

Abstract
Hypertension is a major cause of cardiovascular morbidity and mortality, despite the availability of antihypertensive drugs with different targets and mechanisms of action. Here, we provide evidence that pharmacological inhibition of TMEM16A (ANO1), a calcium-activated chloride channel expressed in vascular smooth muscle cells, blocks calcium-activated chloride currents and contraction in vascular smooth muscle in vitro and decreases blood pressure in spontaneously hypertensive rats. The acylaminocycloalkylthiophene TMinh-23 fully inhibited calcium-activated TMEM16A chloride current with nanomolar potency in Fischer rat thyroid cells expressing TMEM16A, and in primary cultures of rat vascular smooth muscle cells. TMinh-23 reduced vasoconstriction caused by the thromboxane mimetic U46619 in mesenteric resistance arteries of wild-type and spontaneously hypertensive rats, with a greater inhibition in spontaneously hypertensive rats. Blood pressure measurements by tail-cuff and telemetry showed up to a 45-mmHg reduction in systolic blood pressure lasting for four-six hours in spontaneously hypertensive rats after a single dose of TMinh-23. A minimal effect on blood pressure was seen in wild-type rats or mice treated with TMinh-23. Five-day twice daily treatment of spontaneously hypertensive rats with TMinh-23 produced sustained reductions of 20–25 mmHg in daily mean systolic and diastolic blood pressure. TMinh-23 action was reversible, with blood pressure returning to baseline in spontaneously hypertensive rats by three days after treatment discontinuation. Thus, our studies provide validation for TMEM16A as a target for antihypertensive therapy and demonstrate the efficacy of TMinh-23 as an antihypertensive with a novel mechanism of action.
Keywords: antihypertensives, hypertension, vasodilators
Hypertension, affecting >25% of the global adult population and 1 in 3 US adults, is a major risk factor for coronary artery disease, stroke, heart failure, and renal failure.1,2 Drug treatment of hypertension reduces the probability of cardiovascular events, with more intensive blood pressure (BP) control correlating with reduced cardiovascular morbidity and mortality.1,3 Hypertension remains a major problem despite the availability of antihypertensive drugs with different targets and mechanisms of action. Of patients receiving antihypertensive medications, 12% to 15% have resistant disease and 8% have uncontrolled hypertension despite being treated with ≥3 different classes of antihypertensives.4,5 There is an unmet need for antihypertensive drugs with novel mechanisms of action to better control hypertension and reduce cardiovascular morbidity and mortality.
TMEM16A (transmembrane member 16A or anoctamin1) is a Ca2+-activated Cl− channel expressed in vascular smooth muscle cells,6–9 as well as in airway smooth muscle, gastrointestinal pacemaker cells, and various epithelial cell types.10 Because vascular smooth muscle cells actively accumulate Cl−,11 TMEM16A activation leads to Cl− efflux and membrane depolarization that results in secondary activation of ion channels that modulate vasoconstriction, including voltage-dependent Ca2+ channels (VDCCs).12 TMEM16A knockout in mice decreases in vitro contractile responses to angiotensin II in aortic strips, reduces BP, and attenuates the hypertensive response to angiotensin II.13 In vitro studies using a low-potency TMEM16A inhibitor14 showed relaxation in mouse arteries in response to methoxamine.7 Whether pharmacologic TMEM16A inhibition may be efficacious for treatment of hypertension in vivo has not been investigated.
Our group recently identified, and optimized by medicinal chemistry, TMEM16A inhibitor TMinh-23 (Figure 1a), which showed nanomolar potency and good selectivity and pharmacologic properties.15,16 Herein, we investigated its efficacy in blocking vascular smooth muscle contraction in vitro and reducing BP in a rat model of hypertension.
Figure 1 |. 2-Bromodifluoroacetylamino-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxylic acid o-tolylamide (TMinh-23) pretreatment blocks Ca+2-activated Cl− secretion in Fischer rat thyroid (FRT) cells and rat primary vascular smooth muscle cells.
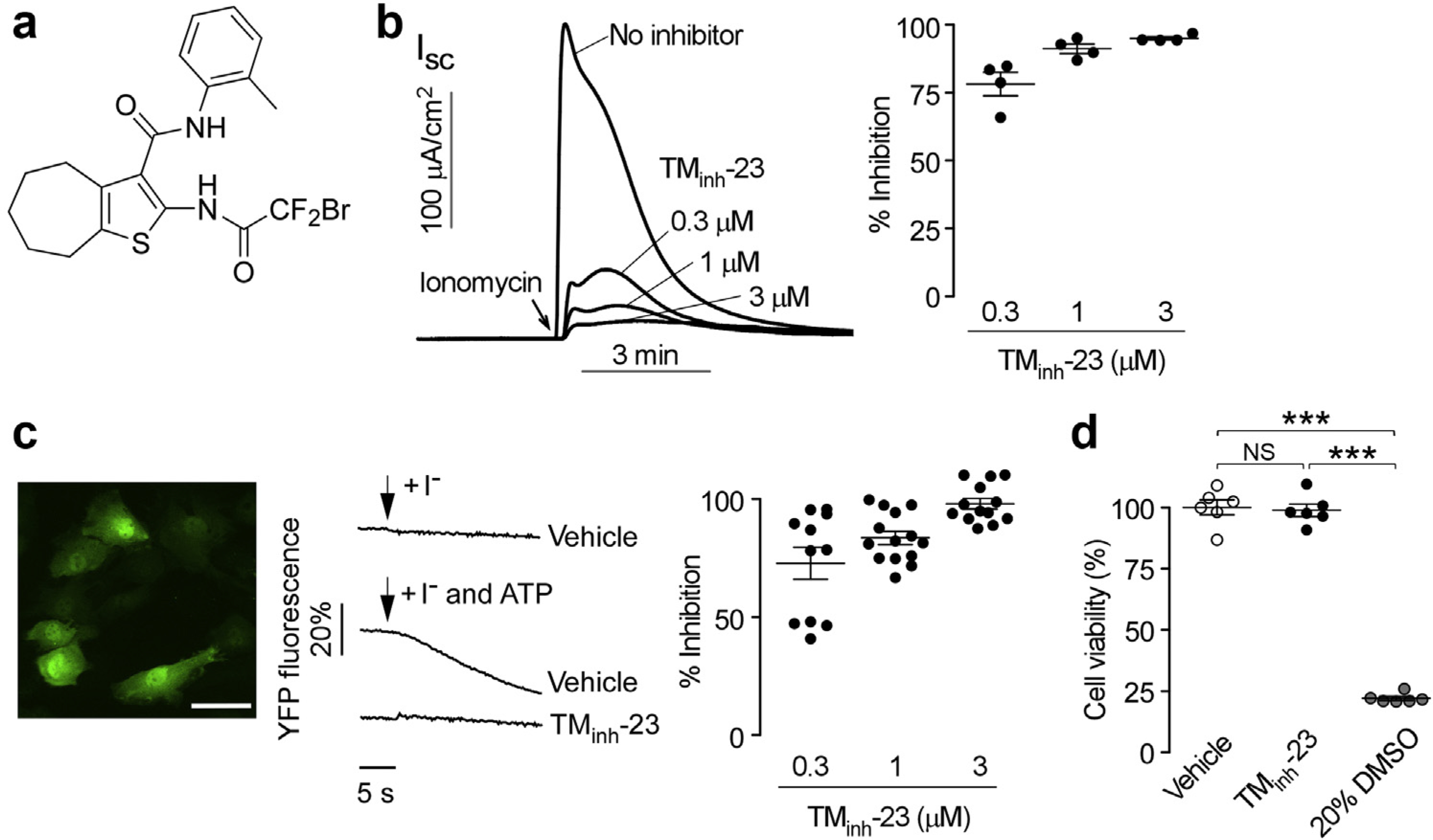
(a) Chemical structure of TMinh-23. (b) FRT cells expressing transmembrane member 16A or anoctamin-1 (TMEM16A) were pretreated with indicated concentrations of TMinh-23 for 10 minutes before the addition of ionomycin (1 μM). Original data are shown on the left, and averaged data (mean ± SEM; n = 4) are shown on the right. (c) (Left) Rat pulmonary artery smooth muscle cells (RPASMCs) transduced with lentivirus to express halide-sensing yellow fluorescent protein (YFP; bar = 50 μm). (Middle) YFP fluorescence quenching in response to the extracellular addition of iodide, with or without adenosine triphosphate (ATP) and 3 μM TMinh-23. (Right) Summary of the percentage inhibition of I- influx by TMinh-23 in RPASMCs (mean ± SEM; n = 11–14). (d) Cell viability assayed by Alamar blue in RPASMCs incubated for 24 hours with vehicle control (0.1% dimethylsulfoxide [DMSO]) or 3 μM TMinh-23. The 20% DMSO was used as positive control. Data are given as mean ± SEM, n = 6 wells per group, 1-way analysis of variance and post hoc Newman-Keuls multiple comparisons test. ***P < 0.001, NS, not significant. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
METHODS
Chemicals
TMinh-23 (2-bromodifluoroacetylamino-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxylic acid o-tolylamide) (Figure 1a, originally named 10bm in a previous study15) was synthesized in 3 steps, as described,15 and had >95% purity determined by high-performance liquid chromatography analysis at 254 nm. Other chemicals were purchased from Sigma-Aldrich, unless indicated otherwise.
Short-circuit current measurements
Short-circuit current measurements were done on Fischer rat thyroid cells stably expressing human TMEM16A. Cells were cultured, as described,15 and grown as tight monolayers on inserts (12-mm diameter, 0.4-μm polyester membrane; Corning Life Sciences). For measurement of short-circuit current, cells were mounted in Ussing chambers and a basolateral-to-apical Cl− gradient was applied, as described.15 Basolateral chambers were filled with bicarbonate-buffered Ringer solution (pH 7.4, in mM: 120 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 D-glucose, 5 HEPES, and 25 NaHCO3). For the apical chamber solution, 120 mM NaCl was replaced by 60 mM NaCl and 60 mM Na-gluconate. The solutions were aerated with 95% O2/5% CO2 and maintained at 37°C. Short-circuit current was measured using an EVC4000 multi-channel voltage clamp (World Precision Instruments) via Ag/AgCl electrodes and 3 M KCl agar bridges. Cells were preincubated with TMinh-23 for 10 minutes before addition of 1 μM ionomycin (Alfa Aesar) to increase cytoplasmic Ca2+ concentration. All compounds were added to both apical and basolateral bathing solutions.
TMEM16A function in rat pulmonary artery smooth muscle cells
Primary cultures of rat pulmonary artery smooth muscle cells (RPASMCs) were purchased from Sigma-Aldrich and cultured according to the manufacturer’s protocols. For experiments, RPASMCs were grown for 48 to 96 hours in a SensoPlate 96 well glass-bottom, black-walled microplate (Greiner Bio-One) and transduced with EYFP-H148Q/I152L/F46L using lentivirus. One day after transduction, RPASMCs were washed twice with phosphate-buffered saline, incubated in 100 ml phosphate-buffered saline, and transferred to Nikon TE2000 microscope (Nikon Inc.). To measure TMEM16A function, cells were imaged for 5 seconds before the addition of 100 μl of phosphate-buffered saline, in which NaCl was replaced by NaI with 100 mM adenosine triphosphate (ATP) to stimulate TMEM16A activity. TMinh-23 inhibition was studied by preincubation with different concentrations of TMinh-23 for 10 minutes before addition of iodide and ATP.
Cellular toxicity
RPASMCs were plated in black-walled, clear-bottom tissue culture plates at a density of 20,000 cells/well. After 48 hours in culture, cells were incubated with 3 μM TMinh-23, 0.1% dimethylsulfoxide (vehicle control), or 20% dimethylsulfoxide (positive control) for 24 hours for assay of cell viability using Alamar blue (ThermoFischer Scientific), according to the manufacturer’s instructions.
Animals
Spontaneously hypertensive rats (SHRs; male and female; aged 16–20 weeks) and wild-type Wistar rats (male and female; aged 16–20 weeks) were purchased from Charles River Laboratories. Wild-type CD1 mice (male and female; aged 10–14 weeks) were bred in house. All animal experiments were approved by the respective Institutional Animal Care and Use Committees at each institution. Rats and mice were housed in communal cages with 12-hour light-dark cycle (light period: 7 am–7 pm). During recording days for telemetry experiments, rats were housed individually with free access to food and water.
Tail-cuff BP measurements
BP was measured by tail cuff in SHRs, wild-type Wistar rats, and wild-type CD1 mice using a Visitech BP-2000 instrument (Visitech Systems Inc.). Animals were acclimatized to tail-cuff measurements by performing daily BP measurements for 5 to 7 days until BP stabilized. After acclimatization, each animal underwent a baseline BP measurement and then was treated with 10 mg/kg TMinh-23 (i.p.; in saline containing 5% dimethylsulfoxide and 10% Kolliphor HS) or vehicle under light isoflurane anesthesia, from which animals recovered quickly (<2 minutes). BP was measured at 15 minutes and 1, 2, and 4 hours after treatments. In SHRs, BP was also measured at 6 and 24 hours after treatment. BP measurements were made in awake animals placed in a restrainer on a heated platform in a quiet room. For each measurement, after 3 initial test measurements (not used for analysis), 20 serial measurements were done and averaged to determine BP. Baseline BP measurements were done at approximately 10:00 am everyday, followed by i.p. injections and subsequent BP measurements.
Telemetry studies
BP and electrocardiogram (ECG) recordings were made in awake, freely moving SHRs and wild-type rats using an implanted radio telemetry device (HD-S11; Data Sciences Intl). The devices were surgically implanted in the abdomen, and the biopotential leads were placed adjacent to the heart in a lead II arrangement, as described.17 The BP catheter was inserted in the abdominal aorta. Data recording began after full recovery via an antenna receiver under the cage interfaced to a computer system. ECG and pressure waveforms were acquired once every minute with the DSI Ponemah V6.2 software (Data Sciences Intl) before, during, and after TMinh-23 or vehicle treatments. Data were subsequently analyzed for arterial BP (systolic, diastolic, and mean), heart rate, and ECG.
After animal recovery and acclimatization for 2 weeks, baseline telemetry recording was done for 1 to 2 days before each experiment. To assess short-term effect of TMinh-23 on BP, SHRs and wild-type rats were treated with a single dose of TMinh-23 (10 mg/kg; i.p.) or vehicle around noon under light isoflurane anesthesia and recordings were made for the next 24 hours. In long-term studies, SHRs were administered 10 mg/kg TMinh-23 twice daily (i.p.; daily injections done approximately at 10 am and 4 pm under light isoflurane anesthesia) for 5 days.
In vitro vascular smooth muscle contraction
Experiments were done on third-order mesenteric arteries from SHRs and wild-type rats or segments of thoracic aorta from wild-type rats (all of Wistar background). Wire or pin myography was used to assess function of mesenteric artery and aorta segments, respectively. After dissection, arterial segments were mounted on tungsten wires in a myograph (Danish MyoTech) in physiological salt solution (pH 7.4), which contained (in mM): 119 NaCl, 4.5 KCl, 1.17 MgSO4, 1.18 NaH2PO4, 25 NaHCO3, 5 glucose, and 1.25 CaCl2. The force elicited by tension of the vessel wall on the wires/pins was recorded using LabChart software (ADInstruments). All vessels were subjected to normalization, where they were incrementally stretched to determine the resting diameter of the vessel lumen under a transmural pressure of 100 mm Hg. The vessels were then set to 90% of this diameter as this was considered the ideal passive stretch for active tension development. Following a 30-minute postnormalization equilibration period, the segments were challenged with physiological salt solution containing 60 mM K+ (replacing Na+) to determine maximum contractility. After washout and restabilization of tension, the concentration-response for U46619 induced contractions was measured. All contraction responses were normalized and reported as percentage contraction induced by physiological salt solution containing 60 mM K+.
Statistical analysis
Experiments with 2 groups were analyzed using Student’s t test; for ≥3 groups, analysis was done with 1-way analysis of variance and post hoc Newman-Keuls multiple comparisons test. P < 0.05 was considered statistically significant.
RESULTS
TMinh-23 inhibition of Ca2+-activated TMEM16A Cl− conductance
As reported previously, the small-molecule inhibitor TMinh-23 (Figure 1a) is a chemically stable compound with drug-like physicochemical properties, which inhibited ATP-activated TMEM16A Cl− conductance with nanomolar potency.15 To confirm that TMEM16A inhibition by TMinh-23 is independent of purinergic signaling, short-circuit current measurements of TMEM16A Cl− conductance were done in Fischer rat thyroid cells expressing TMEM16A in response to elevation of intracellular Ca2+ concentration by the calcium ionophore ionomycin. TMinh-23 pretreatment largely blocked Cl− secretion at 300 nM with near complete inhibition at 1 and 3 μM (Figure 1b). The effects of TMinh-23 were also investigated in primary cultures of rat vascular smooth muscle cells that were transduced with a halide-sensing yellow fluorescent protein using lentivirus. Yellow fluorescent protein fluorescence decreased in response to an I– gradient when in the presence of the Ca2+ agonist ATP (Figure 1c). TMinh-23 pretreatment inhibited the ATP-stimulated I− entry, with complete inhibition at 3 μM. To rule out TMinh-23 toxicity in these experiments, Alamar blue assay showed no effect of 24-hour incubation with 3 μM TMinh-23 on the viability of primary rat vascular smooth muscle cells (Figure 1d). Prior pharmacokinetics measurements in mice showed sustained therapeutic TMinh-23 serum concentrations for at least 4 hours following a single i.p. dose at 10 mg/kg.16 Preliminary studies in rats showed a similar TMinh-23 pharmacokinetic profile following a single i.p. dose at 10 mg/kg (data not shown). These studies provided a rational basis for compound dosing in the animal experiments below.
TMinh-23 efficacy in reducing BP, as measured by tail cuff
To investigate TMinh-23 action on BP, initial measurements were made by tail cuff in SHRs and wild-type rats, as well as in wild-type mice. In SHRs, TMinh-23 at 10 mg/kg administered i.p. produced up to an ~45–mm Hg decrease in systolic BP (SBP) compared with vehicle control, with peak response at 120 minutes after injection (Figure 2a, left). SBP was significantly reduced in SHRs for at least 6 hours after treatment and returned to baseline by 24 hours, indicating reversibility of the TMinh-23 effect. There was also a trend in reduction of diastolic BP (DBP) with TMinh-23 treatment (Figure 2a, right), although the differences were not statistically significant, in part because of large variability in DBP, as the tail-cuff method can have limited accuracy in measuring DBP.18,19
Figure 2 |. Single-dose 2-bromodifluoroacetylamino-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxylic acid o-tolylamide (TMinh-23) lowers blood pressure (BP) in spontaneously hypertensive rats (SHRs), but not in wild-type (WT) rats or mice.
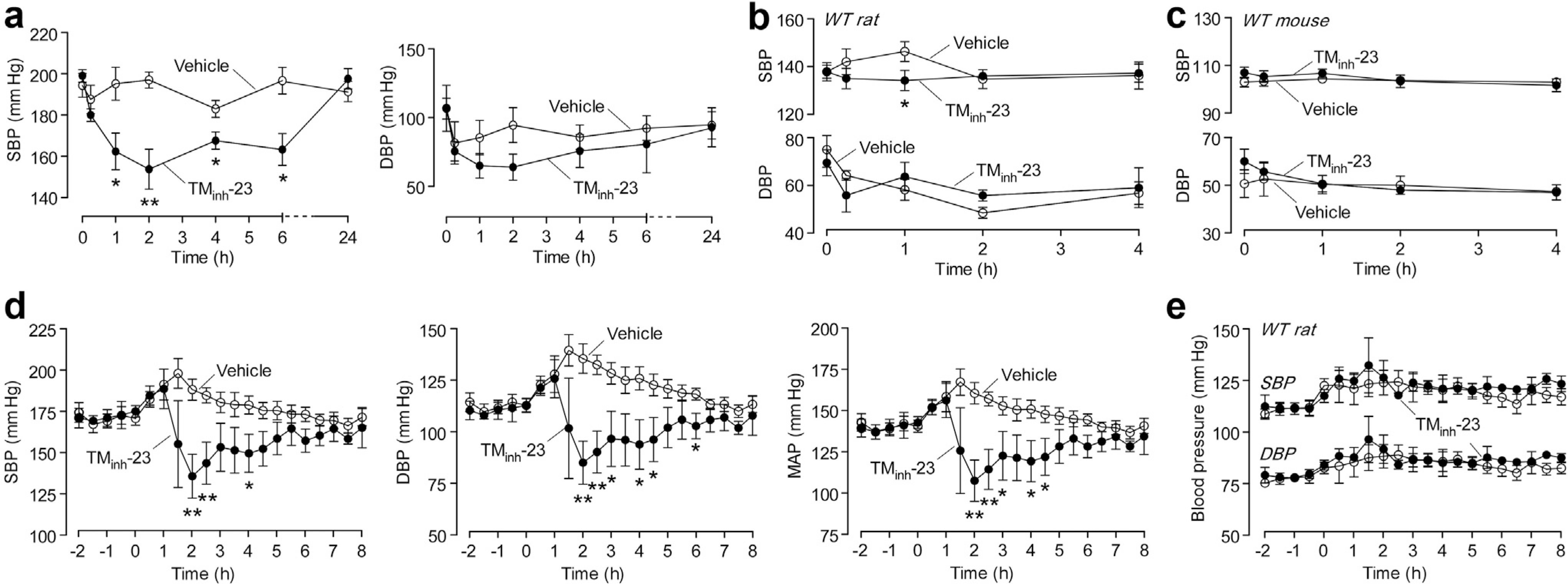
(a) Tail-cuff systolic blood pressure (SBP; left) and diastolic blood pressure (DBP; right) in SHRs administered TMinh-23 or vehicle at time 0 (mean ± SEM; n = 5 rats per group). (b) Tail-cuff BP in WT Wistar rats administered TMinh-23 (10 mg/kg; i.p.) or vehicle at time 0 (mean ± SEM; n = 6 rats per group). (c) Tail-cuff BP in WT CD1 mice administered TMinh-23 (10 mg/kg; i.p.) or vehicle at time 0 (mean ± SEM; n = 3 mice per group). (d) Telemetry BP in SHRs administered TMinh-23 or vehicle at time 0 (mean ± SEM; n = 3–6 rats per group). (e) Telemetry BP in WT Wistar rats administered TMinh-23 or vehicle at time 0 (mean ± SEM; n = 3 rats per group). The Student’s t test was used. *P < 0.05, **P < 0.01 compared with the vehicle group; all other comparisons between groups are not significant. Each data point represents the average of all measurements in the preceding 30-minute period in panels (d) and (e). MAP, mean arterial pressure.
In vehicle-treated wild-type rats, there was a small increase in SBP of ~9 mm Hg at 60 minutes after injection (Figure 2b), which may be due to distress and/or pain related to anesthesia, i.p. injections, and restraining. TMinh-23 slightly lowered SBP at 60 minutes compared with vehicle treatment (P = 0.04), with no effect on DBP. In wild-type mice, TMinh-23 did not significantly affect SBP or DBP (Figure 2c).
Pharmacodynamics of single-dose TMinh-23 in reducing BP, as measured by telemetry
As the tail-cuff method has recognized limitations, it is generally considered as a screening method suitable for detection of relatively large BP changes. Telemetry is the gold standard method for continuous measurement of BP in freely moving rodents, providing unambiguous data without significant confounding variables, such as restraining.20
Telemetric monitoring was done in freely moving SHRs and wild-type rats to assess TMinh-23 pharmacodynamics. After acclimatization and baseline BP measurement, rats were administered a single dose of TMinh-23 (10 mg/kg; i.p.) or vehicle. In SHRs, TMinh-23 produced substantial drops in SBP, DBP, and mean arterial pressure (MAP) compared with vehicle treatment, with peak effect at 120 minutes (Figure 2d), in general agreement with the tail-cuff studies. BP values returned to baseline levels within 6 to 8 hours after treatment. In agreement with results using the tail-cuff method, TMinh-23 had little or no effect on BP in wild-type rats (Figure 2e).
Long-term TMinh-23 treatment in SHRs causes sustained reduction in BP
In long-term studies, SHRs treated with 10 mg/kg TMinh-23 (i.p.) twice daily had sustained reductions in BP that returned to baseline levels at 3 to 4 days after discontinuation of treatment (raw data presented in Figure 3). As can be seen in the raw data, there was significant physiological diurnal variation in BP, which was analyzed by calculating average daily BP (Figure 4a). Further analysis also calculated daily daytime (7 am–7 pm) and nighttime (7 pm–7 am) average BP (Figure 4b). TMinh-23 decreased daily mean SBP, DBP, and MAP by 20 to 25 mm Hg during the 5-day treatment period, with gradual return to baseline values over 3 to 4 days after treatment cessation (Figure 4a). TMinh-23 reduced both average daytime and nighttime BP (Figure 4b), indicating sustained antihypertensive efficacy.
Figure 3 |. Long-term 2-bromodifluoroacetylamino-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxylic acid o-tolylamide (TMinh-23) treatment causes sustained decreases in blood pressure (BP) in spontaneously hypertensive rats (SHRs).
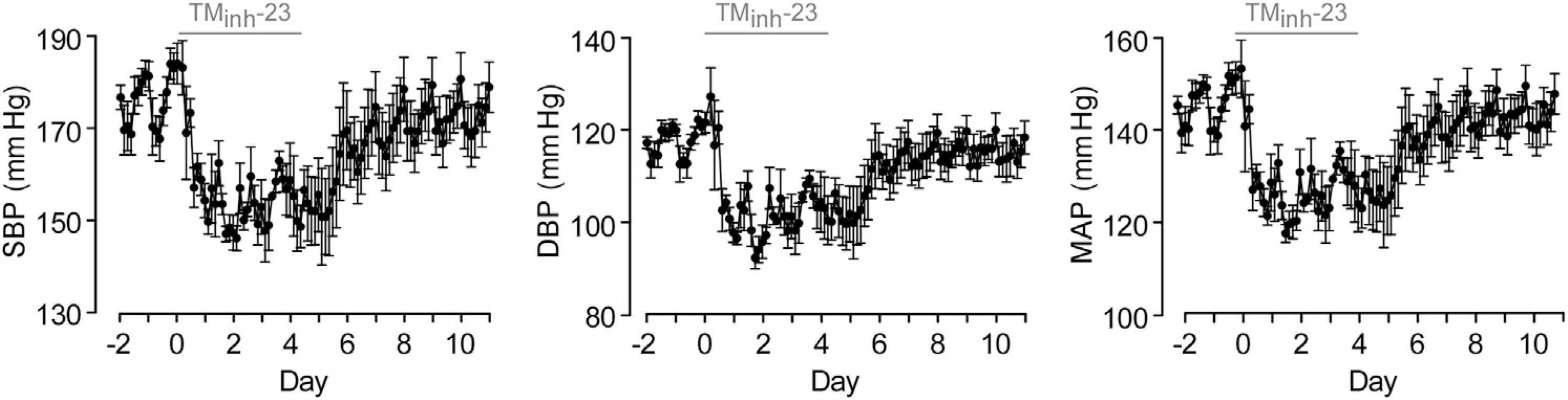
Telemetry BP in SHRs with twice daily TMinh-23 (10 mg/kg; i.p.) treatment for 5 days, starting on day 0. The TMinh-23 treatment period is shown as a horizontal gray bar above each graph. Each data point is the average of all measurements in the preceding 3-hour period (mean ± SEM; n = 4 rats). DBP, diastolic blood pressure; MAP, mean arterial pressure; SBP, systolic blood pressure.
Figure 4 |. Long-term 2-bromodifluoroacetylamino-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxylic acid o-tolylamide (TMinh-23) treatment causes sustained decreases in 24-hour average, daytime, and nighttime blood pressure (BP) in spontaneously hypertensive rats (SHRs).
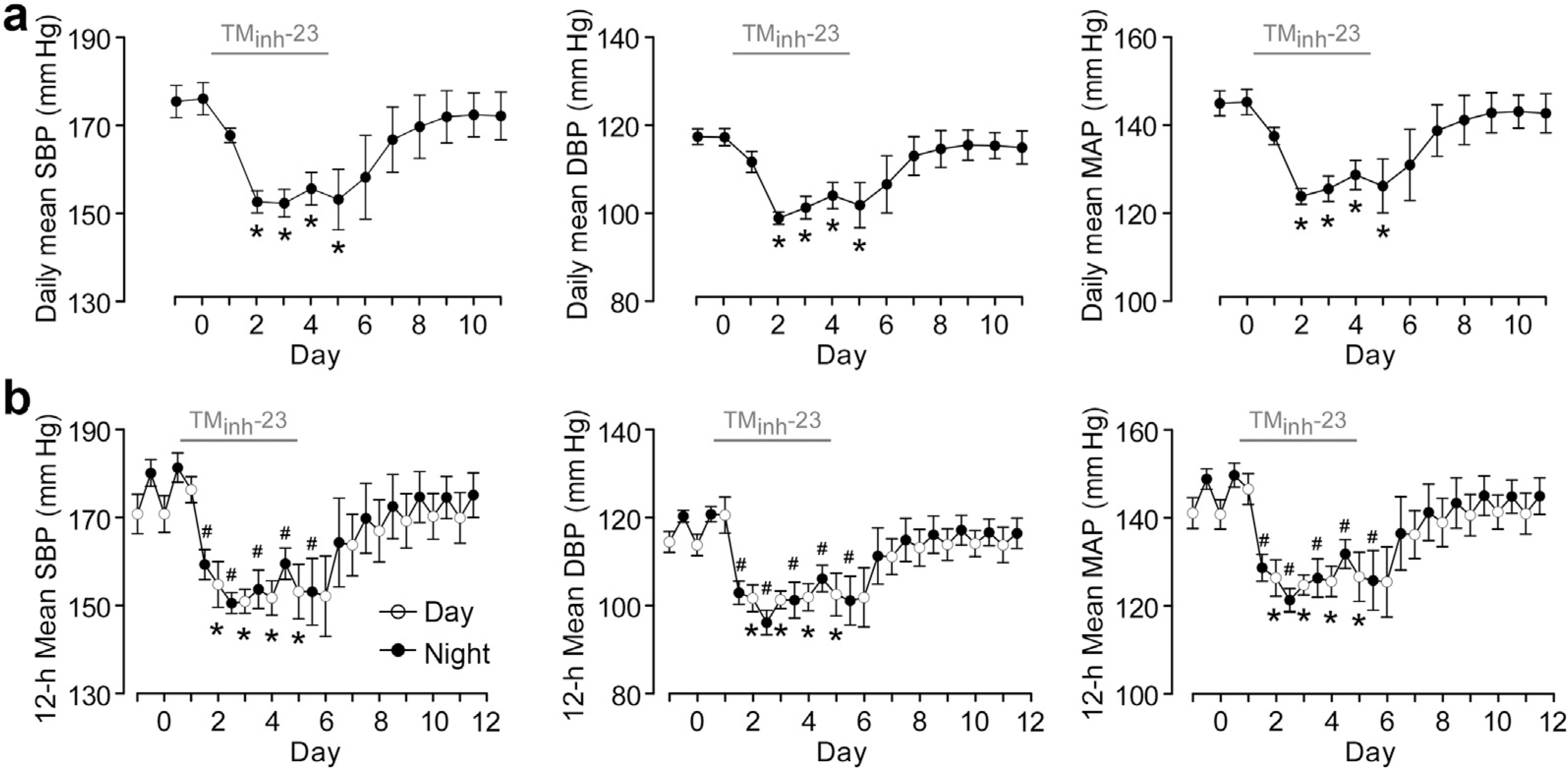
(a) Daily mean BP (measured by telemetry) in SHRs with twice daily TMinh-23 (10 mg/kg; i.p.) treatment for 5 days, starting on day 0 (mean ± SEM; n = 4 rats), Student’s t test, *P < 0.05 compared with day 0. (b) Diurnal variation in blood pressure in SHRs. Open circles show average daytime BP (7 am–7 pm), and closed circles represent average nighttime BP (7 pm–7 am). The TMinh-23 treatment period is shown as a horizontal gray bar above each graph (mean ± SEM; n = 4 rats). Student’s t test was used. *P < 0.05 compared with daytime BP on day 0; #P < 0.05 compared with nighttime BP on day 0. DBP, diastolic blood pressure; MAP, mean arterial pressure; SBP, systolic blood pressure.
The effect of TMinh-23 treatment on heart rate and ECG was also studied. Long-term TMinh-23 treatment resulted in increased heart rate during the first 2 to 3 days of the 5-day treatment period, which then returned to near baseline values (Figure 5a, left). The daily average heart rate after treatment cessation was slightly lower than baseline values (Figure 5a, middle), which was primarily due to slight reduction in nocturnal heart rate (Figure 5a, right). ECG analysis showed slightly reduced PR interval, QRS duration, and QT interval at day 2 of TMinh-23 treatment, as expected from the increased heart rate. Each ECG parameter returned to baseline after treatment cessation (Figure 5b).
Figure 5 |. Heart rate (HR) and electrocardiogram (ECG) with long-term 2-bromodifluoroacetylamino-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxylic acid o-tolylamide (TMinh-23) treatment in spontaneously hypertensive rats (SHRs).
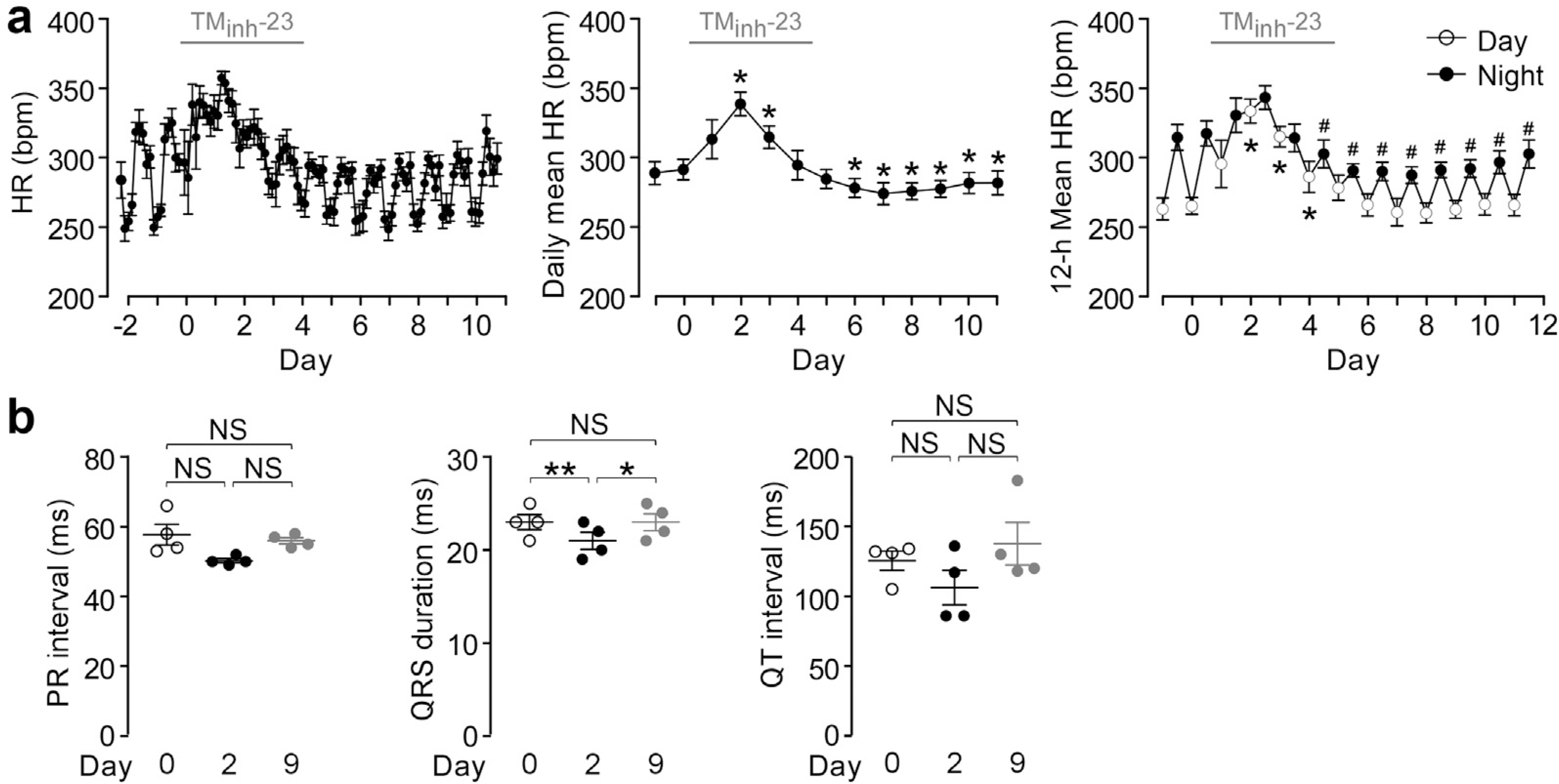
(a) (Left) HR in SHRs with twice daily TMinh-23 (10 mg/kg; i.p.) treatment for 5 days, starting on day 0. Each data point is the average of all measurements in the preceding 3-hour period. (Middle) Daily mean HR in SHRs. *P < 0.05 compared with day 0. (Right) Diurnal variation in HR in SHRs. Open circles show average daytime (7 am–7 pm) HR, and closed circles represent average nighttime (7 pm–7 am) HR (mean ± SEM; n = 4 rats). Student’s t test was used. *P < 0.05 compared with average daytime HR on day 0; #P < 0.05 compared with average nighttime HR on day 0. The TMinh-23 treatment period is shown as a horizontal gray bar above each graph. (b) ECG parameters in SHRs with twice daily TMinh-23 (10 mg/kg; i.p.) treatment for 5 days, starting on day 0 (mean ± SEM; n = 4 rats). One-way analysis of variance with post hoc Newman-Keuls multiple comparison test was used. *P < 0.05, **P < 0.01. Bpm, beats per minute; NS, not significant.
TMinh-23 inhibits vascular smooth muscle contraction in ex vivo rat resistance arteries
To investigate the antihypertensive mechanism of TMinh-23, isometric vascular smooth muscle contractions in response to a vasoconstrictor (thromboxane mimetic U46619) were studied in mesenteric resistance arteries, which are critical for BP regulation. TMinh-23 largely, but incompletely, inhibited the U46619-induced vasoconstriction in mesenteric resistance arteries, even at 3 μM (Figure 6a and b). TMinh-23 effect was also investigated on contractions induced by high K+, which is thought to depolarize vascular smooth muscle cells independent of calcium-activated chloride channels (CaCCs) and cause vasoconstriction by activating VDCCs. The high K+ response in mesenteric arteries was biphasic, with an initial peak followed by a smaller plateau (Figure 6a). Interestingly, the high K+ response was also inhibited by TMinh-23 (Figure 6a and c). The TMinh-23 effect was reversible, as seen from the return of high K+ response to pretreatment values after washout (Figure 6a, bottom trace).
Figure 6 |. 2-Bromodifluoroacetylamino-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxylic acid o-tolylamide (TMinh-23) blocks vascular smooth muscle contractions in mesenteric resistance arteries of wild-type rats.
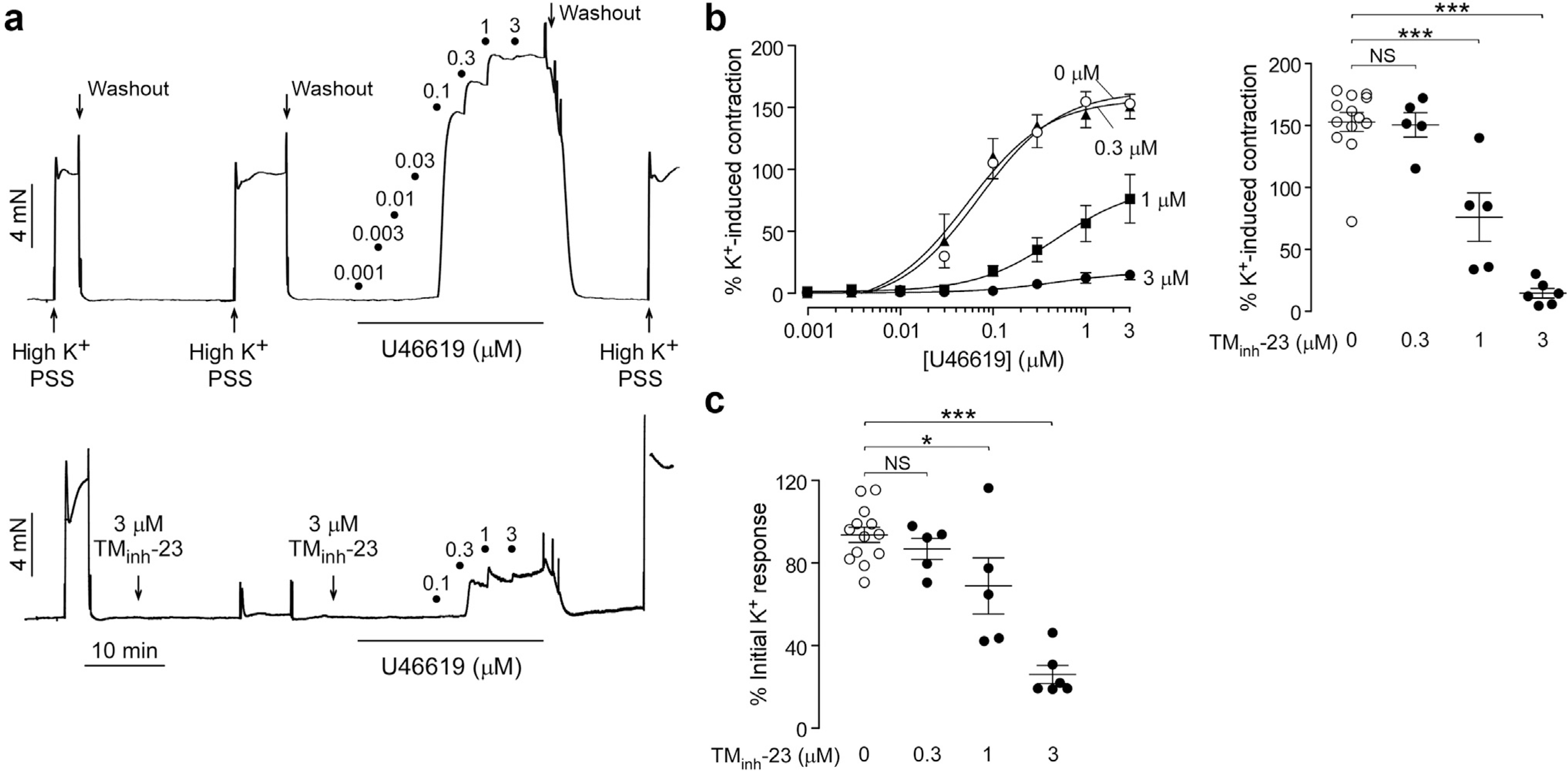
(a) Representative traces showing isometric contractions in response to high K+ (60 mM) and increasing concentrations of U46619 in rat mesenteric arteries treated with vehicle (0.1% dimethylsulfoxide; top) or 3 μM TMinh-23 (bottom). (b) (Left) U46619 concentration-response curves at indicated concentrations of TMinh-23. (Right) Maximum U46619 response in the presence of different concentrations of TMinh-23. (c) High K+-induced peak contraction responses as a function of TMinh-23 concentration. N = 5 to 13 experiments per group; 1-way analysis of variance with post hoc Newman-Keuls multiple comparison test was used. *P < 0.05, ***P < 0.001. NS, not significant; PSS, physiological salt solution.
Contractions were also measured in mesenteric resistance arteries from SHRs. TMinh-23 completely inhibited maximum U46619-induced vasoconstriction in SHR mesenteric arteries at 3 μM (Figure 7a and b). Consistent with the greater BP-lowering effect in vivo in SHRs, TMinh-23 inhibited maximum U46619-induced contractions significantly more in mesenteric arteries from SHRs than wild-type rats (Figure 7c, left). Contractions induced by lower U46619 concentrations (100 nM) in SHR arteries were even more sensitive to TMinh-23, compared with wild-type rats (Figure 7c, right). As seen in wild-type rats, TMinh-23 also inhibited high K+-induced vasoconstriction in SHR mesenteric arteries (Figure 7d).
Figure 7 |. 2-Bromodifluoroacetylamino-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxylic acid o-tolylamide (TMinh-23) blocks vascular smooth muscle contractions in mesenteric resistance arteries of spontaneously hypertensive rats (SHRs).
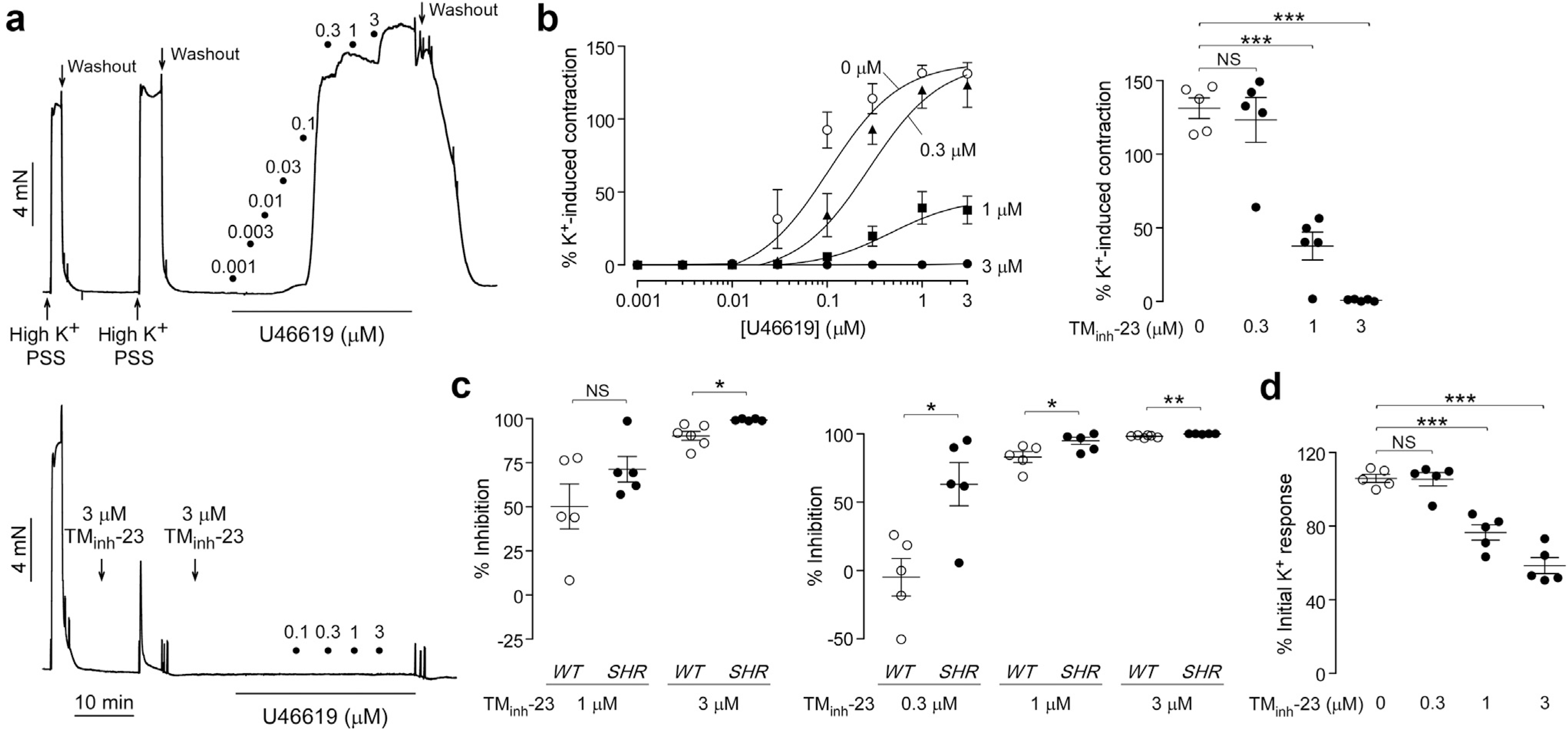
(a) Representative traces showing isometric contractions in response to high K+ (60 mM) and increasing concentrations of U46619 in mesenteric arteries from SHRs treated with vehicle (0.1% dimethylsulfoxide; top) or 3 μM TMinh-23 (bottom). (b) (Left) U46619 concentration-response curves at indicated concentrations of TMinh-23. (Right) Maximum U46619 response in the presence of indicated concentrations of TMinh-23. (c) (Left) Percentage inhibition of maximal U46619 response in the presence of 1 or 3 μM TMinh-23 in wild-type (WT) and SHR mesenteric arteries. (Right) Percentage inhibition of the 100 nM U46619 response in the presence of TMinh-23 in WT and SHR mesenteric arteries. (d) High K+-induced peak contraction responses as a function of TMinh-23 concentration. N = 5 to 6 experiments per group; Student’s t test was used for (c), and 1-way analysis of variance with post hoc Newman-Keuls multiple comparison test was used for (b) and (d). *P < 0.05, **P < 0.01, and ***P < 0.001. NS, not significant; PSS, physiological salt solution.
TMinh-23 has minimal effect on ex vivo rat aorta smooth muscle contraction
In contrast to the results in rat resistance arteries, in aorta, TMinh-23 pretreatment slightly reduced U46619-induced contractions (by ~20%) even at a high concentration of 3 μM (Figure 8a and b; P = 0.14). TMinh-23 pretreatment had no effect on high K+-induced vasoconstriction in aorta (Figure 8a and c). These findings suggest that TMEM16A inhibition preferentially prevents vasoconstriction in rat resistance arteries but not in aorta, noting that the latter have minimal contribution to BP.
Figure 8 |. 2-Bromodifluoroacetylamino-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxylic acid o-tolylamide (TMinh-23) effect on vascular smooth muscle contractions in wild-type rat aorta.
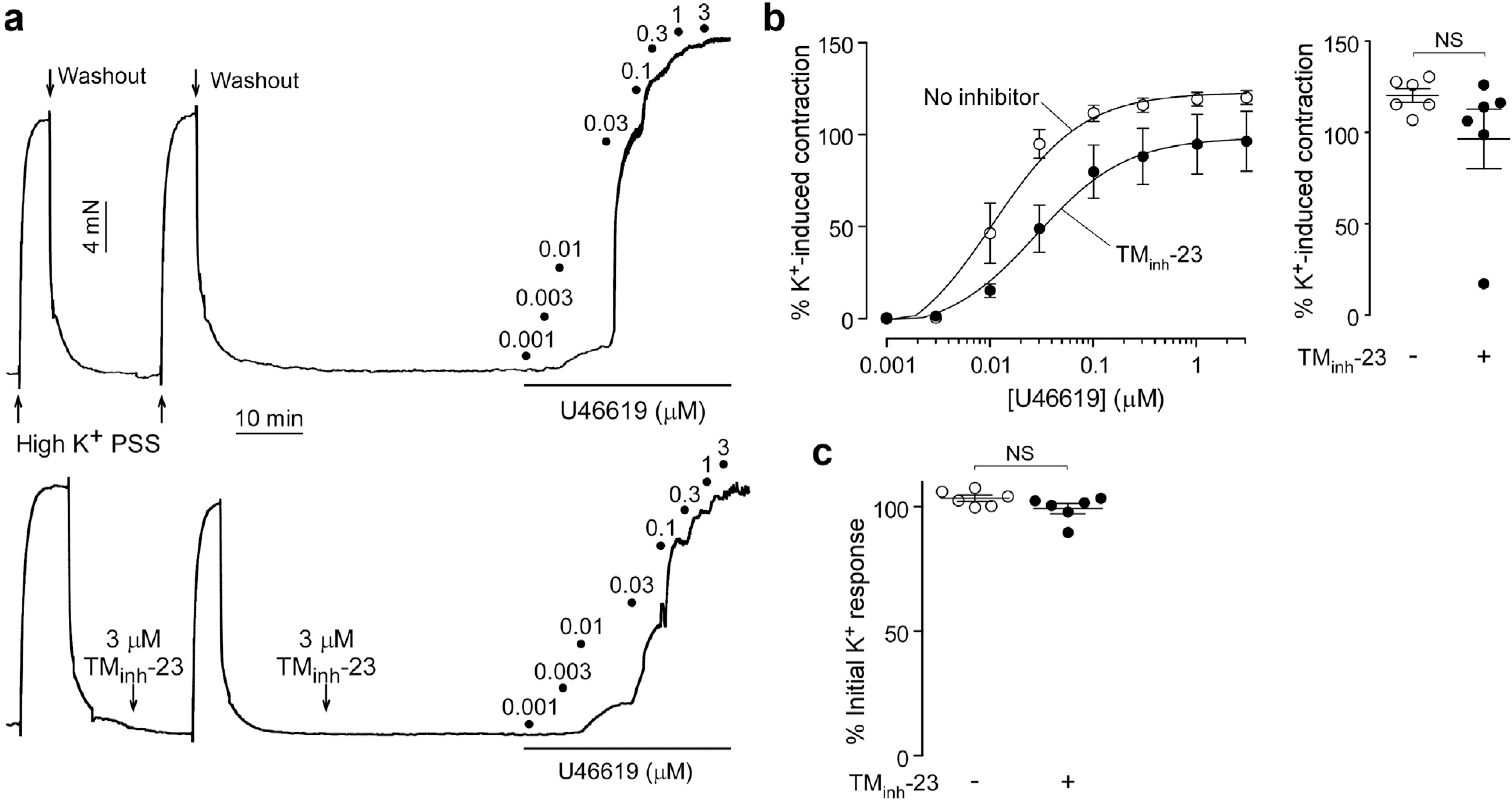
(a) Representative traces showing isometric contractions in response to high K+ (60 mM) and U46619 in rat aorta treated with vehicle (top) or 3 μM TMinh-23 (bottom). (b) (Left) U46619 concentration-response curves in the absence or presence of 3 μM TMinh-23. (Right) Maximum U46619 response in the absence or presence of 3 μM TMinh-23. (c) High K+-induced peak contraction in the absence or presence of 3 μM TMinh-23. N = 6 experiments per group; Student’s t test was used. NS, not significant; PSS, physiological salt solution.
DISCUSSION
TMEM16A inhibition by a small-molecule drug candidate markedly reduced BP in a rat model of hypertension, offering proof of concept for pharmacologic inhibition of TMEM16A by a small molecule for antihypertensive therapy with a novel mechanism of action. An interesting observation was the large reductions in BP of SHRs with TMEM16A inhibition, although there was minimal effect in wild-type rats and mice. This differential effect may in part be a consequence of differences in TMEM16A expression and/or activity between SHRs and wild-type rodents. A recent study showed 4-fold increased TMEM16A expression in smooth muscle cells of coronary arteries in SHRs compared with wild-type rats.21 Herein, we found that mesenteric arteries from SHRs were considerably more sensitive to TMEM16A inhibition than arteries from wild-type rats, which may explain the greater in vivo BP-lowering efficacy of TMinh-23 in SHRs versus wild-type rats. Another explanation for the greater BP-lowering effect of TMinh-23 in SHRs may be the markedly increased peripheral vascular resistance in SHRs compared with wild-type rats.22
TMEM16A expression in vascular smooth muscle is species and artery dependent. In mice, TMEM16A is strongly expressed in aorta, with some studies reporting relatively little expression in small resistance arteries,6,13,23 whereas others reported comparable TMEM16A expression in aorta and small mesenteric resistance arteries.7 In rats, TMEM16A is expressed throughout the arterial system, including small resistance arteries.21,24 We found herein that TMEM16A inhibition by a selective small-molecule inhibitor largely blocked vascular smooth muscle contraction in rat mesenteric resistance arteries, suggesting TMEM16A as the major CaCC in these vessels. Interestingly, TMEM16A inhibition had limited effect on vasoconstriction in rat aorta, which might be due to the presence of alternative, non-TMEM16A CaCC(s) in this segment. This idea is supported by an earlier study showing 50% reduction in norepinephrine-induced vasoconstriction in rat aortic strips using the nonselective CaCC inhibitor niflumic acid.25 Because resistance arteries are key regulators of vascular tone and peripheral arterial resistance,26 our findings suggest that the mechanism of antihypertensive action by TMinh-23 is likely vasodilatation in resistance arteries. Similar to rats, TMEM16A is expressed in small resistance arteries of humans,7 supporting the potential antihypertensive efficacy of TMEM16A inhibition in humans.
Elevation in extracellular K+ results in vascular smooth muscle cell depolarization, which opens VDCCs and causes vasoconstriction. This action has been presumed to be independent of CaCCs. We found that TMinh-23 inhibited high K+-induced vasoconstriction responses in rat mesenteric resistance arteries, but not aorta. Similarly, earlier studies reported 40% to 60% lower high K+-induced contraction responses in rat coronary arteries in the presence of other small-molecule TMEM16A inhibitors21 or in rat mesenteric arteries with TMEM16A knockdown.27 Consistent with lack of TMinh-23 effect in rat aorta herein, previous studies showed no effect of the nonselective CaCC inhibitor niflumic acid on high K+-induced vasoconstriction in rat aortic strips.25 Our findings, together with these earlier studies, suggest a potential functional interplay between TMEM16A and VDCCs in rat resistance arteries but not aorta. There is now evidence that the TMEM16A protein may directly alter VDCC activity, as vascular L-type Ca+2 channels were downregulated by TMEM16A small interfering RNA knockdown or in a constitutive mouse model.23,27 TMEM16A has also been implicated in angiotensin II–mediated vascular effects through increased Rho kinase activity in cerebral arteries.28 TMEM16A inhibition might therefore exert its antihypertensive effects by several different mechanisms in addition to inhibition of Cl− conductance.
TMinh-23 treatment in SHRs produced a transient increase in heart rate, which is likely due to the baroreceptor reflex in response to BP reduction, as reported for other vasodilators, such as calcium channel blockers.29 Earlier studies reported expression of TMEM16A and other putative CaCCs in mouse ventricle30 and human left ventricular cardiomyocytes.31 CaCC inhibition by the nonselective compound 9-anthracene carboxylic acid reduced phase 1 repolarization of the action potential in isolated canine cardiomyocytes, with increased incidence of early after-depolarizations,32 suggesting that CaCC currents might have antiarrhythmic effects. In contrast, earlier TMEM16A inhibitors had no effect on cardiac parameters whilst they increased coronary blood flow in isolated Langendorff experiments.21 Herein, we found that 5-day TMinh-23 treatment did not produce ECG changes other than the expected shortened intervals associated with increased heart rate. Future studies investigating the potential cardiac effects of TMEM16A inhibitors might be informative.
TMEM16A inhibition is a unique antihypertensive mechanism that targets the initial depolarization of vascular smooth muscle cells in response to intracellular Ca+2 elevation. TMEM16A inhibitors can thus potentially have additive effect with other vasodilatory antihypertensives, such as Ca+2 channel blockers, which target downstream mechanisms. In addition, the rapid onset of TMinh-23 action makes TMEM16A inhibitors potentially suitable for treatment of hypertensive emergencies in the hospital setting.
Several limitations of the study herein are noted. Although the data support TMEM16A inhibition to relax mesenteric resistance arteries, possible regional differences in the vasorelaxant response in the systemic circulation remain unknown. Of note, we found vasorelaxant effects of TMinh-23 in rat pulmonary arteries as well, which suggests the potential utility of TMEM16A inhibitors in pulmonary hypertension, because increased TMEM16A expression and activity appear to be an important pathologic mechanism in this condition.33 As TMEM16A is expressed in various tissues in addition vascular smooth muscle, off-target effects of TMEM16A inhibition are possible, depending on inhibitor tissue distribution. Data in knockout or inhibitor-treated mice suggest possible actions of TMEM16A inhibition on airway smooth muscle to cause bronchodilation,34 on gastrointestinal pacemaker cells to inhibit gastric motility,16 and on secretory epithelia to reduce saliva35 and tear fluid secretion.36 Although these off-target actions may be of lesser concern for short-term TMEM16A inhibition therapy of hypertensive emergencies, they warrant careful evaluation for long-term therapy. Further medicinal chemistry (e.g., prodrug strategies) may promote preferential inhibitor targeting to vascular smooth muscle.
In conclusion, the TMEM16A inhibitor TMinh-23 was efficacious in reducing BP in a rat model of hypertension. This compound, or alternative TMEM16A inhibitors, may be useful for treatment of hypertension as first-in-class antihypertensives with a novel mechanism of action.
Translational Statement.
Hypertension is a major risk factor for cardiovascular diseases and renal failure. Despite the availability of antihypertensive drugs with different mechanisms of action, hypertension remains a significant clinical problem. There is an unmet need for novel antihypertensives to control blood pressure, particularly for patients with resistant hypertension. Herein, we characterize and show proof-of-concept efficacy of a small-molecule inhibitor of vascular smooth muscle calcium-activated chloride channel TMEM16A (transmembrane member 16A or anoctamin-1). TMEM16A inhibitor prevented vasoconstriction in isolated arteries, reduced blood pressure in spontaneously hypertensive rats, and had a sustained antihypertensive effect with long-term administration. TMEM16A inhibition is a novel approach for the treatment of hypertension.
ACKNOWLEDGEMENTS
This work was supported by grants from the American Heart Association (18POST33990365 and Stephanie Watts Career Development Award) and the National Institutes of Health (DK126070 and DK072517).
Footnotes
DISCLOSURE
All the authors declared no competing interests.
REFERENCES
- 1.James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311:507–520. [DOI] [PubMed] [Google Scholar]
- 2.Vital signs: awareness and treatment of uncontrolled hypertension among adults - United States, 2003–2010. MMWR Morb Mortal Wkly Rep. 2012;61:703–709. [PubMed] [Google Scholar]
- 3.The SPRINT Research Group. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Persell SD. Prevalence of resistant hypertension in the United States, 2003–2008. Hypertension. 2011;57:1076–1080. [DOI] [PubMed] [Google Scholar]
- 5.Sim JJ, Bhandari SK, Shi J, et al. Characteristics of resistant hypertension in a large, ethnically diverse hypertension population of an integrated health system. Mayo Clin Proc. 2013;88:1099–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis AJ, Forrest AS, Jepps TA, et al. Expression profile and protein translation of TMEM16A in murine smooth muscle. Am J Physiol Cell Physiol. 2010;299:C948–C959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis AJ, Shi J, Pritchard HAT, et al. Potent vasorelaxant activity of the TMEM16A inhibitor T16A(inh) -A01. Br J Pharmacol. 2013;168:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manoury B, Tamuleviciute A, Tammaro P. TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J Physiol. 2010;588:2305–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas-Gatewood C, Neeb ZP, Bulley S, et al. TMEM16A channels generate Ca2⁺-activated Cl− currents in cerebral artery smooth muscle cells. Am J Physiol Heart Circ Physiol. 2011;301:H1819–H1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamaleddin MA. Molecular, biophysical, and pharmacological properties of calcium-activated chloride channels. J Cell Physiol. 2018;233:787–798. [DOI] [PubMed] [Google Scholar]
- 11.Chipperfield AR, Harper AA. Chloride in smooth muscle. Prog Biophys Mol Biol. 2000;74:175–221. [DOI] [PubMed] [Google Scholar]
- 12.Leblanc N, Ledoux J, Saleh S, et al. Regulation of calcium-activated chloride channels in smooth muscle cells: a complex picture is emerging. Can J Physiol Pharmacol. 2005;83:541–556. [DOI] [PubMed] [Google Scholar]
- 13.Heinze C, Seniuk A, Sokolov MV, et al. Disruption of vascular Ca2+− activated chloride currents lowers blood pressure. J Clin Invest. 2014;124: 675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286:2365–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Truong EC, Phuan PW, Reggi AL, et al. Substituted 2-acylaminocycloalkylthiophene-3-carboxylic acid arylamides as inhibitors of the calcium-activated chloride channel transmembrane protein 16A (TMEM16A). J Med Chem. 2017;60:4626–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cil O, Anderson MO, Yen R, et al. Slowed gastric emptying and improved oral glucose tolerance produced by a nanomolar-potency inhibitor of calcium-activated chloride channel TMEM16A. FASEB J. 2019;33:11247–11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jordan MC, Zheng Y, Ryazantsev S, et al. Cardiac manifestations in the mouse model of mucopolysaccharidosis I. Mol Genet Metab. 2005;86: 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng M, Whitesall S, Zhang Y, et al. Validation of volume–pressure recording tail-cuff blood pressure measurements. Am J Hypertens. 2008;21:1288–1291. [DOI] [PubMed] [Google Scholar]
- 19.Zhao X, Ho D, Gao S, et al. Arterial pressure monitoring in mice. Curr Protoc Mouse Biol. 2011;1:105–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luft FC. Men, mice, and blood pressure: telemetry? Kidney Int. 2019;96: 31–33. [DOI] [PubMed] [Google Scholar]
- 21.Askew Page HR, Dalsgaard T, Baldwin SN, et al. TMEM16A is implicated in the regulation of coronary flow and is altered in hypertension. Br J Pharmacol. 2019;176:1635–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mulvany MJ. Do resistance vessel abnormalities contribute to the elevated blood pressure of spontaneously-hypertensive rats? a review of some of the evidence. Blood Vessels. 1983;20:1–22. [DOI] [PubMed] [Google Scholar]
- 23.Jensen AB, Joergensen HB, Dam VS, et al. Variable contribution of TMEM16A to tone in murine arterial vasculature. Basic Clin Pharmacol Toxicol. 2018;123:30–41. [DOI] [PubMed] [Google Scholar]
- 24.Wang B, Li C, Huai R, et al. Overexpression of ANO1/TMEM16A, an arterial Ca+2-activated Cl- channel, contributes to spontaneous hypertension. J Mol Cell Cardiol. 2015;82:22–32. [DOI] [PubMed] [Google Scholar]
- 25.Criddle DN, de Moura RS, Greenwood IA, et al. Effect of niflumic acid on noradrenaline-induced contractions of the rat aorta. Br J Pharmacol. 1996;118:1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tykocki NR, Boerman EM, Jackson WF. Smooth muscle ion channels and regulation of vascular tone in resistance arteries and arterioles. Compr Physiol. 2017;7:485–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dam VS, Boedtkjer DMB, Nyvad J, et al. TMEM16A knockdown abrogates two different Ca(2+)-activated Cl (−) currents and contractility of smooth muscle in rat mesenteric small arteries. Pflugers Arch. 2014;466:1391–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li RS, Wang Y, Chen HS, et al. TMEM16A contributes to angiotensin II-induced cerebral vasoconstriction via the RhoA/ROCK signaling pathway. Mol Med Rep. 2016;13:3691–3699. [DOI] [PubMed] [Google Scholar]
- 29.Behuliak M, Bencze M, Polgárová K, et al. Hemodynamic response to gabapentin in conscious spontaneously hypertensive rats. Hypertension. 2018;72:676–685. [DOI] [PubMed] [Google Scholar]
- 30.Ye Z, Wu MM, Wang CY, et al. Characterization of cardiac anoctamin1 Ca2⁺-activated chloride channels and functional role in ischemia-induced arrhythmias. J Cell Physiol. 2015;230:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horváth B, Váczi K, Hegyi B, et al. l. Sarcolemmal Ca(2+)-entry through L-type Ca(2+) channels controls the profile of Ca(2+)-activated Cl(−) current in canine ventricular myocytes. J Mol Cell Cardiol. 2016;97:125–139. [DOI] [PubMed] [Google Scholar]
- 32.Hegyi B, Horváth B, Váczi K, et al. Ca(2+)-activated Cl(−) current is antiarrhythmic by reducing both spatial and temporal heterogeneity of cardiac repolarization. J Mol Cell Cardiol. 2017;109:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papp R, Nagaraj C, Zabini D, et al. Targeting TMEM16A to reverse vasoconstriction and remodelling in idiopathic pulmonary arterial hypertension. Eur Respir J. 2019;53:1800965. [DOI] [PubMed] [Google Scholar]
- 34.Huang F, Zhang H, Wu M, et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc Natl Acad Sci U S A. 2012;109:16354–16359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Catalán MA, Kondo Y, Peña-Munzenmayer G, et al. A fluid secretion pathway unmasked by acinar-specific Tmem16A gene ablation in the adult mouse salivary gland. Proc Natl Acad Sci U S A. 2015;112:2263–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu D, Thelin WR, Rogers TD, et al. Regional differences in rat conjunctival ion transport activities. Am J Physiol Cell Physiol. 2012;303:C767–C780. [DOI] [PMC free article] [PubMed] [Google Scholar]