

Abstract
Four new compounds (1–4) were isolated from the stem bark of Entandrophragma angolense along with eleven known stuctures (5–15). The chemical structures were elucidated on the basis of spectroscopic and HRMS data, and the absolute configuration was established with the aid of electronic circular dichroism. Compound 5 displayed moderate cytotoxicity against MDA-MB-231, OVCAR3, MDA-MB-435, and HT29 cell lines, with IC50 values ranging from 2.0–5.9 μM.
Keywords: Entandrophragma angolense, Meliaceae, limonoid, triterpenoid, seco-triterpenoid, cytotoxicity, quantitative NMR
Graphical Abstract
1. Introduction
The genus Entandrophragma (Meliaceae) inhabiting in tropical Africa is made up by about 14 species of deciduous trees, commonly known as the mahogany. Apart from being economically important timber plants, members of this genus have been extensively used in African traditional medicine for the treatment of ailments such as malaria, stomach-ache, arthritic pain, and sickle cell disease[1]. Biological evaluations of Entandrophragma plant extracts have demonstrated antiplasmodial, anti-inflammatory, cytotoxicity, antiulcer, antisickling, and antifeedant activities [1]. Chemically, this genus is characterized by the presence of limonoids and its biosynthetic precursors, the protolimonoids, which not only serve as the chemotaxonoic markers of the taxon, but were also found to possess a variety of biological properties, including anticancer, antioxidant, anti-inflammatory, neuroprotective, antimalarial, anti-HIV, and antimicrobial activities [2–4].
Among Entandrophragma plants, E. angolense (Welw.) C. DC. is widely spread in the East and West Africa, and it was the first plant studied for phytochemical composition [5]. The discovery of gedunin from this species has opened the revenue to many other chemical investigations into the genus. E. angolense is now known to contain limonoids, triterpenoids, steroids, and fatty acids [1, 6]. As a traditional medicine, E. angolense has been used for treating pains such as abdominal pain, ear pain, rheumatic/arthritic pain, and peptic ulcer [7]. On the other hand, pharmacological studies of the plant extracts have demonstrated antisickling, antioxidant, anti-inflammatory, antiulcer, antifeedant, antimalarial, and antibacterial activities [8–12].
In a continuing effect to explore ethnobotanicals from African medicinal plants for new bioactive substances, we have studied an extract of the stem bark of E. angolense. In this report, the isolation of a new limonoid (1), a new triterpene (2), and two new seco-triterpenoids (3 and 4), together with eleven known structures, as well as the cytoxicity evaluation of the isolates, is described. In the course of the present study, quantitative NMR (qNMR) was employed to estimate the ratio of the isomer pair of 1, and the electronic circular dichroism (ECD) techniqe was applied to the determination of absolute configuration.
2. Experimental section
2.1. General experimental procedures
Optical rotations at the sodium D line were measured with a Perkin Elmer 241digital polarimeter using a quartz cell with a path length of 100 mm at room temperature. Concentrations (c) are given in g/100 mL. IR spectra were measured on a Nicolet 380 Fourier Transform Infrared Spectrometer loaded with OMNIC software. UV spectra were collected on a Shimadzu UFLC system with a photodiode array detector. The electronic circular dichroism (ECD) spectra were recorded on a J-810 spectropolarimeter (JASCO, Easton, MD, USA). The NMR spectroscopic data were recorded on a Bruker AV-400 spectrometer, and the data were processed on MestReNova (Mnova) software v14.1.2 (Santiago de Compostela, Spain). All chemical shifts were quoted on the δ scale in ppm using residual solvent as internal standard (chloroform-d: δH 7.26 for 1H NMR, δC 77.16 for 13C NMR). Coupling constants (J) are reported in Hz. HRESIMS were obtained on a Waters SYNAPT hybrid quadrupole/time of flight spectroscopy using positive electrospray ionization mode. For HPLC purification, a C18 semi-preparative HPLC column (Agilent SB-C18 column, 250×9.4 mm, 5μm) and a Shimadzu UFLC system were utilized. Human melanoma cancer cells MDA-MB-435, human breast cancer cells MDA-MB-231, human colorectal adenocarcinoma cells HT29, and human ovarian cancer cells OVCAR3 were purchased from the American Type Culture Collection (Manassas, VA, USA).
2.2. Plant material
Dried stem bark of E. angolense was collected by Bamisaye O. Oyawaluja from the Southwest region of Nigeria in April, 2016. The plant was identified by the botanist O. O. Oyebanji in the Department of Botany, University of Lagos, Nigeria, at which the voucher specimen has been deposited (LUH 6977).
2.3. Extraction and isolation
The air-dried and ground stem bark of E. angolense (5 kg) was extracted with 80 % EtOH (11×20 L) and dried under reduced pressure to afford 1.2 kg of crude extract. This extract was partitioned sequentially with hexanes (6×18 L, 11 g), EtOAc (10×18 L, 54 g), and n-BuOH (5×18 L, 700 g), followed by further partition of the EtOAc part with CHCl3 (5×2 L, 12 g). The hexane partition was then fractionated on an ODS column (MeOH−H2O, 10−100% MeOH) to afford 8 fractions, H1−H8. Of these, fraction H3 (200 mg) was chromatographed on an ODS column (MeOH−H2O, 20−100% MeOH) to yield seven subfractions (H31−H37). Subfraction H32 (50 mg) was subjected to purification over a semi-preparative RP C-18 column using CH3CN−H2O (40:60) as solvent to afford 1 (5 mg, tR= 23.9 min), 5 (15 mg, tR= 41.3 min), and 6 (20 mg, tR= 15.4 min). Then, H4 (150 mg) was subjected to a Sephadex LH-20 column to afford 4 subfractions, H41-H44. Of these, subfraction H43 (8 mg) was purified using a semi-preparative RP C-18 column to afford 7 (2 mg, tR= 43.4 min). Fraction H6 (874 mg) was then eluted on a Sephadex LH-20 column to yield five subfractions, H61−H65. One of these fractions, H62 (330 mg), was purified on silica gel (hexane-EtOAc, 80% to 50%) to afford 3 fractions, H621−H623, followed up by further purification of H622 on a silica gel column (CH2Cl2-Acetone, 95 to 50%) and over a semi-preparative RP C-18 column, using gradient conditions (CH3CN−H2O, 83.5 to 85 %). As a result, crops of 2 (2 mg, tR= 32.8 min), 8 (3 mg, tR= 21.1 min), and 9 (5 mg, tR= 35.6 min) were obtained. Similarly, 4 (2 mg, tR= 25.3 min), 10 (2 mg, tR= 25.3 min), and 11 (2 mg, tR= 33.0 min) were obtained from subfraction H623 using a semi-preparative RP-C18 column (CH3CN−H2O, 60:40). On the other hand, H7 (2.0 g) was eluted on a silica gel column (hexane-EtOAc, 90% to 70%) to yield 7 subfractions, H71−H77. Fractions H71 and H72 were purified using a semi-preparative RP C-18 column, with CH3CN−H2O (85:15) to give 3 (3mg, tR= 25.5 min) and 12 (2 mg, tR= 36.0 min).
The CHCl3 partition was then fractionated on a silica gel column (CH2Cl2−MeOH, 99 to 40%) to afford 9 fractions, C1−C9. Fraction C2 (1 g) was chromatographed on Sephadex LH-20 (100% MeOH) to yield seven subfractions (C21−C27), among which C21 yielded methyl angolensate (13, 10 mg) by precipitation. Subfraction C22 was separated on an open MCI gel column (MeOH−H2O, 10−70% MeOH) to yield 7 subfractions, C221-C227. Fraction C227 (130 mg) was subjected to purification over a semi-preparative RP-C8 column using CH3CN−H2O (45:55) as solvent to afford 14 (2 mg, tR= 34.3 min) and 15 (2 mg, tR= 17.3 min).
2.3.1. Compound 1
White amorphous powder; [α]D25 = +27 (c 0.1, MeOH); UVmax 232 nm; IR νmax 3361, 2919, 1743, 1670, 1632, 1440, 1382, 1270, 1137, 1024 cm−1; ECD (MeOH) λ (Δε) 238 (−15.40), 258 (5.24), 330 (10.01) nm; 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; HRESIMS m/z 501.2130 [M + H]+ (calcd for C27H33O9, 501.2125).
Table 1.
1H (400 MHz) NMR Spectroscopic Data of 1–4(δH in ppm, CDCl3).
| position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 7.09 (d, 10.5), 7.14 (d, 10.5) | 1.18, m | 1.63, m | 1.64, m |
| 1.37, m | 1.63, m | 1.64, m | ||
| 2 | 6.06 (d, 10.5), 6.10 (d, 10.5) | 1.60, m | 2.24, m | 2.42, m |
| 1.92, m | 2.37, m | 2.25, m | ||
| 3 | 4.68, t (2.5) | |||
| 5 | 2.66a, m | 1.98, m | 2.41, m | 2.42, m |
| 6 | 2.31 (m), 2.35 (m) | 1.63, m | 2.02, m | 2.20, m |
| 2.49 (m), 2.53 (dd, 3.4, 7.0) | 1.58, m | 2.18, m | 2.05, m | |
| 7 | 3.75, t (2.5) | 5.24, m | 5.26, br s | |
| 9 | 2.46 (m), 2.48 (m) | 1.34, m | 2.43, m | 2.43, m |
| 11 | 1.87a, m | 1.33, m | 1.46, m | 1.49, m |
| 2.02a, m | 1.33, m | |||
| 12 | 1.15a, m | 1.83, m | 1.35, m | 1.23, m |
| 1.92a, m | 1.83, m | 1.60, m | 1.76, m | |
| 15 | 4.03 (s), 4.05 (s) | 1.65, m | 1.51, m | 1.54, m |
| 1.55, m | 1.51, m | |||
| 16 | 0.95, m | 1.23, m | 1.37, m | |
| 0.95, m | 1.96, m | 1.87, m | ||
| 17 | 5.42 (s), 5.43 (s) | 2.01, m | 1.95, m | 2.36, m |
| 18 | 0.99 (s), 1.02 (s) | 0.75, d (4.9) | 0.97, s | 0.89, s |
| 0.51, d (4.9) | ||||
| 19 | 0.96 (s), 0.97 (s) | 0.90, s | 0.82, s | 0.84, s |
| 20 | 2.10, m | 2.78, m | 2.92, dd (4.1, 11.4) | |
| 21 | 4.97, d (4.9) | |||
| 22 | 7.29a, m | 1.88, m | 2.82, m | 5.14, d (4.1) |
| 1.79, m | 2.60, dd (2.3, 16.5) | |||
| 23 | 6.14 (m), 6.21 (m) | 4.27, ddd (1.6, 5.3, 10.2) | ||
| 24 | 3.23, br s | 6.03, m | 6.12, br s | |
| 25 | ||||
| 26 | 1.29, s | 1.87, d (1.1) | 1.95, s | |
| 27 | 1.26, s | 2.12,d(1.1) | 2.15, s | |
| 28 | 1.07a, s | 0.85, s | 4.79, br s | 4.80, s |
| 4.83, br s | 4.84, s | |||
| 29 | 1.09 (s), 1.10 (s) | 0.89, s | 1.78, br s | 1.78, s |
| 30α | 5.28 (s), 5.31 (s) | 1.03, s | 0.98, s | 1.02, s |
| 30β | 5.37 (s), 5.39 (s) | |||
| 31 | 3.71, m | 3.69, s | 3.62, s | |
| 3.45, dq (7.3, 9.6) | ||||
| 32 | 1.22, t (7.1) | 3.67, s | ||
| 33 | 2.18 s | |||
| 2' | 5.77, s | |||
| 3' | ||||
| 4' | 1.90, s | |||
| 5' | 2.18, s | |||
| 7' | 3.71 (s), 3.72 (s) |
peaks overlapped
Table 2.
13C (100 MHz) NMR Spectroscopic Data of 1–4(δC in ppm, CDCl3).
| position | 1a | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 153.2, 154.0 | 34.0 | 32.0 | 31.7 |
| 2 | 125.8, 126.1 | 23.0 | 28.2 | 27.8 |
| 3 | 203.8, 204.7 | 77.1 | 174.8 | 178.3 |
| 4 | 46.2, 46.3 | 36.4 | 147.5 | 147.4 |
| 5 | 42.8, 43.0 | 41.4 | 49.5 | 49.6 |
| 6 | 31.5, 31.6 | 24.3 | 30.3 | 30.6 |
| 7 | 174.4, 174.6 | 74.4 | 118.5 | 118.7 |
| 8 | 138.1, 138.7 | 39.2 | 146.0 | 145.7 |
| 9 | 48.7, 48.9 | 44.1 | 40.7 | 40.7 |
| 10 | 43.2, 43.3 | 37.5 | 36.9 | 36.9 |
| 11 | 21.3, 21.4 | 16.3 | 18.1 | 18.0 |
| 12 | 29.2, 29.4 | 25.8 | 30.0 | 30.3 |
| 13 | 39.3, 39.5 | 28.6 | 43.3 | 43.5 |
| 14 | 67.6, 67.7 | 36.4 | 51.4 | 51.4 |
| 15 | 55.2, 55.3 | 26.0 | 33.8 | 33.9 |
| 16 | 165.5, 166.3 | 26.2 | 27.5 | 26.5 |
| 17 | 75.2, 77.4 | 48.4 | 49.9 | 45.4 |
| 18 | 14.5, 15.2 | 13.9 | 21.8 | 22.3 |
| 19 | 20.3, 20.3 | 15.8 | 16.0 | 16.0 |
| 20 | 133.6, 133.8 | 49.1 | 42.5 | 48.3 |
| 21 | 168.9, 169.0 | 108.2 | 176.5 | 171.7 |
| 22 | 149.7, 150.0 | 32.2 | 47.0 | 78.4 |
| 23 | 97.0, 97.9 | 77.1 | 199.0 | 194.9 |
| 24 | 75.7 | 123.4 | 119.8 | |
| 25 | 73.2 | 156.1 | 160.4 | |
| 26 | 26.7 | 27.8 | 28.3 | |
| 27 | 26.5 | 21.0 | 21.5 | |
| 28 | 22.8, 22.9 | 27.9 | 114.1 | 114.2 |
| 29 | 22.7a | 22.0 | 22.6 | 22.4 |
| 30 | 122.8, 123.2 | 19.6 | 27.7 | 27.7 |
| 31 | 64.2 | 51.7 | 51.7 | |
| 32 | 15.6 | 51.7 | 170.6 | |
| 33 | 20.9 | |||
| 1' | 166.6 | |||
| 2' | 117.1 | |||
| 3' | 156.0 | |||
| 4' | 27.5 | |||
| 5' | 20.4 | |||
| 7' | 52.3, 52.4 |
peaks overlapped
2.3.2. Compound 2
White amorphous powder; [α]D25 = −42 (c 0.1, MeOH); UVmax 216 nm; IR νmax 3334, 2942, 2831, 1662, 1448, 1114, 1022 cm−1; ECD (MeOH) λ (Δε) 208 (−5.58), 243 (−1.20) nm; 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; HRESIMS m/z 571.3998 [M – C2H5O]+ (calcd for C35H55O6, 571.3999).
2.3.3. Compound 3
White amorphous powder; [α]D25 = −56 (c 0.1, MeOH); UVmax 237 nm; IR νmax 3261, 1635, 1407, 1116, 1014 cm-1; ECD (MeOH) λ (Δε) 226 (−1.84) nm; 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; HRESIMS m/z 513.3566 [M + H]+ (calcd for C32H49O5, 513.3580).
2.3.4. Compound 4
White amorphous powder; [α]D25 = +12 (c 0.1, MeOH); UVmax 244 nm; IR νmax 3247, 2944, 2834, 1643, 1448, 1112, 1016 cm-1; ECD (MeOH) λ (Δε) 211 (−14.69) nm; 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; HRESIMS m/z 557.3469 [M + H]+ (calcd for C33H49O7, 557.3478).
2.4. Quantitative HNMR with GSD approach
Compound 1 (1.5 mg) was dissolved in 500 μL of CDCl3 and transferred into a 5 mm NMR tube. The HNMR spectrum was acquired using the following qHNMR conditions: relaxation delay (D1) of 60 s, calibrated 90° pulse (P1), 64 number of scans, receiver gain (RG) of 63, and acquisition time of 8.19 s. The acquired spectrum was then treated with the following postacquisition processing: 256k of zero-filling, Lorentzian−Gaussian window function (exponential factor −0.3, Gaussian factor 0.05 in GF mode), and baseline correction (third order polynomial) [13].
The qHNMR spectrum was processed with the global spectral deconvolution (GSD) approach for 1a and 1b. It was initiated by individual peak fitting using the “edit fit” with five fitting cycles in the global SD function of the Mnova software. The deconvoluted lines and their peak areas were used to obtain the average peak areas and standard deviation. For doublets, the two individual peak areas were combined to give the total area.
2.5. Conformational analysis
The conformational analyses of the plausible stereoisomers of 1–4 were performed by SYBYL-X-2.1.1 program with MMFF94s molecular force field. The energy of optimized structures at the B3LYP/6–31+G(d) level in the Gaussian 09 software was applied to screen stable conformers. The ECD and NMR calculations of all the selected conformers were carried out with the TD/B3LYP/6–31+G(d) mode in the gas phase and the GIAO/mPW1PW91/6–311G(d,p) method in chloroform, respectively. The overall ECD curves were weighed by Boltzmann distribution of each conformer and compared with the experimental results using the SpecDis 1.71 software with UV correction. The theoretical chemical shifts calculation for 13C NMR was analysed by using linear regression and DP4 probability to assign the configurations of C-22 in 4. Subsequently, the simulated ECD curves of all plausible stereoisomers of 1–4 were utilized for the absolute configuration determination.
2.6. Cytotoxicity assay
Cytotoxicity tests were carried as previously described [14]. Paclitaxel was used as positive control (IC50 = 0.1 – 3.1 nM), and IC50 values are expressed in μM relative to the solvent (DMSO) control.
3. Results and discussion
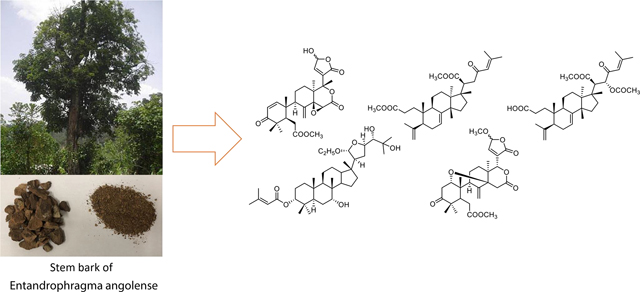
The stem bark of E. angolense was extracted with 80% EtOH and worked up by solvent partitioning and repeated chromatography. During the separation process, column fractions were monitored by thin-layer chromatography (TLC) using a specific visualizing reagent, vanillin sulfuric reagent, for the detection of limonoids and triterpenoids [15]. In general, the limonoid/triterpenoid spots will change color from red/pink to blue after 1 hour on the TLC plates. Major fractions were also subjected to 1H NMR analyses to detect the presence of characteristic proton resonances (such as methyls around δH 0.8–1.2, aliphatic CH2 protons at δH 1.5–3.0, and unsaturated protons at δH 5.0–7.0). Those fractions showing positive results were further separated, leading to the isolation of four new compounds (1–4, Fig. 1), together with 11 known compounds (5–15).
Fig. 1.

Structures of compounds 1–4.
Compound 1 was isolated as a white amorphous powder. Its molecular formula was deduced to be C27H32O9 from the HRESIMS [M + H]+ ion at m/z 501.2130 (calcd for C27H33O9, 501.2125) and 13C NMR data, corresponding to 12 indices of hydrogen deficiency. The UV λmax 232 nm and IR νmax 1743 and 1670 cm−1 indicated the presence of carbonyl, carboxyl, and/or ester groups. The 1H and 13C NMR spectra of 1 (Tables 1 and 2) displayed resonances attributed to four tertiary methyl, a methyl ester, four methylene, and five sp3 methine groups including three oxymethines H-15, H-17, and H-23. Resonance signals of a disubstituted double bond (δH 6.06, 6.10, 7.09, and 7.14), an γ-hydroxy-α,β-unsaturated-γ-lactone ring, four sp3 quaternary carbons including an oxycarbon (δC 67.6 and 67.7) and two ketones (δC 153.2, 154.0, 203.8, and 204.7) were also observed. Careful interpretation of the HMBC and COSY spectra (Fig. 2) pointed to a structure of andirobin-type limonoid similar to andirolide S [16], the only difference being the replacement of the ethoxyl group by a hydroxyl on C-23 in 1. The planar structure of 1 was thus established.
Fig. 2.

1H–1H COSY and HMBC key correlations of 1–4.
The relative configuration of 1 was determined by the NOESY data (Fig. 3). Thus, the correlations between Me-18/H-30, Me-18/H-22, H-30/H-9, and H-9/H-5 suggested cofacial orientation of the groups, and Me-18, H-9, and H-5 were assigned an α-orientation. On the other hand, the cross-peak between H-1/H-19 indicated that Me-19 was β-oriented.
Fig. 3.

NOESY key correlations of 1–4.
The absolute configuration of 1, except C-23, was determined by a comparison of the experimental and calculated ECD spectra using time dependent density functional theory (TDDFT) (Fig. 4) [17]. The ECD spectrum of 1 displayed a negative Cotton effect at 238 nm and a positive Cotton effect at 330 nm, in good agreement with the Boltzmann-averaged ECD result of (5R,9R,10R,13S,14S,15S,17R)-1. All available evidence supported the structure of 1 as depicted, and it was given the trivial name 23-O-deethylanderolide S.
Fig. 4.

Experimental and calculated ECD spectra of 1–4.
In the course of the NMR study, it was noted that, in the 1H NMR spectrum of 1, doubling of some signals occurred, indicating the existence of a pair of C-23 epimers. In order to estimate the ratio of the epimers, quantitative 1H NMR (qHNMR) approach was adopted by employing the GSD method. This approach helped to assess the ratio of the epimers and preserved the compound from the sample loss by avoiding chromatographic separation [18]. Thus, proton signals of H-1, H-15, H-19, and H-30 were designated to epimers 1a and 1b based on the correlations observed in the NOESY spectrum. H-1, H-15, and H-30 were processed using the GSD in the Mnova software and the generated peak table was used to calculated their relative ratio (Fig. 5 and Table S1). As a result, a ratio of 1.1:1.0 was obtained. The two C-23 epimers were therefore estimated to occur in a 1:1 ratio.
Fig. 5.

The global spectral deconvolution of 23-epimers of 1. The colored lines represent the sum (purple), the fitted peaks (green), and the residual (red) of the decolvolution approach.
Compound 2, isolated as an amorphous white solid, was assigned a molecular formula of C37H60O7 based on an HRESIMS [M – C2H5O]+ ion peak at m/z 571.3998 (calcd for C35H55O6, 571.3999). In its 1H NMR spectrum, resonances due to eight tertiary methyls (δH 0.85, 0.89, 0.90, 1.03, 1.26, 1.29, 1.90, and 2.18 ), a cyclopropyl methylene [δH 0.51 (d, J = 4.9 Hz) and 0.75 (d, J = 4.9 Hz)], an acetal methine proton [δH 4.97 (d, J = 4.9)], and an olefinic proton (δH 5.77) were observed (Table 1). The 13C NMR spectrum revealed 37 carbon signals, which could be sorted into nine methyls, ten methylenes, ten methines, and eight quaternary carbons including one carbonyl carbon at δ 166.6 (Table 2). The presence of a senecioyl moiety was obvious from the NMR data. Careful comparison of the NMR data with those of prototiamin F revealed similarities as a glabretal-type triterpenoid [19]. In particular, the presence of three hydroxyl groups at C-7, C-24, and C-25 and a cyclopropane ring at C-13, C-14, and C-18 could be established by the 13C NMR and HMBC data. The presence of a senecioyl ester group at C-3 was confirmed by the HMBC correlation between the carbonyl signal at δC 166.6 (C-1’) and the proton signal at δH 4.68 (H-3) (Fig. 2). A major difference between 2 and prototiamin F was the presence of an O-ethyl group (δH 1.22, 3.45, and 3.71 and δC 15.6 and 64.2) in the former. The location of the ethyl group was assigned to C-21 as evidenced by the HMBC correlation between C-31 (δC 64.2) and H-21 (δH 4.97). The NOESY data supported the same relative configuration as that of prototiamin F, except for C-3 (Fig. 3). To be specific, the NOE correlations between H-3/ Me-19, H-5/H-9, H-7/Me-30, H-9/H-18, H-17/H-21, H-18/H-20, and Me-19/Me-30 confirmed that H-5, OH-7, H-9, H-20, and the 13,14,18-cyclopropane ring were of α-orientation whereas H-3, H-17, Me-19, H-21, and Me-30 were β-oriented. Compound 2 is thus a C-3 epimer of prototiamin F. Indeed, the NMR resonances of CH-3 were consistent with those of 3β-H orientation in similar glabretal structures. Thus, the C-3 signal was in the neighbourhood of δC 77 and the H-3 signal appeared as a triplet with coupling constant of 2.5 Hz [20]. On the other hand, the β orientation of H-24 [δH 3.23 (br s)] was consistent with that of prototiamin F as evidenced by a negligible coupling constant [21]. With all the evidence on hand, the structure of 2 was determined to be as depicted in Fig. 1 and assigned the trivial name 3βH-21-O-ethylprototiamin F. The calculated ECD spectrum of (3R,5R,7R,8R,9R,10S,13R,14S,17S,20S,21R,23R,24S)-2 was in good agreement with the experimental reslts (Fig. 4). It is interesting to note that this is the first report of glabretal-type triterpenoid from E. angolense. Only four structures of this type (including prototiamin F) have been previously found in E. congoense of this genus [19].
Compound 3 was obtained as an amorphous white powder; the HRESIMS [M + H]+ ion at m/z 513.3566 suggested the molecular formula of C32H48O5 (calcd for C32H49O5, 513.3580). The UV spectrum showed an absorption at 237 nm consistent with an α,β-unsaturated carbonyl moiety. With nine indices of hydrogen deficiency, this compound exhibited three double bonds, one conjugated ketone and two carbonyls in the 13C NMR spectrum, thus confirming the presence of three rings. The 1H NMR spectrum of 3 exhibited resonance signals for six methyl groups on quaternary carbons, an olefinic proton coupled to a vicinal methylene, and two methoxy groups. A major difference between 3 and the known structure 23-oxo-3,4-secotirucalla-4(28),7,24-trien3,21-dioic acid 21-methyl ester [22] was the presene of a methyl ester group on C-3. Indeed, CH3-31 displayed correlations with C-3 in the HMBC spectrum (Fig. 2). The relative configuration at C-20 was supported by NOESY correlations between H-9/ H3-18/ H-20 and H3-30/ H-17 (Fig. 3). The available data suggested the structure of 3 to be 23-oxo-3,4-secotirucalla-4(28),7,24-trien-3,21-dioic acid 3,21-dimethyl ester. Its experimental ECD spectrum matched well with the calculated spectrum for (5S,9R,10S,13S,14S,17S,20S)-3 (Fig. 4). It represents the third seco-triterpenoid found in E. angolense.
The HRESIMS of 4 displayed an [M + H]+ ion at m/z 557.3469, suggestiong the molecular formula of C33H48O7. An inspection of the NMR data (Tables 1 and 2) suggested close resemblance with those of 23-oxo-3,4-secotirucalla-4(28),7,24-trien-3,21-dioic acid 21-methyl ester (12) [22], with the exceptions that additional signals of an acetyl (C-32 and CH3-33) were present in 4. The acetyl group could be assigned to C-22 since the chemical shifts of CH-22 (δC 78.4; δH 5.14, d, J = 4.1 Hz) were consistent with those attached to an oxygenated functional group. This was further suggested by a cross peak observed between δC 170.6 (C-32) and δH 5.14 (H-22) in the HMBC spectrum (Fig. 2). To determine the configuration of C-22, theoretical calculations of 13C NMR shifts of the possible isomers 22R* and 22S* were conducted by using the GIAO method with the Gaussian 09 software at the mPW1PW91/6–311G(d,p) level [23]. Comparison of the experimental and calculated 13C chemical shifts suggested that the configuration of 4 is 22R* with a DP4 probability of approximately 99.42 %, compared to 0.58 % for 22S* configuration (Fig. S1 and Table S2). Finally, the experimental ECD spectrum of 4 matched well with the calculated spectrum of (5S,9R,10S,13S,14S,17S,20R,22R)-4 (Fig. 4). Compound 4 was determined to be 22(R)-acetoxy-23-oxo-3,4-secotirucalla-4(28),7,24-trien-3,21-dioic acid 21-methyl ester. It is the fourth seco-triterpenoid found in E. angolense.
Along with the new compounds, eleven limonoids and triterpenoids were isolated; they are, namely, 6-deacetoxydomesticulide D 21-methylether (5) [12], 6-deacetoxydomesticulide D (6) [12], andirobin (7) [24], toonapubesin B (8) [25], piscidinol A (9) [26], 3βH-21-O-methylprototiamin F (10) [20], 3αH-21-O-methylprototiamin F (11) [20], 23-oxo-3,4-secotirucalla-4(28),7,24-trien-3,21-dioic acid 21-methyl ester (12) [22], methyl angolensate (13) [27], swietmanin J (14) [28], and toonin A (15) [29]. Compounds 7, 8, 14, and 15 were found for the first time in this genus and 9–11 for the first time in this species.
Limonoids represent a group of nor-seco-triterpenoids mostly found in plants of the Rutales (Meliaceae, Rutaceae, and Simaroubaceae families). They are derived from tetracyclic triterpenes through a series of oxidation and rearrangement reactions. As such, limonoids and related triterpenoids (the protolimonoids) such as the tirucallane-, apotirucallane-, and glabretal-type serve as chemotaxonomic markers of the Entandrophragma species [1]. Of particular interest is the isolation of 3,4-seco-tirucallanes (3, 4, and 12) from E. angolense. The formation of this type of metabolites where C-4 is oxygenated or part of a double bond has been suggested to involve an ezymatic Baeyer–Villiger oxidative process [30]. In this manner, formation of 3, 4, and 12 could be correlated to a plausible triterpene precursor, 3,23-dioxotirucalla-7,24-dien-21-al, which was found to co-exist in E. angolense [22].
Compounds 1–5, 13, and 14 were evaluated for cytotoxic activity. All compounds showed no activity (IC50 ≥ 25 μM) except 6-deacetoxydomesticulide D 21-methylether (5), which displayed modest activities against MDA-MB-231 (IC50 = 1.9 μM), OVCAR3 (IC50 = 3.1 μM), MDA-MB-435 (IC50 = 3.4 μM) and HT29 (IC50 = 5.9 μM) cell lines. Among all isolated compounds, 7 has been previously reported to be inactive in MDA-MB-231 [31].
To conclude, the present study on E. angolense has resulted in the isolation of secondary metabolites belonging to the limonoid (1, 5, 6, 7, 13, 14, and 15) and both glabretal- (2, 10, and 11) and tirucallane-types (3, 4, 8, 9, and 12) of triterpenoid. This findings highlights the chemotaxonomic significance of these markers for the Entandrophragma genus.
Supplementary Material
Acknowledgements
This work was partially supported by a grant from the National Cancer Institute (P01 CA125066) to carry out the bioassays. We acknowledge Dr. Dejan Nikolic for assisting with the mass spectrometry experiments, Brenna Kirkpatrick and Kimberly Heath for conducting the cytotoxicity assays, and Dr. Junfei Zhou for assisting with the isolation.
Footnotes
Declaration of Competing Interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
NMR spectra for 1–4 are available in Supporting Information. Supplementary data to this article can be found online at [doi: https://doi.org/10.1016/j.fitote.2021.104846]
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Happi GM, Ngadjui BT, Green IR, Kouam FS, Phytochemistry and pharmacology of the genus Entandrophragma over the 50 years from 1967 to 2018: a ‘golden’ overview, J. Pharm. Pharmacol 70 (2018) 1431–1460, doi: 10.1111/jphp.13005. [DOI] [PubMed] [Google Scholar]
- [2].Roy A, Saraf S, Limonoids: overview of significant bioactive triterpenes distributed in plants kingdom, Biol. Pharm. Bull 29 (2006) 191–201, doi: 10.1248/bpb.29.191. [DOI] [PubMed] [Google Scholar]
- [3].Tan Q-G, Luo X-D, Meliaceous limonoids: chemistry and biological activities, Chem. Rev 111 (2011) 7437–7522, doi: 10.1021/cr9004023. [DOI] [PubMed] [Google Scholar]
- [4].Tundis R, Loizzo MR, Menichini F, An overview on chemical aspects and potential health benefits of limonoids and their derivatives, Crit. Rev. Food Sci. Nutr 54 (2014) 225–250, doi: 10.1080/10408398.2011.581400. [DOI] [PubMed] [Google Scholar]
- [5].Akisanya A, Bevan CWL, Hirst J, Halsall TG, Taylor DAH, West African timbers. Part III. Petroleum extracts from the genus Entandrophragma, J. Chem. Soc (1960) 3827–3829, doi: 10.1039/JR9600003827. [DOI] [Google Scholar]
- [6].Happi GM, Wouamba SCN, Ismail M, Kouam SF, Frese M, Lenta BN, Sewald N, Ergostane-type steroids from the Cameroonian ‘white tiama’ Entandrophragma angolense, Steroids 156 (2020) 108584, doi: 10.1016/j.steroids.2020.108584. [DOI] [PubMed] [Google Scholar]
- [7].Tchinda AT, Entandrophragma angolense (Welw.) C. DC https://uses.plantnet-project.org/e/index.php?title=Entandrophragma_angolense_(PROTA)&oldid=325974, 2008 (Accessed 11 November 2020).
- [8].Bickii J, Tchouya GRF, Tchouankeu JC, Tsamo E, The antiplasmodial agents of the stem bark of Entandrophragma angolense (Meliaceae), Afr. J. Tradit. Complement. Altern. Med 4 (2007) 135–139. [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang W-Y, An F-L, Zhou M-M, Chen M-H, Jian K-L, Quasie O, Yang M-H, Luo J, Kong L-Y, Limonoids with diverse frameworks from the stem bark of Entandrophragma angolense and their bioactivities, RSC Advances 6 (2016) 97160–97171, doi: 10.1039/C6RA19532F. [DOI] [Google Scholar]
- [10].Njar VCO, Adesanwo JK, Raji Y, Methyl angolensate: the antiulcer agent of the stem bark of Entandrophragma angolense, Planta Med. 61 (1995) 91–92, doi: 10.1055/s-2006-958015. [DOI] [PubMed] [Google Scholar]
- [11].Shittu GA, Akor ES, Phytochemical screening and antimicrobial activities of the leaf extract of Entandrophragama angolense, Afr. J. Biotechnol 14 (2015) 202–205, doi: 10.5897/AJB2013.13237. [DOI] [Google Scholar]
- [12].Nsiama TK, Okamura H, Hamada T, Morimoto Y, Doe M, Iwagawa T, Nakatani M, D-seco Rings and B,D-seco tetranortriterpenoids from root bark of Entandrophragma angolense, Phytochemistry 72 (2011) 1854–1858, doi: 10.1016/j.phytochem.2011.05.014. [DOI] [PubMed] [Google Scholar]
- [13].Phansalkar RS, Simmler C, Bisson J, Chen S-N, Lankin DC, McAlpine JB, Niemitz M, Pauli GF, Evolution of quantitative measures in NMR: quantum mechanical qHNMR advances chemical standardization of a Red Clover (Trifolium pratense) extract, J. Nat. Prod 80 (2017) 634–647, doi: 10.1021/acs.jnatprod.6b00923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Young AN, Herrera D, Huntsman AC, Korkmaz MA, Lantvit DD, Mazumder S, Kolli S, Coss CC, King S, Wang H, Swanson SM, Kinghorn AD, Zhang X, Phelps MA, Aldrich LN, Fuchs JR, Burdette JE, Phyllanthusmin derivatives induce apoptosis and reduce tumor burden in high-grade serous ovarian cancer by late-stage autophagy inhibition, Mol. Cancer Ther 17 (2018) 2123–2135, doi: 10.1158/1535-7163.MCT-17-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yamasaki RB, Klocke JA, Lee SM, Stone GA, Darlington MV, Isolation and purification of azadirachtin from neem (Azadirachta indica) seeds using flash chromatography and high-performance liquid chromatography, J. Chromatogr. A 356 (1986) 220–226, doi: 10.1016/S0021-9673(00)91483-3. [DOI] [Google Scholar]
- [16].Sakamoto A, Tanaka Y, Inoue T, Kikuchi T, Kajimoto T, Muraoka O, Yamada T, Tanaka R, Andirolides Q–V from the flower of andiroba (Carapa guianensis, Meliaceae), Fitoterapia 90 (2013) 20–29, doi: 10.1016/j.fitote.2013.07.001. [DOI] [PubMed] [Google Scholar]
- [17].Pescitelli G, Di Bari L, Berova N, Application of electronic circular dichroism in the study of supramolecular systems, Chem. Soc. Rev 43 (2014) 5211–5233, doi: 10.1039/C4CS00104D. [DOI] [PubMed] [Google Scholar]
- [18].Selegato DM, Freire RT, Pilon AC, Biasetto CR, de Oliveira HC, de Abreu LM, Araujo AR, da Silva Bolzani V, Castro-Gamboa I, Improvement of bioactive metabolite production in microbial cultures—A systems approach by OSMAC and deconvolution-based 1HNMR quantification, Magn. Reson. Chem 57 (2019) 458–471, doi: 10.1002/mrc.4874. [DOI] [PubMed] [Google Scholar]
- [19].Happi GM, Kouam SF, Talontsi FM, Lamshöft M, Zühlke S, Bauer JO, Strohmann C, Spiteller M, Antiplasmodial and cytotoxic triterpenoids from the bark of the Cameroonian medicinal plant Entandrophragma congoënse, J. Nat. Prod 78 (2015) 604–614, doi: 10.1021/np5004164. [DOI] [PubMed] [Google Scholar]
- [20].Mitsui K, Maejima M, Saito H, Fukaya H, Hitotsuyanagi Y, Takeya K, Triterpenoids from Cedrela sinensis, Tetrahedron 61 (2005) 10569–10582, doi: 10.1016/j.tet.2005.08.044. [DOI] [Google Scholar]
- [21].Zhou Z-F, Taglialatela-Scafati O, Liu H-L, Gu Y-C, Kong L-Y, Guo Y-W, Apotirucallane protolimonoids from the Chinese mangrove Xylocarpus granatum Koenig, Fitoterapia 97 (2014) 192–197, doi: 10.1016/j.fitote.2014.06.009. [DOI] [PubMed] [Google Scholar]
- [22].Orisadipe AT, Adesomoju AA, D’Ambrosio M, Guerriero A, Okogun JI, Tirucallane triterpenes from the leaf extract of Entandrophragma angolense, Phytochemistry 66 (2005) 2324–2328, doi: 10.1016/j.phytochem.2005.07.017. [DOI] [PubMed] [Google Scholar]
- [23].Smith SG, Goodman JM, Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability, J. Am. Chem. Soc 132 (2010) 12946–12959, doi: 10.1021/ja105035r. [DOI] [PubMed] [Google Scholar]
- [24].Cheng Y-B, Chien Y-T, Lee J-C, Tseng C-K, Wang H-C, Lo IW, Wu Y-H, Wang S-Y, Wu Y-C, Chang F-R, Limonoids from the seeds of Swietenia macrophylla with inhibitory activity against Dengue virus 2, J. Nat. Prod 77 (2014) 2367–2374, doi: 10.1021/np5002829. [DOI] [PubMed] [Google Scholar]
- [25].Wang J-R, Liu H-L, Kurtán T, Mándi A, Antus S, Li J, Zhang H-Y, Guo Y-W, Protolimonoids and norlimonoids from the stem bark of Toona ciliata var. pubescens, Org. Biomol. Chem 9 (2011) 7685–7696, doi: 10.1039/C1OB06150J. [DOI] [PubMed] [Google Scholar]
- [26].McChesney JD, Dou J, Sindelar RD, Goins DK, Walker LA, Rogers RD, Tirucallane-type triterpenoids: nmr and X-ray diffraction analyses of 24-epi-piscidinol A and piscidinol A, J. Chem. Crystallogr 27 (1997) 283–290, doi: 10.1007/BF02575975. [DOI] [Google Scholar]
- [27].Kadota S, Marpaung L, Kikuchi T, Ekimoto H, Constituents of the seeds of Swietenia mahagoni JACQ. I.: isolation, structures, and 1H-and 13C-nuclear magnetic resonance signal assignments of new tetranortriterpenoids related to swietenine and swietenolide, Chem. Pharm. Bull 38 (1990) 639–651, doi: 10.1248/cpb.38.639. [DOI] [Google Scholar]
- [28].Lin B-D, Yuan T, Zhang C-R, Dong L, Zhang B, Wu Y, Yue J-M, Structurally diverse limonoids from the fruits of Swietenia mahagoni, J. Nat. Prod 72 (2009) 2084–2090, doi: 10.1021/np900522h. [DOI] [PubMed] [Google Scholar]
- [29].Dong X-J, Zhu Y-F, Bao G-H, Hu F-L, Qin G-W, New limonoids and a dihydrobenzofuran norlignan from the roots of Toona sinensis, Molecules 18 (2013) 2840–2850, doi: 10.3390/molecules18032840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Almeida A, Dong L, Appendino G, Bak S, Plant triterpenoids with bond-missing skeletons: biogenesis, distribution and bioactivity, Nat. Prod. Rep 37 (2020) 1207–1228, doi: 10.1039/C9NP00030E. [DOI] [PubMed] [Google Scholar]
- [31].Sidjui LS, Eyong KO, Hull KG, Folefoc GN, Leddet VM, Herbette G, Ollivier E, Taube J, Klausmeyer K, Romo D, Bioactive seco-lanostane-type triterpenoids from the roots of Leplaea mayombensis, J. Nat. Prod 80 (2017) 2644–2651, doi: 10.1021/acs.jnatprod.7b00210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.