

Abstract
Wild‐type transthyretin amyloid cardiomyopathy (ATTRwt‐CM) is caused by the deposition of wild‐type transthyretin (TTR) amyloid fibrils in the heart. The age at diagnosis of ATTRwt‐CM is reported to be approximately 70–80 years, and patients commonly present with non‐disease‐specific cardiac abnormalities, such as heart failure with preserved ejection fraction and diastolic dysfunction. The disease can be fatal if left untreated, with an approximate survival of 3–5 years from diagnosis. An oral TTR stabilizer, tafamidis, has enabled early intervention for the treatment of ATTRwt‐CM. However, awareness of ATTRwt‐CM remains low, and misdiagnosis and a delay in diagnosis are common. This review discusses the epidemiology, characteristics, treatment strategy, and red‐flag symptoms and signs of ATTRwt‐CM based on the published literature, as well as recent advances in diagnostic modalities that enable early and accurate diagnosis of the disease. We also discuss an algorithm for early and accurate diagnosis of ATTRwt‐CM in daily clinical practice. In our diagnostic algorithm, a suspected diagnosis of ATTRwt‐CM should be triggered by unexplained left ventricular hypertrophy (LVH), which is LVH that cannot be explained by an increased afterload due to hypertension or valvular disease. In addition, heart failure symptoms, laboratory test results (N‐terminal pro‐B‐type natriuretic peptide, high‐sensitivity troponin T, or high‐sensitivity troponin I), electrocardiogram and imaging (echocardiogram or cardiac magnetic resonance) data, age (≥60 years), and medical history suggestive of ATTRwt‐CM (e.g. carpal tunnel syndrome) should be examined. Detailed examinations using bone scintigraphy and monoclonal protein detection tests followed by tissue biopsy, amyloid typing, and TTR genetic testing are warranted for a definite diagnosis of ATTRwt‐CM.
Keywords: ATTRwt‐CM, HFpEF, Scintigraphy, Biopsy, Carpal tunnel syndrome, Tafamidis
Introduction
Amyloidosis is caused by the deposition of insoluble amyloid fibrils formed by misfolded soluble proteins in organs or tissues. 1 Amyloidosis can be systemic (amyloid deposition at sites distant from the site of misfolded protein production) or localized (amyloid deposition in the organ or tissue of misfolded protein production). 1 The most common types of systemic amyloidosis are amyloid light‐chain (AL) amyloidosis, amyloid A amyloidosis, and transthyretin (TTR) amyloidosis, which result from the deposition of immunoglobulin λ or κ free light chains (FLCs), serum amyloid A protein, and TTR, respectively, as amyloid fibrils. 1
Cardiac involvement of amyloidosis (cardiac amyloidosis) is a common presentation in AL and TTR amyloidosis 2 and may occur in individuals with either a mutated [variant transthyretin amyloid cardiomyopathy (ATTRv‐CM)] or a wild‐type [wild‐type transthyretin amyloid cardiomyopathy (ATTRwt‐CM), formerly known as senile systemic amyloidosis] TTR gene. 3 , 4 Several research groups have discussed the early signs and symptoms (red‐flag symptom clusters) of ATTRwt‐CM and proposed diagnostic algorithms. 3 , 5 , 8 With the aim of improving early diagnosis of suspected ATTRwt‐CM in Japan, a medical advisory board meeting was held in Tokyo, Japan, on 3 February 2019. The diagnostic algorithm presented in this article was developed based on discussions held at this meeting. The epidemiology, clinical and paraclinical features including cardiac symptoms, and the diagnostic and treatment algorithm of ATTRv‐CM have been discussed previously 9 and are thus beyond the scope of this article. Of note, the Japanese Circulation Society (JCS) also published guidelines for the diagnosis and treatment of cardiac amyloidosis in August 2020. 4
Wild‐type transthyretin amyloid cardiomyopathy: a life‐threatening but often overlooked disease
Although data are insufficient to discuss the true prevalence of ATTRwt‐CM in the general population, previous findings suggest that the prevalence of ATTRwt‐CM is considerable in the elderly population worldwide (Table 1 ). 10 , 11 , 12 , 13 ATTRwt‐CM is slowly progressive but can be fatal if left untreated; in general, patients with ATTRwt‐CM have an approximate survival of 3–5 years from diagnosis. 7 , 14 , 15 , 16 Nevertheless, awareness of ATTRwt‐CM remains low, with misdiagnosis and a delay in the diagnosis of the disease being relatively common. For example, although a nationwide survey in Japan identified 51 patients with ATTRwt‐CM between 2012 and 2014, a diagnosis was only made in 11 of 2341 departments that responded to the survey. 17 Moreover, 29% and 26% of participants of an online survey by the Amyloidosis Research Consortium (patients with ATTRwt‐CM or their caregivers, n = 55) reported missed diagnoses and misdiagnoses, respectively, and 65% of patients had to see ≥2 cardiologists before diagnosis. 18 In Europe, among 100 patients with ATTRwt‐CM diagnosed at two centres, 34 (34%) had been previously misdiagnosed, with the most common previous diagnoses being hypertensive cardiomyopathy (12 patients) and hypertrophic cardiomyopathy (eight patients). 19
Table 1.
Prevalence of ATTRwt‐CM
| Publication | Study population | Prevalence of ATTRwt‐CM |
|---|---|---|
| Tanskanen (2008) 10 | General Finnish population (age ≥ 85 years) | 63 of 256 individuals (25%) |
| González‐López (2015) 11 | Spanish patients (age ≥ 60 years) admitted owing to HFpEF with LVH | 16 of 120 individuals (13%) |
| Castaño (2017) 12 | US patients (mean age 84 years) with severe calcific aortic stenosis admitted to undergo transcatheter aortic valve replacement | 24 of 151 individuals (16%) |
| Ueda (2011) 13 | Japanese patients (age > 80 years) who had undergone autopsy at the Kumamoto University Hospital | 3 of 26 individuals (12%) |
ATTRwt‐CM, wild‐type transthyretin amyloid cardiomyopathy; HFpEF, heart failure with preserved ejection fraction; LVH, left ventricular hypertrophy.
Characteristics of wild‐type transthyretin amyloid cardiomyopathy
Evidence suggests that patients with ATTRwt‐CM are primarily men (male:female ratio = 25–50:1) 20 who present with dominant cardiac involvement at an older age (Table 2 ). 15 , 17 , 19 , 21 , 22 , 23 The symptoms of ATTRwt‐CM are often vague and non‐specific, because of which a delay in diagnosis or a misdiagnosis is not uncommon. Patients commonly present with cardiac abnormalities, such as heart failure with preserved ejection fraction (HFpEF) and diastolic dysfunction, which are also characteristic of an elderly population. 16 In addition, carpal tunnel syndrome (CTS; i.e. compression of the median nerve of the carpal tunnel), 24 lumbar canal stenosis, or rotator cuff tears, which are caused by the deposition of amyloid fibrils in the ligaments and tendons, are also common initial symptoms of ATTRwt‐CM. 17 , 25 , 26 , 27
Table 2.
Clinical characteristics of ATTRwt‐CM
| Publication | Study location or setting | Number of patients | Age at diagnosis | Number of male patients | Common cardiac findings | Number of patients with CTS |
|---|---|---|---|---|---|---|
| Connors (2011) 15 | 1 centre in the USA | 82 | Median 73.8 years | 79 (96%) |
Abnormal echocardiogram (99%) Abnormal ECG (93%) |
5/80 (6%) |
| González‐López (2017) 19 | 2 centres in Spain and Italy | 108 | Mean 78.6 years | 88 (81%) |
Atrial fibrillation (56%) Pseudo‐infarct pattern (63%) |
36 (33%) |
| Minamisawa (2016) 21 | 1 centre in Japan | 21 | Mean 74 years | 17 (81%) |
Heart failure (66%) Low QRS voltage (19%) |
5 (24%) |
| Sekijima (2018) 17 | 11 departments across 10 institutes in Japan | 51 | Mean 73.6 years | 41 (82%) |
Cardiac failure (76%) Cardiac conduction defects/arrhythmia (59%) |
23 (45%) |
| Tsutsui (2019) 22 | 1 centre in Japan | 18 | Mean 82.3 years | 12 (67%) |
HFpEF (61%) Low voltage (6%) Atrial fibrillation (50%) |
Not specified |
| Yamada (2020) 23 | 1 centre in Japan | 129 | Mean 78.5 years | 110 (85%) |
LVEF < 50% (41%) Low voltage (36%) Bundle branch block (36%) |
57/106 (54%) |
ATTRwt‐CM, wild‐type transthyretin amyloid cardiomyopathy; CTS, carpal tunnel syndrome; ECG, electrocardiogram; HFpEF, heart failure with preserved ejection fraction; LVEF, left ventricular ejection fraction.
What are the treatment options available to patients with wild‐type transthyretin amyloid cardiomyopathy in Japan?
Conventional pharmacotherapeutic approaches targeting cardiac damage, including diuretics, are of limited therapeutic benefit for the treatment of ATTRwt‐CM. 28 Tafamidis (Vyndaqel® capsules 20 mg, Pfizer Japan Inc.), an oral TTR stabilizer, demonstrated efficacy in patients with transthyretin amyloid cardiomyopathy (ATTRwt‐CM or ATTRv‐CM) in the Transthyretin Amyloidosis Cardiomyopathy Clinical Trial (ATTR‐ACT; ClinicalTrials.gov identifier NCT01994889) 29 and was approved in 2019 in Japan for the treatment of transthyretin amyloid cardiomyopathy. 30 In this randomized, double‐blind, placebo‐controlled, Phase 3 study, patients receiving tafamidis for 30 months (n = 264) had reductions in all‐cause mortality [hazard ratio 0.70, 95% confidence interval (CI) 0.51–0.96] and cardiovascular‐related hospitalizations (relative risk ratio 0.68, 95% CI 0.56–0.81) compared with those receiving placebo (n = 177). The adverse event profiles were similar between the tafamidis and placebo groups. 29 Moreover, additional long‐term (60 month) safety and efficacy of tafamidis were confirmed in an open‐label extension study of ATTR‐ACT in patients with transthyretin amyloid cardiomyopathy (ClinicalTrials.gov identifier NCT02791230). 31
In Japan, patients with transthyretin amyloid cardiomyopathy can receive an initial prescription for tafamidis only if the criterion of both the site and the physician being certified by the JCS is met. 30 Patients eligible for tafamidis treatment should have a history of hospitalization with heart failure (HF) or symptoms of HF requiring treatment, biopsy‐confirmed amyloid deposits (cardiac or non‐cardiac), immunohistochemically confirmed TTR precursor protein, and end‐diastolic interventricular septum thickness > 12 mm. 30 Once diagnosed with ATTRwt‐CM, patients should be referred to a JCS‐certified physician for initiation of treatment with tafamidis. 30 In addition, patients may be granted a Medical Care Recipient Certificate if they have a definite diagnosis per the intractable disease criteria for ATTRwt‐CM. 32 In Japan, treatment costs for patients with a definite diagnosis are eligible for medical expense deductions.
The need for a differential diagnosis between wild‐type transthyretin amyloid cardiomyopathy and cardiac amyloidosis of other causes
Differentiation between ATTRwt‐CM and AL amyloidosis is crucial because cardiac AL amyloidosis has a poor prognosis if left untreated; without treatment, patients with cardiac AL amyloidosis live for <1 year. 33 For example, the median overall survival from diagnosis in patients with stage IV AL amyloidosis is reported to be 5.8 months. 34 Patients with AL amyloidosis commonly present with HF with multiorgan involvement, such as hepatomegaly, peripheral and autonomic neuropathy, nephrotic syndrome, macroglossia, and periorbital purpura. 33 AL amyloidosis is caused by the deposition of misfolded immunoglobulin light chains produced by clonal plasma cells in organs (mainly caused by plasma cell dyscrasia), 33 meaning that a completely different treatment strategy (high‐dose chemotherapy with stem cell transplant 35 , 36 and referral of patients to a haematologist) than that for ATTRwt‐CM is required.
Moreover, differentiation from monoclonal gammopathy of undetermined significance (MGUS), a pre‐malignant plasma cell disorder that may progress to primary amyloidosis, 37 is also essential in patients developing cardiac amyloidosis. The results of a retrospective analysis of 140 patients with ATTRwt‐CM in Boston, USA, demonstrated that 55 (39%) of them had an MGUS, as indicated by an abnormality in the serum FLC ratio and/or serum immunofixation electrophoresis. 38 A moderate increase in circulating FLCs is not necessarily pathological because up to 5% of adults aged ≥50 years and up to 40% of patients with ATTRwt‐CM have MGUS. 38 , 39 Misdiagnosis of AL cardiac amyloidosis is the most common pitfall as up to 10% of misdiagnoses occur even in referral centres. 40 , 41
How can we notice early signs of wild‐type transthyretin amyloid cardiomyopathy?
As ATTRwt‐CM is caused by amyloid deposition in the heart over time, it is essential that patients are diagnosed at an early stage of the disease, and treatment is initiated promptly before the progression of HF. Generally, in Japan, patients presenting with cardiac symptoms undergo an echocardiogram as an initial step, regardless of HF symptoms; therefore, it is advisable to carefully examine echocardiographic findings for warning signs of ATTRwt‐CM, such as unexplained left ventricular hypertrophy (LVH), which is LVH that cannot be explained by an increased afterload due to hypertension or valvular disease.
In consideration of Japanese daily medical practice, cardiac pump dysfunction (HF), abnormal heart rhythms (arrhythmia), investigation of cardiac involvement in patients already diagnosed as having amyloidosis in other organs, and cardiac findings during medical check‐ups could serve to highlight a suspected diagnosis of ATTRwt‐CM.
Red‐flag symptoms and signs of wild‐type transthyretin amyloid cardiomyopathy in Japan
To ensure early diagnosis of ATTRwt‐CM, we recommend a sequential algorithm (Figure 1 ). A suspected diagnosis of ATTRwt‐CM should be triggered by unexplained LVH. Next, the presence of HF symptoms, elevated laboratory test results [N‐terminal pro‐B‐type natriuretic peptide (NT‐proBNP), high‐sensitivity troponin T (hs‐TnT), or high‐sensitivity troponin I (hs‐TnI)], abnormal electrocardiogram (ECG) findings, imaging [echocardiogram or cardiac magnetic resonance (CMR)] findings characteristic of ATTRwt‐CM, advanced age (≥60 years), and medical history suggestive of ATTRwt‐CM (CTS and spinal canal stenosis) should be examined. If a patient with unexplained LVH meets ≥1 criterion listed earlier, a detailed examination using 99mTechnetium‐pyrophosphate (99mTc‐PYP) bone scintigraphy and monoclonal protein detection tests should be offered to exclude AL amyloidosis and grade the cardiac 99mTc‐PYP uptake. To differentiate between ATTRv‐CM and ATTRwt‐CM, tissue biopsy, amyloid typing, and TTR genetic testing should be considered (Figure 1 ).
Figure 1.
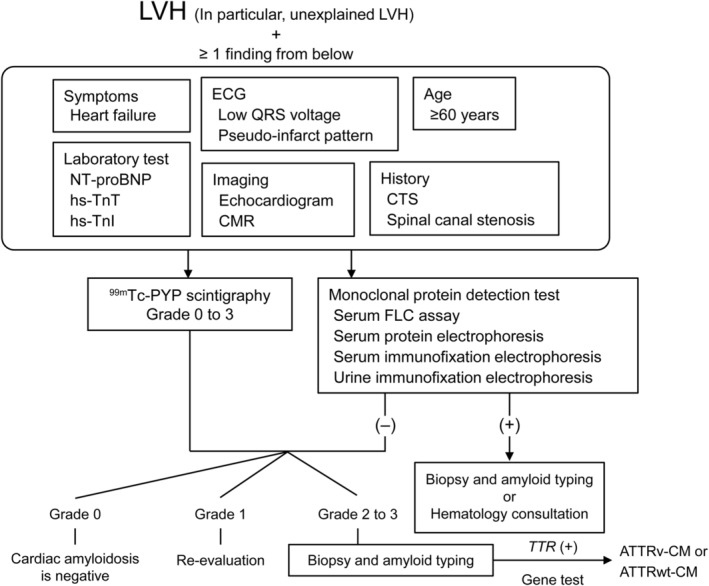
Sequential diagnostic algorithm for ATTRwt‐CM. 99mTc‐PYP, 99mTechnetium‐pyrophosphate; ATTRv‐CM, variant transthyretin amyloid cardiomyopathy; ATTRwt‐CM, wild‐type transthyretin amyloid cardiomyopathy; CMR, cardiac magnetic resonance; CTS, carpal tunnel syndrome; ECG, electrocardiogram; FLC, free light chain; hs‐TnI, high‐sensitivity troponin I; hs‐TnT, high‐sensitivity troponin T; LVH, left ventricular hypertrophy; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; TTR, transthyretin.
Unexplained left ventricular hypertrophy
Because an increase in ventricular wall thickness, resulting from amyloid fibril deposition, is a prominent characteristic of ATTRwt‐CM, 7 , 11 , 42 particular attention should be given to patients presenting with LVH, especially those with unexplained LVH. Previous recommendations have proposed a wall thickness ≥ 14 mm for a suspected diagnosis of ATTRwt‐CM. 7 However, this may not be applicable to Japanese patients because of the smaller body shape of this population compared with Western populations, as illustrated by the lower body mass index range for the definition of obesity 43 and smaller left ventricular (LV) diastolic diameter 44 in the Japanese population. Instead, an LV wall thickness > 12 mm may be more practical, 4 although this warrants further investigation. According to research conducted in Japan, the mean interventricular septal wall thickness (IVST) and posterior wall thickness (PWT) in patients with positive 99mTc‐PYP scintigraphy results were 13.4 and 12.8 mm, respectively, 22 suggesting the validity of setting >12 mm as a cut‐off point for early diagnosis of ATTRwt‐CM without a risk of oversight.
Symptoms of heart failure
As ATTRwt‐CM is an underdiagnosed disease that accounts for a significant number (13%) of HFpEF cases, 11 suspicion of ATTRwt‐CM should be triggered in elderly patients who have been hospitalized for HF with mid‐range ejection fraction. 12 In 108 patients with ATTRwt‐CM in Spain and Italy, HF was reported as the most common (68%) profile that led to a diagnosis despite the heterogeneous clinical manifestation of the disease. 19 Moreover, according to an autopsy study in the USA, the age‐adjusted and sex‐adjusted prevalence of ATTRwt‐CM was significantly higher in patients with HFpEF than in control subjects (odds ratio 3.8, 95% CI 1.5–11.3, P = 0.03). 45
Electrocardiogram
Low QRS voltage is a commonly reported ECG finding in patients with ATTRwt‐CM, 14 , 42 , 46 although its absence does not exclude the possibility of ATTRwt‐CM. 42 However, the usefulness and sensitivity of low QRS voltage are limited in patients with ATTRwt‐CM, and cardiac amyloidosis is commonly hallmarked by a disproportionate QRS voltage to LV wall thickness ratio. 47 Unexplained atrial arrhythmia, conduction system disease, or need for a pacemaker should also prompt further investigation. 47 In addition, abnormal ECG findings, such as atrial fibrillation, atrioventricular block, and pseudo‐infarct pattern, should be monitored as they could be indicative of ATTRwt‐CM (Figure 2 ).
Figure 2.
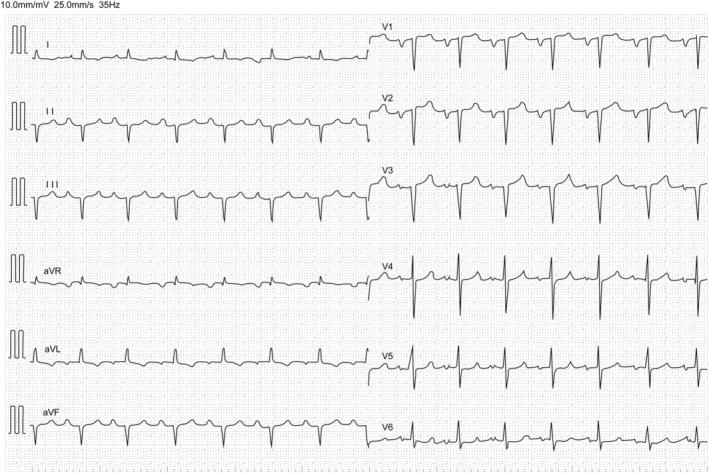
Representative ECG image of a patient with ATTRwt‐CM. ECG showing QS pattern in the right pre‐cordial leads, left atrial loading, and cardiac conduction system disorder, including first‐degree atrioventricular block and left anterior fascicular block. Low voltage in limb leads on ECG may be less common at an early stage of ATTRwt‐CM. ATTRwt‐CM, wild‐type transthyretin amyloid cardiomyopathy; ECG, electrocardiogram.
Echocardiogram
In an echocardiogram (Figure 3 ), in addition to increased wall thickness, 7 , 11 , 42 granular sparkling, the IVST/PWT ratio, and global longitudinal strain (not mandatory) are indicators of ATTRwt‐CM. Acquisition of two‐dimensional (2D), colour, and spectral Doppler imaging (LV wall thickness, myocardial echogenicity, atrial size and function, interatrial septum and valves, pericardial effusion, diastolic function, and estimated pulmonary artery systolic and right atrial pressure); tissue Doppler imaging (tissue Doppler velocities); and strain imaging [longitudinal LV strain and longitudinal LV strain bullseye map (automated function imaging)] is recommended. 48 Echocardiographic signs suggestive of ATTRwt‐CM include infiltrative phenotype (e.g. biventricular hypertrophy, biatrial enlargement, pericardial effusion, valve thickening, and interatrial septal thickening); restrictive LV filling with right ventricular (RV) wall thickening; depressed longitudinal LV function despite normal ejection fraction; low myocardial contraction fraction; aortic stenosis with RV thickening, particularly with paradoxical low flow/low gradient; and reduction in longitudinal strain with apical sparing (not mandatory). 47 Of note, the LV cavity size is generally larger in patients with ATTRwt‐CM than in those with ATTRv‐CM. A combined echocardiographic (IVST/PWT < 1.6) and ECG (QRS voltage < 30 mm) model also assists in the identification of patients with ATTRwt‐CM in daily clinical practice with high sensitivity (93.9%) and specificity (83.3%). 49 However, to detect potential ATTRwt‐CM signals on an echocardiogram, a multiparametric approach (segmental systolic function, RV function, and ventricular diastolic function) may be required. 50
Figure 3.
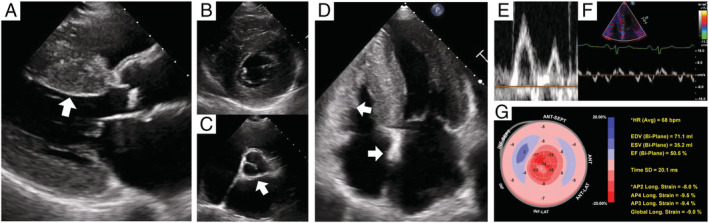
Representative TTE image of a patient with ATTRwt‐CM (A–D). TTE 2D images show asymmetrical LV hypertrophy with granular sparkling appearance, valve thickening, interatrial septum, and right ventricular wall (arrow heads). Left atrium is enlarged despite sinus rhythm, indicating LV diastolic dysfunction. (E, F) Transmitral flow velocity pattern and LV tissue Doppler imaging show severe LV diastolic dysfunction. (G) 2D speckle‐tracking imaging reveals reduced longitudinal strain at basal and mid‐ventricular wall segments, known as the relative apical sparing pattern. (A) Parasternal long‐axis view, (B) mitral valve level, (C) atrioventricular level in parasternal short‐axis view, and (D) apical four‐chamber view. 2D, two‐dimensional; ATTRwt‐CM, wild‐type transthyretin amyloid cardiomyopathy; LV, left ventricular; TTE, transthoracic echocardiography.
A longitudinal strain analysis using speckle‐tracking echocardiography is also useful to differentiate cardiac amyloidosis from other types of LVH (cut‐off value: septal apical to basal segmental longitudinal peak systolic strain ratio > 2.1) 51 or sub‐types of cardiac amyloidosis (cut‐off value: LV apical ratio > 0.96, RV apical ratio > 0.8). 52
Cardiac magnetic resonance
Cardiac magnetic resonance with late gadolinium enhancement (LGE) has emerged as the gold standard to differentiate cardiac amyloidosis from other causes of LVH, such as hypertension and hypertrophic cardiomyopathy. 53 Therefore, it is advisable to perform CMR as part of the diagnosis of ATTRwt‐CM, whenever possible. Acquisition of LV function and morphology (LV function, wall thickness, and mass; stroke volume index; atrial size and function; and pericardial effusion), amyloid imaging (LGE imaging and myocardial signal suppression pattern), and amyloid quantitation [pre‐contrast native T1 mapping and post‐contrast native T1 mapping for extracellular volume (ECV) estimation] are recommended. 48 Patients with ATTRwt‐CM are characterized by diffuse sub‐endocardial and transmural LGE and abnormal nulling time for the myocardium (Figure 4 ), elevated native T1, and ECV on T1 mapping sequences. 47 , 54 , 55 , 56 ATTRwt‐CM and AL amyloidosis can also be differentiated by CMR with LGE, with a sensitivity of 87% and specificity of 96%. 57 Moreover, an increase in native T1 mapping (59 ms increase) and ECV (3% increase) can predict mortality in patients with ATTRwt‐CM. 58
Figure 4.
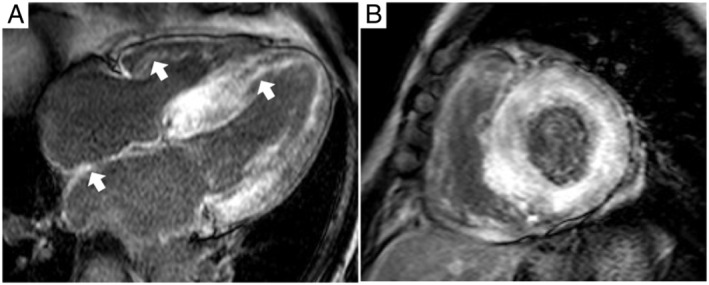
Representative CMR image of a patient with ATTRwt‐CM in (A) short‐axis view and (B) four‐chamber view showing a characteristic pattern of global sub‐endocardial LGE, which also can be located in the atrial wall and right ventricle (arrows). ATTRwt‐CM, wild‐type transthyretin amyloid cardiomyopathy; CMR, cardiac magnetic resonance; LGE, late gadolinium enhancement.
N‐terminal pro‐B‐type natriuretic peptide and high‐sensitivity troponin T
Persistent mild elevation of troponin levels in patients with an echocardiographic hypertrophic phenotype is suggestive of ATTRwt‐CM. 59 Elevation of NT‐proBNP, hs‐TnI, or hs‐TnT should be checked when a diagnosis of ATTRwt‐CM is suspected. High hs‐TnT levels (>0.03 ng/mL) are suggestive of ATTRwt‐CM, with a sensitivity of 74% and specificity of 76%. 59 NT‐proBNP levels that are disproportionate to the degree of HF are suggestive of ATTRwt‐CM. 47 NT‐proBNP is also considered an independent predictor of mortality in ATTRwt‐CM. 60 , 61 In addition, staging systems using NT‐proBNP (>3000 pg/mL), in combination with estimated glomerular filtration rate (<45 mL/min/1.73 m2) 62 or troponin T (>0.05 ng/mL), 63 are also helpful in predicting survival in patients with ATTRwt‐CM.
Medical history
A diagnosis of ATTRwt‐CM should be suspected in elderly patients presenting with bilateral CTS (particularly in men), lumbar canal stenosis, or rotator cuff tears. 27 In particular, bilateral CTS or CTS preceding cardiac symptoms is highly suggestive of ATTRwt‐CM. 26 According to a report from the Netherlands, the overall prevalence rate of CTS in the general population was higher in women than in men (5.8% vs. 0.6%). 64 In contrast, CTS was observed at the time of diagnosis in 23 of 51 (45%) patients with ATTRwt‐CM identified in a Japanese nationwide survey, which included 82% of male patients, 17 and 27 of 107 (25.2%) patients with ATTRwt‐CM visiting a university centre in Italy. 65 The rate of patients with ATTRwt‐CM with co‐morbid CTS reported in previous studies (24–54% in Japan vs. 6–33% in Western countries; Table 2 ) 15 , 17 , 19 , 21 , 23 , 65 suggests that CTS is more frequently observed in Japanese patients with ATTRwt‐CM than in patients in Western countries. Typical findings and symptoms suggestive of CTS are pain and paraesthesia in the hand (in particular, the palmar aspect of the thumb, index and middle fingers, and radial half of the ring finger), which are often exacerbated at night and in the early morning, and the flick sign (shaking of the hand to provide relief), which is 93% sensitive and 96% specific for CTS. 24
Aging
As illustrated by its former name (senile systemic amyloidosis), ATTRwt‐CM is commonly reported in the elderly. The age at diagnosis is reported to be approximately 70–80 years in both Western and Japanese patient populations (Table 2 ). To identify patients with ATTRwt‐CM early in the disease course, a suspected diagnosis should be considered earlier than the perceived age at diagnosis of the disease. Indeed, the youngest age at diagnosis reported in the Japanese nationwide survey of ATTRwt‐CM was 42 years. 17 According to the 2020 JCS guidelines, 99mTc‐PYP scintigraphy for suspected diagnosis is recommended or should be considered in patients (aged ≥70 years) with persistent and mild elevation in hs‐TnT and patients (aged ≥60 years) with low‐flow, low‐gradient aortic stenosis; HF of unidentified cause with LVH; or atrioventricular block, bundle branch block, or atrial fibrillation with cardiac hypertrophy. 4
Diagnostic procedures for wild‐type transthyretin amyloid cardiomyopathy
99mTechnetium‐pyrophosphate scintigraphy
99mTechnetium‐pyrophosphate scintigraphy (Figure 5 ) is helpful for diagnosing ATTRwt‐CM 66 , 67 as well as for assessing the risk of mortality or HF hospitalization. 68 , 69 The sensitivity and specificity of 99mTc‐PYP scintigraphy for ATTRwt‐CM are 58–99% and 79–100%, respectively, and a majority of false positives are AL amyloidosis. 5 , 66 When AL amyloidosis is excluded, the specificity and positive predictive value for ATTRwt‐CM are both 100%. 5 Screening tests using 99mTc‐PYP scintigraphy can also be applied to high‐risk populations as the risk of future cancer is low. 70 Per the JCS 2020 guidelines, 99mTc‐PYP scintigraphy is recommended (recommendation class I: evidence and/or general agreement that a given procedure or treatment is useful and effective) with a four‐grade data assessment of myocardial uptake: Grade 0 (no uptake), 1 (mild uptake less than rib uptake), 2 (moderate uptake equal to rib uptake), or 3 (high uptake greater than rib uptake). Grades 2 and 3 are deemed positive for ATTRwt‐CM. As false positives may occur in planar imaging due to physiological accumulation of 99mTc‐PYP in blood pools, single‐photon emission computed tomography (SPECT) imaging is recommended for Grades 0–2 for a more accurate assessment of uptake in the myocardium and blood pools. 4 Similarly, a two‐step data interpretation (Step 1, visual diagnosis of ATTRwt‐CM using planar and SPECT images; Step 2, semi‐quantitative grading of myocardial uptake) is recommended by an expert group assembled by the American Society of Nuclear Cardiology for evaluating the myocardial uptake of 99mTc‐PYP. 48
Figure 5.
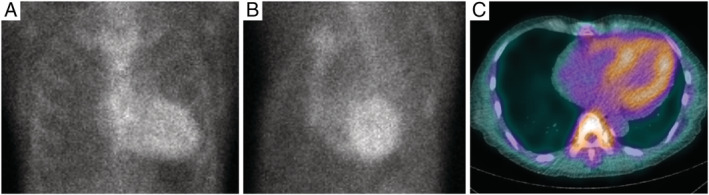
Representative 99mTc‐PYP scintigraphy image of a patient with ATTRwt‐CM. Planar images of (A) anterior and (B) LAO views. (C) SPECT image of axial view. Myocardial uptake of 99mTc‐PYP is greater than bone uptake (Grade 3). 99mTc‐PYP, 99mTechnetium‐pyrophosphate; ATTRwt‐CM, wild‐type transthyretin amyloid cardiomyopathy; LAO, left anterior oblique; SPECT, single‐photon emission computed tomography.
A suspected diagnosis of ATTRwt‐CM using 99mTc‐PYP scintigraphy can be accurately predicted if a patient shows hs‐TnT ≥ 0.0308 ng/mL, LV posterior wall thickness ≥ 13.6 mm, and QRS ≥ 120 ms; the positive scintigraphy rate was 96%, 63%, 21%, and 13% when 3, 2, 1, and none of the criteria were met, respectively. 71 Another integrated approach consisting of hs‐TnT measurement and 99mTc‐PYP scintigraphy also has the potential to increase the diagnostic rate of ATTRwt‐CM. 72 Scintigraphy using 99mTechnetium‐3,3‐diphosphono‐1,2‐propanodicarboxylic acid or 99mTechnetium‐hydroxymethylene diphosphonate is also applicable for the diagnosis of ATTRwt‐CM, 73 , 74 but these techniques are not commonly used in Japan as they are not covered by the Japanese health insurance.
Differentiation from amyloid light‐chain amyloidosis and hypertrophic cardiomyopathy
To detect the presence of monoclonal protein for differentiation between ATTRwt‐CM and AL amyloidosis, three tests should be ordered (serum FLC assay, serum protein electrophoresis with immunofixation, and urine protein electrophoresis with immunofixation); if only one is positive, the patient must be referred to a haematologist. If the results of any of these tests are abnormal, bone scintigraphy should not be used to make the diagnosis of ATTRwt‐CM, and biopsy is recommended. Furthermore, as the presence of monoclonal protein in a patient with suspected cardiac amyloidosis cannot differentiate between AL amyloidosis and ATTRwt‐CM with a concurrent MGUS, a biopsy (organ or fat pad) is also required. 35 , 38 Of note, as up to 40% of patients with ATTRwt‐CM can have an MGUS, scintigraphy alone cannot ensure a specificity of 100%, thus requiring endomyocardial biopsy (EMB) for a definitive diagnosis. 47
In patients with AL amyloidosis, abnormal immunoglobulin light chains produced by clonal plasma cells result in an abnormal ratio of κ and λ FLCs. 75 AL amyloidosis should be suspected when FLC assay results beyond the normal range (3.3–19.4 mg/L for κ FLCs, 5.7–26.3 mg/L for λ FLCs, or a κ/λ ratio of 0.26–1.65) are observed. 76 In addition to these three tests, serum or urine protein electrophoresis can also be applied for the differential diagnosis of AL amyloidosis. 77
Differentiation between ATTRwt‐CM and hypertrophic cardiomyopathy (HCM) is also crucial, as their treatment strategies are completely different. 78 Evidence suggests that ATTRwt‐CM was the most common unrecognized mimic (approximately 9%) among patients initially diagnosed with HCM. 79 Although HCM may most commonly present as asymmetric septal hypertrophy, 80 it is not advisable to rely solely on this phenotype to form a differential diagnosis. For an accurate differential diagnosis, CMR techniques (LGE, T1 mapping, and ECV) 81 and echocardiography (strain analysis) 82 are deemed useful.
Biopsy
To confirm amyloid deposits and the type of amyloidosis, it is advisable to perform a tissue biopsy followed by immunohistochemical staining. For AL amyloidosis and ATTRv‐CM, the rate of amyloid detection is sufficiently high for biopsy at less invasive sites (e.g. lip salivary glands, stomach/duodenum, and skin), while the detection rate for biopsy other than EMB may not be sufficient for ATTRwt‐CM. 4 As the reported sensitivity of abdominal fat pad biopsy is highly variable, 83 , 84 , 85 , 86 , 87 repeated biopsy tests are recommended when a patient's results are strongly suggestive of ATTRwt‐CM in view of clinical features and other laboratory findings. When amyloid deposits are not detected in these tissues, EMB should be considered. 83 , 88 EMB can be safely enforced, with a <1% incidence of serious cardiac complications. 88 In patients with ambiguity in diagnosis, especially for differentiation of ATTRwt‐CM from MGUS or AL amyloidosis, EMB should be performed without hesitation because its sample error rate is low. 83 Of note, although biopsy is useful for a definite diagnosis of ATTRwt‐CM, 99mTc‐PYP scintigraphy is also helpful in diagnosing and differentiating ATTRwt‐CM.
Staining with Congo red should be performed to detect amyloid deposits. 83 , 85 , 89 Stained amyloid deposits show characteristic green birefringence under polarized microscopy. 40 Immunotyping using histochemical staining of amyloid deposits or mass spectroscopy is also helpful to confirm the type of amyloidosis. 40 The Amyloidosis Research Committee of the Ministry of Health, Labour and Welfare of Japan 90 provides an immunohistochemical staining service using an antibody panel consisting of anti‐AL κ, anti‐AL λ, anti‐ATTR, anti‐amyloid A, and anti‐β‐2 microglobulin antibodies.
Genotyping
As the final diagnostic step, TTR genotyping should be performed for patients to differentiate between ATTRwt‐CM and ATTRv‐CM. 40 TTR genotyping is covered by the Japanese health insurance and can be ordered from facilities across the country. Physicians can seek advice from the Amyloidosis Research Committee of Japan regarding the conduct and data interpretation of histological examinations and TTR genotyping. 90 When diagnosed with ATTRv‐CM, genetic counselling should be offered to the patient and relatives. 9
Conclusions
Wild‐type transthyretin amyloid cardiomyopathy is a progressive and fatal disease. As tafamidis is now available for the treatment of ATTRwt‐CM, early diagnosis of the disease and treatment initiation are essential for improved patient outcomes. However, a low awareness of ATTRwt‐CM, together with difficulties in diagnosis, suggests that the condition is undiagnosed in many patients. We hope that the diagnostic algorithm presented in this article will raise awareness of ATTRwt‐CM, resulting in an earlier and more accurate diagnosis of the disease.
Conflict of interest
Takayuki Inomata has received consulting fees or honoraria from Daiichi‐Sankyo Co., Japan Medtronics Co., Mitsubishi Tanabe Pharma Co., Otsuka Pharmaceutical Co., Pfizer Inc., Bristol‐Myers Squibb, and Boehringer Ingelheim GmbH. M.U. has received consulting fees or honoraria, support for travel to meetings, and administrative support for writing assistance, medicines, or equipment from Pfizer Inc. for the submitted work and reports financial relationships outside of the submitted work with Pfizer Inc. and Alnylam Pharmaceuticals Inc. Tomonori Ishii and Y.K. are full‐time employees of Pfizer Pharmaceuticals K.K. J.K. has received consulting fees or honoraria and support for travel to meetings from Pfizer Inc. N.T., K.N., and J.E. report no conflicts of interest.
Funding
The conduct of the medical advisory board meeting was supported by Pfizer Pharmaceuticals K.K.
Acknowledgements
We thank Dr Mitsuaki Isobe (Sakakibara Heart Institute, Tokyo, Japan), Dr Claudio Rapezzi (University of Ferrara, Ferrara, Italy), Dr Olivier Lairez (Centre Hospitalier Universitaire de Toulouse, Toulouse, France), Hahn‐Ey Lee (Pfizer Pharmaceuticals K.K.), and Eleonora Russo (Pfizer Pharmaceuticals K.K.) for their invaluable advice regarding the content of this manuscript. Medical writing and editorial assistance was provided by Mami Hirano, MS, of Cactus Life Sciences (part of Cactus Communications) and funded by Pfizer Pharmaceuticals K.K.
Inomata, T. , Tahara, N. , Nakamura, K. , Endo, J. , Ueda, M. , Ishii, T. , Kitano, Y. , and Koyama, J. (2021) Diagnosis of wild‐type transthyretin amyloid cardiomyopathy in Japan: red‐flag symptom clusters and diagnostic algorithm. ESC Heart Failure, 8: 2647–2659. 10.1002/ehf2.13473.
References
- 1. Nienhuis HL, Bijzet J, Hazenberg BP. The prevalence and management of systemic amyloidosis in Western countries. Kidney Dis (Basel) 2016; 2: 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med 1997; 337: 898–909. [DOI] [PubMed] [Google Scholar]
- 3. Ton VK, Mukherjee M, Judge DP. Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin Med Insights Cardiol 2015; 8: 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kitaoka H, Izumi C, Izumiya Y, Inomata T, Ueda M, Kubo T, Koyama J, Sano M, Sekijima Y, Tahara N, Tsukada N, Tsujita K, Tsutsui H, Tomita T, Amano M, Endo J, Okada A, Oda S, Takashio S, Baba Y, Misumi Y, Yazaki M, Anzai T, Ando Y, Isobe M, Kimura T, Fukuda K, Japanese Circulation Society Joint Working Group . JCS 2020 guideline on diagnosis and treatment of cardiac amyloidosis. Circ J 2020; 84: 1610–1671. [DOI] [PubMed] [Google Scholar]
- 5. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, Wechalekar AD, Berk JL, Quarta CC, Grogan M, Lachmann HJ, Bokhari S, Castano A, Dorbala S, Johnson GB, Glaudemans AW, Rezk T, Fontana M, Palladini G, Milani P, Guidalotti PL, Flatman K, Lane T, Vonberg FW, Whelan CJ, Moon JC, Ruberg FL, Miller EJ, Hutt DF, Hazenberg BP, Rapezzi C, Hawkins PNS. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016; 133: 2404–2412. [DOI] [PubMed] [Google Scholar]
- 6. Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, Berk JL, Plante‐Bordeneuve V, Schmidt HHJ, Merlini G. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol 2015; 66: 2451–2466. [DOI] [PubMed] [Google Scholar]
- 7. Witteles RM, Bokhari S, Damy T, Elliott PM, Falk RH, Fine NM, Gospodinova M, Obici L, Rapezzi C, Garcia‐Pavia P. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail 2019; 7: 709–716. [DOI] [PubMed] [Google Scholar]
- 8. Yamamoto H, Yokochi T. Transthyretin cardiac amyloidosis: an update on diagnosis and treatment. ESC Heart Fail 2019; 6: 1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sekijima Y, Ueda M, Koike H, Misawa S, Ishii T, Ando Y. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red‐flag symptom clusters and treatment algorithm. Orphanet J Rare Dis 2018; 13: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, Singleton A, Kiuru‐Enari S, Paetau A, Tienari PJ, Myllykangas L. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2‐macroglobulin and tau: a population‐based autopsy study. Ann Med 2008; 40: 232–239. [DOI] [PubMed] [Google Scholar]
- 11. González‐López E, Gallego‐Delgado M, Guzzo‐Merello G, de Haro‐Del Moral FJ, Cobo‐Marcos M, Robles C, Bornstein B, Salas C, Lara‐Pezzi E, Alonso‐Pulpon L, Garcia‐Pavia P. Wild‐type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36: 2585–2594. [DOI] [PubMed] [Google Scholar]
- 12. Castaño A, Narotsky DL, Hamid N, Khalique OK, Morgenstern R, DeLuca A, Rubin J, Chiuzan C, Nazif T, Vahl T, George I, Kodali S, Leon MB, Hahn R, Bokhari S, Maurer MS. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J 2017; 38: 2879–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ueda M, Horibata Y, Shono M, Misumi Y, Oshima T, Su Y, Tasaki M, Shinriki S, Kawahara S, Jono H, Obayashi K, Ogawa H, Ando Y. Clinicopathological features of senile systemic amyloidosis: an ante‐ and post‐mortem study. Mod Pathol 2011; 24: 1533–1544. [DOI] [PubMed] [Google Scholar]
- 14. Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, Salvi F, Ciliberti P, Pastorelli F, Biagini E, Coccolo F, Cooke RM, Bacchi‐Reggiani L, Sangiorgi D, Ferlini A, Cavo M, Zamagni E, Fonte ML, Palladini G, Salinaro F, Musca F, Obici L, Branzi A, Perlini S. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation 2009; 120: 1203–1212. [DOI] [PubMed] [Google Scholar]
- 15. Connors LH, Doros G, Sam F, Badiee A, Seldin DC, Skinner M. Clinical features and survival in senile systemic amyloidosis: comparison to familial transthyretin cardiomyopathy. Amyloid 2011; 18 (Suppl 1): 157–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Connors LH, Sam F, Skinner M, Salinaro F, Sun F, Ruberg FL, Berk JL, Seldin DC. Heart failure resulting from age‐related cardiac amyloid disease associated with wild‐type transthyretin: a prospective, observational cohort study. Circulation 2016; 133: 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sekijima Y, Yazaki M, Ueda M, Koike H, Yamada M, Ando Y. First nationwide survey on systemic wild‐type ATTR amyloidosis in Japan. Amyloid 2018; 25: 8–10. [DOI] [PubMed] [Google Scholar]
- 18. Lousada I, Maurer M, Warner M, Guthrie S, Hsu K, Grogan M. Amyloidosis research consortium cardiac amyloidosis survey: results from patients with AL and ATTR amyloidosis and their caregivers. J Am Coll Cardiol 2018; 71: A890. [Google Scholar]
- 19. González‐López E, Gagliardi C, Dominguez F, Quarta CC, de Haro‐Del Moral FJ, Milandri A, Salas C, Cinelli M, Cobo‐Marcos M, Lorenzini M, Lara‐Pezzi E, Foffi S, Alonso‐Pulpon L, Rapezzi C, Garcia‐Pavia P. Clinical characteristics of wild‐type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J 2017; 38: 1895–1904. [DOI] [PubMed] [Google Scholar]
- 20. Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation 2012; 126: 1286–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Minamisawa M, Koyama J, Sekijima Y, Ikeda S, Kozuka A, Ebisawa S, Miura T, Motoki H, Okada A, Izawa A, Ikeda U. Comparison of the standard and speckle tracking echocardiographic features of wild‐type and mutated transthyretin cardiac amyloidoses. Eur Heart J Cardiovasc Imaging 2016; 17: 402–410. [DOI] [PubMed] [Google Scholar]
- 22. Tsutsui Y, Kubota T, Kato S, Nozoe M, Suematsu N, Okabe M, Yamamoto Y, Tsutsui H. Utility of 99mTc‐pyrophosphate scintigraphy in diagnosing transthyretin cardiac amyloidosis in real‐world practice. Circ Rep 2019; 1: 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamada T, Takashio S, Arima Y, Nishi M, Morioka M, Hirakawa K, Hanatani S, Fujisue K, Yamanaga K, Kanazawa H, Sueta D, Araki S, Usuku H, Nakamura T, Suzuki S, Yamamoto E, Ueda M, Kaikita K, Tsujita K. Clinical characteristics and natural history of wild‐type transthyretin amyloid cardiomyopathy in Japan. ESC Heart Fail 2020; 7: 2829–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wipperman J, Goerl K. Carpal tunnel syndrome: diagnosis and management. Am Fam Physician 2016; 94: 993–999. [PubMed] [Google Scholar]
- 25. Sekijima Y, Uchiyama S, Tojo K, Sano K, Shimizu Y, Imaeda T, Hoshii Y, Kato H, Ikeda S. High prevalence of wild‐type transthyretin deposition in patients with idiopathic carpal tunnel syndrome: a common cause of carpal tunnel syndrome in the elderly. Hum Pathol 2011; 42: 1785–1791. [DOI] [PubMed] [Google Scholar]
- 26. Nakagawa M, Sekijima Y, Yazaki M, Tojo K, Yoshinaga T, Doden T, Koyama J, Yanagisawa S, Ikeda S. Carpal tunnel syndrome: a common initial symptom of systemic wild‐type ATTR (ATTRwt) amyloidosis. Amyloid 2016; 23: 58–63. [DOI] [PubMed] [Google Scholar]
- 27. Sueyoshi T, Ueda M, Jono H, Irie H, Sei A, Ide J, Ando Y, Mizuta H. Wild‐type transthyretin‐derived amyloidosis in various ligaments and tendons. Hum Pathol 2011; 42: 1259–1264. [DOI] [PubMed] [Google Scholar]
- 28. Emdin M, Aimo A, Rapezzi C, Fontana M, Perfetto F, Seferović PM, Barison A, Castiglione V, Vergaro G, Giannoni A, Passino C, Merlini G. Treatment of cardiac transthyretin amyloidosis: an update. Eur Heart J 2019; 40: 3699–3706. [DOI] [PubMed] [Google Scholar]
- 29. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington‐Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB, Rapezzi C. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379: 1007–1016. [DOI] [PubMed] [Google Scholar]
- 30. Endo J, Sano M, Izumiya Y, Tsujita K, Nakamura K, Tahara N, Kuwahara K, Inomata T, Ueda M, Sekijima Y, Ando Y, Tsutsui H, Isobe M, Fukuda K. A statement on the appropriate administration of tafamidis in patients with transthyretin cardiac amyloidosis. Circ J 2020; 84: 15–17. [DOI] [PubMed] [Google Scholar]
- 31. Damy T, Garcia‐Pavia P, Hanna M, Judge DP, Merlini G, Gundapaneni B, Patterson TA, Riley S, Schwartz JH, Sultan MB, Witteles R. Efficacy and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR‐ACT) and long‐term extension study. Eur J Heart Fail; 23: 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Japan Intractable Diseases Information Center . Systemic amyloidosis. http://www.nanbyou.or.jp/entry/207. Accessed 8 November 2020 (in Japanese).
- 33. Grogan M, Dispenzieri A, Gertz MA. Light‐chain cardiac amyloidosis: strategies to promote early diagnosis and cardiac response. Heart 2017; 103: 1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, Laumann K, Zeldenrust SR, Leung N, Dingli D, Greipp PR, Lust JA, Russell SJ, Kyle RA, Rajkumar SV, Gertz MA. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012; 30: 989–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gertz MA. Immunoglobulin light chain amyloidosis: 2018 update on diagnosis, prognosis, and treatment. Am J Hematol 2018; 93: 1169–1180. [DOI] [PubMed] [Google Scholar]
- 36. Shimazaki C, Hata H, Iida S, Ueda M, Katoh N, Sekijima Y, Ikeda S, Yazaki M, Fukushima W, Ando Y. Nationwide survey of 741 patients with systemic amyloid light‐chain amyloidosis in Japan. Intern Med 2018; 57: 181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, Melton LJ 3rd. A long‐term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med 2002; 346: 564–569. [DOI] [PubMed] [Google Scholar]
- 38. Phull P, Sanchorawala V, Connors LH, Doros G, Ruberg FL, Berk JL, Sarosiek S. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid 2018; 25: 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dispenzieri A, Katzmann JA, Kyle RA, Larson DR, Melton LJ 3rd, Colby CL, Therneau TM, Clark R, Kumar SK, Bradwell A, Fonseca R, Jelinek DF, Rajkumar SV. Prevalence and risk of progression of light‐chain monoclonal gammopathy of undetermined significance: a retrospective population‐based cohort study. Lancet 2010; 375: 1721–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation 2017; 135: 1357–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, Pepys MB, Hawkins PN. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med 2002; 346: 1786–1791. [DOI] [PubMed] [Google Scholar]
- 42. Damy T, Maurer MS, Rapezzi C, Planté‐Bordeneuve V, Karayal ON, Mundayat R, Suhr OB, Kristen AV. Clinical, ECG and echocardiographic clues to the diagnosis of TTR‐related cardiomyopathy. Open Heart 2016; 3: e000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goda A, Masuyama T. Obesity and overweight in Asian people. Circ J 2016; 80: 2425–2426. [DOI] [PubMed] [Google Scholar]
- 44. Daimon M, Watanabe H, Abe Y, Hirata K, Hozumi T, Ishii K, Ito H, Iwakura K, Izumi C, Matsuzaki M, Minagoe S, Abe H, Murata K, Nakatani S, Negishi K, Yoshida K, Tanabe K, Tanaka N, Tokai K, Yoshikawa J, JAMP Study Investigators . Normal values of echocardiographic parameters in relation to age in a healthy Japanese population: the JAMP study. Circ J 2008; 72: 1859–1866. [DOI] [PubMed] [Google Scholar]
- 45. Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, Roger VL, Gertz MA, Dispenzieri A, Zeldenrust SR, Redfield MM. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail 2014; 2: 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carroll JD, Gaasch WH, McAdam KP. Amyloid cardiomyopathy: characterization by a distinctive voltage/mass relation. Am J Cardiol 1982; 49: 9–13. [DOI] [PubMed] [Google Scholar]
- 47. Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, Fontana M, Grogan M, Kristen AV, Lousada I, Nativi‐Nicolau J, Cristina Quarta C, Rapezzi C, Ruberg FL, Witteles R, Merlini G. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail 2019; 12: e006075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, Fontana M, Gheysens O, Gillmore JD, Glaudemans AWJM, Hanna MA, Hazenberg BPC, Kristen AV, Kwong RY, Maurer MS, Merlini G, Miller EJ, Moon JC, Murthy VL, Quarta CC, Rapezzi C, Ruberg FL, Shah SJ, Slart RHJA, Verberne HJ, Bourque JM. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2‐evidence base and standardized methods of imaging. J Nucl Cardiol 2019; 26: 2065–2123. [DOI] [PubMed] [Google Scholar]
- 49. Gustavsson S, Granåsen G, Grönlund C, Wiklund U, Mörner S, Henein M, Suhr OB, Lindqvist P. Can echocardiography and ECG discriminate hereditary transthyretin V30M amyloidosis from hypertrophic cardiomyopathy? Amyloid 2015; 22: 163–170. [DOI] [PubMed] [Google Scholar]
- 50. Di Bella G, Pizzino F, Minutoli F, Zito C, Donato R, Dattilo G, Oreto G, Baldari S, Vita G, Khandheria BK, Carerj S. The mosaic of the cardiac amyloidosis diagnosis: role of imaging in subtypes and stages of the disease. Eur Heart J Cardiovasc Imaging 2014; 15: 1307–1315. [DOI] [PubMed] [Google Scholar]
- 51. Liu D, Hu K, Niemann M, Herrmann S, Cikes M, Störk S, Gaudron PD, Knop S, Ertl G, Bijnens B, Weidemann F. Effect of combined systolic and diastolic functional parameter assessment for differentiation of cardiac amyloidosis from other causes of concentric left ventricular hypertrophy. Circ Cardiovasc Imaging 2013; 6: 1066–1072. [DOI] [PubMed] [Google Scholar]
- 52. Moñivas Palomero V, Durante‐Lopez A, Sanabria MT, Cubero JS, González‐Mirelis J, Lopez‐Ibor JV, Navarro Rico SM, Krsnik I, Dominguez F, Mingo AM, Hernandez‐Perez FJ, Cavero G, Santos SM. Role of right ventricular strain measured by two‐dimensional echocardiography in the diagnosis of cardiac amyloidosis. J Am Soc Echocardiogr 2019; 32: 845–853.e1. [DOI] [PubMed] [Google Scholar]
- 53. Banypersad SM. The evolving role of cardiovascular magnetic resonance imaging in the evaluation of systemic amyloidosis. Magn Reson Insights 2019; 12: 1178623X19843519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Barison A, Aquaro GD, Pugliese NR, Cappelli F, Chiappino S, Vergaro G, Mirizzi G, Todiere G, Passino C, Masci PG, Perfetto F, Emdin M. Measurement of myocardial amyloid deposition in systemic amyloidosis: insights from cardiovascular magnetic resonance imaging. J Intern Med 2015; 277: 605–614. [DOI] [PubMed] [Google Scholar]
- 55. Martinez‐Naharro A, Treibel TA, Abdel‐Gadir A, Bulluck H, Zumbo G, Knight DS, Kotecha T, Francis R, Hutt DF, Rezk T, Rosmini S, Quarta CC, Whelan CJ, Kellman P, Gillmore JD, Moon JC, Hawkins PN, Fontana M. Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol 2017; 70: 466–477. [DOI] [PubMed] [Google Scholar]
- 56. Fontana M, Banypersad SM, Treibel TA, Maestrini V, Sado DM, White SK, Pica S, Castelletti S, Piechnik SK, Robson MD, Gilbertson JA, Rowczenio D, Hutt DF, Lachmann HJ, Wechalekar AD, Whelan CJ, Gillmore JD, Hawkins PN, Moon JC. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging 2014; 7: 157–165. [DOI] [PubMed] [Google Scholar]
- 57. Dungu JN, Valencia O, Pinney JH, Gibbs SD, Rowczenio D, Gilbertson JA, Lachmann HJ, Wechalekar A, Gillmore JD, Whelan CJ, Hawkins PN, Anderson LJ. CMR‐based differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc Imaging 2014; 7: 133–142. [DOI] [PubMed] [Google Scholar]
- 58. Martinez‐Naharro A, Kotecha T, Norrington K, Boldrini M, Rezk T, Quarta C, Treibel TA, Whelan CJ, Knight DS, Kellman P, Ruberg FL, Gillmore JD, Moon JC, Hawkins PN, Fontana M. Native T1 and extracellular volume in transthyretin amyloidosis. JACC Cardiovasc Imaging 2019; 12: 810–819. [DOI] [PubMed] [Google Scholar]
- 59. Takashio S, Yamamuro M, Izumiya Y, Hirakawa K, Marume K, Yamamoto M, Ueda M, Yamashita T, Ishibashi‐Ueda H, Yasuda S, Ogawa H, Ando Y, Anzai T, Tsujita K. Diagnostic utility of cardiac troponin T level in patients with cardiac amyloidosis. ESC Heart Fail 2018; 5: 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Siepen FAD, Bauer R, Voss A, Hein S, Aurich M, Riffel J, Mereles D, Röcken C, Buss SJ, Katus HA, Kristen AV. Predictors of survival stratification in patients with wild‐type cardiac amyloidosis. Clin Res Cardiol 2018; 107: 158–169. [DOI] [PubMed] [Google Scholar]
- 61. Damy T, Jaccard A, Guellich A, Lavergne D, Galat A, Deux JF, Hittinger L, Dupuis J, Frenkel V, Rigaud C, Plante‐Bordeneuve V, Bodez D, Mohty D. Identification of prognostic markers in transthyretin and AL cardiac amyloidosis. Amyloid 2016; 23: 194–202. [DOI] [PubMed] [Google Scholar]
- 62. Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinez‐Naharro A, Quarta CC, Rezk T, Whelan CJ, Gonzalez‐Lopez E, Lane T, Gilbertson JA, Rowczenio D, Petrie A, Hawkins PN. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2018; 39: 2799–2806. [DOI] [PubMed] [Google Scholar]
- 63. Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, Klarich KW, Miller WL, Maleszewski JJ, Dispenzieri A. Natural history of wild‐type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol 2016; 68: 1014–1020. [DOI] [PubMed] [Google Scholar]
- 64. de Krom MC, Knipschild PG, Kester AD, Thijs CT, Boekkooi PF, Spaans F. Carpal tunnel syndrome: prevalence in the general population. J Clin Epidemiol 1992; 45: 373–376. [DOI] [PubMed] [Google Scholar]
- 65. Milandri A, Farioli A, Gagliardi C, Longhi S, Salvi F, Curti S, Foffi S, Caponetti AG, Lorenzini M, Ferlini A, Rimessi P, Mattioli S, Violante FS, Rapezzi C. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail 2020; 22: 507–515. [DOI] [PubMed] [Google Scholar]
- 66. Castano A, Haq M, Narotsky DL, Goldsmith J, Weinberg RL, Morgenstern R, Pozniakoff T, Ruberg FL, Miller EJ, Berk JL, Dispenzieri A, Grogan M, Johnson G, Bokhari S, Maurer MS. Multicenter study of planar technetium 99m pyrophosphate cardiac imaging: predicting survival for patients with ATTR cardiac amyloidosis. JAMA Cardiol 2016; 1: 880–889. [DOI] [PubMed] [Google Scholar]
- 67. Harb SC, Haq M, Flood K, Guerrieri A, Passerell W, Jaber WA, Miller EJ. National patterns in imaging utilization for diagnosis of cardiac amyloidosis: a focus on Tc99m‐pyrophosphate scintigraphy. J Nucl Cardiol 2017; 24: 1094–1097. [DOI] [PubMed] [Google Scholar]
- 68. Mohamed‐Salem L, Santos‐Mateo JJ, Sanchez‐Serna J, Hernández‐Vicente Á, Reyes‐Marle R, Castellón Sánchez MI, Claver‐Valderas MA, Gonzalez‐Vioque E, Haro‐Del Moral FJ, García‐Pavía P, Pascual‐Figal DA. Prevalence of wild type ATTR assessed as myocardial uptake in bone scan in the elderly population. Int J Cardiol 2018; 270: 192–196. [DOI] [PubMed] [Google Scholar]
- 69. Vranian MN, Sperry BW, Hanna M, Hachamovitch R, Ikram A, Brunken RC, Jaber WA. Technetium pyrophosphate uptake in transthyretin cardiac amyloidosis: associations with echocardiographic disease severity and outcomes. J Nucl Cardiol 2018; 25: 1247–1256. [DOI] [PubMed] [Google Scholar]
- 70. Einstein AJ, Shuryak I, Castaño A, Mintz A, Maurer MS, Bokhari S. Estimating cancer risk from 99mTc pyrophosphate imaging for transthyretin cardiac amyloidosis. J Nucl Cardiol 2020; 27: 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Marume K, Takashio S, Nishi M, Hirakawa K, Yamamoto M, Hanatani S, Oda S, Utsunomiya D, Shiraishi S, Ueda M, Yamashita T, Sakamoto K, Yamamoto E, Kaikita K, Izumiya Y, Yamashita Y, Ando Y, Tsujita K. Combination of commonly examined parameters is a useful predictor of positive 99mTc‐labeled pyrophosphate scintigraphy findings in elderly patients with suspected transthyretin cardiac amyloidosis. Circ J 2019; 83: 1698–1708. [DOI] [PubMed] [Google Scholar]
- 72. Ochi Y, Kubo T, Nakashima Y, Baba Y, Hirota T, Yamasaki N, Yamashita T, Ueda M, Ando Y, Kitaoka H. Integrated diagnostic approach to wild‐type transthyretin cardiac amyloidosis with the use of high‐sensitivity cardiac troponin T measurement and 99mTc‐pyrophosphate scintigraphy. J Cardiol 2020; 75: 12–19. [DOI] [PubMed] [Google Scholar]
- 73. Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, Leone O, Farsad M, Ciliberti P, Bacchi‐Reggiani L, Fallani F, Branzi A, Rapezzi C. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc‐3,3‐diphosphono‐1,2‐propanodicarboxylic acid scintigraphy. J Am Coll Cardiol 2005; 46: 1076–1084. [DOI] [PubMed] [Google Scholar]
- 74. Galat A, Rosso J, Guellich A, Van Der Gucht A, Rappeneau S, Bodez D, Guendouz S, Tissot C‐M, Hittinger L, Dubois‐Randé J‐L, Plante‐Bordeneuve V, Itti E, Meignan M, Damy T. Usefulness of 99mTc‐HMDP scintigraphy for the etiologic diagnosis and prognosis of cardiac amyloidosis. Amyloid 2015; 22: 210–220. [DOI] [PubMed] [Google Scholar]
- 75. Sanchorawala V. Light‐chain (AL) amyloidosis: diagnosis and treatment. Clin J Am Soc Nephrol 2006; 1: 1331–1341. [DOI] [PubMed] [Google Scholar]
- 76. Dispenzieri A, Kyle R, Merlini G, Miguel JS, Ludwig H, Hajek R, Palumbo A, Jagannath S, Blade J, Lonial S, Dimopoulos M, Comenzo R, Einsele H, Barlogie B, Anderson K, Gertz M, Harousseau JL, Attal M, Tosi P, Sonneveld P, Boccadoro M, Morgan G, Richardson P, Sezer O, Mateos MV, Cavo M, Joshua D, Turesson I, Chen W, Shimizu K, Powles R, Rajkumar SV, Durie BGM, International Myeloma Working Group . International Myeloma Working Group guidelines for serum‐free light chain analysis in multiple myeloma and related disorders. Leukemia 2009; 23: 215–224. [DOI] [PubMed] [Google Scholar]
- 77. Jenner E. Serum free light chains in clinical laboratory diagnostics. Clin Chim Acta 2014; 427: 15–20. [DOI] [PubMed] [Google Scholar]
- 78. Kitaoka H, Kubo T, Doi YL. Hypertrophic cardiomyopathy—a heterogeneous and lifelong disease in the real world. Circ J 2020; 84: 1218–1226. [DOI] [PubMed] [Google Scholar]
- 79. Maurizi N, Rella V, Fumagalli C, Salerno S, Castelletti S, Dagradi F, Torchio M, Marceca A, Meda M, Gasparini M, Boschi B, Girolami F, Parati G, Olivotto I, Crotti L, Cecchi F. Prevalence of cardiac amyloidosis among adult patients referred to tertiary centres with an initial diagnosis of hypertrophic cardiomyopathy. Int J Cardiol 2020; 300: 191–195. [DOI] [PubMed] [Google Scholar]
- 80. Parato VM, Antoncecchi V, Sozzi F, Marazia S, Zito A, Maiello M, Palmiero P, Italian Chapter of ISCU . Echocardiographic diagnosis of the different phenotypes of hypertrophic cardiomyopathy. Cardiovasc Ultrasound 2016; 14: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Burrage MK, Ferreira VM. Cardiovascular magnetic resonance for the differentiation of left ventricular hypertrophy. Curr Heart Fail Rep 2020; 17: 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pagourelias ED, Mirea O, Duchenne J, Van Cleemput J, Delforge M, Bogaert J, Kuznetsova T, Voigt JU. Echo parameters for differential diagnosis in cardiac amyloidosis: a head‐to‐head comparison of deformation and nondeformation parameters. Circ Cardiovasc Imaging 2017; 10: e005588. [DOI] [PubMed] [Google Scholar]
- 83. Fine NM, Arruda‐Olson AM, Dispenzieri A, Zeldenrust SR, Gertz MA, Kyle RA, Swiecicki PL, Scott CG, Grogan M. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am J Cardiol 2014; 113: 1723–1727. [DOI] [PubMed] [Google Scholar]
- 84. Ikeda S, Sekijima Y, Tojo K, Koyama J. Diagnostic value of abdominal wall fat pad biopsy in senile systemic amyloidosis. Amyloid 2011; 18: 211–215. [DOI] [PubMed] [Google Scholar]
- 85. Quarta CC, Gonzalez‐Lopez E, Gilbertson JA, Botcher N, Rowczenio D, Petrie A, Rezk T, Youngstein T, Mahmood S, Sachchithanantham S, Lachmann HJ, Fontana M, Whelan CJ, Wechalekar AD, Hawkins PN, Gillmore JD. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. Eur Heart J 2017; 38: 1905–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. van Gameren II, Hazenberg BP, Bijzet J, van Rijswijk MH. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for detecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum 2006; 54: 2015–2021. [DOI] [PubMed] [Google Scholar]
- 87. Giorgadze TA, Shiina N, Baloch ZW, Tomaszewski JE, Gupta PK. Improved detection of amyloid in fat pad aspiration: an evaluation of Congo red stain by fluorescent microscopy. Diagn Cytopathol 2004; 31: 300–306. [DOI] [PubMed] [Google Scholar]
- 88. Isogai T, Yasunaga H, Matsui H, Ueda T, Tanaka H, Horiguchi H, Fushimi K. Hospital volume and cardiac complications of endomyocardial biopsy: a retrospective cohort study of 9508 adult patients using a nationwide inpatient database in Japan. Clin Cardiol 2015; 38: 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nakagawa M, Sekijima Y, Tojo K, Ikeda S. High prevalence of ATTR amyloidosis in endomyocardial biopsy‐proven cardiac amyloidosis patients. Amyloid 2013; 20: 138–140. [DOI] [PubMed] [Google Scholar]
- 90. Amyloidosis Research Committee of the Ministry of Health, Labour and Welfare of Japan. http://amyloidosis‐research‐committee.jp/. Accessed 8 November 2020 (in Japanese).