

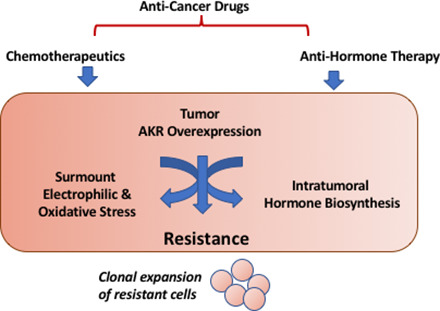
Abstract
Human aldo-keto reductases (AKRs) catalyze the NADPH-dependent reduction of carbonyl groups to alcohols for conjugation reactions to proceed. They are implicated in resistance to cancer chemotherapeutic agents either because they are directly involved in their metabolism or help eradicate the cellular stress created by these agents (e.g., reactive oxygen species and lipid peroxides). Furthermore, this cellular stress activates the Nuclear factor-erythroid 2 p45-related factor 2 (NRF2)-Kelch-like ECH-associated protein 1 pathway. As many human AKR genes are upregulated by the NRF2 transcription factor, this leads to a feed-forward mechanism to enhance drug resistance. Resistance to major classes of chemotherapeutic agents (anthracyclines, mitomycin, cis-platin, antitubulin agents, vinca alkaloids, and cyclophosphamide) occurs by this mechanism. Human AKRs also catalyze the synthesis of androgens and estrogens and the elimination of progestogens and are involved in hormonal-dependent malignancies. They are upregulated by antihormonal therapy providing a second mechanism for cancer drug resistance. Inhibitors of the NRF2 system or pan-AKR1C inhibitors offer promise to surmount cancer drug resistance and/or synergize the effects of existing drugs.
Significance Statement
Aldo-keto reductases (AKRs) are overexpressed in a large number of human tumors and mediate resistance to cancer chemotherapeutics and antihormonal therapies. Existing drugs and new agents in development may surmount this resistance by acting as specific AKR isoforms or AKR pan-inhibitors to improve clinical outcome.
I. Introduction to Aldo-Keto Reductases
Aldo-keto reductases (AKRs) are a superfamily of NAD(P)(H)-dependent oxidoreductases, which are predominately cytosolic and most often exist as monomers of 37 kDa in molecular weight. In most instances, they catalyze the reduction of carbonyl groups to alcohols. Aldehydes are converted to primary alcohols and ketones are converted to secondary alcohols on a variety of substrates (Jez and Penning, 2001; Mindnich and Penning, 2009). This functionalization would categorize these enzymes as phase-1 enzymes. Upon functionalization, the alcohols can be conjugated as glucuronides or as sulfates for elimination.
There are 15 human AKRs, and while their carbonyl substrates may be diverse, there are some common features in these proteins (Penning and Drury, 2007) (Table 1). They share a common (α/β)8 barrel in which there is an alternating pattern of α-helices and β-strands. The β-strands make up the staves of a barrel, and at the base of the barrel a catalytic tetrad exists of Y55, K84, D40, and H117, the exception being AKR1D1, in which a H120E substitution exists to replace the catalytic histidine. At the back of the barrel, three large loops exist which help define substrate specificity, where different amino acids are recruited to the binding pocket based on substrate (Fig. 1).
TABLE 1.
Human AKRs and involvement in drug resistance
| Gene | Protein Names (Aliases) | Chromosomal Localization | Substrate Specificity |
|---|---|---|---|
| AKR1A1 | Aldehyde reductase; dihydrodiol dehydrogenase 1 | 1p33-p32 |
DL-glyceraldehyde Melvadate |
| AKR1B1 | Aldose reductase | 7q35 | Glucose, advanced end glycation products, 4-HNE, reactive carbonyls |
| AKR1B10 | Small intestine like aldose reductase; 9-cis-retinal reductase | 7q33 | Retinal; reactive carbonyls |
| AKR1B15 | 3-Keto- acyl CoA reductase | 7q33 | 3-Keto-acyl-coA; 9-cis-retinaldehyde |
| AKR1C1 | 3α(20α)-Hydroxysteroid dehydrogenase; dihydrodiol dehydrogenase 1; | 10p15-10p14 | Reduction of 20-ketosteroids e.g., progesterone |
| AKR1C2 | 3α-hydroxysteroid dehydrogenase type 3; dihydrodiol dehydrogenase 2; bile-acid binding protein | 10p15-10p14 | Reduction of 3-ketosteroids, dihydroprogesterone, dihydrotestosterone |
| AKR1C3 | 3α-hydroxysteroid dehydrogenase type 2; 17β-hydroxysteroid dehydrogenase type 5; prostaglandin F synthase; dihydrodiol dehydrogenase X | 10p15-10p14 | Reduction of 17-ketosteroids; 4-androstene-3,17-dione; estrone; prostaglandin H2; prostaglandin D2 |
| AKR1C4 | 3α−hydroxysteroid dehydrogenase type 1; dihydrodiol dehydrogenase 4; chlordecone reductase | 10p15-10p14 | 3-Ketosteroids; bile acid precursors; chlordecone |
| AKR1D1 | Steroid 5β-reductase | 7q32-7q33 | Delta4-3-Ketosteroids |
| AKR1E2 | 1,5-Andyhro-D-frucose reductase | 10p15 | 1,5-anhydro-D-fructose |
| AKR6A3 | Potassium voltage gated channel β-subunit-1 | 3q26.1 | |
| AKR6A5 | Potassium voltage gated channel β-subunit-2 | 1p36.3 | |
| AKR6A9 | Potassium voltage gated channel β-subunit-3 | 17p13.1 | |
| AKR7A2 | Aflatoxin aldehyde reductase | 1p35.1- p36.23 |
Reduction of succinic semialdehyde; acrolein 2-carboxybenzlaldehyde; aflatoxin dialdehyde |
| AKR7A3 | Aflatoxin aldehyde reductase | 1p35.1-p36.23 | Aflatoxin dialdehyde |
Fig. 1.

Structure of human AKRs and catalytic tetrad. α/β8-barrel protein fold of AKRs (A); loop structures at back of barrel (B); and (C) catalytic tetrad. From AKR1C2.NADP+ complex (PDB 2hdj). Helices (blue); β-strands (purple); stick model shows position of NADP+ and catalytic tetrad in A, and B. Figure produced in Pymol.
AKRs are overexpressed in a broad range of human cancers, e.g., prostate (Stanbrough et al., 2006), breast (Ma et al., 2012; McNamara and Sasano, 2019), glioma (Park et al., 2010), neuroblastoma (Lyon et al., 2013), nonsmall lung cancer and small cell lung cancer (Hsu et al., 2001; Tian et al., 2016), colon cancer (Kropotova et al., 2014; Zu et al., 2017), cancer of the small intestine (Wang et al., 2009), and acute myelocytic leukemia (AML) (Birtwistle et al., 2009). Tumor-specific expression of the AKR isoforms has been observed, e.g., nonsmall cell lung cancer (AKR1C1, AKR1C2, AKR1C3 and AKR1B10) (Hsu et al., 2001; Cubillos-Tian et al., 2016; Angulo et al., 2020), colon cancer (AKR1B10) (Kropotova et al., 2014), breast and prostate cancer (AKR1C3) (Stanbrough et al., 2006; McNamara and Sasano, 2019), and AML (AKR1C3) (Birtwistle et al., 2009). The abundance of AKR overexpression has led to the proposition that they can be used as tumor biomarkers (Hamid et al., 2013; Tian et al., 2014; Tian et al., 2016; Cubillos-Angulo et al., 2020).
The AKRs are also stress response genes and respond to electrophilic and oxidative stress, osmotic stress, and steroid hormones (Penning and Drury 2007). They are induced by the Nuclear factor-erythroid 2 p45-related factor 2 (NRF2) (NFE2L2)-Kelch-like ECH-associated protein 1 (KEAP1) (KEAP1) pathway (Tebay et al., 2015) by AP-1 transcription factors, a tonictiy responsive enhancer (Ko et al., 1997), and are regulated by steroid hormone receptors via steroid response elements (Penning and Drury 2007). Since they respond to electrophilic and oxidative stress stimuli, which are generated by chemotherapeutic agents, they are upregulated by these agents, and this contributes to drug resistance. Because they also play a role in steroid hormone transformation, they are involved in chemoresistance to hormone ablative therapies.
II. Stress Response and Nuclear Factor-Erythroid 2 p45-Related Factor 2 Induction of Aldo-Keto Reductases
NRF2-KEAP1 is a master regulator of stress response genes (Tebay et al., 2015). Under basal conditions, NRF2 is tethered to KEAP1, which is an adaptor protein for the cullin-3. RealIy Interesting New Gene-box protein 1 ubiquitin ligase, which targets NRF2 for ubiquitination and proteasomal degradation (Dinkova-Kostova et al., 2005). Electrophiles and reactive oxygen species (ROS) modify key cysteine residues in KEAP1, which prevents ubiquitination of NRF2 (Wakabayashi et al., 2004; Dinkova-Kostova et al., 2017). Newly synthesized NRF2 can thus evade ubiquitination, translocate to the nucleus, and bind to its heterodimeric partner small-maf on the antioxidant response element (ARE) (5′-A/G-TGA-C/G-NNN-GC-A/G-3′) of stress response genes to increase gene transcription (Fig. 2). AKR1C1-AKR1C3 genes contain multiple AREs (6–16 based on a consensus sequence search and a positional matrix search) in their promoters (Tebay et al., 2015) (see Table 2).
Fig. 2.

NRF2-Keap1 regulation of human AKR genes. Under basal conditions, the transcription factor Nrf2 is sequestered by Keap1 which acts as an adaptor program for culin 3 (cul 3) ubiquitin ligase which leads to the ubiquitination of Nrf2 and its proteasomal degradation. Upon exposure to oxidative or electrophilic stress, cysteine residues on Keap1 become either oxidized or covalently modified which disrupts the interaction with Nrf2 and prevents its ubiquitination. Under these conditions, newly synthesized Nrf2 evades proteasomal degradation, translocates to the nucleus where it binds to its heterodimeric partner small maf; and the resulting dimer binds to ARE in the promoters of genes that detoxify the stress signal, e.g., AKR1C, NQO1, and γ-glutamylcysteine synthetase (GCLM).
TABLE 2.
Evidence that human aldo-keto reductases are regulated by the NRF2-KEAP1 system
| Increased Expression with NRF2 Activator | Decreased Expression with NFE2L2/KEAP1 k/d | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AKR Gene | AREs in Promotor Region | AREs Based on Positional Matrix | Inducer | Increased RNA | Increased Protein/Activity | Cell Type* | Knockdown Method | Change in RNA | Change in Protein/ Activity | Cell Type* |
| AKR1A1 | 6 | 5 |
KEAP1 sh-RNA KEAP1 si-RNANFE2L2 si-RNA |
yes no yes |
HK-2f HaCaTg HaCaTg |
|||||
| AKR1B1 | 1 | 3 |
KEAP1 si-RNA KEAP1 sh-RNA KEAP1 si-RNA NFE2L2 si-RNA |
yes yes yes yes |
yes | MCF-10Aa HK-2f HaCaTg HaCaTg |
||||
| AKR1B10 | 4 | 2 | SFN | yes yes |
yes yes |
MCF10Aa MCF12Aa |
KEAP1 si-RNA KEAP1 si-RNA KEAP1 sh-RNA KEAP1 si-RNA NFE2L2 si-RNA |
yes yes yes yes yes |
yes yes yes |
MCF-10Aa MCF-12Aa HK-2f HaCaTg HaCaTg |
| AKR1B15 | 4 | 5 | ||||||||
| AKR1C1 | 10 | 16 | Ethacrynic acid, SFN, CDDO | yes yes yes yes yes |
yes yes yes yes yes |
HT29b HepG2c MCF10Aa MCF12Aa HBEC-KTd |
KEAP1 si-RNA KEAP1 si-RNA KEAP1 sh-RNA KEAP1 si-RNA NFE2L2 si-RNA CRISPR/Cas9 |
yes yes yes yes yes yes |
yes yes yes yes yes |
MCF-10Aa MCF-12Aa HK-2f HaCaTg HaCaTg HK-2f A-549d |
| AKR1C2 | 15 | 24 | Ethacrynic acid, SFN, CDDO | yes yes yes yes yes |
yes yes yes yes yes |
HT29b HepG2c MCF10Aa MCF12Aa HBEC-KTd |
NFE2L2 KEAP1 si-RNA KEAP1 si-RNA KEAP1 si-RNA KEAP1 sh-RNA NFE2L2 si-RNA CRISPR/Cas9 NFE2L2 |
yes yes no yes no yes |
yes yes yes no yes |
MCF-10Aa MCF-12Aa HaCaTg HK-2f HaCaTg HK-2f A-549d |
| AKR1C3 | 4 | 6 | Ethacrynic acid, SFN, CDDO | yes yes yes yes |
yes yes yes yes |
HepG2c MCF10Aa MCF12Aa HBEC-KTd |
KEAP1 si-RNA KEAP1 si-RNA KEAP1 sh-RNA KEAP1 si-RNA NFE2L2 si-RNA CRISPR/Cas9 NFE2L2 |
yes yes yes yes yes yes |
yes yes yes |
MCF-10Aa MCF-12Aa HK-2f HaCaTg HaCaTg A-549d |
| AKR1C4 | 2 | 2 | ||||||||
| AKR1D1 | 11 | 27 | ||||||||
| AKR1E2 | 6 | 3 | ||||||||
| AKR7A2 | 4 | 9 | Methyl gloxal | yes | yes | HepG2e | NFE2L2 si-RNA | yes | yes | Hep-G2e |
| AKR7A3 | 1 | 4 | ||||||||
aAgyeman et al. (2012).
bCiaccio et al. (1994).
cBurczynski et al. (1999).
dMurray et al. (2019).
fJung (2013).
gMacLeod (2009).
*Cell lines: HK-2 (renal carcinoma); MCF-10A, MCF-12A (breast carcinoma); A-549 (lung adenocarcinoma); Hep-G2 (hepatoma cells); HaCaT (human keratinocytes).
Prototypic NRF2 activators such as R-sulforaphane (SFN) and the triterpenoid 1-(2-Cyano-3,12,28-trioxooleana-1,9(11)-dien-28-yl)-1H-imidazole (CDDO) have been used as cancer chemo-prevention agents in that they could lead to the inactivation of electrophiles that would damage DNA and initiate cancer (Yates et al., 2006; Kensler et al., 2013). The same activators could also lead to the elimination of cytotoxic species required for the efficacy of the cancer chemotherapeutic response. Thus, the activation of NRF2 leading to chemoresistance is considered the “dark side” of NRF2 activation.
Studies by Ciaccio and Tew originally showed that human colon cancer cells that were resistant to ethacrynic acid had highly elevated levels of dihydrodiol dehydrogenase (Ciaccio et al., 1993; Ciaccio and Tew, 1994; Ciaccio et al., 1994), which at that time was a trivial name given to AKR1C1-AKR1C4 (Burczynski et al., 1998). Further, Northern analysis and RNA protection studies showed that AKR1C1-AKR1C3 transcripts were upregulated by electrophilic and oxidative stress in HepG2 cells (Burczynski et al., 1999) (Table 2). MacLeod et al. (2009) showed that small interfering-RNA (si-RNA) for KEAP1 in human immortal keratinocytes (HaCaT) resulted in upregulation of AKR1B10, AKR1C1/2, and AKR1C3 transcripts by reverse-transcription polymerase chain reaction and AKR1B10 and AKR1C1/2 protein. Similarly, NFE2L2 si-RNA resulted in downregulation of the same AKRs. In another study, it was found that KEAP1 si-RNA resulted in upregulation of AKR1B10, AKR1C1, AKR1C2, and AKR1C3 in human breast cancer cells at the transcript level and at the protein level as well by stable isotope labeling by amino acids in cell culture-labeling followed by mass spectrometry. Furthermore, this transcriptomic and proteomic analysis showed that these genes were the most highly regulated by the NRF2-KEAP1 pathway (Agyeman et al., 2012). Recent CRISPR/Cas9 gene editing of NFE2L2 in A549 lung adenocarcinoma cells showed that high constitutive expression of AKR1C1, AKR1C2, and AKR1C3 was eliminated by NRF2 deletion (Murray et al., 2019b). In A549 cells, KEAP1 is hypermethylated and mutated, leading to high levels of NRF2 (Wang et al., 2008; Fabrizio et al., 2019). It is now known that the majority of the human AKR genes are regulated by the NRF2-KEAP1 pathway and these data are summarized in Table 2. These findings suggest that NRF2 activation would exacerbate drug resistance mediated by AKRs. In addition, NRF2 activation can lead to an increase in GSH biosynthesis via induction of gamma-glutamyl cysteine synthetase light chain and increase cofactor availability for glutathione-S-transferases (Dinkova-Kostova et al., 2005). Increased glutathione-S-transferase activity can eliminate cytotoxic species generated by chemotherapeutic agents (e.g., 4-hydroxy-2-nonenal and acrolein) by glutathione conjugation. Thus, a place exists for both AKR inhibitors and inhibitors of NRF2 to surmount drug resistance. Examples of how AKRs mediate resistance to major classes of chemotherapeutic drugs and antihormonal therapies follow in sections 4 and 5.
III. Somatic Mutations and Epigenetic Regulation of the Nuclear Factor-Erythroid 2 p45-Related Factor 2-Kelch-Like ECH-Associated Protein 1 Pathway
Somatic mutations in KEAP1 that suppress its activity or epigenetic regulation of the NRF2-KEAP1 pathway could lead to high constitutive expression of AKRs and could contribute to cancer drug resistance in multiple tumor types.
There are 174 KEAP1 somatic mutations in human lung cancer which may affect the expression of NRF2. They reside in the intervening region (IVR) domain responsible for the ubiquitination of NRF2, and in the double glycine repeat (DGR) domain which interacts with NRF2 (Fig. 3) (Canning et al., 2015; Grossman et al., 2016). These mutations are often observed in different lung cancer subtypes: for example, the A427V mutant (located in the DGR domain of KEAP1) is seen in small cell lung carcinoma (Ohta et al., 2008), the H200P and R272C mutants (located in the IVR domain of KEAP1) are observed in adenocarcinoma (Ohta et al., 2008), and the G430C, R415G, and G364C mutants (located in the DGR domain of KEAP1) are seen in adenocarcinoma. Early detection of KEAP1 mutations in different subtypes of lung cancer may predict early onset insensitivity to chemotherapeutic agents and could be clinically actionable. Somatic mutations of NFE2L2 in the coding region have also been observed in human cancers (Kerins et al., 2018). NRF2 mis-sense mutations occur in amino acids in the DLG or ETGE motifs which interact with KEAP1, resulting in aberrant cellular accumulation of NRF2. Cells expressing NRF2 mutants display constitutive induction of cytoprotective enzymes and are insensitive to KEAP1-mediated regulation.
Fig. 3.

Domain structures of KEAP1 and NRF2. KEAP1 has four domains, the N-terminal Broad complex, Tramtrack and Bric-à-Brac (BTB) domain which binds cullin 3 ubiquitin ligase, an IVR, and the Kelch domain which binds NRF2. NRF2 contains seven functional domains, Neh1–7. Neh1 contains the DNA binding and heterodimerization domain for small maf, while Neh2 is the regulatory domain which interacts with Keap1 via its DLG and ETGE motifs.
Mutations of KEAP1 and NFE2L2 occur in more than 30% of lung cancer cases and may contribute to drug resistance. The Cancer Genome Atlas database catalogs 226 unique NFE2L2-mutant tumors identified from 10,364 cancer cases (Kerins et al., 2018). NFE2L2 mutations were found in 21 out of 33 different tumor types. A total of 11 hotspots were identified. Of these, amino acids in the DLG motif R25 to R34 were among the common hotspots observed.
The NRF2-KEAP1 pathway is also epigenetically regulated by DNA methylation, histone modification, and long non-coding-RNA (Guo et al., 2015). The methylation pattern of CpG islands in the KEAP1 promoter differ between normal lung and lung cancer and between normal human bronchial epithelial cells and human lung carcinoma cells (Fabrizio et al., 2019), and similar differences were noted in colorectal cancer cells and tissues (Hanada et al., 2012). Treatment of lung cancer cells with the methylation inhibitor 5′-aza-2′-deoxycytidine restored KEAP1 mRNA levels, suggesting an epigenetic mechanism of KEAP1 regulation. Thus, hypermethylation of KEAP1 leads to its suppression. It is unknown if the methylation status of KEAP1, and hence its regulation, is affected by chronic chemotherapeutic drug exposure to impose epigenetic memory.
IV. Resistance to Major Cancer Chemotherapeutics Mediated by Aldo-Keto Reductases
Chemotherapeutic drug resistance is a major mechanism by which tumors can evade drug treatment. Resistance is multifactorial and can include increased drug metabolism, e.g., induction of CYP3A4 (Yao et al., 2000), changes in expression of solute uptake transporters [solute carriers (SLC)] and drug efflux pumps, e.g., ABC transporters (P-glycoprotein and multiple drug resistance proteins) (Gupta et al., 2020), and increased GSH biosynthesis to increase conjugation capacity by glutathione-S-transferases and counter oxidative stress. SLC are less studied as compared with ABC transporters, although about 70% of cytotoxic compounds depend on SLC transporters (Lanthaler et al., 2011). As uptake transporters, they regulate influx of drugs (e.g., SLC29A1 mediates the influx of fluorouracil, SLC35A2 and SLC38A5 mediate the influx of cis-platin, and more than 20 SLC mediate the uptake of methotrexate) and affect their intracellular concentrations. The role of these transporters was identified by genetic screens against 60 different cytotoxic drugs (Girardi et al., 2020). Downregulation of SLC genes may prevent drug uptake, and upregulation may support higher energy and nutrition requirement of cancer cells. Alterations in expression of a number of SLC genes has been observed in ovarian cancer cell lines W1 and A2780 resistant to cis-platin, doxorubicin, topotecan, and paclitaxel (Januchowski et al., 2013, 2014). Since multiple mechanisms of drug resistance can exist, treatment of resistance by targeting a single mechanism is likely to fail since, under the selection pressure of drug treatment, the tumor can adapt and substitute other pathways of resistance.
Numerous studies implicate human AKRs in drug resistance to the major classes of cancer chemotherapeutic agents: the anthracyclines (daunorubicin and doxorubicin); mitomycin; cis-platin, methotrexate, oracin, vinca alkaloids, and cyclophosphamide. Three mechanisms may account for the drug resistance observed: 1) involvement of AKRs in drug inactivation by catalyzing carbonyl reduction to alcohols; 2) involvement of AKRs in the reduction of quinones to hydroquinones (anthracyclines and mitomycin) with the concomitant production of ROS to increase NRF2 activation and elevate AKR expression by a feed-forward mechanism; and 3) upregulation due to the cellular stress created by drug exposure and elimination of that stress, e.g., inactivation of 4-hydroxy-2-nonenal (4-HNE) produced as a byproduct of lipid peroxidation (Fig. 4). When five human recombinant AKRs were compared for the conversion of 4-HNE to 1,4-dihydroxy-2-nonene, the enzyme with the highest catalytic efficiency for this reaction was found to be AKR1C1 (Burczynski et al., 2001). Direct evidence that these AKRs are involved in drug resistance comes from genetic approaches, e.g., RNA interference, transfection studies, and pharmacological inhibition of AKRs. While overexpression of the AKRs is commonly mediated by the NRF2-KEAP1 pathway, they can also be overexpressed as part of disease pathogenesis. In the subsections that follow, we describe the mode-of-action of each therapeutic agent, evidence that AKRs are involved in resistance to that drug based on enzyme activity, transfection studies, forced overexpression, RNA interference, and association studies with overexpression in cancer cell lines. Where possible, we address the relative contribution of AKRs to the drug resistance phenotype. The ability of AKR isoform inhibitors to reverse drug resistance and synergize the effects of chemotherapeutic drugs are described in sections 7 and 8, respectively.
Fig. 4.

Drug resistance mechanisms mediated by AKRs. In mechanism 1, AKRs inactivate carbonyls on chemotherapeutic agents adding in their elimination; in mechanism 2, AKRs redox cycle p-quinones and produce ROS to activate NRF2 to increase AKR gene expression; in mechanism 3, AKRs inactivate breakdown cytotoxic products of lipid peroxides such as 4-HNE.
A. Anthracyclines (Daunorubicin, Doxorubicin, Idarubicin, and Epirubicin)
The anthracycline cancer chemotherapeutic agents intercalate double-stranded DNA and inhibit transcription and replication. They form a heterotrimeric complex with topoisomerase II and DNA. Topoisomerase catalyzes double-strand DNA breaks when uncoiling supercoiled DNA, and the ternary drug complex prevents religation of the broken strands essential for DNA replication (Gewirtz, 1999). The AKRs can eliminate the anthracyclines via carbonyl reduction. The anthracyclines also possess a p-quinone in their structure which can undergo one electron and two electron redox cycling leading to the production of ROS and oxidative stress (Fig. 5). The production of ROS leads to induction of the AKR genes which can eliminate both the anthracycline and the 4-HNE produced as a result of ROS.
Fig. 5.

Structures of daunorubicin and doxorubicin (representative anthracyclines) and mitomycin inactivated by AKRs. Arrow on structures on left show the carbonyl group reduced and the common p-quinone structure. Redox-cycling (right) shows the formation of ROS that could result in the formation of cytotoxic 4-HNE that would be inactivated by AKRs, and the formation of the hydroquinone which can be eliminated as a conjugate.
Aldehyde reductase (AKR1A1) was first identified as “daunorubicin reductase” by Felsted et al. (1974). This activity was important since it eliminated the active drug by forming daunorubicinol, but in doing so formed this cardiotoxic metabolite. Thus, AKR1A1 inhibitors would be chemoprotective and promote chemosensitization (Fig. 5). The involvement of carbonyl reduction in anthracycline drug resistance was supported by transfection studies from the Maser group, which showed that overexpression of carbonyl reductase, AKR1A1, and AKR1B1 were involved in drug resistance in human pancreatic cancer cells (Plebuch et al., 2007). AKRs have since been implicated in resistance to a wide range of anthracyclines including daunorubicin, doxorubicin, idarubicin, and epirubicin.
Daunorubicin now appears to be inactivated by AKR1A1, AKR1C2, and AKR1B1 (Ohara et al., 1995; Plebuch et al., 2007a) and AKR1C3 (Hofman et al., 2014). Examination of catalytic efficiencies showed that AKR1C3 had the highest catalytic efficiency for the reduction of daunorubicin, while reduction of doxorubicin and idarubicin occurred with 2- to 5-fold lower catalytic efficiencies, respectively (Hofman et al., 2014). A dose-dependent relationship existed between the level of AKR1C3 expressed and the ensuing drug resistance observed. AKR1B10 also reduces daunorubicin and idarubicin but is less active toward doxorubicin and epirubicin (Zhong et al., 2011).
AKR1C3 has been implicated in resistance to the anthracyclines in A549 (lung adenocarcinoma), HeLa (cervical carcinoma), MCF-7 (breast cancer cells) and HCT116 (colon cancer) cells by transfection studies and restoration of drug sensitivity with the AKR1C3 inhibitor 2’-hydoxyflavanone (Hofman et al., 2014). The mechanism of AKR1C3-mediated doxorubicin resistance was examined in MCF-7/Dox resistance cells, where overexpression of AKR1C3 led to the loss of the tumor suppressor gene phosphatase and tensin homolog (PTEN) and activated protein kinase B (AKT) signaling (Zhong et al., 2015). In a microfluidic model of a stage IV glioblastoma cell line, in which the microenvironment could be manipulated, transcriptome sequencing identified overexpressed genes responsible for the early appearance of doxorubicin drug resistance, and were found to correspond to AKR1B1, AKR1C1, AKR1C2, and AKR1C3 (Han et al., 2016).
Association of AKR overexpression with daunorubicin resistance has been observed in numerous cancer cell lines, including the overexpression of AKR1A1 and AKR1B1 in pancreatic adenocarcinoma cells (Plebuch, et al., 2007a); the overexpression of AKR1B10 in lung cancer cells (Zhong et al., 2011); and the overexpression of AKR1C3 in lung, colon, breast cancer, and cervical cancer cell lines (Hofman et al., 2014). Similarly, an association of AKR overexpression with doxorubicin resistance has been observed in multiple cell lines including those from cervical cancer involving AKR1B1 (Lee et al., 2002); from nonsmall lung cancer involving AKR1C1, AKR1C2, and AKR1C3 (Hung et al., 2006; Wang et al., 2007); from breast cancer involving AKR1C2 and AKR1C3 (Veitch et al., 2009; Heibein et al., 2012); and from gastric cancer involving AKR1B1 (Morikawa et al., 2015). Resistance to idarubicin and epirubicin has also been linked to the expression of AKR enzymes in breast cancer with involvement from AKR1C1, AKR1C2 (Veitch et al., 2009), and AKR1C3 (Hofman et al., 2014), with involvement of AKR1C3 (Hofman et al., 2014) and AKR1B10 in lung cancer (Zhong et al., 2011), and with involvement of AKR1C3 in colon and cervical cancer cell lines (Hofman et al., 2014). It is uncertain whether the mechanism of resistance mediated by AKRs is due to carbonyl reduction on the side chain, elimination of 4-HNE, or both. The relative contribution of AKR1C2 to doxorubicin resistance was assessed in MCF-7 cells. It was found that MCFDox-2 cells (that were ABC transporter null) could have their resistance reversed by 5β-cholanic acid (an AKR1C2 inhibitor) but, in cells that were resistant to epirubicin and overexpressed an ABC transporter, the AKR1C2 inhibitor was without effect (Veitch et al., 2009).
B. Mitomycin
Mitomycin is widely used for patients with advanced metastatic cancers of the stomach and colon. Mitomycin undergoes both enzymatic and spontaneous rearrangement to form bifunctional or trifunctional alkylating agents (Wellestein, 2018). It cross links DNA at the N6 position of adenine and the O6 and N7 position of guanine inhibiting DNA synthesis. Mitomycin is also a p-quinone which can redox cycle both enzymatically and nonenzymatically to the hydroquinone. The principal enzyme involved in two electron reduction of the p-quinone is NADPH: quinone oxidoreductase (NQO1). Formation of the hydroquinone can lead to its oxidation back to the quinone with consumption of molecular oxygen and the formation of ROS, establishing a futile redox cycle leading to oxidative damage of lipids and DNA. AKRs also have the potential to catalyze two electron quinone reduction to form the hydroquinone and exacerbate mitomycin redox cycling. However, in both instances, formation of the hydroquinone prevents the formation of the more reactive semiquinone anion radical and can lead to elimination of the hydroquinone as conjugates (Fig. 5). Thus, two mechanisms of chemoresistance can exist, formation of the hydroquinone for conjugation and elimination of lipid peroxide breakdown products resulting from ROS formation. 4-HNE is inactivated principally by AKRB10 in the colon and may contribute to mitomycin drug resistance by eliminating the cytotoxic 4-HNE (Matsunaga et al., 2011). AKR1B10 is considered a potential biomarker for colon cancer and tumors of the small intestine and was originally named as “small intestine-like” aldose reductase. Notably, both NQO1 and AKR1B10 are regulated by the NRF2-KEAP1 pathway. The contribution of AKR1B10 to mitomycin drug resistance was assessed in nonresistant HCT-15 colon cells where overexpression of AKR1B10 and silencing of AKR1B10 decreased and increased sensitivity to the drug (Matsunaga et al., 2011).
C. Cis-Platin and Platin-Based Drugs
Cis-platin is transported into cancer cells by a high affinity Cu2+ solute carrier transporter. Upon aquation, it loses its chloride ligands and the activated platinum complexes react with DNA and sulfhydryl groups to form protein-DNA cross links that inhibit transcription (Wellestein, 2018). Simpkins showed that growth of ovarian cancer cells in the presence of cis-platin made them drug-resistant to platin therapy. Gene expression analysis identified dihydrodiol dehydrogenase (AKR1C1) as one the most overexpressed genes in the resistant cells (Deng et al., 2002). To demonstrate causality, transfection of the AKR1C1 cDNA into parental ovarian cancer cells established the drug-resistant phenotype. Subsequent studies showed dihydrodiol dehydrogenase 1 and 2 (AKR1C1 and AKR1C2) were responsible for cis-platin and carboplatin resistance in human ovarian, cervical, lung, and germ cell tumor cell lines (Deng et al., 2004). Transfection of si-RNA against AKR1C1 and AKR1C2 in 2008/C13 cells restored sensitivity to cis-platin with a concomitant production of ROS (Chen et al., 2008). In the cis-platin-resistant HeLa cervical cell line silencing of AKR1C1, AKR1C2, AKR1C3, and AKR1C4 restored drug sensitivity (Shiiba et al., 2017). AKR1B10 was later implicated in cis-platin resistance by eliminating the accumulation of 4-HNE formed. The resistance was also accompanied by downregulation of PPARγ, and sensitivity could be restored by the transfection of PPARγ and the addition of a PPARγ agonist rosiglitazone (Matsunaga et al., 2016b). Thus, exposure to cis-platin produces both ROS and an electrophilic signal (4-HNE) to activate the NRF2-KEAP1 pathway and induce AKR genes. The downregulation of PPARγ is a pro-proliferative response and could counter the cytotoxic effects of cis-platin.
AKR overexpression has since been associated with resistance to cis-platin in many different cancer cells including: AKR1B1 and AKR1C1 in cervical cancer (HeLa) cells (Lee et al., 2002; Shiiba et al., 2017); AKR1C1 and AKR1C3 in colon cancer cells (Matsunaga et al., 2013); AKR1C1 and AKR1C2 in nonsmall cell lung carcinoma (Chen et al., 2010; Wang et al., 2007); AKR1C1, AKR1C2, AKR1C3, and AKR1C4 in oral cancer (Shiiba et al., 2017); AKR1C1 in head and neck squamous carcinomas (Chang et al., 2019); AKR1C1 in nasopharyngeal carcinoma (Zhou et al., 2020), where loss of AKR1C1 expression was a good prognostic indicator of chemosensitivity to cis-platin; and overexpression of AKR1C1, AKR1C2, and AKR1C3 in metastatic bladder cancer were associated with resistance to cis-platin (Matsumoto et al., 2016).
Association of AKR overexpression with resistance to carboplatin has also been seen in cancer cell lines including: AKR1C1 and AKR1C2 in cervical cancer (Deng et al., 2004); AKR1C1 and AKR1C2 in ovarian cancer (Deng et al., 2004); AKR1C1 and AKR1C2 in metastatic lung cancer and germ cell tumors (Deng et al., 2004); and in oxaliplatin resistance in gastric cancer involving AKR1C1 and AKR1C3 overexpression (Chen et al., 2013). Thus, AKR overexpression is part of the cancer chemotherapeutic resistance phenotype to platin-based cancer chemotherapeutics.
D. Methotrexate
Methotrexate is a potent competitive inhibitor of dihydrofolate reductase and prevents the formation of tetrahydrofolate, which is required for the de novo synthesis of thymidine and blocks DNA synthesis in rapidly proliferating cells (Rajagopalan et al., 2002). Methotrexate produces an 80%–90% cure rate in choriocarcinoma, but drug resistance ensues in the remaining patients. JEG-3 choriocarcinoma cells can be made methotrexate drug-resistant by the overexpression of AKR1C3 and cells can be sensitized to the drug by AKR1C3 small hairpin-RNA. Increased drug sensitivity is associated with increased ROS formation, apoptosis, and cell cycle arrest (Zhao et al., 2014). Methotrexate drug resistance has since been observed in HT29 human colon cancer cells and is associated with the overexpression of AKR1C1. AKR1C1 was shown to be transcriptionally regulated by phosphorylated Sp1 and Sp3 in this instance (Selga et al., 2008). It is difficult to assess the contribution of AKR1C isoforms to methotrexate resistance in JEG-3 cells since other mechanisms contribute, which include the overexpression of the chloride intracellular channel protein-1 and multi-drug resistance-associated protein (MRP1) (Wu et al., 2017).
E. Oracin
Oracin is a promising cytostatic drug which is inactivated by carbonyl reduction by stereoselective reduction of its 11-keto group (Fig. 6). The enzyme most implicated in this transformation is AKR1C1 when AKRC1, AKR1C2, and AKR1C4 were compared, where AKR1C1 gave predominately the (+) enantiomer (Wsol et al., 2007). Subsequent studies showed that AKR1C3 was also involved (Novotna et al., 2008). Since the ARK1C genes are upregulated by stress response, it is not surprising that their overexpression may lead to drug resistance resulting in increased metabolism of oracin. The relative contribution of AKRs to oracin drug resistance has yet to be determined.
Fig. 6.

Inactivation of oracin by carbonyl reduction by AKRs.
F. Vincristine
Vincristine is a vinca alkaloid and an antitubulin agent that prevents the proliferation of rapidly growing cells by preventing formation of the mitotic spindle (Jordan, 2002). Vincristine is the drug of choice for T-cell acute lymphoblastic leukemia (T-ALL). T-ALL cells from therapy-resistant patients overexpress AKR1C1-AKR1C3, and decreased expression using si-RNA improved sensitivity to the drug (Bortolozzi et al., 2018). Furthermore, categorization of patients as therapy responders or nonresponders based on minimal residual disease showed that minimal residual disease–positive resistant patients had high levels of AKR1C1, AKR1C2, and AKR1C3. As well as acting as an antitubulin, vinca alkaloids can cause collapse of the mitochondria, elevating ROS production. As the expression of AKR1C isoforms is a component of the antioxidant response in humans, it is not surprising that their upregulation may attenuate the effect of the vinca alkaloids. Other antitubulin-based agents, e.g., taxanes, can upregulate drug efflux pumps, e.g., ATP-Binding Cassette Subfamily B transporters, indicating that different drug-resistant mechanisms may work concurrently to prevent a chemotherapeutic drug response (Matsunaga et al., 2016a).
G. Irinotecan
Irinotecan is a topoisomerase inhibitor often used to treat colon cancer and small lung cell cancer in combination with fluorouracil or cis-platin, respectively. Two dimensional sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by proteomic analysis by in-gel digest with trypsin followed by electrospray ionization-quadrupole time-of-flight mass spectrometry identified 15 proteins that were overexpressed in irinotecan-resistant LoVo (human colon adenocarcinoma) cells versus the parental cells. One of these proteins was assigned as AKR1A1 based on coverage obtained with 12 tryptic peptides (Peng et al., 2010). However, the exact role of AKR1A1 in Irinotecan drug resistance remains to be determined. Recently, Irinotecan resistance was linked to AKR1C3 upregulation. Matsunaga et al. (2020) observed increased AKR1C3 expression and activity in Irinotecan resistant colon cancer cell lines DLD-1, RKO, and LoVo as compared with parental sensitive cells. In these cells, AKR1C3 overexpression decreased and AKR1C3 silencing restored chemosensitivity. Treatment of parental colon cancer cells with Irinotecan leads to formation of cytotoxic 4-HNE. In chemoresistant cells, higher 4-HNE reductase activity and consequently lower 4-HNE levels were seen as compared with parental cells. As AKR1C3 is one of several AKR1C enzymes that can inactivate 4-HNE (Burczynski et al., 2001), the authors concluded that 4-HNE detoxification by AKR1C3 may represent the major resistance mechanism to Irinotecan.
H. Cyclophosphamide
Cyclophosphamide is activated by P450 isoforms (2A6, 2C9, 2C10, and 3A4) to the active metabolite 4-hydroxycyclophosphamide, which exists in its tautomeric form aldophosphamide, and this aldehyde is detoxified by aldehyde dehydrogenase (ALDH) to yield carboxyphosphamide. A portion of aldophosphamide can decompose to yield phosphoramide mustard and acrolein, which are the cytotoxic species responsible for the ability of cyclophosphamide to inhibit the growth of rapidly proliferating cells (Fig. 7). Examination of gene expression profiles in medulloblastoma cell lines resistant to cyclophosphamide showed a 20-fold elevation in AKR1B10 expression. AKR1B10 may work in concert with ALDH1A1 to inactivate the reactive aldehyde aldophosphamide and contribute to cyclophosphamide resistance (Bacolod et al., 2008). Acrolein can also be eliminated by AKR1B1, AKR1B10, and AKR7A2, where AKR1B10 is the major contributor (Shen et al., 2011; Li et al., 2012a) However, because acrolein is a strong electrophile, its major route of metabolism will be GSH conjugation. The elimination of the aldophosphamide by AKR1B10 is likely a major contributing mechanism of cyclophosphamide drug resistance.
Fig. 7.

Competing roles of ALDH1A1 and AKRs in the metabolism of cyclophosphamide.
I. Tyrosine Kinase Inhibitors
Several tyrosine kinase inhibitors such as imatinib, nilotinib, erlotinib, and ponatinib are currently used for the treatment of some cancers. The AKRs do not appear to be involved in resistance to these agents. To the contrary, the tyrosine kinase inhibitor ibrutinib is an inhibitor of AKR1C3 and can counteract anthracycline drug resistance mediated by AKR1C3 (Morell et al., 2020). Apatinib also inhibits the NRF2 pathway which would lead to a downregulation of AKR genes regulated by NRF2 and could surmount drug resistance to other chemotherapeutic agents (Sun et al., 2020). Imatinib, nilotinib, and ponatinib are amides, while erlotinib does not contain a carbonyl group and therefore, these compounds would not be substrates for AKRs.
V. Resistance to Antihormonal Therapies Mediated by Aldo-Keto Reductases
AKR1C enzymes are intimately involved in the biosynthesis and metabolism of steroid hormones. In particular, AKR1C3 is one of the principal enzymes involved in peripheral androgen biosynthesis and is upregulated by androgen deprivation in castration-resistant prostate cancer, the lethal form of prostate cancer (Hofland et al., 2010; Hamid et al., 2013; Mitsiades et al., 2012). As a result, AKR1C3 is implicated in drug resistance to the new androgen receptor signaling inhibitors (ARSI), e.g., abiraterone acetate (an inhibitor of P450c17: 17α-hydroxylase/17,20-lyase) and enzalutamide (a potent androgen receptor antagonist) used in the treatment of castration-resistant prostate cancer (Scher et al., 2010; Fizazi et al., 2012).
The 17-ketosteroid reductase activity of AKR1C3 converts 4-androstene-3,17-dione to testosterone, which can also act as substrate for the peripheral formation of 17β-estradiol via aromatase. By providing a substrate for aromatase, overexpression of AKR1C3 may also play a role in hormonal-dependent breast cancer (Byrns et al., 2010; McNamara and Sasano, 2019). Concurrent overexpression of AKR1C1, which is the principal 20-ketosteroid reductase involved in the elimination of progesterone as 20α-hydroxyprogesterone, can work in concert with AKR1C3 to increase the ratio of 17β-estradiol:progesterone in estrogen-dependent malignancies (Penning et al., 2000; Rizner et al., 2006).
A. Antiandrogenic Therapy
C4-2B prostate cancer cells grown in the presence of either enzalutamide or abiraterone acquire drug resistance to these agents and overexpress AKR1C3 (Liu et al., 2015; Liu et al., 2017). Xenografts of these resistant cells fail to respond to these drugs, but indomethacin (INDO), a potent and selective inhibitor of AKR1C3, can surmount this drug resistance and have antitumor activity. AKR1C3 is repressed by androgens and upregulated by androgen deprivation (Hofland et al., 2010; Hamid et al., 2013; Mitsiades et al., 2012). Enzalutamide is a potent androgen receptor (AR) antagonist and can remove the repressive effects of the AR on the AKR1C3 promoter leading to overexpression of AKR1C3. This repressive effect can also be surmounted by overexpression of the TMPRSS2-ERG, a fusion protein that arises in late-stage prostate cancer. Erythroblast transformation specific related gene replaces the AR on the AKR1C3 promoter, and the resultant increase in AKR1C3 in concert with steroid 5α-reductase leads to more 5α-dihydrotestosterone to bind to AR (Powell et al., 2015). This leads to a feed-forward mechanism to increase TMPRSS2-ERG expression and maintain AKR1C3 levels to sustain ARSI drug resistance (Powell et al., 2015).
Abiraterone may also deprive the AR of its ligand leading to removal of the repressive effects of AR on the AKR1C3 promoter. This could be accomplished by inhibition of P450c17 in both the adrenal and the tumor itself. However, evidence for de novo synthesis of C19 steroids in prostate tissue is poor, and instead, the mechanism may involve inhibition of 3β-hydroxysteroid dehydrogenase type 1 by the delta-4-abiraterone metabolite (Li et al., 2012b).
B. Antiestrogenic Therapy
Exemestane is a steroidal based aromatase inhibitor used in the endocrine therapy of hormonal-dependent breast cancer. Its primary metabolite is 17β-dihydroexemestane, which is produced by carbonyl metabolism (Platt et al., 2016). While 17β-dihydroexemestane can still function as an aromatase inhibitor, it can also undergo glucuronidation by UDP-glucuronosyltransferases leading to its elimination. Carbonyl reductases responsible for the formation of 17β-dihydroexemestane include the AKR1C enzymes (Platt et al., 2016). However, AKR1C3 has very low catalytic efficiency for exemestane making it doubtful that it is a major mechanism of drug resistance.
VI. Inactivators of the Nuclear Factor-Erythroid 2 p45-Related Factor 2-Kelch-Like ECH-Associated Protein 1 System
The preceding sections make the case that NRF2 inactivators would sensitize tumor cells to chemotherapy. Natural products exist that act as NRF2-inactivators that would counter this effect, e.g., the quassinoid brusatol, luteolin, chrysin, and wogonin. Brusatol was identified as a natural product inhibitor of NRF2, but it does not bind to the transcription factor directly. Rather, it is an inhibitor of both cap-dependent and cap-independent translation, which is required for the stability of short-lived proteins such as NRF2 (Harder et al., 2017). By contrast, flavonoids such as chrysin repress NRF2 expression through downregulation of the P13K-AKT and extracellular signal-regulated kinase (ERK) pathway (Gao et al., 2013). High-throughput screening using ARE-luciferase reporters gene constructs could be used to identify compounds that would attenuate NRF2 signaling and act as NRF2 inactivators.
VII. Inhibitors of Aldo-Keto Reductases to Surmount Drug Resistance
The association of AKR forced overexpression with chemotherapeutic drug resistance, AKR overexpression in cancer cell lines, and genetic silencing of AKRs to reverse drug resistance suggests that AKR inhibitors could be exploited to surmount drug resistance. A number of AKR inhibitors have been used in vitro and in vivo for this indication and are summarized in Table 3 and (Fig. 8). However, it is apparent that for many chemotherapeutic agents, a battery of AKR enzymes may be involved, suggesting that if only one AKR is inhibited, others may substitute. Thus, both AKR isoform-selective inhibitors have been used as well as pan-AKR1C inhibitors. One major class of drugs that inhibits the AKR1C isoforms are the nonsteroidal anti-inflammatory drugs (NSAIDs) which can discriminate between the four AKR1C isoforms (Byrns et al., 2008). For example, salicylate is an AKR1C1 isoform-selective inhibitor, and INDO is an AKR1C3 isoform-selective inhibitor, whereas flufenamic acid is a pan-AKR1C inhibitor (Byrns et al., 2008; El-Kabbani et al., 2010; El-Kabbani et al., 2011). NSAIDs and their analogs have played a prominent role in determining the utility of AKR inhibitors to surmount drug resistance, especially in the case of cis-platin.
TABLE 3.
Drug resistance to chemotherapeutic agents reversed by AKR inhibitors
| Platinum-Based Drugs | ||||
|---|---|---|---|---|
| AKR | AKR Inhibitors | Chemotherapeutic Drug Resistance Inhibited | Cancer/Cell Line | Reference |
| AKR1B1 | 1-Benzyl-3-hydroxy-2(5H)-oxopyrrole-4-carboxylate (EBPC) | Cis-platin | Cervical cancer HeLa |
(Lee et al., 2002) |
| AKR1C1, AKR1C2, AKR1C3, AKR1C4 | Mefenamic acid | + mouse xenograft | (Shiiba et al., 2017) | |
| AKR1C1 AKR1C3 |
3-Bromo-5-phenylsalicylic acid (AKR1C1) Tolfenamic acid (AKR1C3) |
Colon cancer HCT15 |
(Matsunaga et al., 2013) | |
| AKR1C1, AKR1C2, AKR1C3, AKR1C4 |
Mefenamic acid | Oral squamous cell carcinoma Sa3 | (Shiiba et al., 2017) | |
| AKR1C1 | 3-Bromo-5-phenylsalicylic acid (Ki = 4 nM) | Head and neck squamous carcinoma 293 T, FaDu Cal-27, HSC-2 and HSC-4 |
(Chang et al., 2019) | |
| AKR1C1 | Flufenamic acid | Metastatic bladder cancer UM-UC-3 |
(Matsunaga et al., 2016a) | |
| AKR1C1, AKR1C2, AKR1C3 |
Phenolphthalein α-methylcinnamic acid, Flufenamic acid |
Oxaliplatin | Gastro-intestinal tract cancer TSGH | (Chen et al., 2013) |
| Anthracyclines | ||||
| AKR1B1 | 1-Benzyl-3-hydroxy-2(5H)-oxopyrrole-4-carboxylate | Doxorubicin | Cervical cancer HeLa |
(Lee et al., 2002) |
| AKR1C2 | 5β-Cholanic acid-3α,7α-diol | Breast cancer MCF-7 |
(Veitch et al., 2009) | |
| AKR1B10, AKR1C3, AKR1C2 | 5β-Cholanic acid-3α,7α-diol | Breast cancer MCF-7 | (Heibein et al., 2012) | |
| AKR1B10 | Oleanoic acid 3-(4-hydroxy-2-methoxyphenyl-1)acrylic acid 3-(3-hydroxyphenyl)propyl ester |
Gastric cancer MKN45 |
(Morikawa et al., 2015) | |
| AKR1C1 AKR1C3 |
3-Bromo-5-phenylsalicylic acid, Tolfenamic acid, |
Daunorubicin | Leukemia (acute monocytic leukemia) U937 |
(Matsunaga et al., 2014) |
| AKR1B10 AKR1C3 |
Emodin | Lung cancer A-549 Liver cancer Hep-G2 |
(Hintzpeter et al., 2016) | |
| AKR1C3 | Purvalanol A Roscovine | Colon cancer HCT 116 |
(Novotná et al., 2018a) | |
| Dinaciclib | (Novotná et al., 2018a, 2018b) | |||
| AKR1C3 | Irutinib Acalabrutinib Buparlisib |
Colon cancer HCT 116 |
(Morrell et al., 2020) (Bukum et al. 2019) |
|
| AKR1B10 | Epalrestat | Daunorubicin, Idarubicin |
Lung cancer NCI-H460 | (Zhong et al., 2011) |
| AKR1C3 | 2-Hydroxyflavanone | Lung cancer A-549 |
(Hofman et al., 2014) | |
| Other drugs | ||||
| AKR1B10 | (Z)-2-(4-methoxyphenyl-imino)-7-hydroxy-N-(pyridin-2-yl)-»H-chromene-3-carbox-amide | Mitomycin-c | Colon cancer HT-29 |
(Matsunaga et al., 2011) |
| AKR1C3 | Baccharin derivatives | Etoposide | Leukemia (acute myeloid leukemia) HL-60, KG-1a |
(Verma et al., 2016) |
| AKR1C1, AKR1C2, AKR1C3 |
6-Medroxy-progesterone acetate | Vincristine | Leukemia (acute lymphoblastic leukemia) CCRF-CEM, DND-41, Loucy |
(Bortolozzi et al., 2018) |
| AKR1C1, AKR1C2, AKR1C3, AKR1C4 | Mefenamic acid | 5-Fluoro uracil | Cervical cancer HeLa + mouse xenograft |
(Shiiba et al., 2017) |
| AKR1C3 | 1,3,5-Trisubstituted amido cinnamic acids Compounds Kv-49a, Kv-49g |
Cytarabine | Leukemia (acute myeloid leukemia) HL-60, TMP-1, KG-1a (T-cell acute lymphoblastic leukemia) primary cells |
(Verma et al., 2019) |
| AKR1C3 | Tolfenamic acid | Irinotecan | Colon cancer cell lines DLD1, RKO, LoVo | (Matsunaga et al., 2020) |
| Antihormone Therapy | ||||
| AKR1C3 | Indomethacin | Enzalutamide Abiraterone Acetate |
Castration resistant prostate cancer | (Liu et al., 2015; 2017) |
| AKR1C3 | Kv-37 and related Baccharin analogs | Enzalutamide | Castration resistant prostate cancer | (Verma et al., 2018) |
| 1,3,5-Trisubstituted amido cinnamic acids Compound Kv-49g |
Apalutamide Darolutamide |
Castration resistant prostate cancer | (Morsy & Trippier, 2020) | |
Fig. 8.

Structures of common AKR inhibitors including KV-37 and the Baccharin analogs.
A. Anthracycline Chemotherapeutics and Aldo-Keto Reductase Inhibitors
AKR inhibitors have been tested in vitro to restore sensitivity to anthracycline drugs in lung, breast, gastric, liver, and colon cancer cell lines and in leukemia (Veitch et al., 2009; Zhong et al., 2011; Heibein et al., 2012; Matsunaga et al., 2013; Hofman et al., 2014; Matsunaga et al., 2014; Morikawa et al., 2015; Hintzpeter et al., 2016; Novotná et al., 2018a, 2018b; Bukum et al., 2019). In the lung adenocarcinoma A549 cell line which has high endogenous expression of AKR1C3, 10 μM 2-hydroxyflavanone, a specific AKR1C3 inhibitor, restored cytotoxicity to daunorubicin and idarubicin (Hofman et al., 2014). In addition, 30 μM emodin, a potent inhibitor of AKR1B10 and AKR1C3, decreased viability of these cells when treated with 0.5 and 1 μM daunorubicin (Hintzpeter et al., 2016). Treatment with 100 μM epalrestat, an AKR1B10 inhibitor, decreased resistance to daunorubicin and idarubicin in the lung cancer NCI-H460 cell line, which has high expression of AKR1B10 but no expression of AKR1B1 (Zhong et al., 2011).
In MCF-7 breast cancer cells made resistant to doxorubicin, high levels of AKR1B10, AKR1C3, and AKR1C2 were expressed, and the specific AKR1C2 inhibitor 5β-cholanic acid-3α,7α-diol restored chemo-sensitivity and prevented nuclear localization of doxorubicin, which is crucial for its action as a topoisomerase II inhibitor. This effect was not seen in epirubicin-resistant or control MCF-7 cells (Veitch et al., 2009; Heibein et al., 2012).
In the MKN45 gastric cancer cell line, Morikawa et al. (2015) observed that exposure to doxorubicin upregulates expression of AKR1B10, AKR1C1, AKR1C2, and AKR1C3, but only the AKR1B10 inhibitors; 20 μM oleanoic acid or 10 μM 3-(4-hydroxy-2-methoxyphenyl-1)acrylic acid 3-(3-hydroxyphenyl)propyl ester significantly sensitized these cells to 60 μM doxorubicin. By contrast, the specific AKR1C1 (3-bromo-5-phenylsalicylic acid), AKR1C2 (ursodeoxycholic acid), and AKR1C3 [E-3-(3-(3-hydroxybenzyl)oxy)-4-(3-phenylpropanoyl)oxy)phenyl)acrylic acid] inhibitors had no effect on cell viability. These results demonstrate that AKR1 isoform specific inhibitors could be used to determine their individual contributions to the drug resistance phenotype.
In the colon carcinoma HCT116 cell line transiently transfected with AKR1C3, the purine based analog cyclin-dependent kinase inhibitors, roscovine and purvalanol A, potently inhibited the metabolism of clinically relevant concentrations of 1.0 μM daunorubicin, yielding IC50 values of 3.7 μM for roscovine and an IC50 value of 6.4 μM for purvalanol A. In the same cells, the cyclin dependent kinase inhibitor dinaciclib inhibited 1 μM daunorubicin metabolism and acted as a potent inhibitor of AKR1C3 and a modest inhibitor of AKR1B10 yielding IC50 values of 0.22 μM and 181 μM respectively for these enzymes. Dinaciclib was found to be a tight-binding inhibitor of AKR1C3 with a Ki 0.07 μM, and sensitivity to daunorubicin was restored when HCT116-AKR1C3 cells were preincubated with dinaciclib (0.1 μM) for 2 hours and then treated with 0.1 to 1.0 mM daunorubicin (Novotná et al., 2018a, b), The 2-hour pretreatment of cells with dinaciclib mimics the 2-hour intravenous infusion used to administer the drug in clinical trial (Flynn et al., 2015).
Recently, a pan-phosphoinositide-3-kinase inhibitor, buparlisib, which is under clinical evaluation for treatment of different cancers, was reported to inhibit AKR1C3 with a Ki = 14 μM. In colon cancer HCT116-AKR1C3 cells, buparlisib inhibited the reduction of 1 μM daunorubicin with an IC50 value = 7.9 μM (Bukum et al., 2019). In human leukemic U937 cells, daunorubicin induced expression of AKR1C1 and AKR1C3 and decreased cell sensitivity to the drug and 4-HNE, while pretreatment with 20 μM 3-bromo-5-phenylsalicylic acid, an AKR1C1 inhibitor, or 20 μM tolfenamic acid, an AKR1C3 inhibitor, restored sensitivity to daunorubicin. The specific AKR1C2 inhibitor ursodeoxycholic acid had no effect on cell viability or drug sensitivity (Matsunaga et al., 2014).
In summary, human enzymes of the AKR1B and AKR1C family mediate resistance to the anthracyclines, and inhibitors of these enzymes can restore drug sensitivity. However, the AKRs involved can vary by cancer cell line depending on their expression level and differing substrate specificity for the anthracycline family of therapeutics, suggesting that precision therapy with these inhibitors would require tumor biopsies for RNA analysis or protein expression.
B. Cis-platin Therapeutics and Aldo-Keto Reductase Inhibitors
The N-phenylanthraniliates, flufenamic acid, and mefenamic acid which are NSAIDs restore sensitivity to cis-platin (Morikawa et al., 2015; Shiiba et al., 2017). Specific or pan-AKR1C inhibitors have been used in colon, cervical, oral squamous, and bladder cancer cell lines to restore sensitivity to cis-platin, where the pan-AKR1C inhibitors flufenamic acid and mefenamic acid also showed effects in explant mouse models (Shiiba et al., 2017; Chang et al., 2019). In the cis-platin-resistant colon cancer cell line HCT15, expression of AKR1C1 and AKR1C3 increased with development of resistance. A 2-hour pretreatment with 40 μM 3-bromo-5-phenylsalicylic acid (a specific AKR1C1 inhibitor) and tolfenamic acid (a specific AKR1C3 inhibitor) or their combination increased cell susceptibility to cis-platin. Use of the selective AKR1C2 inhibitor ursodeoxycholic acid revealed no effect, suggesting that either a pan-AKR1C inhibitor or multiple isoform-selective inhibitors are needed. Sensitivity to cis-platin was enhanced when AKR1C1 and AKR1C3 inhibitors were combined with 0.2 μM proteasomal inhibitor MG132, where a compensatory overexpression of AKR1C1 and AKR1C3 and proteasome subunits was observed (Matsunaga et al., 2013).
In the cis-platin–resistant HeLa cervical cell line and the oral squamous cell carcinoma cell line Sa3, which express high levels of AKR1C1, AKR1C2, AKR1C3 and AKR1C4, the use of 200 and 400 μM of the pan-AKR1C inhibitor mefenamic acid restored chemosensitivity, which was confirmed in vivo after implantation of HeLa resistant cells into nude mice and administration of mefenamic acid (Shiiba et al., 2017).
In the bladder cancer cell lines UM-UC-3, 5637 and J82, treatment with cis-platin increased expression of AKR1C1, AKR1C2, and AKR1C3. The liver and bone metastatic cells originating from the UM-UC-3 xenograft mouse model showed further upregulation of AKR1C1 as well as AKR1C2 and AKR1C3, where AKR1C1 was upregulated by inflammatory cytokines (IL1β and IL-6). The use of the AKR1C inhibitor flufenamic acid (10 μM) decreased UM-UC-3 cell invasion as measured by a Matrigel assay. Combined treatment with 5 μM cis-platin, which is commonly used for treatment of advanced bladder cancer, and 10 μM flufenamic acid restored sensitivity to cis-platin in liver and bone metastatic cancer cells (Matsumoto et al., 2016).
In head and neck squamous carcinoma, AKR1C1 expression is associated with cis-platin resistance, where patients with higher AKR1C1 expression have shorter median survival and recur earlier than those with lower AKR1C1 levels (Chang et al., 2019). In the model cell lines FaDu and HSC-2 with high AKR1C1 expression, silencing of AKR1C1 increased chemosensitivity, but cotreatment with 500 nM 3-bromo-5-phenylsalicilic acid, a potent selective AKR1C1 inhibitor (Ki = 4 nM), showed no effects on the viability of these cells at IC50 concentrations of cis-platin, thus, the authors concluded that AKR1C1 may promote cis-platin resistance in an enzyme-independent manner (Chang et al., 2019).
AKR inhibitors have also been examined to alleviate resistance to oxaliplatin. In the resistant human gastric carcinoma cell line TSGH-S3, which has high expression of AKR1C1 and AKR1C3, use of specific and pan-AKR1C inhibitors [phenolphthalein (0.5 – 5 μM), α-methylcinnamic acid (0.5 -5 μM), and flufenamic acid (10 -50 μM)] restored oxaliplatin resistance. Further knock-down of NFE2L2 decreased the expression of AKR1C1, AKR1C2, and AKR1C3 and reversed the resistance, supporting the crucial role of NRF2 in regulating drug resistance by induction of AKR1C1-AKR1C3 (Zhou et al., 2020).
C. Other Cancer Chemotherapeutics and Aldo-Keto Reductase Inhibitors
AKR inhibitors have also been used to reverse drug resistance to other chemotherapeutic agents. In the colon cancer cell line HT29, which was resistant to mitomycin-c, AKR1B10 was upregulated. High levels of AKR1B10 decreased production of 4-HNE and a 2-hour pretreatment with 5 μM (Z)-2-(4-methoxyphenyl-imino)-7-hydroxy-N-(pyridin-2-yl)-H-chromene-3-carbox-amide, a potent AKR1B10 inhibitor with a Ki = 2.7 nM, restored sensitivity to mitomycin-C (Matsunaga et al., 2011).
In the cis-platin–resistant HeLa cervical cell line, sensitivity could be restored to 5-fluoruracil by 200–400 μM mefenamic acid, a pan-AKR1C inhibitor. Chemosensitivity to 5-fluoruracil was also restored in implants in athymic mice using this strategy (Selga et al., 2008). This represents an example whereby expression of AKR1C can lead to a multidrug resistance phenotype to 5-fluoruracil and cis-platin which can be surmounted by pan-AKR1C inhibition.
Sensitization of T-ALL cells to vincristine could also be achieved by the pan-AKR1C inhibitor 6-medroxyprogesterone acetate. In addition, activation of NRF2 by tert-butyl hydroquinone also increased vincristine resistance (Bortolozzi et al., 2018).
In summary, specific AKR1 and pan AKR1C inhibitors can restore chemosensitivity in cell lines and explant models. Specific AKR1A1, AKR1B10, and AKR1C3 inhibitors can alleviate resistance to individual anthracyclines. AKR1C1 and AKR1C2 inhibitors or pan-AKR1C inhibitors may have roles in alleviating resistance to platinum-based chemotherapeutics. Pan-AKR1C and specific AKR1C inhibitors may also help restore sensitivity to other chemotherapeutics. The translation of these findings to the clinical setting could involve the use of existing drugs, e.g., NSAIDs to chemo-sensitize patients, whereas other isoform specific AKR inhibitors may require further preclinical development.
VIII. Inhibitors of Aldo-Keto Reductases to Potentiate the Effects of Chemotherapeutic Agents
The previous section describes the use of AKR inhibitors to sensitize cells to cancer chemotherapeutic agents. This raises the issue of whether combinations of AKR inhibitors and cancer therapeutic agents will exert a greater effect than either drug alone leading to synergistic effects to inhibit cancer cell growth. Baccharin analogs have been used to target AKR1C3 to obtain synergistic drug responses to enzalutamide, darolutamide, and apalutamide in prostate cancer cells (Verma et al., 2018; Morsy and Trippier, 2020), to etoposide and daunorubicin in AML (Verma et al., 2016), and to daunorubicin and cytarabine in AML and T-ALL (Verma et al., 2019). Synergism was measured using the Chou-Talalay median-effect plot (Chou and Talalay, 1984; Chou, 2010), and in many cases synergism exceeded 200-fold. The observed synergism can be accounted for by the mode-of-action of AKR1C3 in these diseases. In prostate cancer AKR1C3 synthesizes potent androgens for the AR which can bypass the therapeutic efficacy of ARSIs (Adeniji et al., 2013; Knudsen and Penning, 2010). In leukemia, the prostaglandin F synthase activity of AKR1C3 prevents signaling via PPARγ and promotes cellular proliferation.
A. Prostate Cancer
The cinnamic acid derivative KV-37 is a highly selective AKR1C3 inhibitor that has 109-fold selectivity over the AKR1C2 isoform with an IC50 of 66 nM for AKR1C3 inhibition (Fig. 8). KV-37 was found to exert a high degree of synergistic drug interaction upon combination with enzalutamide that results in >200-fold potentiation of cytotoxicity. In the enzalutamide-resistant, high AKR1C3–expressing 22Rv1 prostate cancer cell line (Verma et al., 2018), pretreatment of 22Rv1 cells with KV-37 followed by exposure to enzalutamide, when stimulated to grow in the presence of the AKR1C3 substrate (4-androstene-3,17-dione), resulted in a 208-fold potentiation effect to provide a combination IC50 of 4.3 µM for enzalutamide and KV-37, which is well below the maximum achievable plasma concentration of enzalutamide in patients. Further evidence that AKR1C3 is responsible for enzalutamide resistance was demonstrated in LNCaP cells stably transfected to overexpress AKR1C3 (LNCaP1C3), wherein the cytotoxicity of enzalutamide was significantly reduced (IC50 = 150 µM) compared with AKR1C3-null LNCaP cells (IC50 = 50 μM). Enzalutamide resistance was again reversed upon pretreatment with KV-37 (yielding a combination IC50 = 8.19 µM). The higher combination IC50 values seen in the LNCaP1C3 cells over the 22Rv1 cells is due to the higher expression of AKR1C3 in the LNCaP1C3 cell line. The AKR1C3 inhibitor alone and in combination with enzalutamide resulted in increased apoptosis, and attenuated AR transactivation and PSA expression in both the 22Rv1 and LNCaP1C3 cells.
A second generation AKR1C3 inhibitor developed around the same chemical scaffold, KV-49g, was >2800-fold more selective for AKR1C3 over AKR1C1 and AKR1C2, possessed an IC50 of 70 nM, and was found to potentiate the effects of two newly approved AR antagonists apalutamide (ARN) and darolutamide (ODM) (Morsy and Trippier, 2020). Upon 24-hour pretreatment of cell lines with KV-49g, a potentiation effect of 126-fold and 75-fold was observed with ARN and ODM, respectively.
INDO acts as an AKR1C3 inhibitor (IC50 = 100 nM, with an AKR1C3 selectivity = 365-fold). Its clinically approved status positions the compound to bring about immediate benefits to patients with prostate cancer (Liu et al., 2015) but is likely to lead to gastrointestinal side effects because of its COX-1 inhibition activity. In in vitro model systems, INDO exhibited lower potentiation effects in combination with enzalutamide than the baccharin analogs. In LNCaP1C3 cells, INDO conferred a 6.4-fold potentiation effect affording a combination IC50 = 31.4 µM compared with KV-37 (25-fold potentiation and combination IC50 = 8.19 µM). Thus, more potent AKR1C3 inhibitors confer a greater potentiation effect. This was further confirmed in potentiation studies of ARN and ODM in 22Rv1 cells in which a combination of INDO provided an 8-fold potentiation effect yielding a combination IC50 = 30 µM, whereas KV-49g produced a 126-fold potentiation effect with a combination IC50 = 12.47 µM for ARN and 75-fold potentiation effect with a combination IC50 = 5.64 µM for ODM. Since inhibition of AKR1C3 by INDO has been shown to overcome resistance to enzalutamide and abiraterone acetate both in vitro and in vivo, with the combination resulting in a significant reduction in tumor volume over three weeks in CWR22Rv1 xenografts using just 3 mg/kg i.p. of INDO (Liu et al., 2015), the KV- compounds are expected to provide superior efficacy.
The ability of INDO to potentiate abiraterone acetate and enzalutamide has led to the search for more potent AKR1C3 inhibitors based around its chemical scaffold. In 2013, Liedtke et al. (2013) developed INDO analogs, wherein derivatives exhibited nanomolar potency, with >100-fold selectivity for AKR1C3 over the AKR1C1 and AKR1C2 isoforms. These compounds also inhibited the formation of testosterone in the LNCaP1C3 cell line (Liedtke et al., 2013). However, studies have yet to be conducted to determine their ability to potentiate the effect of AR antagonists.
Even though flufenamic acid is a potent AKR1C3 inhibitor, it causes COX-2–dependent target effects. To circumvent this drawback, a series of N-phenylanthranilic acid derivatives were synthesized that retain nanomolar potency for AKR1C3 and show >100-fold selectivity but no longer inhibit COX-1 or COX-2 (Adeniji et al. 2012, 2013). Like the INDO analogs, these have not been tested for synergistic effects with either enzalutamide or abiraterone acetate. Pippione et al. (2017) replaced the benzoic acid moiety in flufenamic acid to generate hydroxytriazole derivatives with potent and selective AKR1C3 inhibition activity but without COX-2 effects. These derivatives were evaluated for antiproliferative activity in the abiraterone acetate and enzalutamide resistant AKR1C3 expressing 22Rv1 cell line and exhibited moderate antiproliferative activity with IC50 values of 67 and 81 µM, respectively. When one lead compound, or abiraterone acetate and enzalutamide, were used as single agents, cell viability was inhibited by approximately 20%–30%, whereas in combination a synergistic effect on cell viability was observed (Pippione et al., 2017). When the benzoic acid moiety of flufenamic acid was replaced with an acidic hydroxybenzoisoxazole moiety, a new lead compound, possessing an IC50 of 27 µM, was identified which showed approximately 450-fold selectivity for AKR1C3 compared with the AKR1C2 isoform, with minimum COX mediated off-target effects. When 22RV1 cells were exposed for 72 hours to 20 μM lead compound in combination with either abiraterone or enzalutamide, cell viability was decreased from 30% with the compound alone and to 50% for the drug combination (Pippione et al., 2018).
B. Leukemia
AKR1C3 is known to catalyze the synthesis of prostaglandins that sustain growth of myeloid precursors in the bone marrow. The enzyme is overexpressed in several subtypes of AML and in T-ALL. This overexpression results in chemoresistance toward etoposide (Verma et al., 2016), the anthracyclines, and cytarabine (Verma et al., 2019).
The cotreatment of selective AKR1C3 inhibitors KV-49a and KV-49g with daunorubicin did not result in an appreciable potentiation effect in the AML cell lines HL-60, THP-1, and KG1a, which have low, moderate, and high AKR1C3 expression, respectively. However, upon 24-hour pretreatment with either compound, potentiation of the activity of daunorubicin was observed, with >100-fold potentiation effect in the daunorubicin-resistant cell line KG1a. In the THP-1 cell line, a 16-fold potentiation effect was noted while in the HL-60 cell line and the effect of daunorubicin was potentiated by 1.3–1.7-fold. A direct correlation between AKR1C3 expression and potentiation effect was apparent in these studies (Verma et al., 2019).
Cytarabine (araC) is an antimetabolite anticancer agent clinically approved for use in many hematologic cancers including several subtypes of AML and in T-ALL. To the best of our knowledge, AKR1C3 does not metabolize drugs with an amide group. Pretreatment for 24 hours with either KV-49a or KV-49g followed by araC led to a >100-fold potentiation of the effect of araC in the KG1a cell line which shows a high expression of AKR1C3. Similar pretreatments in the HL-60 cell line resulted in 12-fold (with KV-49g) and 33-fold (with KV-49a) potentiation, whereas in the THP-1 cell line, 18-fold (with KV-49g) and 13-fold (with KV-49a) potentiation was observed. These results again show a correlation between AKR1C3 expression and potentiation effect among the various cell lines (Verma et al., 2019).
In the high-risk pediatric T-ALL, a superior drug resistance profile is observed versus the related B-cell acute lymphoblastic leukemia. The expression and enzymatic activity of AKR1C3 was examined in a large cohort of patients with T-ALL which found a significant upregulation in resistant or poorly responding tumors to vincristine (Bortolozzi et al., 2018). In primary patient-derived T-ALL cell lines obtained at patient diagnosis (low AKR1C3 expression) and at treatment relapse (high AKR1C3 expression), cotreatment of KV-49a with daunorubicin resulted in 13-fold and 20-fold increase in potentiation, respectively. Pretreatment of KV-49a followed by daunorubicin greatly increased the potentiation effect. A >100-fold potentiation of daunorubicin was observed in both cell lines and reduced the daunorubicin IC50 from 0.005 µM and 0.07 µM at diagnosis and in relapsed cells, respectively, to < 0.001 µM in both cell lines. Similarly, in primary T-ALL cells taken at patient diagnosis, cotreatment of AKR1C3 inhibitor KV-49a with araC resulted in a 16-fold potentiation effect. In primary T-ALL cells taken at relapse, this effect was increased to >100 potentiation. Upon pretreatment of these cell lines with KV-49a followed by araC exposure, a >100-fold potentiation effect was achieved in both cell lines (Verma et al., 2019). Again, greater AKR1C3 expression was correlated with a greater potentiation effect upon the use of an AKR1C3 inhibitor.
Etoposide is a DNA intercalator and topoisomerase II inhibitor that is clinically approved for the treatment of several forms of leukemia including AML. Pretreatment of an AKR1C3 inhibitor followed by exposure of cells to etoposide resulted in a 5.5-fold and 6.3-fold potentiation effect in HL-60 and KG1a AML cells, respectively, with a greater effect seen in higher AKR1C3-expressing cell lines. The higher potentiation effect seen in KG1a cells may be because of the higher expression of AKR1C3 in these cells (Verma et al., 2016).
Khanim et al. (2009) identified tetracycline as a potent and selective AKR1C3 inhibitor. Further studies showed that 4-methyl(de-dimethylamine)-tetracycline, a breakdown product of tetracycline, was a potent (IC50 = 0.51 µM) and somewhat selective AKR1C3 inhibitor. Despite the presence of 11-keto-prostaglandin reductase activity catalyzed by AKR1C3 in the KG1a AML cell line, 4-methyl(de-dimethylamine)-tetracycline did not exhibit antiproliferative activity up to 50 μM concentration in the AML cell lines HL60, KG1a, and K562. When combined with bezafibrate, no potentiation effect was noted (Khanim et al., 2009). However, medroxyprogesterone acetate, a pan-AKR1C inhibitor, provided enhanced reduction in cell viability when combined with bezafibrate in K562 and KG1a AML cell lines (Khanim et al., 2009). The ability of 6-medroxyprogesterone to have this affect implicates the action of AKR1C isoforms other than AKR1C3 in drug resistance.
C. Colorectal Cancer
As described earlier, purvalanol A and roscovitine exhibit good potency and selectivity for AKR1C3 inhibition, yielding Ki values of 5.5 μM and 1.4 μM, respectively in the AKR1C3 transfected human colon carcinoma (HCT116) cell line. A combination study with 0.1–1 μM daunorubicin and 5 μM purvalanol A or 10 μM roscovitine in AKR1C3 overexpressing HCT116 cells revealed excellent synergy compared with nontransfected HCT116 cells with a potentiation effect of the two drugs of 12.97 ± 6.26 and 17.06 ± 8.36-fold, respectively. These studies indicate that a combination of purine analogs and daunorubicin resulted in excellent potentiation of activity and chemosensitized AKR1C3 expressing cell lines to the action of daunorubicin (Novotná et al., 2018a).
Buparlisib, a pyrimidine analog, is a known phosphoinositide 3-kinase (PI3K) inhibitor and is in clinical trial for the treatment of various cancers including leukemia. Buparlisib inhibits AKR1C3 with an IC50 of 9.5 µM and in AKR1C3 overexpressing HCT116 cells was evaluated for its inhibitory activity of AKR1C3-mediated intracellular reduction of daunorubicin, wherein a Ki value of 14 µM was determined using an enzymatic assay. This combination of buparlisib and daunorubicin results in decreased daunorubicin metabolism in AKR1C3 overexpressing human colon carcinoma cells (Bukum et al., 2019).
When AKR1C inhibitors are used to potentiate the effects of chemotherapeutic agents, some common features emerge. When AKR1C inhibitors and chemotherapeutic agents are given concurrently, potentiation is weak. However, more dramatic effects are seen if cancer cells are “primed by pretreatment” with the AKR1C inhibitors. This ordered addition of drug may have to be considered in the clinic for presensitization of patients. In some instances, the AKR1C inhibitors clearly inhibit the enzyme activity of the protein, but in other instances, this may not be the case, pointing toward a role in drug resistance that may be enzyme independent.
IX. Resistance Versus Disease Progression: Cause or Consequence
AKRs did not evolve to metabolize chemotherapeutic drugs, instead they have evolved to prevent cellular response to oxidative and electrophilic stress and may play a role in disease pathogenesis. For example, AKR1B10 converts all-trans-retinaldehyde to retinal and prevents the formation of retinoic acid and blocks retinoic acid signaling and cellular differentiation (Gallego et al., 2006, 2007). This pathogenic mechanism may account for its overexpression in many tumor types, e.g., nonsmall cell lung carcinoma and colon cancer (Hsu et al., 2001; Kropotova et al., 2014). Similarly, AKR1C3 also acts as a prostaglandin (PG) F2α synthase and converts PGH2 and 11-keto-PGD2 to prostaglandins of the F series (Matsuura et al., 1998), which in turn can activate the FP1 receptor leading to activation of MAP-kinase and a pro-proliferative signal (Desmond et al., 2003; Penning, 2019). This pathogenic mechanism may account for its overexpression in many types of leukemia (Birtwistle et al., 2009; Verma et al., 2016; Bortolozzi et al., 2018). Finally, AKR1C1, AKR1C2 and AKR1C3 regulate the amount of ligands that can bind to steroid hormone receptors, and this prereceptor regulation of hormone action may contribute to the growth of hormone dependent malignancies (Penning et al., 2019). A consequence of this pathology, because of their ability to reduce carbonyl groups, can be chemoresistance.
X. Clinical Use of Nuclear Factor-Erythroid 2 p45-Related Factor 2 and AKR Inhibitors to Surmount Chemotherapeutic Drug Resistance
The ability of existing drugs to inhibit AKR1C isoforms selectively has been tested in some clinical trials. In the Nurses health cohort study, the use of both aspirin and nonaspirin based NSAIDs prolonged the survival of ovarian cancer patients where the primary chemotherapeutic agent was cis-platin and demonstrated the clinical utility of inhibiting an AKR1C isoform (Trabert et al., 2014). The clinically approved COX inhibitor INDO is currently in phase II clinical trials to surmount drug resistance to abiraterone acetate and enzalutamide mediated by AKR1C3 in castrate resistance prostate cancer (see NCT02849990 and NCT02935205). AKR1C3 has also been targeted in AML using a combination of 6-medroxyprogesterone acetate (a pan-AKR1C inhibitor) and bezafibrate (a PPARγ agonist) with the goal of preventing PGD2 conversion to 11β-PGF2α and preventing cellular proliferation (Murray et al., 2010); this approach has led to a phase II trial (Murray et al., 2019a). To move AKR inhibitors into oncologic practice to improve drug response to anticancer drugs, trials are in their infancy. Adaptive clinical trials that exploit overexpression of AKR isoforms could be considered so that personalized treatment based on tumor phenotype and AKR isoform specific inhibitors could be a treatment strategy. The use of NRF2 inhibitors would be another approach, but we need to better understand the mode of action of these agents when they do not bind to NRF2 directly.
XI. Conclusions
Resistance to anticancer drugs, whether they be cancer chemotherapeutic agents or anti-hormone therapeutics, remains a major factor limiting their use. For the former, there can also be dose-limiting toxicity. Identification of targets that mediate resistance and the development of inhibitors for these targets can prolong the benefit of existing agents, limit toxicity, and increase overall survival. This review establishes the case that members of the AKR gene superfamily are intimately involved in drug resistance and inhibitors may have a significant benefit in surmounting resistance and increasing drug sensitivity. In some instances, a battery of AKR genes can be involved, and thus, a balance exists between the use of isoform specific inhibitors and pan-inhibitors. Some of these inhibitors are existing drugs (e.g., indomethacin, 6-medroxyprogesterone acetate, flufenamic acid, purvalanol A, roscovitine, and buparlisib) and could be used for improved outcomes and can be tested in adaptive clinical trials. In other cases where isoform specific inhibitors have been identified from enzyme screening and efficacy in cancer cell models has been established, more preclinical development of these agents is required. Knowing that the AKR genes are regulated by the NRF2-KEAP1 pathway identifies NRF2 inhibitors as another strategy to surmount drug resistance.
Acknowledgments
We acknowledge Dr. Jessica Murray and Nicole Kerstetter for their assistance in the preparation of figures. We would also like to dedicate this article to Dr. Amy Margaret Quinn, who completed her thesis on AKR1B10 in 2008 in Professor Penning’s laboratory and died as a result of AML.
Abbreviations
- ABC
ATP-binding cassette
- AKR
aldo-keto reductase
- AKT
protein kinase B
- ALDH
aldehyde dehydrogenase
- AML
acute myelocytic leukemia
- AR
androgen receptor
- ARSI
androgen receptor signaling inhibitor
- araC
cytarabine
- ARE
antioxidant response element
- ARN
apalutamide
- ARSI
androgen receptor signaling inhibitor
- COX
cyclooxygenase
- DGR
double glycine repeat
- Dox
doxorubicin; GSH, glutahione
- HaCaT
immortal human keratinocyte cell line
- 4-HNE
4-hydroxy-2-nonenal
- INDO
indomethacin
- IVR
intervening region
- KEAP1
Kelch-like ECH-associated protein 1
- NADPH
nicotinamide adenine dinucleotide phosphate
- NQO1
NADPH: quinone oxidoreductase
- NRF2/NFE2L2
nuclear factor-erythroid 2 p45-related factor 2
- NSAID
nonsteroidal anti-inflammatory drug
- ODM
darolutamide
- PPARγ
peroxisome proliferator activated receptor γ
- PI3K
phosphoinositide 3–kinase
- P450
cytochrome P450
- ROS
reactive oxygen species
- si-RNA
short interfering RNA
- SLC
solute carrier
- T-ALL
T-cell acute lymphoblastic leukemia
- TMPRSS2-ERG
transmembrane proetase serine 2 and eryrthroblast transformation specific related gene/protein.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Penning, onnalagadda, Trippier, Rižner.
Footnotes
These studies were supported in part by National Institutes of Health [Grants P30-ES013508 and R01-ES029294] (to T.M.P.) and [Grant R01-CA226436] (to P.C.T.) and by the Slovenian Research Agency [Grants J3-8212 and J3-2535] (to T.L.R.).
References
- Adeniji AO, Chen M, Penning TM (2013) AKR1C3 as a target in castrate resistant prostate cancer. J Steroid Biochem Mol Biol 137:136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeniji AO, Twenter BM, Byrns MC, Jin Y, Chen M, Winkler JD, Penning TM (2012) Development of potent and selective inhibitors of aldo-keto reductase 1C3 (type 5 17β-hydroxysteroid dehydrogenase) based on N-phenyl-aminobenzoates and their structure-activity relationships. J Med Chem 55:2311–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agyeman AS, Chaerkady R, Shaw PG, Davidson NE, Visvanathan K, Pandey A, Kensler TW (2012) Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res Treat 132:175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolod MD, Lin SM, Johnson SP, Bullock NS, Colvin M, Bigner DD, Friedman HS (2008) The gene expression profiles of medulloblastoma cell lines resistant to preactivated cyclophosphamide. Curr Cancer Drug Targets 8:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birtwistle JHayden REKhanim FLGreen RMPearce CDavies NJWake NSchrewe HRide JPChipman JK, et al. (2009) The aldo-keto reductase AKR1C3 contributes to 7,12-dimethylbenz(a)anthracene-3,4-dihydrodiol mediated oxidative DNA damage in myeloid cells: implications for leukemogenesis. Mutat Res 662:67–74. [DOI] [PubMed] [Google Scholar]
- Bortolozzi RBresolin SRampazzo EPaganin MMaule FMariotto EBoso DMinuzzo SAgnusdei VViola G, et al. (2018) AKR1C enzymes sustain therapy resistance in paediatric T-ALL. Br J Cancer 118:985–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukum N, Novotna E, Morell A, Hofman J, Wsol V (2019) Buparlisib is a novel inhibitor of daunorubicin reduction mediated by aldo-keto reductase 1C3. Chem Biol Interact 302:101–107. [DOI] [PubMed] [Google Scholar]
- Burczynski ME, Harvey RG, Penning TM (1998) Expression and characterization of four recombinant human dihydrodiol dehydrogenase isoforms: oxidation of trans-7, 8-dihydroxy-7,8-dihydrobenzo[a]pyrene to the activated o-quinone metabolite benzo[a]pyrene-7,8-dione. Biochemistry 37:6781–6790. [DOI] [PubMed] [Google Scholar]
- Burczynski ME, Lin HK, Penning TM (1999) Isoform-specific induction of a human aldo-keto reductase by polycyclic aromatic hydrocarbons (PAHs), electrophiles, and oxidative stress: implications for the alternative pathway of PAH activation catalyzed by human dihydrodiol dehydrogenase. Cancer Res 59:607–614. [PubMed] [Google Scholar]
- Burczynski ME, Sridhar GR, Palackal NT, Penning TM (2001) The reactive oxygen species--and Michael acceptor-inducible human aldo-keto reductase AKR1C1 reduces the alpha,beta-unsaturated aldehyde 4-hydroxy-2-nonenal to 1,4-dihydroxy-2-nonene. J Biol Chem 276:2890–2897. [DOI] [PubMed] [Google Scholar]
- Byrns MC, Duan L, Lee SH, Blair IA, Penning TM (2010) Aldo-keto reductase 1C3 expression in MCF-7 cells reveals roles in steroid hormone and prostaglandin metabolism that may explain its over-expression in breast cancer. J Steroid Biochem Mol Biol 118:177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrns MC, Steckelbroeck S, Penning TM (2008) An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3alpha-HSD, type 5 17beta-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem Pharmacol 75:484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canning P, Sorrell FJ, Bullock AN (2015) Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med 88 (Pt B):101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WM, Chang YC, Yang YC, Lin SK, Chang PM, Hsiao M (2019) AKR1C1 controls cisplatin-resistance in head and neck squamous cell carcinoma through cross-talk with the STAT1/3 signaling pathway. J Exp Clin Cancer Res 38:245, DOI: 10.1186/s13046-019-1256-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Emara N, Solomides C, Parekh H, Simpkins H(2010) Resistance to platinum-based chemotherapy in lung cancer cell lines. Cancer Chemother Pharmacol 66:1103–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CCChu CBLiu KJHuang CYChang JYPan WYChen HHCheng YHLee KDChen MF, et al. (2013) Gene expression profiling for analysis acquired oxaliplatin resistant factors in human gastric carcinoma TSGH-S3 cells: the role of IL-6 signaling and Nrf2/AKR1C axis identification. Biochem Pharmacol 86:872–887. [DOI] [PubMed] [Google Scholar]
- Chen J, Adikari M, Pallai R, Parekh HK, Simpkins H (2008) Dihydrodiol dehydrogenases regulate the generation of reactive oxygen species and the development of cisplatin resistance in human ovarian carcinoma cells. Cancer Chemother Pharmacol 61:979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55. [DOI] [PubMed] [Google Scholar]
- Chou TC (2010) Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70:440–446. [DOI] [PubMed] [Google Scholar]
- Ciaccio PJ, Tew KD (1994) cDNA and deduced amino acid sequences of a human colon dihydrodiol dehydrogenase. Biochim Biophys Acta 1186:129–132. [DOI] [PubMed] [Google Scholar]
- Ciaccio PJ, Jaiswal AK, Tew KD (1994) Regulation of human dihydrodiol dehydrogenase by Michael acceptor xenobiotics. J Biol Chem 269:15558–15562. [PubMed] [Google Scholar]
- Ciaccio PJ, Stuart JE, Tew KD (1993) Overproduction of a 37.5-kDa cytosolic protein structurally related to prostaglandin F synthase in ethacrynic acid-resistant human colon cells. Mol Pharmacol 43:845–853. [PubMed] [Google Scholar]
- Cubillos-Angulo JM, Fukutan E, Cruz LAB, Arriaga MB, Lima JV, Andrade BB, Queiroz ATL, Fukutani KF (2020) Systems biology analysis of publicly available transcriptomic data reveals a critical link between AKR1B10 gene expression, smoking and occurrence of lung cancer. PLoS One 15:e0222552, DOI: 10.1371/journal.pone.0222552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HB, Adikari M, Parekh HK, Simpkins H (2004) Ubiquitous induction of resistance to platinum drugs in human ovarian, cervical, germ-cell and lung carcinoma tumor cells overexpressing isoforms 1 and 2 of dihydrodiol dehydrogenase. Cancer Chemother Pharmacol 54:301–307. [DOI] [PubMed] [Google Scholar]
- Deng HB, Parekh HK, Chow KC, Simpkins H (2002) Increased expression of dihydrodiol dehydrogenase induces resistance to cisplatin in human ovarian carcinoma cells. J Biol Chem 277:15035–15043. [DOI] [PubMed] [Google Scholar]
- Desmond JC, Mountford JC, Drayson MT, Walker EA, Hewison M, Ride JP, Luong QT, Hayden RE, Vanin EF, Bunce CM (2003) The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res 63:505–512. [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Kensler TW (2005) The role of Keap1 in cellular protective responses. Chem Res Toxicol 18:1779–1791. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Kostov RV, Canning P (2017) Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch Biochem Biophys 617:84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Kabbani O, Dhagat U, Hara A (2011) Inhibitors of human 20α-hydroxysteroid dehydrogenase (AKR1C1). J Steroid Biochem Mol Biol 125:105–111. [DOI] [PubMed] [Google Scholar]
- El-Kabbani O, Scammells PJ, Day T, Dhagat U, Endo S, Matsunaga T, Soda M, Hara A (2010) Structure-based optimization and biological evaluation of human 20α-hydroxysteroid dehydrogenase (AKR1C1) salicylic acid-based inhibitors. Eur J Med Chem 45:5309–5317. [DOI] [PubMed] [Google Scholar]
- Fabrizio FP, Sparaneo A, Centra F, Trombetta D, Storlazzi CT, Graziano P, Maiello E, Fazio VM, Muscarella LA (2019) Methylation Density Pattern of KEAP1 Gene in Lung Cancer Cell Lines Detected by Quantitative Methylation Specific PCR and Pyrosequencing. Int J Mol Sci 20:2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsted RL, Gee M, Bachur NR (1974) Rat liver daunorubicin reductase. An aldo-keto reductase. J Biol Chem 249:3672–3679. [PubMed] [Google Scholar]
- Fizazi KScher HIMolina ALogothetis CJChi KNJones RJStaffurth JNNorth SVogelzang NJSaad F, et al. ; COU-AA-301 Investigators (2012) Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol 13:983–992. [DOI] [PubMed] [Google Scholar]
- Flynn JJones JJohnson AJAndritsos LMaddocks KJaglowski SHessler JGrever MRIm EZhou H, et al. (2015) Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia 29:1524–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego O, Belyaeva OV, Porté S, Ruiz FX, Stetsenko AV, Shabrova EV, Kostereva NV, Farrés J, Parés X, Kedishvili NY (2006) Compartive functional analysis of human medium-chain dehydrogenases, short chain dehydrogenases/reductases and aldo-keto reductases with retinoids. Biochem J 399:101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego O, Ruiz FX, Ardèvol A, Domínguez M, Alvarez R, de Lera AR, Rovira C, Farrés J, Fita I, Parés X (2007) Structural basis for the high all-trans-retinaldehyde reductase activity of the tumor marker AKR1B10. Proc Natl Acad Sci USA 104:20764–20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao A-M, Ke Z-P, Shi F, Sun GC, Chen H (2013) Chrysin enhances sensitivity of BEL-7402/ADM cells to doxorubicin by suppressing PI3K/Akt/Nrf2 and ERK/Nrf2 pathway. Chem Biol Interact 206:100–108. [DOI] [PubMed] [Google Scholar]
- Gewirtz DA (1999) A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 57:727–741. [DOI] [PubMed] [Google Scholar]
- Girardi ECésar-Razquin ALindinger SPapakostas KKonecka JHemmerich JKickinger SKartnig FGürtl BKlavins K, et al. (2020) A widespread role for SLC transmembrane transporters in resistance to cytotoxic drugs. Nat Chem Biol 16:469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, Staudt LM(2016) Toward a shared vision for cancer genomic data. N Engl J Med 375:1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Yu S, Zhang C, Kong AN (2015) Epigenetic regulation of Keap1-Nrf2 signaling. Free Radic Biol Med 88 (Pt B):337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SK, Singh P, Ali V, Verma M (2020) Role of membrane-embedded drug efflux ABC transporters in the cancer chemotherapy. Oncol Rev 14:448, 10.4081/oncol.2020.448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid AR, Pfeiffer MJ, Verhaegh GW, Schaafsma E, Brandt A, Sweep FC, Sedelaar JP, Schalken JA (2013) Aldo-keto reductase family 1 member C3 (AKR1C3) is a biomarker and therapeutic target for castration-resistant prostate cancer. Mol Med 18:1449–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Jun Y, Kim SH, Hoang HH, Jung Y, Kim S, Kim J, Austin RH, Lee S, Park S (2016) Rapid emergence and mechanisms of resistance by U87 glioblastoma cells to doxorubicin in an in vitro tumor microfluidic ecology. Proc Natl Acad Sci USA 113:14283–14288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada NTakahata TZhou QYe XSun RItoh JIshiguro AKijima HMimura JItoh K, et al. (2012) Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC Cancer 12:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder B, Tian W, La Clair JJ, Tan A-C, Ooi A, Chapman E, Zhang DD (2017) Brusatol overcomes chemoresistance through inhibition of protein translation. Mol Carcinog 56:1493–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heibein AD, Guo B, Sprowl JA, Maclean DA, Parissenti AM (2012) Role of aldo-keto reductases and other doxorubicin pharmacokinetic genes in doxorubicin resistance, DNA binding, and subcellular localization. BMC Cancer 12:381, DOI: 10.1186/1471-2407-12-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintzpeter J, Seliger JM, Hofman J, Martin HJ, Wsol V, Maser E (2016) Inhibition of human anthracycline reductases by emodin - A possible remedy for anthracycline resistance. Toxicol Appl Pharmacol 293:21–29. [DOI] [PubMed] [Google Scholar]
- Hofland J, van Weerden WM, Dits NFJ, Steenbergen J, van Leenders GJ, Jenster G, Schröder FH, de Jong FH (2010) Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res 70:1256–1264. [DOI] [PubMed] [Google Scholar]
- Hofman J, Malcekova B, Skarka A, Novotna E, Wsol V (2014) Anthracycline resistance mediated by reductive metabolism in cancer cells: the role of aldo-keto reductase 1C3. Toxicol Appl Pharmacol 278:238–248. [DOI] [PubMed] [Google Scholar]
- Hsu NY, Ho HC, Chow KC, Lin TY, Shih CS, Wang LS, Tsai CM (2001) Overexpression of dihydrodiol dehydrogenase as a prognostic marker of non-small cell lung cancer. Cancer Res 61:2727–2731. [PubMed] [Google Scholar]
- Hung JJ, Chow KC, Wang HW, Wang LS (2006) Expression of dihydrodiol dehydrogenase and resistance to chemotherapy and radiotherapy in adenocarcinoma cells of lung. Anticancer Res 26 (4B):2949–2955. [PubMed] [Google Scholar]
- Januchowski R, Zawierucha P, Andrzejewska M, Ruciński M, Zabel M (2013) Microarray-based detection and expression analysis of ABC and SLC transporters in drug-resistant ovarian cancer cell lines. Biomed Pharmacother 67:240–245. [DOI] [PubMed] [Google Scholar]
- Januchowski R, Zawierucha P, Ruciński M, Andrzejewska M, Wojtowicz K, Nowicki M, Zabel M (2014) Drug transporter expression profiling in chemoresistant variants of the A2780 ovarian cancer cell line. Biomed Pharmacother 68: 447-53. [DOI] [PubMed] [Google Scholar]
- Jez JM, Penning TM (2001) The aldo-keto reductase (AKR) superfamily: an update. Chem Biol Interact 130-132:499–525. [DOI] [PubMed] [Google Scholar]
- Jordan MA (2002) Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr Med Chem Anticancer Agents 2:1–17. [DOI] [PubMed] [Google Scholar]
- Jung KA, Choi BH, Nam CW, Song M, Kim ST, Lee JY, Kwak MK (2013) Identification of aldo-keto reductases as NRF2-target marker genes in human cells. Toxicol Lett 218:39–49, DOI: 10.1016/j.toxlet.2012.12.026. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Egner PA, Agyeman AS, Visvanathan K, Groopman JD, Chen JG, Chen TY, Fahey JW, Talalay P (2013) Keap1-nrf2 signaling: a target for cancer prevention by sulforaphane. Top Curr Chem 329:163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerins MJ, Ooi A (2018) A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci Rep 8:12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanim FLHayden REBirtwistle JLodi ATiziani SDavies NJRide JPViant MRGunther ULMountford JC, et al. (2009) Combined bezafibrate and medroxyprogesterone acetate: potential novel therapy for acute myeloid leukaemia. PLoS One 4:e8147, DOI: 10.1371/journal.pone.0008147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen KE, Penning TM (2010) Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab 21:315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko BC, Ruepp B, Bohren KM, Gabbay KH, Chung SS(1997) Identification and characterization of multiple osmotic response sequences in the human aldose reductase gene. J Biol Chem 272:16431–16437. [DOI] [PubMed] [Google Scholar]
- Kropotova ES, Zinovieva OL, Zyryanova AF, Dybovaya VI, Prasolov VS, Beresten SF, Oparina NY, Mashkova TD (2014) Altered expression of multiple genes involved in retinoic acid biosynthesis in human colorectal cancer. Pathol Oncol Res 20:707–717. [DOI] [PubMed] [Google Scholar]
- Lanthaler K, Bilsland E, Dobson PD, Moss HJ, Pir P, Kell DB, Oliver SG (2011) Genome-wide assessment of the carriers involved in the cellular uptake of drugs: a model system in yeast. BMC Biol 9:70, DOI: 10.1186/1741-7007-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EK, Regenold WT, Shapiro P (2002) Inhibition of aldose reductase enhances HeLa cell sensitivity to chemotherapeutic drugs and involves activation of extracellular signal-regulated kinases. Anticancer Drugs 13:859–868. [DOI] [PubMed] [Google Scholar]
- Li D, Ma S, Ellis EM(2015) Nrf2-mediated adaptive response to methyl glyoxal in HepG2 cells involves the induction of AKR7A2. Chem Biol Interact 234:366–371. [DOI] [PubMed] [Google Scholar]
- Li D, Ferrari M, Ellis EM (2012a) Human aldo-keto reductase AKR7A2 protects against the cytotoxicity and mutagenicity of reactive aldehydes and lowers intracellular reactive oxygen species in hamster V79-4 cells. Chem Biol Interact 195:25–34. [DOI] [PubMed] [Google Scholar]
- Li R, Evaul K, Sharma KK, Chang KH, Yoshimoto J, Liu J, Auchus RJ, Sharifi N (2012b) Abiraterone inhibits 3β-hydroxysteroid dehydrogenase: a rationale for increasing drug exposure in castration-resistant prostate cancer. Clin Cancer Res 18:3571–3579. [DOI] [PubMed] [Google Scholar]
- Liedtke AJ, Adeniji AO, Chen M, Byrns MC, Jin Y, Christianson DW, Marnett LJ, Penning TM (2013) Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (Type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J Med Chem 56:2429–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, Gao AC (2017) Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol Cancer Ther 16:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, Evans CP, Gao AC (2015) Intracrine androgens and AKR1C3 activation confer resistance to Enzalutamide in prostate cancer. Cancer Res 75:1413–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon RC, Li D, McGarvie G, Ellis EM (2013) Aldo-keto reductases mediate constitutive and inducible protection against aldehyde toxicity in human neuroblastoma SH-SY5Y cells. Neurochem Int 62:113–121. [DOI] [PubMed] [Google Scholar]
- Ma JLuo DXHuang CShen YBu YMarkwell SGao JLiu JZu XCao Z, et al. (2012) AKR1B10 overexpression in breast cancer: association with tumor size, lymph node metastasis and patient survival and its potential as a novel serum marker. Int J Cancer 131:E862–E871. [DOI] [PubMed] [Google Scholar]
- MacLeod AK, McMahon M, Plummer SM, Higgins LG, Penning TM, Igarashi K, Hayes JD (2009) Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: demonstration that the KEAP1-NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis 30:1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto R, Tsuda M, Yoshida K, Tanino M, Kimura T, Nishihara H, Abe T, Shinohara N, Nonomura K, Tanaka S (2016) Aldo-keto reductase 1C1 induced by interleukin-1β mediates the invasive potential and drug resistance of metastatic bladder cancer cells. Sci Rep 6:34625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga T, Hojo A, Yamane Y, Endo S, El-Kabbani O, Hara A (2013) Pathophysiological roles of aldo-keto reductases (AKR1C1 and AKR1C3) in development of cisplatin resistance in human colon cancers. Chem Biol Interact 202:234–242. [DOI] [PubMed] [Google Scholar]
- Matsunaga T, Okumura N, Saito H, Morikawa Y, Suenami K, Hisamatsu A, Endo S, Ikari A (2020) Significance of aldo-keto reductase 1C3 and ATP-binding cassette transporter B1 in gain of irinotecan resistance in colon cancer cells. Chem Biol Interact 332:109295, DOI: 10.1016/j.cbi.2020.109295. [DOI] [PubMed] [Google Scholar]
- Matsunaga T, Saito H, Endo S, Iguchi K, Soda M, El-Kabbani O, Hara A, Ikari A (2016a) Roles of aldo-keto reductases 1B10 and 1C3 and ATP-binding cassette transporter in docetaxel tolerance. Free Radic Res 50:1296–1308. [DOI] [PubMed] [Google Scholar]
- Matsunaga TSuzuki AKezuka COkumura NIguchi KInoue ISoda MEndo SEl-Kabbani OHara A, et al. (2016b) Aldo-keto reductase 1B10 promotes development of cisplatin resistance in gastrointestinal cancer cells through down-regulating peroxisome proliferator-activated receptor-γ-dependent mechanism. Chem Biol Interact 256:142–153. [DOI] [PubMed] [Google Scholar]
- Matsunaga T, Yamaguchi A, Morikawa Y, Kezuka C, Takazawa H, Endo S, El-Kabbani O, Tajima K, Ikari A, Hara A (2014) Induction of aldo-keto reductases (AKR1C1 and AKR1C3) abolishes the efficacy of daunorubicin chemotherapy for leukemic U937 cells. Anticancer Drugs 25:868–877. [DOI] [PubMed] [Google Scholar]
- Matsunaga T, Yamane Y, Iida K, Endo S, Banno Y, El-Kabbani O, Hara A (2011) Involvement of the aldo-keto reductase, AKR1B10, in mitomycin-c resistance through reactive oxygen species-dependent mechanisms. Anticancer Drugs 22:402–408. [DOI] [PubMed] [Google Scholar]
- Matsuura K, Shiraishi H, Hara A, Sato K, Deyashiki Y, Ninomiya M, Sakai S (1998) Identification of a principal mRNA species for human 3alpha-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J Biochem 124:940–946. [DOI] [PubMed] [Google Scholar]
- McNamara KM, Sasano H (2019) The role of 17βHSDs in breast tissue and breast cancers. Mol Cell Endocrinol 489:32–44. [DOI] [PubMed] [Google Scholar]
- Mindnich RD, Penning TM (2009) Aldo-keto reductase (AKR) superfamily: genomics and annotation. Hum Genomics 3:362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades N, Sung CC, Schultz N, Danila DC, He B, Eedunuri VK, Fleisher M, Sander C, Sawyers CL, Scher HI (2012) Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res 72:6142–6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morell A, Čermáková L, Novotná E, Laštovičková L, Haddad M, Haddad A, Portillo R, Wsól V (2020) Bruton’s tyrosine kinase inhibitors ibrutinib and acalabrutinib counteract anthracycline resistance in cancer cells expressing AKR1C3. Cancers (Basel) 12:3731, DOI: 10.3390/cancers12123731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa Y, Kezuka C, Endo S, Ikari A, Soda M, Yamamura K, Toyooka N, El-Kabbani O, Hara A, Matsunaga T (2015) Acquisition of doxorubicin resistance facilitates migrating and invasive potentials of gastric cancer MKN45 cells through up-regulating aldo-keto reductase 1B10. Chem Biol Interact 230:30–39. [DOI] [PubMed] [Google Scholar]
- Morsy A, Trippier PC (2020) Reversal of apalutamide and darolutamide aldo-keto reductase 1C3-mediated resistance by a small molecule inhibitor. ACS Chem Biol 15:646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JA, Khanim FL, Hayden RE, Craddock CF, Holyoake TL, Jackson N, Lumley M, Bunce CM, Drayson MT (2010) Combined bezafibrate and medroxyprogesterone acetate have efficacy without haematological toxicity in elderly and relapsed acute myeloid leukaemia (AML). Br J Haematol 149:65–69. [DOI] [PubMed] [Google Scholar]
- Murray J, Pratt G, Jacob, A , Clark F, Blundred R, Fox S, Bishop R, Wheatley K, Khanim F, Bunce C, Drayson M. (2019a). Single arm phase II trials assessing the sfaety, compliance with and activity of bezafibrate and medroxyprogesterone acetate (BaP) therapy against myeloid and lymphoid cancers. Contemp Clin Trials Commun 14:100361, DOI: 10.1016/j.conctc.2019.100361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JR, de la Vega L, Hayes JD, Duan L, Penning TM (2019b) Induction of the antioxidant response by the transcription factor NRF2 increases bioactivation of the mutagenic air pollutant 3-nitrobenzanthrone in human lung cells. Chem Res Toxicol 32:2538–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotná E, Büküm N, Hofman J, Flaxová M, Kouklíková E, Louvarová D, Wsól V (2018a) Roscovitine and purvalanol A effectively reverse anthracycline resistance mediated by the activity of aldo-keto reductase 1C3 (AKR1C3): A promising therapeutic target for cancer treatment. Biochem Pharmacol 156:22–31. [DOI] [PubMed] [Google Scholar]
- Novotná E, Büküm N, Hofman J, Flaxová M, Kouklíková E, Louvarová D, Wsól V (2018b) Aldo-keto reductase 1C3 (AKR1C3): a missing piece of the puzzle in the dinaciclib interaction profile. Arch Toxicol 92:2845–2857. [DOI] [PubMed] [Google Scholar]
- Novotna R, Wsol V, Xiong G, Maser E (2008) Inactivation of the anticancer drugs doxorubicin and oracin by aldo-keto reductase (AKR) 1C3. Toxicol Lett 181:1–6. [DOI] [PubMed] [Google Scholar]
- Ohara H, Miyabe Y, Deyashiki Y, Matsuura K, Hara A (1995) Reduction of drug ketones by dihydrodiol dehydrogenases, carbonyl reductase and aldehyde reductase of human liver. Biochem Pharmacol 50:221–227. [DOI] [PubMed] [Google Scholar]
- Ohta TIijima KMiyamoto MNakahara ITanaka HOhtsuji MSuzuki TKobayashi AYokota JSakiyama T, et al. (2008) Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res 68:1303–1309. [DOI] [PubMed] [Google Scholar]
- Park ALLin HKYang QSing CWFan MMapstone TBGross NLGumerlock MKMartin MDRabb CH, et al. (2010) Differential expression of type 2 3α/type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) in tumors of the central nervous system. Int J Clin Exp Pathol 3:743–754. [PMC free article] [PubMed] [Google Scholar]
- Peng XC, Gong FM, Wei M, Chen X, Chen Y, Cheng K, Gao F, Xu F, Bi F, Liu JY (2010) Proteomic analysis of cell lines to identify the irinotecan resistance proteins. J Biosci 35:557–564. [DOI] [PubMed] [Google Scholar]
- Penning TM (2019) AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase): Roles in malignancy and endocrine disorders. Mol Cell Endocrinol 489:82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning TM, Drury JE (2007) Human aldo-keto reductases: function, gene regulation, and single nucleotide polymorphisms. Arch Biochem Biophys 464:241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning TM, Burczynski ME, Jez JM, Hung C-F, Lin H-K, Ma H, Moore M, Palackal N, Ratnam K (2000) Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J 351:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning TM, Wangtrakuldee P, Auchus RJ (2019) Structural and functional biology of aldo-keto reductase steroid-transforming enzymes. Endocr Rev 40:447–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippione ACCarnovale IMBonanni DSini MGoyal PMarini EPors KAdinolfi SZonari DFestuccia C, et al. (2018) Potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors based on the benzoisoxazole moiety: application of a bioisosteric scaffold hopping approach to flufenamic acid. Eur J Med Chem 150:930–945. [DOI] [PubMed] [Google Scholar]
- Pippione ACGiraudo ABonanni DCarnovale IMMarini ECena CCostale AZonari DPors KSadiq M, et al. (2017) Hydroxytriazole derivatives as potent and selective aldo-keto reductase 1C3 (AKR1C3) inhibitors discovered by bioisosteric scaffold hopping approach. Eur J Med Chem 139:936–946. [DOI] [PubMed] [Google Scholar]
- Platt A, Xia Z, Liu Y, Chen G, Lazarus P (2016) Impact of nonsynonymous single nucleotide polymorphisms on in-vitro metabolism of exemestane by hepatic cytosolic reductases. Pharmacogenet Genomics 26:370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plebuch M, Soldan M, Hungerer C, Koch L, Maser E (2007) Increased resistance of tumor cells to daunorubicin after transfection of cDNAs coding for anthracycline inactivating enzymes. Cancer Lett 255:49–56. [DOI] [PubMed] [Google Scholar]
- Powell KSemaan LConley-LaComb MKAsangani IWu YMGinsburg KBWilliams JSquire JAMaddipati KRCher ML, et al. (2015) ERG/AKR1C3/AR constitutes a feed-forward loop for AR signaling in prostate Cancer Cells. Clin Cancer Res 21:2569–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan PT, Zhang Z, McCourt L, Dwyer M, Benkovic SJ, Hammes GG (2002) Interaction of dihydrofolate reductase with methotrexate: ensemble and single-molecule kinetics. Proc Natl Acad Sci USA 99:13481–13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizner TL, Smuc T, Rupreht R, Sinkovec J, Penning TM (2006) AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Mol Cell Endocrinol 248:126–135. [DOI] [PubMed] [Google Scholar]
- Scher HIBeer TMHigano CSAnand ATaplin MEEfstathiou ERathkopf DShelkey JYu EYAlumkal J, et al. ; Prostate Cancer Foundation/Department of Defense Prostate Cancer Clinical Trials Consortium (2010) Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet 375:1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selga E, Noé V, Ciudad CJ (2008) Transcriptional regulation of aldo-keto reductase 1C1 in HT29 human colon cancer cells resistant to methotrexate: role in the cell cycle and apoptosis. Biochem Pharmacol 75:414–426. [DOI] [PubMed] [Google Scholar]
- Shen Y, Zhong L, Johnson S, Cao D (2011) Human aldo-keto reductases 1B1 and 1B10: a comparative study on their enzyme activity toward electrophilic carbonyl compounds. Chem Biol Interact 191:192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiiba M, Yamagami H, Yamamoto A, Minakawa Y, Okamoto A, Kasamatsu A, Sakamoto Y, Uzawa K, Takiguchi Y, Tanzawa H (2017) Mefenamic acid enhances anticancer drug sensitivity via inhibition of aldo-keto reductase 1C enzyme activity. Oncol Rep 37:2025–2032. [DOI] [PubMed] [Google Scholar]
- Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP (2006) Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res 66:2815–2825. [DOI] [PubMed] [Google Scholar]
- Sun X, Li J, Li Y, Wang S, Li Q (2020) Apatinib, a novel tyrosine kinase inhibitor, promotes ROS-dependent apoptosis and autophagy via the Nrf2/HO-1 pathway in ovarian cancer cells. Oxid Med Cell Longev 2020:3145182, DOI: 10.1155/2020/3145182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraweera L, Vasantha Rupasinhe HP, Dellaire G, Xu G. (2020) Regulation of Nrf2/ARE pathway by dietary flavonoids: a friend or foe for cancer management? Antioxidants (Basel) 11;9:973, DOI: 10.3390/antiox9100973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebay L E, Roberston H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, Hayes JD (2015) Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med 88 (Pt B):108-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, Li X, Jiang W, Lv C, Sun W, Huang C, Chen R (2016) High expression of AKR1C1 is associated with proliferation and migration of small-cell lung cancer cells. Lung Cancer (Auckl) 7:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Zhao L, Zhang H, Liu X, Zhao L, Zhao X, Li Y, Li J (2014) AKR1C3 overexpression may serve as a promising biomarker for prostate cancer progression. Diagn Pathol 9:42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabert BNess RBLo-Ciganic WHMurphy MAGoode ELPoole EMBrinton LAWebb PMNagle CMJordan SJ, et al. ; Australian Ovarian Cancer Study Group, Australian Cancer Study (Ovarian Cancer); Ovarian Cancer Association Consortium (2014) Aspirin, nonaspirin nonsteroidal anti-inflammatory drug, and acetaminophen use and risk of invasive epithelial ovarian cancer: a pooled analysis in the Ovarian Cancer Association Consortium. J Natl Cancer Inst 106:djt431, DOI: 10.1093/jnci/djt431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitch ZW, Guo B, Hembruff SL, Bewick AJ, Heibein AD, Eng J, Cull S, Maclean DA, Parissenti AM (2009) Induction of 1C aldoketoreductases and other drug dose-dependent genes upon acquisition of anthracycline resistance. Pharmacogenet Genomics 19:477–488. [DOI] [PubMed] [Google Scholar]
- Verma K, Gupta N, Zang T, Wangtrakluldee P, Srivastava SK, Penning TM, Trippier PC (2018) AKR1C3 inhibitor KV-37 exhibits antineoplastic effects and potentiates enzalutamide in combination therapy in prostate adenocarcinoma cells. Mol Cancer Ther 17:1833–1845. [DOI] [PubMed] [Google Scholar]
- Verma K, Zang T, Gupta N, Penning TM, Trippier PC (2016) Selective AKR1C3 inhibitors potentiate chemotherapeutic activity in multiple acute myeloid leukemia (AML) cell lines. ACS Med Chem Lett 7:774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma K, Zang T, Penning TM, Trippier PC (2019) Potent and highly selective aldo-keto reductase 1C3 (AKR1C3) inhibitors act as chemotherapeutic potentiators in acute myeloid leukemia and T-cell acute lymphoblastic leukemia. J Med Chem 62:3590–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P (2004) Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA 101:2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Yan R, Luo D, Watabe K, Liao DF, Cao D (2009) Aldo-keto reductase family 1 member B10 promotes cell survival by regulating lipid synthesis and eliminating carbonyls. J Biol Chem 284:26742–26748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HW, Lin CP, Chiu JH, Chow KC, Kuo KT, Lin CS, Wang LS (2007) Reversal of inflammation-associated dihydrodiol dehydrogenases (AKR1C1 and AKR1C2) overexpression and drug resistance in nonsmall cell lung cancer cells by wogonin and chrysin. Int J Cancer 120:2019–2027. [DOI] [PubMed] [Google Scholar]
- Wang R, An J, Ji F, Jiao H, Sun H, Zhou D (2008) Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem Biophys Res Commun 373:151–154. [DOI] [PubMed] [Google Scholar]
- Wellestein A (2018) General principles of pharmacotherapy of cancer, in Goodman & Gilman’s The Pharmacological Basis of Therapeutics (Brunton L, Dandan-Hilal R, Knollmann BC eds) pp 1161-1236, John Wiley, New York. [Google Scholar]
- Wsol V, Szotakova B, Martin HJ, Maser E (2007) Aldo-keto reductases (AKR) from the AKR1C subfamily catalyze the carbonyl reduction of the novel anticancer drug oracin in man. Toxicology 238:111–118. [DOI] [PubMed] [Google Scholar]
- Wu J, Wang D (2017) CLIC1 induces drug resistance in human choriocarcinoma through positive regulation of MRP1. Oncol Res 25:863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D, Ding S, Burchell B, Wolf CR, Friedberg T (2000) Detoxication of vinca alkaloids by human P450 CYP3A4-mediated metabolism: implications for the development of drug resistance. J Pharmacol Exp Ther 294:387–395. [PubMed] [Google Scholar]
- Yates MSKwak MKEgner PAGroopman JDBodreddigari SSutter TRBaumgartner KJRoebuck BDLiby KTYore MM, et al. (2006) Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole. Cancer Res 66:2488–2494. [DOI] [PubMed] [Google Scholar]
- Zhao J, Xiang Y, Xiao C, Guo P, Wang D, Liu Y, Shen Y (2014) AKR1C3 overexpression mediates methotrexate resistance in choriocarcinoma cells. Int J Med Sci 11:1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Shen H, Huang C, Jing H, Cao D (2011) AKR1B10 induces cell resistance to daunorubicin and idarubicin by reducing C13 ketonic group. Toxicol Appl Pharmacol 255:40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong T, Xu F, Xu J, Liu L, Chen Y (2015) Aldo-keto reductase 1C3 (AKR1C3) is associated with the doxorubicin resistance in human breast cancer via PTEN loss. Biomed Pharmacother 69:317–325. [DOI] [PubMed] [Google Scholar]
- Zhou C, Shen G, Yang F, Duan J, Wu Z, Yang M, Liu Y, Du X, Zhang X, Xiao S (2020) Loss of AKR1C1 is a good prognostic factor in advanced NPC cases and increases chemosensitivity to cisplatin in NPC cells. J Cell Mol Med 24:6438–6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu XYan RPan JZhong LCao YMa JCai CHuang DLiu JChung FL, et al. (2017) Aldo-keto reductase 1B10 protects human colon cells from DNA damage induced by electrophilic carbonyl compounds. Mol Carcinog 56:118–129. [DOI] [PubMed] [Google Scholar]