

Abstract
Alveolar epithelial–capillary barrier disruption is a hallmark of acute respiratory distress syndrome (ARDS). Contribution of mitochondrial dysfunction to the compromised alveolar-capillary barrier in ARDS remains unclear. Mesenchymal stromal cells-derived extracellular vesicles (MSC-EVs) are considered as a cell-free therapy for ARDS. Mitochondrial transfer was shown to be important for the therapeutic effects of MSCs and MSC-EVs. Here we investigated the contribution of mitochondrial dysfunction to the injury of alveolar epithelial and endothelial barriers in ARDS and the ability of MSC-EVs to modulate alveolar–capillary barrier integrity through mitochondrial transfer.
Primary human small airway epithelial and pulmonary microvascular endothelial cells and human precision cut lung slices (PCLSs) were stimulated with endotoxin or plasma samples from patients with ARDS and treated with MSC-EVs, barrier properties and mitochondrial functions were evaluated. Lipopolysaccharide (LPS)-injured mice were treated with MSC-EVs and degree of lung injury and mitochondrial respiration of the lung tissue were assessed.
Inflammatory stimulation resulted in increased permeability coupled with pronounced mitochondrial dysfunction in both types of primary cells and PCLSs. Extracellular vesicles derived from normal MSCs restored barrier integrity and normal levels of oxidative phosphorylation while an extracellular vesicles preparation which did not contain mitochondria was not effective. In vivo, presence of mitochondria was critical for extracellular vesicles ability to reduce lung injury and restore mitochondrial respiration in the lung tissue.
In the ARDS environment, MSC-EVs improve alveolar–capillary barrier properties through restoration of mitochondrial functions at least partially via mitochondrial transfer.
Short abstract
This study demonstrates that mitochondrial dysfunction is an important mechanism of ARDS pathogenesis. Mitochondrial transfer is crucial for the ability of MSC extracellular vesicles to restore integrity of the alveolar–capillary barrier. https://bit.ly/2JuqoCY
Introduction
Acute respiratory distress syndrome (ARDS) is a life-threatening condition characterised by widespread uncontrolled inflammation in the lungs. Damage to the alveolar epithelial–endothelial barrier is a key aspect of ARDS pathophysiology implicated in development of oedema and diffuse alveolar damage [1, 2].
No specific pharmacological treatment is available for patients with ARDS, largely due to the heterogeneity of the underlying pathophysiological mechanisms in different subpopulations of patients [3, 4]. Recently, two biological phenotypes have been identified retrospectively in four randomised clinical trials [5–7] and one observational study [8]. These phenotypes had different clinical characteristics, biomarker profiles, clinical outcomes and, more importantly, they responded differently to interventions (positive end-expiratory pressure, fluid management strategy, low-dose macrolide therapy and simvastatin administration). Although the field is still waiting for the confirmation of these findings in prospective studies, when developing novel therapeutics, it will now be important to consider which ARDS phenotype is present and how it may respond.
Mitochondrial dysfunction and its potential mechanistic role in the pathophysiology of lung diseases, such as chronic obstructive pulmonary disease (COPD), asthma, pulmonary arterial hypertension or idiopathic pulmonary fibrosis, is increasingly recognised [9–11]. Clinical observational data suggest that mitochondrial dysfunction has been associated with higher mortality in critically ill patients with sepsis [12] and survivors of the multiple organ dysfunction syndrome had better mitochondrial function with preservation of ATP and biogenesis markers [13]. However, the role of mitochondrial dysfunction in the pathogenesis of ARDS is not sufficiently studied to date.
Mesenchymal stromal cells (MSCs) are being actively investigated as a potential therapy for ARDS. Accumulating evidence suggests that MSCs act primarily through their secretome, key components of which are extracellular vesicles (EVs) [14, 15]. MSC-EVs are being considered as a cell-free therapy for ARDS [16, 17]. Several publications, including our own studies, have previously reported that MSC-EVs contain mitochondria [18–21]. Since the original finding by Spees et al. [22], demonstrating that mitochondrial transfer from MSCs resulted in restoration of aerobic respiration in mitochondria-depleted A549ρ° cells, transfer of MSC-derived mitochondria to pulmonary epithelial cells has been associated with decreased inflammation and injury in the in vivo models of acute lung injury [18] and asthma [23]. We have demonstrated that extracellular vesicle-mediated mitochondrial transfer was critical for MSC modulation of primary human macrophages [20]. Furthermore, we have recently found that extracellular vesicle-mediated mitochondrial transfer is required for MSC ability to promote repair of human distal lung epithelial cells [21].
In this study, we explored the contribution of mitochondrial dysfunction to the injury of alveolar epithelial and endothelial barriers in ARDS and the ability of MSC-EVs to modulate alveolar-capillary barrier functions through mitochondrial transfer. We hypothesised that the beneficial effect of MSC-EVs will depend on their ability to alleviate mitochondrial dysfunction and that extracellular vesicles will have differential effects in the microenvironments of hypo- and hyper-inflammatory ARDS phenotypes. Some of the results of these studies were previously reported in the form of an abstract [24].
Methods
Detailed methods are described in Supplemental Methods.
Cell culture
Human bone marrow mesenchymal stromal cells (MSCs) were from American Type Culture Collection (ATCC PCS-500-012). These cells met all criteria set by the International Society of Cellular Therapy for the definition of MSCs [25]. Primary human pulmonary microvascular endothelial cells (HPMECs) and human small airway epithelial cells (HSAECs) were from PromoCell (Heidelberg, Germany) and cultured according to manufacturer's instructions.
Generation of extracellular vesicles from MSC with normal and dysfunctional mitochondria
MSC-EVs were isolated from MSC conditioned medium (CM) by ultracentrifugation according to Zhu et al. [26] and characterised according to International Society for Extracellular Vesicles guidelines [27]. To generate MSCs with dysfunctional mitochondria, MSCs were treated with 1 µg·mL−1 Rhodamine-6G (Sigma-Aldrich) for 48 h [20] in medium supplemented with 50 ug·mL−1 uridine and 2.5 mM sodium pyruvate (Sigma-Aldrich) to support glycolysis, then washed with PBS and incubated for 48 h in serum free medium. extracellular vesicles were isolated from conditioned medium (Rho-EVs) and handled identically to extracellular vesicles isolated from normal MSCs.
In vivo LPS-induced lung injury model
All animal experiments were approved by Animal Welfare Ethical Review Body of Queen's University Belfast, in accordance with UK Animals (Scientific Procedures) Act 1986. C57BL/6 male mice (8–12 weeks old; Envigo RMS UK Ltd, Blackthorn, UK) were used. Mice were anaesthetised by xylazine/ketamine and LPS was instilled intratracheally (2 mg/kg of body weight). 4 h after injury, PBS, extracellular vesicles or Rho-EVs were given via tail vein. 24 h after injury, mice were euthanised and bronchoalveolar lavage fluid (BALF) or lungs were taken for analysis or preparation of precision cut lung slices (mPCLSs).
Human precision-cut lung slices and plasma samples from ARDS patients
Human lungs from organ donors (where the organ had been unsuitable for transplantation, and next of kin had consented for use in research) were used for lung slices cultured ex vivo. Ethical approval was given by National Review Ethics Service in association with NHS Blood and Transplant for lungs obtained within the UK (REC 14/LO/0250). Human PCLSs were prepared according to protocol by Uhl et al. [28]. Plasma samples used in the study were from patients recruited to HARP-2 study [29]. These samples were previously classified into two phenotypes based on concentrations of plasma inflammatory biomarkers [7]. Only baseline samples obtained prior to intervention were used for experiments. Ten plasma samples representative of each phenotype were pooled and diluted in 1% complete medium to final concentration of 10% before use, plasma from healthy volunteers was used as a control. Ethical approval for use of patient samples for research was granted by the Office for Research Ethics Committees Northern Ireland.
Statistical analysis
Statistical analysis was performed using Prism 6 software (GraphPad Software, La Jolla, CA, USA). Experiments were done at least in triplicate, the average of three technical replicates was taken as a single data point for each donor, and the points were pooled together for statistical analysis. Pooled data were presented as the mean with standard deviation. For parametric data, a student's test or one-way ANOVA with post hoc analysis using Bonferroni's selected comparisons were performed. For nonparametric data, a Kruskal–Wallis test with post hoc analysis using Dunn's selected comparisons was used. The statistical significance level was set at p<0.05.
Results
MSC-EVs characterisation
Extracellular vesicles were isolated by ultracentrifugation from MSC-conditioned medium generated by MSCs serum starved for 48 h. Viability of MSCs after 48 h of serum starvation was >90%. The dose of extracellular vesicles used in the experiments was based on the final cell count of MSCs, which generated the conditioned medium (10 μL per 1×106 cells). The protein and RNA concentrations of this dose of extracellular vesicles were 98.32±34.6 μg and 31.7±10.7 ng, respectively (n=3).
Nanoparticle tracking analyses (NTA) demonstrated a particle size range of 100–700 nm comprised of two populations of particles with sizes at 100–200 nm corresponding to exosomes, 200–400 nm corresponding to microvesicles, and substantial proportion of larger particles with the sizes at 500, 700 nm and above falling into the size range of mitochondria (figure 1a). Consistently, fine structural analysis by electron microscopy showed presence of exosomes, multi-vesicular bodies and vesicles containing mitochondria (figure 1b). Western blotting of the extracellular vesicle pellet demonstrated pronounced expression of a mitochondrial marker, translocase of outer membrane 20 (Tom20) protein (figure 1c). To further corroborate presence of mitochondria in the extracellular vesicle preparation, extracellular vesicles were isolated from conditioned medium generated from MSCs pre-stained with Mitotracker Deep Red FM, incubated with beads conjugated with anti-CD44 or -CD63Ab and analysed by flow cytometry which demonstrated presence of vesicles double positive for MSC-derived mitochondria and surface markers for MSC-EVs. Quantitatively, CD44+Mito+ extracellular vesicles constituted 11.3±7.9% of the total extracellular vesicles and CD63+Mito+ extracellular vesicles constituted 17.6±11.7% of total extracellular vesicles (figure 1d).
FIGURE 1.

Characterisation of MSC-derived extracellular vesicles (MSC-EVs). a) Characterisation of MSC-EVs using nanoparticle tracking analysis (histogram generated from five independent measurements). b) Representative transmission electron microscopy (TEM) images showing: multivesicular bodies (upper left, scale bar=50 nm), exosomes (bottom left, scale bar=100 nm), mitochondria like structures (upper right, scale bar=1 µm) and mitochondria like structures (bottom right, scale bar 200 nm). The images were taken using TEM microscope (JEOL, JEM 1400Plus, Japan). c) Immunoblot for protein expression levels of TOM20 in MSC-EVs and MSC-EVs-Rho lysates. All lanes were loaded with the same amount of total protein. d) Representative flow cytometry plots of MSC-EVs conjugated with 4 µm beads demonstrating presence of EVs populations double positive for CD44 and MitoTracker Deep Red FM (19.1%) and EV populations double positive for CD63 and MitoTracker Deep Red FM (23.8%). e) Representative histogram of nanoparticle tracking analysis of EVs isolated from Rhodamine-6G pre-treated MSC (histogram generated from five independent measurements). f) Immunoblot for protein expression levels of TOM20 in Rho-EVs lysates. All lanes were loaded with the same amount of total protein as in c.
In a previous study, we have used Rhodamine-6G to induce specific irreversible inhibition of MSC mitochondrial function [19]. Importantly, this treatment does not affect MSC viability or capacity to secrete paracrine factors. Here we also tested if Rhodamine-6G treatment would affect MSC-EV particle size distribution. NTA demonstrated that the size distribution profile and concentration of Rho-EVs was comparable to the profile of extracellular vesicles isolated from control MSCs except that particles larger than 400 nm, corresponding to the size range of mitochondria, were absent (figure 1e). Consistently, Western blot analysis of Rho-EVs did not detect presence of TOM20 protein (figure 1f). Rho-EVs were used in all the subsequent experiments to investigate the contribution of mitochondria to the overall extracellular vesicle effect.
Extracellular vesicle mitochondria are readily internalised by HSAECs and HPMECs and restore barrier integrity disrupted by LPS
Mitochondrial uptake from MSC-EVs by pulmonary cells was visualised by Nikon Eclipse Ti-E microscopy. EVs were isolated from MSC pre-treated with Mitotracker Deep Red FM and co-cultured with HSAECs or HPMECs that were pre-stained with Mitotracker Green to visualise endogenous mitochondria (figure 2a–c). Three-dimensional reconstruction of multiple z-stacks and deconvolution of images were done using Nikon 6D Eclipse software. At 24 h, there was evidence of robust uptake of MSC-derived mitochondria by both epithelial and endothelial cells (supplementary videos).
FIGURE 2.
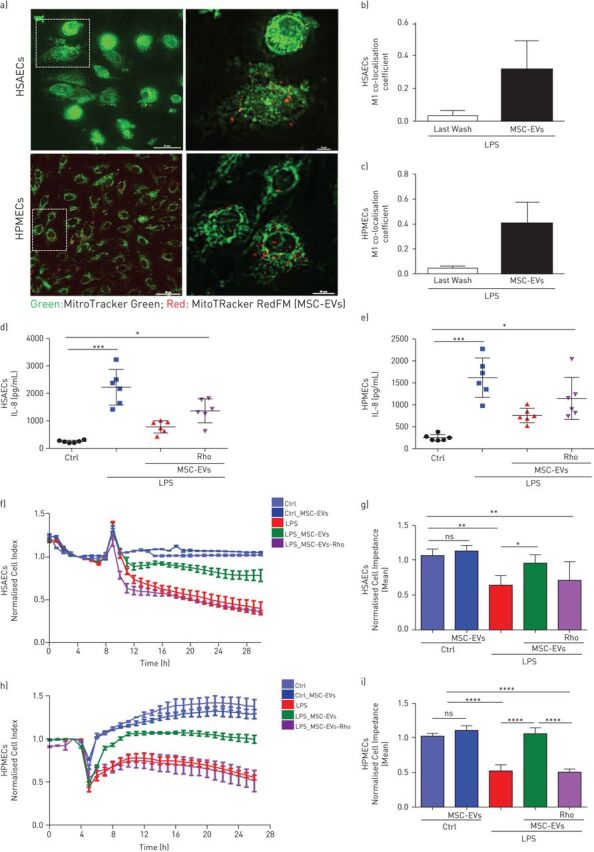
MSC-EVs improve barrier integrity of human primary lung epithelial and endothelial cells through transfer of functional mitochondria. a) Representative live images of MSC-EVs mitochondria internalisation in human small airway epithelial cells (HSAECs) and human pulmonary microvascular endothelial cells (HPMECs) (Scale bars=50 nm (left panel) and 10 nm (right panel)). b and c) M1 coefficient co-localisation in HSAECs (b) and HPMECs (c). The images were taken using a Nikon 6D Eclipse Ti-E inverted microscope with Okolab touch temperature unit and CO2 environmental chamber (Nikon Instruments, Japan) (40× dry super plan fluor ELWD objective with 0.6 NA). Data are mean±sd of ten images frames. d and e) Levels of interleukin (IL)-8 secretion by HSAECs (d) and HPMECs (e). f) Representative real-time impedance analysis of HSAECs exposed to lipopolysaccharide (LPS) and treated with MSC-EVs or MSC-EVs-Rho. g) Cell impedance of HSAECs of XCelligence RTCA measurements. Data are mean±sd (n=3). h) Representative real-time impedance analysis of HPMECs exposed to LPS and treated with MSC-EVs or MSC-EVs-Rho. i) Cell impedance of HPMECs of XCelligence RTCA measurements. Data are mean±sd (n=3). ns: not significant; *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001. Kruskal-Wallis test with post hoc Dunn's test (d and e). One-way ANOVA analysis with post hoc Bonferroni's test (g and i).
As expected, in both HSAECs and HPMECs, LPS stimulation resulted in significant upregulation of interleukin (IL)-8 production. This effect was significantly reversed by both types of extracellular vesicles (figure 2d and e), indicating that mitochondrial transfer is not critical for extracellular vesicle effects on IL-8 production.
LPS stimulation also resulted in significant disruption of both epithelial and endothelial cell barriers as indicated by the alterations in the electrical impedance of the cell monolayer (figure 2f–i). In both cell types, control MSC-EVs were able to restore barrier integrity while Rho-EVs had no effect.
LPS induces mitochondrial dysfunction in HSAECs and HPMECs which is alleviated by extracellular vesicle mitochondria
LPS induced marked decrease in the mitochondrial membrane potential (measured as a ratio between red and green JC-1 dye fluorescence) in both cell types (figure 3a–c). This was accompanied by significant upregulation of the mitochondrial reactive oxygen species (mtROS), (figure 3d–f), collectively demonstrating LPS-induced mitochondrial dysfunction.
FIGURE 3.
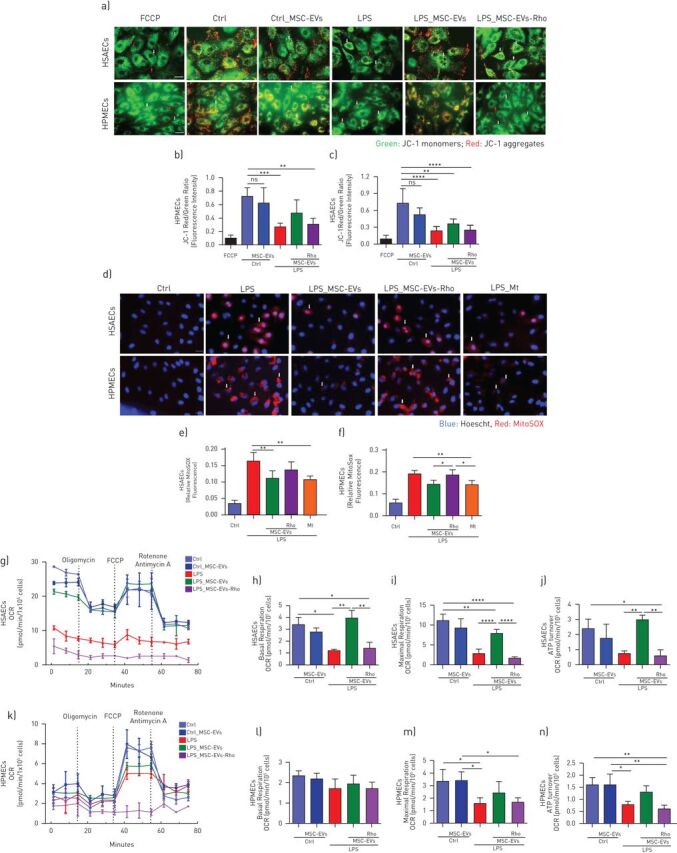
Lipopolysaccharide (LPS) induces mitochondrial dysfunction in human primary lung epithelial and endothelial cells which is alleviated by transfer of functional mitochondria in MSC-EVs. a) Representative live images of JC-1 dye fluorescence as an indicator of mitochondrial membrane potential in human small airway epithelial cells (HSAECs) and human pulmonary microvascular endothelial cells (HPMECs) after exposure to LPS and treatment with MSC-EVs or MSC-EVs-Rho for 24 h. White arrows pointing at the areas of depolarised mitochondrial membranes (JC-1 monomers) and red arrows indicating polarised mitochondrial membranes (JC-1 aggregates). FCCP was used as a control for mitochondria depolarisation. The images were taken using a Nikon 6D Eclipse Ti-E inverted microscope (Scale bar=100 µm). b and c) Quantification of red/green fluorescence intensity ratio in HSAECs (b) and HPMECs (c). Data presented as mean±sd of ten images. d) Representative live images of HSAECs and HPMECs of mitochondrial superoxide production detected with MitoSOX after exposure to LPS and treatment with MSC-EVs or MSC-EVs-Rho for 24 h or pre-treated with MitoTempo (Mt), 4 h prior stimulation. White arrows demonstrating mitochondrial ROS formation in HSAECs and HPMECs. The images were taken using a Nikon 6D Eclipse Ti-E inverted microscope (Scale bar=100 µm). e and f) Quantitative fluorescence intensity of MitoSOX in HSAECs (e) and HPMECs (f). Data presented as mean±sd of ten images. g) Representative Seahorse Mito Stress assay showing oxygen consumption rate (OCR) in HSAECs. h-j) Calculated values for respiratory parameters: basal respiration (h), maximal respiration (i) and ATP production (j), (n=3). k) Representative Seahorse Mito Stress assay showing OCR in HPMECs. l–n) Calculated values for respiratory parameters: basal respiration (l), maximal respiration (m) and ATP production (n), (n=3). Data presented as mean±sd. ns: not significant; *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001. Kruskal-Wallis test with post hoc Dunn's test (c, d, f and g). One-way ANOVA analysis with post hoc Bonferroni's test (h–j and l–n).
EVs were able to partially restore these parameters in both cell types and this effect was dependant on the presence of mitochondria, as Rho-EVs did not have any effect (figure 3a–f).
These findings were further corroborated by assessment of the mitochondrial respiration by measurements of cell oxygen consumption rate, LPS significantly reduced basal and maximum mitochondrial respiration and ATP turnover in both cell types, these effects were partially reversed by control extracellular vesicles but not Rho-EVs (figure 3g–n).
MSC-EVs reverse LPS-induced changes in mitochondrial quality control mechanisms in HSAECS and HPMECs
To evaluate effects on mitochondrial homeostasis we assessed levels of mitophagy and mitochondrial biogenesis in epithelial and endothelial cells. Mitophagy was assessed by quantification of co-localisation of mitochondria (Mitotracker Red) to autophagosomes (LC3-II lysosomal marker, green) using confocal microscopy. LPS stimulation induced significant upregulation in the levels of mitophagy in both cell types which were restored to normal levels by MSC-EVs (figure 4a–c).
FIGURE 4.
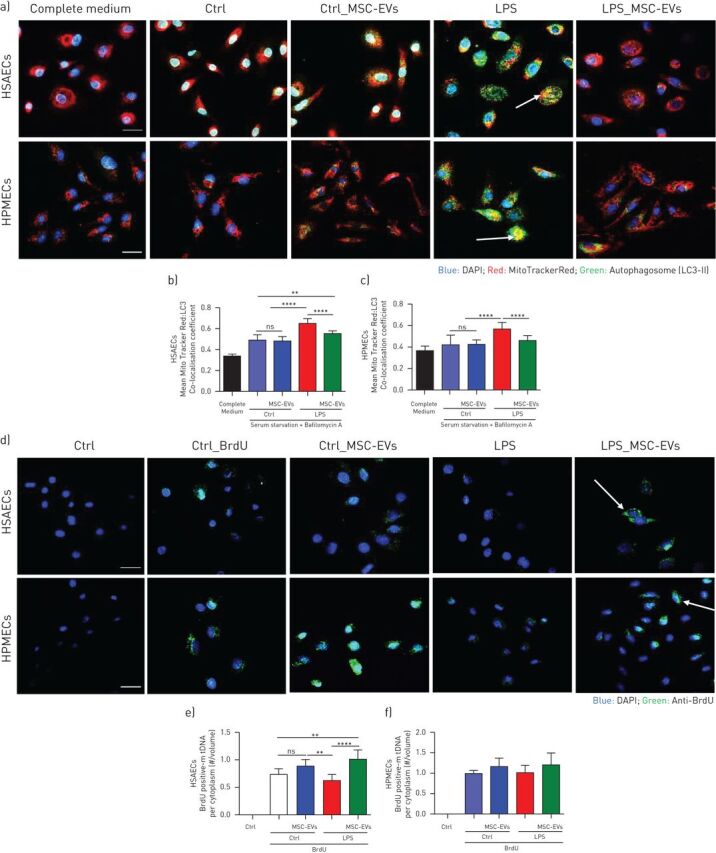
Inflammatory environment perturbs mitochondrial homeostasis in human primary lung epithelial and endothelial cells which is alleviated by MSC-EVs. a) Representative confocal microscopy of human small airway epithelial cells (HSAECs) and human pulmonary microvascular endothelial cells (HPMECs) showing the nucleus (DAPI), mitochondria (red) and LC3-II (green) in different experimental conditions. Arrows indicate co-localisation, indicating incorporation of mitochondria into autophagosome. The images were taken using Leica SP8 confocal microscope with a 40× oil-immersion objective (Scale bar=50 µm). b and c) Mitophagy assessed by co-localisation of mitochondria specific red fluorescence with green autophagosome marker LC3-II in HSAECs (b) and HPMECs (c). Data are mean±sd of ten image frames. d) Representative confocal microscopy showing biogenesis of mitochondria in HSAECs and HPMECs cells. Arrow points at the cytoplasmic accumulation of BrdU-positive mtDNA. The images were taken using Leica SP8 confocal microscope with a 40× oil-immersion objective (Scale bar=50 µm). e and f) Quantification of the ratio of BrdU-positive mtDNA per cytoplasm volume in HSAECs (e) and HPMECs (f). Data are mean±sd of ten image frames. ns: not significant; **: p<0.01; ****: p<0.0001. Kruskal-Wallis test with post hoc Dunn's test.
Mitochondrial biogenesis was assessed by measurements of mtDNA replication levels using BrdU incorporation assay [30]. As opposed to mitophagy, levels of mitochondria biogenesis were significantly inhibited by LPS in HSAECS and treatment with MSC-EVs resulted in restoration of the normal levels of mtDNA replication (figure 4d and e). In HPMECs, although changes in biogenesis levels followed the same trend, effects did not reach statistical significance (figure 4d and f).
MSC-EVs downregulate LPS-induced inflammatory response and attenuate mitochondrial dysfunction in human PCLSs
Next, we investigated the effects of extracellular vesicles on LPS-induced mitochondrial dysfunction in the more physiologically relevant human lung tissue model. Human precision cut lung slices (PCLSs) were exposed to LPS and treated with extracellular vesicles for 24 h. Mitochondrial transfer to PCLSs was visualised by confocal microscopy (supplementary figure S1). LPS stimulation resulted in significant upregulation of IL-8 and tumour necrosis factor (TNF)-α secretion levels in PCLS conditioned medium, which were significantly alleviated by control extracellular vesicles but not Rho-EVs (figure 5a and b). LPS also induced significant upregulation of the receptor for advanced glycation end products (RAGE) levels indicating alveolar epithelial injury [30]. Addition of control but not Rho-EVs restored RAGE levels to the baseline (figure 5c). Importantly, LPS stimulation did not induce cell death (as measured by LDH release) suggesting that observed changes in cytokine secretion were not accountable to passive leakage from necrotic cells (figure 5d). Addition of a specific mtROS inhibitor, MitoTempo, resulted in downregulation of TNF-α, IL-8 and RAGE secretion recapitulating the effect of control extracellular vesicles, suggesting that inhibition of mtROS may be partially responsible for the anti-inflammatory effect of extracellular vesicle mitochondrial transfer (figure 5a–d).
FIGURE 5.

MSC-EVs inhibit inflammatory response and restore mitochondrial function through mitochondrial transfer in human lung tissue in ex vivo cultured Human Precision Cut Lung Slices (PCLSs). a) Levels of interleukin(IL)-8 secretion in PCLSs supernatants at 24 h after exposure to LPS (n=6–8, 3 donors). b) Levels of tumour necrosis factor (TNF)-α secretion in PCLSs supernatants at 24 h after exposure to LPS (n=6–8, 3 donors). c) Levels of receptor for advanced glycation end products (RAGE) secretion in PCLSs supernatants at 24 h after exposure to LPS (n=6–8, 3 donors). d) PCLSs viability measured by lactate dehydrogenase (LDH) release (n=8–10, 3 donors). e) Representative live images of JC-1 fluorescence in PCLSs as an indicator of mitochondrial membrane potential. FCCP was used as positive control for complete mitochondrial membrane depolarisation. White arrows point at the areas with low mitochondrial membrane potential (accumulation of JC-1 monomers) and orange arrows point at the areas of normally polarised mitochondrial membranes (accumulation of JC-1 aggregates) in lung tissue. The images were taken using a Nikon 6D Eclipse Ti-E inverted microscope (Scale bar=1 mm). f) Quantification of red to green JC-1 fluorescence ratio in PCLSs was analysed by ImageJ software. Data represented as mean±sd of ten image frames. g) Representative images of mitochondrial superoxide production detected with MitoSOX in PCLSs. The images were taken using a Nikon 6D Eclipse Ti-E inverted microscope (Scale bar=1 mm). h) Quantitative fluorescence intensity was analysed by ImageJ software. Data represented as mean±sd of ten image frames. i) Representative seahorse mito stress test assay showing oxygen consumption rate (OCR) in PCLSs. j–l) Calculated values were normalised to control PCLSs for respiratory parameters: basal respiration (j), maximal respiration (k) and ATP production (l). (n=3 independent experiments, 3 donors). Data represented as mean±sd. ns: not significant; *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001. Kruskal-Wallis test with post hoc Dunn's test.
Exposure of PCLSs to LPS for 24 h induced significant depolarisation of the mitochondrial membrane potential and upregulation of mtROS. Both of these endpoints were restored by administration of control extracellular vesicles but not Rho-EVs (figure 5e–h). Consistently, analysis of PCLS oxygen consumption rate demonstrated LPS-induced inhibition of basal and maximal mitochondrial respiration and ATP turnover. Addition of control but not Rho-EVs was able to restore mitochondrial respiration (figure 5i–l). Treatment with MitoTempo did not have a significant effect on restoration of mitochondrial respiration in PCLSs suggesting that inhibition of mtROS alone is not enough to alleviate LPS-induced mitochondrial dysfunction at the lung tissue level.
MSC-EVs modulate barrier properties and inflammatory response of the HSAECS and HPMECs when stimulated with plasma samples from patients with ARDS; hypo-inflammatory ARDS environment is more responsive to the extracellular vesicle treatment
To mimic human ARDS environment more closely and to investigate extracellular vesicle effects in the different ARDS phenotypes, HSAECs and HPMECs were cultured for 24 h in the presence of 10% plasma samples of patients with ARDS previously classified into hypo- or hyper-inflammatory phenotypes [7].
Exposure to both types of plasma samples elicited comparable increases in IL-8 secretion in both epithelial and endothelial cells. Addition of both control and Rho-EVs comparably reduced ARDS plasma-stimulated increases (figure 6a and b). Addition of MitoTempo resulted in strong inhibition of IL-8 secretion in both cell types and both ARDS phenotypes.
FIGURE 6.

MSC-EVs modulate inflammatory response and partially restore epithelial and endothelial barrier properties via mitochondrial transfer in ARDS environment in vitro. a, b) Levels of interleukin (IL)-8 secretion by human small airway epithelial cells (HSAECs) (a) and human pulmonary microvascular endothelial cells (HPMECs) (b) at 24 h in both types of acute respiratory distress syndrome (ARDS) plasma (n=3). c) Representative real-time impedance analysis of HSAECs exposed to hypo-inflammatory ARDS plasma. d) Cell impedance analysis of XCelligence RTCA measurements in HSAECs (n=3). e) Representative real-time impedance of HPMECs exposed to hypo-inflammatory ARDS plasma. f) Cell impedance analysis of XCelligence RTCA measurements in HPMECs (n=3). g) Representative real-time impedance of HSAECs exposed to hyper-inflammatory ARDS plasma. h) Cell impedance analysis of XCelligence RTCA measurements in HSAECs (n=3). i) Representative real-time impedance analysis of HPMECs exposed to hyper-inflammatory ARDS plasma samples. j) Cell impedance analysis of XCelligence RTCA measurements in HPMECs (n=3). k and l). Calculated values for mitochondrial ATP production of HSAECs (k) and HPMECs (l) in both ARDS microenvironments (n=3). Data represented as mean±sd. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001. Kruskal-Wallis test with post hoc Dunn's test.
Assessment of the barrier function demonstrated that exposure to each type of ARDS plasma induced comparable disruption of the barrier integrity in HSAECs and HPMECs (figure 6c–j). This was partially restored by extracellular vesicles only in the case of hypo-inflammatory plasma, Rho-EVs had no effect (figure 6c–f).
Exposure to both types of ARDS plasma significantly impaired mitochondrial ATP turnover in HSAECs and HPMECs (figure 6k and l). In both cell types, MSC-EVs were able to significantly attenuate defects in the mitochondrial respiration in the presence of hypo-inflammatory plasma, whereas in hyper-inflammatory plasma extracellular vesicle effect was partial or absent. Rho-EVs did not have any effect on the mitochondrial respiration. Treatment with MitoTempo was not able to significantly improve mitochondrial ATP turnover (figure 6k and l).
Angiopoietin-2
Angiopoietin-2 (Ang-2) is a well-recognised mediator and biomarker of pulmonary and systemic vascular injury [31–33]. Plasma concentrations of Ang-2 have important predictive value for the development of ARDS and robustly predict poor clinical outcomes in adults and children with ARDS [34, 35]. Importantly, statistically significant decrease in plasma Ang-2 concentrations at 6 h after MSC infusion was reported in the START study (a randomised phase 2a safety trial of MSCs in ARDS) [36]. Therefore, we were interested in the effects of extracellular vesicles on Ang-2 secretion levels by endothelial and epithelial cells in our models. LPS stimulation resulted in robust upregulation of Ang-2 secretion by both HPMECs and SAECs (of note, SAECs had numerically almost 10-fold lower levels of Ang-2 secretion compared with HPMECs), this was partially restored by control extracellular vesicles and MitoTempo but not Rho-EVs (figure 7a and b). Consistently, stimulation with ARDS plasma also elicited an increase in Ang-2 secretion by both cell types. In HPMECs, control extracellular vesicles but not Rho-EVs were able to partially reduce its levels in the presence of hypo-inflammatory plasma whereas no effect was detected in the presence of hyper-inflammatory plasma (figure 7c). In SAECs, control extracellular vesicles but not Rho-EVs were able to partially restore Ang-2 levels in the presence of hypo-inflammatory plasma whereas addition of both control and Rho-EVs comparably reduced Ang-2 concentrations in the presence of hyper-inflammatory plasma (figure 7d).
FIGURE 7.

MSC-EVs downregulate levels of angiopoetin (Ang)-2 secretion by human primary lung epithelial and endothelial cells. a) Levels of Ang-2 secretion in human pulmonary microvascular endothelial cells (HPMECs) supernatants at 24 h after exposure to LPS (n=4) and treated with PBS, MSC-EVs, MSC-EVs-Rho or Mitotempo (Mt). b) Levels of Ang-2 secretion in human small airway epithelial cells (HSAECs) supernatants at 24 h after exposure to LPS (n=5) and treated with PBS, MSC-EVs, MSC-EVs-Rho or Mitotempo (Mt). c) Levels of Ang-2 secretion in HPMECs supernatants at 24 h in both types of acute respiratory distress syndrome (ARDS) plasma (n=3) and treated with PBS, MSC-EVs, MSC-EVs-Rho or Mitotempo (Mt). d) Levels of of Ang-2 secretion in HSAECs supernatants at 24 h in both types of ARDS plasma (n=5) and treated with PBS, MSC-EVs, MSC-EVs-Rho or Mitotempo (Mt). Data represented as mean±sd. *: p<0.05; **: p<0.01; ***: p<0.001. Kruskal-Wallis test with post hoc Dunn's test.
MSC-EVs attenuate lung injury and restore lung tissue mitochondrial respiration in the mouse model of LPS-induced lung injury
Given the above results, we hypothesised that extracellular vesicle-mediated mitochondrial transfer would improve mitochondrial dysfunction in an in vivo lung injury model. Effects of two different extracellular vesicle doses were compared: extracellular vesicles generated from 5×105 and 1×106 MSCs. Administration of either dose of control extracellular vesicles comparably decreased the LPS-stimulated increases in BALF total protein, total and differential cell counts, (figure 8a–d). Administration of Rho-EVs (generated from 1×106 MSCs) had no effect. Cytospin preparations of BALF demonstrated substantial inflammatory cell recruitment to the alveolar compartment, consisting predominantly of neutrophils, in the LPS-injured group which was reduced by administration of extracellular vesicles but not Rho-EVs (figure 8e).
FIGURE 8.

MSC-EVs reduce LPS-induced lung injury and restore mitochondrial respiration in the murine lung tissue in vivo. a) Total protein concentrations in the bronchoalveolar lavage fluid (BALF) samples (n=8–10 mice per group). b–d) Total cells counts (b), absolute neutrophil counts (c) and absolute mononuclear cell counts (d) in the BALF samples (n=6 mice per group). e) Representative images of BALF cytospin preparations demonstrating inflammatory cell recruitment to the airspaces were taken using the Leica Epifluorescence DM5500 microscope (original magnification, x20). f) BALF levels of tumour necrosis factor alpha (TNF-α). g) BALF levels of keratinocyte-derived chemokine (KC, murine analogue of interleukin-8). h) Schematic representation of generation of murine precision cut-lung slices (mPCLSs). i) Representative Seahorse Mito Stress test assay showing oxygen consumption rate (OCR) in mPCLSs. j–l) Calculated values for basal respiration (j), maximal respiration (k) and ATP production (l) of mPCLSs. Data represented as mean ±sd. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001. Kruskal-Wallis test with post hoc Dunn's test.
Extracellular vesicle but not Rho-EV treatment demonstrated a trend towards reduction of BALF levels of representative inflammatory cytokines (TNF-α and keratinocyte-derived chemokine) however the effect did not reach statistical significance (figure 8f and g).
To assess mitochondrial function, we measured mitochondrial respiration in the mouse lung tissue in this model using PCLSs (figure 8h). LPS-induced injury resulted in significant downregulation of basal and maximal mitochondrial respiration and of mitochondrial ATP turnover. Systemic administration of both doses of control extracellular vesicles but not Rho-EVs restored both mitochondrial respiration and ATP production (figure 8i–l).
Discussion
The major findings from this study are the following. 1) MSC-EVs contain mitochondria, which are readily internalised by recipient cells and intercalate into the endogenous mitochondrial network (figures 1 and 2). 2) LPS-induced inflammatory response and increase in the epithelial and endothelial barrier permeability coupled with significant mitochondrial dysfunction. The transfer of mitochondria in MSC-EVs was crucial not only for the alleviation of mitochondrial dysfunction but also for the restoration of barrier integrity in both cells types, suggesting important role of mitochondria in the maintenance of alveolar-capillary barrier function (figures 2 and 3). 3) In both cell types, MSC-EVs were able to restore normal levels of mitophagy and mitochondrial biogenesis, which were perturbed by LPS (figure 4). 4) MSC-EVs mitochondrial transfer was able to attenuate mitochondrial dysfunction and restore mitochondrial respiration inhibited by LPS at the lung tissue level in PCLSs (figure 5). 5) Stimulation with ARDS plasma significantly impaired barrier integrity and mitochondrial function in both epithelial and endothelial cells, however therapeutic effect of MSC-EVs was only seen in the presence of plasma from patients with hypo-inflammatory phenotype (figures 6 and 7). 6) Therapeutic effect of MSC-EVs in the in vivo model of LPS-induced lung injury was dependent on the presence of mitochondria and EV mitochondrial transfer improved mitochondrial respiration in the lung tissue in vivo (figure 8).
Once questioned on the reality of their existence, MSC extracellular vesicles are now entering clinical arena as a potent alternative to the whole-cell therapy [16, 37, 38]. Islam et al. [18] was the first to report that MSC-EVs could act as vehicles to transfer functional mitochondria and restore bioenergetics of the host cells, compromised by inflammatory microenvironment. This was followed by a seminal publication by Phinney et al. [19], demonstrating that MSCs actively secrete mitochondria into extracellular vesicles, as a result of unfinished mitophagy. We have previously reported that mitochondrial transfer is important for MSC modulation of macrophages leading to their metabolic reprogramming towards anti-inflammatory phenotype and also promote wound healing capacity of human small airway epithelial cells [20, 21, 39]. Notably, recent publication reports that monocytes also are capable of mitochondrial secretion in the extracellular vesicles and that mitochondrial stress contributes to the inflammatory properties of their secretome [40]. Here we provide characterisation of the mitochondria present in the MSC-EVs by a range of techniques (figure 1). We demonstrate presence of vesicles larger than 400 nm which are not accountable for apoptotic bodies and possessing ultrastructural characteristics of mitochondria, presence of vesicles double-positive for MSC mitochondria and MSC-EV surface markers CD44 and CD63, and high level of expression of mitochondrial outer membrane protein in the extracellular vesicles (figure 1a–d). Importantly, we demonstrate that MSCs in which mitochondria were rendered dysfunctional by pre-treatment with Rhodamine-6G [20] also secrete extracellular vesicles and their concentration and size distribution profiles are comparable with extracellular vesicles secreted by normal MSC, except for the absence of vesicles above 400 nm in size, corresponding to the mitochondria (figure 1e).
Accumulating evidence indicate that alterations in the mitochondrial function play a role in development of many forms of critical illness [13, 41, 42]. However, the data on contribution of mitochondrial dysfunction to the disruption of the alveolar–capillary barrier in ARDS are limited. Islam et al. [18] were the first to consider that MSC therapeutic effect in lung injury is mediated through restoration of mitochondrial bioenergetics and ATP production of alveolar epithelial cells. A recent publication from the same group determined mechanistic links between mitochondrial dysfunction and microvascular hyperpermeability in vivo [43]. Here we demonstrate that LPS stimulation results in significant impairment of barrier integrity of primary human small airway epithelial and microvascular endothelial cells which is coupled with profound mitochondrial dysfunction and disturbance of mitochondrial biogenesis and mitophagy (figures 3 and 4). Donation of the exogenous mitochondria results in the restoration of normal mitochondrial respiration in both cell types to the normal levels and is critical for the therapeutic effect of extracellular vesicles on recovery of the epithelial and endothelial barrier functions.
In line with these findings, we observe that LPS stimulation impairs mitochondrial membrane potential, increases mtROS production and inhibits mitochondrial respiration in the human PCLSs, and mitochondrial transfer from MSC-EVs is crucial to alleviate mitochondrial dysfunction and restore mitochondrial respiration in this model (figure 5).
Overproduction of mtROS is thought to be crucial factor in the development of mitochondrial dysfunction leading to disruption of mitochondrial membrane integrity, dissipation of ΔΨ and further damage to the cell [44–46]. Antioxidant-based therapies are explored for clinical efficacy in multiple disease conditions including critical illness [47–49]. Interestingly, treatment with mitochondrial ROS inhibitor, MitoTempo, was sufficient to reverse inflammatory response but was not able to restore mitochondrial respiration (figures 5 and 6), suggesting that MSC-EVs effect cannot be fully explained by inhibition of mtROS production. It is plausible to hypothesise that upregulation of mitochondrial biogenesis observed with extracellular vesicle mitochondrial transfer could be important for alleviation of LPS-induced mitochondrial dysfunction.
Two biological subphenotypes of ARDS (hypo- and hyper-inflammatory) are retrospectively identified in four large clinical trials, including HARP-2 [7]. Interestingly, a subsequent study using microarray analysis of whole-blood gene expression in an observational cohort of 210 patients with sepsis-related ARDS found that one-third of genes were differentially expressed between phenotypes and patients in the “reactive” or “hyper-inflamed” group were characterised by expression of genes indicative of mitochondrial dysfunction [50]. We found that stimulation of epithelial as well as endothelial cells with ARDS plasma induces inhibition of mitochondrial respiration regardless of the phenotype (figure 6). MSC-EVs however were able to partially restore both mitochondrial function and barrier integrity only in hypo-inflammatory plasma and presence of mitochondria was central to this effect (figure 6), suggesting, that patients with hypo-inflammatory phenotype may be more responsive to the MSC-EVs treatment. It is plausible that in the setting of hyper-inflammatory microenvironment there is a threshold to endothelial and epithelial cell injury, beyond which retrieval of physiological bioenergetic function is hindered.
Ang-2 has been established as a key mediator and biomarker of endothelial injury in ARDS [31–35] and was the single biomarker which levels were significantly reduced after MSC administration in the START study [36]. It is important to highlight that in the present study control but not Rho-EVs demonstrated strong trend towards reduction of Ang-2 secretion levels by both HPMECS and SAECS in the presence of LPS and ARDS plasma, indicating relevance of extracellular vesicle mitochondrial transfer for alleviation of the severity of endothelial injury (figure 7).
To confirm the importance of MSC-EVs mitochondrial transfer in the lung injury in vivo, endotoxin-injured mice were given extracellular vesicles derived from normal or Rhodamine-6G-treated MSCs. Normal extracellular vesicle, but not Rho-EVs, were able to significantly reduce the extent of lung injury after 24 h (figure 8). These data are in line with several previously published reports demonstrating therapeutic effects of MSC-EVs in the in vivo models of ARDS [26, 51–55]. Furthermore, measurements of oxygen consumption rates of the lung tissue in this model using mPCLSs, demonstrated that instillation of endotoxin significantly inhibited mitochondrial respiration and ATP production in the mouse lungs and extracellular vesicle mitochondrial transfer was able to restore mitochondrial respiration to normal levels (figure 8).
This study has limitations. We used ARDS plasma diluted to 10% by volume; while exposing cells to all constituents of the ARDS microenvironment, this effectively reduced the concentrations of the stimuli. We acknowledge that BALF would be more suitable environment to stimulate epithelial cells, unfortunately BALF samples from both phenotypes were not available. The endotoxin-induced lung injury model is relatively mild and does not reflect complexity of human ARDS; the primary aim of the in vivo experiments was to provide a proof of principle that MSC-EVs are capable to alleviate mitochondrial dysfunction in vivo. Additionally, we did not investigate molecular mechanisms by which mitochondria in extracellular vesicles are protective in lung injury and specifically molecular mechanisms responsible for alleviation of endothelial barrier dysfunction, this is the subject of ongoing work. We did not use fibroblasts-derived extracellular vesicles as cell specific control for in vivo experiments, however previous studies have demonstrated that fibroblast-derived extracellular vesicles had no therapeutic effects in LPS and Escherichia coli-induced murine lung injury models [55, 56]. Our primary focus in this study was to investigate the mechanistic role of MSC-EV-mediated mitochondrial transfer, therefore we used Rho-EVs which contain similar profile of vesicles but lack mitochondria, we believe this would constitute a better control.
In conclusion, we for the first time, have demonstrated that ARDS environment induces significant mitochondrial dysfunction in human lung tissue implicated in the impairment of alveolar–capillary barrier functions. Therapeutic effect of MSC-EVs on the restoration of barrier integrity is mediated by mitochondrial transfer.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary methods ERJ-02978-2020.Supplement (300.3KB, pdf)
Supplementary figure S1 ERJ-02978-2020.Figure_S1 (290.3KB, pdf)
Supplementary video S1 ERJ-02978-2020.Video_S1 (2.6MB, m4v)
Supplementary video S2 ERJ-02978-2020.Video_S2 (3.1MB, m4v)
Shareable PDF
Acknowledgements
We thank Ileana Micu for technical assistance with imaging work, John Conlon for technical assistance with processing of clinical samples, and Ke Xu and David Butler for general technical support.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Conflict of interest: J. Dutra Silva has nothing to disclose.
Conflict of interest: Y. Su has nothing to disclose.
Conflict of interest: C.S. Calfee reports grants from NIH, during the conduct of the study; grants and personal fees for consultancy from Roche/Genentech and Bayer, personal fees for advisory board work from Quark Pharmaceuticals and Prometic, personal fees for consultancy from Gen1e Life Sciences and Vasomune, outside the submitted work.
Conflict of interest: K.L. Delucchi has nothing to disclose.
Conflict of interest: D. Weiss has nothing to disclose.
Conflict of interest: D.F. McAuley reports personal fees from consultancy for GlaxoSmithKline, Boehringer Ingelheim and Bayer, outside the submitted work; his institution has received grants from the UK NIHR, Wellcome Trust and others; has a patent issued to his institution for a treatment for ARDS; and is a Director of Research for the Intensive Care Society and NIHR EME Programme Director.
Conflict of interest: C. O'Kane reports grants from Wellcome Trust, MRC and NI HSC R& D, outside the submitted work, and that her spouse has received consultancy fees from GlaxoSmithKline, Boehringer Ingelheim and Bayer.
Conflict of interest: A.D. Krasnodembskaya reports grants from Medical Research Council, UK, during the conduct of the study.
Support statement: This work was supported by UK Medical Research Council Research Awards (MRC MR/R025096/1 and MR/S009426/1 – to AK). Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Huppert LA, Matthay MA, Ware LB. Pathogenesis of acute respiratory distress syndrome. Semin Respir Crit Care Med 2019; 40: 31–39. doi: 10.1055/s-0039-1683996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matthay MA, Zemans RL, Zimmerman GA, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers 2019; 5: 18. doi: 10.1038/s41572-019-0069-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis SR, Pritchard MW, Thomas CM, et al. Pharmacological agents for adults with acute respiratory distress syndrome. Cochrane Database Syst Rev 2019; 7: CD004477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laffey JG, Kavanagh BP. Negative trials in critical care: why most research is probably wrong. Lancet Respir Med 2018; 6: 659–660. doi: 10.1016/S2213-2600(18)30279-0 [DOI] [PubMed] [Google Scholar]
- 5.Calfee CS, Delucchi K, Parsons PE, et al. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med 2014; 2: 611–620. doi: 10.1016/S2213-2600(14)70097-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Famous KR, Delucchi K, Ware LB, et al. Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med 2017; 195: 331–338. doi: 10.1164/rccm.201603-0645OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calfee CS, Delucchi KL, Sinha P, et al. Acute respiratory distress syndrome subphenotypes and differential response to simvastatin: secondary analysis of a randomised controlled trial. Lancet Respir Med 2018; 6: 691–698. doi: 10.1016/S2213-2600(18)30177-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bos LD, Schouten LR, van Vught LA, et al. Identification and validation of distinct biological phenotypes in patients with acute respiratory distress syndrome by cluster analysis. Thorax 2017; 72: 876–883. doi: 10.1136/thoraxjnl-2016-209719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riou M, Alfatni A, Charles AL, et al. New insights into the implication of mitochondrial dysfunction in tissue, peripheral blood mononuclear cells, and platelets during lung diseases. J Clin Med 2020; 9: 1253. doi: 10.3390/jcm9051253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryter SW, Rosas IO, Owen CA, et al. Mitochondrial dysfunction as a pathogenic mediator of chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Ann Am Thorac Soc 2018; 15: S266–S272. doi: 10.1513/AnnalsATS.201808-585MG [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho HY, Kleeberger SR. Mitochondrial biology in airway pathogenesis and the role of NRF2. Arch Pharm Res 2020; 43: 297–320. doi: 10.1007/s12272-019-01182-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360: 219–223. doi: 10.1016/S0140-6736(02)09459-X [DOI] [PubMed] [Google Scholar]
- 13.Carre JE, Orban JC, Re L, et al. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med 2010; 182: 745–751. doi: 10.1164/rccm.201003-0326OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phinney DG, Pittenger MF. Concise review: MSC-derived exosomes for cell-free therapy. Stem Cells 2017; 35: 851–858. doi: 10.1002/stem.2575 [DOI] [PubMed] [Google Scholar]
- 15.Elahi FM, Farwell DG, Nolta JA, et al. Preclinical translation of exosomes derived from mesenchymal stem/stromal cells. Stem Cells 2020; 38: 15–21. doi: 10.1002/stem.3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abraham A, Krasnodembskaya A. Mesenchymal stem cell-derived extracellular vesicles for the treatment of acute respiratory distress syndrome. Stem Cells Transl Med 2020; 9: 28–38. doi: 10.1002/sctm.19-0205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahida RY, Matsumoto S, Matthay MA. Extracellular vesicles: a new frontier for research in acute respiratory distress syndrome. Am J Respir Cell Mol Biol 2020; 63: 15–24. doi: 10.1165/rcmb.2019-0447TR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Islam MN, Das SR, Emin MT, et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 2012; 18: 759–765. doi: 10.1038/nm.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phinney DG, Di Giuseppe M, Njah J, et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat Commun 2015; 6: 8472. doi: 10.1038/ncomms9472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrison TJ, Jackson MV, Cunningham EK, et al. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am J Respir Crit Care Med 2017; 196: 1275–1286. doi: 10.1164/rccm.201701-0170OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fergie N, Todd N, McClements L, et al. Hypercapnic acidosis induces mitochondrial dysfunction and impairs the ability of mesenchymal stem cells to promote distal lung epithelial repair. FASEB J 2019; 33: 5585–5598. doi: 10.1096/fj.201802056R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spees JL, Olson SD, Ylostalo J, et al. Differentiation, cell fusion, and nuclear fusion during ex vivo repair of epithelium by human adult stem cells from bone marrow stroma. Proc Natl Acad Sci USA 2003; 100: 2397–2402. doi: 10.1073/pnas.0437997100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmad T, Mukherjee S, Pattnaik B, et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J 2014; 33: 994–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silva JD, Su Y, McAuley DF, et al. Transfer of mitochondria through MSC-derived extracellular vesicles improves alveolar-capillary barrier integrity and alleviate mitochondrial dysfunction in acute respiratory distress syndrome (ards). Cytotherapy 2020; 22: S25. doi: 10.1016/j.jcyt.2020.03.502 [DOI] [Google Scholar]
- 25.Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006; 8: 315–317. doi: 10.1080/14653240600855905 [DOI] [PubMed] [Google Scholar]
- 26.Zhu YG, Feng XM, Abbott J, et al. Human mesenchymal stem cell microvesicles for treatment of Escherichia coli endotoxin-induced acute lung injury in mice. Stem Cells 2014; 32: 116–125. doi: 10.1002/stem.1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 2018; 7: 1535750. doi: 10.1080/20013078.2018.1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uhl FE, Vierkotten S, Wagner DE, et al. Preclinical validation and imaging of Wnt-induced repair in human 3D lung tissue cultures. Eur Respir J 2015; 46: 1150–1166. doi: 10.1183/09031936.00183214 [DOI] [PubMed] [Google Scholar]
- 29.McAuley DF, Laffey JG, O'Kane CM, et al. Simvastatin in the acute respiratory distress syndrome. N Engl J Med 2014; 371: 1695–1703. doi: 10.1056/NEJMoa1403285 [DOI] [PubMed] [Google Scholar]
- 30.Lentz SI, Edwards JL, Backus C, et al. Mitochondrial DNA (mtDNA) biogenesis: visualization and duel incorporation of BrdU and EdU into newly synthesized mtDNA in vitro. J Histochem Cytochem 2010; 58: 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhandari V, Choo-Wing R, Lee CG, et al. Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat Med 2006; 12: 1286–1293. doi: 10.1038/nm1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parikh SM. Angiopoietins and Tie2 in vascular inflammation. Curr Opin Hematol 2017; 24: 432–438. doi: 10.1097/MOH.0000000000000361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agrawal A, Matthay MA, Kangelaris KN, et al. Plasma angiopoietin-2 predicts the onset of acute lung injury in critically ill patients. Am J Respir Crit Care Med 2013; 187: 736–742. doi: 10.1164/rccm.201208-1460OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calfee CS, Gallagher D, Abbott J, et al. Plasma angiopoietin-2 in clinical acute lung injury: prognostic and pathogenetic significance. Crit Care Med 2012; 40: 1731–1737. doi: 10.1097/CCM.0b013e3182451c87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zinter MS, Spicer A, Orwoll BO, et al. Plasma angiopoietin-2 outperforms other markers of endothelial injury in prognosticating pediatric ARDS mortality. Am J Physiol Lung Cell Mol Physiol 2016; 310: L224–L231. doi: 10.1152/ajplung.00336.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matthay MA, Calfee CS, Zhuo H, et al. Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): a randomised phase 2a safety trial. Lancet Respir Med 2019; 7: 154–162. doi: 10.1016/S2213-2600(18)30418-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiklander OPB, Brennan MA, Lotvall J, et al. Advances in therapeutic applications of extracellular vesicles. Sci Transl Med 2019; 11: eaav8521. doi: 10.1126/scitranslmed.aav8521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JH, Park J, Lee JW. Therapeutic use of mesenchymal stem cell-derived extracellular vesicles in acute lung injury. Transfusion 2019; 59: 876–883. doi: 10.1111/trf.14838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jackson MV, Morrison TJ, Doherty DF, et al. Mitochondrial transfer via tunneling nanotubes is an important mechanism by which mesenchymal stem cells enhance macrophage phagocytosis in the in vitro and in vivo models of ARDS. Stem Cells 2016; 34: 2210–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puhm F, Afonyushkin T, Resch U, et al. Mitochondria are a subset of extracellular vesicles released by activated monocytes and induce type I IFN and TNF responses in endothelial cells. Circ Res 2019; 125: 43–52. doi: 10.1161/CIRCRESAHA.118.314601 [DOI] [PubMed] [Google Scholar]
- 41.Supinski GS, Schroder EA, Callahan LA. Mitochondria and critical illness. Chest 2020; 157: 310–322. doi: 10.1016/j.chest.2019.08.2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harrois A, Huet O, Duranteau J. Alterations of mitochondrial function in sepsis and critical illness. Curr Opin Anaesthesiol 2009; 22: 143–149. [DOI] [PubMed] [Google Scholar]
- 43.Hough RF, Islam MN, Gusarova GA, et al. Endothelial mitochondria determine rapid barrier failure in chemical lung injury. JCI Insight 2019; 4: e124329. doi: 10.1172/jci.insight.124329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X, Fang P, Mai J, et al. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol 2013; 6: 19. doi: 10.1186/1756-8722-6-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piantadosi CA, Suliman HB. Mitochondrial dysfunction in lung pathogenesis. Annu Rev Physiol 2017; 79: 495–515. doi: 10.1146/annurev-physiol-022516-034322 [DOI] [PubMed] [Google Scholar]
- 46.Kozlov AV, Lancaster JR Jr, Meszaros AT, et al. Mitochondria-meditated pathways of organ failure upon inflammation. Redox Biol 2017; 13: 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biswas S, Hwang JW, Kirkham PA, et al. Pharmacological and dietary antioxidant therapies for chronic obstructive pulmonary disease. Curr Med Chem 2013; 20: 1496–1530. doi: 10.2174/0929867311320120004 [DOI] [PubMed] [Google Scholar]
- 48.Apostolova N, Victor VM. Molecular strategies for targeting antioxidants to mitochondria: therapeutic implications. Antioxid Redox Signal 2015; 22: 686–729. doi: 10.1089/ars.2014.5952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dushianthan A, Cusack R, Burgess VA, et al. Immunonutrition for acute respiratory distress syndrome (ARDS) in adults. Cochrane Database Syst Rev 2019; 1: CD012041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bos LDJ, Scicluna BP, Ong DSY, et al. Understanding heterogeneity in biologic phenotypes of acute respiratory distress syndrome by leukocyte expression profiles. Am J Respir Crit Care Med 2019; 200: 42–50. doi: 10.1164/rccm.201809-1808OC [DOI] [PubMed] [Google Scholar]
- 51.Park J, Kim S, Lim H, et al. Therapeutic effects of human mesenchymal stem cell microvesicles in an ex vivo perfused human lung injured with severe E. coli pneumonia. Thorax 2019; 74: 43–50. doi: 10.1136/thoraxjnl-2018-211576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu S, Park J, Liu A, et al. Mesenchymal stem cell microvesicles restore protein permeability across primary cultures of injured human lung microvascular endothelial cells. Stem Cells Transl Med 2018; 7: 615–624. doi: 10.1002/sctm.17-0278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Varkouhi AK, Jerkic M, Ormesher L, et al. Extracellular vesicles from interferon-gamma-primed human umbilical cord mesenchymal stromal cells reduce escherichia coli-induced acute lung injury in rats. Anesthesiology 2019; 130: 778–790. doi: 10.1097/ALN.0000000000002655 [DOI] [PubMed] [Google Scholar]
- 54.Tang XD, Shi L, Monsel A, et al. Mesenchymal stem cell microvesicles attenuate acute lung injury in mice partly mediated by Ang-1 mRNA. Stem Cells 2017; 35: 1849–1859. doi: 10.1002/stem.2619 [DOI] [PubMed] [Google Scholar]
- 55.Monsel A, Zhu YG, Gennai S, et al. Therapeutic effects of human mesenchymal stem cell-derived microvesicles in severe pneumonia in mice. Am J Respir Crit Care Med 2015; 192: 324–336. doi: 10.1164/rccm.201410-1765OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hao Q, Gudapati V, Monsel A, et al. Mesenchymal stem cell-derived extracellular vesicles decrease lung injury in mice. J Immunol 2019; 203: 1961–1972. doi: 10.4049/jimmunol.1801534 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary methods ERJ-02978-2020.Supplement (300.3KB, pdf)
Supplementary figure S1 ERJ-02978-2020.Figure_S1 (290.3KB, pdf)
Supplementary video S1 ERJ-02978-2020.Video_S1 (2.6MB, m4v)
Supplementary video S2 ERJ-02978-2020.Video_S2 (3.1MB, m4v)
This one-page PDF can be shared freely online.
Shareable PDF ERJ-02978-2020.Shareable (379.8KB, pdf)