B cell–derived U-698 cell line mostly overlaps with single-cell clusters annotated to be B cells, but some overlap with T cells. These examples suggest that these in vitro cultivated cell lines may serve as representative models for the corresponding in vivo cell types, while many other cell lines (fig. S5) show less overlap with the expected in vivo cell types, suggesting that caution should be taken when using these cell lines as models for the corresponding cell type.
DISCUSSION
In conclusion, we here present a single–cell type transcriptomics map of human tissues and blood complemented with antibody-based profiling and with comparisons to bulk transcriptomics. An open access HPA Single Cell Type Atlas (www.proteinatlas.org/celltype) has been launched with more than 250,000 interactive UMAP plots and expression bar plots to allow researchers to explore the individual single–cell type data for all protein-coding genes in these tissues. We plan to update the Single Cell Type Atlas in the future as part of the annual update of the HPA taking into account new genome assemblies and addition of new data. Future inclusion of more tissues and datasets with more cells analyzed and sequencing at higher depths will allow this effort to be extended into more specialized tissues and organs helped by international efforts such as the Human Cell Atlas (3) and other efforts to analyze human single cells in tissues (6, 10, 11 23). The analysis may also be extended to include single-nuclei data from tissues that are difficult to obtain using the single-cell approach. Integration of these datasets on both the transcriptomic and proteomic level gives a unique opportunity to validate the exact tissue localization in situ. Thus, the approach described here to combine single-cell data with antibody-based profiling will facilitate efforts to provide a holistic cell-, tissue-, and organ-wide map of the proteins across the human body to act as a basis for research in human biology and disease.
MATERIALS AND METHODS
scRNA-seq dataset selection
The scRNA-seq dataset was retrieved from published studies based on healthy human tissues. We performed meta-analysis of literatures on scRNA-seq and search single-cell databases, including the Single Cell Expression Atlas (https://ebi.ac.uk/gxa/sc/home), the Human Cell Atlas (https://humancellatlas.org), the Gene Expression Omnibus (https://ncbi.nlm.nih.gov/geo/), and the European Genome-phenome Archive (https://ebi.ac.uk/ega/). To avoid technical bias and to ensure that the single-cell dataset can best represent the corresponding tissue, we applied the following criteria for data selection: (i) We limited the single-cell transcriptomic dataset to those based on the Chromium single-cell gene expression platform from 10X Genomics (version 2 or 3); (ii) scRNA-seq was performed on single-cell suspension from tissues without pre-enrichment of cell types; (iii) only studies with >4000 cells and 20 million read counts were included; and (iv) only dataset whose pseudo-bulk gene expression profile is highly correlated with the expression profile of the corresponding HPA tissue bulk sample is included. Note that, for the tissue eye, we do not have the corresponding bulk transcriptome. In addition, the dataset for lung had fewer reads (~7.3 million), while the dataset for pancreas (3719 cells) and rectum (3898 cells) had less than 4000 cells analyzed. However, these datasets were still included because the data provided important insights into cell type–enriched genes in these tissues. In total, we included datasets for 13 tissues plus peripheral blood mononuclear cells (PBMCs) (data S1).
Quantifying transcriptomic expression of clusters and pseudo-bulk
Quantified raw sequencing data were downloaded from the corresponding depository database based on the accession number provided by the study (data S1) in the available format (total cells, read, and feature counts, or count tables). Unfiltered data were used as input for downstream analysis with in-house pipeline using Single-Cell Analysis in Python (Scanpy, version 1.4.4.post1) in Python (version 3.7.3). In the pipeline, the data were filtered using two criteria: A cell is considered as valid if it has at least 200 genes, and a gene is considered as valid if it is expressed in at least 10% of the cells. Subsequently, the cell counts were normalized to have total count per cell of 10,000. Afterward, the valid cells were then clustered using Louvain clustering function within Scanpy and then gene rankings for each cluster were calculated with “rank_genes_group” function. The total read counts for all genes in each cluster were calculated by adding up the read counts of each gene in all cells belonging to the corresponding cluster. Last, the read counts were normalized to pTPM for each of the single-cell clusters. In the case of calculating the expression profile for pseudo-bulk samples based on single-cell transcriptomics, we added the read counts for all genes from all cells of the sample and normalized it to pTPM in the same way as for the cluster ones.
Defining cell types
Each of the 192 different cell type clusters was manually annotated based on an extensive survey of >500 well-known tissue- and cell type–specific markers, including both markers from the original publications and additional markers used in pathology diagnostics. For most cell types, three marker genes were used, and for each cluster, one main cell type was chosen based on the overall expression pattern of all the marker genes. For four clusters, no main cell type could be selected, and these clusters were not used for classification. The most relevant markers (data S2) are presented in a heatmap on the Cell Type Atlas on each organ- and gene-specific page to clarify cluster annotation to visitors.
Bulk RNA-seq analysis and antibody-based protein profiling
Human tissue samples for analysis of bulk RNA-seq and gene expression at protein level in the HPA datasets were collected and handled in accordance with Swedish laws and regulation. Tissues were obtained from the Clinical Pathology Department, Uppsala University Hospital, Sweden and collected within the Uppsala Biobank organization. All samples were anonymized for personal identity by following the approval and advisory report from the Uppsala Ethical Review Board (reference nos. 2002-577, 2005-388, 2007-159, and 2011-473). The RNA extraction and RNA-seq procedure have been described previously (28). For immunohistochemistry, formalin-fixed, paraffin-embedded (FFPE) tissue blocks were collected from the pathology archives based on normal histology using a hematoxylin and eosin–stained tissue section evaluated by a pathologist. For generation of tissue microarrays (TMAs), representative 1-mm-diameter cores were sampled from FFPE blocks and assembled into TMAs. TMA blocks were cut into 4-μm-thick sections using waterfall microtomes (Microm HM 355S, Thermo Fisher Scientific, Freemont, CA, USA), placed on SuperFrost Plus slides (Thermo Fisher Scientific), dried at room temperature overnight, and baked at 50°C for 12 to 24 hours before immunohistochemical staining. Automated immunohistochemistry was performed using the Autostainer 480S Module (Thermo Fisher Scientific).
Immunohistochemical staining and high-resolution digitization of stained TMA slides were performed essentially as previously described (29). Primary antibodies were diluted and optimized based on IWGAV (International Working Group for Antibody Validation) criteria for antibody validation (30). Antibodies used for the immunohistochemical example images are listed in data S4. Protocol optimization was performed on a test TMA containing 20 different normal tissues. The stained slides were digitized with Scanscope AT2 (Leica Aperio, Vista, CA, USA). All images were manually evaluated by two independent observers, comprising a total of 576 images per antibody, covering 15,320 different human genes and publicly available on v20.proteinatlas.org.
Gene classification
Clusters were normalized using trimmed means of M (TMM) using the tmm function from NOISeq (31) with a median column as reference, with the parameters doWeighting = T and logratioTrim = 0.3. Clusters were aggregated per cell type by using the median expression of each gene. Genes were then classified as per standard HPA procedure as described in table S1.
Generation of network plot
The network plot was generated in Cytoscape 3.6.1 (32), and nodes were filtered to remove complexity such that nodes were displayed if they (i) contained cell type–enriched genes, (ii) contained at least five genes, or (iii) ranked top two largest nodes for any connected cell type and contained at least three genes.
Acknowledgments
We thank the entire staff of the Human Protein Atlas program and the Science for Life Laboratory (SciLifeLab) for their valuable contributions. Funding: Main funding was provided from the Knut and Alice Wallenberg Foundation (WCPR) and the Erling Persson Foundation (KCAP). The computations and data handling were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at UPPMAX partially funded by the Swedish Research Council through grant agreement no. 2018-05973. Author contributions: M.U. and C.L. conceived and designed the analysis. M.K., C.Z., W.Z., L.M., E.S., F.E., M.A., O.A., X.L., M.O., A.M., L.F., J.M., Y.L., F.P., and C.L. collected and contributed data to the study. M.K., C.Z., W.Z., C.L., L.M., F.P., and M.U. performed the data analysis. K.v.F., M.Z., and P.O. created the database portal. M.U., M.K., C.Z., and C.L. drafted the manuscript. All authors read and approved the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Single–cell type gene expression data are available to download on the Human Protein Atlas resource download page (https://proteinatlas.org/about/download). Scripts for data analysis can be found at GitHub (https://github.com/maxkarlsson/HPA-SingleCellType).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/31/eabh2169/DC1
References and Notes
- 1.Crosetto N., Bienko M., van Oudenaarden A., Spatially resolved transcriptomics and beyond. Nat. Rev. Genet. 16, 57–66 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Luecken M. D., Theis F. J., Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 15, e8746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Regev A., Teichmann S. A., Lander E. S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M., Clevers H., Deplancke B., Dunham I., Eberwine J., Eils R., Enard W., Farmer A., Fugger L., Göttgens B., Hacohen N., Haniffa M., Hemberg M., Kim S., Klenerman P., Kriegstein A., Lein E., Linnarsson S., Lundberg E., Lundeberg J., Majumder P., Marioni J. C., Merad M., Mhlanga M., Nawijn M., Netea M., Nolan G., Pe’er D., Phillipakis A., Ponting C. P., Quake S., Reik W., Rozenblatt-Rosen O., Sanes J., Satija R., Schumacher T. N., Shalek A., Shapiro E., Sharma P., Shin J. W., Stegle O., Stratton M., Stubbington M. J. T., Theis F. J., Uhlen M., van Oudenaarden A., Wagner A., Watt F., Weissman J., Wold B., Xavier R., Yosef N.; Human Cell Atlas Meeting Participants , The Human Cell Atlas. eLife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uhlen M., Fagerberg L., Hallstrom B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A. K., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P. H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., Ponten F., Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Sjostedt E., Zhong W., Fagerberg L., Karlsson M., Mitsios N., Adori C., Oksvold P., Edfors F., Limiszewska A., Hikmet F., Huang J., Du Y., Lin L., Dong Z., Yang L., Liu X., Jiang H., Xu X., Wang J., Yang H., Bolund L., Mardinoglu A., Zhang C., von Feilitzen K., Lindskog C., Pontén F., Luo Y., Hökfelt T., Uhlén M., Mulder J., An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367, eaay5947 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Stahl P. L., Salmén F., Vickovic S., Lundmark A., Navarro J. F., Magnusson J., Giacomello S., Asp M., Westholm J. O., Huss M., Mollbrink A., Linnarsson S., Codeluppi S., Borg Å., Pontén F., Costea P. I., Sahlén P., Mulder J., Bergmann O., Lundeberg J., Frisén J., Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Renier N., Wu Z., Simon D. J., Yang J., Ariel P., Tessier-Lavigne M., iDISCO: A simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 159, 896–910 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Lun A. T., Bach K., Marioni J. C., Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 17, 75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y., Song W., Wang J., Wang T., Xiong X., Qi Z., Fu W., Yang X., Chen Y. G., Single-cell transcriptome analysis reveals differential nutrient absorption functions in human intestine. J. Exp. Med. 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parikh K., Antanaviciute A., Fawkner-Corbett D., Jagielowicz M., Aulicino A., Lagerholm C., Davis S., Kinchen J., Chen H. H., Alham N. K., Ashley N., Johnson E., Hublitz P., Bao L., Lukomska J., Andev R. S., Björklund E., Kessler B. M., Fischer R., Goldin R., Koohy H., Simmons A., Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 567, 49–55 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Liao J., Yu Z., Chen Y., Bao M., Zou C., Zhang H., Liu D., Li T., Zhang Q., Li J., Cheng J., Mo Z., Single-cell RNA sequencing of human kidney. Sci. Data 7, 4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacParland S. A., Liu J. C., Ma X. Z., Innes B. T., Bartczak A. M., Gage B. K., Manuel J., Khuu N., Echeverri J., Linares I., Gupta R., Cheng M. L., Liu L. Y., Camat D., Chung S. W., Seliga R. K., Shao Z., Lee E., Ogawa S., Ogawa M., Wilson M. D., Fish J. E., Selzner M., Ghanekar A., Grant D., Greig P., Sapisochin G., Selzner N., Winegarden N., Adeyi O., Keller G., Bader G. D., McGilvray I. D., Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 9, 4383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qadir M. M. F., Álvarez-Cubela S., Klein D., van Dijk J., Muñiz-Anquela R., Moreno-Hernández Y. B., Lanzoni G., Sadiq S., Navarro-Rubio B., García M. T., Díaz Á., Johnson K., Sant D., Ricordi C., Griswold A., Pastori R. L., Domínguez-Bendala J., Single-cell resolution analysis of the human pancreatic ductal progenitor cell niche. Proc. Natl. Acad. Sci. U.S.A. 117, 10876–10887 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L., Yu P., Zhou B., Song J., Li Z., Zhang M., Guo G., Wang Y., Chen X., Han L., Hu S., Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 22, 108–119 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Vieira Braga F. A., Kar G., Berg M., Carpaij O. A., Polanski K., Simon L. M., Brouwer S., Gomes T., Hesse L., Jiang J., Fasouli E. S., Efremova M., Vento-Tormo R., Talavera-López C., Jonker M. R., Affleck K., Palit S., Strzelecka P. M., Firth H. V., Mahbubani K. T., Cvejic A., Meyer K. B., Saeb-Parsy K., Luinge M., Brandsma C. A., Timens W., Angelidis I., Strunz M., Koppelman G. H., van Oosterhout A. J., Schiller H. B., Theis F. J., van den Berge M., Nawijn M. C., Teichmann S. A., A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 25, 1153–1163 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Henry G. H., Malewska A., Joseph D. B., Malladi V. S., Lee J., Torrealba J., Mauck R. J., Gahan J. C., Raj G. V., Roehrborn C. G., Hon G. C., MacConmara M. P., Reese J. C., Hutchinson R. C., Vezina C. M., Strand D. W., A cellular anatomy of the normal adult human prostate and prostatic urethra. Cell Rep. 25, 3530–3542.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo J., Grow E. J., Mlcochova H., Maher G. J., Lindskog C., Nie X., Guo Y., Takei Y., Yun J., Cai L., Kim R., Carrell D. T., Goriely A., Hotaling J. M., Cairns B. R., The adult human testis transcriptional cell atlas. Cell Res. 28, 1141–1157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vento-Tormo R., Efremova M., Botting R. A., Turco M. Y., Vento-Tormo M., Meyer K. B., Park J. E., Stephenson E., Polański K., Goncalves A., Gardner L., Holmqvist S., Henriksson J., Zou A., Sharkey A. M., Millar B., Innes B., Wood L., Wilbrey-Clark A., Payne R. P., Ivarsson M. A., Lisgo S., Filby A., Rowitch D. H., Bulmer J. N., Wright G. J., Stubbington M. J. T., Haniffa M., Moffett A., Teichmann S. A., Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563, 347–353 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sole-Boldo L., Raddatz G., Schütz S., Mallm J.-P., Rippe K., Lonsdorf A. S., Rodríguez-Paredes M., Lyko F., Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol. 3, 188 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menon M., Mohammadi S., Davila-Velderrain J., Goods B. A., Cadwell T. D., Xing Y., Stemmer-Rachamimov A., Shalek A. K., Love J. C., Kellis M., Hafler B. P., Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration. Nat. Commun. 10, 4902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J., Cheung F., Shi R., Zhou H., Lu W.; CHI Consortium , PBMC fixation and processing for chromium single-cell RNA sequencing. J. Transl. Med. 16, 198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodge R. D., Bakken T. E., Miller J. A., Smith K. A., Barkan E. R., Graybuck L. T., Close J. L., Long B., Johansen N., Penn O., Yao Z., Eggermont J., Höllt T., Levi B. P., Shehata S. I., Aevermann B., Beller A., Bertagnolli D., Brouner K., Casper T., Cobbs C., Dalley R., Dee N., Ding S. L., Ellenbogen R. G., Fong O., Garren E., Goldy J., Gwinn R. P., Hirschstein D., Keene C. D., Keshk M., Ko A. L., Lathia K., Mahfouz A., Maltzer Z., McGraw M., Nguyen T. N., Nyhus J., Ojemann J. G., Oldre A., Parry S., Reynolds S., Rimorin C., Shapovalova N. V., Somasundaram S., Szafer A., Thomsen E. R., Tieu M., Quon G., Scheuermann R. H., Yuste R., Sunkin S. M., Lelieveldt B., Feng D., Ng L., Bernard A., Hawrylycz M., Phillips J. W., Tasic B., Zeng H., Jones A. R., Koch C., Lein E. S., Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hawrylycz M. J., Lein E. S., Guillozet-Bongaarts A. L., Shen E. H., Ng L., Miller J. A., van de Lagemaat L. N., Smith K. A., Ebbert A., Riley Z. L., Abajian C., Beckmann C. F., Bernard A., Bertagnolli D., Boe A. F., Cartagena P. M., Chakravarty M. M., Chapin M., Chong J., Dalley R. A., Daly B. D., Dang C., Datta S., Dee N., Dolbeare T. A., Faber V., Feng D., Fowler D. R., Goldy J., Gregor B. W., Haradon Z., Haynor D. R., Hohmann J. G., Horvath S., Howard R. E., Jeromin A., Jochim J. M., Kinnunen M., Lau C., Lazarz E. T., Lee C., Lemon T. A., Li L., Li Y., Morris J. A., Overly C. C., Parker P. D., Parry S. E., Reding M., Royall J. J., Schulkin J., Sequeira P. A., Slaughterbeck C. R., Smith S. C., Sodt A. J., Sunkin S. M., Swanson B. E., Vawter M. P., Williams D., Wohnoutka P., Zielke H. R., Geschwind D. H., Hof P. R., Smith S. M., Koch C., Grant S. G. N., Jones A. R., An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K. R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S., Mero P., Dirks P., Sidhu S., Roth F. P., Rissland O. S., Durocher D., Angers S., Moffat J., High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163, 1515–1526 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Wang T., Birsoy K., Hughes N. W., Krupczak K. M., Post Y., Wei J. J., Lander E. S., Sabatini D. M., Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uhlen M., Karlsson M. J., Zhong W., Tebani A., Pou C., Mikes J., Lakshmikanth T., Forsström B., Edfors F., Odeberg J., Mardinoglu A., Zhang C., von Feilitzen K., Mulder J., Sjöstedt E., Hober A., Oksvold P., Zwahlen M., Ponten F., Lindskog C., Sivertsson Å., Fagerberg L., Brodin P., A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 366, eaax9198 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Ang S. Y., Evans B. A., Poole D. P., Bron R., DiCello J., Bathgate R. A. D., Kocan M., Hutchinson D. S., Summers R. J., INSL5 activates multiple signalling pathways and regulates GLP-1 secretion in NCI-H716 cells. J. Mol. Endocrinol. 60, 213–224 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I. M., Edlund K., Lundberg E., Navani S., Al-Khalili Szigyarto C., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P.-H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., Pontén F., Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Kampf C., Olsson I., Ryberg U., Sjöstedt E., Pontén F., Production of tissue microarrays, immunohistochemistry staining and digitalization within the human protein atlas. J. Vis. Exp. 2012, 3620 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uhlen M., Bandrowski A., Carr S., Edwards A., Ellenberg J., Lundberg E., Rimm D. L., Rodriguez H., Hiltke T., Snyder M., Yamamoto T., A proposal for validation of antibodies. Nat. Methods 13, 823–827 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tarazona S., Furió-Tarí P., Turrà D., Pietro A. D., Nueda M. J., Ferrer A., Conesa A., Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 43, e140 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/31/eabh2169/DC1