

Summary
Mitochondria supply ATP essential for neuronal survival and regeneration. Brain injury and ischemia trigger acute mitochondrial damage and a local energy crisis leading to degeneration. Boosting local ATP supply in injured axons is thus critical to meet increased energy demand during nerve repair and regeneration in adult brains, where mitochondria remain largely stationary. Here, we elucidate an intrinsic energetic repair signaling axis that boosts axonal energy supply by reprogramming mitochondrial trafficking and anchoring in response to acute injury-ischemic stress in mature neurons and adult brains. PAK5 is a brain mitochondrial kinase with declined expression in mature neurons. PAK5 synthesis and signaling is spatiotemporally activated within axons in response to ischemic stress and axonal injury. PAK5 signaling remobilizes and replaces damaged mitochondria via the phosphorylation switch that turns off the axonal mitochondrial anchor syntaphilin. Injury-ischemic insults trigger AKT growth signaling that activates PAK5 and boosts local energy supply, thus protecting axon survival and facilitating regeneration in in vitro and in vivo models. Our study reveals an axonal mitochondrial signaling axis that responds to injury and ischemia by remobilizing damaged mitochondria for replacement, thereby maintaining local energy supply to support CNS survival and regeneration.
Keywords: AKT, degeneration, regeneration, energy crisis, axonal injury, ischemia, mitochondrial anchoring, mitochondrial remobilization, neuron maturation, PAK5, syntaphilin
Graphical Abstract
eTOC Blurb
By applying injury and ischemic models combined with genetic reprogramming, Huang et al. elucidate an energetic AKT-PAK5 signaling axis that boosts axonal energy supply in response to injury and ischemia by removing damaged mitochondria and replenishing with healthy ones, thus protecting against energy crisis for neuron survival and regeneration.
Introduction
Mitochondria are the main cellular powerhouses that produce most of adenosine triphosphate (ATP) in the brain, where a constant ATP supply is essential for neuron growth, signaling processing, and regeneration.1–4 Given unique morphological features, neurons face exceptional challenges to maintain energy supply in axons and growth cones. In developing neurons, ~30-50% of axonal mitochondria undergo bi-directional transport driven by the anterograde motor kinesin and retrograde motor dynein.5–8 However, axonal mitochondrial motility declines with neuron maturation9 and the majority of axonal mitochondria remain stationary in adult brains.10–13 Although anchored mitochondria ideally serve as local energy sources, they need to be removed when damaged.9,14–16 Damaged mitochondria not only supply ATP less efficiently, but also release reactive oxygen species and apoptotic factors triggering degeneration.1,17 This is particularly problematic in brain injury and ischemia, where mitochondria are damaged leading to energy crisis. Axonal regeneration requires high levels of energy consumption. Remobilizing and replacing damaged mitochondria with healthy ones will accelerate energetic recovery, and thus meet increased energy demand for nerve repair. These raise a fundamental question of whether anchored axonal mitochondria can be remobilized and replaced when they are damaged during brain injury and ischemia.
Syntaphilin (SNPH) acts as an anchor that holds axonal mitochondria stationary on microtubules (MTs).18,19 SNPH expression is robustly elevated in adult mouse and rat brains,9,20 thus maintaining the majority of axonal mitochondria stationary in disease and after ischemia.14,21 Deleting the snph gene in mice enhances axonal mitochondria motility and thus promotes axon regeneration after injury.9,22 However, genetic deletion is not physiologically relevant to brain injury and ischemia. Local mitochondrial signaling is an effective mechanism that maintains energy homeostasis in distal axons23 and supports regeneration after peripheral nerve injury.24,25 However, it remains obscure whether mature neurons and adult central nervous system (CNS) harbor an axon mitochondrial signaling that responds to local energy crisis following brain injury and ischemia.
Here, we reveal an intrinsic energetic repair program that boosts axonal energy supply by reprogramming mitochondrial motility in response to acute injury-ischemia. PAK5 is a brain kinase of the PAK family.26 By super-resolution imaging, we demonstrate that PAK5 localizes to axonal mitochondrial surfaces in developing neurons and its expression and signaling are declined with neuron maturation and undetectable in adult and aging neurons. However, axonal injury-ischemia spatiotemporally activate local PAK5 synthesis and its signaling. PAK5 mediates a phosphorylation switch that turns off SNPH-anchoring, thus remobilizing SNPH-anchored damaged mitochondria for replacement with healthy ones, and protecting against injury-induced energy crisis. AKT growth signaling further activates PAK5, accelerating the replacement of damaged mitochondria. Reprogramming the AKT-PAK5 axis protects mature neuron survival from ischemia and promotes CNS regeneration. While cell-to-cell mitochondrial transfer has surfaced as a possible avenue for therapeutic development in brain injury and stroke,27,28 our study demonstrates that reprogramming an enhanced ATP-PAK-SNPH axis can robustly reverse energy crises, protect neuron survival, and facilitate CNS regeneration, thus providing a potentially therapeutic strategy for brain injury and ischemia.
Results
Axonal PAK5 synthesis and signaling is activated in response to ischemic stress
Brain injury-ischemia trigger acute mitochondrial damage leading to local energy crisis.1,9,17,21,22 Recovering ATP supply is thus critical to meet increased energy demand to support CNS survival and repair. PAK5 is a mitochondria-targeted P21-activated serine/threonine kinase of the PAK family and expressed in developing neurons.29–31 Deep-sequencing analysis demonstrated that PAK5 mRNA is detected in axons of retinal ganglion cells and declines during postnatal development.32 We asked whether PAK5 signaling within axons is activated in response to ischemia. We visualized newly synthesized PAK5 within axons in situ upon ischemic stress by combining microfluidic devices with the Puro-PLA system33 (Figures 1A and 1B). To model ischemia, we treated mature cortical neurons at 14 days in vitro (DIV14) in oxygen-glucose-deprivation (OGD) media for 30 minutes, followed by reperfusion (R) for 0, 2, 4, 6, and 8 hours. Axonal PAK5 Puro-PLA signals were significantly increased at 4 hours post-OGD-R (P<0.001; Figures 1C and 1D), and then declined at 6 hours. To validate in situ PAK5 synthesis, we preincubated neurons with 40 μM anisomycin to inhibit protein synthesis. PAK5 Puro-PLA signals in soma-dendritic and axonal chambers were abolished (P<0.001) by anisomycin (Figures S1A–S1D). PAK5 is activated by its auto-phosphorylation at Ser-602 (p-PAK5).34 We next confirmed axonal activation of PAK5 by collecting lysates from axon chambers. Both PAK5 (P<0.05) and p-PAK5 (P<0.01) displayed a transient increase at 4 hours post-OGD-R, returned to pre-OGD-R levels after 8 hours (Figures 1E and 1F), suggesting that axonal PAK5 synthesis and signaling is transiently activated in response to ischemia.
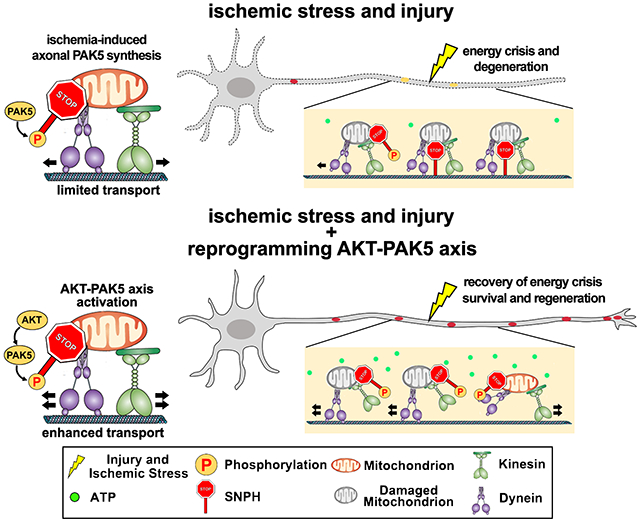
Figure 1. Axonal PAK5 signaling protects against ischemia-induced energy crisis.
(A and B) Schematic of a microfluidic device (A) showing physical and fluidic separation of axons from somas/dendrites, and Puro-PLA assay (B) monitoring newly synthesized PAK5 in axons. Puromycin-labeled PAK5 was detected by antibodies against puromycin (red Y) and PAK5 (black Y). PLA signals (red dots) were detected when PLAplus and PLAminus oligonucleotide-coupled secondary antibodies (blue Y and green Y) are close enough to be ligated and amplified.
(C and D) Puro-PLA analysis showing elevated PAK5 translation within axons of cortical neurons at DIV14 upon OGD-R. Axonal PAK5-Puro-PLA signals were normalized to Tau signal area.
(E and F) Transient elevation of PAK5 and p-PAK5 within axonal compartments upon 4-hour OGD-R. 5-μg axonal lysates were immunoblotted with the indicated antibodies. The intensity of PAK5 and p-PAK5 was quantified (n=3) and normalized to 0-hour of OGD-R.
(G and H) Depletion of PAK5 and p-PAK5 by PAK5-shRNA. Lysates were harvested from axon chambers at DIV14, and 5-μg lysates were immunoblotted with the indicated antibodies. The intensity of PAK5 and p-PAK5 was quantified (n=3) and calibrated with GAPDH levels, and normalized to PAK5 or p-PAK5 in Ctrl-shRNA.
(I-K) Axon-restricted inhibition of PAK5 activity. Neurons at DIV14 were treated with DMSO or PF-3758309 (1 μM) in axon chambers for 3 hours, followed by lysate collection from axonal chambers. The intensity of PAK5 and p-PAK5 was quantified (n=3), calibrated with βIII-tubulin levels, and normalized to PAK5 or p-PAK5 in the DMSO group.
(L and M) Depleting PAK5 with shRNA (L) or inhibiting axonal PAK5 activity by loading PF-3758309 (M) induced a more robust axonal energy crisis following ischemia. The ratio of F560nm (ATP-bound) to F510nm (ATP-free) reflects relative ATP availability.
(N) Depleting PAK5 accelerates axon degeneration following ischemic stress. The percentage of axons displaying fragmentation or bead-like degeneration was quantified.
(O-Q) Enhanced PAK5 activation rescues axonal ATP depletion induced by ischemia (O, P) or Rotenone (Q).
Data were quantified from n=30-31 (D) or n=30 (L-N, P, Q) of axonal images from >3 devices per condition in three experiments, expressed as mean±SEM, and analyzed by Student’s t test (H, K-N) or one-way ANONA with Tukey’s multiple comparisons test (D, F, P, Q). ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars: 25 μm. (See also Figure S1)
We examined whether PAK5 signaling contribute to axonal energetic maintenance after ischemia using two approaches. First, expressing a PAK5-shRNA depleted PAK5 and p-PAK5 expression detected in axonal lysates (Figures 1G and 1H), while loading PF-3758309, a PAK inhibitor, into axon chambers selectively abolished axonal PAK5 signaling (Figures 1I–1K). In contract, axon-restricted loading of the PAK5 inhibitor failed to reduce somatodendritic p-PAK5 (Figure S1E). Second, we applied the FRET-based and red-shifted ATP probe GO-ATeam235 to monitor ATP levels within axons before and post-OGD-R. In mature neurons (DIV14), axonal ATP levels dramatically declined after OGD-R. However, depleting PAK5 or inhibiting axonal PAK5 signaling triggered an even more robust energy crisis (P<0.01; Figures 1L and 1M) and accelerated axon degeneration (Figure 1N). These results suggest that axonal PAK5 signaling can protect energy homeostasis and delay ischemia-induced axon degeneration.
To boost axonal ATP supply, we reprogrammed PAK5 signaling by expressing PAK5-S573N, a constitutively active kinase mutant (caPAK5). Expressing caPAK5 activated PAK5 signaling within axon compartments in mature neurons (Figure S1F) and reduced axonal energy deficits 10 hours post-OGD-R (P < 0.001, Figures 1O and 1P). In contrast, expressing the kinase-dead mutant PAK5-K478M (kdPAK5) failed to activate PAK5 (p-PAK5) and rescue energy crisis. As a negative control, we applied an ATP-insensitive GO-ATeam3 probe (R122K/R126K).35 Expressing caPAK5 did not affect the ratiometric integrated intensity of GO-ATeam3 post-OGD-R (Figures S1G and S1H). Loading axonal chambers with the mitochondrial complex I inhibitor rotenone (5 μM) progressively reduced axonal ATP levels over time (Figure 1Q). However, expressing caPAK5 suppressed ATP decline in axons (P<0.01). These results support our first conclusion: ischemia transiently activates axonal PAK5 signaling that protects against local energy crisis after ischemia and mitochondrial damage.
PAK5 signaling protects against injury-induced energy crisis
To determine whether local PAK5 synthesis is an early axonal energy surveillance in response to injury, we performed axotomy within axonal chambers as the second injury model (Figure 2A). Puro-PLA analysis showed enhanced local in situ PAK5 synthesis along axon bundles 12 hours post-injury, relative to the uninjured group (P<0.001). However, this local PAK5 synthesis declined 36 hours post-injury (P<0.001; Figures 2B and 2C). Lysates isolated from axon bundles were collected before and at various time points post-injury. Consistently, PAK5 and p-PAK5 in axon bundles elevated gradually and peaked at 24 hours post-injury (PAK5: P<0.001; p-PAK5: P<0.001; Figures 2D and 2E), then declined, suggesting that enhanced local PAK5 synthesis and signaling is a transient response to acute injury. Depleting PAK5 expression or inhibiting axonal PAK5 activity abolished the recovery of ATP levels at axon tips 24 hours post-axotomy (Figures 2F and 2G). Furthermore, mature neurons at DIV14 displayed reduced ATP levels along axon bundles 3 hours post-axotomy (Figures 2H and 2I), indicating an injury-induced acute energy crisis. ATP levels progressively increased from 3 to 48 hours post-injury, but did not recover to the pre-injury level after 48 hours (P<0.001). However, reprogramming PAK5 signaling by expressing caPAK5, but not kdPAK5, ameliorated axonal energy crisis (P<0.001) in all three time points and accelerated ATP recovery at axonal tips by 12 hours (Figures 2J and 2K). As a control, applying ATP-insensitive GO-ATeam3 did not produce the similar changes in the ratiometric integrated intensity (Figures S1I and S1J). We further investigated an in vivo response by applying a spinal cord injury (SCI) model in 2-month-old adult mice. Following a thoracic 8 (T8) hemisection, spinal cord tissue anterior to the lesion site (5-mm long within T5-T7 segment) was isolated on day 3, 7, or 14 post-SCI for immunoblot analysis. Total PAK5 (P=0.0012) and p-PAK5 (P<0.001) were increased 14 days post-SCI compared with non-surgery (sham) mice (Figures 2L and 2M). These data support our second conclusion: axonal injury activates PAK5 signaling that rescue injury-induced energy crisis.
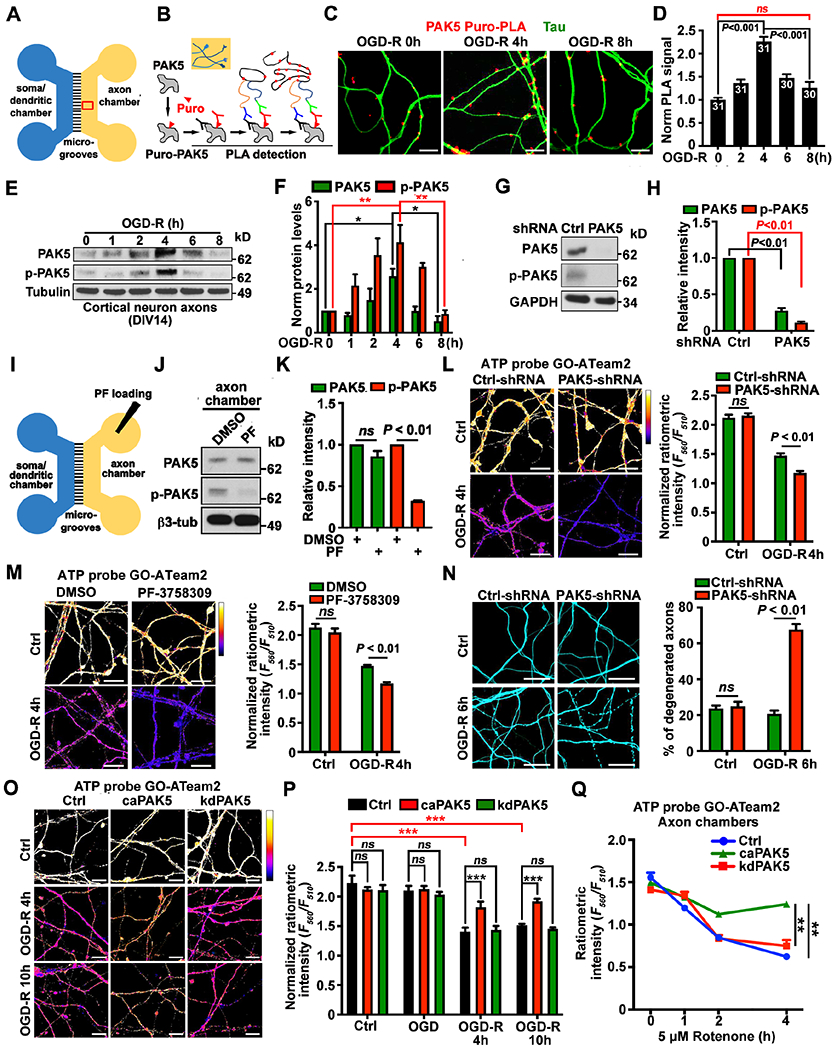
Figure 2. Injury-induced PAK5 signaling protects against axonal energy crisis.
(A) Schematic diagram of a microfluidic device showing axonal bundles within microgrooves (A1), axotomy by vacuum aspiration, and axonal regrowth in the axon chamber (A2).
(B and C) Puro-PLA analysis showing enhanced in situ PAK5 synthesis within axon bundles upon axonal injury. PAK5-Puro-PLA signals (arrows) were normalized to Tau-labeled axonal areas.
(D and E) Axonal injury transiently elevates PAK5 and p-PAK5 within axon compartments. 5-μg axoplasmic lysates were immunoblotted and the intensity of PAK5 and p-PAK5 was quantified (n=3) and normalized to protein levels before injury.
(F and G) Deleting PAK5 with PAK5-shRNA (F) or inhibiting local PAK5 activity by loading PF-3758309 (1 μM) (G) triggers energy crisis at axon tips 24 hours post-axotomy. The F560nm/F510nm ratio reflects relative ATP availability.
(H-K) Activating PAK5 rescues injury-induced axonal energy crisis. Neurons were co-infected with lentiviruses encoding GO-ATeam2 and HA-tagged control, caPAK5, or kdPAK5, followed by imaging at DIV14 of axon bundles (H, I) or tips (J, K) before or at various time points post-injury.
(L and M) In vivo PAK5 activation in spinal cord axons after SCI in adult female mice (2-month-old). 15-μg homogenates were immunoblotted. The intensity of PAK5 and p-PAK5 was quantified (n=3) and normalized to the sham mice.
Data were quantified from n=30-47 axon images (C), or n=30 images (F, G, I, K) from >3 devices per condition in three experiments, and analyzed by Student’s t test (F, G), one-way ANONA with Tukey’s multiple comparisons test (C, E, I, K) or Dunnett’s multiple comparisons test (M). ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001. Scale bars: 10 μm (B, F, G, J) and 25 μm (H). (See also Figure S1)
PAK5 localizes on axonal mitochondria surfaces and declines with neuron maturation
We next characterized PAK5 expression and subcellular localization during neuronal maturation. Confocal microscopy images showed that endogenous PAK5 co-localized with cytochrome c, an inner mitochondrial membrane marker, along axons (Figure 3A). To confirm the distribution under super-resolution levels, we applied diffraction-unlimited stimulated emission and depletion (STED) microscopy, which can achieve a resolution of ~50-90 nm. Cortical neurons at DIV7 were co-transfected with GFP-PAK5 and mitochondrial inner membrane marker DsRed-Mito, followed by STED imaging at DIV10. GFP-PAK5 engulfed DsRed-Mito signals, indicating its localization on mitochondrial surfaces (Figures 3B and 3C). Analyses of Mander’s colocalization coefficient revealed that endogenous PAK5 is more closely colocalized with the mitochondrial outer membrane marker TOM20 than the inner membrane marker Cyto C (P<0.01, Figures 3D and 3E). By measuring the mitochondria/cytosol ratio of PAK5 signal along axons, we found that PAK5 distribution on axonal mitochondria did not significantly changed at 24 hours post-axotomy or 4 hours post-OGD-R (Figures S2A–S2D). Both PAK5 and activated p-PAK5 peaked at early developmental stages between DIV3-7 and rapidly declined when neurons mature after DIV14, a pattern opposite to enhanced SNPH expression during maturation (Figures S2E and S2F).
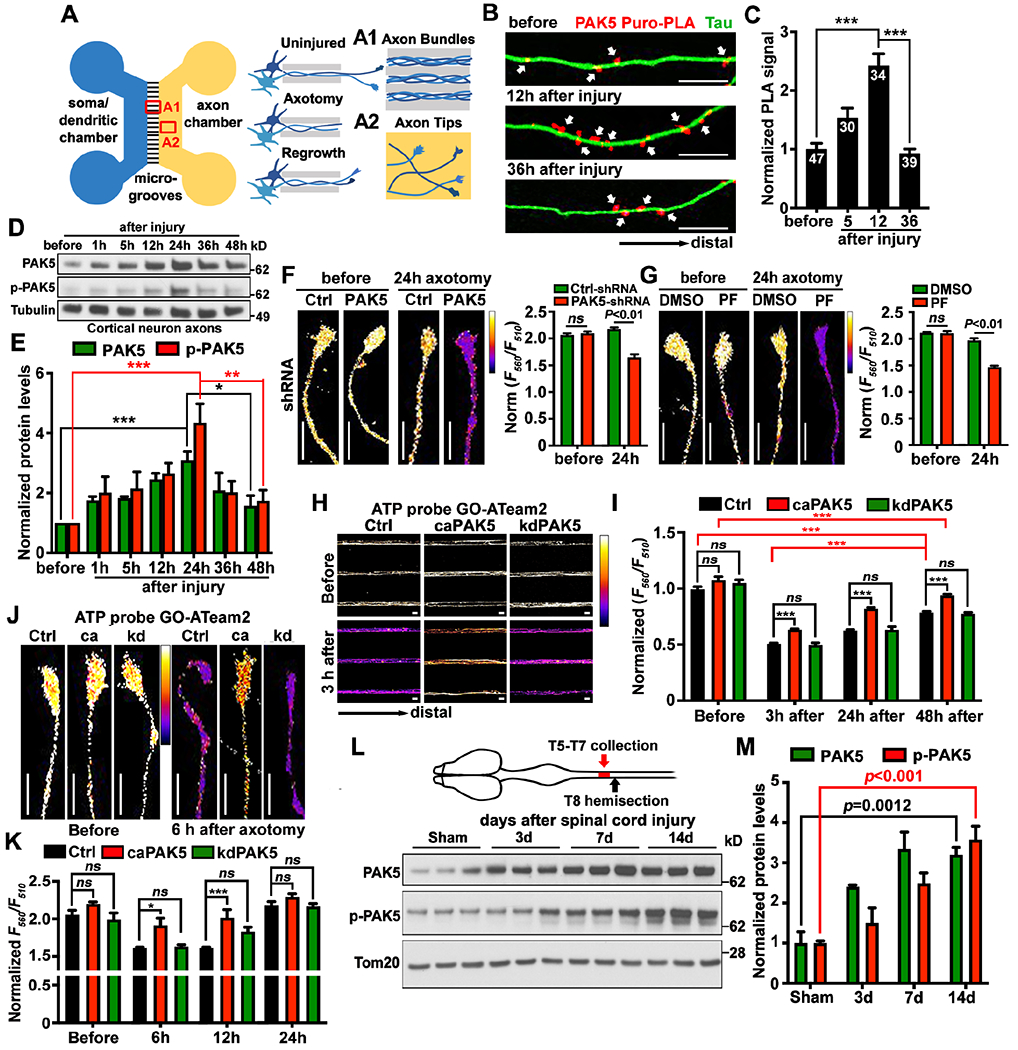
Figure 3. Activating PAK5 signaling remobilizes axonal mitochondria in mature neurons.
(A-E) Confocal (A) and STED (B, D) images, line-scan (C) and quantitative analysis (E) showing PAK5 distribution on the surface of axonal mitochondria. Relative colocalization of PAK5 with Cyto c or TOM20 was analyzed by Mander’s colocalization coefficient using the JACoP plugin in ImageJ (E). Data were quantified from n=15 STED images.
(F and G) Kymographs showing aging-associated decline of axonal mitochondrial transport in DRG neurons (F) and reversed aging-associated decline by activating PAK5 in aging neurons (G). DRG neurons isolated from aging mice (P12M) were nucleofected with DsRed-mito and HA-tag control, HA-tagged caPAK5, or kdPAK5 at DIV0, followed by time-lapse imaging at DIV4 for 90 frames with 5-sec intervals.
(H and I) Representative screenshots (upper), traced images (lower), and analyses of ex vivo mitochondrial motility along the sciatic nerve in young (P30) and adult (P70) Thy1-Mito-CFP mice (H) or mice at P45 were injected with AAV9-2A-GFP or AAV9-caPAK5-2A-GFP (I), followed by imaging at P59. Mitochondrial transport was assessed in sciatic nerve explants after acute dissection from Mito-CFP mice and time-lapse imaging (60 frames with 3-sec intervals). White dashed lines indicate the edge of sciatic nerves. (also see Videos S1–S4).
Data were quantified from n=30-31 neurons (F, G) or 30-60 ex vivo images from three pairs of mice (H, I), expressed as mean±SEM, and analyzed by Mann-Whitney test (E), Student’s t test (F, H, I), or one-way ANONA with Dunnett’s multiple comparisons test (G). Scale bars: 10 μm (A, F, G), 500 nm (B), 1 μm (D), and 5 μm (H, I). (See also Figure S2)
PAK5 signaling remobilizes axonal mitochondria in mature neurons
Mitochondrial transport in axons progressively declines with neuron maturation.9 Axonal injury and ischemia further suppress mitochondrial motility,9,21 therefore the majority of damaged mitochondria are held stationary in mature axons within the CNS. We hypothesized that PAK5 signaling remobilizes damaged mitochondria for replacement, thus maintaining local bioenergetics within injured axons. To test this, we performed the following in vitro and in vivo experiments.
First, we examined axonal mitochondrial motility in developing and mature cortical neurons. In young neurons at DIV7, axonal mitochondrial motility was 37.94±2.14%, almost double that was observed in mature neurons at DIV23 (14.33±1.86%, P<0.001; Figure S2G). Consistently, in dorsal root ganglion (DRG) neurons isolated from young adult mice at postnatal day 30 (P30), axonal mitochondria were significantly more mobile compared to DRG neurons isolated from aging mice at 12-month-old (P12M, P<0.001; Figure 3F). These live imaging analyses support that axonal mitochondrial transport significantly declines in mature and aging neurons.
Second, we examined whether activating PAK5 in mature cortical neurons is required for remobilizing axonal mitochondria. While expressing caPAK5 failed to enhance mitochondrial transport in developing neurons (DIV7) (Figure S2H) where SNPH expression was low (Figure S2E), expressing caPAK5 (P<0.001), but not kdPAK5 (P=0.99), in mature neurons (DIV21) significantly enhanced axonal mitochondrial motility compared to age-matched control neurons (Figure S2I). Similarly, expressing caPAK5 (P<0.001), but not kdPAK5 (P=0.84), increased axonal mitochondrial motility in DRG neurons isolated from aging mice (P12M) relative to age-matched control (Figure 3G). Expressing caPAK5 or kdPAK5 in mature neurons (DIV21) did not change the motility of LAMP1-labeled endolysosomes in axons (Figure S2J), suggesting that PAK5 signaling selectively remobilizes axonal mitochondria.
Third, we further assessed ex vivo mitochondrial transport by imaging sciatic nerve explants from Thy1-Mito-CFP (Mito-CFP) mice, where neuronal mitochondria are labeled with CFP.36 Adult Mito-CFP mice (P70) displayed reduced mitochondrial motility along axon bundles of sciatic nerves (26.84±1.66%) relative to that in young Mito-CFP mice at P30 (55.32±3.43%, P<0.001; Figure 3H, Videos S1 and S2). This model provided an in vivo system for reprogramming PAK5 signaling by injecting AAV9-caPAK5-2A-GFP into mouse sciatic nerves at P45, followed by ex vivo imaging at P59. Expression of AAV9-caPAK5-2A-GFP significantly increased axonal mitochondrial motility (P=0.002; Figure 3I, Videos S3 and S4). To exclude the possibility that PAK5 signaling alters mitochondrial bioenergetics, Seahorse extracellular flux analysis was performed to examine oxygen consumption rate (OCR). No significant change in basal or real-time OCR was observed after caPAK5 or kdPAK5 expression (Figure S2K). Thus, these data support our third conclusion: PAK5 signaling remobilizes axonal mitochondria in mature neurons and adult nervous system.
SNPH is the phosphorylation substrate of PAK5 signaling
Based on the differential expression of PAK5 and SNPH and their opposing roles in axonal mitochondrial motility, we asked whether SNPH is the phosphorylation substrate of PAK5 signaling. We addressed this by performing the following five experiments. First, we co-expressed SNPH and caPAK5 or kdPAK5 in HEK293T cells, followed by analyzing SNPH phosphorylation. A shifted SNPH band was readily detected when it was co-expressed with caPAK5, but not kdPAK5 or caPAK1 (Figure 4A). This shifted SNPH band diminished after λ-phosphatase treatment. Second, by mass spectrometry analysis, a cluster of PAK5 phosphorylation sites (Ser-56, Ser-59, Thr-63, and Ser-64) were mapped at the N-terminus of SNPH (Figure 4B). Third, we constructed a phospho-dead SNPH mutant (SNPH-4A) by replacing these four serine/threonine residues with alanine. Co-expressing caPAK5 induced phosphorylation-shifted SNPH in Phos-tag gels, but not for the SNPH-4A mutant (Figure S3A), confirming the mass spectrometry findings. Fourth, we assessed these phosphorylation events in neurons by generating a phospho-SNPH antibody against Thr-63 and Ser-64 (p-SNPH). Pre-incubation of the anti-p-SNPH antibody with a phospho-peptide effectively blocked p-SNPH signal (Figure S3B). The p-SNPH was readily detected when caPAK5 was co-expressed with SNPH, but not with SNPH-4A (Figure 4C). This phosphorylation status was abolished following λ-phosphatase treatment. Fifth, SNPH phosphorylation in situ on axonal mitochondria was significantly reduced during neuronal maturation from DIV10 to DIV21 (P<0.001; Figures 4D and 4E). Both caPAK5 and kdPAK5 targeted axonal mitochondria and did not affect SNPH distribution (Figures S3C–S3F).
Figure 4. PAK5 signaling remobilizes axonal mitochondrial by phosphorylation of SNPH.
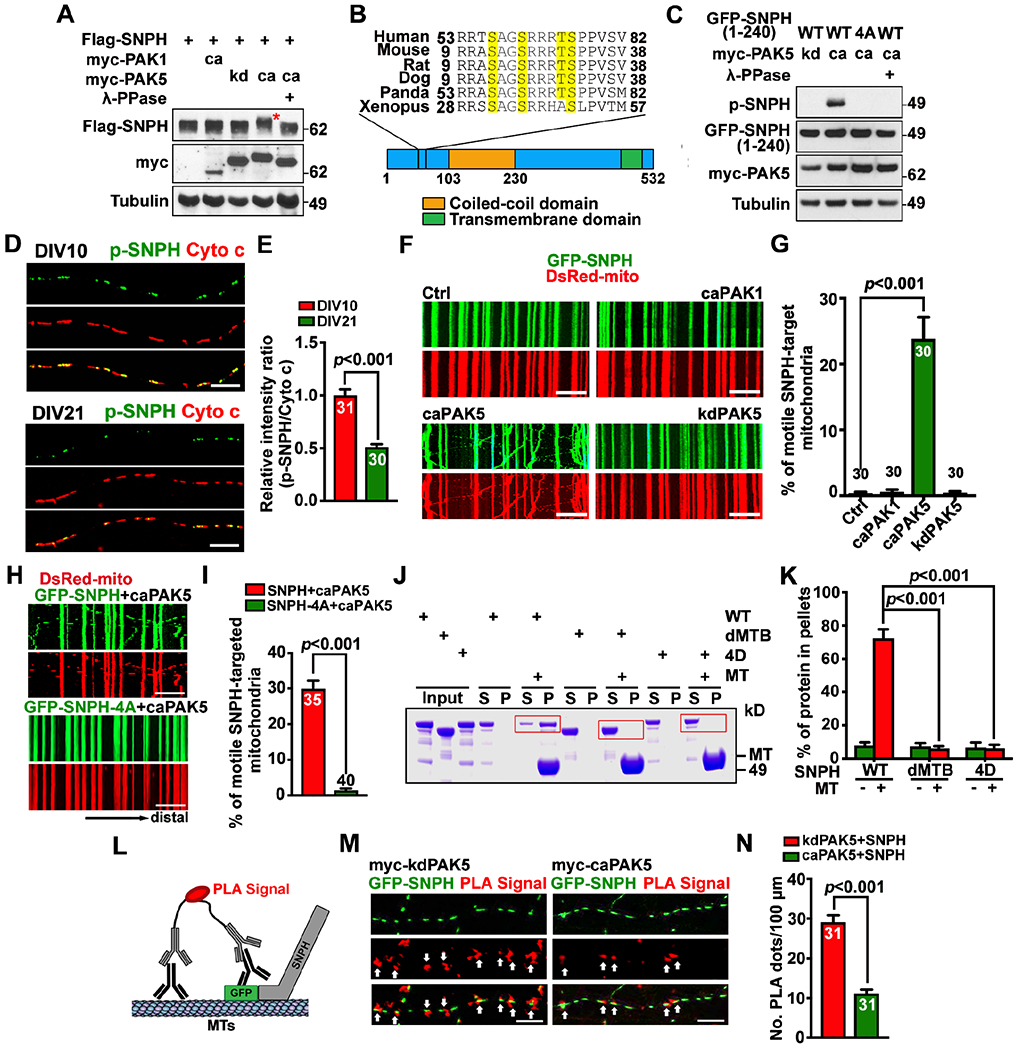
(A and B) PAK5 mediates phosphorylation of SNPH. SNPH band-shift (red asterisk) is indicative of clustered phosphorylation (A). Evolutionarily conserved PAK5 phosphorylation sites (yellow) are clustered at the N-terminus of SNPH and confirmed by mass spectrometry (B).
(C) An anti-p-SNPH antibody detects caPAK5-induced phosphorylation of SNPH.
(D and E) Declined p-SNPH levels on axonal mitochondria with neuron maturation from DIV10 to DIV21. The mean integrated intensity ratio of p-SNPH/Cyto c on individual mitochondria was quantified and normalized to DIV10.
(F-I) Activating PAK5 remobilizes SNPH-anchored axonal mitochondria (F, G) but fails to remobilize mitochondria anchored by phosphorylation-dead SNPH-4A (H, I) in mature cortical neurons. Time-lapse imaging was recorded for 90 frames with 5-sec intervals at DIV10.
(J and K) MT spin-down assays (n=3). After centrifugation, the supernatant (S) and pellets (P) were analyzed by SDS-PAGE and Coomassie Blue staining. Red boxes indicate SNPH.
(L-N) The proximity ligation assay (PLA) shows disrupted SNPH-MT association by PAK5 activation. Arrows mark red fluorescent signals representing in situ SNPH-MT association along axons. The number of PLA signals per 100-μm axon length was quantified.
Data were quantified from n=30-40 neurons (E, G, I, N) per condition as indicated within bars from three experiments, expressed as mean±SEM, and analyzed by Student’s t test (E, I, N) or one-way ANONA with Dunnett’s multiple comparisons test (G, K). Scale bars, 10 μm. (See also Figures S3, S4)
PAK5 remobilizes axonal mitochondria by turning off SNPH anchoring
We next tested our hypothesis that mitochondrial immobilization vs remobilization in mature neuron axons is coordinated by SNPH-mediated anchoring and PAK5-mediated de-anchoring. First, we determined whether activating PAK5 can remobilize SNPH-anchored mitochondria. While SNPH overexpression arrests almost all axonal mitochondria, co-expressing caPAK5 with SNPH remobilized SNPH-anchored mitochondria, thus enhancing mitochondrial motility from 0.23±0.23% to 23.83±3.29% (P<0.001; Figures 4F and 4G). In contrast, co-expression of SNPH with kdPAK5 or caPAK1 failed to remobilize axonal mitochondria. Second, we depleted PAK5 in wild-type (WT) and snph knockout (KO) cortical neurons (Figures S3H and S3I). Axonal mitochondrial motility in PAK5-depleted neurons was significantly decreased (P<0.01; Figures S3J and S3K). However, PAK5 depletion in snph KO neurons displayed no detectable effect on mitochondrial motility, supporting the notion that PAK5 signaling remobilizes axonal mitochondria by turning off the SNPH anchoring mechanism.
To understand the role of SNPH phosphorylation in mitochondrial remobilization, we constructed a phospho-mimicking SNPH (SNPH-4D) by replacing four serine/threonine residues with aspartic acids. Neurons at DIV7 were co-transfected with DsRed-Mito and GFP-tagged SNPH, SNPH-4A, SNPH-4D, or an anchoring loss-of-function mutant SNPH-dMTB, in which MT-binding (MTB) domain was deleted.18,19 Although all three SNPH mutants and WT SNPH similarly distributed on axonal mitochondria (Figures S4A and S4B), their capacity in anchoring mitochondria was remarkably different from each other. While WT SNPH immobilized all axonal mitochondria, SNPH-4D failed to immobilize axonal mitochondria (31.04±1.86%, P=0.32) as compared to SNPH-dMTB (27.51±2.24%; Figures S4C and S4D). Activating PAK5 remobilized SNPH-anchored mitochondria but failed to remobilize SNPH-4A-anchored mitochondria (SNPH+caPAK5: 29.89±2.29%; SNPH-4A+caPAK5: 1.45±0.52%; P<0.001; Figures 4H and 4I). Thus, SNPH-anchored mitochondria can be remobilized upon its phosphorylation at Ser-56, Ser-59, Thr-63, and Ser-64.
SNPH acts as a static anchor that holds axonal mitochondria on MTs through its MTB domain at its N-terminus.18,19 Since these four phosphorylation sites are clustered in the region adjacent to its MTB domain, we asked whether these phosphorylation events disturb SNPH-MT anchoring interaction. To address this, we examined the MT-binding capacity of SNPH both in vitro and in situ within axons. First, we applied a truncated SNPH (1-469) by deleting its C-terminus transmembrane tail that targets the outer mitochondrial membrane (OMM). The majority of SNPH settled down with MTs in the pellet after centrifugation, while SNPH-4D and SNPH-dMTB remained in the supernatant (Figures 4J and 4K). Thus, phospho-mimicking SNPH-4D lost its MT-binding capacity. To further confirm this, we characterized in situ SNPH-MT association in live axons using a proximity ligation assay (PLA) (Figure 4L). SNPH-dMTB was used as an anchoring loss-of-function control. Expressing SNPH-4D, but not SNPH-4A, displayed reduced in situ MT-association along axons (Figures S4E and S4F). PLA signals were significantly decreased in neurons co-expressing SNPH and caPAK5 (P<0.001) compared to kdPAK5 (Figures 4M and 4N). Together, these results support our fourth conclusion: PAK5-mediated phosphorylation of SNPH at the four clustered sites adjacent to its MTB domain turns off its anchoring capacity, thus remobilizing anchored mitochondria in mature neurons where SNPH is highly enriched.
AKT signaling enhances mitochondrial transport by activating PAK5
AKT signaling is involved in neurite outgrowth, axon regeneration, and survival, and may also activates PAK5.37–41 AKT signaling regulates mitochondrial transport, although the underlying mechanism remains unknown.42,43 We proposed that AKT growth signaling acts as an upstream regulator of the PAK5-SNPH axis. To test this, we examined axonal AKT signaling after injury-ischemia. Activated AKT (p-AKT), marked by phosphorylation on Ser-473,44 but not total AKT levels, displayed significant increases within axonal compartments 4 hours post-OGD-R (Figures 5A and 5B), which occurred simultaneously with axonal PAK5 signaling (Figures 1E and 1F). In addition, an increase of p-AKT was readily detectable within axon bundles 12 hours post-axotomy (Figures 5C and 5D), consistent with the time point that axonal PAK5 signaling was activated post-injury (Figures 2D and 2E).
Figure 5. AKT signaling activates PAK5.
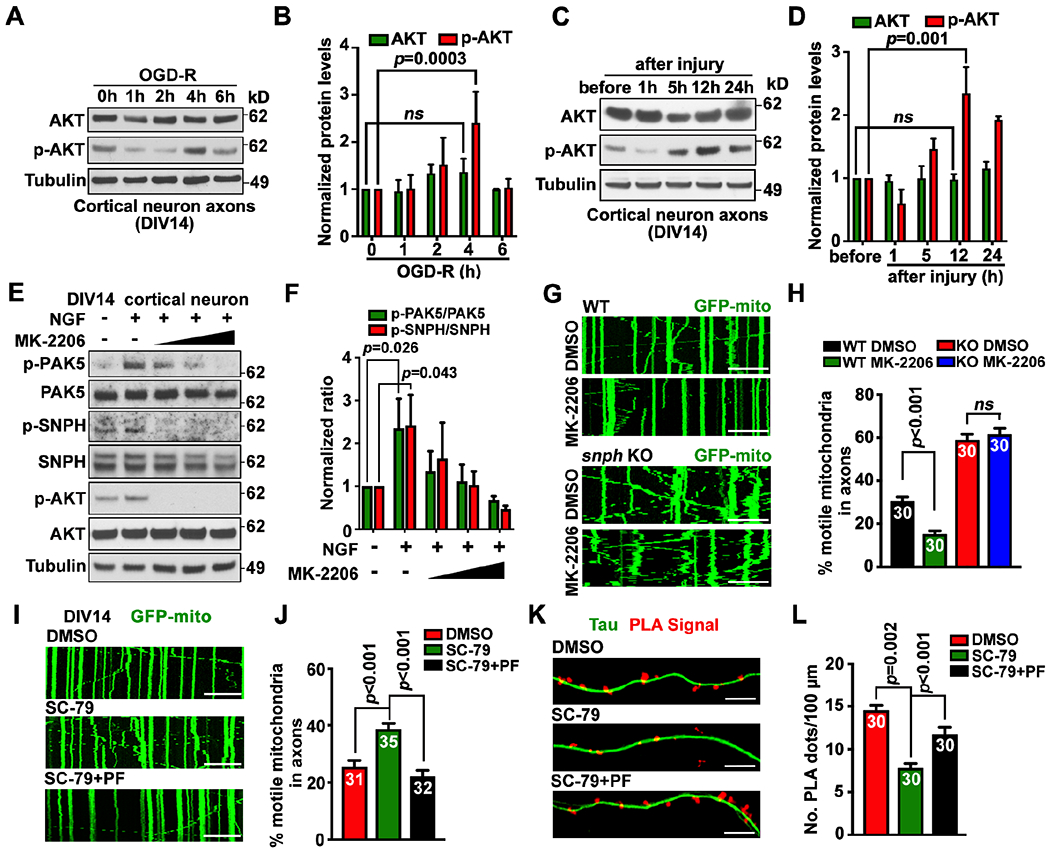
(A-D) Axonal AKT activation upon OGD-R (A, B) or axotomy (C, D) in microfluidic devices. Axoplasmic lysates were collected from axon chambers for immunoblotting. The intensity of AKT and p-AKT was quantified (n=3) and normalized to the 0-hour of OGD-R or before injury.
(E and F) AKT activates PAK5-SNPH signaling. Neurons (DIV14) were treated with NGF (40 ng/ml) and/or an increasing dose of AKT inhibitor MK-2206 (2, 4, or 6 μM) for 1 hour. Cell lysates (8 μg) were immunoblotted. The intensity ratios of p-PAK5/PAK5 and p-SNPH/SNPH were quantified (n=3) and normalized to the ratios in the untreated group.
(G and H) AKT-dependent remobilization of axonal mitochondria. WT or snph KO neurons were transfected with GFP-Mito at DIV10, followed by treatment at DIV14 with DMSO or AKT inhibitor MK-2206 (2 μM for 1 hour) before time-lapse imaging (90 frames with 5-sec intervals).
(I and J) Inhibiting PAK5 abolishes AKT-enhanced axonal mitochondrial transport. Neurons were transfected with GFP-Mito at DIV10, and treated with DMSO, AKT activator SC-79 (2 μg/ml), and/or PAK inhibitor PF-3758309 (PF, 1 μM) for 1 hour at DIV14, followed by time-lapse imaging (90 frames with 5-sec intervals).
(K and L) Activating AKT reduces in situ SNPH-MT association through PAK5. Neurons at DIV10 were treated with AKT activator SC-79 and/or PAK inhibitor PF-3758309 before PLA.
Data were quantified from n=30-40 neurons (H, J, L) per condition as indicated within bars from three experiments, expressed as mean±SEM, and analyzed by Student’s t test (H) or one-way ANONA with Dunnett’s multiple comparisons test (B, D, F, J, L). Scale bars, 10 μm.
We treated cortical neurons at DIV14 with nerve growth factor (NGF, 40 ng/ml, 1 hour) to activate AKT signaling,45 followed by examining p-PAK5, p-SNPH, and p-AKT. After NGF stimulation, the p-PAK5/PAK5 ratio increased (P=0.0268; Figures 5E and 5F); such an increase was abolished by an increasing dose of AKT inhibitor MK-2206 for 1 hour. As the substrate of PAK5, the p-SNPH/SNPH ratio was also elevated upon NGF treatment (P=0.0438) but abolished when AKT signaling was inhibited (Figures 5E and 5F). We next tested whether AKT signaling remobilizes axonal mitochondria in mature neurons by activating the PAK5-SNPH axis. Treating WT neurons with AKT inhibitor MK-2206 reduced axonal mitochondrial motility (P<0.001). However, the same treatment in snph KO neurons failed to reduce mitochondrial transport (Figures 5G and 5H). While activating AKT signaling with AKT activator SC-79 (2 μg/ml, 1 hour) enhanced axonal mitochondrial transport, adding PAK inhibitor PF-3758309 (1 μM, 1 hour) abolished the AKT-induced remobilization of axonal mitochondria (Figures 5I and 5J), thus suggesting a mechanistic link for the AKT-PAK5 axis in remobilizing SNPH-anchored mitochondria. PLA analysis showed reduced in situ SNPH-MT association in axons following AKT activation (P=0.002); blocking PAK5 activity regained axonal PLA signals in AKT-activated neurons (P<0.001; Figures 5K and 5L). These data support our fifth conclusion: AKT signaling activates PAK5, thus remobilizing damaged mitochondria by turning off the SNPH-anchoring switch.
Reprogramming PAK5 signaling protects neuron survival from ischemia
To address whether PAK5 activation enhances mitochondrial transport after acute ischemia, we characterized axonal mitochondrial motility in cortical neurons at DIV14 before, 1, and 4 hours post-OGD-R following expression of caPAK5 or kdPAK5 (Figures 6A and 6B). In the control group, axonal mitochondrial motility declined 1-hour post-OGD-R (P<0.001), and partially recovered 4-hour post-OGD-R (P<0.05). However, reprogramming PAK5 signaling by expressing caPAK5 enhanced mitochondrial motility at all three time points. We next assessed SNPH phosphorylation status in response to ischemic stress. We used snph KO cortical neurons to exclude interference of endogenous SNPH with re-introduced SNPH phosphorylation mutants (Figure S5A). We transduced GO-ATeam2 along with SNPH or SNPH-4A, SNPH-4D, or SNPH-dMTB into snph KO neurons at DIV5, followed by OGD-R at DIV10. While all groups displayed no detectable changes in their axonal ATP levels before OGD-R, snph KO neurons showed better axonal energy maintenance compared to WT neurons (P<0.001) after 4-hour OGD-R (Figures 6C and 6D). When comparing snph KO control group, immobilizing axonal mitochondria by introducing SNPH (P<0.001) or SNPH-4A (P<0.001) reduced axonal energy maintenance, thus supporting the notion that turning off the SNPH-anchoring benefits the maintenance of axonal energy supply after ischemic stress.
Figure 6. Reprogramming AKT-PAK5 protects axon survival from ischemia.
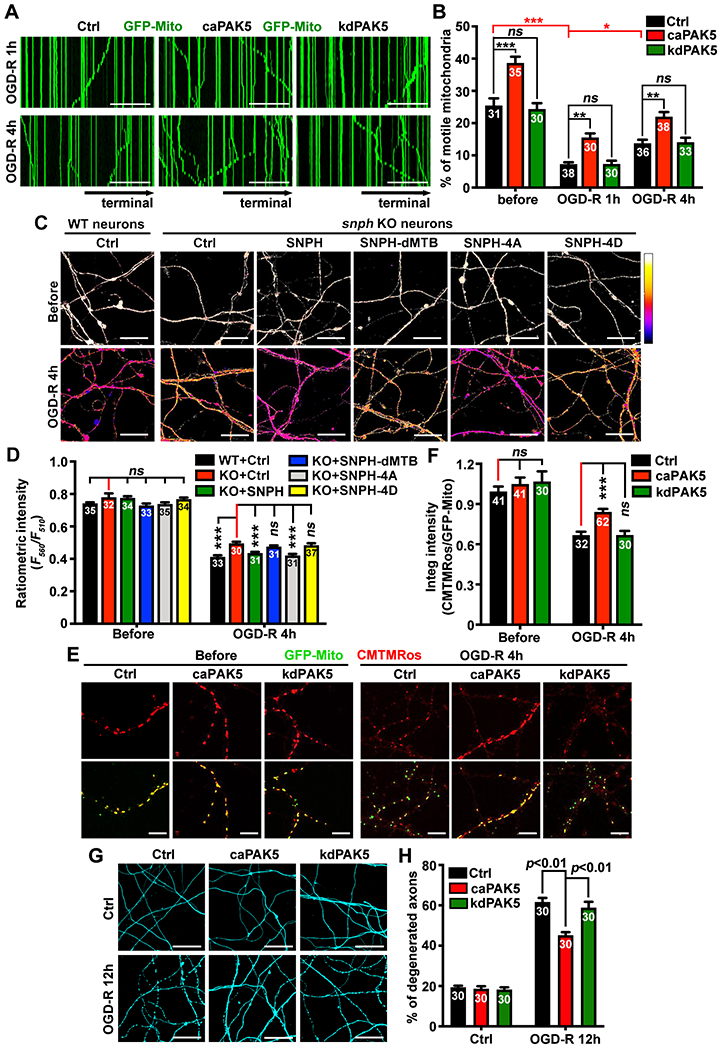
(A and B) Activating PAK5 enhances axonal mitochondrial motility following ischemia. Mitochondrial motility along axonal bundles was recorded before or post-OGD-R at DIV14 (30 frames with 5-sec intervals for a total of 2.5 min).
(C and D) Impairing SNPH-mediated anchoring elevates axonal ATP levels after ischemic stress.
(E and F) CMTMRos staining of axonal mitochondrial membrane potential (ΔΨm) before or 4 hours post-OGD-R. The integrated intensity ratio of CMTMRos/GFP-Mito was measured in individual axonal mitochondria.
(G and H) PAK5 activation reduces ischemia-induced axon degeneration. The percentage of axons with fragmentation or bead-like degeneration was quantified.
Data were quantified from the total number of microgrooves (B) or images (D, F, H) indicated within bars from three experiments, expressed as mean±SEM, and analyzed by one-way ANONA with Tukey’s multiple comparisons test (B) or Dunnett’s multiple comparisons test (D, F, H). ns, not significant; * P < 0.05, ** P < 0.01, *** P < 0.001. Scale bars: 50 μm (A), 25 μm (C, G), 10 μm (E). (See also Figure S5)
To examine whether PAK5-mediated mitochondrial remobilization helps replenish healthy ones, we characterized mitochondrial membrane potential (ΔΨm) in axons before and 4 hours post-OGD-R at DIV14. To ensure that ΔΨm from all neurons were examined at the same time points, we loaded the fixable ΔΨm dye CMTMRos (25 nM) in fixed neurons before and 4 hours post-OGD-R. The integrated intensity ratio of CMTMRos/GFP-Mito was measured in individual axonal mitochondria. Before OGD-R, expressing caPAK5 or kdPAK5 showed no significant change in ΔΨm (Figures 6E and 6F). However, 4-hour OGD-R decreased axonal mitochondrial ΔΨm; expressing caPAK5, but not kdPAK5, partially protected mitochondrial ΔΨm (P<0.001), which correlated with enhanced mitochondrial transport by expressing caPAK5 (Figures 6A and 6B).
We further examined whether reprogramming PAK5 protects axon survival after ischemia. Expressing caPAK5 significantly reduced axon degeneration (P<0.01) as evidenced by the reduced bead-like or fragmented phenotypes compared to OGD-R-treated control neurons or neurons expressing kdPAK5 (Figures 6G and 6H). In addition, reprogramming PAK5 signaling protected axons from degeneration following acute mitochondrial damage by rotenone (Figures S5B and S5C), but failed to protect axons when their MTs were disrupted with vincristine.46,47 These data support our sixth conclusion: reprogramming PAK5 signaling protects axons from ischemia-induced degeneration.
Reprogramming AKT-PAK5 signaling promotes CNS regeneration
CNS neurons typically fail to regenerate after injury, leading to permanent impairments. Since AKT-PAK5 signaling responds to injury, we hypothesized that this transient mechanism is compromised under persistent energy crisis. Thus, reprogramming enhanced AKT-PAK5 signaling may facilitate CNS neuron regeneration. To test this, we performed five lines of in vitro and in vivo studies. First, we assessed AKT-PAK5 signaling on axonal regeneration in cortical neurons after axotomy. Neurons in microfluidic devices at DIV8 were infected with lentiviruses encoding caPAK5 or kdPAK5, ctrl-shRNA or PAK5-shRNA, or combined caAKT1 with PAK5-shRNA, followed by axotomy at DIV12 and imaging at DIV18 (Figure 7A). While control neurons showed little regeneration, neurons expressing caPAK5 displayed robust regeneration (P<0.001; Figures 7B and 7C). Similarly, expressing active caAKT1 enhanced regeneration compared to control neurons (P<0.001; Figures 7D, 7E, and S6A). Such AKT-enhanced regeneration was suppressed (P=0.035) upon depletion of PAK5.
Figure 7. Reprogramming AKT-PAK5 signaling boosts axonal regeneration.
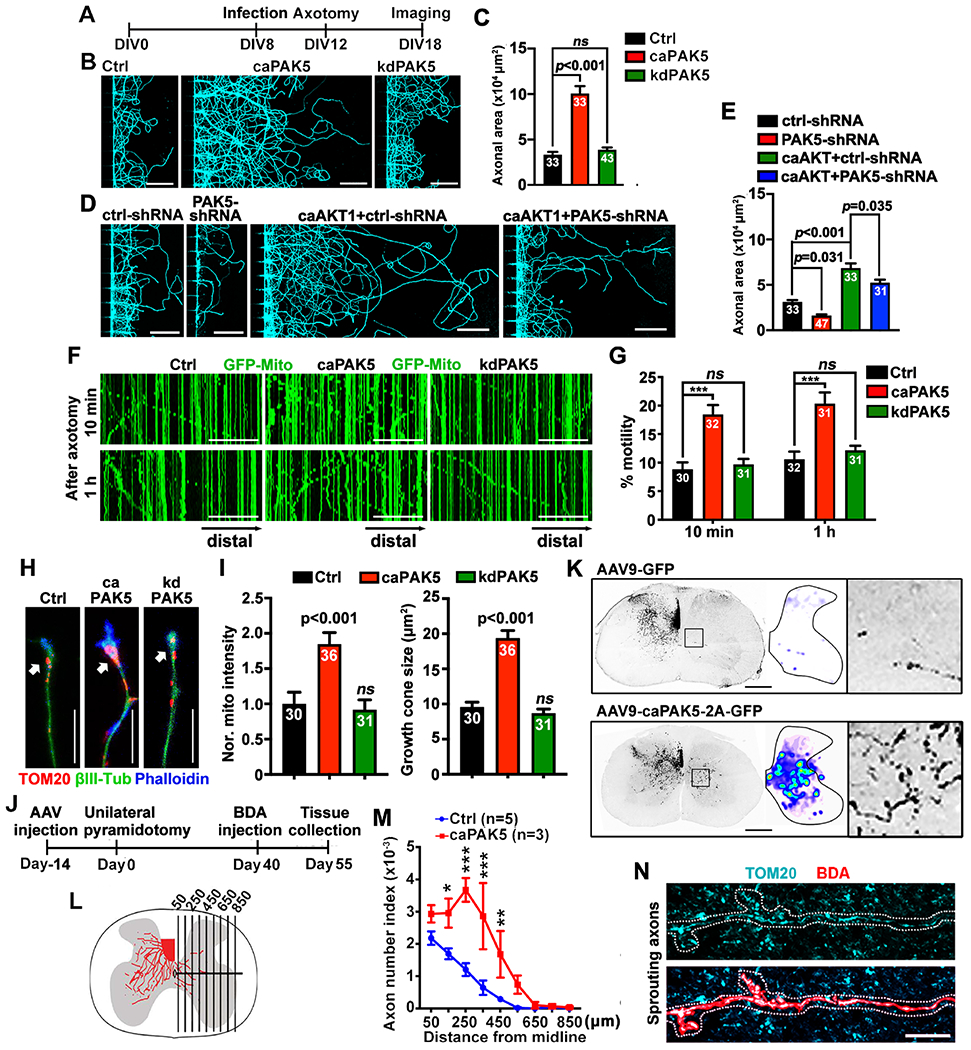
(A-E) A diagram (A) showing the timeline of infection, injury, and imaging. Neurons in microfluidic devices at DIV8 were infected with lentiviruses encoding HA-tagged caPAK5 or kdPAK5 (B), or PAK5-shRNA, or combined caAKT1 with PAK5-shRNA (D), followed by axotomy at DIV12 and βIII-tubulin labeling at DIV18. The regenerated axon area was normalized to the HA control (C) or ctrl-shRNA (E).
(F and G) PAK5-enhanced axonal mitochondrial transport after axotomy. Neurons were co-infected with lentiviruses encoding GFP-Mito and HA-tagged control, caPAK5, or kdPAK5 at DIV8, axotomized at DIV12, followed by time-lapse imaging (30 frames with 5-sec intervals).
(H and I) Expressing caPAK5 induces mitochondrial delivery to axon tips (arrows) and enhances growth cone reformation after axotomy.
(J and K) Timeline of unilateral pyramidotomy (J) and cross-sectional images of BDA-labeled CST axons that sprouted across the midline to the denervated (right) side (K). The distribution of axonal sprouting into the denervated side was converted to heatmaps. The boxed area show BDA-labeled CST axons sprouting into the denervated region.
(L and M) Reprogramming PAK5 signaling facilitates mediolateral distribution of BDA-labeled CST axons crossing vertical lines in the denervated region of the right hemicord. The number of axons crossing a specific distance in the denervated region was normalized to the number of CST axons in the intact dorsal funiculus, and expressed as axon number index.
(N) Cross-sectional image showing mitochondrial delivery into CST sprouting axons on the denervated side in PAK5-activated mouse spinal cord. The high-magnification was selected from three mice expressing AAV9-caPAK5-2A-GFP, where of a total of 28 BDA-positive CST axons, 26 terminals (93%) contained mitochondria, while there were few CST axons sprouting to the denervated side in control mice.
Data were quantified from the total number of axons (I), axonal chamber images (C, E) or microgrooves (G) indicated within bars, or mice (M) indicated above bar graphs from three experiments. Data were expressed as mean±SEM, and analyzed by one-way ANONA with Dunnett’s multiple comparisons test (C, G, I), Tukey’s multiple comparisons test (E), or two-way ANOVA test (M). * P < 0.05, ** P < 0.01, *** P < 0.001, ns: not significant. Scale bars, 10 μm (H, N), 50 μm (F), 100 μm (B, D), 500 μm (K). (See also Figures S6, S7)
Our previous studies showed that snph KO neurons displayed enhanced regeneration capacity.9,22 Second, we tested whether SNPH is an effector of AKT-PAK5-enhanced regeneration. Reintroducing SNPH or phospho-dead SNPH-4A into snph KO neurons abolished regeneration. In contrast, reintroducing phospho-mimicking SNPH-4D did not suppress axonal regeneration (Figures S6B and S6C). Expressing caPAK5 rescues regeneration in snph KO neurons (P<0.001) when SNPH was co-introduced. Thus, reprogramming PAK5 signaling facilitates axon regeneration through the phosphorylation of SNPH. Third, we examined whether enhanced regeneration by reprogramming PAK5 signaling is mediated by mitochondrial transport. To this end, we knocked down Miro1, a motor adaptor driving mitochondrial transport.48,49 Expressing caPAK5 facilitated regeneration in control neurons but not in miro1 knockdown neurons (P=0.001; Figures S6D and S6E). Thus, PAK5-enhanced axon regeneration requires mitochondrial transport. Fourth, we determined whether reprogramming PAK5 signaling remobilizes SNPH-anchored mitochondria after axotomy. Expressing caPAK5, but not kdPAK5, significantly enhanced mitochondrial transport as early as 10 min (P<0.001) and 1 hour post-axotomy (P<0.001; Figures 7F and 7G). By co-labelling TOM20 and phalloidin, we demonstrated that expressing caPAK5 enhanced delivery of mitochondria to axon growing tips (P<0.001) and increased growth cone size (P<0.001) in neurons at DIV18 after injury (Figures 7H and 7I).
Finally, we investigated whether reprogramming PAK5 stimulates a compensatory sprouting response in an in vivo SCI model in adult mice. In addition to axon regeneration from the severed end, axonal growth could be stimulated from fiber tracts that were not injured but rather positioned adjacent or parallel to injured axons.50,51 We performed a unilateral pyramidotomy in which the corticospinal tract (CST) axons were transected in the medulla of the brainstem just above the pyramidal decussation, leading to complete loss of CST innervation in the contralesional spinal cord.22 To reprogramming PAK5, we injected AAV9-2A-GFP or AAV9-caPAK5-2A-GFP into the motor cortex at 14 days pre-injury. An anterograde tracer biotinylated dextran amine (BDA) was bilateral injected into motor cortex at 40 days post-injury, and animals were sacrificed at 55 days post-injury (Figure 7J). Injured mice expressing AAV9-2A-GFP control displayed limited BDA-labeled CST axons sprouted across the midline, with most axons remaining ipsilateral to the lesion site (Figures 7K–7M). Strikingly, mice expressing AAV9-caPAK5-2A-GFP exhibited enhanced CST axons sprouting across the midline and into the denervated side of the spinal cord. Mitochondria were delivered into sprouted axons on the denervated side in the PAK5-reprogrammed group (Figure 7N). Altogether, studies from in vitro and in vivo injury models support our final conclusion: reprogramming AKT-PAK5 signaling protects neuronal survival and promotes axonal regeneration after injury and ischemia, and stimulates axonal regenerative sprouting after spinal cord injury in adult mice.
Discussion
In this study, we reveal an injury-induced AKT-PAK5-SNPH signaling axis that maintains axonal energy supply after acute injury-ischemia in mature neurons and adult brains. Reprogramming PAK5 signaling removes SNPH-anchored damaged mitochondria and replenishes with healthy ones in injured axons, thus reversing energy crisis (Figure S7). Our study suggests that this intrinsic energy repair signaling is efficient in protecting neuron survival and facilitating regeneration after brain injury and ischemia.
To survive an injury, neurons require high levels of energy consumption to support regeneration. In mature neurons, enriched SNPH expression9 results in the vast majority of axonal mitochondria in adult brains remaining stationary.10,11 Brain injury-ischemia induces acute mitochondrial damage.17,21,52–54 In addition, ATP has a limited diffusion capacity within long axons.55,56 These extrinsic insults and intrinsic restrictions lead to an energy crisis in injured axons. These prompted us to investigate into an intrinsic energy repair program that responds to injury-ischemia by remobilizing and replacing damaged mitochondria, maintaining energy supply for regeneration.
Our study demonstrated that axonal PAK5 signaling is transiently activated in response to injury-ischemia and accelerates removal of damaged mitochondria. Such mitochondrial remobilization in mature neurons is achieved by clustered phosphorylation of SNPH by PAK5 and thus turns off the SNPH anchoring mode. In addition, PAK5 is a downstream effector of AKT growth signaling cascade. Reprogramming the AKT-PAK5 axis protects neuron survival and accelerates mitochondrial delivery into sprouted axons on the denervated side of the mouse spinal cord and facilitates axon regeneration.
Enhancing mitochondrial trafficking has emerged as an effective cellular target in facilitating neural regeneration. Misgeld et al. examined proximal segments of transected intercostal nerves and found an “injury response” of mitochondrial re-distribution at the growth cone.36 In the C. elegans ric-7 mutant with impaired mitochondrial transport, injured axons degenerate rapidly; such degeneration is suppressed by forcing mitochondria into axons.57 WldS-induced mitochondrial flux protects axons from Wallerian degeneration after injury.58 In C. elegans, axotomy triggers energy stress in GABA motor neurons; recruiting axonal mitochondria sustains regeneration by supplying ATP.59 In adult retinal ganglion cells, expressing mitochondrial protein Armcx1 promotes axon regeneration by recruiting mitochondria into motile pools.60 In mouse DRG neurons, HDAC6-mediated deacetylation of Miro-1 inhibits axon growth by blocking mitochondrial transport.61 Therefore, activating an intrinsic regrowth program requires the recovery of energy supply in injured axons.1,62 Our previous studies revealed enhanced axon regeneration in the mouse PNS and CNS by genetic deletion of snph9,22 In the current study, we identify the AKT-PAK5-SNPH axis as the first line of energy surveillance that responds to injury-ischemia-induced energy crisis and recovers axonal energy supply in support of CNS survival and regeneration.
SNPH anchors axonal mitochondria through its MT-binding domain18,19 and captures presynaptic mitochondria via an interplay with myosin VI.63 Clustered serine/threonine residues at the N-terminus of SNPH have been predicted as potential phosphorylation targets.64 By mass spectrometry analysis combined with mutation assays, we identified an evolutionarily conserved motif with four serine/threonine residues as PAK5 phosphorylation sites.65,66 We found that the phospho-mimicking SNPH lost its MT-binding capacity and in situ SNPH-MT association in live neurons. Interestingly, in situ SNPH-MT proximity was suppressed in live neurons co-expressing caPAK5. MT-binding is thought to occur through an electrostatic force in the interface, where MT surface carries negatively charged residues and binding proteins hold a region enriched with positively charged residues.67 Consistently, the clustered phosphorylation sites at the N-terminus of SNPH are surrounded by positively charged arginines. We assume that SNPH phosphorylation events neutralize this arginine-rich region and reduce electrostatic-based SNPH-MT association, thus effectively turning off SNPH-anchoring swtich and remobilizing SNPH-anchored after acute injury-ischemia.
Our previous study demonstrated that the generation of SNPH cargo vesicles from chronically stressed mitochondria ensures that SNPH is removed and therefore dysfunctional mitochondria are remobilized.16 However, brain injury-ischemia induces acute mitochondrial damage leading to local energy crisis. Thus, neurons need to adopt an urgent “energy repair program” to support regeneration. The injury-induced AKT-PAK5 signaling can thus quickly boost axonal energy supply by turning off a SNPH-anchoring switch. Therefore, reprogramming an enhanced AKT-PAK5-SNPH axis represents a potential therapeutic target for effective recovery from acute mitochondrial damage during brain injury and ischemic stroke.
The PAK family is classified into two groups based on their sequence homology,68 and PAK5 is a brain-specific Group II PAK that promotes neurite outgrowth.26,69 While Group I PAKs are activated upon Rho binding, PAK5 is activated in a Rho-independent manner.26,69 Expressing caPAK1 (Group I member) fails to mediate SNPH phosphorylation and remobilize axonal mitochondria, suggesting that PAK5 but not PAK1 contributes to energy repairing program.
AKT function in neuron development and regeneration.37–41 AKT activation was observed in a rat ischemia-reperfusion model.70 In our ischemic model, the AKT-PAK5 axis was activated briefly at 4 hours post-reperfusion, thus providing an interpretation for previous observations of partial recovery of axonal mitochondrial transport during the reperfusion process.21 We assume that while brief AKT activation is sufficient to remobilize damaged mitochondria, surviving a severe nerve damage requires an enhanced AKT-PAK5 signaling. This assumption is supported by studies demonstrating that AKT activation rescues stroke-induced neuronal death.70,71 While the AKT-PAK5-SNPH axis facilitates axonal regeneration by remobilizing damaged mticohondria and by rescuing energy crisis, AKT has also been described in growth and regrowth pathways, including the AKT-GSK3B-eIF2ε axis and AKT-Nrf2/ARE axis.41,72,73 Axon regeneration is enhanced upon depleting or oxidizing PTEN, an inhibitor of AKT signaling.74–76 Thus, the AKT-PAK5-SNPH energy repair axis may function in coordination with other AKT growth pathways to protect neuron survival and facilitate axon regeneration. Our findings are consistent with the emerging concept of bioenergetic failures after acute brain injury-ischemia.
STAR * METHODS
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zu-Hang Sheng.
Materials availability
Plasmids generated in this study are available upon request to the lead contact.
Data and code availability
This study did not generate any unique datasets or code. Original/source data for figures in the paper are available from the corresponding author on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse lines and animal care
The snph−/− mouse line was generated by targeted gene replacement in embryonic stem cells as previously described.19 For preparing snph−/− cortical neuron cultures, WT and snph−/− E18 embryonic mice (sex: random) were used. The Tg (Thy1-CFP/COX8A) S2Lich/J transgenic mouse line (No. 007967) was purchased from The Jackson Laboratory. Mice were maintained in the National Institute of Neurological Disorders and Stroke (NINDS) animal facility and housed in a 12-h light/dark cycle. Animal care and use were carried out in accordance with NIH guidelines and approved by the NIH, NINDS/NIDCD Animal Care and Use Committee (ASP1303-21) and the Indiana University Institutional Animal Care and Use Committee (Protocol #18081).
Primary neuron culture
Cortical neurons were collected from E18 embryonic mice (sex: random) as previously described.19 After papain (Worthington) dissociation, neurons were resuspended in plating medium (Neurobasal medium supplemented with 2% B-27, 0.5 mM GlutaMAX, 0.1% 2-Mercaptoenthanol (Thermo Fisher Scientific), 10% FBS (HyClone) and 0.25 μg/ml insulin (Sigma-Aldrich)), and seeded onto 12-mm coverslips (Deckgläser) double-coated with poly-ornithine (Sigma-Aldrich; 1:4 in PBS) and laminin (Roche; 1:400 in PBS). After 24-36 hours of growth, half of the plating medium was replaced by the same amount of neuronal feeding medium (Neurobasal medium supplemented with 2% B-27, 0.5 mM GlutaMAX, and 5 μM glia inhibitor 5-Fluoro-2-deoxyuridine). Neurons were fed every three days by replacing half of the medium with the same amount of fresh neuronal feeding medium. Neurons were transfected with various constructs using the calcium phosphate method or infected with lentiviruses, followed by live imaging with a Zeiss LSM880 Airyscan confocal microscope at the indicated time points.
For oxygen-, glucose-deprivation (OGD) treatment, cortical neurons at DIV14 were refreshed with oxygen-, glucose-free Neurobasal-A medium (Thermo Fisher Scientific), and immediately placed in an incubator (Thermo Fisher Scientific) loaded with 1% O2, 5% CO2 and 94% N2 for 30 min. For reperfusion, neurons were refreshed with normal culture medium.
For DRG neuron cultures, neurons were isolated from P20 or P12M male mouse spinal cords in Hank’s buffered salt solution and digested in 2.5 units/ml dispase II (Roche) and 200 units/ml collagenase (Worthington) at 37°C for 30 min, followed by a 35-min rotation at room temperature. Neurons were filtered with a 70-μm nylon cell strainer (Falcon). Before plating, DRG neurons were transfected with various DNA constructs using a Nucleofector device according to the manufacturer’s specifications (Lonza), then plated on coverslips coated with poly-ornithine (1:3 in PBS) and laminin (1:400 in PBS). DRG neurons were maintained in Neurobasal-A medium supplemented with 2.5% FBS, 0.5 mM GlutaMAX, and B-27, and imaged at DIV4 with a Zeiss LSM880 Airyscan confocal microscope.
METHOD DETAILS
DNA constructs
Lentiviral constructs of caPAK5 and kdPAK5, which contain only the coding sequence (CDS) of the mRNAs, were generated by PCR and cloned into the entry vector pDONR221, followed by transferring into destination lentiviral vectors pHAGE-CMV-n-HA-FLAG as previously reported 9. DsRed-Mito and GFP-Mito were gifts from R. Youle (NINDS, NIH). GO-ATeam2 and GO-ATeam3 were gifts from H. Imamura (Kyoto University, Japan). PAK5-shRNAs and Miro-1-shRNA were purchased from Dharmacon. Scramble shRNA, mApple-LAMP-1, caPAK1, caPAK5, kdPAK5, and caAKT1 constructs were purchased from Addgene. SNPH truncated mutants were generated in a previous study.19
Immunofluorescent staining
Cortical neurons were fixed with PBS containing 4% formaldehyde and 4% sucrose for 20 min at room temperature and washed with PBS several times. After permeabilizing with 0.1% Triton X-100 in PBS for 20 min, neurons were blocked with 1% BSA and 4% normal goat serum in PBS for 1 hour, and incubated with various primary antibodies diluted in blocking buffer at 4°C overnight. Primary antibodies were used as follows: rabbit antibodies against PAK5 (1:200, Novus Biologicals), SNPH (1:200, home-made or Abcam), and p-SNPH (1:200, home-made); mouse antibodies against cytochrome C (1:100, BD Biosciences), TOM20 (1:200, Santa Cruz), and βIII tubulin (1:5000, Sigma-Aldrich). After washing with PBS three times, neurons were incubated with secondary antibodies (Alexa 488, 546, or 633 conjugated, Thermo Fisher Scientific) diluted in blocking buffer for 1 hour at room temperature, rewashed with PBS, and finally mounted with Fluoro-Gel mounting medium (Electron Microscopy Sciences) for imaging.
To measure mitochondrial membrane potential (Ψm), the axonal chamber of microfluidic devices was loaded with 25 nM MitoTracker™ Orange CMTMRos (Thermo Fisher Scientific), a fixable Ψm-dependent dye, for 30 min at 37°C. Neurons were then fixed and imaged on a Zeiss 880 confocal microscope. The relative integrated intensity ratio of CMTMRos signal against GFP-Mito was measured within individual mitochondria in axon chambers.
Live neuron imaging analysis
Transfected or infected neurons were transferred to chambers containing pre-warmed Hibernate E low fluorescence medium (BrainBits) supplemented with 2% B27 and 0.5 mM GlutaMAX. Live neurons were imaged with a 40x 1.3 NA oil immersion objective on a Zeiss LSM 880 Airyscan confocal microscope at 37°C. To image axonal mitochondrial transport, images were taken consecutively every 5 sec for a total duration of 7.5 min. To image axonal LAMP-1 transport, images were taken consecutively every 2 sec for a total duration of 3 min. Kymographs were generated using ImageJ (NIH) as previously described.19 For each experimental group, all visible vesicles on kymographs were pooled together. Mitochondria were considered stationary if they remained immobile during the recording period; a mobile one was counted if the net displacement was > 5 μm. To measure axonal ATP levels, two emission images were collected at 505-550 nm and long pass above 545 nm along axons expressing GO-ATeam2, as described previously.9 Ratiometric images were generated using ImageJ.
Ex vivo imaging of mouse sciatic nerves
Ex vivo imaging was performed as previously described.9 Briefly, dissected sciatic nerve explants from Thy1-mito-CFP mice were quickly transferred into a chamber containing pre-warmed, oxygenated Neurobasal-A medium (Thermo Fisher Scientific). Time-lapse images of axon bundles in the nerve explants were acquired using a Zeiss LSM 880 Airyscan confocal microscope at 37°C. Mitochondrial transport was imaged consecutively every 3 sec. Mitochondrial flux was measured using the Manual Track plugin in ImageJ.
STED super-resolution imaging
STED imaging was performed as described previously.16 An inverted STED microscope with a resolution of ~50-90 nm (TCS SP8 STED 3X, Leica, Germany) was used. Alexa Fluor 488 was excited by a tunable white light laser (70% of maximum power) at 470 nm (10%) with the STED depletion laser at 592 nm (30% of the maximum power); its fluorescence at 480-560 nm was collected using time-gated detection (1.5-6.5 nsec). Alexa Fluor 594 was excited by the tunable white light laser at 594 nm (10%) with the STED depletion laser at 660 nm (30% of the maximum power); its fluorescence between 600-640 nm was collected using time-gated detection (1.5-6.5 nsec). SiR was excited by a tunable white light laser at 652 nm (20%) with the STED depletion laser at 750 nm (20% of the maximum power); its fluorescence at 660-700 nm was collected using time-gated detection (1.5-6.5 nsec). For dual-color imaging, channels at the higher wavelength range were first imaged to avoid bleaching with a frame-scanning mode. Images were subject to deconvolution processing using Huygens software (Scientific Volume Imaging).
Lentivirus production and infection
HEK293T cells were cultured in DMEM (Thermo Fisher Scientific) with 10% FBS (Hyclone) and 0.5 mM GlutaMAX (Thermo Fisher Scientific), and transfected with the vector, psPAX2, and pMD2G at a 4:2:1 ratio to produce the lentivirus. After a 24-hour transfection, the culture medium was replaced with UltraCULTURE™ medium containing 1% Sodium Pyruvate (1% w/v), 1% Sodium Bicarbonate (1% w/v), and 1% GlutaMAX (1% w/v) (Lonza). Virus-containing medium was collected 48 and 72 hours after transfection, filtered through a 0.45 μm filter, and centrifuged at 90,000 x g for 90 min at 4°C. The viral pellets were fully resuspended in PBS, then aliquoted and stored at −80°C until use. 2 x 105 cortical neurons were infected at the indicated time points with 1-2 μl of concentrated virus.
Immunoblots
Mouse cortical neurons, axon compartments, spinal cord tissues, and HEK293T cells with different treatments were collected and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% DOC, 2 mM EDTA) supplemented with a Protease Inhibitor Cocktail (Sigma-Aldrich) and phosphatase inhibitor PhosSTOP™ (Roche). Equal amounts of proteins were loaded and analyzed using 4%–12% Bris-Tris NuPAGE gels (Thermo Fisher Scientific) and immunoblotting. For phosphorylation detection, SuperSep™ Phos-tag™ 7.5% SDS PAGE (FIJIFILM Wako) was used to isolate phosphorylated and non-phosphorylated protein bands. Primary antibodies were used as follows: rabbit antibodies against PAK5 (1:1000, Novus Biologicals), p-PAK4/5/6 (1:500, Cell Signaling), SNPH (1:4000, home-made), p-SNPH (1:2000, home-made), AKT (1:2000, Cell Signaling), and p-AKT (S473) (1:1000, Cell Signaling); mouse antibodies against TOM20 (1:500, Santa Cruz), Miro-1 (1:500, Sigma-Aldrich), Flag (1:2000, Sigma-Aldrich), GFP (1:500, Santa Cruz), myc (1:2000, Sigma-Aldrich), tubulin (1:2000, Abcam), βIII-tubulin (1:2000, Sigma-Aldrich), and GAPDH (1:2000, Millipore).
Proximity ligation assay (PLA)
Fixed neurons were utilized for PLA as described by the manufacturer (Sigma-Aldrich). In brief, fixed neurons were permeabilized in 0.1% Triton X-100 for 20 min, incubated in Duolink blocking buffer for 1 hour, and incubated with primary antibodies (rabbit anti-GFP, 1:200, Abcam; rabbit anti-SNPH, 1:200, Abcam; mouse anti-βIII-tubulin, 1:5000, Sigma-Aldrich) in Duolink antibody diluent at 4°C overnight, then washed with PBS three times for 10 min each, incubated with Duolink secondary antibodies (plus probes and minus probes) at 37°C for 1 hour, washed again with PBS three times for 10 min each, incubated in Duolink PLA ligation solution at 37°C for 30 min, rewashed with PBS three times for 10 min each, incubated with Duolink PLA amplification solution at 37°C for 100 min, washed with PBS twice for 5 min, and mounted with Fluoro-Gel mounting medium (Electron Microscopy Sciences) for imaging. To label newly synthesized PAK5, Puro-PLA was performed as previously reported.33 Briefly, for protein puromycylation, neurons or axons within axon chambers were incubated with 3 μM puromycin in neuronal feeding medium for 15 min. After fixation, neurons were permeabilized with 0.1% Triton X-100 in PBS, and incubated with antibodies against puromycin (1:1000, Millipore) and PAK5 (1:200, Novus Biologicals), followed the PLA process as described above.
Microtubule spin-down assay
GST-SNPH (1-469), SNPH-dMTB, or SNPH-4D were expressed in BL21 Escherichia coli cells, purified by binding to glutathione-Sepharose beads (GE Healthcare), eluted with elution buffer (50 mM Tris-HCl, pH 8.0,10 mM reduced glutathione, and 4 mM DTT), and dialyzed in general tubulin buffer (80 mM PIPES, pH 7.0, 5 mM MgCl2, 1 mM EGTA, and 4 mM DTT). The purified GST-tagged SNPH proteins were used for the microtubule spin-down assay following instructions provided in the manual of the kit (Cytoskeleton). Briefly, 3 μg of GST-tagged SNPH proteins were incubated in the absence or presence of 10 μg tubulin protein with 300 μM taxol at room temperature for 30 min. Each reaction was placed on the top of the cushion buffer (80 mM PIPES, pH 7.0, 5 mM MgCl2, 1 mM EGTA, and 50% glycerol) and centrifuged at 100,000 × g for 40 min at room temperature. The supernatants were collected, and the pellets were resuspended in SDS-PAGE sample buffer with 10 mM DTT in each reaction. Both supernatant and pellet samples were resolved by 4-12% Bis-Tris NuPAGE (Thermo Fisher Scientific), followed by Coomassie Blue staining.
Microfluidic chamber preparation and axon regrowth assay after axotomy
A silicon wafer with a pattern made out of SU-8 by photolithography was used to cast the PDMS microfluidic chamber devices. Briefly, SYLGARD 184 silicone elastomer base was mixed with the curing agent at a ratio of 10:1. The PDMS was then mixed using a THINKY mixer in a two-step program: mixing at 2,000 rpm for 4 min and de-foaming at 2,200 rpm for 4 min. The mixed PDMS was poured onto the silicon wafer and then placed in a Bel-Art vacuum desiccator for 3 hours to remove the air bubbles from the PDMS. The wafer with PDMS was placed in the oven at 80°C for 2 hours to cure. After cooling down at room temperature for 1 hour, the cured PDMS was punched out, washed in 50% ethanol, and dried before use. For plasma-bonding microfluidic chambers, the coverslips and microfluidic chambers were exposed to the plasma cleaning treatment for 1 min in a PDC-32G plasma cleaner and bonded together before plating neurons. For axon regrowth assays, axons in the axon chambers were axotomized by vacuum aspiration at DIV7-10 or DIV12. The two wells on the axon sides were aspirated three times to ensure complete axotomy. After the first and second aspiration, pre-warmed culture medium was quickly filled into the axon chambers, followed by the third aspiration. The chambers were then refilled with fresh culture medium and incubated for the indicated times. Axon regrowth after axotomy in the axon terminal chambers was analyzed by labeling axons with βIII-tubulin and imaged under the same settings using a Zeiss LSM 880 Airyscan confocal microscope with a 40× objective. The absolute axonal area (μm2) of βIII-tubulin signal with the same threshold within a 1024 × 1024 pixel was measured using ImageJ. More than 30 images from multiple chambers were analyzed from at least three independent experiments.
T8 dorsal hemisection
With adequate sedation, a vertical incision was made to expose the 8th-10th thoracic spinous process which serves as a reliable landmark. Using a U-shape channel for mouse spinal stabilizer, the laminectomy was then performed at the 8th thoracic level (T8) and the underlying dura was exposed. Between the interlaminar space, a 32-gauge needle was used to create a durotomy across the width of the cervical spinal cord. A 1.0 mm dorsal hemisection lesion was made by using a 2.4-mm flat blade attached to the Louisville Injury System Apparatus (LISA). After laceration, the mouse was released from spinal stabilizer and the muscle and skin were then closed in layers.
Unilateral pyramidotomy model
Adult mice were anesthetized with a ketamine/xylazine mixture (120 mg/3.3 mg). With adequate sedation, mice in a supine position received a 2-3 cm long midline incision from the chin to the caudal end of the larynx using a sterile scalpel. Blunt dissection of the upper layers of tissue such as glands (e.g. submaxillary gland and parotid gland) and muscles covering the trachea was performed using reverse action with blunted scissors and toothed forceps until the ventral surface of the basioccipital bone was reached. The left craniotomy was made to remove part of the basioccipital bone to gain access to the underlying pyramid. After durotomy to open the outer membrane covering the pyramid, a cut was made in the left pyramid approximately 1.0 mm wide spanning the width of the pyramid and 0.5 mm deep with a modified fine scalpel perpendicular to the basilar artery to interrupt the descending corticospinal tract (CST) axons. After surgery, the skin was sutured with 5-0 Vicryl sutures and the animal kept in a new cage with a heating pad.
AAV injection
AAV9-GFP and AAV9-caPAK5-2A-GFP were purchased from Vigene Biosciences. Sciatic nerve injection was performed as described in a previous study.77 Briefly, after the mouse was fully anesthetized, the sciatic nerve was exposed on the left side. 3 μl of virus mixed with trypan blue was injected into the sciatic nerves with an insulin syringe until the sciatic nerves appeared blue. For motor cortex injection: once fully anesthetized, the mouse head was fixed on a stereotaxic apparatus. The bilateral windows (5 mm in length and 2 mm in width) were performed with the medial edges of the windows 0.5 mm lateral to the bregma to expose the bilateral cortical motor area. Virus (5 μl was injected with a digital stereotactic injector (Item: 51709, Stoelting Co. USA) into the motor cortex at a total of 10 sites (5 sites/side). Mediolateral (ML) coordination: 1.5 mm lateral to the bregma; anteroposterior (AP) coordination from the bregma: −1.0, −0.5, 0, −0.75 and 1.5 mm; dorsoventral (DV) coordination: 0.5 mm from the cortical surface; rate: 0.1 μl/min. After each injection was completed, the injector tip was left in place for an additional 5 min to ensure that the virus solution adequately penetrated the tissue.
Corticospinal tract tracing
To trace the corticospinal tract, the anterograde tracer, BDA, was injected into the mouse motor cortex using the same technique used for AAV virus injection. Briefly, after craniotomy, BDA (5 μl) was stereotaxically injected into the motor cortex at a total of 10 sites (5 sites/side). Mediolateral (ML) coordination: 1.5 mm lateral to the bregma; anteroposterior (AP) coordination from the bregma: −1.0, −0.5, 0, −0.75 and 1.5 mm; dorsoventral (DV) coordination: 0.5 mm from the cortical surface; rate: 0.1 μl/min. Mice were sacrificed for histological examination at 15 days after the BDA injection.
Tissue section and staining
To amplify and visualize GFP signals, 35-μm thick brain sections were incubated with a rabbit anti-GFP antibody (1:2000, Abcam). Signal was detected with a mouse Alexa Fluor 488 fluorescent secondary antibody (1:1000, Thermo Fisher Scientific). To visualize the CST projection fibers, 35-μm transverse spinal cord sections (C5–C8) were prepared from mice that received the BDA tracing. Solution containing streptavidin-horseradish peroxidase (Vectastain R.T.U. Elite ABC Reagent) was added to the sections for 1 hour followed by a 30 min incubation with PerkinElmer Biotinyl tyramide (1:100 in amplification diluent). Signals were detected following a 1-hour incubation with Extra-Avidin@ TRITC (1:200, Sigma-Aldrich).
QUANTIFICATION AND STATISTICAL ANALYSIS
All quantifications were performed not blinded. Statistical parameters including the definitions and exact value of n (e.g., total number of experiments or replications, axons, neurons, mitochondria, images, animals, etc), deviations, p values, and the types of the statistical tests are shown in Figures and Figure Legends. Statistical analysis was performed using Prism 8 (GraphPad Software). Two groups were compared using Student’s t test (sample size n ≥ 30) or Mann-Whitney test (sample size n < 30). Comparisons between three or more groups were performed by one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons test or Tukey’s multiple comparisons test (sample size n ≥ 30), or two-way ANOVA with Bonferroni post hoc correction. Data were expressed as mean ± SEM. Differences were considered significant with P < 0.05.
Supplementary Material
Video S1. Ex vivo imaging of mitochondrial transport along the sciatic nerves from a P30 Mito-CFP mouse. Related to Figure 3H. Mitochondrial transport was assessed in sciatic nerve explants after rapid dissection and immediate immersion in prewarmed oxygenated Neurobasal-A medium. Time-lapse images of multiple axons in nerve explants were acquired using a Zeiss 880 Airyscan confocal microscope with a P-Apochromat 40×/1.3 oil objective at 3-sec intervals for a total of 60 frames. Image stacks were processed using ImageJ software.
Video S2. Ex vivo imaging of mitochondrial transport along the sciatic nerves from a P70 Mito-CFP mouse. Related to Figure 3H. Mitochondrial transport was assessed in sciatic nerve explants after rapid dissection and immediate immersion in prewarmed oxygenated Neurobasal-A medium. Time-lapse images of multiple axons in nerve explants were acquired using a Zeiss 880 Airyscan confocal microscope with a P-Apochromat 40×/1.3 oil objective at 3-sec intervals for a total of 60 frames. Image stacks were processed using ImageJ software.
Video S3. Ex vivo imaging of mitochondrial transport along the sciatic nerves from a Mito-CFP mouse injected with AAV9-2A-GFP. Related to Figure 3I. AAV9-2A-GFP was injected into sciatic nerves at the left-side at P45. Two weeks after injection at P59, mitochondrial transport was assessed in sciatic nerve explants after rapid dissection and immediate immersion into prewarmed oxygenated Neurobasal-A medium. Time-lapse images of multiple axons in nerve explants were acquired using a Zeiss 880 Airyscan confocal microscope with a P-Apochromat 40×/1.3 oil objective at 3-sec intervals for a total of 60 frames. Image stacks were processed using ImageJ software.
Video S4. Ex vivo imaging of mitochondrial transport along the sciatic nerves from a Mito-CFP mouse injected with AAV9-caPAK5-2A-GFP. Related to Figure 3I. AAV9-caPAK5-2A-GFP was injected into sciatic nerves at the left-side at P45. Two weeks after injection at P59, mitochondrial transport was assessed in sciatic nerve explants after rapid dissection and immediate immersion into prewarmed oxygenated Neurobasal-A medium. Time-lapse images of multiple axons in nerve explants were acquired using a Zeiss 880 Airyscan confocal microscope with a P-Apochromat 40×/1.3 oil objective at 3-sec intervals for a total of 60 frames. Image stacks were processed using ImageJ software.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-TOM20 | Santa Cruz | Cat# sc-1616, RRID: AB_630836 |
| Mouse anti-TOM20 | Millipore | Cat# MABT166, RRID: N/A |
| Rabbit anti-PAK5 | Novus Biologicals | Cat# NBP1-77237, RRID: AB_11015582 |
| Rabbit anti-Phospho-PAK4/5/6 | Cell Signaling | Cat# 3241, RRID: AB_2158623 |
| Rabbit anti-SNPH | [19] | N/A |
| Rabbit anti-SNPH | Abcam | Cat# ab192605, RRID: N/A |
| Rabbit anti-Phospho-SNPH | Genescript | N/A |
| Mouse anti-Flag | Sigma-Aldrich | Cat# F1804, RRID: AB_262044 |
| Mouse anti-βIII-tubulin | Sigma-Aldrich | Cat# T8578, RRID: AB_1841228 |
| Mouse anti-Tau1 | Millipore | Cat# MAB3420, RRID: AB_94855 |
| Mouse anti-Cytochrome c | BD Biosciences | Cat# 556432, RRID: AB_396416 |
| Mouse anti-GFP | Santa Cruz | Cat# sc-9996, RRID: AB_628795 |
| Rabbit anti-HA | Sigma-Aldrich | Cat# H9658, RRID: AB_260092 |
| Rabbit anti-myc | Sigma-Aldrich | Cat# M4439, RRID: AB_439694 |
| Rabbit anti-Phospho-AKT (Ser-473) | Cell Signaling | Cat# 4060, RRID: AB_2315049 |
| Rabbit anti-AKT | Cell Signaling | Cat# 4691, RRID: AB_915783 |
| Rabbit anti-alpha-Tubulin | Abcam | Cat# ab7291, RRID: AB_2241126 |
| Rabbit anti-GAPDH | Millipore | Cat# CB1001, RRID: AB_2107426 |
| Mouse anti-Miro-1 | Sigma-Aldrich | Cat# HPA010687, RRID: AB_1079813 |
| Mouse anti-puromycin | Millipore | Cat# MABE341, RRID: N/A |
| Mouse IgG, HRP-linked | GE Healthcare | Cat# NA931, RRID: AB_772210 |
| Rabbit IgG, HRP-linked | GE Healthcare | Cat# na934, RRID: AB_772206 |
| Goat anti-mouse, Alexa 488 Conjugate | Thermo Fisher Scientific | Cat# 11017, RRID: AB_2534084 |
| Donkey anti-rabbit, Alexa 488 Conjugate | Thermo Fisher Scientific | Cat# 21206, RRID: AB_2535792 |
| Goat anti-mouse, Alexa 546 Conjugate | Thermo Fisher Scientific | Cat# 11018, RRID: AB_2534085 |
| Goat anti-rabbit, Alexa 546 Conjugate | Thermo Fisher Scientific | Cat# 11035, RRID: AB_2534093 |
| Goat anti-rabbit, Alexa 594 Conjugate | Thermo Fisher Scientific | Cat# 11037, RRID: AB_2534095 |
| Goat anti-rabbit, Alexa 633 Conjugate | Thermo Fisher Scientific | Cat# 21052, RRID: AB_2535720 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Prolong Diamond Antifade Mountant with DAPI | Thermo Fisher Scientific | Cat# P36971 |
| MitoTracker Orange CMTMRos | Thermo Fisher Scientific | Cat# M7510 |
| Phalloidin, DyLight 633 | Thermo Fisher Scientific | Cat# 21840 |
| Rotenone | Selleckchem | Cat# S2348 |
| Vincristine sulfate salt | Sigma-Aldrich | Cat# V8879 |
| Picrotoxin | Sigma-Aldrich | Cat# P1675 |
| Mouse beta-NGF | Shenandoah Biotechnology | Cat# 200-11 |
| MK-2206 | Selleckchem | Cat# S1078 |
| SC-79 | Selleckchem | Cat# S7863 |
| PF3758309 | Selleckchem | Cat# S7094 |
| DMEM | Thermo Fisher Scientific | Cat# 11995-065 |
| Neurobasal | Thermo Fisher Scientific | Cat# 21103-049 |
| Fetal Bovine Serum | HyClone | Cat# SH30071.03 |
| B27 | Thermo Fisher Scientific | Cat# 17504-044 |
| GlutaMAX | Thermo Fisher Scientific | Cat# 35050-61 |
| Low Fluorescence Hibernate E media | BrainBits | Cat# HELF |
| 2-Mercaptoethanol | Thermo Fisher Scientific | Cat# 21985-023 |
| UltraCULTURE | Lonza | Cat# 12-725F |
| Papain | Worthington | Cat# LS003122 |
| Insulin | Sigma-Aldrich | Cat# I5500 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11836170001 |
| PhosSTOP | Roche | Cat# 04906845001 |
| Sodium Bicarbonate | Lonza | Cat# 17-613E |
| Sodium Pyruvate | Lonza | Cat# 13-115E |
| Poly-ornithine | Sigma-Aldrich | Cat# P4957 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 111668019 |
| IPTG | Thermo Fisher Scientific | Cat# 15529019 |
| Glutathione Sepharose 4 Fast Flow | GE Healthcare | Cat# 17-0756-01 |
| Protein G Sepharose 4 Fast Flow | GE Healthcare | Cat# 17-0618-01 |
| Fluoro-Gel, with Tris Buffer | Electron Microscopy Sciences | Cat# 1798510 |
| Goat Serum | Sigma-Aldrich | Cat# G9023 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat# A2153 |
| Non-Fat Dry Milk | Labscientific | Cat# M0841 |
| 5-Fluoro-2-deoxyuridine | Sigma-Aldrich | Cat# F0503 |
| ExtrAvidin@ TRITC | Sigma-Aldrich | Cat# E3011 |
| Critical Commercial Assays | ||
| Microtubule Binding Protein Spin-Down Assay Kit | Cytoskeleton, Inc. | Cat# BK029 |
| Duolink In Situ Detection Reagents Orange | Sigma-Aldrich | Cat# DUO92007 |
| Duolink In Situ PLA Probe Anti-Mouse MINUS | Sigma-Aldrich | Cat# DUO92004 |
| Duolink In Situ PLA Probe Anti-Rabbit PLUS | Sigma-Aldrich | Cat# DUO92002 |
| Lambda Protein Phosphatase | New England BioLabs | Cat# P0753 |
| TSA Biotin Tyramide Reagent Pack | PerkinElmer | Cat# SAT700001EA |
| Seahorse XFe96 FluxPak mini | Agilent Technologies | Cat# 102601-100 |
| SuperSep Phos-tag | FIJIFILM Wako | Cat# 198-17981 |
| BP Clonase II Enzyme Mix | Thermo Fisher Scientific | Cat# 11789020 |
| LR Clonase II Enzyme Mix | Thermo Fisher Scientific | Cat# 11791020 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat# CRL-3216, RRID: CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse snph−/− | [19] | N/A |
| Tg(Thy1-CFP/COX8A)S2Lich | The Jackson Laboratory | JAX: 007967 |
| Bacterial and Virus Strains | ||
| AAV9-2A-GFP | Vigene Biosciences | N/A |
| AAV9-caPAK5-2A-GFP | Vigene Biosciences | N/A |
| Recombinant DNA | ||
| DsRed-Mito | A gift from Dr. Richard Youle | N/A |
| GFP-Mito | A gift from Dr. Richard Youle | N/A |
| mApple-Lamp1 | A gift from Dr. Michael Davidson | Addgene#54918 |
| GO-ATeam2 | A gift from Dr. Hiromi Imamura | N/A |
| GO-ATeam3 | A gift from Dr. Hiromi Imamura | N/A |
| pGEX-4T-1-SNPH-(1-469) | [19] | N/A |
| pGEX-4T-1-SNPH-dMTB (1-469) | [19] | N/A |
| pGEX-4T-1-SNPH-4D (1-469) | This study | N/A |
| TRC Mouse PAK5 shRNAs | Dharmacon | Clone ID: TRCN0000025169, TRCN0000025170 |
| TRC Mouse Miro-1 shRNA | Dharmacon | Clone ID: TRCN0000077739 |
| Scramble shRNA | Addgene | Addgene#1864 |
| pCMV6-Myc-PAK5-K478M (kinase-dead) | A gift from Dr. Jonathan Chernoff | Addgene#16020 |
| pCMV6-Myc-PAK5-S573N (constitutively active) | A gift from Dr. Jonathan Chernoff | Addgene#16021 |
| pCMV6-Myc-PAK1-T423E (constitutively active) | A gift from Dr. Jonathan Chernoff | Addgene#12208 |
| pCDH-puro-myr-HA-AKT1 (constitutively active) | A gift from Dr. Jialiang Wang | Addgene#46969 |
| pEGFP-PAK5-K478M | This study | N/A |
| pEGFP-PAK5-S573N | This study | N/A |
| pHAGE HA-PAK5-K478M | This study | N/A |
| pHAGE HA-PAK5-S573M | This study | N/A |
| pHAGE HA-SNPH | [16] | N/A |
| pHAGE HA-SNPH-dMTB | [16] | N/A |
| pHAGE HA-SNPH-4A | This study | N/A |
| pHAGE HA-SNPH-4D | This study | N/A |
| Flag-SNPH | [18] | N/A |
| GFP-SNPH | [18] | N/A |
| GFP-SNPH-dMTB | [18] | N/A |
| GFP-SNPH-4A | This study | N/A |
| GFP-SNPH-4D | This study | N/A |
| GFP-SNPH (1-240) | This study | N/A |
| GFP-SNPH-4A (1-240) | This study | N/A |
| pMD2.G | A gift from Dr. Didier Trono | Addgene #12259 |
| psPAX2 | A gift from Dr. Didier Trono | Addgene #12260 |
| Software and Algorithms | ||
| Image J | NIH | https://imagej.nih.gov/ij, RRID: SCR_003070 |
| Prism 7 | GraphPad Software | https://www.graphpad.com/, RRID: SCR_002798 |
| Adobe Photoshop CC | Adobe | http://www.adobe.com/, RRID: SCR_014199 |
| Huygens Software | Scientific Volume Imaging | https://svi.nl/HuygensSoftware, RRID: SCR_014237 |
Highlights:
Axonal PAK5 synthesis and signaling is activated in response to ischemia-injury
Activating PAK5 signaling protects against axonal energy crisis for neuron survival
AKT-PAK5 axis turns off SNPH anchor accelerating damaged mitochondria replacement
Reprogramming AKT-PAK5 boosts local energy metabolism supporting CNS regeneration
Acknowledgments
We thank members of the Sheng lab for assistance and discussion; Y. Li (NINDS Proteomics Core Sequencing Facility) for mass spectrometry analysis; N. Morgan (NIBIB, NIH) for design and fabrication of microfluidic device templates; C. Smith (NINDS Light Imaging Facility) and V. Schram (NICHD Microscopy and Imaging Core) for imaging support; R. Youle (NINDS/NIH) for constructs; J.C. Roney and K. Chamberlain for critical reading and editing. This work was supported by the Intramural Research Program of NINDS, NIH ZIA NS003029 and ZIA NS002946 (Z-H. Sheng) and NIH 1R01NS100531, 1R01NS103481, and 1R01NS111776 (X-M. Xu).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing financial interests.
REFERENCES
- 1.Sheng ZH (2017). The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends Cell Biol 27, 403–416. 10.1016/j.tcb.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mishra P, and Chan DC (2016). Metabolic regulation of mitochondrial dynamics. J Cell Biol 212, 379–387. 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris JJ, Jolivet R, and Attwell D (2012). Synaptic energy use and supply. Neuron 75, 762–777. 10.1016/j.neuron.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 4.Hall CN, Klein-Flugge MC, Howarth C, and Attwell D (2012). Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J Neurosci 32, 8940–8951. 10.1523/JNEUROSCI.0026-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devine MJ, and Kittler JT (2018). Mitochondria at the neuronal presynapse in health and disease. Nat Rev Neurosci 19, 63–80. 10.1038/nrn.2017.170. [DOI] [PubMed] [Google Scholar]
- 6.Misgeld T, and Schwarz TL (2017). Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 96, 651–666. 10.1016/j.neuron.2017.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirokawa N, Niwa S, and Tanaka Y (2010). Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68, 610–638. 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- 8.Sheng ZH, and Cai Q (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13, 77–93. 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou B, Yu P, Lin MY, Sun T, Chen Y, and Sheng ZH (2016). Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J Cell Biol 214, 103–119. 10.1083/jcb.201605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis TL Jr., Turi GF, Kwon SK, Losonczy A, and Polleux F (2016). Progressive Decrease of Mitochondrial Motility during Maturation of Cortical Axons In Vitro and In Vivo. Curr Biol 26, 2602–2608. 10.1016/j.cub.2016.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smit-Rigter L, Rajendran R, Silva CA, Spierenburg L, Groeneweg F, Ruimschotel EM, van Versendaal D, van der Togt C, Eysel UT, Heimel JA, et al. (2016). Mitochondrial Dynamics in Visual Cortex Are Limited In Vivo and Not Affected by Axonal Structural Plasticity. Curr Biol 26, 2609–2616. 10.1016/j.cub.2016.07.033. [DOI] [PubMed] [Google Scholar]
- 12.Takihara Y, Inatani M, Eto K, Inoue T, Kreymerman A, Miyake S, Ueno S, Nagaya M, Nakanishi A, Iwao K, et al. (2015). In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proc Natl Acad Sci U S A 112, 10515–10520. 10.1073/pnas.1509879112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vagnoni A, and Bullock SL (2018). A cAMP/PKA/Kinesin-1 Axis Promotes the Axonal Transport of Mitochondria in Aging Drosophila Neurons. Curr Biol 28, 1265–1272 e1264. 10.1016/j.cub.2018.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai Q, Zakaria HM, Simone A, and Sheng ZH (2012). Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol 22, 545–552. 10.1016/j.cub.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang DT, and Reynolds IJ (2006). Mitochondrial trafficking and morphology in healthy and injured neurons. Prog Neurobiol 80, 241–268. 10.1016/j.pneurobio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Lin MY, Cheng XT, Tammineni P, Xie Y, Zhou B, Cai Q, and Sheng ZH (2017). Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 94, 595–610 e596. 10.1016/j.neuron.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vosler PS, Graham SH, Wechsler LR, and Chen J (2009). Mitochondrial targets for stroke: focusing basic science research toward development of clinically translatable therapeutics. Stroke 40, 3149–3155. 10.1161/STROKEAHA.108.543769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, and Sheng ZH (2013). Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J Cell Biol 202, 351–364. 10.1083/jcb.201302040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, and Sheng ZH (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das S, Boczan J, Gerwin C, Zald PB, and Sheng ZH (2003). Regional and developmental regulation of syntaphilin expression in the brain: a candidate molecular element of synaptic functional differentiation. Brain Res Mol Brain Res 116, 38–49. 10.1016/s0169-328x(03)00212-2. [DOI] [PubMed] [Google Scholar]
- 21.Zheng Y, Zhang X, Wu X, Jiang L, Ahsan A, Ma S, Xiao Z, Han F, Qin ZH, Hu W, and Chen Z (2019). Somatic autophagy of axonal mitochondria in ischemic neurons. J Cell Biol 218, 1891–1907. 10.1083/jcb.201804101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han Q, Xie Y, Ordaz JD, Huh AJ, Huang N, Wu W, Liu N, Chamberlain KA, Sheng ZH, and Xu XM (2020). Restoring Cellular Energetics Promotes Axonal Regeneration and Functional Recovery after Spinal Cord Injury. Cell Metab 31, 623–641 e628. 10.1016/j.cmet.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holt CE, Martin KC, and Schuman EM (2019). Local translation in neurons: visualization and function. Nat Struct Mol Biol 26, 557–566. 10.1038/s41594-019-0263-5. [DOI] [PubMed] [Google Scholar]
- 24.Terenzio M, Schiavo G, and Fainzilber M (2017). Compartmentalized Signaling in Neurons: From Cell Biology to Neuroscience. Neuron 96, 667–679. 10.1016/j.neuron.2017.10.015. [DOI] [PubMed] [Google Scholar]
- 25.Sahoo PK, Smith DS, Perrone-Bizzozero N, and Twiss JL (2018). Axonal mRNA transport and translation at a glance. J Cell Sci 131. 10.1242/jcs.196808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dan C, Nath N, Liberto M, and Minden A (2002). PAK5, a new brain-specific kinase, promotes neurite outgrowth in N1E-115 cells. Mol Cell Biol 22, 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, and Lo EH (2016). Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. 10.1038/nature18928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lightowlers RN, Chrzanowska-Lightowlers ZM, and Russell OM (2020). Mitochondrial transplantation-a possible therapeutic for mitochondrial dysfunction?: Mitochondrial transfer is a potential cure for many diseases but proof of efficacy and safety is still lacking. EMBO Rep 21, e50964. 10.15252/embr.202050964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wells CM, and Jones GE (2010). The emerging importance of group II PAKs. Biochem J 425, 465–473. 10.1042/BJ20091173. [DOI] [PubMed] [Google Scholar]
- 30.Cotteret S, Jaffer ZM, Beeser A, and Chernoff J (2003). p21-Activated kinase 5 (Pak5) localizes to mitochondria and inhibits apoptosis by phosphorylating BAD. Mol Cell Biol 23, 5526–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cotteret S, and Chernoff J (2006). Nucleocytoplasmic shuttling of Pak5 regulates its antiapoptotic properties. Mol Cell Biol 26, 3215–3230. 10.1128/MCB.26.8.3215-3230.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shigeoka T, Jung H, Jung J, Turner-Bridger B, Ohk J, Lin JQ, Amieux PS, and Holt CE (2016). Dynamic Axonal Translation in Developing and Mature Visual Circuits. Cell 166, 181–192. 10.1016/j.cell.2016.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.tom Dieck S, Kochen L, Hanus C, Heumuller M, Bartnik I, Nassim-Assir B, Merk K, Mosler T, Garg S, Bunse S, et al. (2015). Direct visualization of newly synthesized target proteins in situ. Nat Methods 12, 411–414. 10.1038/nmeth.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eswaran J, Lee WH, Debreczeni JE, Filippakopoulos P, Turnbull A, Fedorov O, Deacon SW, Peterson JR, and Knapp S (2007). Crystal Structures of the p21-activated kinases PAK4, PAK5, and PAK6 reveal catalytic domain plasticity of active group II PAKs. Structure 15, 201–213. 10.1016/j.str.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakano M, Imamura H, Nagai T, and Noji H (2011). Ca(2)(+) regulation of mitochondrial ATP synthesis visualized at the single cell level. ACS Chem Biol 6, 709–715. 10.1021/cb100313n. [DOI] [PubMed] [Google Scholar]
- 36.Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, and Lichtman JW (2007). Imaging axonal transport of mitochondria in vivo. Nat Methods 4, 559–561. 10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- 37.Manning BD, and Toker A (2017). AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405. 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dajas-Bailador F, Bantounas I, Jones EV, and Whitmarsh AJ (2014). Regulation of axon growth by the JIP1-AKT axis. J Cell Sci 127, 230–239. 10.1242/jcs.137208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diez H, Benitez MJ, Fernandez S, Torres-Aleman I, Garrido JJ, and Wandosell F (2016). Class I PI3-kinase or Akt inhibition do not impair axonal polarization, but slow down axonal elongation. Biochim Biophys Acta 1863, 2574–2583. 10.1016/j.bbamcr.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 40.Diez H, Garrido JJ, and Wandosell F (2012). Specific roles of Akt iso forms in apoptosis and axon growth regulation in neurons. PLoS One 7, e32715. 10.1371/journal.pone.0032715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Namikawa K, Honma M, Abe K, Takeda M, Mansur K, Obata T, Miwa A, Okado H, and Kiyama H (2000). Akt/protein kinase B prevents injury-induced motoneuron death and accelerates axonal regeneration. J Neurosci 20, 2875–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen S, Owens GC, Crossin KL, and Edelman DB (2007). Serotonin stimulates mitochondrial transport in hippocampal neurons. Mol Cell Neurosci 36, 472–483. 10.1016/j.mcn.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 43.Chen S, Owens GC, and Edelman DB (2008). Dopamine inhibits mitochondrial motility in hippocampal neurons. PLoS One 3, e2804. 10.1371/journal.pone.0002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, and Cohen P (1997). Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7, 261–269. 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 45.Crowder RJ, and Freeman RS (1998). Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci 18, 2933–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukuda Y, Li Y, and Segal RA (2017). A Mechanistic Understanding of Axon Degeneration in Chemotherapy-Induced Peripheral Neuropathy. Front Neurosci 11, 481. 10.3389/fnins.2017.00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Press C, and Milbrandt J (2008). Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J Neurosci 28, 4861–4871. 10.1523/JNEUROSCI.0525-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, and Kittler JT (2009). Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61, 541–555. 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X, and Schwarz TL (2009). The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cafferty WB, McGee AW, and Strittmatter SM (2008). Axonal growth therapeutics: regeneration or sprouting or plasticity? Trends Neurosci 31, 215–220. 10.1016/j.tins.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tuszynski MH, and Steward O (2012). Concepts and methods for the study of axonal regeneration in the CNS. Neuron 74, 777–791. 10.1016/j.neuron.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bradke F, Fawcett JW, and Spira ME (2012). Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat Rev Neurosci 13, 183–193. 10.1038/nrn3176. [DOI] [PubMed] [Google Scholar]
- 53.Cavallucci V, Bisicchia E, Cencioni MT, Ferri A, Latini L, Nobili A, Biamonte F, Nazio F, Fanelli F, Moreno S, et al. (2014). Acute focal brain damage alters mitochondrial dynamics and autophagy in axotomized neurons. Cell Death Dis 5, e1545. 10.1038/cddis.2014.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Donnell KC, Vargas ME, and Sagasti A (2013). WldS and PGC-1alpha regulate mitochondrial transport and oxidation state after axonal injury. J Neurosci 33, 14778–14790. 10.1523/JNEUROSCI.1331-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hubley MJ, Locke BR, and Moerland TS (1996). The effects of temperature, pH, and magnesium on the diffusion coefficient of ATP in solutions of physiological ionic strength. Biochim Biophys Acta 1291, 115–121. 10.1016/0304-4165(96)00053-0. [DOI] [PubMed] [Google Scholar]
- 56.Sun T, Qiao H, Pan PY, Chen Y, and Sheng ZH (2013). Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep 4, 413–419. 10.1016/j.celrep.2013.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rawson RL, Yam L, Weimer RM, Bend EG, Hartwieg E, Horvitz HR, Clark SG, and Jorgensen EM (2014). Axons degenerate in the absence of mitochondria in C. elegans. Curr Biol 24, 760–765. 10.1016/j.cub.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Avery MA, Rooney TM, Pandya JD, Wishart TM, Gillingwater TH, Geddes JW, Sullivan PG, and Freeman MR (2012). WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr Biol 22, 596–600. 10.1016/j.cub.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han SM, Baig HS, and Hammarlund M (2016). Mitochondria Localize to Injured Axons to Support Regeneration. Neuron 92, 1308–1323. 10.1016/j.neuron.2016.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cartoni R, Norsworthy MW, Bei F, Wang C, Li S, Zhang Y, Gabel CV, Schwarz TL, and He Z (2016). The Mammalian-Specific Protein Armcx1 Regulates Mitochondrial Transport during Axon Regeneration. Neuron 92, 1294–1307. 10.1016/j.neuron.2016.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kalinski AL, Kar AN, Craver J, Tosolini AP, Sleigh JN, Lee SJ, Hawthorne A, Brito-Vargas P, Miller-Randolph S, Passino R, et al. (2019). Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J Cell Biol 218, 1871–1890. 10.1083/jcb.201702187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaasik A (2016). Mitochondrial Mobility and Neuronal Recovery. N Engl J Med 375, 1295–1296. 10.1056/NEJMcibr1607955. [DOI] [PubMed] [Google Scholar]
- 63.Li S, Xiong G-J, Huang N, and Sheng Z-H (2020). The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nature Metabolism. 10.1038/s42255-020-00289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, and Burlingame AL (2012). Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol Cell Proteomics 11, 215–229. 10.1074/mcp.O112.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Strochlic TI, Concilio S, Viaud J, Eberwine RA, Wong LE, Minden A, Turk BE, Plomann M, and Peterson JR (2012). Identification of neuronal substrates implicates Pak5 in synaptic vesicle trafficking. Proc Natl Acad Sci U S A 109, 4116–4121. 10.1073/pnas.1116560109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Amano M, Hamaguchi T, Shohag MH, Kozawa K, Kato K, Zhang X, Yura Y, Matsuura Y, Kataoka C, Nishioka T, and Kaibuchi K (2015). Kinase-interacting substrate screening is a novel method to identify kinase substrates. J Cell Biol 209, 895–912. 10.1083/jcb.201412008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cooper JR, and Wordeman L (2009). The diffusive interaction of microtubule binding proteins. Curr Opin Cell Biol 21, 68–73. 10.1016/j.ceb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arias-Romero LE, and Chernoff J (2008). A tale of two Paks. Biol Cell 100, 97–108. 10.1042/BC20070109. [DOI] [PubMed] [Google Scholar]
- 69.Pandey A, Dan I, Kristiansen TZ, Watanabe NM, Voldby J, Kajikawa E, Khosravi-Far R, Blagoev B, and Mann M (2002). Cloning and characterization of PAK5, a novel member of mammalian p21-activated kinase-II subfamily that is predominantly expressed in brain. Oncogene 21, 3939–3948. 10.1038/sj.onc.1205478. [DOI] [PubMed] [Google Scholar]
- 70.Luan Q, Pan L, He D, Gong X, and Zhou H (2018). SC79, the AKT Activator Protects Cerebral Ischemia in a Rat Model of Ischemia/Reperfusion Injury. Med Sci Monit 24, 5391–5397. 10.12659/MSM.910191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jo H, Mondal S, Tan D, Nagata E, Takizawa S, Sharma AK, Hou Q, Shanmugasundaram K, Prasad A, Tung JK, et al. (2012). Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc Natl Acad Sci U S A 109, 10581–10586. 10.1073/pnas.1202810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo X, Snider WD, and Chen B (2016). GSK3beta regulates AKT-induced central nervous system axon regeneration via an eIF2Bepsilon-dependent, mTORC1-independent pathway. Elife 5, e11903. 10.7554/eLife.11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xia B, Liu H, Xie J, Wu R, and Li Y (2015). Akt enhances nerve growth factor-induced axon growth via activating the Nrf2/ARE pathway. Int J Mol Med 36, 1426–1432. 10.3892/ijmm.2015.2329. [DOI] [PubMed] [Google Scholar]
- 74.Cartoni R, Pekkurnaz G, Wang C, Schwarz TL, and He Z (2017). A high mitochondrial transport rate characterizes CNS neurons with high axonal regeneration capacity. PLoS One 12, e0184672. 10.1371/journal.pone.0184672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, and He Z (2008). Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322, 963–966. 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hervera A, De Virgiliis F, Palmisano I, Zhou L, Tantardini E, Kong G, Hutson T, Danzi MC, Perry RB, Santos CXC, et al. (2018). Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat Cell Biol 20, 307–319. 10.1038/s41556-018-0039-x. [DOI] [PubMed] [Google Scholar]
- 77.Gonzalez S, Fernando RN, Perrin-Tricaud C, and Tricaud N (2014). In vivo introduction of transgenes into mouse sciatic nerve cells in situ using viral vectors. Nat Protoc 9, 1160–1169. 10.1038/nprot.2014.073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Ex vivo imaging of mitochondrial transport along the sciatic nerves from a P30 Mito-CFP mouse. Related to Figure 3H. Mitochondrial transport was assessed in sciatic nerve explants after rapid dissection and immediate immersion in prewarmed oxygenated Neurobasal-A medium. Time-lapse images of multiple axons in nerve explants were acquired using a Zeiss 880 Airyscan confocal microscope with a P-Apochromat 40×/1.3 oil objective at 3-sec intervals for a total of 60 frames. Image stacks were processed using ImageJ software.
Video S2. Ex vivo imaging of mitochondrial transport along the sciatic nerves from a P70 Mito-CFP mouse. Related to Figure 3H. Mitochondrial transport was assessed in sciatic nerve explants after rapid dissection and immediate immersion in prewarmed oxygenated Neurobasal-A medium. Time-lapse images of multiple axons in nerve explants were acquired using a Zeiss 880 Airyscan confocal microscope with a P-Apochromat 40×/1.3 oil objective at 3-sec intervals for a total of 60 frames. Image stacks were processed using ImageJ software.
Video S3. Ex vivo imaging of mitochondrial transport along the sciatic nerves from a Mito-CFP mouse injected with AAV9-2A-GFP. Related to Figure 3I. AAV9-2A-GFP was injected into sciatic nerves at the left-side at P45. Two weeks after injection at P59, mitochondrial transport was assessed in sciatic nerve explants after rapid dissection and immediate immersion into prewarmed oxygenated Neurobasal-A medium. Time-lapse images of multiple axons in nerve explants were acquired using a Zeiss 880 Airyscan confocal microscope with a P-Apochromat 40×/1.3 oil objective at 3-sec intervals for a total of 60 frames. Image stacks were processed using ImageJ software.
Video S4. Ex vivo imaging of mitochondrial transport along the sciatic nerves from a Mito-CFP mouse injected with AAV9-caPAK5-2A-GFP. Related to Figure 3I. AAV9-caPAK5-2A-GFP was injected into sciatic nerves at the left-side at P45. Two weeks after injection at P59, mitochondrial transport was assessed in sciatic nerve explants after rapid dissection and immediate immersion into prewarmed oxygenated Neurobasal-A medium. Time-lapse images of multiple axons in nerve explants were acquired using a Zeiss 880 Airyscan confocal microscope with a P-Apochromat 40×/1.3 oil objective at 3-sec intervals for a total of 60 frames. Image stacks were processed using ImageJ software.
Data Availability Statement
This study did not generate any unique datasets or code. Original/source data for figures in the paper are available from the corresponding author on request.