

Abstract
Aims
Heart failure with preserved ejection fraction (HFpEF) is frequently (30%) associated with right ventricular (RV) dysfunction, which increases morbidity and mortality in these patients. Yet cellular mechanisms of RV remodelling and RV dysfunction in HFpEF are not well understood. Here, we evaluated RV cardiomyocyte function in a rat model of metabolically induced HFpEF.
Methods and results
Heart failure with preserved ejection fraction‐prone animals (ZSF‐1 obese) and control rats (Wistar Kyoto) were fed a high‐caloric diet for 13 weeks. Haemodynamic characterization by echocardiography and invasive catheterization was performed at 22 and 23 weeks of age, respectively. After sacrifice, organ morphometry, RV histology, isolated RV cardiomyocyte function, and calcium (Ca2+) transients were assessed. ZSF‐1 obese rats showed a HFpEF phenotype with left ventricular (LV) hypertrophy, LV diastolic dysfunction (including increased LV end‐diastolic pressures and E/e′ ratio), and preserved LV ejection fraction. ZSF‐1 obese animals developed RV dilatation (50% increased end‐diastolic area) and mildly impaired RV ejection fraction (42%) with evidence of RV hypertrophy. In isolated RV cardiomyocytes from ZSF‐1 obese rats, cell shortening amplitude was preserved, but cytosolic Ca2+ transient amplitude was reduced. In addition, augmentation of cytosolic Ca2+ release with increased stimulation frequency was lost in ZSF‐1 obese rats. Myofilament sensitivity was increased, while contractile kinetics were largely unaffected in intact isolated RV cardiomyocytes from ZSF‐1 obese rats. Western blot analysis revealed significantly increased phosphorylation of cardiac myosin‐binding protein C (Ser282 cMyBP‐C) but no change in phosphorylation of troponin I (Ser23, 24 TnI) in RV myocardium from ZSF‐1 obese rats.
Conclusions
Right ventricular dysfunction in obese ZSF‐1 rats with HFpEF is associated with intrinsic RV cardiomyocyte remodelling including reduced cytosolic Ca2+ amplitudes, loss of frequency‐dependent augmentation of Ca2+ release, and increased myofilament Ca2+ sensitivity.
Keywords: Right ventricular dysfunction, Heart failure with preserved ejection fraction, Myofilament sensitivity, Calcium, ZSF‐1 obese rats
Translational perspective
Right ventricular (RV) dysfunction (RVD) in patients suffering from heart failure with preserved ejection fraction (HFpEF) is of paramount clinical relevance given its strong association with increased mortality and hospitalization rates in this patient population. Here, we analysed RVD at the level of single isolated RV cardiomyocytes and detected profound changes in RV excitation–contraction coupling with altered Ca2+ homeostasis and a compensatory increase in myofilament sensitivity, likely due to hyperphosphorylation of cMyBP‐C. Targeting RV cardiomyocyte Ca2+ homeostasis may be an approach to alleviate RVD to improve prognosis in patients with HFpEF and RVD.
Introduction
Right ventricular dysfunction is a frequent complication in patients suffering from HFpEF with an average prevalence of 30%. The diagnosis of RVD is based on echocardiographic assessment of RV morphology (RV hypertrophy and RV dilatation) and function [reduced tricuspid annular plane systolic excursion (TAPSE), fractional area change, or RV ejection fraction (EF)]. 1 , 2 , 3 , 4 , 5 , 6 RVD in HFpEF is closely related to worse clinical outcome as evident by increased hospitalization rates and overall mortality as compared with HFpEF patients without RVD. 4 , 7 , 8 , 9 There are currently no specialized treatment options available for HFpEF and RVD, and the underlying pathomechanisms of both conditions are not understood.
A widely accepted concept concerning the emergence of RVD in HFpEF relates to passive backward congestion from the left ventricle (LV). Key features of HFpEF are diastolic abnormalities that refer to stiffening (passive) and/or impaired relaxation (active) of the LV, ultimately leading to increased LV filling pressures and left atrial enlargement. 10 , 11 Subsequent congestion will first affect the pulmonary circulation and ultimately the RV. 12 Wall tension induced by increased RV afterload may to a certain extent be alleviated by RV hypertrophy. However, once pump capacity and impaired diastolic coronary perfusion no longer match the increased RV oxygen demand, the RV eventually dilates and fails. 13 Obesity, as a commonly observed co‐morbidity in HFpEF, has been linked to a reduced RV functional reserve. 14
In the present study, we investigated RV functional changes in a well‐established HFpEF rat model with metabolic syndrome 15 , 16 with the aim to determine the aetiology of these changes. Specifically, we hypothesized that besides the classic concept of RVD as a consequence of increased RV afterload, RV systolic dysfunction in HFpEF may also result from altered intrinsic RV contractile function at the level of the cardiomyocytes.
To this end, we evaluated RV function in an HFpEF rat model in vivo by echocardiography and invasive haemodynamics as well as in vitro using isolated RV cardiomyocytes to assess Ca2+ homeostasis, contractile function, and regulation of myofilament Ca2+ sensitivity.
Methods
All experiments were approved by the local ethics committee (G0317/17) and performed in agreement with the ARRIVE guidelines and guidelines for the Care and Use of Laboratory Animals (National Institute of Health, USA). Animals were housed in a 12 h light/dark regime in the local animal facility with free access to water. The ZSF‐1 obese rat model is based on a homozygous leptin receptor mutation resulting in a metabolic syndrome‐related HFpEF phenotype with LV hypertrophy, diastolic dysfunction, and preserved EF. 15 Wistar Kyoto rats, referred to as Control, and ZSF‐1 obese rats were obtained at 10 weeks of age (Charles River Laboratories, MA, USA) and fed a high‐caloric diet (Purina 5008: 26.6% protein, 16.5% fat, 56.9% carbohydrates; LabDiet, MO, USA). At 22 weeks, transthoracic echocardiography was performed prior to invasive haemodynamic measurements at 23 weeks. Afterwards, animals were sacrificed, organs were harvested, and morphometry and isolation of RV cardiomyocytes were performed.
Echocardiography
Study individuals were subject to transthoracic echocardiography. Animals were initially anaesthetized with 4% isoflurane, reduced afterwards to 1.5% supplemented with 1.5 L/min oxygen. Body temperature and electrocardiogram were continuously monitored to keep normal physiological and comparable ranges of temperature, heart (HR), and respiratory rate. Eyes were covered with a moisturizing ointment to avoid occular dehydration. The chest was shaven and depilated. Image acquisition was performed using an ultra‐high‐frequency linear array transducer (MS250, 13–24 MHz) in combination with a 2100 Vevo® high‐resolution imaging system (all FUJIFILM VisualSonics, Toronto, Ontario, Canada). Imaging of the LV was performed according to standard guidelines and formulas. 17 Image acquisition for the RV was conducted as follows: (i) in the parasternal long‐axis views (PSLAX), M‐mode recordings of the RV outflow tract just above the aortic root were obtained for measurement of RV diastolic and systolic diameters as well as calculation of RV fractional shortening (FS) and measurement of end‐diastolic RV free wall (RVFW) thickness. (ii) Four‐chamber view (4CV) B‐mode cine loops were recorded for tracing of the RV dimensions, RV area, and RVEF. M‐mode images were acquired from the lateral tricuspid valve annulus for determination of TAPSE. All images and cine loops were stored for offline analysis using the V3.1 Vevolab Software (FUJIFILM VisualSonics). Data from a minimum of three cardiac cycles were evaluated for TAPSE and M‐mode dimensional measurements.
Invasive haemodynamic measurements (pressure‐volume loops)
Invasive haemodynamic measurements and the analysis of pressure‐volume (PV) loops were performed as terminal procedure in all groups using a PV conductance system (MPVS Ultra, Millar Instruments, USA) connected to the PowerLab 8/35 data acquisition system and analysed using LabChart7pro software system (both ADInstruments, Dunedin, New Zealand). Rats were anaesthetized with 4% isoflurane and 1.5 L/min oxygen for 3 min, intubated with an orotracheal tube, and ventilated by a rodent ventilator (SAR‐1000 Small Animal Ventilator, CWE, USA). The animals were placed on a controlled heating platform to maintain core temperature at 37.5°C. Animals were attached to a surface electrocardiogram. Anaesthesia was reduced to 1.5% isoflurane while oxygen remained at 1.5 L/min until haemodynamics reached a steady state. A polyethylene catheter was inserted into the left external jugular vein for fluid administration (0.9% NaCl 10 mL/kg/h), and another polyethylene catheter was inserted into the right external jugular vein for saline calibration (10% NaCl) at the end of the experiment. The 2.0‐Fr PV conductance catheter (SPR‐838, Millar, USA) was inserted into the right carotid artery and advanced into the ascending aorta. The catheter was advanced through the aortic valve into the LV under pressure control. After a stabilization period, baseline measurements were performed. Finally, HR and aortic pressure were recorded. Parameters of systolic and diastolic function including LV end‐systolic and end‐diastolic pressures (LVESP and LVEDP) were measured and calculated according to standard formulas.
Cardiac morphometry
After haemodynamic measurements, a subset of animals was used for analysis of cardiac morphometry. The heart was harvested immediately after sacrifice, and the RV was surgically separated from the LV. After documentation of the RV weight, tissue samples were frozen in pre‐cooled 2‐methylbutane and stored at −80°C.
Histology and fluorescence imaging
Cardiac specimens were fixed in 4% paraformaldehyde and embedded in paraffin. Sections of 4–8 μm were cut using a microtome and transferred onto superfrost slides. Haematoxylin/eosin as well as picrosirius red staining was performed according to common standards. Fluorescent staining was performed using wheat‐germ agglutinin conjugated to Alexa‐488 at a concentration of 5 μg/mL diluted in 1× phosphate buffered saline. Staining for 15 min was followed by two wash steps with Hank's balanced salt solution. Slides were mounted using Fluoromount‐G with DAPI. Analyses were performed by Fiji (ImageJ). 18 For analysis of RV fibrosis, five images of the RVFW were analysed per specimen to calculate relative fibrosis area (%) using the ‘MRI fibrosis tool’ macro provided by Montpellier Ressources. 19 , 20 RV cardiomyocyte cross‐sectional area (CSA) was assessed by measuring 100 cells per specimen/animal. Evaluated specimens originate from a separate cohort at 21 weeks.
Right ventricular cardiomyocyte isolation and Ca2+ measurements
Animals were sacrificed by isoflurane overdose and blood withdrawal, and explanted hearts were enzymatically digested in a Langendorff set‐up as previously described. 21 After digestion, the RV was separated with surgical scissors, chopped, filtered, and washed. Cells were dispersed by agitation. Ca2+ was reintroduced using Tyrode's solutions (130 mM NaCl, 4 mM KCl, 1 mM MgCl2, 10 mM d‐glucose, 10 mM HEPES; pH 7.4 with NaOH) with stepwise increasing Ca2+ concentrations (0.1–1.0 mM). Cells were plated on laminin‐coated glass cover‐slips and incubated with fura‐2 AM (1 μmol/L) (both Thermo Fisher Scientific, Waltham, MA, USA) in 1 mM Ca2+‐supplemented Tyrode's solution for 12 min at room temperature. Cells were washed once, and the cover‐slip was transferred for Ca2+ measurements to a heated, temperature‐controlled stage (37°C) on a ratiometric fluorescence microscope (PMT400 photomultiplier; Axiovert 200 microscope, Zeiss, Jena, Germany; MyoPacer EP and Fluorescence System Interface, both IonOptix, Westwood, MA, USA). The set‐up was additionally equipped with a CCD camera for detection and subsequent analysis of sarcomere shortening (MyoCam‐S, IonOptix, Westwood, MA, USA). Field stimulation with 1 Hz was initially applied until a steady‐state amplitude of the Ca2+ transients was maintained. Afterwards, 2 and 4 Hz were applied, and transients recorded after transient amplitudes reached again a steady state. Measurements were background and autofluorescence corrected by measuring fluorescence intensity of unloaded cells of the same isolation. Data analysis was performed using the IonWizard and Transient Analysis Tool software (both IonOptix).
Myofilament Ca2+ sensitivity
A ratio of peak intracellular [Ca2+] to systolic sarcomere length was calculated to assess Ca2+ sensitivity. In addition, sarcomere shortening as measure of contractility was plotted against intracellular [Ca2+], creating a hysteresis loop. A linear regression line was fitted to calculate the slope between 20% and 80% return from the peak sarcomere shortening. 22 , 23 Only curves with a goodness of linear fit (R 2) > 0.9 were included for this analysis.
Western blot
Tissue samples were homogenized at 4°C in lysis buffer with the following composition: 20 mM Tris–HCl, pH 7.4, 137 mM NaCl, 10% glycerol, 1% NP 40, 20 mM NaF, 1 mM sodium pyrophosphate, 50 mM β‐glycerophosphate, 10 mM EDTA, 1 mM EGTA, 1 mM PMSF, 4 μg/mL aprotinin, 4 μg/mL pepstatin A, and 4 μg/mL leupeptin. For Western blotting, 40 μg of tissue homogenates were run on 4–12% Bis‐Tris polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were probed with anti‐phospho Ser 23/24 troponin I (1/1000, PhosphoSolutions, Aurora, CO, USA) and anti‐troponin (1/1000, Novus, Centennial, CO, USA) or anti‐phosphoSer282 cMyBP‐C (1/1000, Enzo) and anti‐cMyBP‐C (1/1000, Novus, Centennial, CO, USA) overnight at 4°C. Anti‐rabbit IgG linked with IRDye 800CW or anti‐mouse IgG linked with 680RD (LI‐COR, Lincoln, NE, USA) was used as a secondary antibody. The signal was detected with an Odyssey CLx System, and band intensities were determined by Image Studio software (LI‐COR, Lincoln, NE, USA).
Blood glucose
Glucose levels in blood samples from the tail vein were measured using a commercially available blood glucose analyser (Bayer, Leverkusen, Germany).
Statistics
Data are presented as means ± standard error of the mean. Each data point represents one biological replicate or animal. For analysis of cellular data, figure legends include the number of cells evaluated to obtain a mean value per animal. Statistical analysis was performed by Mann–Whitney test, unpaired t‐test, or two‐way analysis of variance with Fisher's least significant difference. Statistical significance was assumed at P < 0.05. Data were analysed using GraphPad Prism V8.0.2 (GraphPad Software, Inc, La Jolla, CA, USA).
Results
Heart failure with preserved ejection fraction phenotype confirmed
Twenty‐three‐week‐old ZSF‐1 obese rats showed characteristic signs of LV hypertrophy (Control 0.98 ± 0.05 vs. ZSF‐1 obese 1.62 ± 0.08, g; P < 0.005) and preserved LVEF (Control 73.2 ± 2.2 vs. ZSF‐1 obese 72.0 ± 1.1, %) (Figure 1 A ). LV end‐diastolic volumes (LVEDV) (Control 370 ± 18 vs. ZSF‐1 obese 507 ± 27, μL; P < 0.005) as well as end‐systolic volumes (LVESV) (Control 98 ± 8 vs. ZSF‐1 obese 141 ± 7, μL; P < 0.005) were significantly increased, whereas cardiac output was not (Control 91 ± 6 vs. ZSF‐1 obese 106 ± 6, mL/min). Cardiac index, however, was significantly reduced in ZSF‐1 obese rats (Control 1.6 ± 0.1 vs. ZSF‐1 obese 1.0 ± 0.01, P < 0.005). HR during echocardiography (Figure 1 B ) was approximately 10% higher in the Control group (Control 335 ± 5 vs. ZSF‐1 obese 292 ± 4, b.p.m.; P < 0.005). Diastolic marker E/e′ ratio was significantly higher in ZSF‐1 obese rats (Control 18.4 ± 1.3 vs. ZSF‐1 obese 25.0 ± 1.2; P < 0.005) (Figure 1 C ). Haemodynamic analysis revealed increased LVEDP (Control 6.4 ± 0.6 vs. ZSF‐1 obese 10.8 ± 0.7 mmHg; P < 0.005) and LVESP (Control 110.7 ± 8.4 vs. ZSF‐1 obese 167.3 ± 2.2 mmHg; P < 0.005). Relaxation time constant tau did not differ significantly between groups (Control 11.5 ± 0.5 vs. ZSF‐1 obese 12.50 ± 0.3, ms) (Figure 1 D ). Blood glucose levels were also significantly higher in ZSF‐1 obese animals (Control 138 ± 13 vs. ZSF‐1 obese 330 ± 23 mg/dL; P < 0.05).
Figure 1.
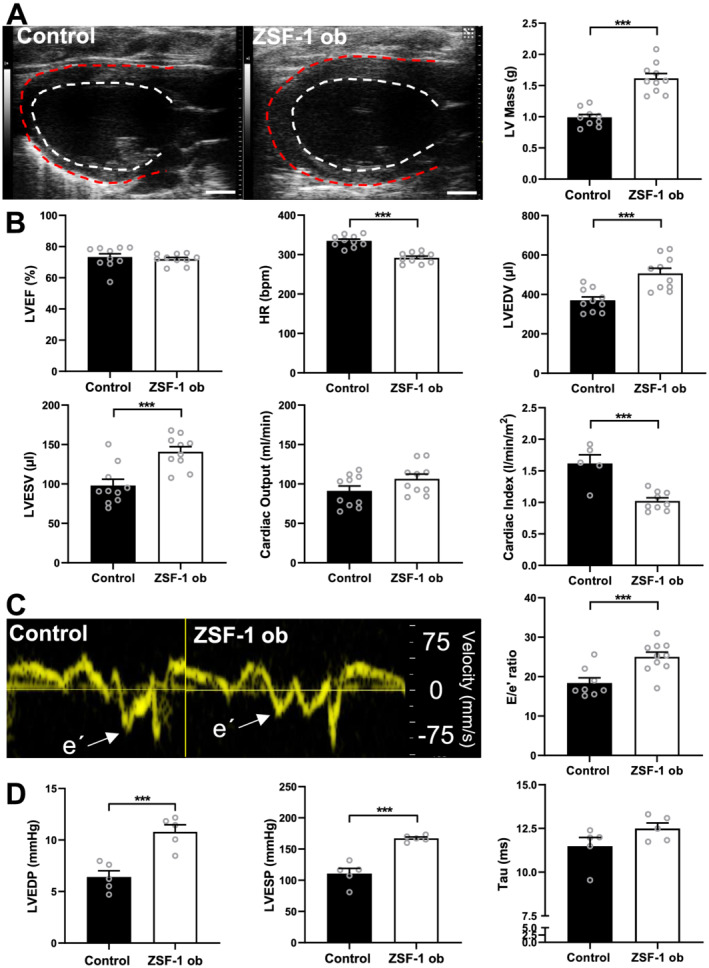
Echocardiographic and haemodynamic confirmation of HFpEF phenotype in ZSF‐1 obese rats. (A) Representative parasternal long‐axis B‐mode images of the left ventricle and quantification of left ventricular (LV) mass in grams (g). (B) Quantification of left ventricular ejection fraction (LVEF) in per cent (%), heart rate (HR) in beats per minute (b.p.m.), LV end‐diastolic and end‐systolic volumes in μL (LVEDV and LVESV), cardiac output (CO) in mL/min, and cardiac index (CI) in L/min/m2. (C) Representative tissue Doppler recordings from the septal mitral valve annulus depicted as velocity in mm/s and quantification of E/e′ ratio. (D) Haemodynamic data quantification of left ventricular end‐systolic/end‐diastolic pressures (LVESP and LVEDP) in mmHg as well as Tau in ms. n = 10 per group for echocardiography (except for CI: Control n = 5) and n = 5 per group for haemodynamics. Control, Wistar Kyoto rats; ZSF‐1 ob, ZSF‐1 obese. Scale bar = 3 mm; data are presented as mean ± standard error of the mean; statistics were performed by Mann–Whitney test or unpaired t‐test with *P < 0.05, **P < 0.01, ***P < 0.005.
Right ventricular dysfunction in heart failure with preserved ejection fraction
Echocardiographic assessment of the RV revealed a significant dilatation in M‐mode recordings from ZSF‐1 obese animals in the parasternal long‐axis view (Figure 2 A,B ) with increased RV systolic diameter (Control 2.0 ± 0.1 vs. ZSF‐1 obese 2.5 ± 0.1, mm; P < 0.01) and diastolic diameter (M‐mode: Control 3.2 ± 0.1 vs. ZSF‐1 obese 4.1 ± 0.1, mm; P < 0.01). This was confirmed in B‐mode recordings from the long‐axis four‐chamber view (Figure 2 C,D ), also showing increased diameters (diastole; Control 3.2 ± 0.3 mm vs. ZSF‐1 obese 4.6 ± 0.2, mm; P < 0.005) and additionally detecting an increase in RV luminal size by approximately 50% in ZSF‐1 obese animals (Control 32.9 ± 2.2 vs. ZSF‐1 obese 49.7 ± 2.6, mm2, P < 0.005). RV functional performance measured by FS was preserved in ZSF‐1 obese rats compared with Control (Control 35.8 ± 1.9 vs. ZSF‐1 obese 38.4 ± 2.8, %). Ratio of RV FS to LV FS was unchanged in ZSF‐1 obese rats compared with controls (Control 0.59 ± 0.03 vs. ZSF‐1 obese 0.61 ± 0.07). TAPSE was unchanged in ZSF‐1 obese animals (Control 1.9 ± 0.2 vs. ZSF‐1 obese 1.9 ± 0.2, mm) whereas RVEF was reduced by about 10% (Control 52.0 ± 3.3 vs. ZSF‐1 obese 42.4 ± 1.9, %; P < 0.05, Figure 2 E ).
Figure 2.
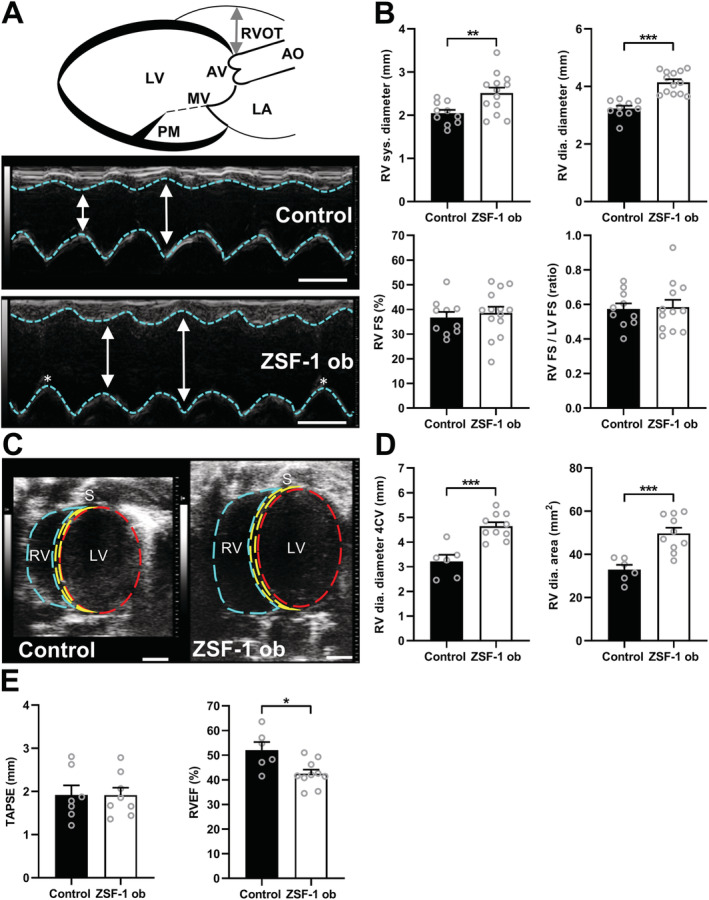
Right ventricular dilation in HFpEF. (A) Schematic of parasternal long‐axis M‐mode measurements of the right ventricular diameter (RVD) with corresponding sample images; scale bar = 3 mm. Double‐headed arrow indicates RVD (B) Mean RVD in systole (sys.) and diastole (dia.), RV fractional shortening (FS), and ratio of RV and LV FS (n = 10–13 per group). (C) Representative B‐mode four‐chamber view (4CV) images; scale bar = 3 mm. (D) Quantification of the RVD and RV area from the 4CV (n = 6–10 per group). (E) Tricuspid annular plane systolic excursion (TAPSE) and RV ejection fraction (EF) measured from the 4CV (n = 6–10 per group). AO, aorta; AV, aortic valve; Control, Wistar Kyoto rats; LA, left atrium; LV, left ventricle; MV, mitral valve; PM, papillary muscle; RV, right ventricle; RVOT, right ventricular outflow tract; S, septum; ZSF‐1 ob, ZSF‐1 obese. Data are presented as mean ± standard error of the mean; statistics were performed using unpaired t‐test with *P < 0.05, **P < 0.01, ***P < 0.005.
Right ventricular hypertrophy in heart failure with preserved ejection fraction
Gravimetric analysis of RV weight to tibia length ratio (Figure 3 A ) yielded a significant difference between ZSF‐1 and controls subjects (Control 3.7 ± 0.3 vs. ZSF‐1 obese 5.3 ± 0.3; P > 0.05). RVFW thickness assessed by echocardiography (Figure 3 B ) was increased by approximately 50% in ZSF‐1 obese rats (Control 0.42 ± 0.02 vs. ZSF‐1 obese 0.65 ± 0.06, mm; P > 0.01). Measurement of RV cardiomyocyte CSA in cardiac histology (Figure 3 C ) also detected a significant increase in the ZSF‐1 obese group (Control 270 ± 11 vs. ZSF‐1 obese 412 ± 14, μm; P > 0.005) whereas determination of relative RV fibrosis revealed no changes (Control 2.6 ± 0.1 vs. ZSF‐1 obese 3.2 ± 0.2, %).
Figure 3.
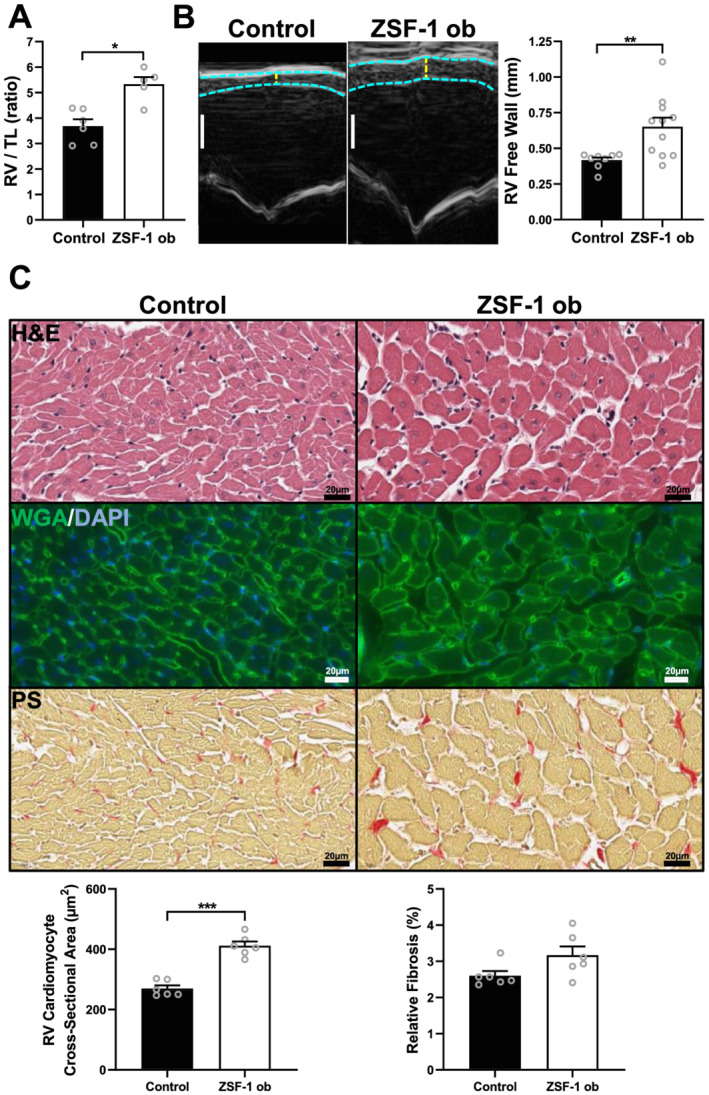
ZSF‐1 obese rats exhibit right ventricular hypertrophy. (A) Gravimetric ratio of RV weight to tibia length (TL); n = 5–6 per group. (B) Representative right ventricular (RV) M‐mode images depicting increased RV free wall thickness with quantification and n = 8–11 per group; scale bar = 1 mm. (C) Representative haematoxylin/eosin (H&E), wheat‐germ agglutinin/DAPI (WGA/DAPI), and picrosirius red (PS) staining images with RV cardiomyocyte cross‐sectional area (CSA) and relative fibrosis quantification; n = 6 per group with 100 cells being measured per data point/animal for CSA assessment and five images of the RV free wall were evaluated per data point/animal for fibrosis analysis. Control, Wistar Kyoto rats; ZSF‐1 ob, ZSF‐1 obese. Data are presented as mean ± standard error of the mean; statistics were performed by Mann–Whitney test or unpaired t‐test with *P < 0.05, **P < 0.01, ***P < 0.005.
Altered Ca2+ handling and sensitivity in isolated right ventricular cardiomyocytes from ZSF‐1 obese rats
Ca2+ transient analysis of isolated RV cardiomyocytes (Figure 4 A, Table 1 ) revealed a frequency‐dependent increase in systolic Ca2+ transient amplitudes in Control whereas in ZSF‐1 obese rats, the frequency response was blunted, and peak systolic [Ca2+] was significantly lower (F 340/F 380 ratio) at 1 Hz (−16%), 2 Hz (−19%), and 4 Hz (−24%) field stimulation. In contrast, diastolic [Ca2+] did not differ between groups. As a result, the Ca2+ amplitude was also significantly decreased by approximately 45% at 1 Hz, 46% at 2 Hz, and 50% at 4 Hz. Sarcomere analysis (Figure 4 B, Table 1 ) did neither detect changes in cardiomyocyte contractility as determined by sarcomere shortening nor in diastolic sarcomere length between control and ZSF‐1 obese animals. Ca2+ transient and sarcomere kinetics were largely unchanged in ZSF‐1 obese rats compared with controls, with exception of Ca2+ transient time to peak being significantly prolonged at 1, 2, and 4 Hz as well as Ca2+ time to 50% baseline at 1 Hz in ZSF‐1 obese rats compared with controls (Table 1 ). Analysis of myofilament Ca2+ sensitivity from [Ca2+]–sarcomere length relationships (Figure 4 C, Table 1 ) revealed a significant increase in slope of the returning Ca2+ hysteresis loop in ZSF‐1 obese rats and a significant reduction of the peak Ca2+ to systolic sarcomere length ratio by approximately 20%.
Figure 4.
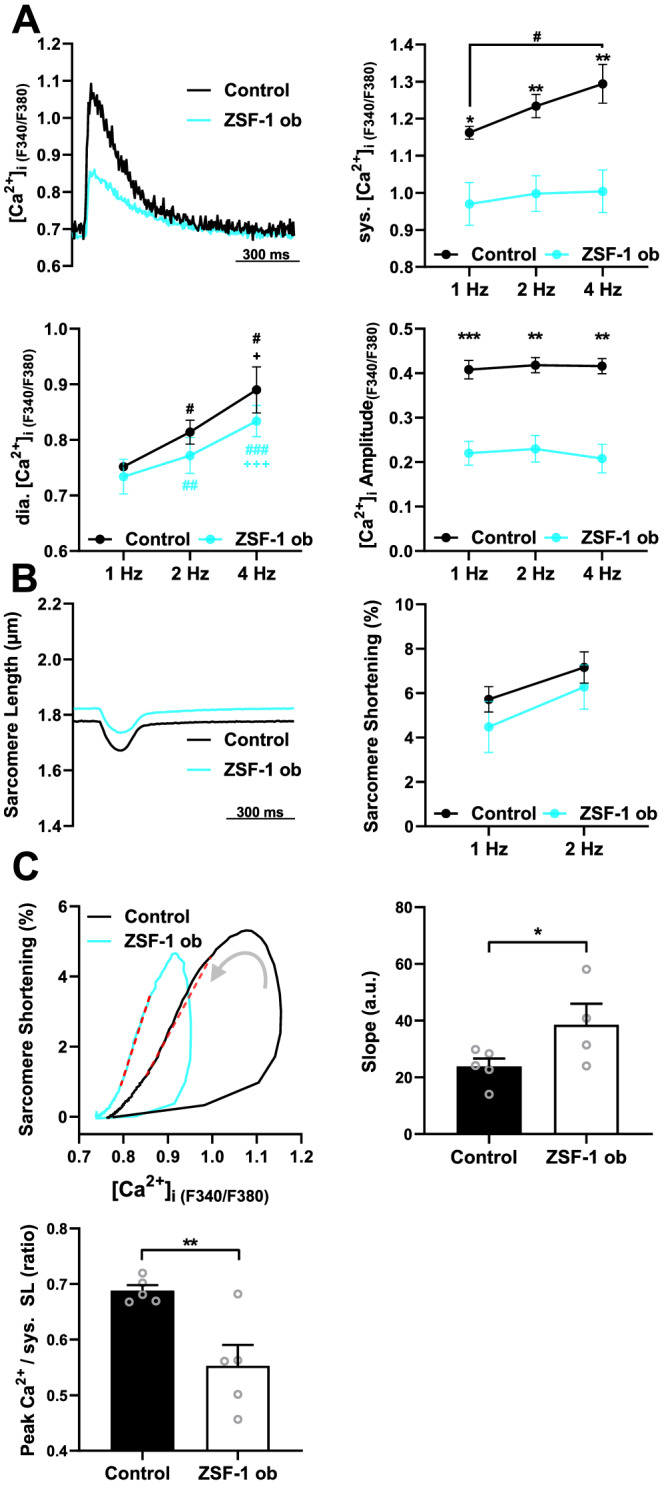
Isolated right ventricular cardiomyocytes from HFpEF rats exhibit low intracellular [Ca2+] and preserved contractility. (A) Representative Ca2+ transients at 1 Hz (left, top), and mean values for systolic (sys.) and diastolic (dia.) [Ca2+], and Ca2+ amplitudes at 1, 2, and 4 Hz field stimulation. (B) Representative example of sarcomere shortening at 1 Hz (left) and mean sarcomere shortening amplitude (right) at 1 and 2 Hz. (C) Representative loops of sarcomere shortening as a function of [Ca2+]i in an RV cardiomyocyte from ZSF‐1 obese and Control rat (left). The linear fit between 20% and 80% relaxation is marked; mean values for the slope of the linear fit (right) and peak [Ca2+]i to syst. SL ratio (bottom); N = 4–5 animals per group (symbols) with an average of 16 cells per animal. Control, Wistar Kyoto rats; ZSF‐1 ob, ZSF‐1 obese. Error bars represent standard error of the mean; statistics were performed by two‐way analysis of variance with Fisher's least significant difference for (A) and (B) and unpaired t‐test for (C) with *P < 0.05, **P < 0.01, ***P < 0.005. # and + in (A) indicate differences compared with 1 and 2 Hz, respectively, within groups.
Table 1.
Right ventricular cardiomyocyte characterization
| Parameter | Control | HFpEF | ||
|---|---|---|---|---|
| Stimulation frequency | Mean ± SEM | P‐values | ||
| Cell shortening (CS) | ||||
| CS amplitude (%) | 1 Hz | 5.72 ± 0.51 | 4.48 ± 1.03 | 0.3646 |
| 2 Hz | 7.16 ± 0.63 | 5.72 ± 0.86 | 0.2163 | |
| Sarcomere length (μm) | 1 Hz | 1.79 ± 0.01 | 1.84 ± 0.04 | 0.1724 |
| 2 Hz | 1.77 ± 0.01 | 1.79 ± 0.01 | 0.6040 | |
| CS TTP (ms) | 1 Hz | 145.40 ± 11.35 | 162.80 ± 5.35 | 0.1656 |
| 2 Hz | 126.00 ± 9.27 | 142.5 ± 6.29 | 0.2122 | |
| CS RT50 (ms) | 1 Hz | 241.20 ± 25.25 | 285.20 ± 25.73 | 0.1513 |
| 2 Hz | 192.00 ± 15.38 | 200.30 ± 9.30 | 0.7929 | |
| CS RT90 (ms) | 1 Hz | 401.20 ± 25.47 | 518.80 ± 68.16 | 0.0526 |
| 2 Hz | 262.80 ± 23.42 | 290.00 ± 3.67 | 0.6528 | |
| CS Tau (s) | 1 Hz | 0.11 ± 0.02 | 0.19 ± 0.05 | 0.0603 |
| 2 Hz | 0.08 ± 0.02 | 0.09 ± 0.01 | 0.8703 | |
| Calcium transients (CaT)—epifluorescence | ||||
| CaT amplitude (F 340/F 380 ratio) | 1 Hz | 0.41 ± 0.02 | 0.22 ± 0.02 | <0.005 |
| 2 Hz | 0.42 ± 0.02 | 0.23 ± 0.03 | <0.005 | |
| 4 Hz | 0.42 ± 0.02 | 0.21 ± 0.03 | <0.005 | |
| Systolic cytosolic [Ca2+] | 1 Hz | 1.16 ± 0.02 | 0.97 ± 0.05 | <0.05 |
| 2 Hz | 1.23 ± 0.03 | 0.99 ± 0.05 | <0.005 | |
| 4 Hz | 1.29 ± 0.05 | 1.00 ± 0.06 | <0.01 | |
| Diastolic cytosolic [Ca2+] | 1 Hz | 0.75 ± 0.01 | 0.73 ± 0.03 | 0.6003 |
| 2 Hz | 0.81 ± 0.02 | 0.77 ± 0.03 | 0.3112 | |
| 4 Hz | 0.89 ± 0.04 | 0.83 ± 0.03 | 0.2991 | |
| CaT TTP (ms) | 1 Hz | 40.20 ± 2.04 | 61.00 ± 3.54 | <0.005 |
| 2 Hz | 40.00 ± 4.47 | 60.00 ± 3.16 | <0.01 | |
| 4 Hz | 42.00 ± 2.00 | 52.00 ± 2.00 | <0.01 | |
| CaT RT50 (ms) | 1 Hz | 204.20 ± 9.48 | 233.60 ± 13.67 | <0.05 |
| 2 Hz | 158.60 ± 9.50 | 177.40 ± 4.47 | 0.1174 | |
| 4 Hz | 119.20 ± 2.78 | 122.80 ± 2.69 | 0.7585 | |
| CaT RT90 (ms) | 1 Hz | 555.60 ± 29.20 | 603.00 ± 56.16 | 0.2263 |
| 2 Hz | 338.20 ± 6.56 | 366.60 ± 13.74 | 0.4735 | |
| 4 Hz | 191.20 ± 10.22 | 211.00 ± 5.34 | 0.6087 | |
| CaT Tau (s) | 1 Hz | 0.25 ± 0.02 | 0.28 ± 0.05 | 0.5464 |
| 2 Hz | 0.18 ± 0.02 | 0.18 ± 0.02 | 0.9156 | |
| 4 Hz | 0.14 ± 0.02 | 0.23 ± 0.08 | 0.0945 | |
| Cardiomyocyte myofilament sensitivity | ||||
| Peak Ca2+ to systolic sarcomere length ratio | 1 Hz | 0.69 ± 0.01 | 0.55 ± 0.03 | 0.0087 |
| Slope (a.u.) | 1 Hz | 23.83 ± 2.78 | 38.60 ± 7.36 | 0.0396 |
Cell shortening (CS); CS amplitude, %; diastolic sarcomere length, μm; CS time to peak (TTP), ms; CS time to 50% baseline (RT50), ms; CS time to 90% baseline (RT90), ms; CS relaxation time constant (Tau), s; calcium transients amplitude (CaT), F 340/F 380 ratio; systolic cytosolic [Ca2+] concentration; diastolic cytosolic [Ca2+] concentration; CaT time to peak (TTP), ms; CaT time to 50% baseline (RT50), ms; CaT time to 90% baseline (RT90), ms; CaT time constant (Tau), s; peak Ca2+ to systolic sarcomere length ratio; slope of cardiomyocyte myofilament sensitivity, a.u.; cell shortening measurements were performed at 1 and 2 Hz stimulation. Epifluorescence measured calcium transients were performed at 1, 2, and 4 Hz stimulation. Control, Wistar Kyoto rats. Four to five animals per group with an average of 16 cells per animal. Error bars represent standard error of the mean (SEM).
Western blot phosphorylation analysis of sarcomeric proteins involved in myofilament Ca2+ sensitivity and contractility from right ventricular tissue specimens
Phosphorylation of cTnI S‐23, 24 (Figure 5 A ) did not differ significantly between RV tissue samples from ZSF‐1 obese rats compared with controls (albeit a trend towards higher phosphorylation in ZSF‐1 obese F was seen: Control 1.00 ± 0.20 vs. ZSF‐1 obese 1.88 ± 0.41, a.u.), whereas cMyBP‐C P‐282 (Figure 5 B ) was significantly increased by 1.35‐fold (Control 1.00 ± 0.04 vs. ZSF‐1 obese 1.35 ± 0.09; P < 0.01).
Figure 5.
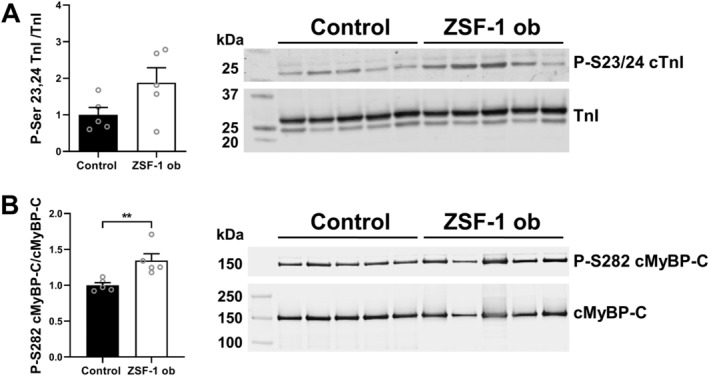
Hyperphosphorylation of cMyBP‐C S282 but not TnI S23, 24 was present in right ventricular tissue from HFpEF rats. (A) Western blot analysis of phosphorylation of cardiac troponin I (cTnI) at serine residues 23, 24 normalized to total cTnI levels. (B) Western blot analysis of phosphorylation of cMyBP‐C serine residue 282 normalized to total cMyBP‐C levels. n = 5 animals per group. Control, Wistar Kyoto rats; ZSF‐1 ob, ZSF‐1 obese. Data are presented as mean ± standard error of the mean; statistics were performed by unpaired t‐test. *P < 0.05, **P < 0.01, ***P < 0.005.
Discussion
In this study, we explored RV function in an established rat model of metabolic syndrome with HFpEF. 15 , 16 , 24 , 25 , 26 , 27 We show that RV remodelling in HFpEF is associated with profound alterations in RV cardiomyocyte Ca2+ homeostasis, as well as increased myofilament Ca2+ sensitivity, likely linked to hyperphosphorylation of cMyBP‐C.
Heart failure with preserved ejection fraction accounts for approximately 50% of all HF patients with no specific treatment currently available to improve clinical outcomes. 24 Involvement of the RV has been reported in about 30% of HFpEF patients and frequently constitutes a major cause of death in this subgroup. 1 , 2 , 3 , 4 , 5 , 6 The role of RV cardiomyocyte remodelling and adaptation of intrinsic cardiomyocyte function, however, has not yet been explored. The ZSF‐1 rat has recently been established as a model of HFpEF, 15 , 16 , 25 , 26 , 27 , 28 and our present findings of LV concentric hypertrophy with preserved LVEF conclusively recapitulate a common clinical phenotype (Figure 1 A–D ). Cardiac volumes (Figure 1 B ) were increased in the HFpEF group in agreement with clinical features of obese HFpEF patients. 29 Cardiac index was significantly reduced suggesting reduced tissue perfusion. HR tended to be lower in some of our measurements, with no relevant effects on myocardial contractile function as LVEF remained preserved and did not differ from Control. We as well as others deliberately chose WKY animals as Control because ZSF‐1 lean rats do show a pathological cardiac phenotype, including increased mean arterial blood pressure and LV hypertrophy. 16 As a result of diastolic dysfunction, filling pressures in ZSF‐1 obese animals were greatly elevated as indicated by an increased E/e′ ratio, and elevated systolic and diastolic LV pressures, whereas relaxation constant tau only showed a trend towards slower relaxation (Figure 1 B–D ). Mechanical load is considered the main pathophysiological trigger for initially compensatory RV concentric hypertrophy and ultimately dilation and RV failure. 4 , 7 , 8 , 9 The model is characterized by a metabolic phenotype, as confirmed by highly elevated blood glucose levels. This is especially interesting in the context of the globally increasing prevalence of type‐2 diabetes and obesity, both largely preventable conditions that are major risk factors for developing HFpEF. 31 , 32 As such, the ZSF‐1 obese rat presents a clinically relevant model for an in‐depth analysis of underlying pathomechanisms in HFpEF. Here, we report that HFpEF in this model is also associated with RVD (Figure 2 A–D ), matching clinical conditions. RVEF was significantly reduced to approximately 42% (Figure 2 E ) in ZSF‐1 obese animals. This matches clinical data by Tadic and colleagues suggesting mild RVD in HFpEF with an RVEF below 45%. 33 Interestingly, unchanged RV FS (measured in PSLAX) and TAPSE (Figure 2 B,E ) did not correspond to the reduced EF. Of note, the RV PSLAX M‐mode measurements do not assess the longitudinal but circumferential shortening of the RV, thereby assessing shortening of transverse muscle fibres that are only pledgable for about 20% of the overall RV output. 34 It may thus be speculated that helical muscle fibres responsible for longitudinal shortening and lengthening, which make up 80% of the RV output, are affected in our model. 34 A different aspect is that our M‐mode measurements were recorded towards the RV base. RV strain analyses show that the RV basal segment is less kinetically active compared with middle and apical segments, and as a result, recordings of the RV base are expected to be less sensitive for detection of subtle functional changes. 35 In addition, previous studies have pointed out lower accuracy of TAPSE in assessment of RV systolic function compared with fractional area change and RVEF. 36 Finally, our data agree with clinical observations that TAPSE is dependent on RV load, as in individuals with mildly dilated RV higher TAPSE cut‐off values may be needed to detect RVD. 37 In post‐mortem gravimetric analysis of the RV, we detected RV hypertrophy (Figure 3 A ), which is expected in functional compensatory states of the RV. 34 Further gravimetric analysis of liver and kidney are supplied in Figure S1 . In support of this finding, echocardiographic data revealed an increased RVFW thickness in the ZSF‐1 obese group (Figure 3 B ), a marker that is also routinely used in the clinics to assess RV hypertrophy. 38 Analysis of histological and fluorescent staining (Figure 3 C ) further strengthened our characterization of RV hypertrophy, showing a distinct increase in RV cardiomyocyte CSA. Whereas general cardiomyocyte hypertrophy in this HFpEF model has previously been described by others, 27 the present study is to our knowledge the first to confirm this finding also specifically for the RV. Overt fibrotic changes in the RV could, on the other hand, not be detected, albeit a trend towards higher relative fibrosis in the ZSF‐1 obese group was seen.
Previous studies on cardiac Ca2+ handling and sensitivity in HFpEF have primarily relied on assessing cardiomyocytes isolated from the LV, 39 whereas RV cardiomyocyte function has not been studied. Here, ratiometric Ca2+ imaging in isolated RV cardiomyocytes revealed a considerably altered cytosolic Ca2+ homeostasis (Figure 4 , Table 1 ) with an almost 50% decrease of the Ca2+ amplitude but unchanged diastolic resting calcium (Figure 4 A, Table 1 ). In addition, the frequency‐dependent augmentation of Ca2+ release observed in the control was lost in ZSF‐1 obese rats. While a reduced Ca2+ transient amplitude is reminiscent of Ca2+ handling in failing cardiomyocytes from the dilated LV in HFrEF, 40 the unchanged diastolic Ca2+ and differentially altered Ca2+ transient kinetics (Figure 4 A, Table 1 ) point towards a unique RV cardiomyocyte phenotype. Interestingly, RV cardiomyocyte sarcomere shortening and resting sarcomere length were not significantly affected by RVD (Figure 4 B, Table 1 ). The marked reduction in the ratio of peak Ca2+ relative to systolic sarcomere length suggests increased myofilament Ca2+ sensitivity (Figure 4 C, Table 1 ). An extended kintetics evaluation can found be Figure S2 . This again is in contrast to failing LV cardiomyocytes, where Ca2+ myofilament sensitivity is characteristically reduced. Increased Ca2+ sensitivity in intact cardiomyocytes was also evident by performing analysis of Ca2+/sarcomere shortening hysteresis loops as previously described. 22 , 23 The curve revealed a left shift of the hysteresis loop with an increased slope during the cardiomyocyte relaxation phase in ZSF‐1 obese animals, in agreement with increased myofilament Ca2+ sensitivity 22 , 23 (Figure 4 C, Table 1 ). To further explore potential molecular mechanisms of altered RV myofilament Ca2+ sensitivity in ZSF‐1 obese rats, we performed western blot analyses of the phosphorylation status of S‐23, 24 TnI and S‐282 cMyBP‐C (Figure 5 ), two myofilament‐interacting proteins known for their involvement in Ca2+ sensitization. 41 , 42 Phosphorylation of TnI S‐23, 24 is primarily mediated via PKA activity secondary to adrenergic stimulation and has proven particularly important in regulating the myofilament response to Ca2+ towards a lowered responsiveness and enhanced relaxation. 42 A previous study assessed TnI phosphorylation in LV samples of the current HFpEF model and revealed a significant reduction in total TnI phosphorylation, in agreement with other studies. 43 In contrast, our present data from RV tissue showed no significantly altered phosphorylation of TnI in ZSF‐1 obese animals, albeit there was a trend towards higher phosphorylation. Previous studies also suggest a modulatory function of myosin‐binding protein C (cMyBP‐C) towards the interaction of myosin and actin. 41 Phosphorylated cMyBP‐C, for example, as a result of PKA activation, favours actin binding over myosin S2 binding, which is reported to contribute to Ca2+ sensitization of the thin filament. 41 , 44 We chose to evaluate the S‐282 phosphorylation site of cMYBP‐C, because it has been described as a requirement for further phosphorylation at S‐273 and S‐302 and is therefore crucial to cMyBP‐C's overall phosphorylation status. 45 In RV myocardium from ZSF‐1 obese animals, we detected increased phosphorylation of cMyBP‐C, suggesting PKA‐dependent cMYBP‐C phosphorylation as one mechanism for the observed increased RV myofilament Ca2+ sensitivity. Interestingly, a recent publication by Kolijn et al. 46 showed in a genetically related model of HFpEF, the ZDF rat, that PKA activity and c‐MyBPc phosphorylation status in the LV were not altered, suggesting an RV‐specific phenotype in our study. Functionally, increased myofilament sensitivity acts compensatory to reduced Ca2+ transient amplitudes, thus explaining unchanged cell shortening in isolated cardiomyocytes. The mechanisms leading to reduced Ca2+ transient amplitudes remain to be determined. While increased Ca2+ buffering by the sensitized myofilaments itself may directly affect cytosolic [Ca2+], 47 the observed unchanged diastolic [Ca2+] but reduced frequency‐dependent Ca2+ release in ZSF‐1 obese animals favours impaired Ca2+‐induced Ca2+ release to be explored in more detail in future studies. Albeit it seems improbable that the observed effects are at large attributable to genetic variance between Wistar Kyoto and ZSF‐1 obese rats used as controls and HFpEF animals, respectively, we cannot exclude a potential partial contribution of this factor to the results and acknowledge this as a potential limitation of the present study. In addition, we would like to mention that great efforts were made to perform 3D imaging with echocardiography and cardiac magnetic resonance imaging (MRI), which both yielded unsatisfying results. During echocardiography, the amount of fat tissue in ZSF‐1 obese rats surrounding the heart greatly limited image quality, a phenomenon also described in the literature. 48 For cardiac MRI recordings, similarly, the obesity of ZSF‐1 obese animals made them incompatible with the bore of the machine available to us (MRsolutions MRS3017), a system already fitted to be compatible with overweight small animals. On this account, M‐mode imaging of the RV in the parasternal long‐axis view is the preferred method in studies featuring small animal echocardiography 49 , 50 , 51 and—given its high temporal and spatial resolution—viewed as the accepted approach to assess relative RV dimensions and function.
In conclusion, in a small animal model of metabolic HFpEF, we identified distinct modifications in RV cardiomyocyte function in RVD. While HFpEF has been extensively studied, we believe to be the first to evaluate RVD at the level of single isolated RV cardiomyocytes, reporting profound changes in RV excitation–contraction coupling with altered Ca2+ homeostasis and a compensatory increase in myofilament sensitivity, likely in part attributable to hyperphosphorylation of cMyBP‐C. The underlying mechanisms of this endogenous adaptation of reduced cardiomyocyte Ca2+ release and increased RV myofilament sensitivity should be further investigated to possibly identify a novel therapeutic target, tailored for patients suffering from RVD in conjunction with HFpEF.
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Funding
This project was funded by a research grant from Sanofi‐Aventis Deutschland GmbH under the Cooperation Framework Agreement of Sanofi‐Aventis, Berlin Institute of Health, and Max‐Delbrück Centrum für Molekulare Medizin.
Author contributions
F.R.H., D.M., and N.H. devised the initial main conceptual ideas. N.H. performed echocardiography, cell experiments, and fluorescence staining; related data analysis; and revised the manuscript. U.P. performed invasive haemodynamics, related data analysis, and supervised echocardiography as well as cell experiments. N.H., U.P., and F.R.H. wrote the manuscript. D.B. provided histological specimens and technical assistance with cell isolation/analysis. P.W. conducted western blots and related data analysis. N.B. performed blood glucose analysis. R.K. performed histological staining. L.M. helped to re‐evaluate and discuss western blot data. F.R.H., F.H., J.G., W.M.K., and B.P. aided in providing critical feedback and the final discussion.
Data availability statement
The data underlying this article will be shared on reasonable request to the corresponding author.
Supporting information
Figure S1. Expanded organ gravimetry. A) Liver weight and B) kidney weight to tibia length (TL) ratios; n = 8–9 animals per group; Control = Wistar Kyoto rats; ZSF‐1 ob = ZSF‐1 obese; data are presented as mean ± SEM; statistics were performed by Mann‐test. *P < 0.05, **P < 0.01, ***P < 0.005.
Figure S2. Ca2+ and contractile kinetics. A) Ca2+ time‐to‐peak, time to 50% relaxation, time to 90% relaxation and relaxation constant tau in seconds (s). B) Contractility (Contr.) time‐to‐peak, time to 50% relaxation, time to 90% relaxation and relaxation constant tau in seconds (s); Control = Wistar Kyoto rats; ZSF‐1 ob = ZSF‐1 obese; data are presented as mean ± SEM; statistics were performed by 2‐Way Anova with Fisher's LSD test. *P < 0.05, **P < 0.01, ***P < 0.005.
Acknowledgements
We would like to thank Brian Danzer and Bettina Bublath for their support in the lab and animal facility.
Open Access funding enabled and organized by Projekt DEAL.
Hegemann, N. , Primessnig, U. , Bode, D. , Wakula, P. , Beindorff, N. , Klopfleisch, R. , Michalick, L. , Grune, J. , Hohendanner, F. , Messroghli, D. , Pieske, B. , Kuebler, W. M. , and Heinzel, F. R. (2021) Right‐ventricular dysfunction in HFpEF is linked to altered cardiomyocyte Ca2+ homeostasis and myofilament sensitivity. ESC Heart Failure, 8: 3130–3144. 10.1002/ehf2.13419.
References
- 1. Puwanant S, Priester TC, Mookadam F, Bruce CJ, Redfield MM, Chandrasekaran K. Right ventricular function in patients with preserved and reduced ejection fraction heart failure. Eur J Echocardiogr 2009; 10: 733–737. [DOI] [PubMed] [Google Scholar]
- 2. Damy T, Kallvikbacka‐Bennett A, Goode K, Khaleva O, Lewinter C, Hobkirk J, Nikitin NP, Dubois‐Rande JL, Hittinger L, Clark AL, Cleland JG. Prevalence of, associations with, and prognostic value of tricuspid annular plane systolic excursion (TAPSE) among out‐patients referred for the evaluation of heart failure. J Card Fail 2012; 18: 216–225. [DOI] [PubMed] [Google Scholar]
- 3. Morris DA, Gailani M, Vaz Perez A, Blaschke F, Dietz R, Haverkamp W, Ozcelik C. Right ventricular myocardial systolic and diastolic dysfunction in heart failure with normal left ventricular ejection fraction. J Am Soc Echocardiogr 2011; 24: 886–897. [DOI] [PubMed] [Google Scholar]
- 4. Mohammed SF, Hussain I, AbouEzzeddine OF, Takahama H, Kwon SH, Forfia P, Roger VL, Redfield MM. Right ventricular function in heart failure with preserved ejection fraction: a community‐based study. Circulation 2014; 130: 2310–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ghio S, Guazzi M, Scardovi AB, Klersy C, Clemenza F, Carluccio E, Temporelli PL, Rossi A, Faggiano P, Traversi E, Vriz O, Dini FL, All Investigators . Different correlates but similar prognostic implications for right ventricular dysfunction in heart failure patients with reduced or preserved ejection fraction. Eur J Heart Fail 2017; 19: 873–879. [DOI] [PubMed] [Google Scholar]
- 6. Melenovsky V, Hwang SJ, Lin G, Redfield MM, Borlaug BA. Right heart dysfunction in heart failure with preserved ejection fraction. Eur Heart J 2014; 35: 3452–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gorter TM, Hoendermis ES, van Veldhuisen DJ, Voors AA, Lam CS, Geelhoed B, Willems TP, van Melle JP. Right ventricular dysfunction in heart failure with preserved ejection fraction: a systematic review and meta‐analysis. Eur J Heart Fail 2016; 18: 1472–1487. [DOI] [PubMed] [Google Scholar]
- 8. Platt MJ, Huber JS, Romanova N, Brunt KR, Simpson JA. Pathophysiological mapping of experimental heart failure: left and right ventricular remodeling in transverse aortic constriction is temporally, kinetically and structurally distinct. Front Physiol 2018; 9: 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cenkerova K, Dubrava J, Pokorna V, Kaluzay J, Jurkovicova O. Right ventricular systolic dysfunction and its prognostic value in heart failure with preserved ejection fraction. Acta Cardiol 2015; 70: 387–393. [DOI] [PubMed] [Google Scholar]
- 10. Bhuiyan T, Maurer MS. Heart failure with preserved ejection fraction: persistent diagnosis, therapeutic enigma. Curr Cardiovasc Risk Rep 2011; 5: 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gorter TM, van Veldhuisen DJ, Bauersachs J, Borlaug BA, Celutkiene J, Coats AJS, Crespo‐Leiro MG, Guazzi M, Harjola VP, Heymans S, Hill L, Lainscak M, Lam CSP, Lund LH, Lyon AR, Mebazaa A, Mueller C, Paulus WJ, Pieske B, Piepoli MF, Ruschitzka F, Rutten FH, Seferovic PM, Solomon SD, Shah SJ, Triposkiadis F, Wachter R, Tschope C, de Boer RA. Right heart dysfunction and failure in heart failure with preserved ejection fraction: mechanisms and management. Position statement on behalf of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2018; 20: 16–37. [DOI] [PubMed] [Google Scholar]
- 12. Rosenkranz S, Lang IM, Blindt R, Bonderman D, Bruch L, Diller GP, Felgendreher R, Gerges C, Hohenforst‐Schmidt W, Holt S, Jung C, Kindermann I, Kramer T, Kubler WM, Mitrovic V, Riedel A, Rieth A, Schmeisser A, Wachter R, Weil J, Opitz C. Pulmonary hypertension associated with left heart disease: recommendations of the Cologne Consensus Conference 2016. Dtsch Med Wochenschr 2016; 141: S48–S56. [DOI] [PubMed] [Google Scholar]
- 13. Thenappan T, Shah SJ, Gomberg‐Maitland M, Collander B, Vallakati A, Shroff P, Rich S. Clinical characteristics of pulmonary hypertension in patients with heart failure and preserved ejection fraction. Circ Heart Fail 2011; 4: 257–265. [DOI] [PubMed] [Google Scholar]
- 14. Wong CY, O'Moore‐Sullivan T, Leano R, Hukins C, Jenkins C, Marwick TH. Association of subclinical right ventricular dysfunction with obesity. J Am Coll Cardiol 2006; 47: 611–616. [DOI] [PubMed] [Google Scholar]
- 15. Hohendanner F, Bode D, Primessnig U, Guthof T, Doerr R, Jeuthe S, Reimers S, Zhang K, Bach D, Wakula P, Pieske BM, Heinzel FR. Cellular mechanisms of metabolic syndrome‐related atrial decompensation in a rat model of HFpEF. J Mol Cell Cardiol 2018; 115: 10–19. [DOI] [PubMed] [Google Scholar]
- 16. Hamdani N, Franssen C, Lourenco A, Falcao‐Pires I, Fontoura D, Leite S, Plettig L, Lopez B, Ottenheijm CA, Becher PM, Gonzalez A, Tschope C, Diez J, Linke WA, Leite‐Moreira AF, Paulus WJ. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail 2013; 6: 1239–1249. [DOI] [PubMed] [Google Scholar]
- 17. Primessnig U, Schonleitner P, Holl A, Pfeiffer S, Bracic T, Rau T, Kapl M, Stojakovic T, Glasnov T, Leineweber K, Wakula P, Antoons G, Pieske B, Heinzel FR. Novel pathomechanisms of cardiomyocyte dysfunction in a model of heart failure with preserved ejection fraction. Eur J Heart Fail 2016; 18: 987–997. [DOI] [PubMed] [Google Scholar]
- 18. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open‐source platform for biological‐image analysis. Nat Methods 2012; 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Romano G, Reggi S, Kutryb‐Zajac B, Facoetti A, Chisci E, Pettinato M, Giuffre MR, Vecchio F, Leoni S, De Giorgi M, Avezza F, Cadamuro M, Crippa L, Leone BE, Lavitrano M, Rivolta I, Barisani D, Smolenski RT, Giovannoni R. APOA‐1Milano muteins, orally delivered via genetically modified rice, show anti‐atherogenic and anti‐inflammatory properties in vitro and in Apoe−/− atherosclerotic mice. Int J Cardiol 2018; 271: 233–239. [DOI] [PubMed] [Google Scholar]
- 20. Rabacal W, Schweitzer F, Rayens E, Tarantelli R, Whang P, Jimenez VC, Outwater JA, Norris KA. Statin treatment prevents the development of pulmonary arterial hypertension in a nonhuman primate model of HIV‐associated PAH. Sci Rep 2019; 9: 19832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bode D, Lindner D, Schwarzl M, Westermann D, Deissler P, Primessnig U, Hegemann N, Blatter LA, van Linthout S, Tschope C, Schoenrath F, Soltani S, Stamm C, Duesterhoeft V, Rolim N, Wisloff U, Knosalla C, Falk V, Pieske BM, Heinzel FR, Hohendanner F. The role of fibroblast—cardiomyocyte interaction for atrial dysfunction in HFpEF and hypertensive heart disease. J Mol Cell Cardiol 2019; 131: 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bailey BA, Dipla K, Li S, Houser SR. Cellular basis of contractile derangements of hypertrophied feline ventricular myocytes. J Mol Cell Cardiol 1997; 29: 1823–1835. [DOI] [PubMed] [Google Scholar]
- 23. Soppa GK, Lee J, Stagg MA, Felkin LE, Barton PJ, Siedlecka U, Youssef S, Yacoub MH, Terracciano CM. Role and possible mechanisms of clenbuterol in enhancing reverse remodelling during mechanical unloading in murine heart failure. Cardiovasc Res 2008; 77: 695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cilia L, Saeed A, Ganga HV, Wu WC. Heart failure with preserved ejection fraction: prevention and management. Am J Lifestyle Med 2019; 13: 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang L, Halliday G, Huot JR, Satoh T, Baust JJ, Fisher A, Cook T, Hu J, Avolio T, Goncharov DA, Bai Y, Vanderpool RR, Considine RV, Bonetto A, Tan J, Bachman TN, Sebastiani A, McTiernan CF, Mora AL, Machado RF, Goncharova EA, Gladwin MT, Lai YC. Treatment with treprostinil and metformin normalizes hyperglycemia and improves cardiac function in pulmonary hypertension associated with heart failure with preserved ejection fraction. Arterioscler Thromb Vasc Biol 2020; 40: 1543–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schauer A, Draskowski R, Jannasch A, Kirchhoff V, Goto K, Mannel A, Barthel P, Augstein A, Winzer E, Tugtekin M, Labeit S, Linke A, Adams V. ZSF1 rat as animal model for HFpEF: development of reduced diastolic function and skeletal muscle dysfunction. ESC Heart Fail 2020; 7: 2123–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nguyen ITN, Brandt MM, van de Wouw J, van Drie RWA, Wesseling M, Cramer MJ, de Jager SCA, Merkus D, Duncker DJ, Cheng C, Joles JA, Verhaar MC. Both male and female obese ZSF1 rats develop cardiac dysfunction in obesity‐induced heart failure with preserved ejection fraction. PLoS ONE 2020; 15: e0232399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xia F, Gao F, Yao H, Zhang G, Gao B, Lu Y, Wang X, Qian Y. Identification of angiogenesis‐inhibiting peptides from Chan Su. Protein Expr Purif 2019; 163: 105445. [DOI] [PubMed] [Google Scholar]
- 29. Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation 2017; 136: 6–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hagdorn QAJ, Bossers GPL, Koop AC, Piek A, Eijgenraam TR, van der Feen DE, Sillje HHW, de Boer RA, Berger RMF. A novel method optimizing the normalization of cardiac parameters in small animal models: the importance of dimensional indexing. Am J Physiol Heart Circ Physiol 2019; 316: H1552–H1557. [DOI] [PubMed] [Google Scholar]
- 31. Altara R, Giordano M, Norden ES, Cataliotti A, Kurdi M, Bajestani SN, Booz GW. Targeting obesity and diabetes to treat heart failure with preserved ejection fraction. Front Endocrinol (Lausanne) 2017; 8: 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hruby A, Hu FB. The epidemiology of obesity: a big picture. Pharmacoeconomics 2015; 33: 673–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tadic M, Pieske‐Kraigher E, Cuspidi C, Morris DA, Burkhardt F, Baudisch A, Hassfeld S, Tschope C, Pieske B. Right ventricular strain in heart failure: clinical perspective. Arch Cardiovasc Dis 2017; 110: 562–571. [DOI] [PubMed] [Google Scholar]
- 34. Buckberg G, Hoffman JI. Right ventricular architecture responsible for mechanical performance: unifying role of ventricular septum. J Thorac Cardiovasc Surg 2014; 148: 3166–3171 e3161–3164. [DOI] [PubMed] [Google Scholar]
- 35. Mukherjee M, Chung SE, Ton VK, Tedford RJ, Hummers LK, Wigley FM, Abraham TP, Shah AA. Unique abnormalities in right ventricular longitudinal strain in systemic sclerosis patients. Circ Cardiovasc Imaging 2016; 9: e003792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee JZ, Low SW, Pasha AK, Howe CL, Lee KS, Suryanarayana PG. Comparison of tricuspid annular plane systolic excursion with fractional area change for the evaluation of right ventricular systolic function: a meta‐analysis. Open Heart 2018; 5: e000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao H, Kang Y, Pickle J, Wang J, Han Y. Tricuspid annular plane systolic excursion is dependent on right ventricular volume in addition to function. Echocardiography 2019; 36: 1459–1466. [DOI] [PubMed] [Google Scholar]
- 38. Lang RM, Badano LP, Mor‐Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski L, Spencer KT, Tsang W, Voigt JU. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 2015; 28: 1–39 e14. [DOI] [PubMed] [Google Scholar]
- 39. Rouhana S, Farah C, Roy J, Finan A, Rodrigues de Araujo G, Bideaux P, Scheuermann V, Saliba Y, Reboul C, Cazorla O, Aimond F, Richard S, Thireau J, Fares N. Early calcium handling imbalance in pressure overload‐induced heart failure with nearly normal left ventricular ejection fraction. Biochim Biophys Acta Mol Basis Dis 1865; 2019: 230–242. [DOI] [PubMed] [Google Scholar]
- 40. Roe AT, Frisk M, Louch WE. Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr Pharm Des 2015; 21: 431–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carrier L, Mearini G, Stathopoulou K, Cuello F. Cardiac myosin‐binding protein C (MYBPC3) in cardiac pathophysiology. Gene 2015; 573: 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Salhi HE, Hassel NC, Siddiqui JK, Brundage EA, Ziolo MT, Janssen PM, Davis JP, Biesiadecki BJ. Myofilament calcium sensitivity: mechanistic insight into TnI Ser‐23/24 and Ser‐150 phosphorylation integration. Front Physiol 2016; 7: 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pabel S, Wagner S, Bollenberg H, Bengel P, Kovacs A, Schach C, Tirilomis P, Mustroph J, Renner A, Gummert J, Fischer T, Van Linthout S, Tschope C, Streckfuss‐Bomeke K, Hasenfuss G, Maier LS, Hamdani N, Sossalla S. Empagliflozin directly improves diastolic function in human heart failure. Eur J Heart Fail 2018; 20: 1690–1700. [DOI] [PubMed] [Google Scholar]
- 44. Moss RL, Fitzsimons DP, Ralphe JC. Cardiac MyBP‐C regulates the rate and force of contraction in mammalian myocardium. Circ Res 2015; 116: 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang L, Sadayappan S, Kawai M. Cardiac myosin binding protein C phosphorylation affects cross‐bridge cycle's elementary steps in a site‐specific manner. PLoS ONE 2014; 9: e113417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kolijn D, Pabel S, Tian Y, Lodi M, Herwig M, Carrizzo A, Zhazykbayeva S, Kovacs A, Fulop GA, Falcao‐Pires I, Reusch PH, Linthout SV, Papp Z, van Heerebeek L, Vecchione C, Maier LS, Ciccarelli M, Tschope C, Mugge A, Bagi Z, Sossalla S, Hamdani N. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro‐inflammatory‐oxidative pathways and protein kinase Gα oxidation. Cardiovasc Res 2021; 117: 495–507. [DOI] [PubMed] [Google Scholar]
- 47. Diaz ME, Trafford AW, Eisner DA. The effects of exogenous calcium buffers on the systolic calcium transient in rat ventricular myocytes. Biophys J 2001; 80: 1915–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Uppot RN. Technical challenges of imaging & image‐guided interventions in obese patients. Br J Radiol 2018; 91: 20170931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cheng HW, Fisch S, Cheng S, Bauer M, Ngoy S, Qiu Y, Guan J, Mishra S, Mbah C, Liao R. Assessment of right ventricular structure and function in mouse model of pulmonary artery constriction by transthoracic echocardiography. J Vis Exp 2014: e51041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Egemnazarov B, Schmidt A, Crnkovic S, Sydykov A, Nagy BM, Kovacs G, Weissmann N, Olschewski H, Olschewski A, Kwapiszewska G, Marsh LM. Pressure overload creates right ventricular diastolic dysfunction in a mouse model: assessment by echocardiography. J Am Soc Echocardiogr 2015; 28: 828–843. [DOI] [PubMed] [Google Scholar]
- 51. Sydykov A, Luitel H, Mamazhakypov A, Wygrecka M, Pradhan K, Pak O, Petrovic A, Kojonazarov B, Weissmann N, Seeger W, Grimminger F, Ghofrani HA, Kosanovic D, Schermuly RT. Genetic deficiency and pharmacological stabilization of mast cells ameliorate pressure overload‐induced maladaptive right ventricular remodeling in mice. Int J Mol Sci 2020; 21: 9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Expanded organ gravimetry. A) Liver weight and B) kidney weight to tibia length (TL) ratios; n = 8–9 animals per group; Control = Wistar Kyoto rats; ZSF‐1 ob = ZSF‐1 obese; data are presented as mean ± SEM; statistics were performed by Mann‐test. *P < 0.05, **P < 0.01, ***P < 0.005.
Figure S2. Ca2+ and contractile kinetics. A) Ca2+ time‐to‐peak, time to 50% relaxation, time to 90% relaxation and relaxation constant tau in seconds (s). B) Contractility (Contr.) time‐to‐peak, time to 50% relaxation, time to 90% relaxation and relaxation constant tau in seconds (s); Control = Wistar Kyoto rats; ZSF‐1 ob = ZSF‐1 obese; data are presented as mean ± SEM; statistics were performed by 2‐Way Anova with Fisher's LSD test. *P < 0.05, **P < 0.01, ***P < 0.005.
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.