

Blocking collaborative pathways within the integrated signaling network synergistically disrupts the migration of cells.
Abstract
Integrating signals is essential for cell survival, leading to the concept of synthetic lethality. However, how signaling is integrated to control cell migration remains unclear. By conducting a “two-hit” screen, we revealed the synergistic reduction of cell migration when serine-threonine kinase 40 (STK40) and mitogen-activated protein kinase (MAPK) were simultaneously suppressed. Single-cell analyses showed that STK40 knockdown reduced cell motility and coordination by strengthening focal adhesion (FA) complexes. Furthermore, STK40 knockdown reduced the stability of yes-associated protein (YAP) and subsequently decreased YAP transported into the nucleus, while MAPK inhibition further weakened YAP activities in the nucleus to disturb FA remodeling. Together, we unveiled an integrated STK40-YAP-MAPK system regulating cell migration and introduced “synthetic dysmobility” as a novel strategy to collaboratively control cell migration.
INTRODUCTION
The cell is a robust signaling system. Within it, various pathways interact with each other for survival and cellular functions (1–3), creating an impeccable integrated system for signaling. Thus, current antimicrobial and anticancer treatments used combination therapies to simultaneously disrupt multiple pathways as a practice of “synthetic lethality” (4–7). This integrated signaling system is critical for cell survival, yet whether it also contributes to cell migration and other cellular functions remains elusive. Moreover, there are extensive studies on cytoskeletal dynamics in cell migration (8–12). However, how different pathways are integrated within the cell signaling network to control cytoskeleton for cell migration remains unclear. To address these questions, we conducted a short hairpin RNA (shRNA)–drug two-hit screen to study signaling cross-talk during cell migration in human umbilical vein endothelial cells (HUVECs). Results from this screen provided a proof of concept for the proposal of “synthetic dysmobility.” Furthermore, in-depth investigations of the interactions identified from this screen lead to the discovery of an integrated serine-threonine kinase 40 (STK40)–yes-associated protein (YAP)–mitogen-activated protein kinase (MAPK) system driving cell migration.
RESULTS
Two-hit screen to elucidate signaling interaction in cell migration
In this screen, HUVECs were doubly “hit” by one shRNA and one small-molecule inhibitor and evaluated by the standardized scratch-wound healing assay (Fig. 1A) (13). We designed a hypothesis-driven targeted screen using selected shRNAs and inhibitors to primarily provide the proof of concept on synthetic dysmobility. For the first hit, we aimed to use shRNAs suppressing genes from different functional modules during cell migration (13). Therefore, we purposefully elected all 119 genes identified from both of the previously reported comprehensive cell migration screens (13, 14) instead of applying a genome-wide search. For the second hit, three effective and well-defined drugs inhibiting store-operated Ca2+ entry (15), Rho-associated kinase (ROCK) (16, 17), or MAPK signaling (18) were used for we aimed to cover characterization in major signaling regulators, structural components, and their coordinators during cell migration. Concentrations of shRNAs and drugs were carefully titrated to minimize alterations on cell viability (fig. S1A), while their effects on wound healing were maintained (Fig. 1, B and C).
Fig. 1. Two-hit screen revealed STK40-MAPK interaction in sheet migration.
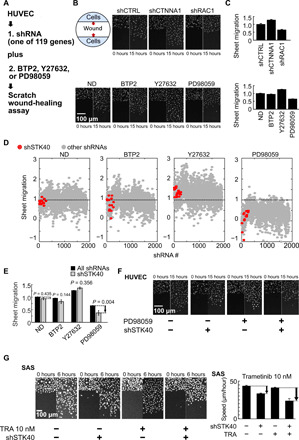
(A) Scheme for the two-hit sheet migration screen. (B) Representative images showing sheet migration of HUVECs treated with shRNAs: shCTRL, shCTNNA1, and shRAC1; and drug inhibitors: ND [no drug; i.e., dimethyl sulfoxide (DMSO)], 2 μM Ca2+ inhibitor BTP2, 5 μM ROCK inhibitor Y27632, and 10 μM MAPK inhibitor PD98059. Hoechst stain marked cell nucleus. (C) Quantification of the sheet migration effects by shRNAs and drugs in (B). (D) Dot plot demonstrates sheet migration effects by individual shRNAs. Red circles resemble shSTK40s, while gray circles resemble other individual shRNAs. (E) Black bars resemble average sheet migration of all shRNAs (n = 1890), while gray bars resemble average sheet migration of shSTK40 (n = 15) upon specific drug treatments. Notice that shSTK40 markedly decreased sheet migration under PD98059. (F) Representative images of shSTK40- and/or PD98059-treated sheet migration in our HUVEC screen. (G) SAS cells treated with shSTK40 and another MAPK inhibitor trametinib (TRA) also revealed synergistic suppression in sheet migration. Left: Representative images. Right: Quantification. Error bars denote means ± SEM. n = 6 biological repeats for each group.
Proceeding with the screening experiments, raw data of sheet migration were processed by cell density correction (fig. S1B) and normalization using control shRNAs within each plate (Fig. 1D). We then calculated z scores of each shRNA based on the values of control shRNAs within each group (data S1), followed by comparing the effects of shRNAs with or without drugs to evaluate their interaction (fig. S1C). When the efficacy of an shRNA was magnified or reduced by a drug, we defined the shRNA-drug interaction to be synergistic (fig. S1D) or antagonistic (fig. S1E), respectively. This strategy enabled us to identify many interesting shRNA-drug interaction pairs, including 20 pairs in the BTP2 group (11 synergistic and 9 antagonistic), 23 pairs in the Y27632 group (11 synergistic and 12 antagonistic), and 14 pairs in the PD98059 group (fig. S1, C to E). Comprehensive literature search further revealed that many of these identified pairs have been reported before (data S2), including the antagonistic interaction between Ste20-like serine/threonine protein kinase (SLK) and ROCK via suppressive phosphorylation of RhoA by SLK (19–21), the synergistic interaction between fibroblast growth factor receptor 1 (FGFR1)/insulin-like growth factor 1 receptor and MAPK via classical receptor signaling cascade (22–26), and the antagonistic interaction between Rho GTPase Activating Protein 29 (ARHGAP29) and ROCK (27–29) via regulators of guanosine triphosphatase activities. Identification of these interaction pairs with literature support indicates that our screen has sufficient power to find important and specific shRNA-drug interactions.
We further examined these shRNA-drug pairs and focused on the STK40-MAPK interaction (Fig. 1D and fig. S1, C and D). Previous studies suggested potential involvement of STK40 on MAPK modulation in tissue development (30), but its mechanistic linkage remained unclear. We hence began to characterize the role of STK40-MAPK interaction in cell migration. As shown in the screen, shSTK40, together with MAPK suppressor PD98059, synergistically reduced sheet migration in HUVECs (Fig. 1, D to F, and fig. S1, C and D). To verify STK40-MAPK interaction in sheet migration, we further treated different cell lines with shSTK40 and another MAPK inhibitor trametinib using HUVECs (fig. S2E), head and neck cancer SAS cells (Fig. 1G and fig. S2D), trophoblast 3A-Sub E cells (fig. S2F), and keratinocyte HSC3 cells (fig. S2G). Moreover, concentrations of trametinib were carefully titrated to generate the dose-response curves and/or median inhibitory concentration (IC50) values without and with shSTK40 in each cell line. On the basis of the concept of synthetic interaction (fig. S2, A to C), if synergistic interactions exist between shSTK40 and trametinib, then we would notice the decrease in IC50 in trametinib by shSTK40 (fig. S2A) and leftward shift of dose-response curves in trametinib by shSTK40 (fig. S2C). In addition, synergistic suppressive effects by shSTK40 would be more prominent in low rather than high concentration of trametinib (fig. S2C). Results with all four cell lines supported all above speculations, suggesting robust synergistic suppression between shSTK40 and trametinib (fig. S2, D to G). Therefore, STK40 interacts with MAPK signaling to control sheet migration. Furthermore, using HUVECs in the transwell dextran permeability assay, we demonstrated that shSTK40, together with trametinib, synergistically disrupted the barrier capacity of endothelial cells (fig. S2H), indicating the importance of STK40-MAPK synergism in physiological functions.
STK40 changes cell migration by mediating adhesion cytoskeletons
Next, we explored the mechanism underlying STK40-MAPK interaction by focusing on the role of STK40. Since previous screens showed that STK40 knockdown reduced cell-cell coordination (13, 14), we analyzed migration behaviors of individual SAS cells by automatic single-cell tracing (Fig. 2 and figs. S3 and S4). Both random migration (Fig. 2A and movies S1 and S2) and wound healing (fig. S3A and movies S3 and S4) assays were used because they belong to different modes of cell migration. Cell speed is determined by motility and adhesion in random migration but by polarity in addition to motility for wound healing. We observed markedly impaired cell speed and cell-cell coordination by STK40 knockdown in random migration (Fig. 2, A and B). However, in wound healing, cell speed was mildly reduced by shSTK40, and we did not observe consistent change in directionality (fig. S3, A and C). These differential effects of shSTK40 implied that shSTK40 regulated cell adhesion rather than cell migration polarity. Therefore, the role of STK40 is to regulate cell adhesion during cell migration.
Fig. 2. STK40 knockdown reduced cell migration by enhancing FA complexes.
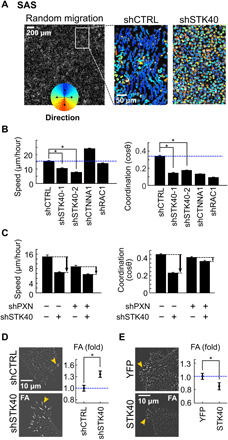
(A) Random migration experiments in SAS cells. Left: Cell nuclei were labeled with Hoechst 33342 for tracing of their migration. Right: Colored lines depicted traces and directions of individual migrating cells treated with shCTRL or shSTK40 within the 2-hour period. (B) Well-based quantification showed prominent reduction of cell speed and cell-cell coordination by shSTK40. (C) Double inhibition experiment in random migration showed the reduction of shSTK40 effect by PXN knockdown (shPXN) in cell speed, and the reversal of the shSTK40 effect by shPXN in coordination. (D and E) Left: Representative images of FA complexes transduced with (D) STK40 shRNAs and (E) control [yellow fluorescent protein (YFP)] and STK40 constructs. Right: Quantification of the integrated FA signals (described in fig. S5). Error bars denote means ± SEM. *P < 0.05.
We further explored the specific adhesion cytoskeleton structure regulated by STK40 with a double-inhibition assay. SAS cells were simultaneously treated with shSTK40 and shRNAs/inhibitors suppressing molecules in the adhesion cytoskeleton structure [Rac1 for F-actin (31), α-catenin for adherens junction (32), paxillin (PXN) for focal adhesion (FA) complexes (33, 34), and myosin light-chain kinase for myosin (35)] (fig. S3D). We characterized the cell migration behaviors of these treated cells through wound healing (quantified by speed and directionality) (fig. S3, E to H) and random migration (quantified by speed and coordination) (Fig. 2C; fig. S4, B to E; and data S3). In wound healing, shSTK40 reduced cell speed and cell directionality in the absence of shRNA targeting Ras-related C3 botulinum toxin substrate 1 (shRAC1), but it failed to affect both parameters with the presence of shRAC1 (fig. S3E). In random migration, the capacity of shSTK40 to reduce cell speed and coordination also disappeared under shRAC1 (fig. S4B). These results indicated that shRAC1 abolished the effect of shSTK40 on cell speed, coordination, and directionality (figs. S3E and S4B). Similarly, the capacity of ML9 to increase cell speed (figs. S3F and S4C) and coordination (fig. S4C) disappeared under the presence of shSTK40, indicating that shSTK40 abolished the effect of myosin light chain kinase inhibitor ML9 on the same parameters (figs. S3F and S4C). These epistatic effects implied that actin and myosin were both involved in the regulation of STK40-mediated adhesion structure. In contrast, shRNA targeting catenin alpha 1 (shCTNNA1) showed additive effects to shSTK40 on cell migration parameters (figs. S3G and S4D), indicating that the adherens junction was independent from STK40-mediated structure. Notably, shPXN not only reduced effects on speed and directionality of shSTK40 (Fig. 2C and fig. S3H) but also reversed its suppression in coordination (Fig. 2C). This implied that PXN knockdown antagonized the effect of shSTK40. Since PXN is the major component of the FA complex, our results suggested that shSTK40 might disrupt cell migration by strengthening FA complexes.
To answer whether shSTK40 enhanced FA complexes, we examined the signals of the anti-PXN antibody–labeled FA in SAS cells. shSTK40 increased both PXN-labeled FA area and mean PXN signals overlaying the area (Fig. 2D and fig. S5), indicating that shSTK40 not only increased the size of FA in two-dimensional manners but also increased the height or strengthened the structure of FA. Moreover, the average number of FA in individual cells was not notably altered by shSTK40 (fig. S6A), suggesting that STK40 was not involved in the initiation process of FA but was responsible for its turnover. These STK40 effects on FA were further confirmed by the results that shSTK40 also increased integrated FA signals (total signals from individual FAs) in HepG2 cells (fig. S6B) and HUVECs (fig. S6C) and that overexpression of STK40 reduced integrated FA signals in SAS cells (Fig. 2E). Thus, our results confirmed that shSTK40 disrupted cell migration by strengthening FA complexes.
Next, we explored how shSTK40 altered FA turnover, to see whether shSTK40 strengthened FA by increasing formation or decreasing degradation of FA complexes. We used SAS cells overexpressing enhanced green fluorescent protein (EGFP)–tagged PXN in live-cell imaging for the measurement (fig. S7A and movie S5) and analyses (Fig. 3, A and B, and fig. S7, B and C) of FA dynamics in SAS cells. STK40 knockdown increased the rates of both FA formation and degradation (Fig. 3C and fig. S7C) but did not notably shorten the duration of an average FA turnover cycle (Fig. 3D). Therefore, we inferred that shSTK40 increased FA by facilitating its formation, while increased FA degradation probably reflected compensatory responses. Because force-mediated FA strengthening is the major mechanism of FA formation (36–38), we examined whether this mechanism also applied to shSTK40-mediated FA enhancement. When we labeled stress fibers with phalloidin and monitored the contractile activities by overlying phalloidin with the phosphorylated myosin light chain (p-MLC) (17), shSTK40 increased the number of stress fibers and their contractile activities (Fig. 3E). These results indicated that shSTK40 altered FA via force-mediated FA strengthening, which was further supported by the elimination of shSTK40 effects on FA using ROCK inhibitor or myosin inhibitor (Fig. 3, F and G, and fig. S7D). In contrast, although calpain is the major mechanism of FA degradation (39, 40) with its suppression increasing average FA sizes (fig. S7E) by creating huge FAs (Fig. 3H), its effect was not altered by the presence of shSTK40 (Fig. 3H). These independent effects between calpain knockdown and shSTK40 on FA dynamics further support the involvement of STK40 on force-mediated FA remodeling.
Fig. 3. STK40 knockdown enhanced force-mediated FA formation.
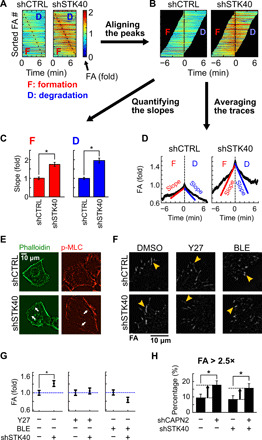
(A to D) Quantification of FA dynamics using SAS cells expressing GFP-tagged PXN. (A) Color maps of FAs sorted by the timing of peak FA signals. (B) Sorted FAs were realigned by the timing of peak signals. (C) Quantification of slopes [derivatives of FA signals over time, depicted in (D)] during FA formation and degradation. (D) Average traces of FA signals based on aligned FA signals in (B). (E) shSTK40 increased the number of stress fibers (labeled by phalloidin) and their overlaying p-MLC in SAS cells. (F) Representative images of FA in SAS cells treated with shSTK40 plus DMSO, 5 μM Y27632 (Y27), or 5 μM blebbistatin (BLE) overnight. (G) Quantification of FA in (F). Note the abolishment of shSTK40 effect by both drugs. (H) Bar graph of the percentage of “huge FAs” in cells treated with STK40 knockdown (shSTK40) and/or calpain knockdown (shCAPN2). Huge FAs were defined as the integrated FA intensity of >2.5× of the average FA signal in shCTRL. Note the increased percentage of huge FAs by shCAPN2 but not by shSTK40. Error bars denote means ± SEM. *P < 0.05.
We further studied the mode of action in STK40-FA remodeling using three approaches. We first dissected the role of STK40 by altering functions and locations of STK40 proteins and examining their effects on FA. STK40 was reported to regulate cell differentiation as a putative nuclear kinase (30, 41–43) and to maintain protein stability as a cytoplasmic pseudo-kinase (44, 45). Thus, we want to know either STK40 location or kinase activity determined FA remodeling. Again, SAS cells overexpressing STK40 protein of full length (FL) had reduced FA (Fig. 4, A and B) and increased cell migration speed (fig. S8A) compared to cells overexpressing the yellow fluorescent protein (YFP) plasmid. In contrast, truncated STK40 with merely its kinase domain (KD) failed to reduce FA. Truncated STK40 without its KD (ΔKD) reduced FA and increased cell speed as the FL protein did (fig. S8A), indicating that the KD of STK40 was not mandatory for FA remodeling. Moreover, translocating STK40 from the nucleus to the cytosol by tagging it with a nuclear exportation sequence did not significantly alter its reduction effect on FA (Fig. 4, C and D). Therefore, STK40 effects on FA dynamics were potentially not directly related to its kinase function or nuclear localization.
Fig. 4. STK40 and MAPK signaling regulated YAP-mediated FA remodeling in collaborative manners.
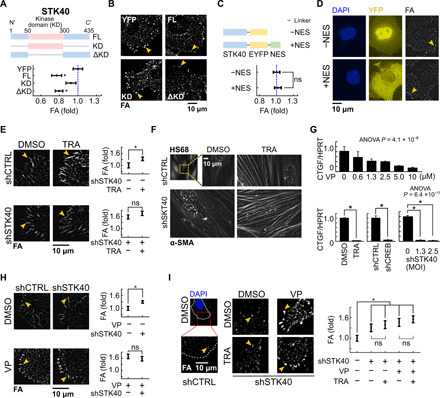
(A) Top: Diagram of FL STK40, STK40 with KD only, and ΔKD constructs expressed in SAS cells. Bottom: Quantification of integrated FA intensity in SAS cells overexpressing above vectors. YFP, control vector. (B) Representative images of FAs of (A). (C) Top: Diagram of STK40-EYFP and STK40-EYFP-NES constructs expressed in SAS cells. Bottom: Quantification of integrated FA intensity in SAS cells overexpressing above vectors. (D) Representative images of FA of (C). 4of (Diamidino-2-phenylindole (DAPI) channel revealed locations of cell nuclei. YFP channel denotes subcellular distribution of above vectors. Note the distinct cytosolic translocation of STK40 by the NES. (E) Representative images and quantification of FA in SAS cells without or with shSTK40 treated with 100 nM trametinib overnight. Note that the trametinib effect was masked by STK40 knockdown in high efficiency. (F) Representative images of α-SMA in fibroblast HS68 cells without or with shSTK40 treated with 100 nM trametinib overnight. Note that the lower knockdown efficiency of STK40 in HS68 cells than in SAS cells resulted in marked synergistic enrichment of α-SMA on stress fibers of HS68 cells under combination treatment. (G) YAP activity [labeled by connective tissue growth factor (CTGF) expression] was decreased in SAS cells treated with (top) verteporfin (VP) and (bottom) trametinib, shRNA targeting cyclic adenosine 3′,5′-monophosphate response element–binding protein (shCREB), or shSTK40. HPRT, hypoxanthine-guanine phosphoribosyltransferase. (H) Representative images and quantification of FA in SAS cells without or with VP treated with shCTRL or shSTK40. (I) Representative images and quantification of FA in SAS cells with shSTK40 and treated with VP and/or trametinib. Note that shSTK40 and VP abolished effects of trametinib. Error bars denote means ± SEM. *P < 0.05. ns, not significant.
We then resorted to literature and examined whether STK40-mediated FA dynamics was related to known functions of STK40 and/or FA-mediated signaling. Although STK40 was reported to regulate the differentiation of several cell types, our explorations revealed that shSTK40 did not alter cell size, cell cycle, and major markers of epithelial-mesenchymal transition (fig. S8, B to D), further suggesting that STK40 did not alter FA by regulating cell proliferation or differentiation in the nucleus. Next, we investigated whether STK40 affected signaling regulators of FA, specifically FA kinase (FAK) signaling (46–48). Our immunochemistry data showed that shSTK40 did not change the expression and phosphorylation of FAK (fig. S8E), indicating that STK40 exerted no direct effect on FAK signaling. In sum, our first two approaches indicated that, in STK40-mediated FA remodeling, the participation of kinase activity from STK40, STK40 functions in the nucleus, indirect effects from cell differentiation and FAK signaling were all excluded.
STK40 collaborates with MAPK on YAP-mediated FA remodeling
We took a third approach by turning back to our screen results and examined whether MAPK interacted with STK40 for this force-mediated FA strengthening. The MAPK kinase (MEK) inhibitor trametinib enhanced FA in SAS cells similarly to shSTK40 (Fig. 4E). Moreover, high efficiency of STK40 knockdown reduced the effects of trametinib on FA and cell speed, and vice versa (Fig. 4E and fig. S9A). These epistatic effects of trametinib in high concentration to STK40 knockdown in high efficiency showed that STK40 and MAPK signaling were involved in the same pathway for FA remodeling and cell migration. Furthermore, immunofluorescence of α–smooth muscle actin (α-SMA) revealed its prominent enrichment on stress fibers under simultaneous shSTK40 plus trametinib treatment (Fig. 4F). Together, our results confirmed the participation of STK40-MAPK synergism in force-mediated FA remodeling.
STK40 was suggested to regulate tissue development by MAPK signaling (30, 49). We also observed that shSTK40 mildly reduced MEK and extracellular signal–regulated kinase (ERK) activities (fig. S9B) and increased the sensitivities in MAPK suppression by trametinib (fig. S9C), while the c-RAF protein levels decreased (fig. S9D). These data supported the role of STK40 in MAPK signaling. However, while shSTK40 only slightly reduced MAPK activities, trametinib completely suppressed it (fig. S9B). However, both shSTK40 and trametinib induced a marked and comparable increase in FA (Fig. 4E). These disproportionate results between shSTK40-induced MAPK suppression and FA enhancement suggested the existence of missing links between STK40 and MAPK signaling.
We investigated the missing links. The increase in FA by trametinib was seen 16 hours after treatment (Fig. 4E) but was not visible within the first 8 hours (fig. S9E), indicating potential involvement of transcriptional control in MAPK-mediated FA remodeling. Thus, we explored whether there existed any transcriptional machinery specifically responsible for FA remodeling. Recent reports demonstrated that suppression of the YAP pathway increased FA formation (29, 50, 51) similarly to our results of STK40 knockdown or MAPK inhibition. These similarities in phenotypes encouraged us to examine whether the YAP pathway was involved in STK40-MAPK interaction. Using the expression levels of connective tissue growth factor (CTGF) (52), cysteine-rich angiogenic inducer 61 (CYR61) (53), ankyrin repeat domain 1 (ANKDR1) (54), and 8XGTIIC luciferase assay (55) as the readout, we demonstrated that the activity of YAP was suppressed by STK40 knockdown and also by MAPK suppression (Fig. 4G and fig. S9, F to I) (56). The YAP inhibitor verteporfin (VP) not only increased FA as reported previously (51) but also fully abolished the effects of STK40 knockdown (Fig. 4H) or MAPK inhibition (Fig. 4I). Moreover, YAP knockdown also fully masked the effect of shSTK40 on FA (fig. S9J). Therefore, we inferred that the YAP pathway took a dominant role on STK40-MAPK interaction for FA remodeling.
Next, we worked on the mechanism of how STK40 and MAPK were involved in YAP-mediated FA remodeling. Western blots suggested that STK40 regulated YAP levels as it remarkably abolished trametinib-mediated compensatory YAP accumulation (fig. S10B). Literature showed that YAP localization was critical for its activity (57), as YAP in the nucleus regulated gene expression, while YAP in the cytosol was prone to be degraded (58). We thus examined whether STK40 altered the expression and spatial distribution of YAP at the subcellular level. Immunofluorescence revealed that shSTK40 reduced YAP level both in the nucleus and cytosol (fig. S10A), together with the reduction of its nucleus-to-cytosol (N/C) ratio (Fig. 5A). The exportin inhibitor leptomycin B (59), which blocked nuclear exportation to cause YAP accumulation in the nucleus (fig. S10C), abolished shSTK40-induced FA increase (Fig. 5B). Furthermore, the decrease in YAP level by shSTK40 was rescued by overexpression of STK40 in FL and in its KD-truncated form (Fig. 5C and fig. S10D), compatible with their rescue effects on FA dynamics (Fig. 4A). These results indicated that shSTK40 suppressed YAP activities via reducing YAP expression. This suppression might occur by reducing its expression or by promoting its degradation. The YAP reduction subsequently exerted less effect in cell nucleus for FA remodeling. Previous reports showed that STK40 bound to Constitutive Photomorphogenic Protein 1 (COP1) (44), which functioned as the E3 ubiquitin ligase for protein degradation. Hence, shSTK40 might alter YAP stability via the degradation machinery. STK40 knockdown was also reported to increase the expression of histone deacetylase 5 (41), which in turn might increase YAP expression (60) and its effect on cytoskeletal remodeling. In our experiments, shSTK40 did not significantly alter mRNA levels of YAP (fig. S10E). In contrast, shSTK40 enhanced the degradation of YAP under cycloheximide treatment (Fig. 5D). Therefore, STK40 probably enhances protein stability of YAP, subsequently altering YAP effect for FA remodeling.
Fig. 5. STK40 and MAPK signaling regulated YAP-mediated FA remodeling in distinct mechanisms.
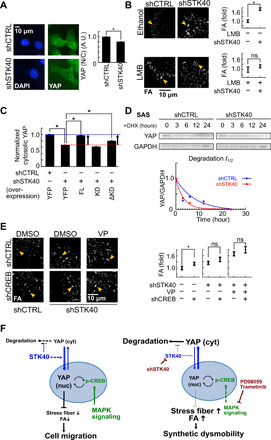
(A) Representative images and quantification of subcellular YAP distribution. Notice the decreased N/C ratio of YAP by shSTK40. A.U., arbitrary units. (B) Representative images and quantification of FA in SAS cells without or with leptomycin B (LMB) treated with shCTRL or shSTK40. Note the vanishment of STK40 effect under leptomycin B. (C) SAS cells received STK40 knockdown (shSTK40) followed by rescue experiments using STK40 of FL, KD, and ΔKD constructs. Their cytosolic YAP levels were measured using immunofluorescence. Notice that FL and ΔKD rescued YAP levels that were reduced by shSTK40. shCTRL, control shRNA; YFP, control vector. (D) Top: SAS cells with STK40 knockdown (shSTK40) were treated with cycloheximide. Their YAP protein was collected at different time points after cycloheximide treatment and measured using Western blots. Bottom: Quantification of the above results. Note that shSTK40 facilitated YAP degradation. (E) Representative images and quantification of FA in SAS cells with shSTK40 and treated with VP and/or shCREB. Note that shSTK40 and VP abolished effects of shCREB. Error bars denote means ± SEM. *P < 0.05. (F) Working models depict how STK40 and MAPK collaboratively regulate YAP activities (left) and how STK40 plus MAPK suppression causes synthetic dysmobility (right).
Last, we examined how MAPK inhibition regulated YAP effects on FA remodeling. Since both trametinib and shRNA targeting cyclic adenosine 3′,5′-monophosphate response element–binding protein (shCREB) suppressed YAP activity (Fig. 4G) and increased FA (Figs. 4G and 5E), we proposed that MAPK signaling augmented YAP effects on FA via its downstream target CREB (61). Similarly to trametinib treatment (Fig. 4I), the FA-enhancing effect by shCREB was also reduced under STK40 knockdown or YAP inhibition (Fig. 5E), confirming the involvement of CREB to augment YAP activities. Furthermore, with careful titration of shCREB levels, synergistic suppression of shSTK40 and shCREB on YAP activities was revealed as indicated by CTGF expression (fig. S10F). Notably, although decreasing YAP activity as indicated by CTGF reduction (Fig. 4G), trametinib increased intracellular YAP levels (fig. S10B). These results supported strong feedback interactions between CREB and YAP as reported in the literatures (61, 62), so MAPK inhibition resulted in compensatory increase in YAP levels. Together, we found an integrated YAP-centered system regulating cell adhesion and cell migration. Within this system, STK40 promoted YAP stability and, hence, its activity, which was further augmented by MAPK signaling via CREB to control adhesion cytoskeleton. Therefore, simultaneous suppression of STK40 and MAPK collaboratively disrupted FA turnover resulting in synthetic dysmobility (Fig. 5F).
DISCUSSION
Previous studies unveiled potential involvement of STK40 on cell differentiation and tissue development (30, 41–43, 45, 49), but its molecular mechanism has remained elusive. Our research resolved this issue by confirming the kinase-independent effect of STK40 on force-mediated FA turnover. We further elucidated that STK40 exerted effects on FA by altering YAP protein stability, as well as its subsequent N/C localization and activities. Hence, STK40 likely regulates cell differentiation and tissue development via its kinase-independent action on YAP signaling. Although direct molecular targets of STK40 remain to be identified, recent reports suggested that it altered protein activities via scaffold properties (44, 45). We thus speculate that STK40 may directly bind to proteins responsible for YAP stability. Candidate proteins include YAP itself, the machinery of YAP ubiquitination and degradation, and the machinery of its N/C shuttling, all of which are currently under our investigations.
Our results also identified the missing links between STK40 and MAPK (30) by unraveling how STK40 collaborated with MAPK on different levels of YAP control, as STK40 regulates YAP protein stability, while MAPK uses its downstream transcription factor CREB to enhance YAP signaling in mutual feedback loops (61, 62). Via this STK40-YAP-MAPK interaction, we demonstrated how different signaling pathways constituted an integrated system to drive cytoskeletons. First and foremost, our screen disclosed that cell migration could be blocked by multitargeting strategies mimicking the concept of synthetic lethality, which will lead to the development of combination drug therapies to stop cell migration in collaborative manners.
MATERIALS AND METHODS
Cell culture
SAS cells and HSC3 cells were gifts from J. S. Chia’s Lab (National Taiwan University, Taipei, Taiwan). 3A-Sub E cells were gifts from H.-N. Kung’s lab (National Taiwan University, Taipei, Taiwan). HepG2, HS68, and human embryonic kidney (HEK) 293T cells were purchased from American Type Culture Collection (Manassas, VA, USA), while HUVECs were purchased from Lonza (Lonza, Basel Stücki, Switzerland). Cells were maintained at 37°C and 5% CO2. SAS, 3A-Sub E, HepG2, HSC3, HS68, and HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Thermo Fisher Scientific, Waltham, MA, USA and HyClone, Logan, UT, USA). HUVECs were grown in endothelial cell growth medium-2 (EGM2) (Lonza). Culture medium was supplemented with 1% of penicillin/streptomycin (P/S) (Gibco) and 10% fetal bovine serum (FBS; HyClone). Cells were passaged using 0.5% trypsin-EDTA (Gibco) and washed with phosphate-buffered saline (PBS) (Corning, New York, USA) within 10 passages.
Generation of knockdown plasmids and overexpression constructs
To knockdown STK40 or other genes [RAC1, CTNNA1, PXN, YAP, CREB1, and calpain 2 (CAPN2)], we transfected shRNA plasmids bought from National RNAi Core (NRC; Academia Sinica, Taipei, Taiwan) into HEK293T cells to prepare lentiviruses for infection (data S4). For knockdown control, we directly bought shRNA lentiviruses (shLuc, shGFP, shLacZ, and shRFP) from NRC (Academia Sinica, Taipei, Taiwan) and mixed these viruses in the same quantity as our shControl. To overexpress STK40 or PXN, we created seven different constructs (data S5).
1) Control construct pLAS2w.eYFP.Pbsd was created by digesting the vector pLAS2w.Pbsd (Academia Sinica) using restriction enzymes Nhe I [New England Biolabs (NEB), MA, USA] and Eco RI (NEB) and then ligating cloned YFP using T4 ligase (NEB) to the vector. YFP sequence was cloned from primers eYFP_NheI and eYFP-EcoRI and pEX-SP-eYFP.STIM1 (gift from T. Meyer’s lab, Stanford, CA, USA) and was cloned to pLAS2w.Pbsd vector.
2) pLAS2w.STK40-P2A-EYFP was created by digesting vector pLAS2w.Pbsd using restriction enzymes Nhe I and Xba I (Takara, Shiga, Kyoto, Japan) and ligating two inserts, STK40 and porcine teschovirus-1 2A (P2A)-EYFP, to the vector. Cloned STK40 section was created by performing nested polymerase chain reaction (PCR) with two sets of primers: STK40_NstPCR1/F, STK40_NstPCR1/R and STK40 cds RE Nhe I F, STK40 cds Xba I R, using extracted cDNA from HepG2 cells as template. P2A-EYFP was cloned by performing PCR with primers P2A-EYFP RE Xba I F and P2A-EYFP RE Sbf I R using pLAS2w.FGFRIIIb-P2A-EYFP as template.
3) pLAS2w.STK40-EYFP was generated from pLAS2w.STK40-P2A-EYFP by performing PCR, site-directed mutagenesis, and self-ligation. Primers RE Xba I EYFP-F and STK40 cds Xba I R, template pLAS2w.STK40-P2A-EYFP, and reagents from QuikChange site-directed mutagenesis kit (Agilent, Santa Clara, CA, USA) were used for PCR. The enzyme DpnI from the kit was added to the PCR product to degrade the original template. Restriction enzyme Xba I was further used for digestion of the vector, and self-ligation was performed to create the final construct.
4) pLAS2w.STK40-EYFP-nuclear export signal (NES) was created by replacing STK40-EYFP from pLAS2w.STK40-EYFP with STK40-EYFP-NES. pLAS2w.STK40-EYFP was digested using restriction enzymes Nhe I and Sbf I. Cloned insert STK40-EYFP-NES was then ligated to the vector. STK40-EYFP-NES was generated by performing PCR with primers STK40 cds RE Nhe I F and STK40-EYFP-NES primer R pLAS2w.Sbf I, using pLAS2w.STK40-EYFP as template.
5) pLAS2w.STK40-ΔKD(51 to 299)-P2A-EYFP was generated by truncating amino acids 51 to 299 of STK40 from pLAS2w.STK40-P2A-EYFP. PCR was carried out using primers Kinase domain truncate 300 aa Xma I primer F and Kinase domain truncate 50 aa Xma I primer R and pLAS2w.STK40-P2A-EYFP as template to clone the sequence excluding of the KD. DPN1 (Agilent) was used to degrade template pLAS2w.STK40-P2A-EYFP. Restriction enzyme Xma I (NEB) was used for digestion, and self-ligation was then performed to create the final construct.
6) pLAS2w.STK40–KD(50 to 300 amino acids) only–P2A-EYFP comprises only the KD of STK40, which is the amino acids 50 to 300 of STK40. pLAS2w.STK40-P2A-EYFP was digested with restriction enzymes Nhe I and Sbf I with the insert STK40-KD ligated to it. STK40-KD was generated by performing in-fusion PCR cloning using primers STK40 35 ATG infusion F and STK40 331 infusion R, Phusion DNA polymerase (Thermo Fisher Scientific) and the template pLAS2w.STK40-EYFP.
7) To create a cell line SAS-GFP-PXN for FA dynamics, we created pLKO.AS3W-GFP-PXN.bsd. pLKO.AS3W-GFP-PXN.bsd was created by digesting vector pLKO.AS3w.bsd (Academia Sinica) using restriction enzymes Nhe I and Sbf I and then ligating cloned GFP-PXN to the vector. GFP-PXN was cloned using primers GFP-PXN-F and GFP-PXN-R and template GFP-PXN (gift from T. Meyer’s lab). pLKO.AS3W-GFP-PXN.bsd was later expressed in SAS cells and sorted to create cell line SAS-GFP-PXN to label FAs in FA dynamic experiments.
Lentivirus preparation and infection
HEK293T cells were plated on 6- or 10-cm dish (Jet Biofil, Guanzhou, China) and transfected with shRNA plasmid or cloned construct, pMD2.G and pCMV Δ8.91 (gifts from C.-C. Chen’s lab, National Taiwan University) using Lipofectamine 3000 (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) and P3000 enhancer (Invitrogen) in Opti-MEM (Gibco). After 6 hours of transfection, the medium was removed and replaced with DMEM with 1% bovine serum albumin (BSA). Viral supernatant was collected for 48 hours after transfection, centrifuged, and concentrated using Lenti-X (ClonTech, Takara, Shiga, Kyoto, Japan). SAS, HUVEC, 3A-Sub E, or HepG2 cells were infected with lentiviruses, including polybrene (8 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA, USA) for enhancement of infection. Cells were then selected using puromycin (2 μg/ml; Sigma-Aldrich, St. Louis, MI, USA) for shRNAs or blasticidin (10 μg/ml; InvivoGen) for overexpression constructs for 24 to 48 hours.
“Two-hit” migration screen
High-throughput screening [high content screening (HCS)] service and shRNA provided by NRC in Academia Sinica were used to accelerate the process of the two-hit migration screen. HUVECs were plated manually on 96-well plates (Corning, Costar 3599) coated with collagen I (30 μg/ml; rat tail, Invitrogen). A total of 119 genes were selected from two previous screens (13, 14) as our candidates in the two-hit migration screen. A total of 588 shRNAs targeting the 119 genes were used to infect HUVECs operated by robotic arms. On the day of wound healing assay, HUVECs were stained with Hoechst 33342 (Invitrogen) in EGM2 medium at room temperature. After nuclei staining, the medium was changed to EGM2 containing dimethyl sulfoxide (DMSO; Sigma-Aldrich), 2 μM BTP2 (Sigma-Aldrich), 5 μM Y27632 (Sigma-Aldrich), or 10 μM PD98059 (Sigma-Aldrich). A scratcher (13) was then used to create wounds for wound healing migration. Plates were sent to ArrayScar VTI HCS Reader (Thermo Fisher Scientific, Cellomics) to take images at 0 and 15 hours of incubation at 37°C.
Data from our two-hit migration screen were analyzed using MATLAB (MathWorks, Natick, MA, USA) in the following flow (data S7):
1) Generation of wound healing area. The wound-healing area of the sheet migration images between 0 and 15 hours in the two-hit migration screen on HUVECs was analyzed. (data S1). The extent of wound healing after wound-scratch was demonstrated as the wound healing area.
2) Density correction. Density correction (fig. S1B) was carried out by generating a linear regression curve of sheet migration versus cell density. The estimated sheet migration per density was generated on the basis of the regression line. Density corrected speed was calculated as the following formula
3) Normalization. Normalized sheet migration was calculated as the following formula (Fig. 1D and data S1).
4) z scores. z scores of sheet migration were calculated as the following formula (fig. S1, C to E, and data S1)
Positive z score values imply greater wound healing, while negative values imply less healing. Take STK40 as an example: The z score of the N column of shSTK40 is close to 0, while shSTK40 plus PD98059 generated a negative value. This implies a synergistic reduction in sheet migration when coinhibiting STK40 and MAPK.
Single-cell tracing migration (random migration and wound healing assays)
SAS, HUVEC, HSC3, and 3A-Sub E cells were plated on collagen I (rat tail, Invitrogen)–coated 96-well plates (Corning, CoStar 3599). Cells were infected with lentiviruses for 24 hours and selected with antibiotics for 24 to 48 hours (see the “Lentivirus preparation and infection” section). On the day performing migration assays, SAS, HSC3, and 3A-Sub E cells were stained with Hoechst 33342 (1 μg/ml) for 1 hour at 37°C, while HUVECs were stained with Hoechst 33342 (1 μg/ml) for 15 to 30 min at the same temperature. After nucleus staining, medium of SAS and 3A-Sub E cells was changed to serum-free medium containing 20 mM Hepes (Gibco), 0.1% BSA (BioShop, Ontario, Canada), and epidermal growth factor (0.5 ng/ml; PeproTech, Rockyhill, NJ, USA) or supplemented with FGF1 (25 ng/ml; Invitrogen) plus 10 U of heparin (Sigma-Aldrich). Medium of HUVECs was changed to EGM2 medium. Addition of other drugs such as 100 nM trametinib (LC Laboratories, Woburn, MA, USA) or DMSO was added into the medium together with the supplements. For wound healing assays, cells were scratched by a scratcher, while random migration assays do not require wound scratch. After wound scratch (or unscratched), the cells were then placed in an acrylic incubator at 32°C for SAS cells and 37°C for HUVEC and 3A-Sub E cells using Nikon Eclipse Ti microscope (Nikon, Tokyo, Japan) for live-cell imaging. Images were taken every 10 min for a duration of 6 to 10 hours. Parameters such as speed, coordination, and directionality were analyzed using MATLAB (data S7).
Double-inhibition experiment
SAS cells were plated on collagen I (rat tail)–coated 96-well plates. For double-inhibition experiments, cells were coinhibited with (i) shSTK40 plus (ii) shRNAs or drug targeting migration-related molecules: Rac1 for F-actin, α-catenin for adherens junction, and PXN, FA, and myosin light-chain kinase for myosin. Cells were infected with shSTK40 plus shCTNNA1, shRAC1, or shPXN for 24 hours and selected with puromycin for 24 hours before performing migration. Ten micromolars of ML9 (Sigma-Aldrich), myosin-light chain kinase inhibitor, was added into serum-free DMEM with supplements after nucleus staining on the day performing migration. Random migration or wound healing migration was carried out after infection and selection [see the “Single-cell tracing migration (random migration and wound healing assays)” section]. Nucleus-stained or drug-treated SAS cells, scratched or unscratched, were then placed in an incubator at 32°C for migration using Nikon Eclipse Ti microscope for live-cell imaging. Images were taken every 10 min for a duration of 10 hours, followed by analysis using MATLAB.
Immunofluorescent staining
SAS, HUVEC, and HepG2 cells were plated on collagen (100 μg/ml; collagen I, bovine, Gibco)–coated chamber slides (Thermo Fisher Scientific, 155411 or 155383 Nunc Lab-TEK) or 96-well plate (Thermo Fisher Scientific, Nunc 165305) and infected with shRNA viruses or prepared viruses of overexpression constructs, followed by aforementioned antibiotic selection. SAS cells were treated with or without drugs before fixation. Drugs include 5 μM Y27632, 5 μM blebbistatin, 100 nM trametinib treatment overnight, 2.5 μM VP (MedChemExpress, Monmouth Junction, NJ, USA) for 8 hour, and 100 nM leptomycin B (Cayman Chemical Company, Ann Arbor, MI, USA) for 2 to 3 hours.
Before fixation, cells were processed to a 2-hour migration. Cells were washed with PBS, and wounds were created using a tip for chamber slides or a scratcher for 96-well plates. The cells were incubated in serum-free DMEM supplemented with 20 mM Hepes, 0.1% BSA, FGF1 (25 ng/ml), and 10 U of heparin, including or excluding drug treatment at 37°C for 2 hours to perform migration. After 2 hours of migration, cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) in PBS at room temperature for 15 min, permeated with 0.25% Triton X-100 (J. T. Baker, Phillipsburg, NJ, USA) at room temperature for 10 min, and blocked with 5% BSA at room temperature for 1 hour. Cells were then incubated overnight at 4°C with primary antibodies with 1% BSA in PBS. The cells were then stained with secondary antibodies (see the “Antibodies” section), phalloidin dyes, and 4′,6-diamidino-2-phenylindole (DAPI; 10 μg/ml; Invitrogen) with 1% BSA in PBS. Phalloidin dyes, Alexa Fluor 488 phalloidin (Invitrogen, catalog no. A12379; 1:500 to 1:1000 dilution), and Alexa Fluor 594 phalloidin (Invitrogen, catalog no. A12381; 1:500 to 1:1000 dilution) were used to stain stress fibers. We then used Nikon Eclipse Ti to take images of the fixed cells.
In vitro permeability assay
HUVECs were plated on 24-well transwell inserts with a 0.4-mm pore size (Corning) and attached overnight. Then, cells were infected with lentiviruses for 24 hours and selected with puromycin for 24 hours (see the “Lentivirus preparation and infection” section). After that, we treated the cells with trametinib (100 nM) or DMSO overnight. On the day performing permeability assay, 70-kDa fluorescein isothiocyanate (FITC)–conjugated dextran (1 mg/ml; Sigma-Aldrich) was added to the upper compartment of the inserts at specific time points (60 and 120 min). Fluorescence of the medium of the lower compartment was measured with spectrophotometer (Synergy HT, BioTek) excitation of 485 nm and emission of 528 nm. FITC-dextran concentrations were calculated on the basis of the standard curve of FITC-conjugated dextran in extracellular matrix ranging from 0 to 1 mg/ml using MATLAB script (data S7).
YAP activity assay
SAS cells were infected with lentiviruses for 24 hours and selected with antibiotics for 24 hours (see the “Lentivirus preparation and infection” section). Cells (treated with or without shRNAs) were then seeded 2 × 105 cells per well on a 6-well plate and attached overnight. Next, 8XGTIIC-luciferase plasmid (Addgene) and pRL-TK plasmid (Promega) were cotransfected to the cells with Lipofectamine 3000 (Invitrogen) for at least 14 hours before being harvested. VP or DMSO was treated to the cells 6 hours after transfection for drug treatment experiment. Cell lysate was then harvested and transferred to a 96-well-plate for analysis using the Luc-Pair Duo-Luciferase Assay Kit (GeneCopoeia). Luminescent signals were detected using spectrophotometer (Synergy HT, BioTek).
Real-time reverse transcription PCR
Total RNAs of cells were extracted using Iso-RNA lysis reagent (Five Prime Therapeutics, South San Francisco, CA, USA) or NucleoZol (MACHEREY-NAGEL, Düren, Germany) and converted into cDNAs using SuperScript Transcriptase II or SuperScript Transcriptase IV (Invitrogen). Sample reactions containing SYBR Green Supermix (Bio-Rad), primers (data S6), and cDNA templates were then loaded onto 96-well PCR plates (Bio-Rad) and then set up for real-time PCR using the CFX Connect Real-Time PCR Detection System (Bio-Rad). MATLAB was used to analyze PCR cycles, annealing temperature, and quantification of mRNA levels (data S7).
FA analysis
Immunofluorescent images of FA were analyzed using MATLAB (data S7). FA quantification was performed using the following flow: (i) FA background subtraction: Script FA_background_20200313.m was used for FA background subtraction. The background signal of each of the individual pixels was separately determined by the median value of its surrounding pixels within the radius of 1.46 μm. (ii) Thresholding: Script FA_identification_FAclearer_20200316.m was used. The threshold for the mask of FA was determined as two SDs above the mode of the background value. (iii) Segmentation: Function of watershed segmentation in MATLAB was used for FA segmentation. (iv) FA selection: FAs were identified and selected in a semiautomatic manner. FAs within 15 μm of the lamellipodia edge were further analyzed. For each FA, we acquire its area and mean FA signal. The integrated FA signal (fig. S5) was generated by
FAs and cells from multiple sites were analyzed, and data were pulled up using Microsoft Excel (Redmond, WA, USA) for statistical analysis. Here is a list of the cells and FAs chosen in each figure:
1) FAs of eight cells at the wound edge from one site of each condition were analyzed for STK40 knockdown and overexpression (Fig. 2, D and E) in SAS cells. Number of FAs per cell was also analyzed (fig. S6A). FAs of 23 cells at wound edge from one site in cells infected with shControl and 31 cells from one site in cells infected with shSTK40 were analyzed in HepG2 cells (fig. S6B). FAs of 14 cells at the wound edge from one site in cells infected with shControl and 13 cells from one site in cells infected with shSTK40 were analyzed in HUVECs (fig. S6C).
2) FAs of eight cells at the wound edge from one site of each treatment were analyzed in the experiment removing traction force upon knockdown of STK40 (Fig. 3, F and G, and fig. S7D).
3 FAs of 8 to 10 cells at the wound edge from one to three sites of each treatment were analyzed for shSTK40 and shCAPN2 double-treatment experiment. (Fig. 3G and fig. S7E)
4) FAs of eight cells at the wound edge from one to two sites of each treatment were analyzed for the overexpression and KD truncation of STK40 (Fig. 4, A and B).
5) FAs of 24 cells at the wound edge from three sites of each condition were analyzed in the experiment translocating STK40 from nucleus to cytosol (Fig. 4, C and D).
6) FAs of eight cells at the wound edge from one site of each condition were analyzed in the experiment of shSTK40 plus trametinib (Fig. 4E).
7) FAs of eight cells at the wound edge from one site of each condition were analyzed in the experiment of shSTK40 plus VP (Fig. 4H).
8) FAs of eight cells at the wound edge from two to four sites of each condition were analyzed in the experiment of shSTK40 plus trametinib and VP (Fig. 4I).
9) FAs of eight cells at the wound edge from one site for each condition were analyzed in the experiment of shSTK40 plus leptomycin B (Fig. 5B).
10) FAs of eight cells at the wound edge from one site for each condition were analyzed in the experiment of shSTK40 plus VP and shCREB (Fig. 5E).
11) FAs of eight cells at wound edge from one to two sites for each condition were analyzed in the experiment of shSTK40 plus shYAP (fig. S9J).
Images of FA shown in figures were processed using MATLAB (data S7). Script Generate_FA_demo_20200831.m was used.
FA dynamic assay
Sorted cells were used to create cell line SAS-GFP-PXN for FA dynamics experiments (Fig. 3, A to D, and fig. S7, A to C). We infected SAS cells with viruses packaged pLKO.AS3w-GFP-PXN.bsd and cultured the cells for cell sorting. After trypsinizing and centrifuging infected cells, the supernatant was removed, and cells were treated and resuspended with 1 ml of sorting buffer that includes 1× PBS (Ca2+/Mg2+-free), 1 mM EGTA (Sigma-Aldrich), 2 mM Hepes, 1 to 2% FBS, and 0.1 to 0.2% BSA at a pH of 7.0. Cells were filtered and prepared with a sterilized sorting tube (BD Biosciences, BD Falcon, 352235) before cell sorting using flow cytometer BD LSRFortessa (The Flow Cytometric Analyzing and Sorting Core of the First Core Laboratory, College of Medicine, National Taiwan University).
Coverglass base plates (96-well plates; Thermo Fisher Scientific, Nunc 164588) were coated with 100 μM poly-d-lysine hydrobromide (Sigma-Aldrich) for 5 min of incubation at 37°C and then coated with collagen (10 μg/ml; collagen I, rat tail) overnight. GFP-PXN-SAS cells were then plated for FA dynamic assay. On the day of FA dynamic assay, the medium of cells was removed and replaced with phenol red-free serum-free medium DMEM (Gibco) supplemented with 10% of FBS and 100 μM l–ascorbic acid (Sigma-Aldrich, catalog no. A4544). Movies of live cells were recorded using Nikon Eclipse Ti microscope with a frequency of 5 s per image and a total duration of 8 min. FA dynamics were analyzed using MATLAB (data S5). All FAs of all cells in one site of each treatment (shControl or shSTK40) were analyzed.
Western blotting
Cells were infected with viruses and treated with or without drugs such as PD98059, trametinib, Y27632, and blebbistatin (Sigma-Aldrich) for 24 hours. Cells were washed in ice-cold PBS and then harvested in radioimmunoprecipitation assay lysis buffer (Cell Signaling Technology, Danvers, MA, USA) and supplemented with phenylmethylsulfonyl fluoride (MD Bio Inc., Rockville, MD, USA) and protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Lysates were centrifuged for 15 min at 15,000 rpm. Samples that were boiled at 95°C for 5 min were loaded into wells of SDS–polyacrylamide gel electrophoresis gel, transferred onto polyvinylidene difluoride (PVDF) membrane (Merck Millipore, Burlington, MA, USA, Immobilon-P PVDF Membrane), blocked with 3% BSA in 0.05% Tris Buffered Saline with Tween-20 (TBST) (BioLink, Lisle, IL, USA), and incubated at 4°C overnight with primary antibodies and secondary antibodies at room temperature for 1 hour (see the “Antibodies” section). Enhanced chemiluminescent substrates (T-Pro Biotechnology, New Taipei City, Taiwan) were used to visualize conjugated proteins, followed by imaging using Bio-Rad Gel Doc2000 (Bio-Rad, Hercules, CA, USA). Image Lab software (Bio-Rad) was used for protein quantification.
Antibodies
Antibodies in immunofluorescent staining were as follows: purified mouse anti-PXN (BD Biosciences, San Jose, CA, USA, catalog nos. 610061 and 610062) at 1:150; p-MLC2 (Ser-19) antibody (Cell Signaling Technology, catalog no. 3671) at 1:1000; YAP (D8H1X) XP rabbit monoclonal antibody (mAb) (Cell Signaling Technology, catalog no. 14074) at 1:1000; goat anti-mouse immunoglobulin G (IgG) (H + L) cross-adsorbed secondary antibody and Alexa Fluor 488 (Invitrogen, catalog no. A11001) at 1:500; goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody and Alexa Fluor 594 (Invitrogen, catalog no. A11005) at 1:500; goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody and Alexa Fluor 488 (Invitrogen, catalog no. A11008) at 1:500; and goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody and Alexa Fluor 594 (Invitrogen, catalog no. A11012) at 1:500. Antibodies in Western blots were as follows: phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody (Cell Signaling Technology, catalog no. 9101) at 1:5000; p44/42 MAPK (Erk1/2) antibody (Cell Signaling Technology, catalog no. 9102) at 1:5000; phospho-MEK1/2 (Ser217/Ser221) (41G9) rabbit mAb (Cell Signaling Technology, catalog no. 9154) at 1:1000; MEK1/2 (L38C12) mouse mAb (Cell Signaling Technology, catalog no. 4694) at 1:1000; phospho-c-Raf (Ser259) antibody (Cell Signaling Technology, catalog no. 9421) at 1:1000; c-Raf antibody (Cell Signaling Technology, catalog no. 9422) at 1:1000; phospho-FAK (Tyr397) antibody (Cell Signaling Technology, catalog no. 3283) at 1:1000; FAK antibody (Cell Signaling Technology, catalog no. 3285) at 1:1000; phospho-YAP (Ser127) (D9W2I) rabbit mAb (Cell Signaling Technology, catalog no. 13008) at 1:1000; YAP (D8H1X) XP rabbit mAb (Cell Signaling Technology, catalog no. 14074) at 1:1000; α-tubulin (DM1A) mouse mAb (Cell Signaling Technology, catalog no. 3873) at 1:5000; E-cadherin antibody [N3C2], internal (GeneTex, Irvine, CA, USA, catalog no. GTX124178) at 1:1000; anti–α-SMA antibody (Abcam, Cambridge, UK, catalog no. ab5694) at 1:1000 dilution; GAPDH (glyceraldehyde-3-phosphate dehydrogenase) loading control mAb (GA1R) (Thermo Fisher Scientific, catalog no. MA5-15738) at 1:5000; horseradish peroxidase (HRP)–conjugated goat anti-rabbit IgG (H + L) mouse serum adsorbed (Abgent, San Diego, CA, USA, catalog no. LP1001c) at 1:2000; and HRP-conjugated goat anti-mouse IgG (H + L) human serum–adsorbed (Abgent, catalog no. LP1002c) at 1:2000.
Myofibroblastic differentiation assay
To assess myofibroblastic differentiation, fibroblasts (6 × 103 cells per well) were plated on 96-well plate coated with collagen (3 μg/μl) and allowed to attach overnight. Cells were infected with lentiviruses for 24 hours and selected with antibiotics for 24 hour (see the “Lentivirus preparation and infection” section). The medium of the cell was changed to DMEM with 0.1% FBS, 1% P/S, and transforming growth factor–β (10 ng/ml; PeproTech, Rocky Hill, NJ, USA) to induce myofibroblastic differentiation of fibroblasts. Cells were incubated for 48 hours and with the addition of trametinib (100 nM) in the night before fixation. Cells were fixed with acetone/methanol (1:1) (acetone, methanol, Sigma-Aldrich) in −20°C fridge for 5 min and then blocked with 5% BSA in PBS for 1 hour. To detect α-SMA, a marker of myofibroblastic differentiation, we treated cells with α-SMA antibodies (Cell Signaling Technology; 1:100) and 5% BSA in PBS at 4°C overnight and then stained with Alexa Fluor 594 dye–conjugated secondary antibody (1:300) and DAPI (10 μg/ml) in 5% BSA in PBS for 1 hour. A fluorescence microscope was used to detect fluorescence from α-SMA (594 nm) and nuclei, respectively.
Flow cytometry
SAS cells were knocked down with shControl and shSTK40 for the flow cytometry experiment to investigate whether shSTK40 alters cell size (fig. S8B). Samples were trypsinized and centrifuged, followed by resuspension with a staining buffer: 1% FBS, (Biological Industries, Beit-Haemek, Israel), 2 mM of EDTA (J. T. Baker), and 0.05% sodium azide (Sigma-Aldrich) in PBS on ice. Flow cytometry was performed on flow cytometer BD LSRFortessa.
YAP analysis
Immunofluorescent images of YAP were analyzed using MATLAB (data S7) in our experiments to investigate nuclear and cytosolic YAP level (Fig. 5, A and C, and fig. 10, A and D). Script N_C_calculation_20190627_YFP_20191026_YAP.m was used for YAP analysis. To determine YAP signals of a single cell, the image of its DAPI signal was used as a nuclear mask. Its nuclear YAP signals were determined by the YAP signals overlying the nuclear mask. Its cytosolic YAP signals were determined by the YAP signals on the ring area of 0.73 μm in width surrounding the nuclear mask. YAP levels of shControl or shSTK40 treated cells were analyzed from 12 sites for each treatment (shControl or shSTK40) (Fig. 5A and fig. S10). YAP levels of shControl- or shSTK40-treated cells and then overexpressed with YFP or FL, ΔKD, or KD-only STK40 construct were analyzed from 20 sites for each treatment.
Cycloheximide assay
In the cycloheximide assay (Fig. 5D), SAS cells were infected with lentiviruses for 24 hours and selected with puromycin for 24 hours (see the “Lentivirus preparation and infection” section). After that, cells were treated with cycloheximide (200 μg/ml; Cayman Chemical Company) at specific time points and then harvested. Western blot was then conducted to determine the protein degradation level.
Double knockdown experiment of shSTK40 and titrations of shCREB using different multiplicities of infection
Double knockdown experiment of shSTK40 and shCREB using titrated multiplicity of infection (MOI) viruses (fig. S10F) was carried out to investigate the relationship between STK40, YAP, and CREB to confirm our model (Fig. 5F). SAS cells were infected with shControl or shSTK40 viruses using an MOI of 0.31 or 10 overnight and selected with puromycin for 24 hours. Next, we infected the cells with shCREB viruses using MOIs of 0, 1, 2, 4, and 8. After 24 hours, these infected cells were harvested for quantitative PCR analysis of CTGF level.
Statistical analysis
Experimental data such as bar graphs and error bars are presented as means ± SE. Two-tailed Student’s t tests and one-way analysis of variance (ANOVA) were performed with Microsoft Excel and MATLAB. Differences were considered statistically significant when P values were less than 0.05.
Acknowledgments
We thank T. Meyer (Cornell University, New York, NY, USA), S. Collins (University of California, Davis, CA, USA), and A. Hayer (McGill University, Montréal, QC, Canada) for critical discussions about the design of the two-hit screen; T.-S. Jou (National Taiwan University, Taipei, Taiwan), K.-H. Lin (Academia Sinica, Taipei, Taiwan), and RNAi Core (Academia Sinica, Taipei, Taiwan) for technical assistance about the screen; and I. T.-L. Lee, H. Y.-C. Chen, J. Yu, and C.-L. A. Yang for critical comments. Funding: This work was supported by grants from The Ministry of Science and Technology in Taiwan (MOST 107-2320-B-002-038-MY3 and MOST 108-2926-I-002-002-MY4), National Taiwan University Hospital (NTUH 106-T02, NTUH 107-T13, NTUH 108-T13, and VN 109-14), and The Liver Disease Prevention and Treatment Research Foundation in Taiwan. Author contributions: Conceptualization: F.-C.T. and C.-J.T. Methodology: F.-C.T., L.-Y.Y., T.-J.T., H.-C.L., C.-L.H., T.-X.L., Y.-C.L., C.-T.P., I.C., and H.-J.C. Investigation: F.-C.T., L.-Y.Y., T.-J.T., H.-C.L., C.-L.H., and T.-X.L. Visualization: F.-C.T., L.-Y.Y., and C.-L.H. Supervision: F.-C.T. Writing—original draft: F.-C.T., L.-Y.Y., and C.-L.H. Writing—review and editing: F.-C.T., L.-Y.Y., C.-L.H., and T.-X.L. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/31/eabg2106/DC1
REFERENCES AND NOTES
- 1.Costanzo M., Baryshnikova A., Bellay J., Kim Y., Spear E. D., Sevier C. S., Ding H., Koh J. L. Y., Toufighi K., Mostafavi S., Prinz J., Onge R. P. S., VanderSluis B., Makhnevych T., Vizeacoumar F. J., Alizadeh S., Bahr S., Brost R. L., Chen Y., Cokol M., Deshpande R., Li Z., Lin Z.-Y., Liang W., Marback M., Paw J., Luis B.-J. S., Shuteriqi E., Tong A. H. Y., van Dyk N., Wallace I. M., Whitney J. A., Weirauch M. T., Zhong G., Zhu H., Houry W. A., Brudno M., Ragibizadeh S., Papp B., Pál C., Roth F. P., Giaever G., Nislow C., Troyanskaya O. G., Bussey H., Bader G. D., Gingras A.-C., Morris Q. D., Kim P. M., Kaiser C. A., Myers C. L., Andrews B. J., Boone C., The genetic landscape of a cell. Science 327, 425–431 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nair A., Chauhan P., Saha B., Kubatzky K. F., Conceptual evolution of cell signaling. Int. J. Mol. Sci. 20, 3292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.H. F. Lodish, A. Berk, C. Kraiser, M. Krieger, A. Bretscher, H. Ploegh, A. Amon, K. C. Martin, Molecular Cell Biology (W. H. Freeman, 8th ed., 2016). [Google Scholar]
- 4.Farmer H., McCabe N., Lord C. J., Tutt A. N. J., Johnson D. A., Richardson T. B., Santarosa M., Dillon K. J., Hickson I., Knights C., Martin N. M. B., Jackson S. P., Smith G. C. M., Ashworth A., Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Turner N. C., Lord C. J., Iorns E., Brough R., Swift S., Elliott R., Rayter S., Tutt A. N., Ashworth A., A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 27, 1368–1377 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang A., Garraway L. A., Ashworth A., Weber B., Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 19, 23–38 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Klobucar K., Brown E. D., Use of genetic and chemical synthetic lethality as probes of complexity in bacterial cell systems. FEMS Microbiol. Rev. 42, fux054 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Giannone G., Dubin-Thaler B. J., Döbereiner H.-G., Kieffer N., Bresnick A. R., Sheetz M. P., Periodic lamellipodial contractions correlate with rearward actin waves. Cell 116, 431–443 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Iskratsch T., Wolfenson H., Sheetz M. P., Appreciating force and shape—The rise of mechanotransduction in cell biology. Nat. Rev. Mol. Cell Biol. 15, 825–833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang H. W., Collins S. R., Meyer T., Locally excitable Cdc42 signals steer cells during chemotaxis. Nat. Cell Biol. 18, 191–201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayer A., Shao L., Chung M., Joubert L.-M., Yang H. W., Tsai F.-C., Bisaria A., Betzig E., Meyer T., Engulfed cadherin fingers are polarized junctional structures between collectively migrating endothelial cells. Nat. Cell Biol. 18, 1311–1323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamada K. M., Sixt M., Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 20, 738–752 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Vitorino P., Meyer T., Modular control of endothelial sheet migration. Genes Dev. 22, 3268–3281 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simpson K. J., Selfors L. M., Bui J., Reynolds A., Leake D., Khvorova A., Brugge J. S., Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat. Cell Biol. 10, 1027–1038 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Tsai F.-C., Seki A., Yang H. W., Hayer A., Carrasco S., Malmersjö S., Meyer T., A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 16, 133–144 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timpson P., Jones G. E., Frame M. C., Brunton V. G., Coordination of cell polarization and migration by the Rho family GTPases requires Src tyrosine kinase activity. Curr. Biol. 11, 1836–1846 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Worthylake R. A., Burridge K., RhoA and ROCK promote migration by limiting membrane protrusions. J. Biol. Chem. 278, 13578–13584 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Zhan H., Bhattacharya S., Cai H., Iglesias P. A., Huang C.-H., Devreotes P. N., An excitable Ras/PI3K/ERK signaling network controls migration and oncogenic transformation in epithelial cells. Dev. Cell 54, 608–623.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guilluy C., Rolli-Derkinderen M., Loufrani L., Bourgé A., Henrion D., Sabourin L., Loirand G., Pacaud P., Ste20-related kinase SLK phosphorylates Ser188 of RhoA to induce vasodilation in response to angiotensin II Type 2 receptor activation. Circ. Res. 102, 1265–1274 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Fokin A. I., Klementeva T. S., Nadezhdina E. S., Burakov A. V., SLK/LOSK kinase regulates cell motility independently of microtubule organization and Golgi polarization. Cytoskeleton 73, 83–92 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Zaman R., Lombardo A., Sauvanet C., Viswanatha R., Awad V., Bonomo L. E.-R., McDermitt D., Bretscher A., Effector-mediated ERM activation locally inhibits RhoA activity to shape the apical cell domain. J. Cell Biol. 220, e202007146 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y., Moerkens M., Ramaiahgari S., de Bont H., Price L., Meerman J., van de Water B., Elevated insulin-like growth factor 1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res. 13, R52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Criswell T., Beman M., Araki S., Leskov K., Cataldo E., Mayo L. D., Boothman D. A., Delayed activation of insulin-like growth factor-1 receptor/Src/MAPK/Egr-1 signaling regulates clusterin expression, a pro-survival factor. J. Biol. Chem. 280, 14212–14221 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Gao L., Wang X., Wang X., Zhang L., Qiang C., Chang S., Ren W., Li S., Yang Y., Tong D., Chen C., Li Z., Song T., Zhi K., Huang C., IGF-1R, a target of let-7b, mediates cross-talk between IRS-2/Akt and MAPK pathways to promote proliferation of oral squamous cell carcinoma. Oncotarget 5, 2562–2574 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomlinson D. C., Baxter E. W., Loadman P. M., Hull M. A., Knowles M. A., FGFR1-induced epithelial to mesenchymal transition through MAPK/PLCγ/COX-2-mediated mechanisms. PLOS ONE 7, e38972 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma D. K., Ponnusamy K., Song M.-R., Ming G., Song H., Molecular genetic analysis of FGFR1 signalling reveals distinct roles of MAPK and PLCγ1 activation for self-renewal of adult neural stem cells. Mol. Brain 2, 16 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Q., Duan H., Gan L., Liu X., Chen F., Shen X., Tang Y.-Q., Wang S., MicroRNA-1291 promotes endometrial fibrosis by regulating the ArhGAP29-RhoA/ROCK1 signaling pathway in a murine model. Mol. Med. Rep. 16, 4501–4510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Post A., Pannekoek W. J., Ponsioen B., Vliem M. J., Bos J. L., Rap1 spatially controls ArhGAP29 to inhibit Rho signaling during endothelial barrier regulation. Mol. Cell. Biol. 35, 2495–2502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiao Y., Chen J., Lim Y. B., Finch-Edmondson M. L., Seshachalam V. P., Qin L., Jiang T., Low B. C., Singh H., Lim C. T., Sudol M., YAP regulates actin dynamics through ARHGAP29 and promotes metastasis. Cell Rep. 19, 1495–1502 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Li L., Sun L., Gao F., Jiang J., Yang Y., Li C., Gu J., Wei Z., Yang A., Lu R., Ma Y., Tang F., Kwon S. W., Zhao Y., Li J., Jin Y., Stk40 links the pluripotency factor Oct4 to the Erk/MAPK pathway and controls extraembryonic endoderm differentiation. Proc. Natl. Acad. Sci. U.S.A. 107, 1402–1407 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ridley A. J., Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 16, 522–529 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Kobielak A., Fuchs E., α-Catenin: At the junction of intercellular adhesion and actin dynamics. Nat. Rev. Mol. Cell Biol. 5, 614–625 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaller M. D., Paxillin: A focal adhesion-associated adaptor protein. Oncogene 20, 6459–6472 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Kanchanawong P., Shtengel G., Pasapera A. M., Ramko E. B., Davidson M. W., Hess H. F., Waterman C. M., Nanoscale architecture of integrin-based cell adhesions. Nature 468, 580–584 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khapchaev A. Y., Shirinsky V. P., Myosin light chain kinase MYLK1: Anatomy, interactions, functions, and regulation. Biochemistry 81, 1676–1697 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Burridge K., Guilluy C., Focal adhesions, stress fibers and mechanical tension. Exp. Cell Res. 343, 14–20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai F.-C., Meyer T., Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Curr. Biol. 22, 837–842 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giannone G., Dubin-Thaler B. J., Rossier O., Cai Y., Chaga O., Jiang G., Beaver W., Döbereiner H.-G., Freund Y., Borisy G., Sheetz M. P., Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 128, 561–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franco S. J., Rodgers M. A., Perrin B. J., Han J., Bennin D. A., Critchley D. R., Huttenlocher A., Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 6, 977–983 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Cortesio C. L., Boateng L. R., Piazza T. M., Bennin D. A., Huttenlocher A., Calpain-mediated proteolysis of paxillin negatively regulates focal adhesion dynamics and cell migration. J. Biol. Chem. 286, 9998–10006 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He K., Hu J., Yu H., Wang L., Tang F., Gu J., Ge L., Wang H., Li S., Hu P., Jin Y., Serine/Threonine kinase 40 (Stk40) functions as a novel regulator of skeletal muscle differentiation. J. Biol. Chem. 292, 351–360 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu H., He K., Wang L., Hu J., Gu J., Zhou C., Lu R., Jin Y., Stk40 represses adipogenesis through translational control of CCAAT/enhancer-binding proteins. J. Cell Sci. 128, 2881–2890 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Wang L., Yu H., Cheng H., He K., Fang Z., Ge L., Cheng T., Jin Y., Deletion of Stk40 impairs definitive erythropoiesis in the mouse fetal liver. Cell Death Dis. 8, e2722 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Durzynska I., Xu X., Adelmant G., Ficarro S. B., Marto J. A., Sliz P., Uljon S., Blacklow S. C., STK40 is a pseudokinase that binds the E3 ubiquitin ligase COP1. Structure 25, 287–294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu J., Li S., Sun X., Fang Z., Wang L., Xiao F., Shao M., Ge L., Tang F., Gu J., Yu H., Guo Y., Guo X., Liao B., Jin Y., Stk40 deletion elevates c-JUN protein level and impairs mesoderm differentiation. J. Biol. Chem. 294, 9959–9972 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitra S. K., Hanson D. A., Schlaepfer D. D., Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Pasapera A. M., Schneider I. C., Rericha E., Schlaepfer D. D., Waterman C. M., Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 188, 877–890 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kleinschmidt E. G., Schlaepfer D. D., Focal adhesion kinase signaling in unexpected places. Curr. Opin. Cell Biol. 45, 24–30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu H., He K., Li L., Sun L., Tang F., Li R., Ning W., Jin Y., Deletion of STK40 protein in mice causes respiratory failure and death at birth. J. Biol. Chem. 288, 5342–5352 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nardone G., Oliver-De La Cruz J., Vrbsky J., Martini C., Pribyl J., Skládal P., Pešl M., Caluori G., Pagliari S., Martino F., Maceckova Z., Hajduch M., Sanz-Garcia A., Pugno N. M., Stokin G. B., Forte G., YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun. 8, 15321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mason D. E., Collins J. M., Dawahare J. H., Nguyen T. D., Lin Y., Voytik-Harbin S. L., Zorlutuna P., Yoder M. C., Boerckel J. D., YAP and TAZ limit cytoskeletal and focal adhesion maturation to enable persistent cell motility. J. Cell Biol. 218, 1369–1389 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao B., Ye X., Yu J., Li L., Li W., Li S., Yu J., Lin J. D., Wang C.-Y., Chinnaiyan A. M., Lai Z.-C., Guan K.-L., TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lai D., Ho K. C., Hao Y., Yang X., Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 71, 2728–2738 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Esteves de Lima J., Bonnin M.-A., Birchmeier C., Duprez D., Muscle contraction is required to maintain the pool of muscle progenitors via YAP and NOTCH during fetal myogenesis. eLife 5, e15593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S., Elvassore N., Piccolo S., Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Reddy B. V. V. G., Irvine K. D., Regulation of Hippo signaling by EGFR-MAPK signaling through Ajuba family proteins. Dev. Cell 24, 459–471 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuge S., Jones N., Nomoto A., Regulation of yAP-1 nuclear localization in response to oxidative stress. EMBO J. 16, 1710–1720 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim Y., Jho E., Regulation of the Hippo signaling pathway by ubiquitin modification. BMB Rep. 51, 143–150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kudo N., Matsumori N., Taoka H., Fujiwara D., Schreiner E. P., Wolff B., Yoshida M., Horinouchi S., Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. U.S.A. 96, 9112–9117 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han H., Yang B., Nakaoka H. J., Yang J., Zhao Y., Le Nguyen K., Bishara A. T., Mandalia T. K., Wang W., Hippo signaling dysfunction induces cancer cell addiction to YAP. Oncogene 37, 6414–6424 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen L., Feng P., Peng A., Qiu X., Zhu X., He S., Zhou D., cAMP response element-binding protein and Yes-associated protein form a feedback loop that promotes neurite outgrowth. J. Cell. Mol. Med. 22, 374–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J., Ma L., Weng W., Qiao Y., Zhang Y., He J., Wang H., Xiao W., Li L., Chu Q., Pan Q., Yu Y., Sun F., Mutual interaction between YAP and CREB promotes tumorigenesis in liver cancer. Hepatology 58, 1011–1020 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Korkiamäki T., Ylä-Outinen H., Leinonen P., Koivunen J., Peltonen J., The effect of extracellular calcium concentration on calcium-mediated cell signaling in NF1 tumor suppressor-deficient keratinocytes. Arch. Dermatol. Res. 296, 465–472 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Korkiamäki T., Ylä-Outinen H., Koivunen J., Karvonen S.-L., Peltonen J., Altered calcium-mediated cell signaling in keratinocytes cultured from patients with neurofibromatosis type 1. Am. J. Pathol. 160, 1981–1990 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arinobu D., Tachibanaki S., Kawamura S., Larger inhibition of visual pigment kinase in cones than in rods. J. Neurochem. 115, 259–268 (2010). [DOI] [PubMed] [Google Scholar]
- 66.Han X., Yang J., Li L., Huang J., King G., Quarles L. D., Conditional deletion of Fgfr1 in the proximal and distal tubule identifies distinct roles in phosphate and calcium transport. PLOS ONE 11, e0147845 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pall G. S., Johnson K. J., Smith G. L., Abnormal contractile activity and calcium cycling in cardiac myocytes isolated from dmpk knockout mice. Physiol. Genomics 13, 139–146 (2003). [DOI] [PubMed] [Google Scholar]
- 68.Flowers B. M., Rusnak L. E., Wong K. E., Banks D. A., Munyikwa M. R., McFarland A. G., Hinton S. D., The pseudophosphatase MK-STYX induces neurite-like outgrowths in PC12 cells. PLOS ONE 9, e114535 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsuzawa K., Ohga H., Shigetomi K., Shiiya T., Hirashima M., Ikenouchi J., MAGIs regulate aPKC to enable balanced distribution of intercellular tension for epithelial sheet homeostasis. Commun. Biol. 4, 337 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ogawa K., Wada H., Okada N., Harada I., Nakajima T., Pasquale E. B., Tsuyama S., EphB2 and ephrin-B1 expressed in the adult kidney regulate the cytoarchitecture of medullary tubule cells through Rho family GTPases. J. Cell Sci. 119, 559–570 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Huang Q., Sheibani N., High glucose promotes retinal endothelial cell migration through activation of Src, PI3K/Akt1/eNOS, and ERKs. Am. J. Physiol. Cell Physiol. 295, C1647–C1657 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Menges C. W., Sementino E., Talarchek J., Xu J., Chernoff J., Peterson J. R., Testa J. R., Group I p21-activated kinases (PAKs) promote tumor cell proliferation and survival through the AKT1 and Raf-MAPK pathways. Mol. Cancer Res. 10, 1178–1188 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huynh N., Liu K. H., Baldwin G. S., He H., P21-activated kinase 1 stimulates colon cancer cell growth and migration/invasion via ERK- and AKT-dependent pathways. Biochim. Biophys. Acta 1803, 1106–1113 (2010). [DOI] [PubMed] [Google Scholar]
- 74.Wang J., Wu H.-F., Shen W., Xu D.-Y., Ruan T.-Y., Tao G.-Q., Lu P.-H., SRPK2 promotes the growth and migration of the colon cancer cells. Gene 586, 41–47 (2016). [DOI] [PubMed] [Google Scholar]
- 75.Zaremba-Czogalla M., Hryniewicz-Jankowska A., Tabola R., Nienartowicz M., Stach K., Wierzbicki J., Cirocchi R., Ziolkowski P., Tabaczar S., Augoff K., A novel regulatory function of CDKN1A/p21 in TNFα-induced matrix metalloproteinase 9-dependent migration and invasion of triple-negative breast cancer cells. Cell. Signal. 47, 27–36 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Chen T., Huang H., Zhou Y., Geng L., Shen T., Yin S., Zhou L., Zheng S., HJURP promotes hepatocellular carcinoma proliferation by destabilizing p21 via the MAPK/ERK1/2 and AKT/GSK3β signaling pathways. J. Exp. Clin. Cancer Res. 37, 193 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Balko J. M., Schwarz L. J., Bhola N. E., Kurupi R., Owens P., Miller T. W., Gómez H., Cook R. S., Arteaga C. L., Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer Res. 73, 6346–6358 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Vriendt V., De Roock W., Di Narzo A. F., Tian S., Biesmans B., Jacobs B., Budinska E., Sagaert X., Rossi S., D’Ario G., Delorenzi M., Simon I., Vecchione L., Tejpar S., DUSP 4 expression identifies a subset of colorectal cancer tumors that differ in MAPK activation, regardless of the genotype. Biomarkers 18, 516–524 (2013). [DOI] [PubMed] [Google Scholar]
- 79.Kirchner A., Bagla S., Dachet F., Loeb J. A., DUSP4 appears to be a highly localized endogenous inhibitor of epileptic signaling in human neocortex. Neurobiol. Dis. 145, 105073 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gremer L., De Luca A., Merbitz-Zahradnik T., Dallapiccola B., Morlot S., Tartaglia M., Kutsche K., Ahmadian M. R., Rosenberger G., Duplication of Glu37 in the switch I region of HRAS impairs effector/GAP binding and underlies Costello syndrome by promoting enhanced growth factor-dependent MAPK and AKT activation. Hum. Mol. Genet. 19, 790–802 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/31/eabg2106/DC1