

Graphical abstract
Keywords: Rehmannia glutinosa, Genome sequence, Oxford Nanopore Technology, Medicinal plant, Genome evolution
Abstract
Rehmannia glutinosa is a potent medicinal plant with a significant importance in traditional Chinese medicine. Its root is enriched with various bioactive molecules mainly iridoids, possessing important pharmaceutical properties. However, the molecular biology and evolution of R. glutinosa have been largely unexplored. Here, we report a reference genome of R. glutinosa using Nanopore technology, Illumina and Hi-C sequencing. The assembly genome is 2.49 Gb long with a scaffold N50 length of 70 Mb and high heterozygosity (2%). Since R. glutinosa is an autotetraploid (4n = 56), the difference between each set of chromosomes is very small, and it is difficult to distinguish the two sets of chromosomes using Hi-C. Hence, only one set of the genome size was mounted to the chromosome level. Scaffolds covering 52.61% of the assembled genome were anchored on 14 pseudochromosomes. Over 67% of the genome consists of repetitive sequences dominated by Copia long terminal repeats and 48,475 protein-coding genes were predicted. Phylogenetic analysis corroborates the placement of R. glutinosa in the Orobanchaceae family. Our results indicated an independent and very recent whole genome duplication event that occurred 3.64 million year ago in the R. glutinosa lineage. Comparative genomics analysis demonstrated expansion of the UDP-dependent glycosyltransferases and terpene synthase gene families, known to be involved in terpenoid biosynthesis and diversification. Furthermore, the molecular biosynthetic pathway of iridoids has been clarified in this work. Collectively, the generated reference genome of R. glutinosa will facilitate discovery and development of important pharmacological compounds.
1. Introduction
Plants are source of rich natural products representing tremendous resources for drug development. There are over 200,000 plant secondary metabolites known to date with several compounds heavily used as drugs [1]. Besides their applications in drug discovery, plants are exploited by human for centuries as herbal drugs and traditional knowledge of plant-based therapies have been transmitted from generation to generation. Population in India, China, Japan, Korea and several African and South American countries considerably rely on traditional herbal medicine [2]. Among the 21,000 medicinal plants listed by the World Health Organization, Rehmannia glutinosa Libosch. Ex Fisch. & C.A. Mey represents one of the most important. It is widely cultivated in Asian countries such as China, Korea, Japan and Vietnam. R. glutinosa has been listed as one of the 50 fundamental herbs in traditional Chinese medicine [3]. It plays a wide range of long lasting pharmacological activities on human health with very less side effects [4]. The main medicinal tissue of R. glutinosa is the tuberous root (Fig. 1), which is used for the treatment of heat and strengthening body tonicity. It has also significant effects on the cardiovascular system, central nervous system, immune system, and visceral system based on in-vitro assays [4], [5], [6], [7], [8], [9], [10].
Fig. 1.

Phenotype of Rhemannia glutinosa cultivar Qinhuai used in this study. Left: whole plant in flowering stage, Right above: leaf, Right below: tuberous root. Scale = 1 cm.
Several bioactive compounds with important pharmaceutical activities have been reported in R. glutinosa root including iridoids (catalpol, rehmannioside A, B, C and D, geniposide, aucubin), ionones and phenylethanoids [3], [4], [11], [12]. Despite the beneficial properties of these bioactive compounds, the molecular mechanisms of their formation are yet to be elucidated mainly because of the lack of high-quality genome resources in R. glutinosa [13], [14], [15], [16], [17].
The exact phylogenetic placement of R. glutinosa in the Lamiales order has been elusive. Initially, based on morphological traits it was placed in the Scrophulariaceae family but after several revisions, it has been classified as a species of the Orobanchaceae family [18], [19], [20], [21]. Nonetheless, the genome structure and evolution of R. glutinosa is yet to be elucidated.
Sequencing and investigating genome evolution in medicinal plants is essential for elucidating their phylogenetic relationship, promoting their sustainable exploitation and facilitating plant-based drug discovery [22]. Genome sequences of several species in the Lamiales order are now available [23], [24], [25], [26], [27], [28], which will facilitate comparative evolutionary studies in R. glutinosa. Recently, the rapid development in new sequencing technologies (Oxford Nanopore Technologies (ONT), PacBio sequencing) and complementary long-range scaffolding technologies (Hi-C sequencing and Bionano optical maps) have facilitated the generation of chromosome-scale genome assemblies in various plant species with large and complex genomes [29], [30].
In this report, we de novo sequenced and assembled a reference genome of R. glutinosa by combining ONT long reads, Illumina NovaSeq short reads and Hi-C sequencing. We annotated and characterized the structure and evolutionary history of the large and complex genome of R. glutinosa. We further performed a genome-wide prediction and expression analysis of terpene synthase and UDP-dependent glycosyltransferases gene families known to be involved in the biosynthesis and diversification of terpenoids. Our work elucidates the genomic evolution of R. glutinosa and provides essential genomic resources for decoding the synthetic pathways of bioactive compounds to facilitate molecular breeding of cultivars with improved medicinal attributes.
2. Material and methods
2.1. Plant materials, DNA library construction and sequencing
Rehmannia glutinosa Libosch. Ex Fisch. & C.A. Mey cultivar Qinhuai is one of the ten main cultivars grown in China. Qinhuai is rich in catalpol, verbascoside, iridoids, and is mainly grown in Jiaozuo, Daohuang district, Henan province, China. Healthy tissue-cultured plants of Qinhuai were obtained from the College of Pharmacy, Henan University of Chinese Medicine, China. High-quality genomic DNA was isolated from fresh leaves using the conventional cetyltriethylammonium bromide method [31]. Agarose gel electrophoresis was used to check the integrity of DNA (DNA Integrity Number > 8) /RNA (RNA Integrity Number > 6). In order to obtain longer reads, it is generally necessary to screen large fragment sequences through gel cutting instead of sequence fragmentation. Illumina sequencing pair-end libraries with insert size of 150 bp were prepared using Nextera DNA Flex Library Prep Kit (Illumina, San Diego, CA, USA). Sequencing was performed using the Illumina NovaSeq platform (Illumina, San Diego, CA, USA). Raw reads (170.32 Gb) were cleaned to discard low-quality reads (reads with adaptors and unknown nucleotides and reads with>20% low-quality bases) using the FastQC (v.0.11.8) tool (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and, after data filtering, 157.73 Gb clean data were used for subsequent analyses (Table S1).
For Oxford Nanopore sequencing, the libraries were prepared with the standard protocol from Oxford Nanopore Technologies previously detailed by Song et al. [32]. The purified library was loaded onto primed R9.4 Spot-On Flow Cells and sequenced using a PromethION sequencer (Oxford Nanopore Technologies, Oxford, UK) with 48-h runs at Wuhan Benagen Tech Solutions Company Limited, Wuhan, China. Base calling analysis of raw data was performed using the Oxford Nanopore GUPPY software (v0.3.0). A total of 297.86 Gb raw data was generated with 230.83 Gb ‘passed’ reads after quality control.
2.2. RNA library construction, sequencing and data processing
Total RNA was extracted from young leaves, mature leaves, jasmonic acid-treated leaves and roots, salicylic acid-treated leaves and roots of cultivars Qinhuai, Huaifeng and Wen85-5 using the HiPure Plant RNA Kit according to the manufacturer’s instructions (Magen, Guangzhou, China). RNA samples were pooled and used for library preparation following the standard protocol from Oxford Nanopore Technologies with the library prep kit (SQK-PCS109 + SQK-PBK004). The library was loaded onto R9.4 SpotON Flow Cells (Oxford Nanopore Technologies, Oxford, UK) and sequenced using a 48-h run time. In addition, Illumina RNA-seq (Illumina NovaSeq, San Diego, CA, USA) short reads were generated based on tissue culture seedlings (triplicate samples) from the cultivars Huaifeng and Qinhuai. RNA-seq reads were remapped to the reference genome assembly using STAR (v.2.7.0; parameters: --twopassMode None) [33] and the FPKM was calculated to evaluate the expression level of each gene using the RSEM (v.1.2.15) tool [34].
2.3. Genome assembly
Based on the sequencing data, the K-mer analysis method [35] was used to estimate the genome size and heterozygosity using the kmer_freq program in the gce package (v.1.0.0) [36]. Genomic assembly was performed using SMARTdenovo software (https://github.com/ruanjue/smartdenovo; parameter: -p jvh -k 17 -J 2000 -t 32 -c 1). Two rounds of error correction were performed on the assembly result based on the Nanopore sequencing data using Racon (v.1.4.11) (https://github.com/isovic/racon). Two rounds of error correction were performed on the assembly result based on the Illumina Novaseq sequencing data using Pilon (v.1.22; parameters: default) [37]. Finally, the genome was de-hybridized using the Purge_haplotigs pipeline (v.1.0.4) [38] to obtain the final assembly result.
2.4. Pseudochromosome level assembly using Hi-C
High-quality DNA extracted from young leaves of healthy tissue-cultured plants was used for Hi-C sequencing. Formaldehyde was used for fixing chromatin. In situ Hi-C chromosome conformation capture was performed according the DNase-based protocol described by Ramani et al. [39]. The libraries were sequenced using 350 bp paired-end mode on an Illumina NovaSeq (Illumina, San Diego, CA, USA). For pseudochromosome level scaffolding, we used the assembly software ALLHIC (v. 0.9.12) [40] and 3D-DNA (v.180419) [41] for stitching, and then we imported the final files (.hic and .assembly) generated by the software into Juicebox (v1.11.08) [42] for plotting.
2.5. Genome annotation
2.5.1. a) repeat sequence annotation
We used the RepeatModeler (v.1.0.4) (https://github.com/rmhubley/RepeatModeler) software to build our own repeat library. After merging the repbase library, we used RepeatMasker (v.4.0.5) (http://www.repeatmasker.org/) for genome repeat annotation.
2.5.2. b) gene prediction
Gene prediction was performed using MAKER software (v.2.31.8) [43] and Augustus software (v.3.0.3) [44]. Protein coding sequences from 15 species including, Cuscuta australis, Ipomea nil, Capsicum annuum, Solanum melongena, Solanum lycopersicum, Chrysanthenum nankingense, Helianthus annuus, Sesamum indicum, Erythranthe guttata, Olea europeae subsp. sylvestris, Utricularia gibba, Antirrhinum majus, Arabidopsis thaliana, Oryza sativa and Vitis vinifera (Table S2), were mapped to the assembly of R. glutinosa using BLAST (v.2.6.0+; parameter: -evalue 1e-5) [45]. The predicted genes were corrected with the de novo assembled RNA-seq long and short reads using Trinity (v.2.6.6) [46]. Next, we employed the BUSCO software (v.4.1.2) [47] for evaluating the quality of the prediction based on the eukaryotic database.
2.5.3. c) gene function annotation
Using BLAST (v.2.6.0+; parameters: -evalue 1e-5 -max_target_seqs 5 -num_threads 10) [45], the predicted protein sequences were compared with the transposable element (TE) protein library. After removing the TE protein genes, the protein-coding genes were functionally annotated based on seven publicly available databases including, Uniprot [48], Pfam [49], GO [50], KEGG [51], Swissprot [48], Interpro [52] and NR [53].
2.5.4. d) Non-coding RNA annotation
Based on the structural features of tRNA, tRNAscan-SE (v.1.23) [54] was used to find tRNA sequences in the R. glutinosa genome. Furthermore, rRNA prediction was performed using RNAmmer (v.1.2) [55]. The miRNA and snRNA was predicted using Rfam_scan.pl (v1.0.4) by inner calling using Infernal (v1.1.1) [56].
2.6. Gene family and evolutionary analysis
2.6.1. a) Gene family clustering
All amino acid sequences of the 15 selected species in addition to R. glutniosa were aligned using BLASTP (v.2.6.0; parameters: -evalue 1e-5 -outfmt 6) [45], and the gene family clustering was performed using OrthoMCL software (v.2.0.9; parameters: percentMatchCutoff = 30, evalueExponentCutoff = 1e-5, expansion coefficient 1.5) [57].
2.6.2. b) Phylogenetic tree construction
A single copy gene family shared by the 16 selected species was screened to construct a phylogenetic tree. First, the protein sequence of each single copy gene family was subjected to alignments in MUSCLE (v.3.8.31) [58] and finally, the maximum likelihood tree was built using RAxML (v.8.2.10) software [59].
2.6.3. c) Gene family contraction and expansion
Gene family contraction and expansion analysis was performed using CAFÉ (v.2.1; parameter: --filter) software [60] based on gene family clustering results.
2.6.4. d) Divergence time analysis
Based on the phylogenetic tree result, the mcmctree of PAML (v.4.9) (parameters: nsample = 1000000; burnin = 200000; seqtype = 0; model = 4) [61] was used to estimate the differentiation time of the different species. Published divergence times for Vitis vinifera-Oryza sativa: 125–150 million years ago (Mya) and Solanum lycopersicum-Helianthus annuus: 95–106 Mya were used to calibrate the divergence time.
2.6.5. e) Whole gene duplication analysis
Whole genome duplication analysis was performed based on five species: Solanum lycopersicum, Cuscuta australis, Ipomoea nil, Vitis vinifera and R. glutinosa. First, we compared the protein sequences of different species using the all-to-all search in BLASTP (v.2.6.0+; parameters: -evalue 1e-5 -outfmt 6) [45], and then used MCScanX (https://github.com/wyp1125/MCScanx; parameters: -a -e 1e-5 -s 5) [62] to analyze the genomic collinear block, and finally calculate the synonymous mutation frequency (Ks) of the collinear gene pairs based on the NG method of Yang implemented in PAML (v.4.9) [61]. The synonymous mutation rate distribution (Ks) was plotted using ggplot2 (v.2.2.1) package in R v.2.15 (www.r-project.org). The synonymous substitution rate of 8.25 X 10-9 mutations per site per year was applied to calculate the ages of the WGDs.
2.7. Investigations of terpene synthase (TPS), UDP-dependent glycosyltransferase (UGT) and iridoid biosynthetic pathway associated genes
The Hidden Markov Model profiles of the TPS domains (PF01397 and PF03936) and UGT domain (PF00201) were obtained from Pfam v.32.0 database (http://Pfam.sanger.ac.uk/) [63] and searched against the genomes of R. glutinosa and related species using HMMER V.3.0 program with “trusted cutoff” as threshold [64]. Candidate genes were further confirmed using the SMART tool [65]. Redundant sequences and sequences without conserved motifs were removed. TPS genes were grouped into subfamilies according to Chen et al. [66]. Similarly, UGT genes were assigned to different families based on the previous work of Yonekura-Sakakibara and Hanada [67]. The protein sequences of genes were subjected to alignments in MUSCLE (v.3.8.31) [58] and the maximum likelihood tree was built using RAxML (v.8.2.10) software [59].
Iridoids are derived from either the plastidial 2-C-methyl-d-erythritol-4-phosphate (MEP) pathway or the cytosolic mevalonic acid (MVA) pathway. Using the KEGG annotation, we searched for all genes MEP and MVA pathways genes and reconstructed the iridoids biosynthesis pathway [11], [68]. Heatmaps displaying gene expression profiles were plotted using pheatmap (v.1.0.12) in R v.2.15 (www.r-project.org) based on various RNA-seq data (Table S3).
3. Results and discussion
3.1. Genome sequencing and assembly
In order to estimate the genome size and heterozygosity of Rehmannia glutinosa cultivar ‘Qinhuai’, genome survey sequencing was performed. A total of 170.32 Gb of Illumina NovaSeq reads was generated and the estimated genome size was 2.35 Gb according to the 19-mer depth distribution (Table S4, Figure S1). Compared to other genome sequenced plant species in the Lamiales order (Sesamum indicum, Erythranthe guttata, Olea europeae subsp. sylvestris, Olea europaea subsp. europaea, Salvia splendens, Utricularia gibba, Antirrhinum majus, Scutellaria baicalensis), R. glutinosa features the largest genome [69], [23], [24], [25], [26], [27], [28]. High heterozygosity (2%) was estimated in R. glutinosa genome and the 19-mer distribution indicated a large proportion of repeat sequences (Table S4), denoting a challenging de novo genome assembly of this species [70].
The hybrid sequencing approach combining long reads and short reads technologies has proven to be efficient for plant species with large or complex genome [32], [69], [71], [72]. Here, we also employed a hybrid genome sequencing approach by combining Oxford Nanopore Technologies (ONT) and Illumina NovaSeq platform (Figure S2). The ONT yielded 297.86 Gb raw data from 3 flow cells composed of 14 million reads (read N50 length, 28.5 Kb) (Table S5). After data filtering (remove reads of quality score < 7), 230.82 Gb data were kept for downstream analyses, representing 98-fold genome coverage (Table S5). A total of 157.73 Gb Illumina clean reads were generated (Table S1), representing 67-fold genome coverage. The genome assembly was conducted on a computer with 250 G CPU memory and 80 threads (Table S6). We assembled and corrected the Nanopore long reads prior using Illumina reads for polishing. In addition, Hi-C data were used for scaffold extension and chromosome mount. Globally, the final assembled genome had a total length of 2.49 Gb and a contig N50 of 658 Kb (Table 1). The assembled genome size is slightly larger than the estimated size of 2.35 Gb, probably due to the high heterozygosity. Similar observations were previously reported in Olea europaea subsp. europaea and Carya illinoinensis [25], [73]. In de novo genome sequencing, the application of Hi-C scaffolding facilitates sequence continuity to reach a chromosome scale [74]. Since R. glutinosa is an autotetraploid (4n = 56) [75], the difference between each set of chromosomes is very small, and it is difficult to distinguish the two sets of chromosomes using Hi-C. Hence, only one set of the genome size was assembled to the chromosome level. Herein, Hi-C assisted assembly helped to anchor a total of 52.61% of R. glutinosa genome on 14 pseudochromosomes named as chr1 ~ chr14 (Fig. 2A, Figure S3, Table S6, Table S7, Table S8), which is consistent with the karyotype (4n = 56) previously reported by Xu [75]. We aligned the Illumina short reads to the assembled genome using BWA, resulting in a mapping rate of 93.7%.
Table 1.
Summary of R. glutinosa genome assembly.
| Genome features | Contig | Scaffold |
|---|---|---|
| Total_length (bp) | 2,498,530,510 | 2,498,814,810 |
| Number of contigs | 5,290 | 3,733 |
| GC_content (%) | 36.52 | 36.52 |
| N50 (bp) | 658,887 | 70,280,146 |
| N90 (bp) | 244,881 | 262,126 |
| Average (bp) | 472,312.01 | 669,385.16 |
| Median (bp) | 363,729 | 239,898 |
| Min (bp) | 30,576 | 25,000 |
| Max (bp) | 2,924,441 | 166,779,478 |
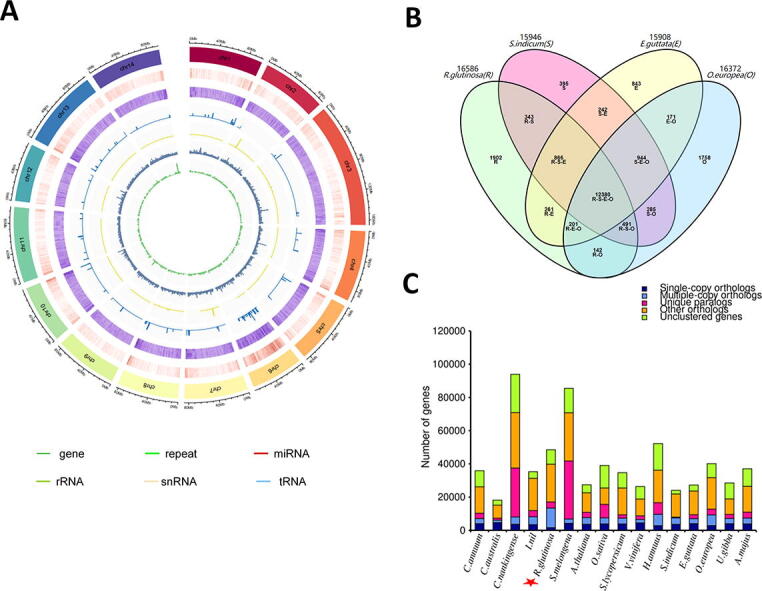
Fig. 2.

De novo genome assembly of R. glutinosa and comparative analysis with related species. (A) Summary of the de novo genome assembly and sequencing analysis of R. glutinosa cultivar Qinhuai (from outside to inside: 1. pseudochromosome number; 2. gene density; 3. repeat density; 4. miRNA density; 5. rRNA density; 6. snRNA density; 7. tRNA density). (B) Venn diagram showing the shared and unique gene families among four Lamiales species. (C) Distribution of genes and gene families across 16 plant species. The red star marks the position of the sequenced species in this study. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. Gene annotation
Analysis of the repeat sequences in Qinhuai genome revealed over 67% repetitive sequences, with the dominant type (>75%) being transposable elements (TE) (Table S9). This is higher than the 29, 43, 55, 58% of repeat sequences reported in genomes of sesame, Olea europeae subsp. sylvestris, Antirrhinum majus and Salvia splendens [24], [26], [27], [28]. TEs could be roughly classified into four types: long terminal repeat (LTR), long interspersed nuclear element, short interspersed nuclear element and DNA transposons. We found that LTRs represent the major repetitive sequences (45%). It has been evidenced that abundance of LTRs contributes to genome size expansion in plants with varying proportions of LTR superfamilies’ [76]. For instance, higher Gypsy than Copia elements was observed in cowpea, Salvia splendens, walnut, Polygonum cuspidatum and Helianthus annus [27], [72], [77], [78], [79]. On the opposite, in plant species such as tomato, Chrysanthemum nankingense and sesame, Copia elements are more dominant than Gypsy elements [24], [32], [80]. In the present study, we noticed that Gypsy elements and Copia elements contribute to 13% and 31% in the genome, respectively (Table S9), implying that the large genome size of R. glutinosa is mainly attributable to the amount of Copia retrotransposons.
An integrated gene prediction protocol was employed based on a combination of de novo assembly of transcriptomes from various cultivars, tissues and stress conditions (whole seedlings (Qinhuai, Wen85-5 and Huaifeng cultivars), young leaves, mature leaves, jasmonic acid-treated leaves and roots, salicylic acid-treated leaves and roots (Table S3)), ab initio prediction and homology search with protein sequences from 16 related species. In total, 48,475 protein-coding genes were obtained in R. glutinosa genome with an average mRNA length, coding sequence length and exon number of 5.01 Kb, 1,291.21 bp and 5.89 per gene, respectively (Table S10). The predicted gene was evaluated based on the embryophyta database using the BUSCO software [47], which delivered a score of completeness of 97.83% including complete and fragmented BUSCOs (Table S11). This result suggests that a near completion genome sequence has been generated for R. glutinosa in this study, similar to published genomes of Arabidopsis and rice [81], [82]. A total of 48,475 protein-coding genes were obtained in R. glutinosa genome, ranking this species as the most gene-enriched in the Lamiales order. Nonetheless, it is worth mentioning that the number of predicted genes can enormously vary depending on the tools and parameters used in gene prediction. We obtained 99% of the predicted protein-coding genes matching entries in seven publicly available databases (Table S12). With regard to non-coding genes, we identified 351 miRNA, 1,800 tRNA, 1,199 rRNA and 2,945 snRNA fragments from the total assembly (Table S13).
3.3. Gene family and phylogenetic analysis
The genome assembly for R. glutinosa was compared with 15 genome-sequenced plant species, including five Solanales species (Cuscuta australis, Convolvulaceae; Ipomea nil Convolvulaceae; Capsicum annuum, Solanaceae; Solanum melongena, Solanaceae; Solanum lycopersicum, Solanaceae), two Asterales species (Chrysanthenum nankingense, Asteraceae; Helianthus annuus, Asteraceae), five Lamiales species (Sesamum indicum, Pedaliaceae; Erythranthe guttata, Prhymaceae; Olea europeae subsp. sylvestris, Oleaceae; Utricularia gibba, Lentibulariaceae; Antirrhinum majus, Plantaginaceae), one Brassicales species (Arabidopsis thaliana, Brassicaceae), one Poales species (Oryza sativa, Poaceae), and one Vitales species (Vitis vinifera, Vitaceae) (Table S2). Based on gene family clustering analysis, 44,335 gene families (689,787 genes) were detected, including 5,309 core gene families (126,451 genes) shared by all the 16 species and 12,380 gene families shared by the four Lamiales species (Fig. 2B; Table S14). Specific genes found in R. glutinosa genome were enriched in various gene ontology (GO) functional categories with the most enriched being the general GO term GO:0016799 (hydrolase activity) and specific GO terms GO:0047938 (glucose-6-phosphate 1-epimerase activity), GO:0016114 (terpenoid biosynthetic process), GO:0047924 (geraniol dehydrogenase activity), GO:0046029 (mannitol dehydrogenase activity), suggesting a high enzymatic activity in R. glutinosa (Table S15).
We constructed a phylogenetic tree based on 266 gene families shared by all species as single-copy orthologous gene families (Fig. 2C). Our goal was to clarify whether R. glutinosa belongs to Scrophulariaceae or Orobanchaceae. Based on previous phylogenetic analyses, Scrophulariaceae and Plantaginaceae are closely related while Orobanchaceae is very close to Prhymaceae [16], [28], [83]. Therefore, we integrated A. majus (Plantaginaceae) and E. guttata (Prhymaceae) in our analysis. We found that R. glutinosa was clustered together with Lamiales species, which is consistent with its phylogenetic placement (Fig. 3). Remarkably, E. guttata was the closest species to R. glutinosa. Since Phrymaceae is close to Orobanchaceae, we deduce that R. glutinosa belongs well to the family of Orobanchaceae. The Orobanchaceae family is essentially composed of parasitic plant species [84], [85]. Therefore, the integration of some non-parasitic plant species such as R. glutinosa in this family still needs further clarification. Recent progresses on genome sequencing of parasitic plants in Orobanchaceae family such as Striga asiatica [86] will be helpful to elucidate the genome evolution and systematics of this family.
Fig. 3.

Phylogenetic analysis and divergence time estimations among 16 plant species including R. glutinosa. The tree was constructed based on all single-copy orthologous genes using Oryza sativa as outgroup. A total of 500 bootstrap replicates was performed and bootstrap values lower than 100 are not presented. The star marks the species used for genome sequencing in this study. Divergence times estimated in million years ago are indicated by the blue lines over the nodes. The span of the blue lines shows 95% confidence interval of the divergence time. The divergence time was estimated for each node in million years (MY). The number of gene-family contraction and expansion events is indicated by green and red numbers, respectively. The number at the root (11,726) denotes the total number of gene families predicted in the most recent common ancestor (MRCA). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
R. glutinosa was estimated to have diverged from E. guttata 21.1 million years ago (Mya) and from the Solanales approximately 86.3 Mya (Fig. 3), which is in accordance with estimations that the Lamiales order has diverged from the Solanales order between 89.8 and 185.8 Mya [24]. Evolutionary driven modifications of gene family size are a natural phenomenon providing selective advantages and contributing to organizational and regulatory diversity in a variety of organisms [87], [88]. In this study, we investigated the gene family expansion and contraction in R. glutinosa lineage. The analysis revealed that 6,237 gene families underwent expansion, while a significant number of gene families (8 4 8) underwent contraction (Fig. 3). Interestingly, the expanded gene families were mainly enriched in GO:0008299 (isoprenoid biosynthetic process) (Table S16), which may have contributed to the high content of bioactive metabolites in this important medicinal plant [4], [89].
3.4. Whole genome duplication event
Whole-genome duplication (WGD) is a main causal agent in diversification, phenotypic and developmental innovation in organisms [90]. Novel lineage-specific WGD events have been reported and dated in all Lamiales species with published genomes. For example, sesame, A. majus and E. europea subsp. sylvestris experienced an independent WGD at 71, 46 and 28 Mya, respectively [24], [26], [28]. WGD event was examined in R. glutinosa in comparison with four other species (Solanum lycopersicum, Cuscuta australis, Ipomoea nil and Vitis vinifera). We identified the syntenic blocks within genomes through intragenome comparisons (Fig. 4A). There was a prominent peak for calculated synonymous substitution rates (Ks) of gene pairs at 0.06 in the R. glutinosa lineage (Fig. 4B), indicative of a very recent independent WGD event which occurred 3.64 Mya after splitting from E. guttatus (21.1 Mya) [91].
Fig. 4.

Comparative genomic and evolutionary analysis. (A) Intragenome comparison showing syntenic relationship between R. glutinosa pseudochromosomes. (B) Distribution of synonymous substitution rates (Ks) for pairs of syntenic paralogs in R. glutinosa and four other species (Rglu: Rhemannia glutinosa, Slyc: Solanum lycopersicum, Caus: Cuscuta australis, Inil: Ipomoea nil and Vvin: Vitis vinifera).
3.5. Molecular biosynthetic pathway of iridoids
The major bioactive molecule in R. glutinosa is iridoid [4], [9]. It has been well documented that iridoids possess beneficial antitumor, antioxidant, diuretic, neuroprotective and anti-inflammatory effects on human health [5], [92], [93], [94]. >30 kinds of iridoids have been isolated from R. glutinosa including, catalpol, aucubin, rehmannioside A, B, C and D [4]. Iridoids are monoterpenes and two general synthetic pathways of terpenoids biosynthesis have been recognized in plants: mevalonate pathway (MVP) and 2C-methyl-D-erythritol-4-phosphate pathway (MEP) [95]. Based on the extensive investigations on Catharanthus roseus and R. glutinosa, two main routes for iridoids biosynthesis have been proposed (Fig. 5A) [68], [96], [97], however structural genes catalyzing steps of this complex pathways have not yet been fully identified [11], [98].
Fig. 5.

Genes involved in iridoids biosynthesis and glycosylation. (A) Biosynthesis pathways of iridoids in R. glutinosa. Log2 fold change of the differentially expressed genes involved in the biosynthesis of iridoids between root samples at various growth stages (SP2/SP1; SP3/SP1) and between leaf and root samples (HF/SP). HF: leaf samples from Huaifeng cultivar; SP3: 2 months after sprouting root (root samples); SP2: 1 month after sprouting root (root samples); SP1: 15–20 days after sprouting root (root samples). (B) The phylogenetic tree showing a total of 116 terpene synthase (TPS) with the different TPS subfamilies labeled. TPSs are highlighted in orange while A. thaliana TPSs are highlighted in black. (C) The phylogenetic tree showing a total of 191 UDP-dependent glycosyltransferases (UGTs) with the different UGT subfamilies labeled. R. glutinosa UGTs are highlighted in red while A. thaliana UGTs are highlighted in black. DMAPP: dimethylally diphosphate; DXP: 1-deoxy-d-xylulose 5-phosphate; GGPP: geranylgeranyl diphosphate; FPP: farnesyl diphosphate; GPP: geranyl diphosphate; HMBPP: 1-Hydroxy-2-methyl-2-butenyl-4 diphosphate; IPP: ispentenyl diphosphate; MEP: 2-C-methyl-d-erythritol 4-phosphate; MVA: mevalonic acid; G10H: geraniol 8-hydroxylase; 10HGO: 10-hydroxygeraniol oxidoreductase; IS: iridoid synthase; 7-DLS/CYP76A26: 7-deoxyloganetic acid synthase; 7-DLH:7-deoxyloganic acid hydroxylase; 7-DLGT: 7-deoxyloganetic acid glucosyltransferase; LAMT: loganic acid methyltransferase; SLS: secologanin synthase; STR: strictosidine synthase; DXS: 1-deoxy-D-xylulose-5-phosphate synthase; IDI: isopentenyl diphosphate isomerase; DXR: 1-deoxy-D-xylulose 5-phosphate reductoisomerase; GES: geraniol synthase; UGD: UDP-glucuronic acid decarboxylase; HMGR: 3-hydroxy-3-methylglutaryl-CoA synthase; GPS: geranyl diphosphate synthase; GGPS: geranylgeranyl pyrophosphate synthase; FPPS: farnesyl diphosphate synthase; HDR: 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase; CPR: cytochrome P450 reductase; CPM: cytochrome P-450 monooxygenase; ALDH: aldehyde dehydrogenase; UPD: uroporphyrinogen decarboxylase; F3H: flavanone 3-dioxygenase; SQLE: squalene monooxygenase. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In this study, we searched for all genes involved in the iridoid biosynthesis pathway and identified a total of 313 candidate genes belonging to 25 enzyme families (Table S17). We employed transcriptome data from root and leaf tissues in order to identify key genes differentially expressed genes (DEG) between tissues and growth stages that could be target of further functional analyses. In total, 137 DEGs were detected and our analysis showed that most of them were up-regulated in older root samples (SP2: 1 month and SP3: 2 months after sprouting) as compared to young root samples (SP1: 15–20 days after sprouting). Duan et al. [98] reported that the content of iridoid in R. glutinosa root increased continuously during growth stages up to 2 months and this correlates well with the expression patterns of the identified DEGs in this study. Furthermore, by comparing expression patterns in root and leaf tissues, we observed that most of the DEGs were down-expressed in the leaf tissues, confirming the fact that iridoids are mainly enriched in R. glutinosa root [99]. Further studies aiming at identifying the transcription factors regulating these DEGs through gene co-expression network analysis [100], will provide important tools for increasing iridoid content not only in root but also in leaf.
3.6. Prediction and expression analysis of terpene synthase gene family
Terpene synthase (TPS) gene family members are key enzymes generating the huge variety of terpene structures [101]. We searched for all annotated TPS genes within R. glutinosa genome and compared with six other species (A. thaliana, A. majus, C. australis, I. nil, S. lycopersicum and V. vinifera). In total, 87 TPS genes were identified, largely surpassing the number of TPS members in other species (Table S18). This indicates an expansion of TPS gene family in R. glutinosa mainly the TPS-a and TPS-b sub-families. Furthermore, a comparison with A. thaliana revealed several clusters of TPS genes unique to R. glutinosa (Fig. 5B), which may play preponderant role in iridoid biosynthesis. We analyzed the expression of TPS genes in various cultivars (Qinghuai, Wen85 and Huaifeng), growth periods (15–20 days, 1 month and 2 months after sprouting root), tissues (root and seedling), root parts (radial striation and non-radial striation) and salicylic acid (SA) treatments (Table S3). Overall, most of TPS genes were preferentially expressed in root tissues than in seedling, which correlates well with the higher content of iridoids in R. glutinosa root [99] (Figure S4). In addition, we found that SA treatment stimulates TPS gene expression and could contribute to high accumulation of iridoids [13]. Finally, we observed a variation of TPS gene expression among cultivars and growth periods (Figure S4), which may provide prospects for increasing iridoid content in R. glutinosa root [13], [14].
3.7. Prediction and expression analysis of UDP-dependent glycosyltransferase gene family
Glycosylation represents the last step in the biosynthesis of numerous natural compounds, including terpenes [102], [103]. In R. glutinosa, glycosylation plays a crucial role in iridoids biosynthesis since most of the iridoids are mainly present as glycosides [4], [104]. UDP-dependent glycosyltransferases (UGTs) belong to the largest family of the glycosyltransferase superfamily and catalyzes glycosylation process [105]. We examined the UGT gene family in R. glutinosa genome in comparison with A. thaliana and V. vinifera. In total, 333 UGT genes were detected in R. glutinosa with members of the groups A and G being the most dominant. Similar to TPS, we observed that UGT gene family has been expanded in R. glutinosa as its members were higher than V. vinifera and A. thaliana (Table S18; Table S19; Fig. 5C). Gene expression profiling in root and seedling showed globally a tissue-preferential activity of UGTs in R. glutinosa (Figure S5). The UGTs highly active in root tissues represent important candidate gene resources for further functional analyses to uncover their specific roles in iridoid decoration.
4. Conclusions
We employed a hybrid sequencing approach to resolve the large and highly complex genome of the potent medicinal plant R. glutinosa. The newly generated reference genome sequence of R. glutinosa increases the genomic resources in the Lamiales order. With the diversity of plant species with special medicinal attributes in the Lamiales order, we anticipate that our results will be cardinal for comparative genomics studies to improve our understanding of the metabolic pathways of specialized bioactive molecules. We provide a strong molecular evidence for the placement of R. glutinosa in the Orobanchaceae, a family dominated by parasitic plants. Lineage specific expansion of gene families involved in terpenoid biosynthesis and their preferential expression in root tissue may have contributed to the diversity and enrichment of iridoids in R. glutinosa root. Altogether, the released genomic resources of R. glutinosa will be essential for gene functional characterization and molecular breeding of high-yielding cultivars.
Author contribution
Ligang Ma and Chi Song: Designed, executed the experiment, performed bioinformatics analysis and drafted the manuscript. Chengming Dong, Xiaolan Wang, Yan Niu and Xiaoke Zheng: Executed the experiment and contributed in data analysis and interpretation. Weisheng Feng and Shilin Chen: Conceived, and supervised the study; provided the plant materials, financial support and revised the first drafts. All authors have read and approved the final version of this manuscript.
Funding
This work was financially supported by the National Key Research and Development Project (The Major Project for Research of the Modernization of TCM: 2017YFC1702800, 2019YFC1708802), the Major Science and Technology Projects in Henan Province (171100310500) and the Henan province high-level personnel special support “ZhongYuan One Thousand People Plan” - Zhongyuan Leading Talent (ZYQR201810080). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
7. Data availability
Rhemannia glutinosa genome assembly sequences have been deposited at the NCBI GenBank under BioProject PRJNA631301 and BioSample Accession SAMN15052190. Raw data of RNAseq used in the present study have been deposited at the NCBI GenBank and the details of the SRA accession numbers are presented in Table S3.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
Not applicable.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.07.006.
Contributor Information
Shilin Chen, Email: slchen@icmm.ac.cn.
Weisheng Feng, Email: fwsh@hactcm.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Dixon R.A., Strack D. Phytochemistry meets genome analysis, and beyond. Phytochemistry. 2003;62(6):815–816. doi: 10.1016/s0031-9422(02)00712-4. [DOI] [PubMed] [Google Scholar]
- 2.Chakraborty P. Herbal genomics as tools for dissecting new metabolic pathways of unexplored medicinal plants and drug discovery. Biochim Open. 2018;6:9–16. doi: 10.1016/j.biopen.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y., Dong L., Luo H., Hao Z., Wang Y., Zhang C. Chemical constituents from root tubers of Rehmannia glutinosa. Chin Tradit Herb Drugs. 2014;45:16–22. [Google Scholar]
- 4.Zhang R.-X., Li M.-X., Jia Z.-P. Rehmannia glutinosa: Review of botany, chemistry and pharmacology. J Ethnopharmacol. 2008;117(2):199–214. doi: 10.1016/j.jep.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 5.Zhang X., Zhang A., Jiang B.o., Bao Y., Wang J., An L. Further pharmacological evidence of the neuroprotective effect of catalpol from Rehmannia glutinosa. Phytomedicine. 2008;15(6-7):484–490. doi: 10.1016/j.phymed.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Gong W. Rehmannia glutinosa Libosch Extracts Prevent Bone Loss and Architectural Deterioration and Enhance Osteoblastic Bone Formation by Regulating the IGF-1/PI3K/mTOR Pathway in Streptozotocin-Induced Diabetic Rats. Int J Mol Sci. 2019;20:3964. doi: 10.3390/ijms20163964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan Y., Kang N., Li Q., Zhang Y., Liu Y., Tan P. Study of the Effect of Neutral Polysaccharides from Rehmannia glutinosa on Lifespan of Caenorhabditis elegans. Molecules. 2019;24(24):4592. doi: 10.3390/molecules24244592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen X. Effects of total saponins extracted from leaves of Rehmannia on accelerated nephrotoxic nephritis induced by rabbit IgG in rat. Chin J Exp Tradit Med Form. 2010;16:179–181. [Google Scholar]
- 9.Liu C.-L., Cheng L., Kwok H.-F., Ko C.-H., Lau T.-W., Koon C.-M. Bioassay-guided isolation of norviburtinal from the root of Rehmannia glutinosa, exhibited angiogenesis effect in zebrafish embryo model. J Ethnopharmacol. 2011;137(3):1323–1327. doi: 10.1016/j.jep.2011.07.060. [DOI] [PubMed] [Google Scholar]
- 10.Hong L., Guo Z., Huang K., Wei S., Liu B.o., Meng S. Ethnobotanical study on medicinal plants used by Maonan people in China. J Ethnobiol Ethnomed. 2015;11(1) doi: 10.1186/s13002-015-0019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li M., Wang X., Zheng X., Wang J., Zhao W., Song K. A new ionone glycoside and three new rhemaneolignans from the roots of Rehmannia glutinosa. Molecules. 2015;20(8):15192–15201. doi: 10.3390/molecules200815192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li M., Wang X., Zhang Z., Zhang J., Zhao X., Zheng X. Three new alkaloids and a new iridoid glycoside from the roots of Rehmannia glutinosa. Phytochem Lett. 2017;21:157–162. [Google Scholar]
- 13.Sun P., Song S., Zhou L., Zhang B., Qi J., Li X. Transcriptome analysis reveals putative genes involved in iridoid biosynthesis in Rehmannia glutinosa. Int J Mol Sci. 2012;13(12):13748–13763. doi: 10.3390/ijms131013748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Y., Wang X., Wang W., Duan H.D. novo transcriptome sequencing-based discovery and expression analyses of verbascoside biosynthesis-associated genes in Rehmannia glutinosa tuberous roots. Mol Breed. 2016;36:139. [Google Scholar]
- 15.Wang F. Transcriptome Analysis of Salicylic Acid Treatment in Rehmannia glutinosa Hairy Roots Using RNA-seq Technique for Identification of Genes Involved in Acteoside Biosynthesis. Front Plant Sci. 2017;8:787. doi: 10.3389/fpls.2017.00787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhi J. Molecular Regulation of Catalpol and Acteoside Accumulation in Radial Striation and non-Radial Striation of Rehmannia glutinosa Tuberous Root. Int J Mol Sci. 2018;19:3751. doi: 10.3390/ijms19123751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiao Z., Cheng Y., Wang H., Lei C., Wang G.G., Han L. Isolation and characterization of microsatellite loci in Rehmannia glutinosa (Scrophulariaceae), a medicinal herb. Appl Plant Sci. 2015;3(10):1500054. doi: 10.3732/apps.1500054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oxelman B., Kornhall P., Olmstead R.G., Bremer B. Further disintegration of Scrophulariaceae. Taxon. 2005;54(2):411–425. [Google Scholar]
- 19.A.H. Wortley P.J. Rudall D.J. Harris R.W. Scotland P. Linder How Much Data are Needed to Resolve a Difficult Phylogeny? 54 5 2005 2005 697 709. [DOI] [PubMed]
- 20.The Angiosperm Phylogeny Group An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot J Linn Soc. 2016;181:1–20. [Google Scholar]
- 21.Zeng S., Zhou T., Han K., Yang Y., Zhao J., Liu Z.-L. The Complete Chloroplast Genome Sequences of Six Rehmannia Species. Genes. 2017;8(3):103. doi: 10.3390/genes8030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao D.C., Xiao P.G. Genomics and Evolution in Traditional Medicinal Plants: Road to a Healthier Life. Evolutionary Bioinformatics. 2015;11:197–212. doi: 10.4137/EBO.S31326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ibarra-Laclette E., Lyons E., Hernández-Guzmán G., Pérez-Torres C.A., Carretero-Paulet L., Chang T.-H. Architecture and evolution of a minute plant genome. Nature. 2013;498(7452):94–98. doi: 10.1038/nature12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L., Yu S., Tong C., Zhao Y., Liu Y., Song C. Genome sequencing of the high oil crop sesame provides insight into oil biosynthesis. Genome Biol. 2014;15(2):R39. doi: 10.1186/gb-2014-15-2-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cruz F., Julca I., Gómez-Garrido J., Loska D., Marcet-Houben M., Cano E. Genome sequence of the olive tree. Olea europaea Gigascience. 2016;5(1) doi: 10.1186/s13742-016-0134-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Unver T. Wild olive genome and oil biosynthesis. Proc Natl Acad Sci. 2017;114:E9413–E9422. doi: 10.1073/pnas.1708621114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.A.-X. Dong H.-B. Xin Z.-J. Li H. Liu Y.-Q. Sun S. Nie et al. High-quality assembly of the reference genome for scarlet sage, Salvia splendens, an economically important ornamental plant 7 7 2018 2018 10.1093/gigascience/giy068. [DOI] [PMC free article] [PubMed]
- 28.Li X., Feng T., Randle C., Schneeweiss G.M. Phylogenetic Relationships in Orobanchaceae Inferred From Low-Copy Nuclear Genes: Consolidation of Major Clades and Identification of a Novel Position of the Non-photosynthetic Orobanche Clade Sister to All Other Parasitic Orobanchaceae. Front Plant Sci. 2019;10:902. doi: 10.3389/fpls.2019.00902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiao W.-B., Schneeberger K. The impact of third generation genomic technologies on plant genome assembly. Curr Opin Plant Biol. 2017;36:64–70. doi: 10.1016/j.pbi.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Amarasinghe S.L. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020;21:30. doi: 10.1186/s13059-020-1935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doyle J.J., Doyle J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–15. [Google Scholar]
- 32.Song C., Liu Y., Song A., Dong G., Zhao H., Sun W. The Chrysanthemum nankingense Genome Provides Insights into the Evolution and Diversification of Chrysanthemum Flowers and Medicinal Traits. Molecular Plant. 2018;11(12):1482–1491. doi: 10.1016/j.molp.2018.10.003. [DOI] [PubMed] [Google Scholar]
- 33.A. Dobin C.A. Davis F. Schlesinger J. Drenkow C. Zaleski S. Jha et al. STAR: ultrafast universal RNA-seq aligner 29 1 2013 2013 15 21. [DOI] [PMC free article] [PubMed]
- 34.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manekar S.C., Sathe S.R. A benchmark study of k-mer counting methods for high-throughput sequencing. GigaScience. 2018;7:giy125. doi: 10.1093/gigascience/giy125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu B. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quantitative Biology. 2013;35:62–67. [Google Scholar]
- 37.Ruan J., Li H. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 2020;17:155–158. doi: 10.1038/s41592-019-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roach M.J., Schmidt S.A., Borneman A.R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinf. 2018;19:460. doi: 10.1186/s12859-018-2485-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramani V., Cusanovich D.A., Hause R.J., Ma W., Qiu R., Deng X. Mapping 3D genome architecture through in situ DNase Hi-C. Nat Protoc. 2016;11(11):2104–2121. doi: 10.1038/nprot.2016.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X., Zhang S., Zhao Q.M., R.,, Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat Plants. 2019;5:833–845. doi: 10.1038/s41477-019-0487-8. [DOI] [PubMed] [Google Scholar]
- 41.Dudchenko O. De novo assembly of the genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356:92–95. doi: 10.1126/science.aal3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Durand N.C., Robinson J.T., Shamim M.S., Machol I., Mesirov J.P., Lander E.S. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016;3(1):99–101. doi: 10.1016/j.cels.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cantarel B.L., Korf I., Robb S.M.C., Parra G., Ross E., Moore B. MAKER: an easy to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18(1):188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stanke M., Keller O., Gunduz I., Hayes A., Waack S., Morgenstern B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34(Web Server):W435–W439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 46.Grabherr M.G. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotech. 2011;29:644. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 48.Apweiler R. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32(90001):115D–119D. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ashburner M. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finn R.D. Pfam: the protein families database. Nucleic Acids Res. 2013;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanehisa M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32(90001):277D–280D. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deng Y. Integrated nr database in protein annotation system and its localization. Comput Eng. 2006;32:71–72. [Google Scholar]
- 54.Todd M. Lowe Sean R. Eddy tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence 25 5 1997 1997 955 964. [DOI] [PMC free article] [PubMed]
- 55.Karin Lagesen Peter Hallin Einar Andreas Rødland Hans-Henrik Stærfeldt Torbjørn Rognes David W. Ussery RNAmmer: consistent and rapid annotation of ribosomal RNA genes 35 9 2007 2007 3100 3108. [DOI] [PMC free article] [PubMed]
- 56.Nawrocki E.P., Eddy S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29(22):2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li L., Stoeckert C.J., Roos D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robert C. Edgar Quality measures for protein alignment benchmarks 38 7 2010 2010 2145 2153. [DOI] [PMC free article] [PubMed]
- 59.Alexandros Stamatakis RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies 30 9 2014 2014 1312 1313. [DOI] [PMC free article] [PubMed]
- 60.De Bie T., Cristianini N., Demuth J.P., Hahn M.W. CAFÉ: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22(10):1269–1271. doi: 10.1093/bioinformatics/btl097. [DOI] [PubMed] [Google Scholar]
- 61.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 62.Tang H., Bowers J.E., Wang X., Ming R., Alam M., Paterson A.H. Synteny and collinearity in plant genomes. Science. 2008;320(5875):486–488. doi: 10.1126/science.1153917. [DOI] [PubMed] [Google Scholar]
- 63.Sara El-Gebali Jaina Mistry Alex Bateman Sean R Eddy Aurélien Luciani Simon C Potter et al. The Pfam protein families database in 2019 47 D1 2019 2019 D427 D432. [DOI] [PMC free article] [PubMed]
- 64.Finn R.D., Clements J., Eddy S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(suppl):W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ivica Letunic Peer Bork 20 years of the SMART protein domain annotation resource 46 D1 2018 2018 D493 D496. [DOI] [PMC free article] [PubMed]
- 66.Feng Chen Dorothea Tholl Jörg Bohlmann Eran Pichersky The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom 66 1 2011 212 229. [DOI] [PubMed]
- 67.Yonekura-Sakakibara K., Hanada K. An evolutionary view of functional diversity in family 1 glycosyltransferases. Plant J. 2011;66:182–193. doi: 10.1111/j.1365-313X.2011.04493.x. [DOI] [PubMed] [Google Scholar]
- 68.Jensen S.R. Plant iridoids, Their Biosynthesis and Distribution in Angiosperms. In: Harborne J.B., Tomas-Barberan F.A., editors. Ecological Chemistry and Biochemistry of Plant Terpenoids. Clarendon Press; Oxford; UK: 1991. pp. 133–158. [Google Scholar]
- 69.Zhao Q., Yang J., Cui M.-Y., Liu J., Fang Y., Yan M. The Reference Genome Sequence of Scutellaria baicalensis Provides Insights into the Evolution of Wogonin Biosynthesis. Mol Plant. 2019;12(7):935–950. doi: 10.1016/j.molp.2019.04.002. [DOI] [PubMed] [Google Scholar]
- 70.Du H., Liang C. Assembly of chromosome-scale contigs by efficiently resolving repetitive sequences with long reads. Nat Commun. 2019;10:5360. doi: 10.1038/s41467-019-13355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mochida K., Sakurai T., Seki H., Yoshida T., Takahagi K., Sawai S. Draft genome assembly and annotation of Glycyrrhiza uralensis, a medicinal legume. Plant J. 2017;89(2):181–194. doi: 10.1111/tpj.13385. [DOI] [PubMed] [Google Scholar]
- 72.Lonardi S., Muñoz‐Amatriaín M., Liang Q., Shu S., Wanamaker S.I., Lo S. The genome of cowpea (Vigna unguiculata [L.] Walp.) Plant J. 2019;98(5):767–782. doi: 10.1111/tpj.14349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Youjun Huang Lihong Xiao Zhongren Zhang Rui Zhang Zhengjia Wang Chunying Huang et al. The genomes of pecan and Chinese hickory provide insights into Carya evolution and nut nutrition 8 5 2019 2019 10.1093/gigascience/giz036. [DOI] [PMC free article] [PubMed]
- 74.Mitsutaka Kadota Osamu Nishimura Hisashi Miura Kaori Tanaka Ichiro Hiratani Shigehiro Kuraku Multifaceted Hi-C benchmarking: what makes a difference in chromosome-scale genome scaffolding? 9 1 2020 2020 10.1093/gigascience/giz158. [DOI] [PMC free article] [PubMed]
- 75.Xu Z.-H. Rehmannia glutinosa: Tissue Culture and Its Potential for Improvement. Medicinal and Aromatic Plants I. 1988;501–512 [Google Scholar]
- 76.MORGANTE M., DEPAOLI E., RADOVIC S. Transposable elements and the plant pan-genomes. Curr Opin Plant Biol. 2007;10(2):149–155. doi: 10.1016/j.pbi.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 77.Lucia Natali Rosa Cossu Elena Barghini Tommaso Giordani Matteo Buti Flavia Mascagni et al. 14 1 2013 686 10.1186/1471-2164-14-686. [DOI] [PMC free article] [PubMed]
- 78.Martínez-García P.J., Crepeau M.W., Puiu D., Gonzalez-Ibeas D., Whalen J., Stevens K.A. The walnut (Juglans regia) genome sequence reveals diversity in genes coding for the biosynthesis of non-structural polyphenols. Plant J. 2016;87(5):507–532. doi: 10.1111/tpj.13207. [DOI] [PubMed] [Google Scholar]
- 79.Zhang Y. Assembly and Annotation of a Draft Genome of the Medicinal Plant Polygonum cuspidatum. Front Plant Sci. 2019;10:1274. doi: 10.3389/fpls.2019.01274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sato S. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Du H. Sequencing and de novo assembly of a near complete indica rice genome. Nat Commun. 2017;8:15324. doi: 10.1038/ncomms15324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Michael T.P. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat Commun. 2018;9:541. doi: 10.1038/s41467-018-03016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tomoyuki Kado Hideki Innan Tal Dagan Horizontal gene transfer in five parasite plant species in Orobanchaceae 10 12 2018 2018 3196 3210. [DOI] [PMC free article] [PubMed]
- 84.McNeal J.R., Bennett J.R., Wolfe A.D., Mathews S. Phylogeny and origins of holoparasitism in Orobanchaceae. Am J Bot. 2013;100(5):971–983. doi: 10.3732/ajb.1200448. [DOI] [PubMed] [Google Scholar]
- 85.Schneeweiss, G.M. “Phylogenetic relationships and evolutionary trends in Orobanchaceae,” in Parasitic Orobanchaceae: Parasitic Mechanisms and Control Strategies, eds D. M. Joel, J. Gressel, and L. J. Musselman (Berlin: Springer), 243–265 (2013).
- 86.Yoshida S., Kim S., Wafula E.K., Tanskanen J., Kim Y.-M., Honaas L. Genome Sequence of Striga asiatica Provides Insight into the Evolution of Plant Parasitism. Curr Biol. 2019;29(18):3041–3052.e4. doi: 10.1016/j.cub.2019.07.086. [DOI] [PubMed] [Google Scholar]
- 87.Lespinet O., Wolf Y.I., Koonin E.V., Aravind L. The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res. 2002;12:1048–1059. doi: 10.1101/gr.174302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Demuth J.P., Hahn M.W. The life and death of gene families. BioEssays. 2009;31(1):29–39. doi: 10.1002/bies.080085. [DOI] [PubMed] [Google Scholar]
- 89.Zhou Y. Metabolite accumulation and metabolic network in developing roots of Rehmannia glutinosa reveals its root developmental mechanism and quality. Sci Rep. 2018;8:14127. doi: 10.1038/s41598-018-32447-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.MacKintosh C., Ferrier D.E.K. Recent advances in understanding the roles of whole genome duplications in evolution. F1000Res. 2017;6:1623. doi: 10.12688/f1000research.11792.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fishman L., Willis J.H., Wu C.A., Lee Y.-W. Comparative linkage maps suggest that fission, not polyploidy, underlies near-doubling of chromosome number within monkeyflowers (Mimulus; Phrymaceae) Heredity. 2014;112:562–568. doi: 10.1038/hdy.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luo Y.Y., Zhang S.Q., Suo J.Z., Sun D.Y., Cui X.C. Determination of catalpol in rehmannia root by high performance liquid chromatography. Chin Pharm J. 1994;29:38–40. [Google Scholar]
- 93.Tundis R., Loizzo M.R., Menichini F., Statti G.A., Menichini F. Biological and pharmacological activities of iridoids: recent developments. Mini Rev Med Chem. 2008;8:399–420. doi: 10.2174/138955708783955926. [DOI] [PubMed] [Google Scholar]
- 94.Bi J., Wang X.-bo., Chen L., Hao S., An L.-jia., Jiang B. Catalpol protects mesencephalic neurons against MPTP induced neurotoxicity via attenuation of mitochondrial dysfunction and MAO-B activity. Toxicol In Vitro. 2008;22(8):1883–1889. doi: 10.1016/j.tiv.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 95.Laule O., Furholz A., Chang H.S., Zhu T., Wang X., Heifetz P.B. Crosstalk between cytosolic and plastidial pathways of isoprenoid biosynthesis in Arabidopsis thaliana. P Nat Acad Sci. 2003;100(11):6866–6871. doi: 10.1073/pnas.1031755100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Damtoft Søren. Biosynthesis of catalpol. Phytochemistry. 1994;35(5):1187–1189. [Google Scholar]
- 97.Jensen S.R., Franzyk H., Wallander E. Chemotaxonomy of the Oleaceae: Iridoids as taxonomic markers. Phytochemistry. 2002;60:213–231. doi: 10.1016/s0031-9422(02)00102-4. [DOI] [PubMed] [Google Scholar]
- 98.Duan H., Liu W., Zeng Y., Jia W., Wang H., Zhou Y. Expression analysis of key enzymes involved in the accumulation of iridoid in Rehmannia glutinosa. Plant Omics Journal. 2019;(12(02):2019):102–108. [Google Scholar]
- 99.Wang Y., Liao D., Qin M., Li X. Simultaneous determination of Catalpol, Aucubin, and Geniposidic acid in different developmental stages of Rehmannia glutinosa leaves by high performance liquid chromatography. Journal of Analytical Methods in Chemistry. 2016;2016:1–6. doi: 10.1155/2016/4956589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dossa K., Mmadi M.A., Zhou R., Zhang T., Su R., Zhang Y. Depicting the core transcriptome modulating multiple abiotic stresses responses in sesame (Sesamum indicum L.) Int J Mol Sci. 2019;20(16):3930. doi: 10.3390/ijms20163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Degenhardt Jörg, Köllner T.G., Gershenzon J. Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry. 2009;70(15-16):1621–1637. doi: 10.1016/j.phytochem.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 102.Yu J., Hu F., Dossa K., Wang Z., Ke T. Genome-wide analysis of UDP-glycosyltransferase super family in Brassica rapa and Brassica oleracea reveals its evolutionary history and functional characterization. BMC Genom. 2017;18:474. doi: 10.1186/s12864-017-3844-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang X. Identification of anthocyanin biosynthesis genes in rice pericarp using PCAMP. Plant Biotechnol J. 2019;17:1700–1702. doi: 10.1111/pbi.13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oshio H., Inouye H. Iridoid glycosides of Rehmannia glutinosa. Phytochemistry. 1982;21(1):133–138. [Google Scholar]
- 105.Mackenzie P.I., Owens I.S., Burchell B., Bock K.W., Bairoch A., Belanger A. The UDP glycosyltransferase gene superfamily: recommended nomenclature update based on evolutionary divergence. Pharmacogenetics. 1997;7(4):255–269. doi: 10.1097/00008571-199708000-00001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Rhemannia glutinosa genome assembly sequences have been deposited at the NCBI GenBank under BioProject PRJNA631301 and BioSample Accession SAMN15052190. Raw data of RNAseq used in the present study have been deposited at the NCBI GenBank and the details of the SRA accession numbers are presented in Table S3.