

Abstract
Aims
Skeletal muscle (SkM) abnormalities may impact exercise capacity in patients with heart failure with preserved ejection fraction (HFpEF). We sought to quantify differences in SkM oxidative phosphorylation capacity (OxPhos), fibre composition, and the SkM proteome between HFpEF, hypertensive (HTN), and healthy participants.
Methods and results
Fifty‐nine subjects (20 healthy, 19 HTN, and 20 HFpEF) performed a maximal‐effort cardiopulmonary exercise test to define peak oxygen consumption (VO2, peak), ventilatory threshold (VT), and VO2 efficiency (ratio of total work performed to O2 consumed). SkM OxPhos was assessed using Creatine Chemical‐Exchange Saturation Transfer (CrCEST, n = 51), which quantifies unphosphorylated Cr, before and after plantar flexion exercise. The half‐time of Cr recovery (t1/2, Cr) was taken as a metric of in vivo SkM OxPhos. In a subset of subjects (healthy = 13, HTN = 9, and HFpEF = 12), percutaneous biopsy of the vastus lateralis was performed for myofibre typing, mitochondrial morphology, and proteomic and phosphoproteomic analysis. HFpEF subjects demonstrated lower VO2,peak, VT, and VO2 efficiency than either control group (all P < 0.05). The t1/2, Cr was significantly longer in HFpEF (P = 0.005), indicative of impaired SkM OxPhos, and correlated with cycle ergometry exercise parameters. HFpEF SkM contained fewer Type I myofibres (P = 0.003). Proteomic analyses demonstrated (a) reduced levels of proteins related to OxPhos that correlated with exercise capacity and (b) reduced ERK signalling in HFpEF.
Conclusions
Heart failure with preserved ejection fraction patients demonstrate impaired functional capacity and SkM OxPhos. Reductions in the proportions of Type I myofibres, proteins required for OxPhos, and altered phosphorylation signalling in the SkM may contribute to exercise intolerance in HFpEF.
Keywords: HFpEF, Exercise, Skeletal muscle
Introduction
The incidence and prevalence of heart failure (HF) with preserved ejection fraction (HFpEF) is increasing as the population ages and grows more obese. 1 Although the pathophysiology of HFpEF is incompletely understood and likely heterogeneous, 2 exercise intolerance is a virtually universal feature. While central haemodynamic abnormalities contribute to exercise limitations, the decreased arterio‐venous oxygen content difference (ΔAVO2) identified in recent studies of HFpEF patients suggests abnormalities in oxygen utilization within the skeletal muscle (SkM). 3
During exercise, free (unphosphorylated) Cr is released to generate ATP as part of the phosphocreatine shuttle ( PCr + ADP ↔ Cr + ATP ). 4 During recovery, the reaction is driven towards the regeneration of PCr, as ATP is produced by the mitochondria. Accordingly, the rate at which free Cr falls during recovery is proportional to SkM mitochondrial oxidative phosphorylation capacity (SkM OxPhos). A recently developed hydrogen‐based MRI technique, known as creatine (Cr) chemical exchange‐saturation transfer (CrCEST), 4 , 5 , 6 tracks changes in free Cr within the SkM. When combined with plantar flexion exercise, a small muscle mass movement that does not tax the limits of cardiac output, CrCEST provides an opportunity to assess SkM OxPhos, independent of central haemodynamic reserve.
In this report, we demonstrate that SkM OxPhos is abnormal in HFpEF subjects as compared to healthy and similarly aged hypertensive controls. Muscle biopsies identified a lower proportion of Type I myofibres in HFpEF SkM. SkM proteomic analysis identified global reductions in the levels of proteins related to mitochondrial OxPhos, providing a structural basis for the functional abnormalities. Our work suggests that intrinsic SkM abnormalities contribute to the impaired exercise capacity seen in HFpEF patients.
Methods
Participants
The general criteria for participant selection have been reported previously. 7 In brief, inclusion criteria for HFpEF subjects included symptomatic HF (NYHA Class II/III) with a preserved ejection fraction (≥ 50%), stable medical management for at least 1 month, and objective evidence of elevated intramyocardial filling pressures. 7 Healthy controls were individuals who did not have a history of hypertension or HF, although treated hypercholesterolaemia was permitted in order to allow representation of elderly subjects. Given the near ubiquitous presence of hypertension in HFpEF patients, we enrolled hypertensive individuals without HF symptoms as an additional control group. Hypertensive individuals included those who were treated with antihypertensive medications, had been on stable medical therapy for at least 1 month, and had no evidence of HF. One subject, without a known history of hypertension, was initially enrolled into the healthy group but was found to be hypertensive during the study visit and in the subsequent period afterwards. This subject was therefore included in the Hypertensive group, prior to data analysis. Key exclusion criteria included current atrial fibrillation, inability to exercise, moderate or greater aortic or mitral valve disease, and significant lung disease. Additional inclusion and exclusion criteria are listed in the Supporting Information, Data S1 . This study complied with the Declaration of Helsinki. The University of Pennsylvania Institutional Review Board approved the study, and all subjects provided written informed consent prior to entry.
Study procedures
Participants presented in the fasted state. Body composition was assessed with dual energy X‐ray absorptiometry (DEXA; Hologic, Inc., Bedford, MA). Appendicular lean mass index was calculated as the sum of two‐arm lean mass and two‐leg lean mass divided by height2. 8
Exercise protocol
As described previously, 7 subjects underwent a maximal effort supine cycle ergometry exercise test with gas exchange measurements (Parvo Medics, Sandy, UT) until exhaustion. Peak oxygen consumption (VO2,peak) and other gas exchange measurements were defined as the average value obtained during the last 30 s of exercise. Peak predicted VO2 was calculated using the Wasserman/Hansen equations. 9 The ventilatory threshold (VT) was determined using the V‐slope and ventilatory equivalents methods, with the results averaged. 10 , 11 Exercise efficiency was estimated as the ratio of total work performed (kilojoules) to total oxygen consumed (litres) above baseline during exercise (‘net efficiency’). Peak VO2 and VT were indexed to body mass and two‐leg lean mass (Peak VO2, leg lean and VTleg lean) derived from DEXA. Exercise efficiency was indexed to two‐leg lean mass. Post‐exercise VO2 recovery kinetics were modelled according to a monoexponential decay (decay constant, τ); VO2 recovery half‐time was also assessed (time required for VO2 to be reduced by 50% of the amplitude of the change from baseline to peak).
Subjects underwent resting and exercise echocardiography using a GE Vivid E9 machine (GE Healthcare, Fairfield, CT), with quantification of stroke volume (SV) from the left‐ventricular outflow tract Doppler velocity‐time integral. 7 , 10 The peak ΔAVO2 was determined as the ratio of oxygen consumption (VO2) to cardiac output (SV*heart rate). Additional details regarding the exercise protocol can be found in the Supplementary Materials.
Creatine chemical exchange saturation transfer procedure
Details of the creatine chemical exchange saturation transfer (CrCEST) imaging protocol have been reported previously. 4 In brief, CrCEST images were obtained on a 3T whole‐body scanner (Siemens Medical Systems, Erlangen, Germany) using a 15‐channel 1H Tx/Rx knee coil (QED, Mayfield Village, OH, USA). Plantar flexion exercise was performed inside the MRI scanner using a pneumatically controlled MRI‐compatible foot pedal against 7 PSI of pressure. The cadence was set at 90 repetitions over 2 min (0.75 Hz). Baseline images were taken for 2 min prior to exercise and for 8 min following exercise at 30‐s (s) intervals (Figure 1 ). Two parameters of SkM OxPhos were defined: the half‐time of Cr recovery (t1/2, Cr) and the slope of Cr decline during early recovery (first 2 min post‐exercise). Additional details regarding the CrCEST protocol and data on popliteal artery blood flow, intramuscular fat, and lower leg lean muscle volume on MRI can be found in the Supplementary Materials.
Figure 1.
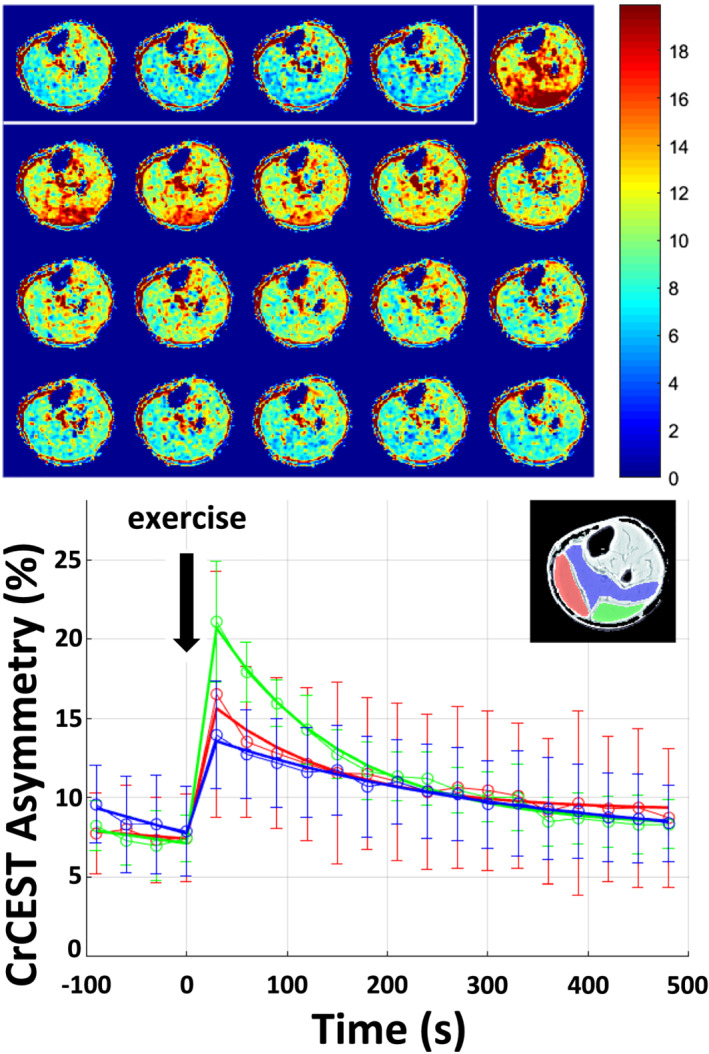
Creatine chemical exchange saturation transfer (CrCEST) maps of exercise‐induced changes in skeletal muscle creatine (Cr) concentration. Data displayed are from one healthy 36‐year‐old male following 2 min of plantar flexion exercise against 7 PSI pressure at 0.75 Hz. Top panel: CrCEST maps before (enclosed by white box) and after exercise show an increase in CrCEST asymmetry (increase in red shading), indicative of increased free Cr, liberated as part of the phosphocreatine shuttle to generate ATP ( PCr + ADP ↔ Cr + ATP ). During recovery, free Cr decreases as the ATP produced by oxidative phosphorylation shifts the reaction towards PCr regeneration. Bottom panel: Changes in CrCEST asymmetry before and after exercise are highlighted in prescribed regions of interest: lateral gastrocnemius (green), medial gastrocnemius (red), and the soleus (blue) muscles. Data are displayed as mean ± SD at each time point. The mean at each time point represents the average value from all activated voxels, and its standard deviation, from within each muscle.
Vastus lateralis muscle biopsy
As part of a separate study, subjects were invited to undergo a resting percutaneous muscle biopsy of the vastus lateralis. As detailed in the Supplementary Materials, muscle tissue was assessed for myofibre typing using immunofluorescence, citrate synthase activity, and subjected to liquid chromatography with tandem mass spectrometry (LC–MS/MS) for proteomic and phosphoproteomic analysis. Electron microscopy (EM) was also performed to assess mitochondrial morphology.
Proteomic and phosphoproteomic methods in brief
Protein extraction and digestion
Frozen tissue biopsy samples (~20 mg per sample) were cut into 0.5‐mm cubes and dry‐ground using a ball mill (MM400 Retsch, Germany). 12 Proteins were extracted in SDC buffer containing protease and phosphatase inhibitors (Roche). Protein concentration was estimated by in‐gel staining with Coomassie Blue G250 solution and Image J analysis. Proteolysis was initiated by sequential addition of LysC, followed by trypsin. Resulting peptides were desalted using an OasisHLB plate (Waters).
Liquid chromatography with tandem mass spectrometry sample preparation
Of the total peptides generated, 5% of each sample was used for whole proteome analysis, 85% was used for immobilized metal affinity chromatography (IMAC) enrichment of phosphopeptides, and the rest was set aside for producing spectral libraries. 13 , 14 , 15 Spectral libraries were generated by pooling the peptides from each sample analysed, followed by chromatographic separation on high pH reversed phase HPLC. 13 , 14 Six fractions were collected, and 5% of each fraction was used to generate spectra libraries for the proteome. The remainder was individually subjected to IMAC enrichment to generate the phosphopeptide library. Prior to LC‐MS analysis, each peptide sample was dried then solubilized in 0.1% TFA containing iRT peptides (Biognosys). Additional details regarding the muscle biopsy procedure, as well as the proteomic and phosphoproteomic preparation and analyses, can be found in the Supplementary Materials.
Statistical analysis
The SkM CrCEST, muscle biopsy, and proteomic/phosphoproteomic data that generate the main findings in this manuscript have not been published previously. 7 Demographic data are presented as n (%), means (SD, standard deviation), or medians (IQR, interquartile range). The Shapiro–Wilk test was used to assess distribution normality. Medians from variables with a non‐normal distribution were compared using the Kruskal–Wallis test, and means from normally distributed variables were compared using ANOVA. When the overall test was significant, post‐hoc comparisons for between group differences were performed with Bonferroni correction. Correlations between variables of interest were performed using Spearman's correlation. Analyses were performed on STATA (Stata/SE, Version 13.1; StataCorp, College Station, TX), with a P < 0.05 taken to be significant. Principal components analysis (PCA) was performed in order to reduce the high‐dimensional proteomic data into a smaller number of meaningful factors that were then compared among clinical groups and to the main clinical outcomes (Peak VO2, leg lean, VTleg lean, t1/2, Cr , and the slope of Cr decline during early recovery). To account for multiple hypothesis testing, a Benjamini‐Hochberg false discovery rate (FDR) below 0.05 was considered statistically significant for each separate hypothesis. Full statistical methods for the proteomic and phosphoproteomic analyses can be found in the Supplementary Materials. Individual proteins that demonstrated different relative levels between groups were entered into the String Database (string-db.org; Version 11; String Consortium 2020, last analyses performed in August 2020) to identify enrichment of biologic pathways using Gene Ontology. 16 , 17 , 18 A similar procedure was used to identify biologic pathway enrichment for the PCA factors. Prism 8 (GraphPad Software, LLC) was used to generate bar graphs of key endpoints. One author (PZ) had access to all data and takes responsibility for its integrity and data analysis. The data underlying this article are available in the article and in its online supplementary material. The sharing of data by the authors may be considered upon reasonable request to the corresponding author. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD025092. 19
Results
Study population
Fifty‐nine subjects (20 healthy, 19 hypertensive, and 20 subjects with symptomatic HFpEF) enrolled in the exercise and MRI studies. Demographic and medical histories are presented in Table 1 . Subjects with HFpEF were older, more obese, and had typical co‐morbidities such as hypertension (100%), diabetes (55%), hyperlipidaemia (90%), and obstructive sleep apnoea (60%). HFpEF subjects exhibited higher NT‐pro‐BNP levels, tricuspid regurgitant jet velocities, and mitral E/septal e′ ratios (Table 1 ).
Table 1.
Subject characteristics for participants who underwent the exercise and CrCEST studies
| Median (IQR) | Healthy | Hypertensive | HFpEF | P‐value |
|---|---|---|---|---|
| Mean ± SD | (n = 20) | (n = 19) | (n = 20) | |
| Age, years | 54 (39, 63) | 66 (50, 71) | 67 a (62, 76) | 0.001 |
| Female, n (%) | 6 (30) | 7 (37) | 13 (65) | 0.07 |
| Ethnicity, n (%) | 0.001 | |||
| White | 20 (100) | 14 (73.7) | 12 (60) | |
| African‐American | 0 (0) | 3 (15.8) | 8 (40) | |
| Asian | 0 (0) | 2 (10.5) | 0 (0) | |
| Height, cm | 171.9 ± 6.8 | 171.6 ± 9.7 | 165.3 ± 9.9 | 0.037 |
| Weight, kg | 81.4 (68.7, 85.7) | 80.4 (73.0, 89.0) | 99.1 a (78.3, 113.5) | 0.02 |
| BMI, kg/m2 | 26.7 (23.6, 28.7) | 27.7 (24.6, 31.5) | 32.1 a , b (28.7, 44.4) | <0.001 |
| Whole body lean mass, kg | 53.4 ± 10.3 | 53.5 ± 8.4 | 53.9 ± 11.1 | 0.98 |
| Calf lean mass, kg | 2.3 ± 0.5 | 2.2 ± 0.6 | 2.2 ± 0.5 | 0.70 |
| Appendicular lean mass index, kg/m2 | 8.0 ± 1.6 | 7.9 ± 1.0 | 8.3 ± 1.3 | 0.62 |
| Hypertension, n (%) | 0 (0) | 19 (100) | 20 (100) | <0.001 |
| Diabetes, n (%) | 0 (0) | 3 (15.8) | 11 (55.0) | <0.001 |
| Insulin | 0 (0) | 0 (0) | 4 (20) | 0.03 |
| Hyperlipidaemia, n (%) | 5 (25) | 11 (57.9) | 18 (90) | <0.001 |
| OSA, n (%) | 1 (5) | 4 (21.1) | 12 (60) | <0.001 |
| Beta‐blocker, n (%) | 0 (0) | 6 (31.6) | 16 (80) | <0.001 |
| CCB, n (%) | 0 (0) | 7 (36.8) | 11 (55) | <0.001 |
| ACEi/ARB, n (%) | 0 (0) | 10 (52.6) | 14 (70) | <0.001 |
| Loop diuretic, n (%) | 0 (0) | 0 (0) | 10 (50) | <0.001 |
| Thiazide diuretic, n (%) | 0 (0) | 3 (15.8) | 4 (20) | 0.13 |
| Statin, n (%) | 3 (15) | 8 (42.1) | 14 (70) | 0.002 |
| NYHA class, n (%) | ||||
| II | 18 (90) | |||
| III | 2 (10) | |||
| eGFR, mL/min | 87.3 ± 12.6 | 79.5 ± 17.8 | 71.1 ± 19.9 a | 0.015 |
| Haemoglobin, g/dL | 13.9 ± 1.3 | 14.0 ± 1.0 | 12.7 ± 1.1 a , b | <0.001 |
| NTproBNP, pg/mL | 35.0 (17.0, 63.5) | 65.0 (34.0, 127.0) | 119.0 a (49.0, 241.5) | 0.002 |
| LV ejection fraction, % | 59.6 ± 6.8 | 59.8 ± 4.5 | 61.9 ± 5.6 | 0.39 |
| Tricuspid regurgitant jet velocity, cm/s | 199.8 ± 26.8 | 233.2 ± 40.8 c | 265.5 ± 28.8 a , b | <0.001 |
| Mitral inflow early velocity (E), cm/s | 67.4 (54.1, 79.4) | 75.2 (64.1, 84.4) | 80.3 a (66.5, 97.3) | 0.035 |
| Septal TD e′ velocity, cm/s | 9.6 ± 2.3 | 8.9 ± 1.9 | 7.6 ± 2.4 a | 0.016 |
| Mitral E/septal e′ ratio | 6.6 (5.9, 8.5) | 9.0 (7.0, 10.5) | 11.9 a , b (8.4, 13.4) | <0.001 |
Data are presented as mean ± SD, median (25–75% interquartile range), or count (%).
ACEi/ARB, angiotensin converting enzyme inhibitor/angiotensin receptor blocker, CCB, calcium channel blocker, OSA, obstructive sleep apnoea, SD, standard deviation, TD, tissue Doppler.
HFpEF vs. healthy, adjusted P < 0.05,
HFpEF vs. HTN, adjusted P < 0.05,
Healthy vs. HTN, adjusted P < 0.05.
Cycle ergometry exercise data
Cycle ergometry data are presented in Figure 2 and Table 2 . As reported previously, 7 HFpEF subjects exercised for a significantly shorter duration than either HTN or healthy controls, with a lower peak VO2, driven primarily by a reduced ΔAVO2. The VT and VO2 efficiency were lower in HFpEF subjects. Following exercise, VO2 recovered more slowly in HFpEF participants than healthy controls (Table 2 ).
Figure 2.
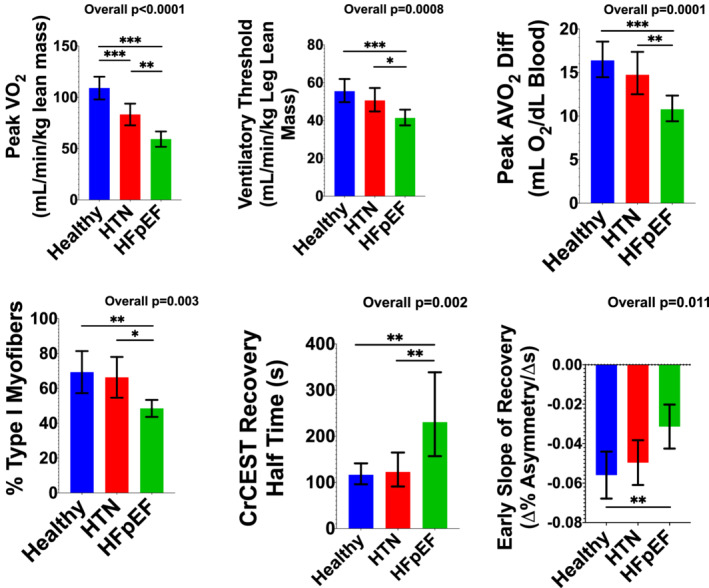
Group data for key endpoints. Data are presented as mean with 95% confidence intervals. When the data were not normally distributed, log‐transformation was performed for statistical analyses, and geometric means with their 95% confidence intervals are plotted to retain native units. ANOVA was used for the overall comparison, with post hoc intergroup comparisons performed with Bonferroni correction. *Adjusted P < 0.05, **adjusted P ≤ 0.01, ***adjusted P ≤ 0.001.
Table 2.
Maximal effort cardiopulmonary exercise test
| Healthy | Hypertensive | HFpEF | P‐value | |
|---|---|---|---|---|
| (n = 20) | (n = 19) | (n = 20) | ||
| Ventilatory threshold | ||||
| VT, L/min | 0.85 (0.77, 1.17) | 0.90 (0.71, 0.97) | 0.76 (0.60, 0.88) | 0.038 |
| VT, mL/kg body weight/min | 11.6 (9.8, 16.1) | 9.9 (8.5, 12.8) | 7.4 a , b (6.4, 9.4) | <0.001 |
| VT, mL/kg leg lean mass/min | 53.5 (47.8, 67.4) | 48.5 (41.7, 57.1) | 42.6 a (37.7, 48.0) | 0.002 |
| Peak | ||||
| Heart rate, b.p.m. | 148.7 ± 18.4 | 136.6 ± 26.6 | 114.6 ± 24.9 a , b | <0.001 |
| RER | 1.19 ± 0.12 | 1.17 ± 0.13 | 1.06 ± 0.13 a , b | 0.004 |
| Arterial lactate, mmol/L | 9.4 (7.5, 11.8) | 7.3 (5.3, 9.1) | 6.1 (5.1, 10.3) | 0.07 |
| Exercise time, min | 24.2 (20.1, 32.3) | 20.0 (13.9, 28.5) | 5.5 a , b (3.3, 10.8) | <0.001 |
| Work rate, Watts | 208.9 (160.1, 312.5) | 150.0 (100.0, 275.0) | 25.0 a , b (19.8, 75.0) | <0.001 |
| VO2, L O2/min | 1.90 (1.31, 2.31) | 1.49 (1.10, 1.68) | 0.95 a , b (0.75, 1.34) | <0.001 |
| % Predicted VO2 | 89.8 ± 19.7 | 76.9 ± 15.1 | 64.0 ± 14.1 a | <0.001 |
| Indexed VO2, mL/min/kg body weight | 23.9 (19.9, 31.5) | 16.8 c (12.7, 20.7) | 10.1 a , b (7.9, 14.7) | <0.001 |
| Indexed VO2, mL/min/kg leg lean mass | 109.0 ± 23.7 | 83.2 ± 21.9 c | 59.2 ± 16.0 a , b | <0.001 |
| AVO2 diff, mL O2/dL of blood | 15.8 (14.3, 19.1) | 14.9 (11.2, 17.5) | 11.2 a , b (8.7, 13.0) | <0.001 |
| Cardiac output, L/min | 11.5 (10.0, 13.8) | 9.7 (7.6, 11.9) | 9.8 (7.5, 11.5) | 0.08 |
| VO 2 net efficiency | ||||
| Net efficiency: total work performed/total O2 consumed during exercise, kJ/L O2 | 5.1 (4.2, 6.3) | 5.5 (3.6, 7.0) | 3.0 a , b (2.4, 3.4) | <0.001 |
| Net efficiency, indexed to leg lean mass, kJ/L O2/kg | 0.27 (0.23, 0.39) | 0.32 (0.24, 0.37) | 0.18 a , b (0.15, 0.23) | <0.001 |
| VO 2 recovery kinetics | ||||
| τ, s | 53.0 (44.1, 56.4) | 66.4 c (53.9, 75.7) | 72.4 a (59.3, 83.2) | <0.001 |
| Recovery half‐time, s | 53.0 (47.5, 57.5) | 63.0 c (55.0, 70.0) | 71.5 a (60.0, 78.0) | <0.001 |
Data are presented as mean ± SD or median (25–75% interquartile range).
MAP, mean arterial pressure, RER, respiratory exchange ratio, SD, standard deviation, VT, ventilatory threshold.
HFpEF vs. healthy, adjusted P < 0.05,
HFpEF vs. HTN, adjusted P < 0.05,
Healthy vs HTN, adjusted P < 0.05.
CrCEST data
Subjects returned for the CrCEST examination within 1 month (median 10 [IQR 2, 21] days) from the supine cycle ergometry exercise study. CrCEST imaging was successfully completed in 51 patients, with summary data presented in Table 3 and Figure 2 . Data were not available from eight subjects due to technical issues with the scanner (n = 1), poor data quality (n = 3), metal implants including pacemakers (n = 3), and claustrophobia (n = 1). The lateral gastrocnemius muscle was most activated by our plantar flexion exercise protocol. The half‐time of CrCEST recovery (t1/2,Cr) was significantly longer in HFpEF subjects. Similarly, the average slope of CrCEST recovery during the initial 2 min post‐exercise was significantly different among groups, with less negative slopes indicating more prolonged recovery in HFpEF. The t1/2,Cr correlated modestly with both Peak VO2, leg lean ( ρ = −0.28, P = 0.047) and VTleg lean ( ρ = −0.34, P = 0.013), whereas the slope of early Cr recovery correlated with the ΔAVO2 ( ρ = −0.45, P = 0.002).
Table 3.
Group data for CrCEST asymmetry, an index of free creatine (Cr), in the lateral gastrocnemius following a standardized plantar flexion exercise protocol
| Healthy | Hypertensive | HFpEF | P‐value | |
|---|---|---|---|---|
| (n = 20) | (n = 17) | (n = 14) | ||
| Baseline CrCEST asymmetry, % | 6.2 ± 0.9 | 6.0 ± 1.2 | 6.3 ± 1.4 | 0.73 |
| Increase in CrCEST asymmetry post‐exercise, % | 8.3 ± 3.5 | 7.8 ± 2.9 | 6.2 ± 2.3 | 0.16 |
| Half‐time CrCEST recovery, s | 127.6 (101.1, 147.5) | 114.4 (87.9, 154.1) | 167.4 a , b (140.9, 458.9) | 0.005 |
| Slope of early Cr recovery, %Asymmetry/s | −0.06 ± 0.03 | −0.05 ± 0.02 | −0.03 ± 0.02 a | 0.01 |
Data are presented as mean ± SD or median (25–75% interquartile range).
IQR, interquartile range; SD, standard deviation.
HFpEF vs. healthy, adjusted P < 0.05.
HFpEF vs. HTN, adjusted P < 0.05.
Healthy vs HTN, adjusted P < 0.05.
Muscle biopsy data
Percutaneous biopsy of the vastus lateralis was performed in 34 (healthy n = 13, HTN n = 9, and HFpEF n = 12) subjects. Demographics of the muscle biopsy participants are broadly consistent with the overall study population (Supporting Information, Table S1 ).
Myofibre typing
The median number of fibres assessed was 339 [25–75% IQR 209, 560], with no difference among groups. HFpEF subjects exhibited a lower percent of Type I fibres (Figure 2 ). There was no difference in the percent of Type IIa fibres among groups (healthy: 31.1 ± 14.1%, HTN 31.8 ± 14.7%, HFpEF 36.8 ± 10.7%, P = 0.57); however, there was a greater percent of Type IIx fibres in HFpEF SkM (healthy median 0 [25–75% IQR 0, 3.5]%, HTN median 0 [25–75% IQR 0, 0.5]%, HFpEF median 8.8 [25–75% IQR 2.8, 21.6]%, P = 0.029, see Supplemental Materials for additional data).
Mitochondrial volume density and morphology
Morphometric analysis of the EM images explored ultrastructural changes in the SkM mitochondria. The surface area occupied by mitochondria, as well as their individual shape and geometry, were not different among groups. Biochemically, quantification of citrate synthase activity was not different among groups, corroborating the EM data (Supporting Information, Table S2 ).
Proteomics analysis
Changes in the relative levels of proteins were quantified using mass spectrometry in muscle biopsy samples from 12 healthy, 9 HTN, and 13 HFpEF participants.
HFpEF vs. HTN
Of the 2832 proteins detected in both HFpEF and HTN, levels of 327 were significantly different between groups (P < 0.05). Bioinformatic analyses of these 327 proteins revealed marked enrichment for proteins participating in metabolic processes (Figure 3 ). The levels of proteins with functional annotations in electron transport system complexes, tricarboxylic acid cycle, and fatty acid and ketone metabolism were reduced in HFpEF as compared with HTN. A group of 37 proteins among the 327 showed a relative decrease or increase by two‐fold (Supporting Information, Table S3 ). These proteins participate in the assembly of complex I of the mitochondrial electron transport chain, in the metabolism of ketones, and in oxidation–reduction processes, suggesting alterations in mitochondrial bioenergetics and metabolism in HFpEF (Supporting Information, Table S4 ).
Figure 3.
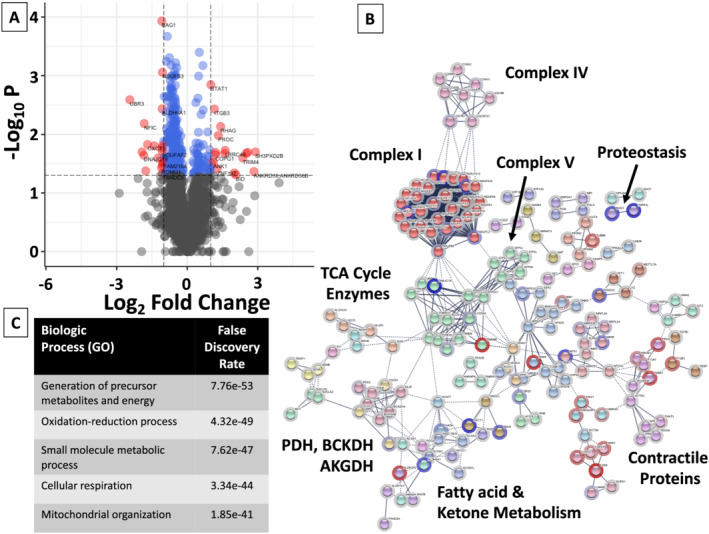
Volcano plot, network map, and biologic enrichment of proteins significantly different between HFpEF and HTN participants. (A) Volcano plots were constructed with blue dots representing proteins with significantly different relative levels between HFpEF and HTN participants (P < 0.05), and red dots and the gene names listed for proteins that had significantly different levels with an absolute log2 fold‐change (FC) > 1. (B) All proteins with significantly different relative levels between HFpEF and HTN participants (P < 0.05), and their log2 fold‐change (FC), were entered into the String Database (string-db.org). Interrelated proteins are displayed along with the connection between proteins and groups. Related proteins are shaded a similar colour. The halo around each protein represents the log2 FC, with blue indicating a relative decrease in protein level in HFpEF participants as compared with HTN participants, and red representing an increase. Although not inclusive, specific clusters of proteins related to energy fuel metabolism are enumerated. (C) Enrichment of the top 5 biologic (GO) processes, along with the false‐discovery rate are listed. AKGDH, alpha‐ketoglutarate dehydrogenase complex; BCKDH, branched‐chain alpha‐keto dehydrogenase complex; PDH, pyruvate dehydrogenase; TCA, tricarboxylic acid cycle.
HFpEF vs. healthy controls
Of the 2844 proteins detected in both HFpEF and healthy subjects, 317 demonstrated a significant difference (P < 0.05) in their relative levels. Bioinformatic analysis of these 317 proteins revealed enrichment in the pathways that relate to oxidation–reduction processes (Figure 4 , Supporting Information, Table S5 ), similar to the findings for the HFpEF vs. HTN comparison. Proteins with either a doubling or halving of relative levels in the skeletal muscle of HFpEF vs. healthy participants are listed in Supporting Information, Table S6 .
Figure 4.
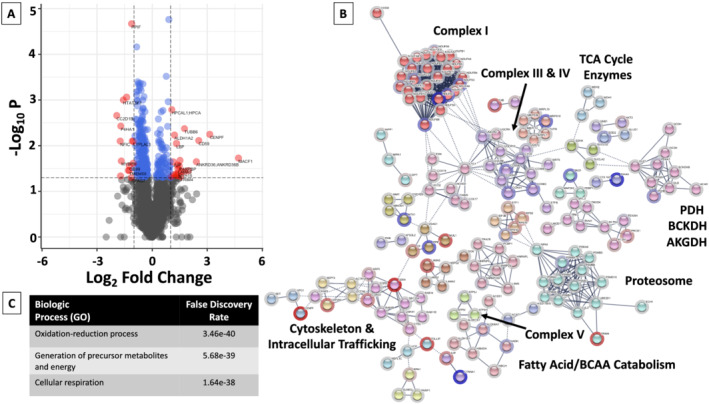
Volcano plot and network map of proteins significantly different between HFpEF and healthy participants. (A) Volcano plots were constructed with blue dots representing proteins with significantly different relative levels between HFpEF and healthy participants (P < 0.05), and red dots and the gene names of proteins that had significantly different levels with an absolute log2 fold‐change (FC) > 1. (B) All proteins with significantly different relative levels between HFpEF and healthy participants (P < 0.05), and their log2 fold‐change (FC), were entered into the String Database (string-db.org). Interrelated proteins are displayed, along with the connection between proteins and groups. Related proteins are shaded a similar colour. The halo around each protein represents the log2 FC, with blue indicating a relative decrease in protein level in HFpEF participants as compared with healthy participants, and red representing an increase. Although not inclusive, specific clusters of proteins have been enumerated. (C) Enrichment of the top biologic (GO) processes and the false‐discovery rate are listed. AKGDH, alpha‐ketoglutarate dehydrogenase complex; BCAA, branched chain amino acids; BCKDH, branched‐chain alpha‐keto dehydrogenase complex; PDH, pyruvate dehydrogenase, TCA, tricarboxylic acid cycle.
Principal component analysis
Principal component analysis was performed to reduce the high‐dimensional proteomic data into a smaller number of meaningful factors. Of these, only Factor 3 was significantly decreased in HFpEF subjects, as compared with either HTN (FDR = 0.01) or healthy (FDR = 0.005) participants (Supplementary Materials, Figures S1 and S2 ), explaining 10.9% of the overall proteomic variance (Supplementary Materials, Table S7 ). Two hundred thirty‐five proteins loaded onto Factor 3, and 171 of these proteins localized to the mitochondrion (FDR = 1.72e10‐129). Biologic analysis of the Factor 3 proteins demonstrated significant enrichment in pathways related to small molecule metabolic processes (FDR = 3.32e10‐75), the generation of precursor metabolites and energy (FDR = 4.12e10‐71), and oxidation–reduction processes (FDR = 1.19e‐70). Associations between Factor 3 score and clinical outcomes are presented in Table 4 and in the Supplementary Materials, Figure S3 . In a model that adjusted for arterial oxygen delivery at peak exercise (product of cardiac output, haemoglobin concentration, and arterial oxygen saturation), Factor 3 remained significantly correlated to Peak VO2, leg lean (beta = 14.56 [95% CI 7.01–22.10], P = 0.001, model R 2 = 0.53).
Table 4.
Association of Factor 3 with clinical outcomes
| Outcome | β | Unadjusted | False discovery rate |
|---|---|---|---|
| (95% CI) | P‐value | ||
| VO2, leg lean | 16.4 (7.7, 25.1) | <0.001 | 0.01 |
| VTleg lean | 7.0 (3.1, 10.9) | 0.001 | 0.017 |
| t1/2, Cr | −53.7 (−102.9, −4.6) | 0.033 | 0.56 |
| Slope of early Cr recovery | −0.008 (−0.02, 0.001) | 0.07 | 0.95 |
VO2, leg lean, peak VO2, indexed to leg lean mass; VTleg lean, ventilatory threshold indexed to leg lean mass; t1/2, Cr, half‐time of CrCEST recovery following plantar flexion exercise.
Phosphoproteomic analysis
In an exploratory analysis, we identified 5551 phosphopeptides in both HFpEF and HTN muscle. Of these, the relative levels of 522 phosphopeptides assigned to 241 proteins were different between HFpEF vs. HTN (P < 0.05, Figure 5 ). Statistical analysis of these phosphopeptides identified a cluster of phosphopeptides that were significantly increased in HTN as compared with HFpEF SkM. Kinase motif analysis, using the human phosphopeptide proteome as a reference, identified a 17.2‐fold enrichment for a P‐X‐[phospho‐S/phospho‐T]‐P motif, within the cluster of phosphopeptides that were increased in HTN participants (P < 0.000001 for each amino acid position). 20 This motif is a known substrate for ERK1/ERK2 kinases, 21 , 22 , 23 suggesting an increase in ERK1/ERK2 signalling in HTN SkM as compared with HFpEF. Similar findings were demonstrated when comparing the phosphoproteome between HFpEF and healthy participants, identifying a 17.3‐fold increase in phosphorylation of the P‐X‐[phospho‐S/phospho‐T]‐P motif in healthy as compared with HFpEF SkM (data not shown). While we did not detect differences in either ERK1/ERK2 relative levels, we found decreased relative ERK2 phosphorylation at Threonine‐185, a known activating site, 24 in HFpEF as compared with both HTN and healthy participants. Taken together, these results suggest reduced ERK signalling in the SkM of HFpEF subjects.
Figure 5.
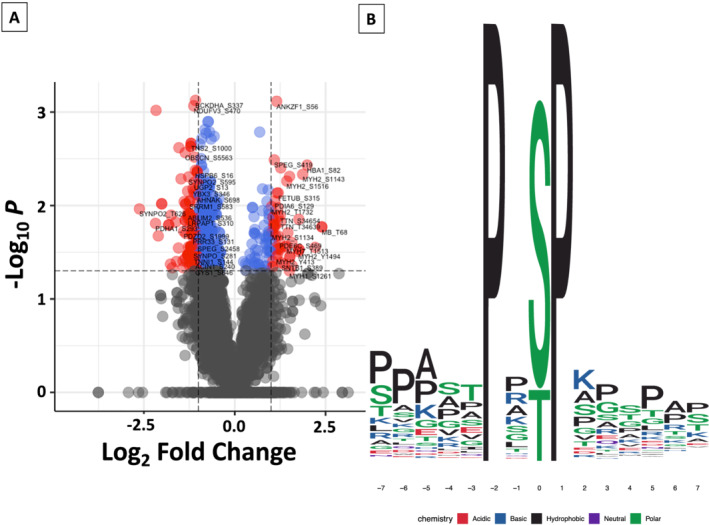
HFpEF vs. HTN muscle biopsy samples demonstrate relative differences in the phosphoproteome. The volcano plot (A) compares the differences in the relative levels of specific phosphopeptides, identifying differences in 522 phosphopeptides (P < 0.05 for each). Blue dots represent peptides with significantly different relative levels (P < 0.05); whereas, red dots indicate peptides with significantly different relative levels and an absolute log2 fold‐change >1. Peptides and the site of phosphorylation are listed. In (B), kinase motif analysis using the human phosphopeptide proteome as a reference identified increased enrichment for a P‐X‐(S/T)‐P motif in HTN as compared with HFpEF SkM. This motif is recognized by ERK1/ERK2, among other kinases.
Discussion
The main findings of our study are (1) HFpEF subjects exhibited a marked reduction in exercise capacity; (2) SkM OxPhos was reduced in HFpEF using a novel MRI‐based technique; (3) HFpEF subjects exhibited a reduction in the percentage of fatigue‐resistant/oxidative Type I fibres; (4) proteomics revealed global reductions in proteins related to oxidative energy fuel metabolism in HFpEF SkM that correlated with exercise capacity, independent of peak oxygen delivery; and (5) in an exploratory analysis, phosphopeptide signatures suggest reductions in ERK1/ERK2 signalling in HFpEF SkM.
Consistent with prior studies, exercise capacity was reduced in our HFpEF subjects, in conjunction with a reduced ΔAVO2, suggesting abnormalities in SkM oxygen utilization. 3 , 7 To explore potential mechanisms for this observation, we used a novel MRI technique, CrCEST, to spatially resolve free Cr levels during post‐exercise recovery and assess in vivo SkM OxPhos. Following plantar flexion exercise, a small muscle mass exercise paradigm not expected to tax the limits of cardiac output, we observed a marked prolongation in Cr recovery in HFpEF subjects, indicative of impaired PCr regeneration due to slowed aerobic metabolism. These data are consistent with accumulating evidence identifying abnormal SkM OxPhos in HFpEF. Using 31P spectroscopy, Weiss et al. demonstrated reduced oxidative capacity in HFpEF subjects, as compared with healthy younger controls. 25 Our data extend the findings by Weiss et al., identifying abnormalities in SkM OxPhos in HFpEF, as opposed to similarly aged hypertensives, and finding key relationships between SkM OxPhos and systemic exercise capacity.
We performed SkM biopsies to further our investigations. We identified a marked difference in myofibre type present in HFpEF subjects, with a much lower percentage of Type I fibres than either healthy controls or hypertensive subjects of similar age. Previously, Kitzman et al. found a similar reduction in Type I fibre percentage in HFpEF subjects as compared with age‐matched healthy controls. 26 Our study demonstrates that this fibre type switch does not occur to the same degree in similarly aged hypertensive subjects without heart failure and provides additional specificity, demonstrating an increase in more glycolytic Type IIx fibres, alongside the reduction in oxidative and fatigue‐resistant Type I fibres.
In addition to the changes in muscle fibre composition, we found global reductions in the relative levels of proteins participating in mitochondrial energy transduction and oxidative phosphorylation in HFpEF participants, suggesting a defect in the SkM independent of lean muscle mass, which was not different among groups. Using principal components analysis, we identified one factor (Factor 3), comprised mainly of mitochondrial proteins, that was significantly lower in HFpEF participants as compared to the other groups. Factor 3 score correlated with peak VO2, leg lean, even after adjustment for arterial oxygen delivery, suggesting an independent relationship between SkM mitochondrial proteins and exercise tolerance. 27
Few published data exist that provide molecular characterization of HFpEF SkM. In addition to the studies described above, Molina et al. compared HFpEF SkM with age‐matched healthy controls, reporting reductions in citrate synthase activity, and the levels of porin and mitofusin 2, two proteins that are localized on the mitochondrial outer membrane. 28 While our study did not identify abnormalities in mitochondrial morphology or content, a recent report by Bekfani et al. identified smaller mitochondria and reduced mitochondrial volume density in HFpEF SkM as compared with similarly aged controls. 29 The reason for the discrepancy between our findings and those of the other studies with regard to mitochondrial content and morphology is not readily apparent, although the small sample size of all of these studies bears mention.
The Bekfani et al. study described reductions in gene expression of key proteins involved in fatty acid oxidation (e.g. peroxisome proliferator‐activated receptor alpha [PPAR‐∝]) and carbohydrate metabolism (e.g. pyruvate dehydrogenase kinase 4 [PDK4]), alongside increased gene expression of proteins associated with muscle atrophy (e.g. Atrogin‐1, Myostatin‐2, and Ubiquitin B). 29 Our data extend these findings, demonstrating impairments in ATP generation in vivo following plantar flexion exercise. Our proteomic analyses demonstrate broad reductions in the proteins and complexes involved in energy fuel metabolism, including TCA cycle enzymes and the mitochondrial complexes that make up the electron transport chain. Given the similar age of participants who underwent the SkM biopsy, we conclude that our findings do not simply reflect the aging process. 30 Rather, these data identify a global impairment in energy fuel metabolism in HFpEF SkM, which associate with the impaired functional capacity in these patients.
Phosphoproteomic analyses have precisely identified thousands of phosphorylation sites across the proteome. Our understanding of the specific kinases involved in these phosphorylation events has increased, although it still remains in its infancy. We see our results as an important dataset for the field so that collectively, we may conduct future studies aimed at deciphering the relevance of the observed changes in protein phosphorylation. Prior study of acute resistance exercise demonstrates an increase in SkM protein phosphorylation by mitogen‐activated protein kinases (MAPK), such as ERK1 (MAPK3) and ERK2 (MAPK1). 31 Our data, obtained in resting individuals who refrained from exercise prior to the biopsy, indicates decreased ERK signalling in HFpEF SkM as compared to both healthy and HTN controls. A recent report demonstrated that increased ERK signalling mediates a fast‐to‐slow myofibre transition. 32 While speculative, our findings of decreased Type I myofibre percentage in HFpEF could be consistent with decreased ERK signalling. It is important to note that other MAPKs, such as p38 and JNK, also increase their activity with exercise. 33 Collectively, MAPKs have been shown to alter energy fuel metabolism following exercise, for example, by increasing fatty acid uptake and oxidation. 33 Furthermore, activation of p38 stimulates PGC‐1∝ (peroxisome‐proliferator‐activated receptor‐γ coactivator‐1∝), a master regulator of muscle mitochondrial function and angiogenesis, leading to a coordinated response that increases SkM mitochondrial oxidative capacity and vascularity. 34 , 35 , 36 Little is known about PGC‐1∝ in HFpEF SkM; however, its combined effects to increase mitochondrial and capillary density, and alter energy fuel substrate metabolism, all suggest that PGC‐1∝ signalling may be decreased in HFpEF SkM. We did not detect PGC‐1∝ in our proteomic analysis, as it was likely below the level of detection. This should be a high priority for future investigation.
There are limitations to this study. Given the detailed phenotyping, our study included a relatively small number of subjects, limiting our power. The plantar flexion exercise employed a common resistance (7 PSI), which may have led to different relative intensities between subjects. There was a delay between the implementation of the muscle biopsy protocol and enrollment in our exercise studies. However, individual findings obtained from SkM biopsies, MRI imaging, and exercise studies are concordant, and jointly demonstrate evidence for functional and structural impairments in SkM OxPhos. As typically seen in global proteomics datasets, a total of 1268 proteins (44%) had at least one missing value, 37 requiring that protein to be excluded from PCA. Given the limitations and uncertainties among different imputation methods in proteomic analyses, 37 we elected to not impute missing data. However, we note that the findings from the PCA analysis are consistent with those using other analytic methods. While our phosphoproteomic analysis suggests a decrease in ERK1/ERK2 signalling in HFpEF participants, we recognize the significant overlap between kinase motifs reported in the literature. Direct in vitro measurements of ERK activity would be confirmatory. We did not identify a reduction in mitochondrial content, either on EM or as reflected by citrate synthase activity, although we recognize the possibility of a Type II error. 28 Key findings were obtained via interrogation of different muscle beds including the vastus lateralis for the muscle biopsy and the gastrocnemius for the in vivo SkM MRI‐based OxPhos estimation. While it would be preferable to make all assessments from the same location, prior work demonstrates histologic and biochemical similarities between the vastus lateralis and gastrocnemius muscles, 38 , 39 which is not surprising given that both are involved in locomotion. 40 We did not alter background medications prior to the exercise test or the SkM biopsy, and beta‐blocker use was more prevalent in our HFpEF participants. However, adjustment for beta‐blocker use did not attenuate the differences in peak VO2 between the groups (data not shown). A recent report specifically in HFpEF subjects noted that beta‐blockers were not associated with a lower peak VO2. 41 Additionally, disuse is known to cause reductions in mitochondrial proteins and respiration. 42 , 43 , 44 Our data cannot answer whether the impairments in SkM OxPhos are causative of the impaired exercise capacity or vice versa. Finally, while we did not find a decrease in SkM mass in HFpEF participants, the reduced SkM OxPhos on CrCEST, the reductions in the relative levels of OxPhos proteins, and the impaired cycle ergometry exercise capacity suggest an impairment in HFpEF muscle quality. 45 This should be the topic of future research.
In conclusion, we quantified a marked reduction in SkM OxPhos in HFpEF subjects, using a highly novel MRI imaging technique that has previously not been applied to cardiovascular disease patients. The imaging findings are supported by muscle tissue characterization, which collectively demonstrate a reduction in Type I myofibre percentage and global reductions in the levels of proteins involved in oxidative metabolism in HFpEF participants. Further research into interventions that modulate SkM OxPhos in HFpEF are warranted.
Conflict of interest
Dr. Zamani has consulted for Vyaire (modest). Dr. Mazurek has received advisory board honoraria from Actelion Pharmaceuticals (modest) and United Therapeutics (modest). Dr. Margulies receives research funding from Sanofi‐Aventis (significant), Merck (significant), and GlaxoSmithKline (significant). Dr. Kelly received advisory board honoraria from Pfizer (significant) and Amgen (modest). Dr. Ischiropoulos is the Gisela and Dennis Alter endowed chair. Dr. Chirinos has received consulting honoraria from Sanifit (significant), Microsoft (modest), Fukuda‐Denshi (modest), Bristol‐Myers Squibb (modest), OPKO Healthcare (modest), Ironwood Pharmaceuticals (modest), Pfizer (modest), Akros Pharma (modest), Merck (modest), Edwards Lifesciences (modest), and Bayer (significant). Dr. Chirinos has received research grants from Microsoft, Fukuda‐Denshi and Bristol‐Myers Squibb (all significant). He is named as inventor in an UPenn patent for the use of inorganic nitrates/nitrites for the treatment of HFpEF and a patent application for the use of novel neoepitope biomarkers of tissue fibrosis in heart failure.
Funding
Dr. Zamani is supported by National Institutes of Health grant (5‐K23‐HL130551). Dr. Margulies is supported by National Institutes of Health grants (U10‐HL110338, R01HL121510, and R01HL133080). Dr. Kelly is supported by National Institutes of Health grants (R01 DK045416, R01 HL058493, and R01 HL128349). Dr. Ischiropoulos is supported by National Institutes of Health grant (RO1 HL054926). Dr. Elrod is supported by National Institutes of Health grants (R01HL142271, R01HL136954, R01HL123966, and P01HL134608‐sub‐5483). Dr. Langham is supported by National Institutes of Health grants (U01 HD087180, R01 HL139358, R01 HL122754, and R21 EB022687). Dr. Poole is supported by National Institutes of Health grant (HL‐2‐108328). Dr. Reddy is supported by National Institutes of Health grant (P41 EB015893). Dr. Chirinos is supported by National Institutes of Health grants (R01‐HL 121510‐01A1, R61‐HL‐146390, R01‐AG058969, 1R01‐HL104106, P01‐HL094307, R03‐HL146874‐01, and R56‐HL136730).
Supporting information
Data S1. Supplemental Materials.
Figure S1. Factor 3 Protein Loadings. Principal component analysis identified 235 proteins that loaded predominately onto Factor 3 after varimax rotation. As in the figure, most proteins correlated positively (loading ≥0.4) with Factor 3. 171 of these proteins localized to the mitochondria, as determined by biologic pathway enrichment analysis, suggesting that Factor 3 largely represents mitochondrial properties.
Figure S2. Distribution of Factor 3 score by group. Participants with HFpEF had significantly lower Factor 3 scores compared to both Healthy and HTN participants. Pathway analysis of the proteins that load onto Factor 3 demonstrate significant mitochondrial localization (171 of the 235 proteins, FDR = 1.72e10−129) and enrichment in biologic pathways related to metabolic processes (FDR = 3.32e10−75) and energy production (FDR = 4.12e10−71). Data are presented as mean with 95% confidence intervals. ANOVA was used for the overall comparison, with post‐hoc intergroup comparisons performed with Bonferroni correction. * adjusted P < 0.05, ** adjusted P≤0.01, *** adjusted P≤0.001.
Figure S3. Association between Factor 3 score and Peak VO2, indexed to leg lean mass. Linear regression identified a significant association between Factor 3 score and Peak VO2, leg lean (beta coefficient = 16.4 [95% CI 7.7–25.1], P = 0.001). Factor 3 explained 33% of the variance in Peak VO2, leg lean.
Table S1. Demographics of participants (n = 34) who underwent the muscle biopsy procedure.
Table S2. – Electron Microscopy Quantification of Mitochondrial Content and Morphologic Features and Citrate Synthase Activity.
Table S3. Proteins with either a doubling or halving (abs log2 FC>1) of relative levels in the skeletal muscle of HFpEF vs. HTN participants.
Table S4. GeneOntology processes associated with the proteins with significantly different relative levels (abs log2FC > 1 and P < 0.05) between HFpEF and HTN participants.
Table S5. Top 10 biologic processes associated with all muscle proteins that demonstrated significantly different concentrations (P < 0.05) between HFpEF and Healthy participants.
Table S6. Proteins with either a doubling or halving of relative levels (abs log2 FC>1) in the skeletal muscle of HFpEF vs. Healthy participants.
Table S7. Principal Component Analysis Factors and Proteomic Variance Explained after Orthogonal Rotation.
Acknowledgements
This work was supported in part by the McCabe Fund at the University of Pennsylvania. The monoclonal antibody developed by Dr. Schiaffino (BA‐F8, SC‐71, BF‐35; University of Padova) and Dr. Lucas (6H1, University of Sydney) were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA. We would like to thank our subjects for volunteering for the studies described herein and the Center for Human Phenomic Sciences at the University of Pennsylvania where the studies were performed. The project described was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1TR001878. We would like to thank Dr. Franzini‐Armstrong, the CHOP Pathology Core Lab, and the UPenn Electron Microscopy Resource Lab for their collective assistance.
Zamani, P. , Proto, E. A. , Wilson, N. , Fazelinia, H. , Ding, H. , Spruce, L. A. , Davila, A. Jr. , Hanff, T. C. , Mazurek, J. A. , Prenner, S. B. , Desjardins, B. , Margulies, K. B. , Kelly, D. P. , Arany, Z. , Doulias, P.‐T. , Elrod, J. W. , Allen, M. E. , McCormack, S. E. , Schur, G. M. , D'Aquilla, K. , Kumar, D. , Thakuri, D. , Prabhakaran, K. , Langham, M. C. , Poole, D. C. , Seeholzer, S. H. , Reddy, R. , Ischiropoulos, H. , and Chirinos, J. A. (2021) Multimodality assessment of heart failure with preserved ejection fraction skeletal muscle reveals differences in the machinery of energy fuel metabolism. ESC Heart Failure, 8: 2698–2712. 10.1002/ehf2.13329.
References
- 1. Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 2017; 14: 591–602. [DOI] [PubMed] [Google Scholar]
- 2. Cohen JB, Schrauben SJ, Zhao L, Basso MD, Cvijic ME, Li Z, Yarde M, Wang Z, Bhattacharya PT, Chirinos DA, Prenner S, Zamani P, Seiffert DA, Car BD, Gordon DA, Margulies K, Cappola T, Chirinos JA. Clinical phenogroups in heart failure with preserved ejection fraction: detailed phenotypes, prognosis, and response to spironolactone. JACC Heart Fail 2020; 8: 172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Houstis NE, Eisman AS, Pappagianopoulos PP, Wooster L, Bailey CS, Wagner PD, Lewis GD. Exercise intolerance in heart failure with preserved ejection fraction: diagnosing and ranking its causes using personalized O2 pathway analysis. Circulation 2018; 137: 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kogan F, Haris M, Debrosse C, Singh A, Nanga RP, Cai K, Hariharan H, Reddy R. In vivo chemical exchange saturation transfer imaging of creatine (CrCEST) in skeletal muscle at 3T. J Magn Reson Imaging: JMRI. [Research Support, N.I.H., Extramural] 2014; 40: 596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haris M, Singh A, Cai K, Kogan F, McGarvey J, Debrosse C, Zsido GA, Witschey WR, Koomalsingh K, Pilla JJ, Chirinos JA, Ferrari VA, Gorman JH, Hariharan H, Gorman RC, Reddy R. A technique for in vivo mapping of myocardial creatine kinase metabolism. Nat Med. [Research Support, N.I.H., Extramural Research Support, Non‐U.S. Gov't] 2014; 20: 209–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DeBrosse C, Nanga RP, Wilson N, D'Aquilla K, Elliott M, Hariharan H, Yan F, Wade K, Nguyen S, Worsley D, Parris‐Skeete C, McCormick E, Xiao R, Cunningham ZZ, Fishbein L, Nathanson KL, Lynch DR, Stallings VA, Yudkoff M, Falk MJ, Reddy R, McCormack SE. Muscle oxidative phosphorylation quantitation using creatine chemical exchange saturation transfer (CrCEST) MRI in mitochondrial disorders. JCI Insight 2016; 1: e88207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zamani P, Proto EA, Mazurek JA, Prenner SB, Margulies KB, Townsend RR, Kelly DP, Arany Z, Poole DC, Wagner PD, Chirinos JA. Peripheral determinants of oxygen utilization in heart failure with preserved ejection fraction: central role of adiposity. JACC Basic Transl Sci 2020; 5: 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baumgartner RN, Koehler KM, Gallagher D, Romero L, Heymsfield SB, Ross RR, Garry PJ, Lindeman RD. Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol 1998; 147: 755–763. [DOI] [PubMed] [Google Scholar]
- 9. Guazzi M, Adams V, Conraads V, Halle M, Mezzani A, Vanhees L, Arena R, Fletcher GF, Forman DE, Kitzman DW, Lavie CJ, Myers J. European Association for Cardiovascular P, Rehabilitation, American Heart A. EACPR/AHA Scientific Statement. Clinical recommendations for cardiopulmonary exercise testing data assessment in specific patient populations. Circulation 2012; 126: 2261–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zamani P, Rawat D, Shiva‐Kumar P, Geraci S, Bhuva R, Konda P, Doulias PT, Ischiropoulos H, Townsend RR, Margulies KB, Cappola TP, Poole DC, Chirinos JA. Effect of inorganic nitrate on exercise capacity in heart failure with preserved ejection fraction. Circulation 2015; 131: 371–380 discussion 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mezzani A. Cardiopulmonary exercise testing: Basics of methodology and measurements. Ann Am Thorac Soc 2017; 14: S3–S11. [DOI] [PubMed] [Google Scholar]
- 12. Kuster DW, Merkus D, Jorna HJ, Dekkers DH, Duncker DJ, Verhoeven AJ. Nuclear protein extraction from frozen porcine myocardium. J Physiol Biochem 2011; 67: 165–173. [DOI] [PubMed] [Google Scholar]
- 13. Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, Burgess MW, Gillette MA, Jaffe JD, Carr SA. Integrated proteomic analysis of post‐translational modifications by serial enrichment. Nat Methods 2013; 10: 634–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dominguez S, Rodriguez G, Fazelinia H, Ding H, Spruce L, Seeholzer SH, Dong H. Sex differences in the phosphoproteomic profiles of APP/PS1 mice after chronic unpredictable mild stress. J Alzheimers Dis 2020; 74: 1131–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McNulty DE, Annan RS. Hydrophilic interaction chromatography reduces the complexity of the phosphoproteome and improves global phosphopeptide isolation and detection. Mol Cell Proteomics 2008; 7: 971–980. [DOI] [PubMed] [Google Scholar]
- 16. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta‐Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV. STRING v11: protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res 2019; 47: D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel‐Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium Nat Genet 2000; 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. The Gene Ontology C . The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 2019; 47: D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Pérez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yılmaz Ş, Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A, Vizcaíno JA. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019; 47: D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang S, Cai Y, Cheng J, Li W, Liu Y, Yang H. motifeR: an integrated web software for identification and visualization of protein posttranslational modification motifs. Proteomics 2019; 19: e1900245. [DOI] [PubMed] [Google Scholar]
- 21. Miller CJ, Turk BE. Homing in: mechanisms of substrate targeting by protein kinases. Trends Biochem Sci 2018; 43: 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Amanchy R, Periaswamy B, Mathivanan S, Reddy R, Tattikota SG, Pandey A. A curated compendium of phosphorylation motifs. Nat Biotechnol 2007; 25: 285–286. [DOI] [PubMed] [Google Scholar]
- 23. Gonzalez FA, Raden DL, Davis RJ. Identification of substrate recognition determinants for human ERK1 and ERK2 protein kinases. J Biol Chem 1991; 266: 22159–22163. [PubMed] [Google Scholar]
- 24. Roskoski R Jr. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 2012; 66: 105–143. [DOI] [PubMed] [Google Scholar]
- 25. Weiss K, Schar M, Panjrath GS, Zhang Y, Sharma K, Bottomley PA, Golozar A, Steinberg A, Gerstenblith G, Russell SD, Weiss RG. Fatigability, exercise intolerance, and abnormal skeletal muscle energetics in heart failure. Circ Heart Fail 2017; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kitzman DW, Nicklas B, Kraus WE, Lyles MF, Eggebeen J, Morgan TM, Haykowsky M. Skeletal muscle abnormalities and exercise intolerance in older patients with heart failure and preserved ejection fraction. Amer J Physiol Heart Circ Physiol. [Research Support, N.I.H., Extramural] 2014; 306: H1364–H1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gifford JR, Garten RS, Nelson AD, Trinity JD, Layec G, Witman MA, Weavil JC, Mangum T, Hart C, Etheredge C, Jessop J, Bledsoe A, Morgan DE, Wray DW, Rossman MJ, Richardson RS. Symmorphosis and skeletal muscle VO2 max: in vivo and in vitro measures reveal differing constraints in the exercise‐trained and untrained human. J Physiol 2016; 594: 1741–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Molina AJ, Bharadwaj MS, Van Horn C, Nicklas BJ, Lyles MF, Eggebeen J, Haykowsky MJ, Brubaker PH, Kitzman DW. Skeletal muscle mitochondrial content, oxidative capacity, and Mfn2 expression are reduced in older patients with heart failure and preserved ejection fraction and are related to exercise intolerance. JACC Heart Fail 2016; 4: 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bekfani T, Bekhite Elsaied M, Derlien S, Nisser J, Westermann M, Nietzsche S, Hamadanchi A, Frob E, Westphal J, Haase D, Kretzschmar T, Schlattmann P, Smolenski UC, Lichtenauer M, Wernly B, Jirak P, Lehmann G, Mobius‐Winkler S, Schulze PC. Skeletal muscle function, structure, and metabolism in patients with heart failure with reduced ejection fraction and heart failure with preserved ejection fraction. Circ Heart Fail 2020; 13: e007198. [DOI] [PubMed] [Google Scholar]
- 30. Ubaida‐Mohien C, Lyashkov A, Gonzalez‐Freire M, Tharakan R, Shardell M, Moaddel R, Semba RD, Chia CW, Gorospe M, Sen R, Ferrucci L. Discovery proteomics in aging human skeletal muscle finds change in spliceosome, immunity, proteostasis and mitochondria. Elife 2019; 23: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Potts GK, McNally RM, Blanco R, You JS, Hebert AS, Westphall MS, Coon JJ, Hornberger TA. A map of the phosphoproteomic alterations that occur after a bout of maximal‐intensity contractions. J Physiol 2017; 595: 5209–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boyer JG, Prasad V, Song T, Lee D, Fu X, Grimes KM, Sargent MA, Sadayappan S, Molkentin JD. ERK1/2 signaling induces skeletal muscle slow fiber‐type switching and reduces muscular dystrophy disease severity. JCI Insight 2019; 9: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kramer HF, Goodyear LJ. Exercise, MAPK, and NF‐κB signaling in skeletal muscle. J Appl Physiol 2007; 103: 388–395. [DOI] [PubMed] [Google Scholar]
- 34. Arany Z, Foo SY, Ma Y, Ruas JL, Bommi‐Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, Baek KH, Rosenzweig A, Spiegelman BM. HIF‐independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC‐1α. Nature 2008; 451: 1008–1012. [DOI] [PubMed] [Google Scholar]
- 35. Rowe GC, Jiang A, Arany Z. PGC‐1 coactivators in cardiac development and disease. Circ Res 2010; 107: 825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, Williams RS, Yan Z. Exercise stimulates Pgc‐1α transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem 2005; 280: 19587–19593. [DOI] [PubMed] [Google Scholar]
- 37. Webb‐Robertson BJ, Wiberg HK, Matzke MM, Brown JN, Wang J, McDermott JE, Smith RD, Rodland KD, Metz TO, Pounds JG, Waters KM. Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry‐based label‐free global proteomics. J Proteome Res 2015; 14: 1993–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Green HJ, Daub B, Houston ME, Thomson JA, Fraser I, Ranney D. Human vastus lateralis and gastrocnemius muscles. A comparative histochemical and biochemical analysis. J Neurol Sci 1981. ‐Dec; 52: 201–210. [DOI] [PubMed] [Google Scholar]
- 39. Gollnick PD, Sjodin B, Karlsson J, Jansson E, Saltin B. Human soleus muscle: a comparison of fiber composition and enzyme activities with other leg muscles. Pflugers Arch 1974; 348: 247–255. [DOI] [PubMed] [Google Scholar]
- 40. Brandell BR. Functional roles of the calf and vastus muscles in locomotion. Am J Phys Med 1977; 56: 59–74. [PubMed] [Google Scholar]
- 41. Maldonado‐Martin S, Brubaker PH, Ozemek C, Jayo‐Montoya JA, Becton JT, Kitzman DW. Impact of beta‐blockers on heart rate and oxygen uptake during exercise and recovery in older patients with heart failure with preserved ejection fraction. J Cardiopulm Rehabil Prev 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Trevino MB, Zhang X, Standley RA, Wang M, Han X, Reis FCG, Periasamy M, Yu G, Kelly DP, Goodpaster BH, Vega RB, Coen PM. Loss of mitochondrial energetics is associated with poor recovery of muscle function but not mass following disuse atrophy. Am J Physiol Endocrinol Metab 2019; 317: E899–E910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Standley RA, Distefano G, Trevino MB, Chen E, Narain NR, Greenwood B, Kondakci G, Tolstikov VV, Kiebish MA, Yu G, Qi F, Kelly DP, Vega RB, Coen PM, Goodpaster BH. Skeletal muscle energetics and mitochondrial function are impaired following 10 days of bed rest in older adults. J Gerontol A Biol Sci Med Sci 2020; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ubaida‐Mohien C, Gonzalez‐Freire M, Lyashkov A, Moaddel R, Chia CW, Simonsick EM, Sen R, Ferrucci L. Physical activity associated proteomics of skeletal muscle: being physically active in daily life may protect skeletal muscle from aging. Front Physiol 2019; 10: 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barbat‐Artigas S, Rolland Y, Zamboni M, Aubertin‐Leheudre M. How to assess functional status: a new muscle quality index. J Nutr Health Aging 2012; 16: 67–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Materials.
Figure S1. Factor 3 Protein Loadings. Principal component analysis identified 235 proteins that loaded predominately onto Factor 3 after varimax rotation. As in the figure, most proteins correlated positively (loading ≥0.4) with Factor 3. 171 of these proteins localized to the mitochondria, as determined by biologic pathway enrichment analysis, suggesting that Factor 3 largely represents mitochondrial properties.
Figure S2. Distribution of Factor 3 score by group. Participants with HFpEF had significantly lower Factor 3 scores compared to both Healthy and HTN participants. Pathway analysis of the proteins that load onto Factor 3 demonstrate significant mitochondrial localization (171 of the 235 proteins, FDR = 1.72e10−129) and enrichment in biologic pathways related to metabolic processes (FDR = 3.32e10−75) and energy production (FDR = 4.12e10−71). Data are presented as mean with 95% confidence intervals. ANOVA was used for the overall comparison, with post‐hoc intergroup comparisons performed with Bonferroni correction. * adjusted P < 0.05, ** adjusted P≤0.01, *** adjusted P≤0.001.
Figure S3. Association between Factor 3 score and Peak VO2, indexed to leg lean mass. Linear regression identified a significant association between Factor 3 score and Peak VO2, leg lean (beta coefficient = 16.4 [95% CI 7.7–25.1], P = 0.001). Factor 3 explained 33% of the variance in Peak VO2, leg lean.
Table S1. Demographics of participants (n = 34) who underwent the muscle biopsy procedure.
Table S2. – Electron Microscopy Quantification of Mitochondrial Content and Morphologic Features and Citrate Synthase Activity.
Table S3. Proteins with either a doubling or halving (abs log2 FC>1) of relative levels in the skeletal muscle of HFpEF vs. HTN participants.
Table S4. GeneOntology processes associated with the proteins with significantly different relative levels (abs log2FC > 1 and P < 0.05) between HFpEF and HTN participants.
Table S5. Top 10 biologic processes associated with all muscle proteins that demonstrated significantly different concentrations (P < 0.05) between HFpEF and Healthy participants.
Table S6. Proteins with either a doubling or halving of relative levels (abs log2 FC>1) in the skeletal muscle of HFpEF vs. Healthy participants.
Table S7. Principal Component Analysis Factors and Proteomic Variance Explained after Orthogonal Rotation.