

Abstract
Aims
Myeloid differentiation protein 1 (MD1) was shown to ameliorate pressure overload‐induced cardiac hypertrophy and fibrosis by negatively regulating the MEK–ERK1/2 and NF‐κB pathways. However, whether MD1 modulates cardiac function and whether the Akt pathway mediates the benefits of MD1 in pressure overload‐induced cardiac remodelling remain unclear.
Methods and Results
Male cardiac‐specific transgenic MD1 (MD1‐TG) mice, MD1‐knockout (KO) mice and wild‐type (WT) littermates aged 8–10 weeks were subjected to sham operation and aortic banding (AB) for 4 weeks. Then, left ventricular (LV) hypertrophy, fibrosis and function of the mice were assessed. When compared with WT‐AB mice, MD1‐TGs showed decreased cross‐sectional area (CSA) of cardiomyocytes (P < 0.001), mRNA expression of β‐myosin heavy chain (β‐MHC) (P < 0.02), ratios of heart weight/body weight and heart weight/tibia length (P < 0.04) and collagen volume fraction (P < 0.001). The LV end‐diastolic diameter was reduced, and LV ejection fraction and fractional shortening were improved in MD1‐TG‐AB mice than in WT‐AB mice (P < 0.05). In cultured H9C2 cells, adenovirus vector‐mediated MD1 overexpression decreased angiotensin II‐induced mRNA expression of brain natriuretic peptide (BNP) and β‐MHC and cell CSA (P < 0.002), whereas knockdown of MD1 by shRNA exhibited opposite effects (P < 0.04). Mechanistically, MD1 suppressed pathological cardiac remodelling at least partly by blocking Akt pathway. Akt inactivation by MK2206 largely offset the pro‐hypertrophic effects of MD1 deficiency in angiotensin II‐stimulated cardiomyocytes.
Conclusions
The Akt pathway mediates the protective effects of MD1 in pressure overload‐induced cardiac remodelling in mice. Targeting MD1 may provide therapeutic strategy for the treatment of pathological cardiac remodelling and heart failure.
Keywords: Myeloid differentiation protein 1, Toll‐like receptor 4, Pathological cardiac remodelling, Heart failure, Akt signalling pathway
Introduction
Arrhythmias and heart failure (HF) caused by pathological cardiac remodelling are major underlying causes of increased cardiovascular morbidity and mortality. Therefore, a better understanding of the factors and mechanisms that modulate pathological cardiac remodelling is crucial. It has been suggested that pathological cardiac remodelling is a self‐perpetuating response of the injured and structurally altered myocardium and cardiac vasculature to long‐standing noxious haemodynamic, metabolic and inflammatory stimuli. 1 There is accumulating evidence to suggest that the Toll‐like receptor 4 (TLR4) participates in the initiation, integration and perpetuation of myocardial injury. 2 Thus, regulating the TLR4 signalling pathway may be an effective strategy for the prevention and treatment of pathological cardiac remodelling.
Myeloid differentiation protein 1 (MD1) is a secreted glycoprotein that forms a complex with radioprotective 105 (RP105), 3 , 4 which is abundantly present in cardiac tissue. MD1‐RP105 can directly interact with MD2‐TLR4 through lateral binding or by forming a tetrameric complex (MD2‐TLR4/MD1‐RP105) with MD2‐TLR4, resembling the usual ligand‐induced TLR homodimers. 5 , 6 These complexes act as physiologically negative regulators of TLR4 signalling. 5 , 6 Our previous research demonstrated that MD1 is downregulated in failing human and murine hearts and can ameliorate aortic banding (AB)‐induced cardiac hypertrophy and fibrosis during the remodelling process by negatively regulating the MEK–ERK 1/2 and NF‐κB signalling pathways. 7 Besides, a recent study indicated that MD1 deficiency exacerbates maladaptive left atrial fibrosis and inflammation. 8 These findings suggest a beneficial effect of MD1 in adverse cardiac remodelling. The Akt pathway, in addition to the MAPK and NF‐κB pathways, is also a downstream signalling cascade of TLR4. 9 , 10 However, whether the Akt pathway is involved in the regulation of MD1 in cardiac remodelling and whether MD1 regulates cardiac function under chronic pressure overload conditions remain unclear.
In the present study, we demonstrated that the pressure overload‐induced cardiac remodelling was limited and cardiac dysfunction was significantly improved in cardiac‐specific MD1 transgenic (MD1‐TG) mice, whereas these were exaggerated in global MD1‐knockout (KO) mice. 11 We further discovered that MD1‐mediated effects were dependent, at least partly, on the negative regulation of the Akt pathway both in vivo and in vitro. Overall, our data suggest potential therapy strategy and target for the treatment of adverse cardiac remodelling and HF.
Methods
Experimental animals
Male MD1‐TG mice (n = 25) and their wild‐type (WT) littermates (n = 20) as well as male global MD1‐KO mice (n = 20) and their WT littermates (n = 20) aged 8–10 weeks were used for experiments. Detailed information about the experimental animals ( Data S1 ) is described elsewhere. 7 , 11 All animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (Publication No. 85‐23, revised 1996) and approved by the Animal Care and Use Committee of the University of South China.
Animal model and echocardiography
AB surgery and sham operations ( Data S1 ) were performed as described elsewhere. 11 We performed echocardiography ( Data S1 ) as described previously 11 to obtain the following values: LV ejection fraction (LVEF), LV fractional shortening (LVFS), LV end‐diastolic diameter (LVEDD), LV end‐systolic diameter (LVESD), interventricular septum diameter in diastole and systole (IVSd and IVSs, respectively) and LV posterior wall diameter in diastole and systole (LVPWd and LVPWs, respectively). At the end of these procedures, some mice in the indicated groups were euthanized using an overdose of pentobarbital sodium (150 mg/kg, intraperitoneal injection), and ratios of heart weight (HW)/body weight (BW), HW/tibia length (TL) and lung weight (LW)/TL of the sacrificed mice from the tested groups were assessed. The LV tissues from some hearts were dissected, snap‐frozen in liquid nitrogen and stored at −80°C for biochemical studies. All procedures were performed in a blinded fashion for all groups.
Histological analysis
Hearts were excised, washed with saline solution and arrested in diastole (10%) KCl, fixed in 4% paraformaldehyde solution and embedded in paraffin. Paraffin‐embedded hearts were sectioned transversely at the level of the LV papillary muscles. Several myocardial slices (4–5 mm thick) were prepared and stained with picrosirius red (PSR) for evaluation of myocardial fibrosis and haematoxylin–eosin (H&E) for morphometric analysis. All micrographs were obtained using a high‐resolution optical microscope. Image‐Pro Plus 6.0 software was used to determine the cross‐sectional area (CSA) of cardiomyocytes and the volume of LV collagen deposition.
Adenoviral vector infection and H9C2 culture
Four kinds of adenoviruses (AdGFP, AdMD1, AdshRNA and AdshMD1) were produced by HanBio, as previously described. 7 We then infected H9C2 cardiomyocytes (purchased from Shanghai Institute of Biochemistry and Cell Biology, Shanghai, China) with AdshRNA or AdshMD1, as well as AdGFP or AdMD1, at a multiplicity of infection of 100. The results showed that more than 95% of the cells expressed the transgenes with no detectable toxic effects.
H9C2 cells were cultured in DMEM/F12 medium containing 10% FBS, 100 U/mL penicillin and 100 mg/mL streptomycin in a humidified atmosphere of 5% CO2, at 37°C. After 48 h, the culture medium was replaced with serum‐free DMEM/F12 containing 0.1% FBS, and the cells were incubated for 12 h. Then, cells were infected with the adenoviruses and simultaneously stimulated with human Ang II (# 05‐23‐0101‐5MGCN; Merck; 1 μM) in DMEM/F12 medium for 24 h or 3 h. In addition, the cells infected with AdshMD1 and stimulated with Ang II (1 μM) simultaneously in DMEM/F12 medium or in DMEM/F12 medium containing MK‐2206 (# S1078; Selleckchem; 50 μM) for 3 h. The expression of specific mRNAs in the cultured cells was determined by quantitative real‐time PCR (qRT‐PCR), and protein synthesis was evaluated by western blotting. To obtain CSA measurements, the cells were stained with α‐actin and DAPI. Micrographs were obtained, and the surface areas of the cells were measured using Image‐Pro Plus 6.0 software.
QRT‐PCR
Total RNA was extracted from mouse LV tissues and reverse transcribed to generate cDNA. qRT‐PCR was performed using an ABI‐PRISM 7900 Sequence Detection System and the SYBR Green One‐Step qRT‐PCR Kit (Beyotime Biotechnology, D7268M). The primers used for the detection of brain natriuretic peptide (BNP) and β‐myosin heavy chain (β‐MHC) were as follows: BNP: forward, 5′‐GAGGTCACTCCTATCCTCTGG‐3′; BNP: reverse, 5′‐GCCATTTCCTCCGACTTTTCTC‐3′; β‐MHC: forward, 5′‐CCGAGTCCCAGGTCAACAA‐3′; β‐MHC: reverse, 5′‐CTTCACGGGCACCCTTGGA‐3′. Gene expression was normalized relative to the housekeeping gene GAPDH, and data were analysed according to the 2−∆∆Ct method. 12 The method details ( Data S1 ) were described in the previous literature. 11
Western blotting
The western blotting method details ( Data S1 ) are described in the previous literature. 11 Primary antibodies were purchased as follows: AKT (Cell Signalling, 4691); p‐Ser473‐AKT (Cell Signalling, 4060); mTOR (Bioworld, BS3611); p‐Ser2448‐mTOR (Bioworld, BS4706); GSK3β (Abcam, ab32391); p‐Ser9‐GSK3β (Abcam, ab131097); and GAPDH (Cell Signalling, 5174S).
Statistical analyses
Statistical analyses were performed using SPSS19.0 or GraphPad Prism software. Data are expressed as mean ± SEM. Statistical significance was determined by one‐way analysis of variance (ANOVA). Multiple comparisons were performed using the Bonferroni‐corrected t‐test when the variance was equal, whereas Dunnett's T3 test was used when the variance was unequal. A P < 0.05 was considered statistically significant.
Results
Effects of MD1 overexpression in mice with AB‐induced hypertrophy
Under basal conditions, MD1‐TG mice showed no alterations in the cardiac phenotype (Table S2 ). However, 4 weeks after AB, MD1‐TG mice exhibited less LV hypertrophy as compared with their WT littermates. This was confirmed by the measurement of a relatively smaller CSA in the TG mice, as revealed by H&E staining (Figure 1 A,H ), and lower HW/BW and HW/TL ratios (Figure 1 F,G ; Table S1 ). The mRNA levels of β‐MHC was markedly lower in the LV tissues of TG mice than in those of the WT mice (Figure 1 B ). Furthermore, MD1‐TG mice exhibited decreased LV dilation and dysfunction, as shown by measurements of the following echocardiographic parameters: LVEDD, LVEF and LVFS (Figure 1 C,E ; Table S1 ). Cardiac fibrosis, a major feature of adverse cardiac remodelling, was less visible in MD1‐TG‐AB mice than in WT‐AB mice (Figure 1 D,I ). Collectively, these data indicate that cardiac MD1 inhibits pressure overload‐induced cardiac remodelling.
Figure 1.
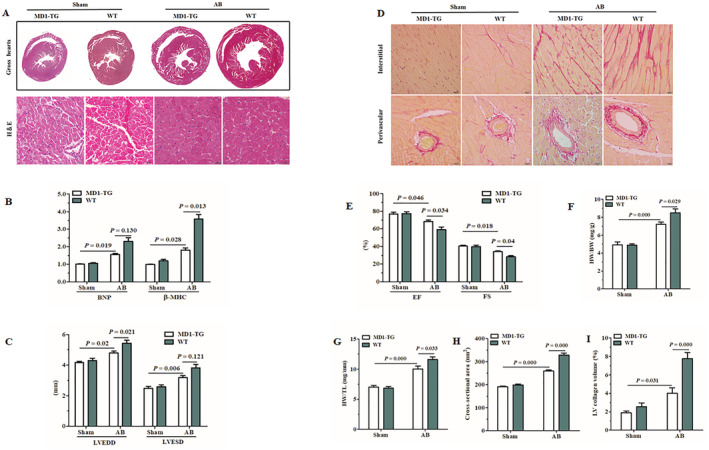
Effects of MD1 overexpression in mice with AB‐induced cardiac pathological remodelling. (A) Gross hearts and H&E staining were performed 4 weeks after sham or AB surgery (n = 6–7). (B) Expression of the hypertrophic markers was determined by qRT‐PCR in MD1‐TG and WT left ventricles 4 weeks after surgery (n = 4). (C,E) Echocardiographic results of the indicated groups (n = 8–9). (D) Staining of PSR on the histological sections of the LV were performed on indicated groups 4 weeks after surgery (n = 6–7). (F,G) HW/BW and HW/TL values of the indicated groups (n = 8–9). (H) LV myocyte CSAs (n = 150 + cells) after sham or AB surgery. (I) Fibrotic areas from histological sections were quantified using an image‐analysis system (n = 22–24 fields). The P‐values are presented above the connection lines.
MD1 negatively regulates the cardiac Akt pathway in response to chronic pressure overload
The involvement of the MAPK and NF‐κB signalling pathways in MD1‐mediated cardioprotective effects has been illustrated in our previous work. 7 We therefore investigated Akt signalling, which is known to be involved in pathological cardiac hypertrophy. Our western blot analyses showed that AB‐induced activation of the Akt pathway was more pronounced in KO mice than in WT mice, as evidenced by a significant increase in Akt, GSK3β and mTOR phosphorylation levels in KO hearts compared with WT hearts (Figure 2 A ). Conversely, overexpression of MD1 dramatically reduced the levels of Akt, GSK3β and mTOR phosphorylation, compared with those of WTs after AB (Figure 2 B ). These results suggest that MD1 may exert its anti‐cardiac remodelling effects by suppressing Akt signalling activation.
Figure 2.
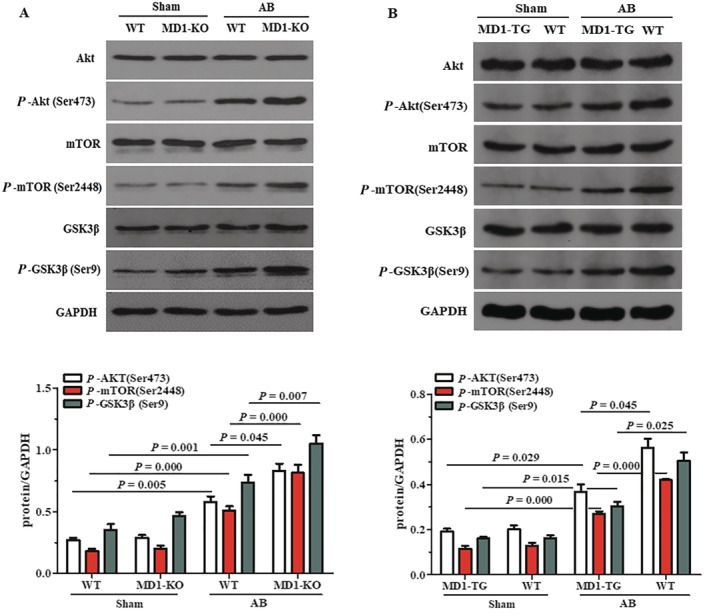
Effects of MD1 on the AKT signalling in response to pressure overload. (A) Representative western blots and quantitative results showing the phosphorylation and total protein levels of AKT, mTOR and GSK3β 4 weeks after sham or AB surgery in WT and MD1‐KO mice (n = 5). (B) Representative western blots and quantitative results showing the phosphorylation and total protein levels of AKT, mTOR and GSK3β 4 weeks after sham or AB surgery in MD1‐TG and WT mice (n = 4). The P‐values are presented above the connection lines.
MD1 partially suppressed Ang II‐induced cardiomyocyte hypertrophy
Next, we determined the anti‐hypertrophic effect of MD1 in vitro using cultured H9C2 cells. Because the levels of MD1 and RP105 were reduced by infection of neonatal rat cardiomyocytes with AdshMD1 or elevated by infection with AdMD1, 7 we omitted this experiment in the present study. The experiments revealed that knockdown or overexpression of MD1 did not alter the size of H9C2 cells under basal conditions (PBS). However, upon exposure to Ang II for 24 h, we observed that Ang II‐induced hypertrophy was aggravated in MD1‐knockdown cells (Figure 3 A , above), whereas it was alleviated in MD1‐overexpressing cells (Figure 3 A , below), as measured by CSA (Figure 3 C,B ) and the mRNA levels of hypertrophy markers (BNP and β‐MHC) (Figure 3 E,D ). These results indicate that MD1 partially suppressed Ang II‐induced cardiomyocyte hypertrophy in vitro.
Figure 3.
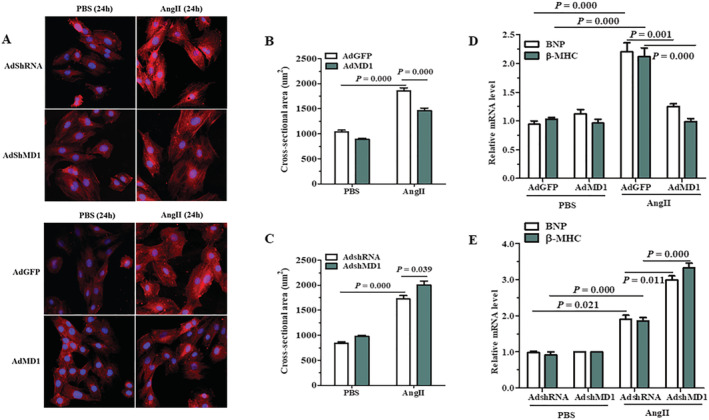
Effects of MD1 on cardiomyocyte hypertrophy in vitro. (A) Representative images of H9C2 cells infected with AdshRNA or AdshMD1, as well as AdGFP or AdMD1, in response to Ang II. (B,C) Quantitative results of CSA in the indicated groups. More than 100 cells were analysed for each group. (D,E) Expression of the hypertrophic markers was determined by qRT‐PCR in H9C2 cells infected with AdGFP or AdMD1 (D) and AdshRNA or AdshMD1 (E) following treatment with PBS or Ang II for 3 h. n = 3, P‐values are shown above the connection lines between compared columns.
MD1 negatively regulates the Akt pathway in Ang II‐stimulated cardiomyocytes
The in vitro experiments confirmed the inhibitory effects of MD1 on Akt signalling. As shown in Figure 4 A,B , the overexpression of MD1 in H9C2 cells, by AdMD1, markedly suppressed the phosphorylation of Akt, GSK3β and mTOR. However, knockdown of MD1 in H9C2 cells, by AdshMD1, enhanced levels of Akt, GSK3β and mTOR phosphorylation in response to Ang II, compared with control H9C2 cells.
Figure 4.
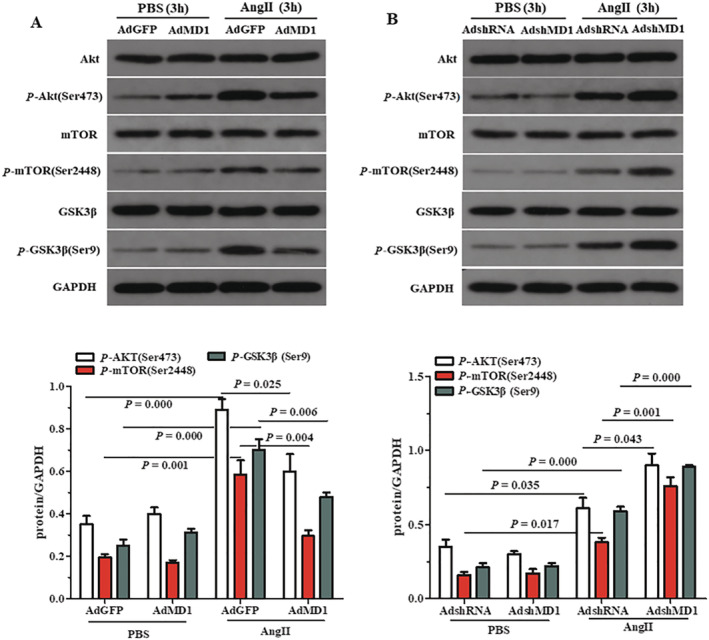
Effects of MD1 on AKT signalling in Ang II‐stimulated cardiomyocytes. (A) Representative blots and quantitative results showing the phosphorylation and total protein levels of AKT, mTOR and GSK3β after infection with AdGFP or AdMD1 in response to PBS or Ang II. (B) Representative blots and quantitative results showing the phosphorylation and total protein levels of AKT, mTOR and GSK3β after infection with AdshRNA or AdshMD1 in response to PBS or Ang II. n = 3, P‐values are shown above the connection lines between compared columns.
Inactivation of Akt signalling reverses the adverse effects of MD1 deficiency in Ang II‐stimulated cardiomyocytes
These aforementioned results indicate that inactivation of Akt reverses the accelerated hypertrophy induced by MD1 knockdown. To test this hypothesis, we exposed cultured H9C2 cells, which had been previously co‐cultivated with AdshMD1, to an Akt inhibitor, MK2206, and then added Ang II for 3 h. Our data revealed that Akt signalling in AdshMD1‐infected H9C2 cells was significantly downregulated by MK2206 treatment, under basal conditions (PBS) and Ang II stimulation, as evidenced by a significant decrease in Akt, GSK3β and mTOR phosphorylation levels in the MK2206 treatment group, compared with the control (Figure 5 B,C ). Analysis of the mRNA levels of hypertrophy markers (BNP and β‐MHC) suggested that MK2206 treatment completely reversed the accelerative effects of MD1 deficiency on hypertrophy of Ang II‐treated H9C2 cells, compared with that of PBS‐treated controls (Figure 5 A ). Collectively, these data indicate that the regulatory role of MD1 in pathological cardiac remodelling may be dependent, at least partly, on the inactivation of Akt.
Figure 5.
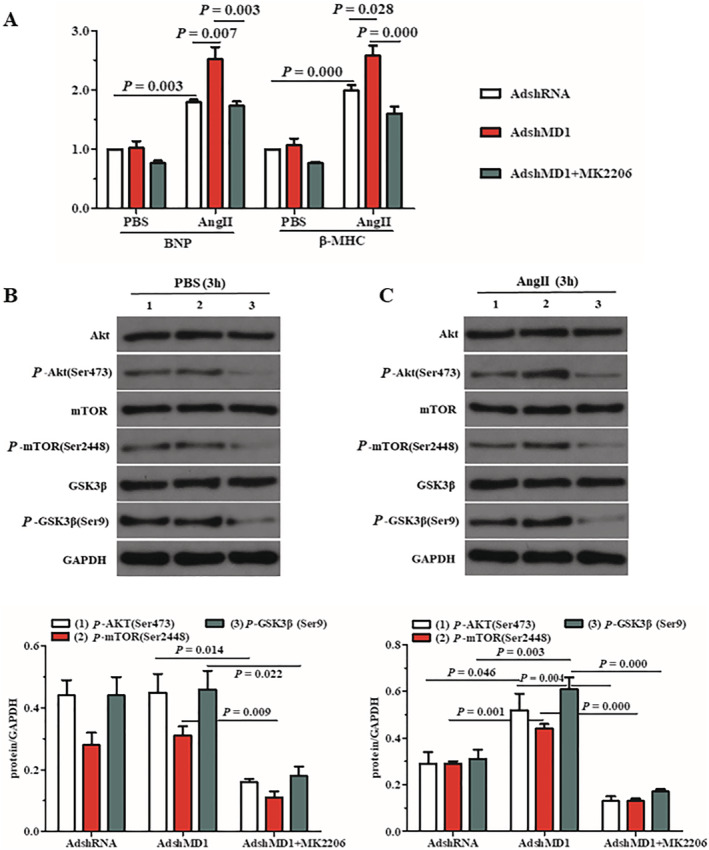
Inactivation of AKT signalling rescues the pro‐hypertrophic effects of MD1 deficiency in Ang II‐treated cardiomyocytes. (A) mRNA levels of hypertrophy markers in H9C2 cells treated with AdshMD1 and MK2206, in response to Ang II, were determined using qRT‐PCR. PBS was used as the control solution. (B,C) Representative blots and quantitative results showing the phosphorylation and total protein levels of AKT, mTOR and GSK3β following infection with AdshMD1, in PBS (B) and Ang II (C) treatments, in the absence or presence of MK2206. Cells infected with AdshRNA were used as controls. n = 3, P‐values are shown above the connection lines between compared columns.
Discussion
In the present study, we revealed that AB‐treated MD1‐TG hearts showed less cardiac hypertrophy and a significant attenuation of cardiac fibrosis and dysfunction as compared with their WTs, whereas these were exaggerated in AB‐treated MD1‐KO hearts. 11 Moreover, both in vivo and in vitro data consistently indicate that MD1‐mediated inhibition of adverse cardiac remodelling may be dependent, at least partly, on the inactivation of the Akt pathway. Thus, MD1 may be a modulator of adverse cardiac remodelling and HF and that upregulation of MD1 in heart would be a good therapeutic strategy for treating these pathological conditions. However, these findings need to be evaluated under other stressful insults such as ischemia, ageing and hyperglycaemia.
Sustained activation of TLR4 signalling following cardiac injury and noxious stress is harmful and can lead to adverse cardiac remodelling and finally to an HF phenotype. 2 Upon stimulation, TLR4 serves as a docking site for downstream signalling cascades, resulting in inflammation, cell apoptosis or survival, cellular growth and hypertrophy. 2 , 10 , 13 , 14 More importantly, disruption of or therapies targeting TLR4 attenuate cardiac remodelling and dysfunction following numerous pathological stimuli. 2 , 15 , 16 , 17 , 18 , 19 These studies suggest that blocking TLR4 signalling suppresses maladaptive cardiac remodelling and improves cardiac function. MD1, an endogenous negative modulator of TLR4 pathway, was shown to protect against AB‐induced cardiac hypertrophy and fibrosis. 7 Consistent with this, our present results indicate that ratios of HW/BW, HW/TL, mRNA level of β‐MHC, CSA, LVEDD and LV collagen volumes of MD1‐TG mice were significantly decreased compared with those of WT mice 4 weeks after AB. Moreover, the LVEF and LVFS of MD1‐TG mice were markedly increased compared with those of WT mice 4 weeks after AB, indicating that MD1 improves cardiac dysfunction. On the other hand, MD1‐KO mice displayed the opposite phenotype. 11 In the in vitro experiments, we also observed that MD1 attenuated Ang II‐induced myocyte hypertrophy. Taken together, these data suggest that MD1 negatively regulates cardiac remodelling and ameliorates cardiac function in response to long‐standing pressure overload.
Besides the MAPK and NF‐κB pathways, the Akt–mTOR pathway also plays a role in the progression of cardiac hypertrophy, and it seems to be a potential therapeutic target for treating human diseases associated with cardiac hypertrophy. 20 , 21 In addition, activation of Akt leads to inhibition of GSK3β by phosphorylating serine 9 residues of GSK3β, which also causes pathological hypertrophy. 14 , 22 , 23 Therefore, to investigate whether the Akt pathway, a downstream signalling cascade of TLR4, mediates the anti‐cardiac hypertrophy effects of MD1 in response to chronic pressure overload, we examined the status of the Akt pathway. The finding is that Akt–mTOR activation and GSK3β inactivation were almost completely blocked by cardiac expression of human MD1, whereas the phosphorylation of Akt, mTOR and GSK3β was enhanced by the loss of MD1 expression in mice after 4 weeks of AB. In the in vitro experiments, we observed similar results in MD1‐overexpressing and MD1‐knockdown H9C2 cells upon exposure to Ang II. Furthermore, inactivation of Akt signalling by its specific inhibitor, MK2206, reversed the pro‐hypertrophic effects of MD1 deficiency on Ang II‐stimulated H9C2 cells in vitro. Collectively, these results indicate that inactivation of Akt is one mechanism underlying the MD1‐elicited protective effects against adverse cardiac hypertrophy.
Cardiac fibrosis is a classic feature of pathological cardiac hypertrophy. In the present study, we demonstrated that MD1 inhibits cardiac fibrosis induced by chronic pressure overload. The mechanism by which MD1 inhibits cardiac fibrosis needs to be elucidated. Previous studies indicate that Akt signalling is a key pathway involved in fibrosis. 14 , 21 , 22 , 23 For example, many different profibrotic stimuli led to Akt–mTOR activation in cardiac fibroblasts, resulting in an increase in collagen synthesis, fibroblast proliferation and transformation of fibroblasts into myofibroblasts. 20 , 21 In addition, reduction in the expression or activity of GSK3β leads to cardiac fibrosis through TGF‐β1‐Smad3‐dependent and β‐catenin‐dependent mechanisms. 23 More importantly, pharmacological inhibition of Akt/GSK3β signalling eliminates Smad2 phosphorylation and Smad2/3 translocation and reduces cardiac fibrosis in hypertrophied hearts. 24 Thus, in this study, the blockade of Akt signalling as a consequence of MD1 overexpression likely contributes, at least in part, to the lesser degree of cardiac fibrosis that was observed in chronic pressure overload hearts.
In conclusion, our data demonstrate that MD1 prevents pressure overload‐triggered cardiac remodelling (hypertrophy, fibrosis) and dysfunction in mice, at least partly by blocking the Akt pathway (Figure 6 ). This study advances our understanding of the molecular mechanisms in cardiac remodelling and therefore provides potential strategy to prevent/treat pathological cardiac remodelling and HF.
Figure 6.
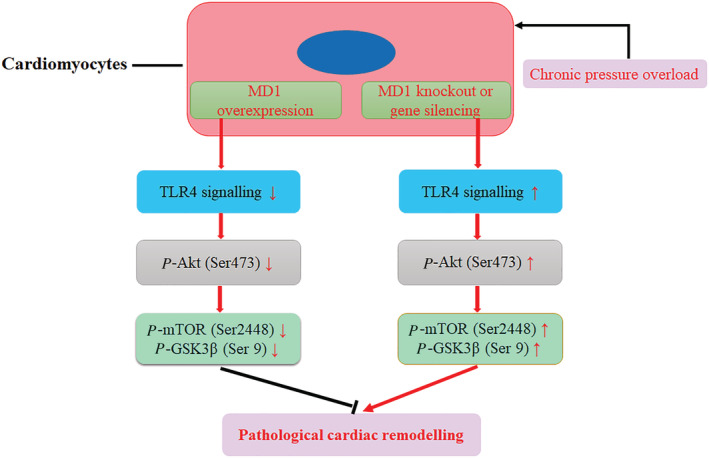
The schematic diagram of the molecular mechanisms of this study.
Conflict of interest
None declared.
Funding
This work was supported by the financial support from The Second Affiliated Hospital of the University of South China (Grant Number: 2018B02).
Supporting information
Table S1. Anatomic, echocardiographic parameters in MD1‐TG and WT mice at 4 weeks after Sham or AB operation.
Table S2. Echocardiographic parameters in WT mice, MD1‐KO mice, and MD1‐TG mice at 8–9 weeks old.
Data S1. Supporting Information.
Acknowledgement
We thank Renmin Hospital of Wuhan University for providing the MD1‐KO mice and MD1‐TG mice freely.
Peng, J. , Zeng, G. , Zhong, P. , Wang, G. , Lei, C. , Tian, G. , Chen, J. , Wu, J. , and Shen, C. (2021) The Akt pathway mediates the protective effects of myeloid differentiation protein 1 in pathological cardiac remodelling. ESC Heart Failure, 8: 3214–3222. 10.1002/ehf2.13447.
References
- 1. Heusch G, Libby P, Gersh B, Yellon D, Böhm M, Lopaschuk G, Opie L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 2014; 383: 1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Topkara VK, Evans S, Zhang W, Epelman S, Staloch L, Barger PM, Mann DL. Therapeutic targeting of innate immunity in the failing heart. J Mol Cell Cardiol 2011; 51: 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miyake K, Shimazu R, Kondo J, Niki T, Akashi S, Ogata H, Yamashita Y, Miura Y, Kimoto M. Mouse MD‐1, a molecule that is physically associated with RP105 and positively regulates its expression. J Immunol 1998; 161: 1348–1353. [PubMed] [Google Scholar]
- 4. Divanovic S, Trompette A, Atabani SF, Madan R, Golenbock DT, Visintin A, Finberg RW, Tarakhovsky A, Vogel SN, Belkaid Y, Kurt‐Jones EA, Karp CL. Negative regulation of toll‐like receptor 4 signaling by the toll‐like receptor homolog RP105. Nat Immunol 2005; 6: 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoon SI, Hong M, Wilson IA. An unusual dimeric structure and assembly for TLR4 regulator RP105‐MD‐1. Nat Struct Mol Biol 2011; 18: 1028–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ohto U, Miyake K, Shimizu T. Crystal structures of mouse and human RP105/MD‐1 complexes reveal unique dimer organization of the toll‐like receptor family. J Mol Biol 2011; 413: 815–825. [DOI] [PubMed] [Google Scholar]
- 7. Xiong X, Liu Y, Mei Y, Peng J, Wang Z, Kong B, Zhong P, Xiong L, Quan D, Li Q, Wang G, Huang H. Novel protective role of myeloid differentiation 1 in pathological cardiac remodelling. Sci Rep 2017; 7: 41857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shuai W, Kong B, Yang H, Fu H, Huang H. Loss of myeloid differentiation protein 1 promotes atrial fibrillation in heart failure with preserved ejection fraction. ESC Heart Fail 2020; 7: 626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of toll‐like receptor signalling complexes. Nat Rev Immunol 2014; 14: 546–558. [DOI] [PubMed] [Google Scholar]
- 10. Li X, Jiang S, Tapping RI. Toll‐like receptor signaling in cell proliferation and survival. Cytokine 2010; 49: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peng J, Liu Y, Xiong X, Huang C, Mei Y, Wang Z, Tang Y, Ye J, Kong B, Liu W, Wang T, Huang H. Loss of MD1 exacerbates pressure overload‐induced left ventricular structural and electrical remodelling. Sci Rep 2017; 7: 5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐delta delta C(T)) method. Methods 2001; 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 13. Zhang Y, Huang Z, Li H. Insights into innate immune signalling in controlling cardiac remodelling. Cardiovasc Res 2017; 113: 1538–1550. [DOI] [PubMed] [Google Scholar]
- 14. Wu QQ, Xiao Y, Yuan Y, Ma ZG, Liao HH, Liu C, Zhu JX, Yang Z, Deng W, Tang QZ. Mechanisms contributing to cardiac remodelling. Clin Sci (Lond) 2017; 131: 2319–2345. [DOI] [PubMed] [Google Scholar]
- 15. Zhang Y, Peng T, Zhu H, Zheng X, Zhang X, Jiang N, Cheng X, Lai X, Shunnar A, Singh M, Riordan N, Bogin V, Tong N, Min WP. Prevention of hyperglycemia‐induced myocardial apoptosis by gene silencing of toll‐like receptor‐4. J Transl Med 2010; 8: 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hu N, Zhang Y. TLR4 knockout attenuated high fat diet‐induced cardiac dysfunction via NF‐κB/JNK‐dependent activation of autophagy. Biochim Biophys Acta Mol Basis Dis 2017; 1863: 2001–2011. [DOI] [PubMed] [Google Scholar]
- 17. Echem C, Bomfim GF, Ceravolo GS, Oliveira MA, Santos‐Eichler RA, Bechara LR, Veras MM, Saldiva PH, Ferreira JC, Akamine EH, Fortes ZB, Dantas AP, de Carvalho MH. Anti‐toll like receptor 4 (TLR4) therapy diminishes cardiac remodeling regardless of changes in blood pressure in spontaneously hypertensive rats (SHR). Int J Cardiol 2015; 187: 243–245. [DOI] [PubMed] [Google Scholar]
- 18. Ehrentraut H, Weber C, Ehrentraut S, Schwederski M, Boehm O, Knuefermann P, Meyer R, Baumgarten G. The toll‐like receptor 4‐antagonist eritoran reduces murine cardiac hypertrophy. Eur J Heart Fail 2011; 13: 602–610. [DOI] [PubMed] [Google Scholar]
- 19. Dange RB, Agarwal D, Masson GS, Vila J, Wilson B, Nair A, Francis J. Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II‐induced hypertension. Cardiovasc Res 2014; 103: 17–27. [DOI] [PubMed] [Google Scholar]
- 20. Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res 2014; 114: 549–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sciarretta S, Forte M, Frati G, Sadoshima J. New insights into the role of mTOR signaling in the cardiovascular system. Circ Res 2018; 122: 489–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Y, Jiang XL, Liu Y, Jiang DS, Zhang Y, Zhang R, Chen Y, Yang Q, Zhang XD, Fan GC, Li H. Toll‐interacting protein (Tollip) negatively regulates pressure overload‐induced ventricular hypertrophy in mice. Cardiovasc Res 2014; 101: 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lal H, Ahmad F, Woodgett J, Force T. The GSK‐3 family as therapeutic target for myocardial diseases. Circ Res 2015; 116: 138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bian ZY, Wei X, Deng S, Tang QZ, Feng J, Zhang Y, Liu C, Jiang DS, Yan L, Zhang LF, Chen M, Fassett J, Chen Y, He YW, Yang Q, Liu PP, Li H. Disruption of mindin exacerbates cardiac hypertrophy and fibrosis. J Mol Med (Berl) 2012; 90: 895–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Anatomic, echocardiographic parameters in MD1‐TG and WT mice at 4 weeks after Sham or AB operation.
Table S2. Echocardiographic parameters in WT mice, MD1‐KO mice, and MD1‐TG mice at 8–9 weeks old.
Data S1. Supporting Information.