

Abstract
The transcription factors NFAT and AP-1 (Fos-Jun) cooperate to promote the effector functions of T cells, but NFAT in the absence of AP-1 imposes a negative feedback program of T cell hyporesponsiveness (“exhaustion”). Here we show that BATF and IRF4 cooperate to counter T cell exhaustion in mouse tumor models. Overexpression of BATF in CD8+ T cells expressing a chimeric antigen receptor (CAR) promoted the survival and expansion of tumor-infiltrating CAR T cells, increased the production of effector cytokines, decreased the expression of inhibitory receptors and the exhaustion-associated transcription factor TOX, and supported the generation of long-lived memory T cells that controlled tumor recurrence. These responses were dependent on BATF-IRF interaction, since cells expressing a BATF mutant unable to interact with IRF4 did not survive in tumors and did not effectively delay tumor growth. BATF may improve the anti-tumor responses of CAR T cells by skewing their phenotypes and transcriptional profiles away from exhaustion and towards increased effector function.
Introduction
CD8+ T cells that encounter antigen together with effective costimulatory signals mount strong effector responses that are able to clear pathogen-infected cells and tumor cells. In contrast, CD8+ T cells that infiltrate solid tumors and are exposed to prolonged antigen stimulation in the absence of adequate costimulation enter a hyporesponsive (“exhausted” or “dysfunctional”) state in which they do not effectively destroy tumor cells1–3. Exhausted T cells express high levels of inhibitory receptors including PD-1, TIM3 and LAG3, low levels of effector proteins including cytokines and granzymes, and transcription factors of the NR4A and TOX families that act to impose exhaustion4–10.
The effector and “exhaustion” responses of CD8+ T cells are both initiated by TCR signalling, and the transcription factor NFAT plays a pivotal role in both responses, with the balance between them depending on the transcriptional partners of NFAT. During acute immune responses, NFAT induces predominantly the effector program by cooperating with its partner transcription factor AP-111,12; the effector program also requires the independent binding of AP-1 and NFκB transcription factors to other sites13. Classical AP-1 is comprised of heterodimers of the basic region-leucine zipper (bZIP) transcription factors FOS and JUN14, but heterodimers of other FOS/ JUN-family proteins can also cooperate with NFAT13. In contrast, whereas NFAT remains nuclear for many hours in antigen-stimulated T cells15, the expression and activity of FOS- andJUN-family members is not sustained14; under these conditions, NFAT acts predominantly in the negative feedback program to induce genes associated with exhaustion/dysfunction4–10.
Recent attention has focused on two downstream targets of NFAT — the NR4A family of orphan nuclear receptors and the TOX family of high-mobility group (HMG)-box DNA-binding proteins16. Depletion of NR4A14, all three NR4A family members5, TOX10, or TOX and TOX2 proteins6 confers robust anti-tumor responses on CD8+ tumor-infiltrating T lymphocytes (CD8+ TILs). Mechanistically, TOX/TOX2 or NR4A depletion prevents some of the chromatin and transcriptional changes characteristic of exhaustion, and partially rescues the effector activity of TILs4–10. The onset of exhaustion coincides with decreased chromatin accessibility of regions enriched for AP-1/bZIP motifs and a decrease in the expression of many bZIP proteins5,6. Based on these findings, we hypothesized that restoring AP-1 expression and function in CD8+ TILs would allow the formation of activating NFAT:AP-1 complexes and prevent CD8+ T cell exhaustion5,6,12,17. Recently, CD8+ T cells expressing a chimeric antigen receptor (CAR) for a tumor antigen and overexpressing JUN were shown to be more effective than control CAR T cells at slowing tumor growth18.
The transcription factors BATF and its partners IRF4 and IRF8 are also induced by TCR signalling19–24. Like NFAT, BATF can contribute both to effector function and to exhaustion, depending on the biological context12,19,25,26. Here we show that overexpressed BATF can cooperate with IRF4 to counteract the development of T cell exhaustion. Overexpression of BATF in CD8+ CAR T cells led to a marked increase in the survival and expansion of TILs; increased the ability of the CAR TILs to produce cytokines and granzymes after stimulation; and reduced their expression of inhibitory cell surface receptors and the exhaustion-associated transcription factor TOX. Tumor-bearing mice that had previously received BATF-transduced CD8+ T cells and rejected the tumor developed long-lived memory T cells that controlled tumor recurrence. There is substantial interest in manipulating CAR T cells to control tumors more effectively, and BATF overexpression potentially represents a simple and therapeutically effective method for achieving this desired outcome.
Results
BATF-transduced CAR T cells exhibit enhanced tumor rejection
A preliminary screen for TFs that could enhance NFAT:AP-1 activity in CD8+ T cells led us to JUN, MAFF, and BATF (Extended Data Fig. 1), and raised the question whether JUN, MAFF, or BATF could confer a functional anti-tumor advantage on CD8+ CAR T cells in vivo.
CD8+ T cells were retrovirally transduced with a CAR directed against human CD19 (hCD19)5,6 together with a retroviral expression vector for JUN, MAFF, or BATF, or an empty (pMIG) retrovirus control, and adoptively transferred 7 days after tumor inoculation into C57BL/6J mice bearing the B16F0-hCD19 tumor. Transduction yielded very high expression of each TF compared to endogenous protein, but did not alter expression of the Myc-tagged CAR (Extended Data Fig. 2). Mice adoptively transferred with control pMIG- or MAFF-transduced CAR T cells showed tumor sizes similar to those of mice treated with PBS alone, whereas mice receiving JUN-transduced CAR T cells showed a variable delay in tumor growth (Fig. 1a,b). Mice injected with BATF-transduced CAR T cells showed a notable delay in tumor growth, as well as a significant improvement in long-term survival compared to all other groups (Fig. 1a–c). The findings with BATF-transduced CAR T cells were confirmed in replicate B16 melanoma experiments and in experiments with an MC38-hCD19 colon adenocarcinoma (Extended Data Fig. 2).
Figure 1. Anti-tumor effects of CAR T cells ectopically expressing bZIP transcription factors.
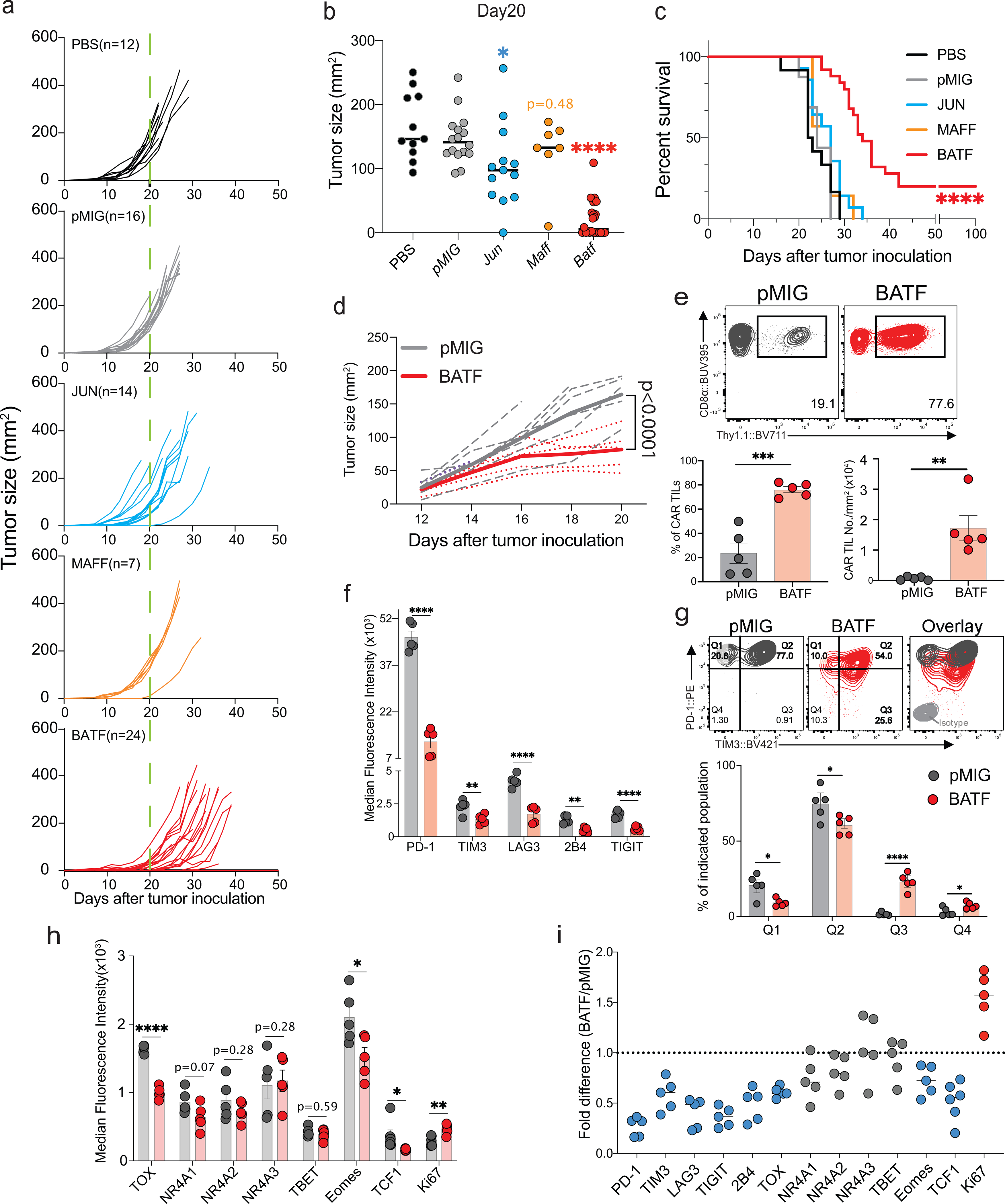
a-c,1×105 B16F0-human CD19 (B16F0-hCD19) tumor cells were injected subcutaneously into the left flank of C57BL/6 mice at day 0 (D0) in 100 μl phosphate-buffered saline (PBS, n=12); 3×106 control pMIG(n=16)-, JUN(n=14)-, MAFF(n=7)- or BATF(n=24)-transduced CAR T cells were adoptively transferred by retro-orbital injection at day 7.
a, b, Tumor growth rates (a) and tumour sizes (b) at day 20 for individual mice.
c, Mouse survival curves up to 100 days after tumor inoculations.
d-i. 1×105 B16F0-hCD19 tumor cells were subcutaneously injected into the left flank of C57BL/6 mice at day 0 (D0); 1.5×106 pMIG(n=5)- or BATF(n=5)-transduced CAR T cells were adoptively transferred at day 12. Tumor-infiltrating lymphocytes were isolated at day 20.
d, Tumor growth curves for individual mice (dashed lines) and average of all tumor growth curves in a group (bold lines).
e, Top, Contour plot of flow cytometry data for the CAR TILs. Bottom, Percentage of CAR TILs relative to total CD8+ TILs in the tumor (left); normalized number of CAR TILs per tumour, obtained by dividing the absolute number of CAR TILs by the tumor area (right).
f, Median fluorescence intensity (MFI) of the entire flow plot for the indicated inhibitory receptors from each group of CAR TILs.
g, Top, Representative contour plots of PD-1 and Tim3 expression on CAR TILs. Bottom, percentage of cells in each of the indicated quadrants (Q1=PD-1highTIM3low, Q2=PD-1highTIM3high, Q3=PD-1intTIM3high and Q4=PD-1intTIM3low).
h, MFI for expression of indicated TFs from each group of CAR TILs.
i, MFI fold change between pMIG- and BATF-transduced CAR TILs.
Each circle in b, e, f, g, h and i represents one mouse, and the bar graphs represent the mean ± standard error of mean (s.e.m.). Data in a-c and d-i were obtained from three and two independent experiments respectively. Data in b were analyzed by one-way ANOVA test; data in c, using a log-rank Mantel-Cox test; data in d, by two-way ANOVA test; and data in e, f, g, and h, by two-tailed unpaired Student’s t-test. *p≤0.05; **p≤0.01; ***p≤0.001; ****p≤0.0001.
To further explore the anti-tumor responses of BATF-transduced CAR T cells, we transferred pMIG- or BATF-transduced CAR T cells into tumor-bearing recipient mice 12 days after tumor inoculation, at which time the tumor is large and well established, and harvested TILs 8 days after CAR T cell transfer. Mice given BATF-transduced CAR T cells showed substantially slower tumor growth compared to mice given control pMIG-transduced CAR T cells (Fig. 1d). BATF-transduced CAR TILs, identified by expression of the Thy1.1 reporter, showed a striking increase in frequency in the tumor compared to control pMIG-transduced cells (Fig. 1e).
BATF overexpression directs CAR TILs away from exhaustion
Consistent with their expansion and function in the tumor microenvironment, BATF-transduced CAR TILs showed decreased immunochemical staining of all the inhibitory receptors tested; a marked increase in the proliferation marker Ki67; decreased expression of naïve/ memory markers CD127 and CD62L; increased expression of CD44 and expression of KLRG1 in a subpopulation of cells; and decreased expression of TOX, a TF strongly associated with CD8+ T cell exhaustion6–10 (Fig. 1f–i; Extended Data Fig. 2). Induction of interferon-γ (IFN-γ) and expression of granzyme B and CD107a were significantly increased after PMA/ ionomycin stimulation in BATF-transduced compared to control pMIG CAR TILs (Extended Data Fig. 2).
Mass cytometry confirmed these findings and provided evidence that additional markers of previously activated or effector CD8+ T cells were upregulated (Fig. 2). TOX and PD-1 were coexpressed in control pMIG-transduced CAR TILs, as in other exhausted CD8+ T cells6–10, but the PD-1high TOXhigh population was absent in BATF-transduced CAR TILs (Fig. 2c). Conversely, ICOS and granzyme B expression were strongly correlated in BATF-transduced but not in pMIG-transduced CAR TILs (Fig. 2d), suggesting the presence of an “effector-like” TIL subset elicited in part by BATF overexpression.
Figure 2. High-dimensional single-cell characterization of pMIG- and BATF-transduced CAR TILs by mass cytometry (CyTOF).
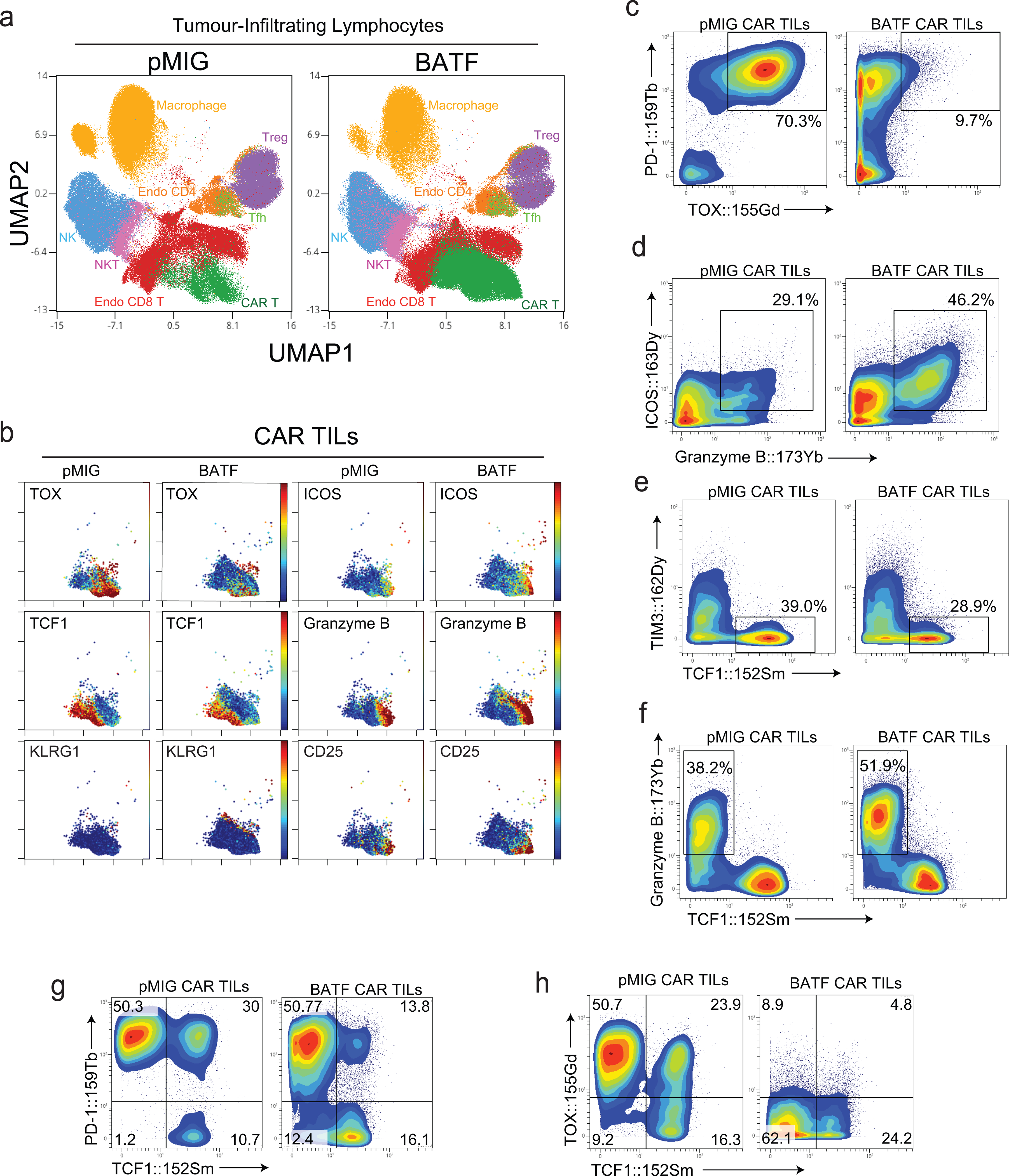
a-h, 1×105 B16F0-hCD19 tumor cells were injected subcutaneously into the left flank of C57BL/6 mice at day 0 (D0). 1.5×106 pMIG- or BATF-transduced CAR T cells were adoptively transferred at day 12. TILs were isolated at day 20 and stained with metal-conjugated antibodies for mass cytometry, performed at day 21 using a CyTOF mass spectrometer. The gating strategy is detailed in Extended Data Fig. 3a.
a, Plots show UMAP views that provide comprehensive single-cell analysis and distinguish TIL subpopulations.
b, Detection of indicated markers on pMIG or BATF CAR TILs is visualized by UMAP.
c-h, Contour plot of indicated markers on pMIG or BATF CAR TILs.
Data are representative of two biological experiments. Each group of samples was pooled from 10 mice.
A progenitor-like T cell population expressing the transcription factor TCF1 sustains the immune response against both tumors and chronic viral infections, and underlies the proliferative response to checkpoint blockade immunotherapy27–31. Among both pMIG- and BATF-transduced CAR TILs, TCF1+ cells remained TIM3low and granzyme Blow (Fig. 2e,f), consistent with a progenitor-like role. The TCF1+ subset constituted a reduced percentage of BATF-overexpressing TILs, but still an ample number of cells to account for their survival and effector function in the tumor (Fig. 2e–h).
BATF-transduced CAR T cells persist after tumor regression
We asked whether CAR TILs persisted in mice that had rejected an initial tumor, and, if so, whether they conferred protection against rechallenge with the same tumor. B16F0-hCD19 tumor cells were injected on the opposite flank of the five surviving mice from the previous experiment, with a corresponding tumor-naïve cohort of 5 age-matched C57BL/6 mice as controls. Tumors grew in the tumor-naïve group as expected, but did not develop (4 mice) or quickly regressed (1 mouse) in the previously challenged group (Fig. 3a). Thy1.1+ CD8+ CAR T cells were recovered from the draining lymph nodes and spleen of all five surviving mice of the BATF-transduced CAR group (Fig. 3b). The recovered CAR T cells displayed characteristics similar to central memory CD8+ T cells, including expression of CD27, CD44, TCF1, CD62L and CD127 (Fig. 3c,d; Extended Data Fig. 3). Tumor rechallenge gave a similar result in the replicate B16-hCD19 experiment (Extended Data Fig. 3). Thus, BATF-transduced CAR T cells persisted for many weeks after tumor clearance and acquired features of memory T cells.
Figure 3. BATF-transduced CAR T cells confer a memory response against tumor rechallenge and exhibit a memory phenotype.
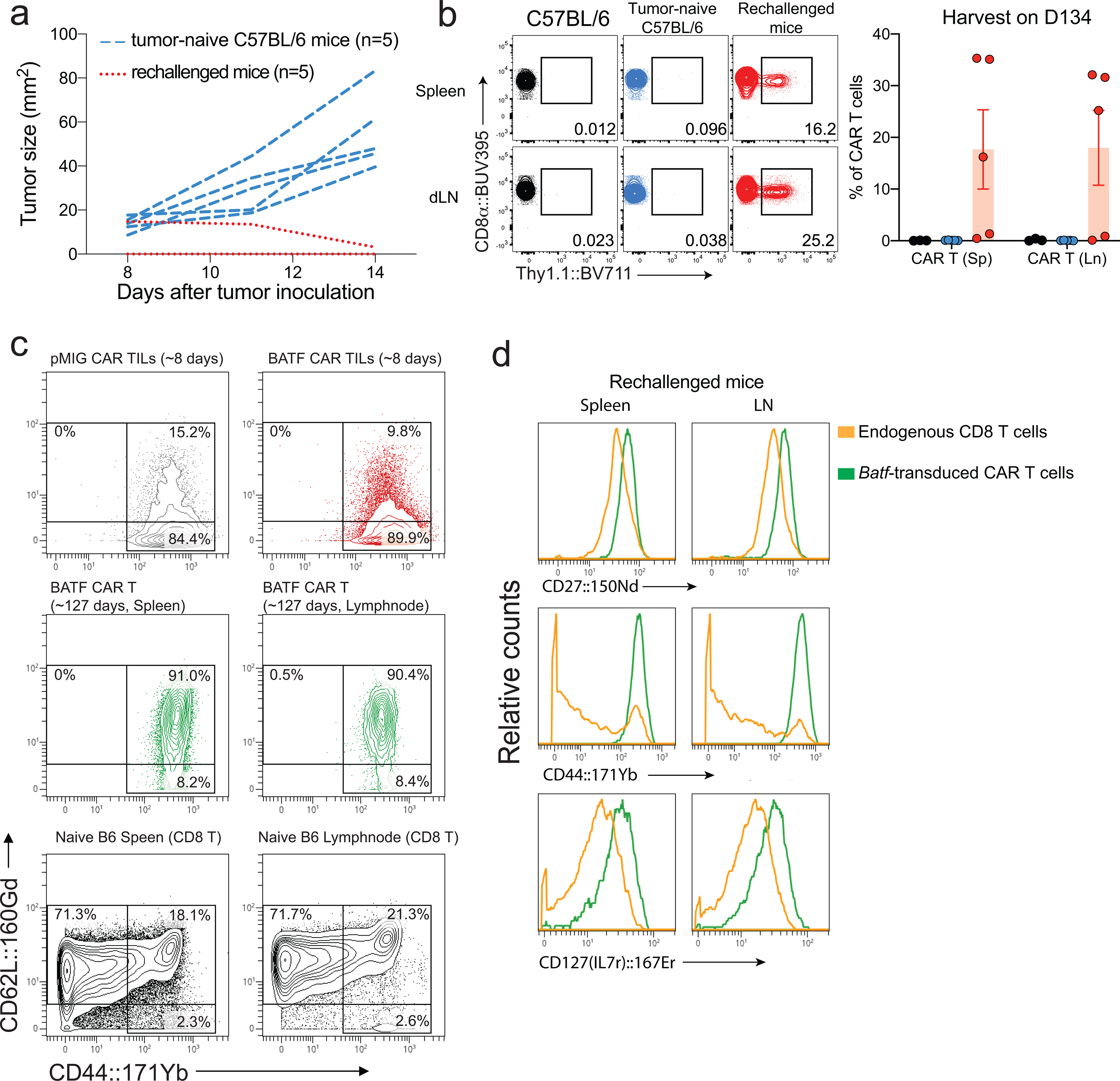
a-d, 1×105 B16F0-hCD19 tumor cells were injected subcutaneously into the right flank of C57BL/6 mice (n=5) to yield the “tumor-naïve” control group, or into tumor-free mice from the experiment in Fig. 1c (n=5) that had rejected an initial B16F0-hCD19 tumor and had survived until day 120 after the first tumor injection (rechallenged group). Spleens and draining lymph nodes were harvested 14 days after tumor inoculation or tumor rechallenge.
a, Tumor growth curves for individual mice (tumor-naïve C57BL/6 mice, blue dashed lines; rechallenged mice, red dotted lines). No tumor growth was detected in four of the rechallenged mice.
b, Left, Representative contour plots showing frequencies of CAR T cells in splenocytes and draining lymph node cells from a fresh control C57BL/6 mouse that did not receive tumor cells, a tumor-bearing C57BL/6 mouse (“tumor-naïve” control group), and a rechallenged mouse. Right, Percentage of CAR TILs relative to total CD8+ TILs in the tumor. The mouse with the lowest frequency of CAR T cells was the one in which the rechallenge tumor had been present initially but then regressed.
c, Contour plots for CD62L (y-axis) and CD44 (x-axis) expression. Top, CD8+ T cells from BATF- and pMIG-transduced CAR TILs 8 days after transfer of CAR T cells from the CyTOF experiment of Fig. 2 and Extended Data Fig. 3; middle, BATF-transduced CAR T cells from spleen and draining lymph nodes of rechallenged mice, ~127 days after CAR T cell adoptive transfer; bottom, splenocytes and lymphocytes from draining lymph nodes of fresh control C57BL/6 mice.
d, Histogram plotting CyTOF signals of the indicated markers in endogenous CD8+ T cells and in BATF-transduced CAR T cells from rechallenged mice.
Each circle in b represents one mouse, and the bar graph represents the mean ± standard error of mean (s.e.m.). Data in b are representative of two biological experiments (see Extended Data Fig. 3e). Samples for each group analyzed in c and d were pooled from 5 mice.
BATF directs human CAR T cells towards an effector-like phenotype
An important question is whether BATF overexpression exerts similar effects in human T cells. We transduced human CD8+ T cells with a human CD19 CAR construct32 and with a vector encoding human BATF or its empty-vector control (Extended Data Fig. 4). The levels of CAR expression were comparable in the experimental and control cells. When cultured together with hCD19-bearing tumor cells to assess effector function, BATF-overexpressing human CAR T cells proliferated more than control CAR T cells, and showed enhanced cytokine expression, granzyme B expression, and cytotoxicity.
BATF-IRF4 interaction is essential for anti-tumor responses
A prominent effect of BATF in CD4+ T cells is to recruit IRF to composite AP1-IRF (AICE) sites in DNA, where a heterodimer of BATF and a JUN-family transcription factor binds cooperatively with IRF4 or IRF820–25,33. We introduced the H55Q/K63D/E77K (HKE) mutations, which suppress the interaction between BATF and IRF420,24,33, into the BATF expression plasmid. BATF-HKE was expressed in CD8+ T cells at levels similar to wildtype BATF (Fig. 4a), and retained DNA binding, as reported previously20,33 and confirmed in ChIP-seq analyses described below. Tumors developing in mice given HKE mutant-transduced CAR T cells at day 7 after tumor inoculation were significantly larger than those in mice given wildtype BATF-transduced CAR T cells (Fig. 4b, Extended Data Fig. 5), and survival of the mice was significantly lower (Fig. 4c). Thus, selectively impairing the interaction of BATF with IRF4 strongly attenuated the ability of BATF-overexpressing CAR T cells to control the tumor.
Figure 4. The BATF-IRF interaction is required for CAR T cell survival, expansion, and anti-tumor responses.
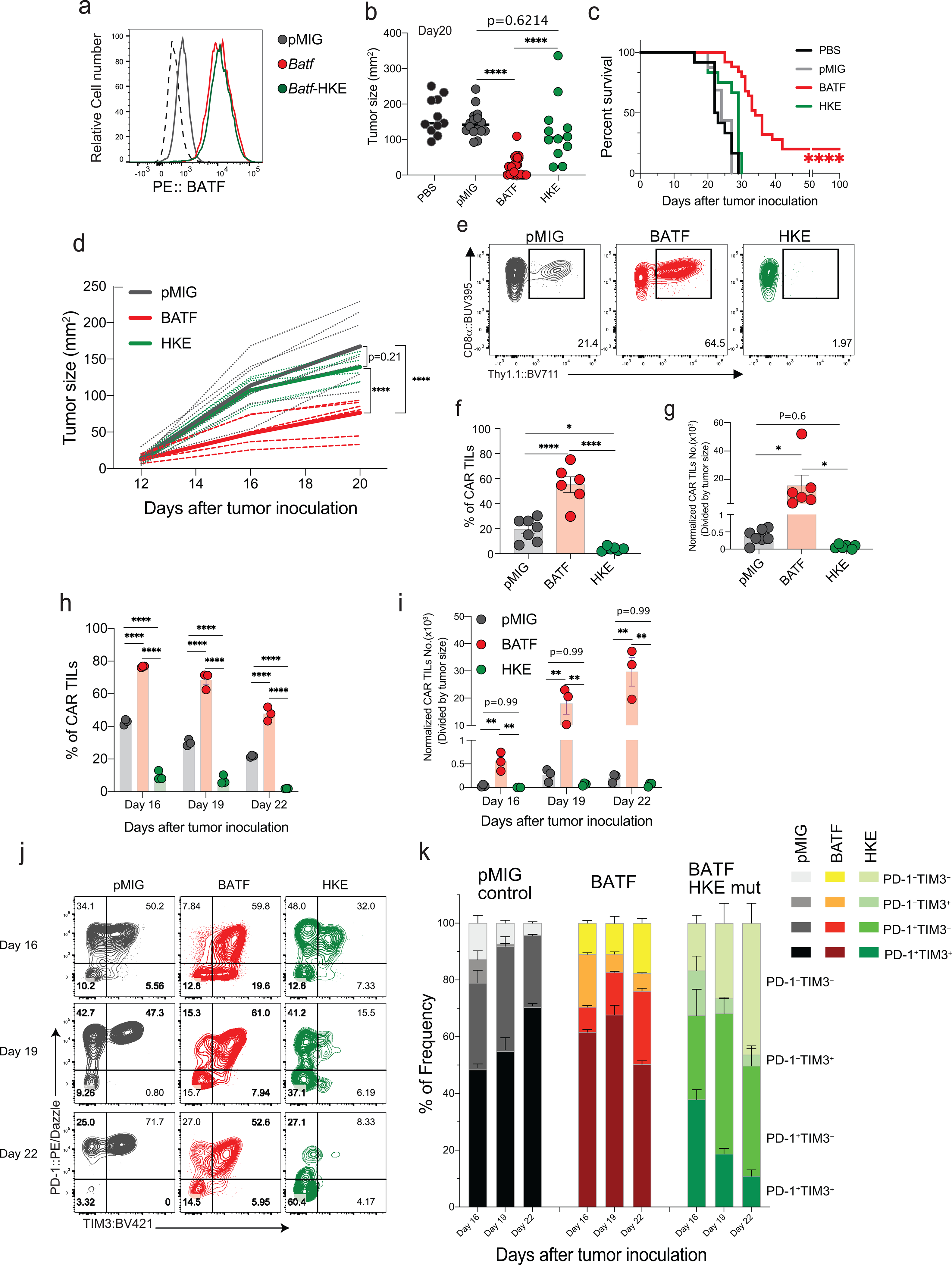
a, Expression of endogenous BATF in pMIG-transduced cells, and of BATF and BATF-HKE in retrovirally transduced CD8+ T cells.
b-c, Experimental protocol as in Fig. 1a–c.
b, Tumor sizes in individual mice at day 20.
c, Survival curves. Data for PBS, pMIG, and BATF are replotted from Fig. 1c, since the BATF-HKE mutant (n=12) was analyzed in the same experimental series.
d-g, Experimental protocol as in Fig. 1d–i, except with pMIG(n=7)-, BATF(n=6)-, or BATF-HKE(n=6)-transduced CAR T cells.
d, Tumor growth curves for individual mice (dashed lines) and the averages for all mice in a group (bold lines).
e, Representative contour plots of CD8α and Thy1.1 expression in the isolated TILs. The Thy1.1 reporter marks CAR T cells.
f, Percentage of CAR TILs among CD8+ T cells.
g, Number of CAR TILs normalized to tumor size.
h-k, 1×105 B16F0-hCD19 tumor cells were injected subcutaneously into the left flank of C57BL/6 mice at day 0 (D0), and indicated CAR T cells were adoptively transferred by retro-orbital injection on day 12. TILs were isolated on Days 13, 16, 19, and 22. No CAR TILs were observed on day 13, one day after adoptive transfer.
h,i, Percentages of CAR TILs (h) and normalized numbers of CAR TILs (i) on the indicated days.
j, Representative contour plots of PD-1 and TIM3 expression on the CAR TILs, assessed by flow cytometry.
k, Frequencies of the indicated PD-1- and TIM3-expressing populations.
Each circle in b, f, g, h, and i represents one mouse, and the bar graphs represent the mean ± standard error of mean (s.e.m.). Data in a and h-k are representative of two independent experiments. Data in b and c were obtained from three, and data in d-g from two, independent biological experiments. Data in b, f, and g were analyzed by one-way ANOVA test; data in c, using a log-rank Mantel-Cox test; and data in d, h, and i, by two-way ANOVA test. *p≤0.05; **p≤0.01; ***p≤0.001; ****p≤0.0001.
HKE mutant-transduced CAR T cells adoptively transferred at day 12 after tumor inoculation were likewise ineffective in controlling tumor growth, and this ineffectiveness was associated with a marked decrease in the frequency and number of CAR TILs (Fig. 4d–g). To follow TIL expansion in vivo, we transferred CAR T cells into tumor-bearing mice at day 12 after tumor inoculation and monitored TIL numbers and phenotypes on days 13, 16, 19, and 22 (Fig. 4h–k; Extended Data Fig. 5). The striking expansion of BATF-transduced CAR TILs compared to control TILs and the contrasting low numbers of HKE mutant-transduced TILs were already obvious four days after transfer and became even more pronounced at later times. The fraction of PD-1+TIM3+ cells among the few CAR TILs expressing the HKE mutant was low in comparison to controls on day 16, and progressively declined from days 16–22 in parallel with an increase in PD-1–TIM3– cells.
CAR T cells carrying a germline deletion of BATF (Batf KO) likewise had no effect on tumor growth and exhibited a striking paucity of TILs (Extended Data Fig. 5). PD-1 expression in the few recovered cells was substantially diminished compared to WT CAR TILs, and PD-1+TIM3+ CAR TILs were almost absent. Moreover, Batf KO CAR TILs displayed a decreased frequency of PD-1+TOXhigh cells and a prominent group of naive-like TCF1+TIM3–cells. Collectively, these data reveal that BATF and the BATF-IRF4 interaction are absolutely required for the survival and expansion of BATF-transduced CAR T cells in tumors, and that endogenous BATF3 does not compensate for the germline loss of BATF.
Coexpression of IRF4 with BATF hampers the anti-tumor response
Given these data, we asked whether coexpressing IRF4 with BATF would further improve the anti-tumor responses of CD8+ TILs. OT-I cells expressing BATF alone, IRF4 alone, or BATF+IRF4 were injected on either day 7 or day 12 after tumor inoculation, and tumor size was measured subsequently (Extended Data Fig. 6). All three types of transduced cells initially slowed tumor growth. Overexpression of either BATF alone or BATF with IRF4 resulted in striking TIL expansion, decreased expression of the exhaustion markers PD-1, TIM3, and TOX, and increased expression of granzymes and effector cytokines. However, the effects of overexpressing BATF or IRF4 individually were distinct: Overexpressing BATF alone was more effective than overexpressing IRF4 alone at promoting TIL expansion and downregulating TOX, PD-1, and TIM3; whereas overexpressing IRF4 alone led to more cytokine and granzyme B expression upon stimulation.
Because BATF binds DNA as a heterodimer with JUN family members, we compared anti-tumor responses in OT-1 cells transduced with BATF alone, JUN alone, or BATF+JUN (Extended Data Fig. 6). JUN overexpression in OT-I cells did not substantially slow the growth of B16F10-OVA tumors beyond that observed with control pMIG-transduced OT-I cells. In contrast, mice given OT-I cells transduced with BATF alone showed a strong reproducible delay in tumor growth, and mice given OT-I cells transduced with both BATF and Jun showed, surprisingly, a less impressive delay.
Distinctive transcriptional profile of BATF-transduced CAR TILs
Transcriptional profiling (Supplementary Table 1) highlighted Ifnar1 and its downstream signalling effectors Stat1 and Stat3, as well as Il12rb2, as genes differentially upregulated in BATF-overexpressing TILs compared to control TILs (Fig. 5a). These differences may render the BATF-overexpressing cells more receptive to IFNα/β and IL12 signals that promote effector and effector/memory function34,35, and may account for the enhanced effector function of BATF-overexpressing T cells in the tumor, and the ability to generate memory CAR T cells. Other genes related to effector function (Icos, Gzma/b/c) showed increased mRNA expression (Fig. 5a), consistent with increased protein levels of ICOS and of granzyme B after stimulation. We also observed increased expression of mRNAs encoding CCL5, CCR2, CXCR3, and CXCR6, chemokines and chemokine receptors that are upregulated in activated/effector CD8+ T cells and that promote trafficking of CD8 T+ cells to tumors and sites of inflammation (Fig. 5a); and decreased expression of mRNA encoding CCR7, a chemokine receptor that is typically downregulated in effector CD8+ T cells. Perhaps most importantly, and again in line with the protein data, BATF-transduced CAR TILs displayed decreased expression of Tox mRNA, indicating a break in a crucial transcriptional link on the pathway to exhaustion. These observed changes are consistent with a transcriptional bias of the BATF-transduced CAR TILs towards increased tumor infiltration, increased expansion within tumors, increased effector function, and decreased propensity to exhaustion.
Figure 5. Genome-wide analysis of differences in transcription and chromatin accessibility between pMIG and BATF-transduced cells.
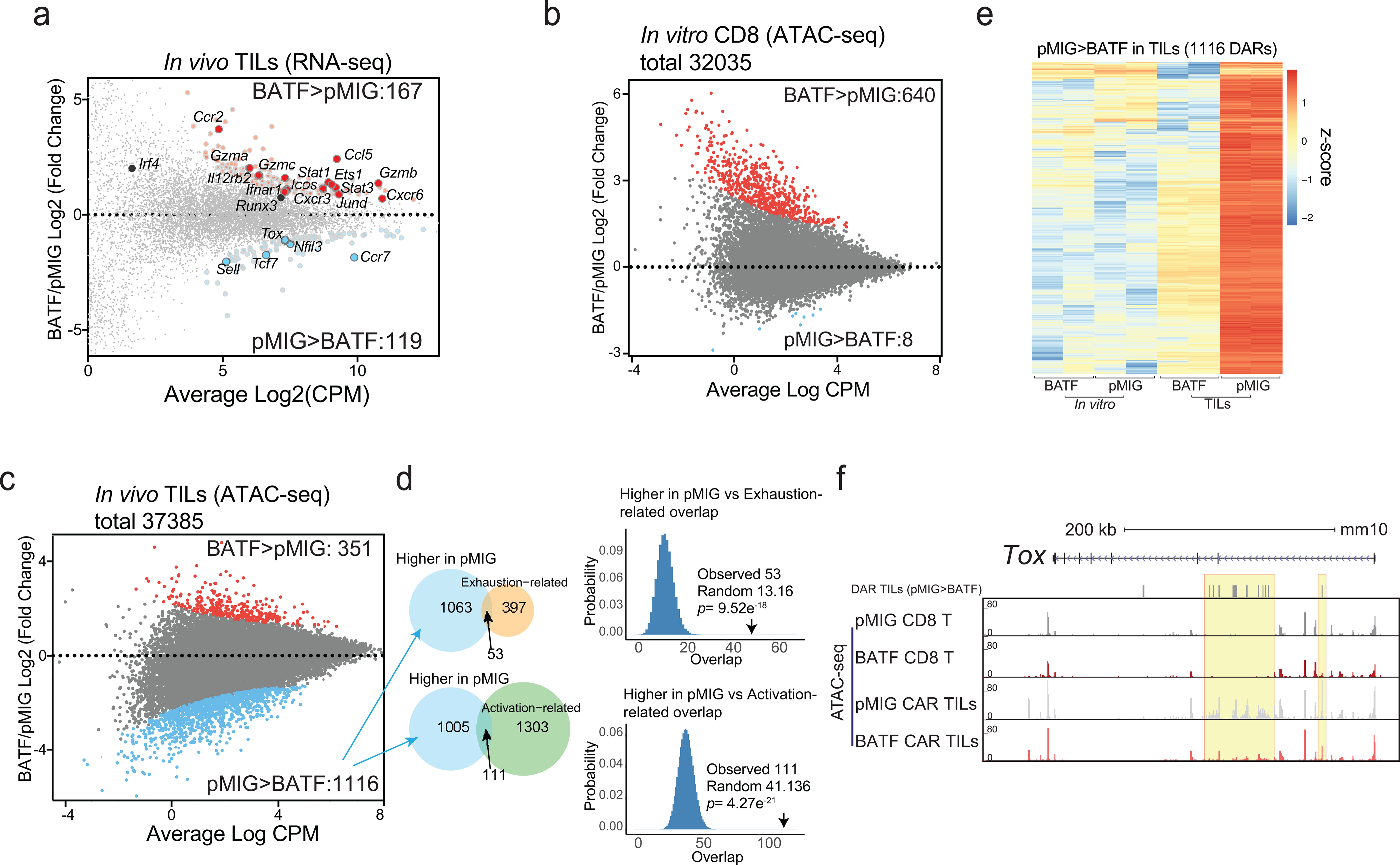
a, MA plot of genes differentially expressed in BATF-transduced versus pMIG-transduced CAR TILs. Differentially expressed genes (adjusted p-value < 0.1, log2(fold-change) ≥ 0.5 or ≤ −0.5) are highlighted; selected genes are labelled.
b, MA plot of ATAC-seq data from CD8+ T cells in vitro. Differentially accessible regions (DARs, log2(fold-change) ≥ 2 and adjusted p-value ≤ 0.05) are shown as red and blue dots for regions more accessible in BATF- or pMIG-transduced CD8+ T cells, respectively. 551 of the 640 regions that were more accessible in BATF-overexpressing cells overlapped a BATF ChIP-seq peak.
c, MA plot of ATAC-seq data from BATF-transduced versus pMIG-transduced CAR TILs. Differentially accessible regions (log2(fold-change) ≥ 2 and adjusted p-value ≤ 0.05) are shown as red and blue dots for regions more accessible in BATF-transduced and pMIG-transduced TILs, respectively.
d, Left, Venn diagrams showing the overlap of the 1116 regions more accessible in pMIG- versus BATF-transduced TILs with the exhaustion-related (top) or activation-related (bottom) regions from Mognol et al36. Right, Histograms illustrate the significance calculation by one-tailed Fisher’s exact test.
e, Heatmap (z-score) of ATAC-seq signals from BATF- and pMIG-transduced CD8+ T cells or CAR TILs, for the 1116 regions more accessible in pMIG TILs compared to BATF TILs. Each column represents a biological replicate.
f, Tox locus with normalized ATAC-seq signals for CD8+ T cells and CAR TILs. The top track marks DARs (pMIG>BATF) in the TILs. Yellow highlights call attention to peaks that differ most strikingly between pMIG and BATF TILs.
Data in a-c, e, and f were obtained from two independent biological experiments.
Chromatin changes elicited by BATF overexpression
To distinguish early changes initiated by overexpressed BATF in transduced cells prior to transfer from subsequent changes induced in BATF-overexpressing TILs within the tumor environment, we carried out parallel ATAC-seq and RNA-seq analyses on transduced T cells just prior to adoptive transfer and on BATF-overexpressing and control TILs harvested from tumors 8 days after adoptive transfer. The very limited alterations in chromatin accessibility in vitro in BATF-overexpressing cells compared to pMIG cells were strongly biased toward greater accessibility in BATF-overexpressing cells. Of 32,035 accessible chromatin regions mapped, 640 regions were more accessible in BATF-overexpressing cells, and just 8 regions were less accessible (Fig. 5b; Supplementary Table 2).
In TILs, in contrast, a solid majority of the differentially accessible regions were more accessible in control pMIG TILs than in BATF-overexpressing TILs (Fig. 5c; Supplementary Table 3). The ATAC-seq peak regions showing greater accessibility in pMIG-transduced cells overlapped significantly with both ‘exhaustion-related’ and ‘activation-related’ peaks identified in TILs by Mognol et al 36 (Fig. 5d). (The peaks from Mognol et al were defined by comparing OT-I tumor antigen-specific CD8+ cells with P14 bystander CD8+ cells, and therefore were directly dependent on TCR signalling in the tumor.) In contrast, the regions more accessible in BATF-overexpressing cells were not enriched for the exhaustion-related subset, and were depleted for the activation-related subset (Extended Data Fig. 7).
The regions of differentially higher accessibility in pMIG TILs reflected almost exclusively chromatin rearrangements occurring in pMIG TILs after adoptive transfer (Fig. 5e). A telling example is the Tox locus, which exhibited similar accessibility in control pMIG- and BATF-transduced cells prior to adoptive transfer, but showed selective increases in accessibility of several regions in pMIG TILs (Fig. 5f). The data of Figs. 5d–f indicate that TCR-dependent signals that ordinarily alter the accessibility of characteristic chromatin regions in tumor-responsive CD8+ TILs are blunted in BATF-overexpressing TILs.
The early patterns of differential accessibility between BATF-expressing and control cells in vitro were superseded by distinct patterns of differential accessibility in TILs (Fig. 5b,c; Extended Data Fig. 7). Binding motifs for ETS, RUNT, bZIP, and IRF transcription factors, as well as composite ETS-RUNT and bZIP-IRF motifs, were substantially enriched in accessible regions of BATF-overexpressing TILs compared to control TILs (Extended Data Fig. 7). Enrichment of ETS-binding motifs is notable, because Ets1 mRNA was significantly upregulated in BATF-transduced CAR TILs (Fig. 5a); ETS1 contributes to T cell development and homeostatic proliferation, and ETS motifs are enriched in the accessible chromatin of effector/memory T cells37–39, consistent with the ability of BATF-transduced CAR TILs to expand and display effector function.
Early changes in transcription factor binding
ChIP-seq data underline the close cooperation of IRF4 with BATF, since IRF4 binds predominantly at BATF peaks in BATF-overexpressing cells (Fig. 6a). However, the cooperation is not symmetrical, as a large fraction of BATF reads map to peaks where there is no significant IRF4 binding (Fig. 6a). While the majority of these latter peaks have low numbers of reads and may represent nonphysiological binding of BATF, a minor subset shows appreciable BATF occupancy (Fig. 6a, right, shoulder in red histogram). The peaks with highest BATF occupancy, when compared against the peaks with lowest occupancy, are enriched in motifs for ETS-family proteins— for example, the HOMER ETS1-binding motif is present in 61.57% of target sequences and in only 10.59% of background sequences, p-value 1e-3124— and in motifs comprising short G-rich tracts. Further attention to these peaks is warranted in light of the upregulation of Ets1 mRNA and the differential enrichment of ETS motifs in accessible regions in BATF-overexpressing CAR TILs.
Figure 6. BATF and IRF4 binding and gene expression changes in pMIG- and BATF-transduced cells.
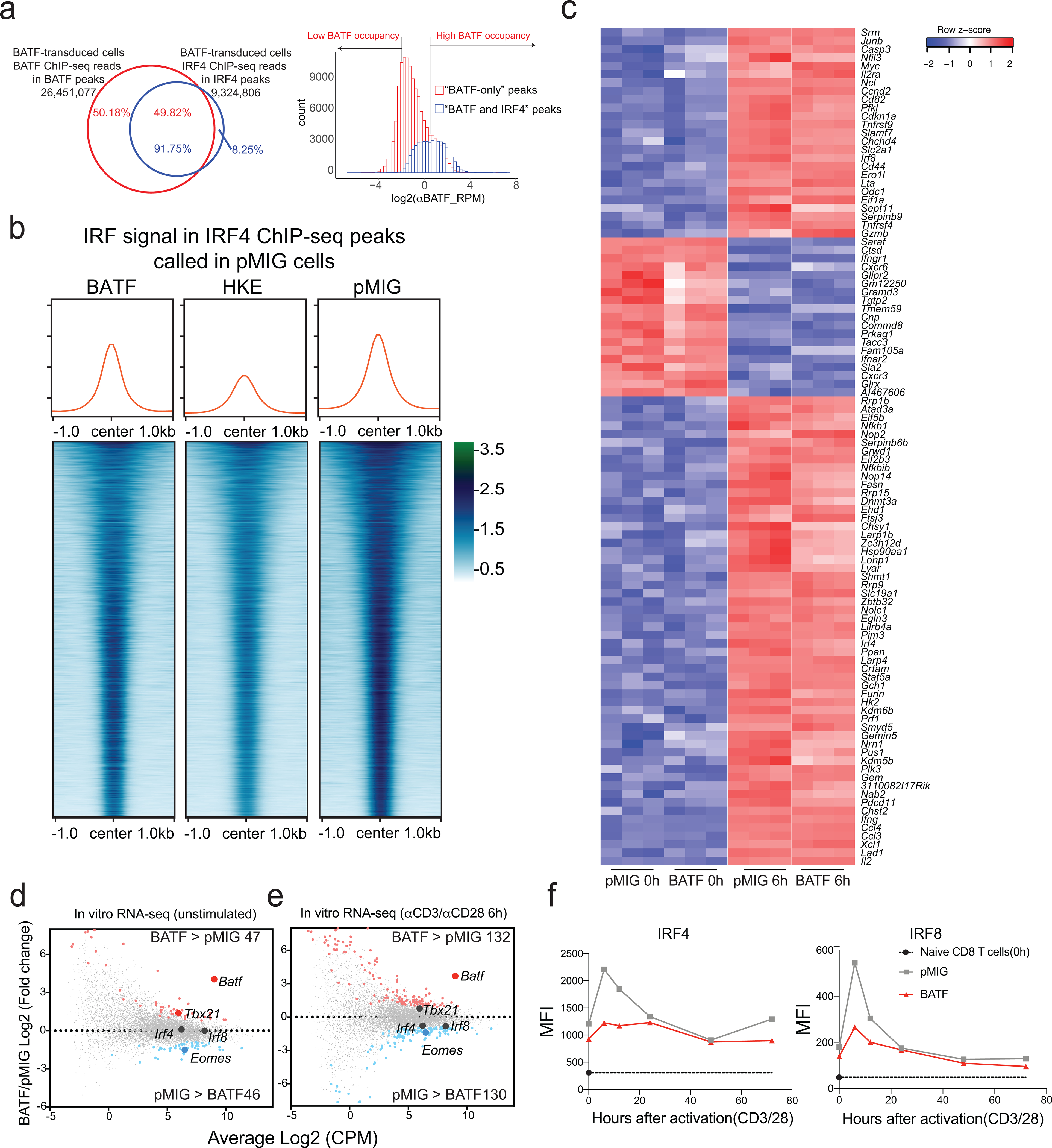
a, Left, Distribution of BATF (red) and IRF4 (blue) ChIP-seq reads in peaks from BATF-transduced cells. Almost all IRF4 reads are located in BATF ChIP-seq peaks, whereas half of BATF reads map to regions that do not coincide with IRF4 peaks. Right, BATF ChIP-seq signal distribution in the shared “BATF and IRF4” peaks (blue) and in “BATF-only” peaks (red). Regions in the histogram corresponding to the peaks of high BATF occupancy (Log2(αBATF_RPM) > 0.5) and low BATF occupancy (Log2(αBATF_RPM) < −2), as discussed in the text, are indicated.
b, Heatmaps of the IRF4 ChIP-seq signal in BATF-transduced, BATF-HKE-transduced, and pMIG-transduced cells, at IRF4 peak locations called in pMIG-transduced cells. Curves at the top show the average signal taken over all peaks in the respective heatmap. The average signal is modestly decreased in BATF-overexpressing cells compared to pMIG control cells, and substantially reduced in BATF-HKE-overexpressing cells.
c, Heatmap of normalized RNA-seq reads (as z-scores) under the indicated conditions, for the top 100 genes differentially expressed in pMIG-transduced cells after αCD3/αCD28 stimulation.
d, MA plot of RNA-seq data from BATF-transduced versus pMIG-transduced CD8+ T cells expanded in vitro as for adoptive transfer, without restimulation. Differentially expressed genes more highly expressed in BATF-transduced cells (red dots) or in pMIG-transduced cells (blue dots) are indicated. Selected genes are labelled.
e, MA plot of RNA-seq data from BATF-transduced versus pMIG-transduced CD8+ T cells expanded in vitro, and restimulated with αCD3/αCD28 for 6 h. Differentially expressed genes more highly expressed in BATF-transduced cells (red dots) or in pMIG-transduced cells (blue dots) are indicated. Selected genes are labelled.
f, IRF4 (left) and IRF8 (right) expression detected by flow cytometry (MFI) in pMIG- and BATF-transduced CD8+ T cells that had been expanded in vitro, at the indicated times after restimulation with αCD3/αCD28. The black symbol on the y-axis shows expression in naïve CD8+ T cells.
Data in a and b were obtained from two, and data in c-e from three, independent biological experiments. Data in f are representative of two independent biological experiments.
The IRF4 ChIP-seq profiles at BATF-IRF4 peaks were qualitatively similar in pMIG control cells and in BATF-overexpressing cells, implying that endogenous levels of BATF and BATF3 are sufficient to recruit IRF4 in most cases (Fig. 6b). On close examination, though, IRF4 binding at peaks called in pMIG cells was decreased, on average, in BATF-overexpressing cells (Fig. 6b); and IRF4 redistributed within the smaller subset of IRF4 peaks called in BATF-overexpressing cells (Extended Data Fig. 8).
It is known that BATF-HKE can bind adjacent to IRF4 at AICE sites, but that it does not cooperate with IRF4 to stabilize IRF4 binding33. In our experiments, despite binding at the same sites as wildtype BATF, and increasing total BATF binding (BATF-HKE plus endogenous BATF) at IRF4 peaks over that in pMIG control cells (Extended Data Fig. 8), overexpressed BATF-HKE decreased the average IRF4 signal substantially below the level in control cells (Fig. 6b). The most likely mechanism is competitive displacement of endogenous BATF and BATF3. Correspondingly, gene expression in BATF-HKE-overexpressing cells deviated from the pattern common to BATF-overexpressing and pMIG control cells, especially in unstimulated cells (Extended Data Fig. 8).
Early changes in gene expression
The pattern of gene expression was very similar in BATF-transduced and pMIG-transduced cells in vitro, whether considering the subset of mRNAs that exhibited the most significant upregulation or downregulation upon αCD3/αCD28 stimulation (Fig. 6c) or all mRNAs at rest and upon activation (Fig. 6d,e). The congruence in gene expression is consistent with the similar patterns of chromatin accessibility and similar IRF4 binding at BATF-IRF4 peaks in BATF-overexpressing and control pMIG-transduced cells. The overall similarity does not imply that the patterns of gene expression in BATF-transduced and pMIG-transduced cells are identical (Supplementary Table 4). It is particularly notable that Tbx21 (encoding T-bet) is upregulated in BATF-overexpressing cells at the time of adoptive transfer, and Eomes is downregulated, which could well predispose the cells toward effector function and against exhaustion2,40–42.
Induction of IRF4 and IRF8 proteins upon stimulation is reduced in BATF-overexpressing cells compared to control cells (Fig. 6f). This is a clear indication that some aspects of TCR signalling have been rewired in BATF-overexpressing cells, and it may have special relevance in light of the finding that high IRF4 expression can antagonize the beneficial effects of BATF on tumor control.
Newly accessible chromatin sites
We asked whether overexpressed BATF might act as a pioneer factor to open new chromatin sites19,22,43,44. BATF ChIP-seq peaks with a substantially higher signal in BATF-overexpressing than in control cells, as a group, do not display correspondingly elevated local chromatin accessibility (Fig. 7a). However, when the peaks are subgrouped into quartiles based on the ATAC-seq signal in pMIG cells, increased BATF binding in BATF-overexpressing cells is correlated with opening of chromatin for regions comprising the lowest quartile of ATAC-seq signal (Fig. 7a). Whether BATF binding is causative for increased chromatin accessibility can only be tested directly by engineered mutation of these sites.
Figure 7. Relation of BATF binding to chromatin accessibility and gene expression in BATF-transduced cells.
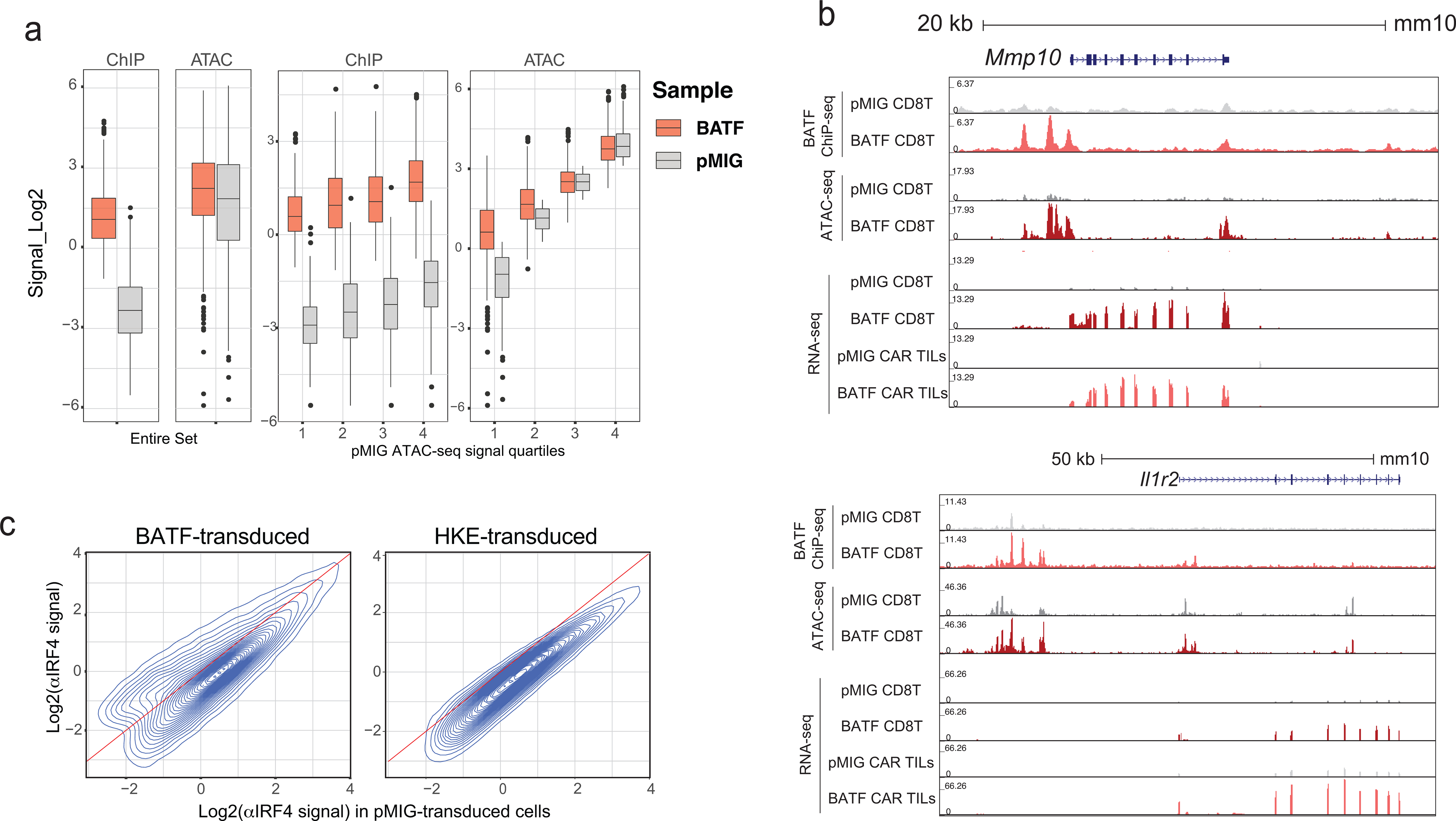
a, Box-and-whisker plots showing the distribution of CPM-normalized ATAC-seq and BATF ChIP-seq signals in the collection of BATF ChIP-seq peaks (2504 peak regions) with a substantial increase in signal (log2FC ≥ 3) in BATF- compared to pMIG-transduced cells. Left, for the entire set; right, subdivided into quartiles based on the ATAC-seq signals from pMIG-transduced cells. The box plots represent the minimum, 1st quartile, median, 3rd quartile, and maximum of their respective samples, excluding outliers
b, Examples of gene loci where increased BATF binding and increased chromatin accessibility correlate with increased gene expression. Genome browser views of the Mmp10 (top) and Il1r2 (bottom) loci, showing BATF ChIP-seq, ATAC-seq, and RNA-seq signals from pMIG- and BATF-transduced CD8+ T cells expanded in vitro, as well as RNA-seq signals from pMIG- and BATF-transduced CAR TILs.
c, Contour plots relating the IRF4 ChIP-seq signals (log2(CPM)) in BATF-transduced (left) or BATF-HKE-transduced (right) CD8+ T cells to the signals from the corresponding peaks in pMIG-transduced cells.
Data in a-c were obtained from two or three independent biological experiments.
Natural questions are, What genes are nearby? Are any of them upregulated? A full list of the neighboring genes is given in (Supplementary Table 5). At least some of these genes are upregulated both pre-transfer and in TILs— examples are Mmp10 and Il1r2 (Fig. 7b) — suggesting that increased chromatin accessibility may contribute to increased gene expression in the relatively small number of loci where BATF binding and chromatin accessibility are sharply higher in BATF-overexpressing cells. The main conclusion, though, is that overexpressed BATF binds predominantly within chromatin regions that are accessible in control pMIG cells, comprising regions that were already accessible in naïve CD8+ T cells and regions that became accessible when the cells were activated prior to retroviral transduction.
Redistribution of IRF4 among its binding sites
We established that normalized αIRF4 ChIP-seq reads report accurately on IRF4 binding at individual sites, for comparisons between BATF-overexpressing cells and control cells (Methods; Extended Data Fig. 9). Quantitative examination of the data then led to two substantive conclusions. First, echoing the finding for the average IRF4 signal at its peaks in Figure 6b, IRF4 binding was measurably decreased at most peaks in BATF-overexpressing cells (Figure 7c, left). Second, there was a redistribution of IRF4 among its binding sites, since IRF4 binding was unchanged or increased at a minority of peaks (Figure 7c, left; Extended Data Fig. 9).
The reduced induction of IRF4 in wildtype BATF-overexpressing cells can account for the decrease in IRF4 binding at most peaks. But why was IRF4 binding to certain sites higher in BATF-overexpressing cells than in pMIG cells? Stabilization of IRF4 binding by increased BATF occupancy at AICE sites is probably one contributing factor, and its contribution will vary from site to site, because the binding energies of BATF-IRF4-DNA complexes vary with the DNA sequence at the individual site45. In a motif-enrichment analysis of peaks that were farther off the diagonal— either above or below— in Fig. 7c than would be expected just from scatter in the data, the top two ‘known’ motifs in the set above the diagonal were different versions of the BATF-IRF element AICE (Fig. 8a,b). These motifs were seen in a substantial fraction of the peaks. The top de novo enriched sequence was very similar to the alternative BATF-IRF element AICE2, and was detected in nearly half of the peaks (Fig. 8c). As a stringent test, we searched for exact matches to four recognized strong AICE2 sequences45 and for exact matches to a specialized version of AICE233. We found massive enrichment for strong AICE2 sequences in the set above the diagonal (Fig. 8d)— even though the peaks in the comparison set below the diagonal were also identified as IRF4-binding peaks by ChIP-seq, and most of them overlapped BATF-binding peaks. Consistent with their increased BATF and IRF4 binding, αIRF4 peaks above the diagonal tended to exhibit greater chromatin accessibility in BATF-overexpressing cells (Extended Data Fig. 9). Stabilization of IRF4 binding by neighboring transcription factors that are differentially expressed or differentially activated in BATF-overexpressing cells is another likely contributing factor and will also depend on the local DNA context.
Figure 8. Regions of high IRF4 binding in BATF-overexpressing cells are enriched in strong consensus AP1-IRF (AICE) and AICE2 motifs.
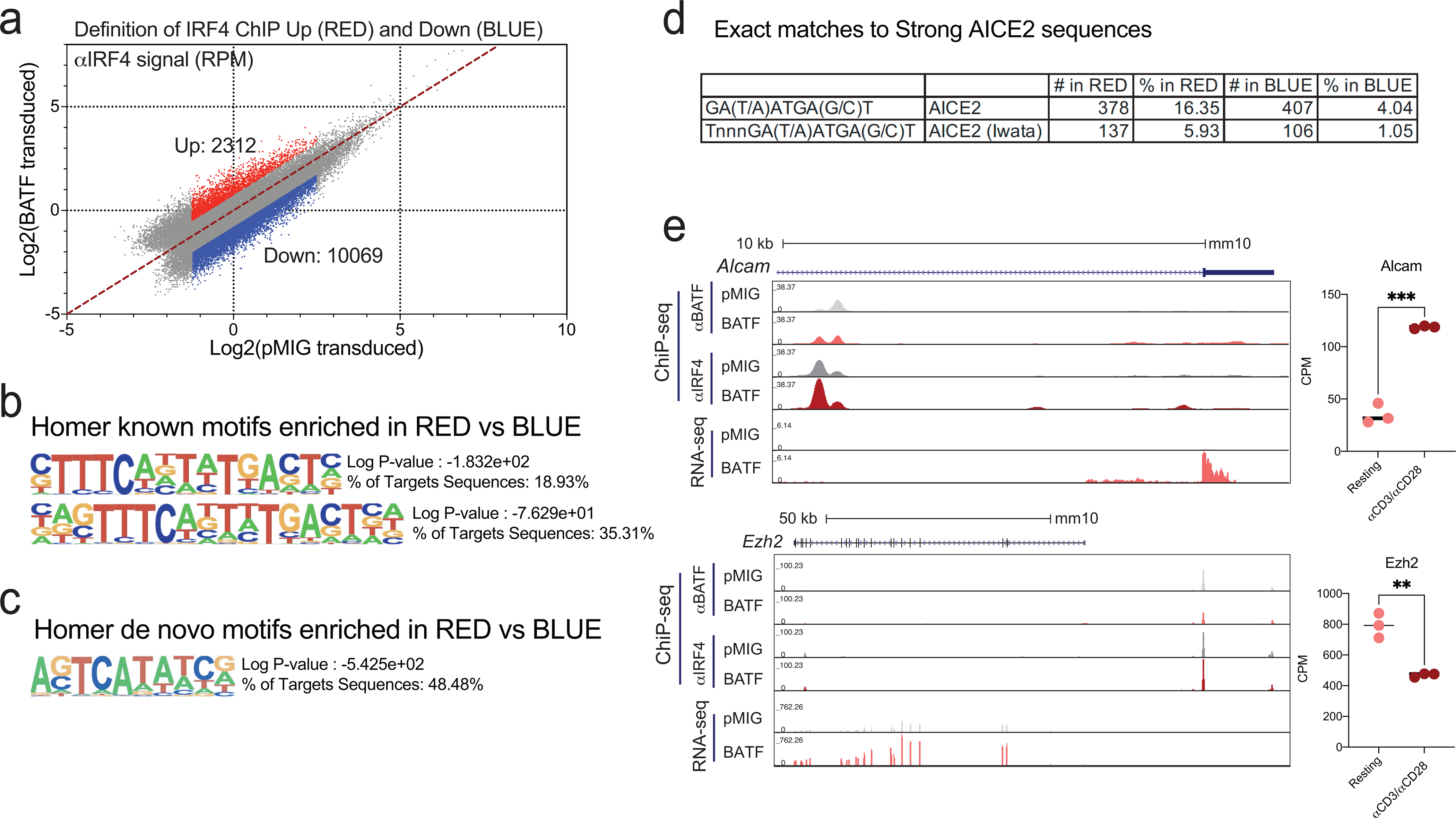
a, Dot plot highlighting regions of the IRF4 ChIP-seq data from Fig. 7c, left, where IRF4 binding increases (log2FC ≥ 0.75, red dots) or decreases (log2FC ≤ −0.75, blue dots) in BATF-overexpressing relative to pMIG-transduced cells. Peaks with very low (log2(signal) < −1.25) or high (log2(signal) > 2.5) IRF4 binding in pMIG cells were judged unlikely to be informative and were omitted from the analysis.
b, Top enriched known motifs reported by HOMER for IRF4 peak regions with increased binding (red dots in a), using as background the peak regions with decreased binding (blue dots in a).
c, Top enriched de novo motifs reported by HOMER in the same comparison.
d, Exact matches to strong AICE2 sequences in peaks from the highlighted regions, using the consensus AICE2 sequence or a novel, specialized, AICE2 sequence from Iwata et al33.
e, Examples of gene loci where increased IRF4 binding in BATF-overexpressing cells correlates with increased gene expression. Left, Genome browser views of Alcam (top) and Ezh2 (bottom) loci, showing BATF ChIP-seq, IRF4 ChIP-seq, and RNA-seq signals from pMIG- and BATF-transduced CD8+ T cells. Right, Quantification of RNA-seq data for Alcam (top) and Ezh2 (bottom) shows expression changes in opposite directions after stimulation with αCD3/αCD28.
Data in a were obtained from two independent biological experiments. Each circle in e, right panel, represents cells expanded in vitro from one mouse. Data in e were analyzed by two-tailed unpaired Student’s t-test. **p≤0.01; ***p≤0.001.
BATF-HKE-overexpressing cells showed a consistent decrease in IRF4 binding at individual peaks, which was not due to reduced IRF4 protein (Extended Data Fig. 9), and no redistribution of IRF4 (Figure 7c, right; Extended Data Fig. 9). The major factor affecting IRF4 binding in BATF-HKE-overexpressing cells is likely to be the replacement of endogenous BATF and BATF3 at AICE sites by BATF-HKE, resulting in a lower affinity for IRF4. The consistent decrease in IRF4 binding elicited by BATF-HKE overexpression is compelling evidence that nearly all IRF4 binding in pMIG control cells depends on the interaction with BATF.
IRF4 binding and gene expression
The evidence indicates that IRF4 binding is tempered by other inputs in determining gene expression. Alcam and Ezh2 are known BATF-IRF4 target genes that exhibit both enhanced IRF4 binding and significantly higher expression in BATF-overexpressing cells (Figure 8e, left panels), but, in both cases, mRNA levels change appreciably and in opposite directions upon αCD3/αCD28 stimulation, indicating that other transcription factors have a role in determining the transcriptional output (Figure 8e, right panels). Moreover, the quantitative changes in IRF4 binding in BATF-overexpressing cells are in general small— the shift in the modal value is ~0.4 Log2 units over a broad range of ChIP-seq signals in pMIG cells, which translates to ~25% decrease in bound IRF4— and the extent of variability is restricted in most cases to a range of 1 Log2 unit around the modal value (Figure 7c). We propose that alterations in IRF4 binding may predominate in controlling the transcriptional output in some cases, while in other cases IRF4 binding only sets a bias, and other transcription factors whose levels or activities differ between BATF-overexpressing and pMIG cells determine the final output.
Discussion
The progressive development of CD8+ T cell exhaustion in tumor-infiltrating T cells and during chronic viral infection occurs through the concerted actions of transcription factors, which impose exhaustion through changes in chromatin structure and gene transcription. One approach to defeating exhaustion is to interfere with the transcription factors that drive it, and we and others have demonstrated that depletion of NR4A or TOX transcription factors – two downstream targets of NFAT that are induced by NFAT and cooperate functionally with NFAT to drive CD8+ T cell exhaustion – allows CD8+ TILs to maintain robust effector function4–10. Here we approach the same objective from a different angle, by asking whether the onset of exhaustion might be prevented by maintaining the expression of transcription factors that favor full T cell effector function. We show that overexpressing BATF in CD8+ CAR TILs confers enhanced effector function and robust anti-tumor responses, and prevents the progressive exhaustion that would otherwise occur in the tumor environment. Notably, some BATF-transduced CAR T cells remain after tumor clearance as memory-like cells that are fully capable of making a subsequent anti-tumor response. Thus BATF overexpression corrects the two cardinal features of T cell exhaustion: the immediate limitation on effector function and the long-term limitation on memory formation.
Elements influencing BATF-overexpression-induced CD8+ TIL function are the early differential expression of Tbx21, Eomes, and other key genes in the T cells prior to adoptive transfer; alterations in signalling leading to less upregulation of IRF4 in response to TCR stimulation; consequent redistribution of IRF4 among its target sites in chromatin; blunted TCR signalling to chromatin in the tumor, with a failure to open many exhaustion-related chromatin regions that normally become accessible in CD8+ TILs; and a failure of the sustained upregulation of Tox that ordinarily occurs in the tumor.
The observed redeployment of IRF4, and the observed decreased IRF4 binding at many sites, are at first counterintuitive. Overexpressed BATF would ordinarily favor increased IRF4 binding at all BATF-IRF sites, except at sites that were fully occupied in pMIG cells. However, because of altered signalling, IRF4 levels are lower in restimulated BATF-overexpressing cells than in restimulated control cells. When IRF4 is limiting, IRF4 binds preferentially to the higher-affinity sites at the expense of lower-affinity sites, parallel to what was shown for BATF-IRF binding in CD4+ T cells subjected to brief or weak stimulation33.
The heightened effector response of BATF-transduced cells depends on BATF-IRF interaction. Previous work in Th2 and Th17 T cells established the importance of a subset of BATF sites in DNA, termed AP1-IRF composite elements (AICE), where JUN-BATF, JUNB-BATF, or JUN-BATF heterodimers bind in a complex with IRF4 or IRF820,23,24,33. The recruitment of IRF4 to these AICE sites is substantially weakened by the HKE mutations in BATF, and the HKE mutations are known to compromise IRF4-mediated transcription in Th2 and Th17 cells20,21,23,33. In our study, CD8+ CAR TILs overexpressing the BATF-HKE mutant failed to survive and expand in tumors, consistent with the known requirements for BATF and IRF4 in early effector CD8+ T cell expansion19,46.
BATF and IRF4 are both induced by TCR activation, and there is ample evidence that BATF and IRF4 are essential for metabolic reprogramming and clonal expansion of effector CD8+ T cells19,25,46,47. The modest upregulation of BATF in chronic viral infections and certain other observations led to the view that BATF and IRF4 might help to induce T cell exhaustion25,26. However, another report for chronic LCMV clone 13 infection closely paralleled our findings, demonstrating that overexpressing BATF in virus-specific P14 TCR-transgenic CD8+ T cells increased their proliferation, expression of effector markers, and control of the viral infection48. The straightforward interpretation of these varied findings is that BATF and IRF4, like NFAT, are ‘ambivalent’ transcription factors that can contribute to either effector or exhaustion programs in CD8+ T cells depending on the signalling context.
In summary, engineered expression of BATF at high levels supports effective antitumor responses in CD8+ T cells. BATF overexpression yielded CAR TILs that were skewed towards an effector phenotype, underwent striking expansion in tumors, secreted large amounts of effector cytokines, and expressed decreased amounts of TOX, a transcription factor notably associated with exhaustion. Additional experiments will be needed to completely elucidate the transcriptional networks involved. From a therapeutic point of view, however, BATF overexpression in CAR TILs has a markedly beneficial effect on both immediate and long-term anti-tumor responses, since it promotes the formation of long-lived memory cells that can control tumor recurrence.
Methods
This research complies with all relevant ethical regulations.
Approval for vertebrate animal studies: La Jolla Institute Animal Care Committee (ACC), protocol number AP00001025.
Approval for human studies: La Jolla Institute Institutional Review Board (IRB), protocol number SCRO_002. Informed consent was obtained from all human blood donors. Donors were compensated per La Jolla Institute policy.
Mice
C57BL6/J, B6.SJL-PtprcaPepcb/BoyJ (CD45.1), and C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I) mice were obtained from Jackson Laboratory. CD45.1+ OT-I mice were obtained by crossbreeding. Male mice were used for experiments on anti-tumor effects in vivo, and both male and female mouse were used for in vitro studies. 6 week-old mice were purchased from Jackson Laboratories to be used as recipient mice, and rested for at least one week after delivery before being used for experiments. Mice were age-matched and between 7–12 weeks old when used for experiments. All mice were bred and/or managed in the animal facility at the La Jolla Institute for Immunology. All experiments were performed in compliance with study protocol number AP00001025 approved by the La Jolla Institute Animal Care Committee. The approved protocol specified a maximal tumor size of 1.77cm3 or greater for three days without signs of regression. This maximal tumour size was not exceeded.
Human peripheral CD8+ T cell isolation
Whole blood samples from healthy subjects were collected by a staff phlebotomist in the Clinical Studies Core at the La Jolla Institute for Immunology, and peripheral blood mononuclear cells (PBMCs) were isolated by Lymphoprep™ (Stem cell), according to the manufacturer’s protocol. Peripheral CD8+ T cells were negatively isolated using a human CD8+ T cell isolation kit (Stem cell) following the manufacturer’s protocol.
Construction of retroviral and lentiviral vectors
CAR expression plasmid.
The sequence of the retroviral vector (MSCV-myc-CAR-2A-Thy1.1) encoding the Myc epitope-tagged chimeric antigen receptor (CAR) has been reported previously49,50; it contains the human CD19 single-chain variable fragment49 and the murine CD3ζ and CD28 sequences. The CAR cDNA was cloned into an MSCV-puro murine retroviral vector in place of PGK- puro. Human CD19 (hCD19) retroviral expression plasmid. A PCR-amplified DNA fragment encoding hCD19 was cloned into an MSCV-puro (Clontech) murine retroviral vector as we describe in previous papers5,6. Retroviral vectors (MSCV-bZIP-IRES-Thy1.1 and MSCV-bZIP-IRES-eGFP). To generate pMIG-Batf, the Batf coding sequence was amplified from pMSCV- Batf -IRES-Thy1.1 (Lio, unpublished; derived from pcDNA3.1-Batf, Addgene #34575) and cloned into pMSCV-IRES-eGFP (Addgene #27490). DNA fragments encoding Jun, Maff, and the Batf HKE-mutant were PCR amplified or synthesized as gBlocks (Integrated DNA Technologies) and cloned into the MSCV-IRES-eGFP (Addgene plasmid #27490), kindly provided by W. S. Pear (University of Pennsylvania). pMIG-IRF4 was purchased from Addgene (Addgene #58987). Lentiviral vectors (pTRPE-19.28z-P2A-NGFR and pTRPE-IRES-eGFP). The plasmid pTRPE-19.28z, which contains the human CD19 single chain variable fragment and the human CD3ζ and CD28 sequences, was kindly provided by A. D. Posey Jr. (University of Pennsylvania). A fragment containing the P2A and NGFR sequences was PCR-amplified and cloned into the pTRPE-19.28z vector to yield pTRPE-19.28z-P2A-NGFR. A fragment containing the IRES and eGFP sequences was PCR-amplified and cloned into the pTRPE-19.29z vector in place of 19.28z to yield pTRPE-IRES-eGFP. DNA fragments encoding human BATF were synthesized as gBlocks (Integrated DNA technologies) and cloned into pTRPE-IRES-eGFP.
Cloning of NFAT:AP1 reporter plasmids
A retroviral reporter plasmid containing six tandem NFAT:AP-1 sites driving GFP expression on a self-inactivating retroviral backbone was kindly provided by H. Spits51. Mouse Thy1.1 was cloned into this plasmid in place of the GFP reporter, using Gibson Assembly. The mouse genes for Jun, Maff, Batf, Batf3, Jund, Fosl2, and Nfil3 were synthesized as gBlocks (Integrated DNA Technologies) and cloned downstream of Thy1.1 with a P2A linker in between using Gibson Assembly.
Cell lines
The B16F0 mouse melanoma cell line was purchased from the American Type Culture Collection (ATCC). The B16F0-humanCD19 (B16F0-hCD19) cell line was generated by transduction with amphotropic virus encoding human CD19, followed by sorting for cells expressing high levels of human CD19. The B16F10-OVA mouse melanoma cell line was kindly provided by S. Schoenberger (La Jolla Institute for Immunology). The Platinum-E Retroviral Packaging Ecotropic (PlatE) cell line was purchased from Cell Bio Labs. All tumor cell lines were tested frequently to be sure they were negative for mycoplasma contamination and were used at passage 4 after thawing from stock.
Transfections
3 × 106 Plat-E cells were seeded in 10-cm dishes in media (DMEM with 10% FBS, 1% L-glutamine, 1% penicillin/streptomycin) the day before transfection, and the medium was changed just before transfection. For retroviral transduction, we used a mixture of 10 μg retroviral plasmid + 3.4 μg pCL-Eco packaging vectors or PCL10A1; for lentiviral transduction, the mixture contained 10 μg Lentiviral plasmid +7.5 μg Gag pol + 5 μg Rev + 2.5 μg VSV-G packaging vectors. The plasmid mixtures were incubated with 40 μl TranslT-LT1 Transfection Reagent (Mirus Bio LLC) at ~22 °C for 20 min in 1.5 ml Opti-MEM media and then added to the PlatE cells, after which the cells were incubated at 37 °C in a 10% CO2 incubator for 30–40 h. The supernatant was filtered through a 40 μm filter before being used for transduction of CD8+ T cells.
Tumor Experiments
Preparation of B16F0-hCD19 or B16F10-OVA melanoma cells for tumor inoculation:
Tumor cells (B16F0-hCD19 or B16F10-OVA) were thawed and cultured in DMEM with 10% FBS, 1% L-glutamine, 1% penicillin/streptomycin at 37 °C in a 5% CO2 incubator, and were split and passaged at days 1, 3, and 5 after thawing before inoculation. Day 0: At the time of tumor inoculation, cells were trypsinized and resuspended in phosphate-buffered saline (PBS) solution, then injected subcutaneously into 7–12 week-old C57BL/6J mice. Generation and transfer of CAR T cells. Splenic CD8+ T cells from C57BL/6, B6.SJL-PtprcaPepcb/BoyJ, C57BL/6-Tg(TcrαTcrβ)1100Mjb/J or CD45.1xOT-I mice were isolated by negative selection using a CD8 isolation kit (Invitrogen or Stem Cell), activated with 1 μg/ml anti-CD3 and anti-CD28 for 1 day, then removed from the plates and retrovirally transduced using 15 μg/ml of polybrene at 37 °C followed by centrifugation at 2000 × g for 1–2 hours. After transduction, cells were cultured in house-made T cell medium containing 100 U/ml human IL-2. A second transduction was performed the next day using the same protocol, after which cells were cultured in T cell media containing 100 U/ml human IL-2 for three days. On the day of adoptive transfer, cells were analyzed by flow cytometry to check transduction efficiency (typically 90% for single retroviral transduction and 80% for double retroviral transductions), and cell counts were obtained by using the Accuri flow cytometer. Cells were washed with PBS, and resuspended in PBS before adoptive transfer into recipient mice. Assessing anti-tumor responses: On day 0, 7–12 week-old C57BL/6J mice were injected subcutaneously with 1×105 B16F0-hCD19 or 2.5×105 B16F10-OVA cells. When tumors were palpable, tumor measurements were recorded with a caliper 3–4 times a week and tumor size was calculated as millimeter squared (length × width). On day 7, 3×106 CAR T cells or 1×106 OT-I T cells were adoptively transferred into tumor-bearing mice. For all survival experiments, tumor growth was monitored until an experimental endpoint of day 100 after tumor inoculation or until IACUC-approved endpoint of a maximal tumor size measurement exceeding a diameter greater than 225 mm2 for more than three days without signs of regression. If mice were pale, had scars or ulcerations, adopted a hunched position, or if their body temperature was low, we euthanized the mice under the guidance of the staff of the Department of Laboratory Animal Care (DLAC) at LJI. In most cases, tumor sizes were measured in a blinded manner by DLAC staff except during the holiday season or when the institute was under restricted access due to the COVID-19 shut-down. Harvesting tumor-infiltrating lymphocytes: On day 0, 7–12 week-old C57BL/6J mice were injected subcutaneously with 1×105 B16F0-hCD19 or 2.5×105 B16F10-OVA cells in PBS. When tumors were palpable, tumor measurements were recorded with a caliper 3–4 times a week and tumor size was calculated as millimeter squared (length × width). On day 12, 1.5 ×106 CAR T cells or 1×106 OT-I T cells were adoptively transferred into tumor-bearing mice. On day 20, tumors were collected from the mice and placed into C tubes (Miltenyi Biotec) containing RPMI 1640 with 10% FBS and Collagenase D (1 mg/mL; Roche), hyaluronidase (30 unit/mL; Sigma-Aldrich), and DNase I (100 μg/mL; Sigma-Aldrich). Tumors were dissociated using the gentle MACS dissociator (Milteny Biotech), incubated with shaking at 2000 rpm for 60 min at 37 °C, filtered through a 70-μM filter and spun down. Lymphocytes were separated using lymphocyte separation medium (MP Biomedicals, cat. no.: 0850494).
NFAT:AP1 reporter assays
Primary mouse CD8+ T cells were isolated from spleens of C57BL/6J mice (Jax #000664) by negative selection (EasySep #19853). Up to 5×106 freshly isolated CD8+ cells were activated with plate-bound anti-CD3 (145–2C11) and anti-CD28 (37.51) at final 1 μg/mL in TCM in a 6-well plate. After 24 hours, cells were transduced with retroviral supernatant at 32 °C for 2 hours at 2000g with 8 μg/mL of polybrene. After transduction, cells were cultured in T cell media containing 100 U/mL IL-2. On day 2, the same transduction was performed. On day 3, cells were surface stained for live CD8+ Thy1.1+ cells as a measure of reporter activity.
Flow cytometry analysis
BD Fortessa, BD LSR III, or BD Celesta flow cytometers were used for cell analysis. Cells were resuspended in FACS buffer (PBS, 1% FBS, 2.5 mM EDTA) and filtered using a 70 μm filter before running the flow cytometer. Fluorochrome-conjugated antibodies were purchased from BD Bioscience, Thermo Scientific, Miltenyi Biotech, and Biolegend. For surface staining, cells were stained with 1:100~ 1:200 dilution of antibodies in FACS buffer (PBS+1% FBS, 2.5 mM EDTA) for 15 min with FC block (BioLegend). For cytokine staining, cells were activated with 10 nM PMA, 500 nM ionomycin and 1 μg/ml Golgi plug and/or Golgi Stop in T Cell Media at 37 °C in a 10% CO2 incubator for 4 hours. After stimulation, cells were stained for surface markers and resuspended with Fix/perm (BD bioscience) buffer for 20 min, washed with FACS buffer twice and stained for cytokines at a final concentration of 1:200 in 1× BD per /wash buffer. For detection of transcription factors, cells were stained for surface markers first, after which the Foxp3/transcriptional staining kit was used according to the manufacturer’s protocol. All transcription factor antibodies were used at 1:200 dilution. All flow data were analyzed with FlowJo (v 10.6.2).
Mass cytometry (CyTOF) analysis
On day 0, 7–12 week-old C57BL/6J mice were injected subcutaneously with 1×105 B16F0-hCD19. When tumors were palpable, tumor measurements were recorded with a caliper 3–4 times a week and tumor size was calculated as millimeter squared (length × width). On day 12, 1.5×106 CAR T cells were adoptively transferred into tumor-bearing mice. On day 20, tumors were collected from the mice and placed into C tubes (Miltenyi Biotec) containing RPMI 1640 with 10% FBS and Collagenase D (1 mg/mL; Roche), hyaluronidase (30 unit/mL; Sigma-Aldrich), and DNase I (100 μg/mL; Sigma-Aldrich). Tumors were dissociated using the gentle MACS dissociator (Milteny Biotech), incubated with shaking at 2000 rpm for 60 min at 37 °C, filtered through a 70 μm filter and spun down. Lymphocytes were separated using lymphocyte separation medium (MP Biomedicals, cat. no.: 0850494), and sorted by flow cytometry based on FSC/SSC gating to get highly purified lymphocytes. After sorting, lymphocytes were rested in T cell media for 4 hours. Cells were washed with PBS, centrifuged at 400 g for 5 min and the supernatant was discarded by aspiration. Cells were resuspended in PBS with Cell-ID™ Cisplatin (5 μM), incubated at ~22 °C for 5 min, and washed with MACS staining buffer (2 mM EDTA, 2% FBS in PBS) using 5× the volume of the cell suspension. Cells were stained with a cocktail of antibodies to surface proteins with FC blocking for 15 min at ~22 °C, washed with MACS staining buffer, then fixed and permeabilized using FoxP3 staining buffer kit (eBioscience) and stained for 1 h at ~22 °C with a cocktail of antibodies to intracellular proteins. Cells were washed twice with perm/wash buffer, fixed with 1.6% paraformaldehyde for 10 min at ~22 °C, and washed twice with perm/wash buffer. Cells were stained with Cell-ID Intercalator-Ir in Fix/perm buffer overnight at 4 °C before analysis of the sample using a CyTOF mass spectrometer. All CyTOF data were analyzed with flowJO(v10.6.2) or the OMIQ.ai analysis platform.
Cell sorting
Cell sorting was performed by the LJI flow cytometry core, using FACS ARIA-I, FACS ARIA-II, or FACS Aria-fusion (BD Biosciences) flow cytometers. For transcriptional profiling using Smart-seq, 10,000 cells were sorted from the Live/Dead dye-negative CD8+Thy1.1+GFP+ population of the isolated tumor-infiltrating lymphocytes or cultured CD8+ T cells. The cells were resuspended in FACS buffer and filtered with a 70 μm filter before sorting. For ATAC-seq, 50,000 live cells were sorted using the same procedure as for Smart-seq. Cells were sorted into 1.5 ml microfuge tubes containing 500 μl 50% FBS. The sorted cells were washed with cold PBS twice before further procedures.
Cell sorting: Antibodies
The following antibodies were used: BUV 395 rat anti-mouse CD8α, clone 53–6.7 (BD Bioscience 563786); BV711 anti-rat CD90/mouseCD90.1 (Thy1.1), clone OX-7 (BioLegend 202539).
Primary cell culture
Splenic CD8+ T cells from C57BL/6 mice were isolated by using Dynabeads™ Untouched™ Mouse CD8 Cells Kit (IN vitrogen) or EasySep™ Mouse CD8+ T Cell Isolation Kit (Stem cell) following the manufacturer’s protocols, following which 3×106 CD8+ T cells/well were stimulated with 1 μg/ml anti-CD3 and anti-CD28 in T cell media at 6 well plate for 1 day, then removed from the plates and retrovirally transduced using 15 μg/ml of polybrene at 37 °C followed by centrifugation at 2000 × g for 1 h. After transduction, cells were cultured in house-made T cell media containing 100 U/ml human IL-2. A second transduction was performed the next day using the same protocol, after which the cells were cultured in T cell media with 100 U/ml human IL-2 for 3 days.
Human CAR T cell experiments
Human CD8+ T cells were stimulated with Dynabeads™ Human T-Activator CD3/CD28 (Gibco) in X-Vivo (Lonza) medium. 2 days later, Dynabeads™ were removed from the cells and the cells were lentivirally transduced using retronectin-coated plates (20 μg/ml) at 32 °C followed by centrifugation at 2000 × g for 2 h. Cells were expanded for 2 days with 500 U/ml IL-2 X-Vivo medium. Human CAR T cells were enriched by positive selection for NGFR (nerve growth factor receptor) using MACS columns and beads (Miltenyi Biotech). In vitro cytotoxicity assay: CAR T cells were labeled with CellTrace Violet dye (Invitrogen) and cocultured with NALM6 tumor cells for 5 h. % cytotoxicity was calculated as 1- (R5/R0)) × 100, R5 = (target cells (% of total) at 5 h)/ (effector cells (% of total) at 5h), R0 = (target cells (% of total) at 0 h)/ (effector cells (% of total) at 0 h). In vitro proliferation assay: CellTrace Violet-labeled CAR T cells were cultured in X-Vivo media with 500 U/ml human IL-2 for 4 days
Chromatin immunoprecipitation (ChIP)-seq library preparation
pMIG- or BATF-transduced CD8+ T cells (1×106 cells/ml in culture media) were fixed with 1% formaldehyde at ~22 °C for 10 min with nutation. To quench the fixation, 0.5 ml 2.5 M glycine was added per 10 ml, the cells were incubated on ice for 5 min, and washed twice with cold PBS. Fixed cells were transferred to low-binding tubes with 1 ml cold PBS and spun down at 2000 rpm at 4 °C for 10 min. Cells were pelleted, snap-frozen with liquid nitrogen, and stored at −80 °C until further processing. To isolate nuclei, cell pellets were thawed on ice and the pellets were resuspended in 1 ml Bioruptor lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP40, 0.25% Triton X-100), and incubated for 10 min at 4 °C with nutation. After centrifugation at 1700 × g at 4 °C for 5 min, the resulting nuclear pellets were washed twice with washing buffer (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA). The pellets were resuspended in 100 μl shearing buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 1% SDS), and sonicated using a Bioruptor in 1.5 ml bioruptor tubes (10 cycles, 30 seconds on, 30 seconds off). After sonication, the supernatants were transferred to 1.5 ml low-binding tubes, and insoluble debris was removed by centrifugation at 20,000 × g. Pellets were resuspended in 100 μl shearing buffer, and 9 volumes of conversion buffer (10 mM Tris-HCl pH 7.5, 255 mM NaCl, 1 mM EDTA, 0.55 mM EGTA. 0.11% Na deoxycholate, 0.11 % Triton X-100) was added. Chromatin was precleared with washed protein A and protein G Dynabeads for 1 hour, and the chromatin concentration was measured by qubit. 5% of chromatin was saved as input, and chromatin was incubated with anti-BATF (Brookwood Biomedical) or anti-IRF4 (clone D9P5H, Cell Signaling Technology, USA) antibodies and protein A and protein G Dynabeads overnight at 4 °C with rotation. The following day, bead-bound chromatin was washed twice with RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% NP40, 0.1% SDS, 0.5% Na deoxycholate), and then with high salt buffer (50 mM Tris-HCl pH8.0, 500 mM NaCl, 1 mM EDTA, 1% NP40, 0.1% SDS), LiCl buffer (50 mM Tris-HCl pH 8.0, 250 mM LiCl, 1 mM EDTA, 1% NP40, 1% Na deoxycholate), and TE buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA). Chromatin was eluted with 100 μl elution buffer (100 mM NaHCO3, 1% SDS, 1 mg/ml RNase A) twice for 30 min at 37 °C using a 1000 rpm shaking heat block. 5 μl proteinase K (20 mg/ml, Ambion) and 8 μl of 5 M NaCl were added to the eluted DNA, and samples were incubated at 65 °C with shaking (1,200 rpm) for de-crosslinking. DNA was purified with Zymo ChIP DNA Clean & Concentrator (Zymo Research). Libraries were prepared using NEB Ultra II library Prep kits (NEB) following the manufacturer’s instructions, and sequenced using an Illumina Novaseq 6000 sequencer (paired-end 50-bp reads).
ATAC-seq and RNA-seq library preparation
ATAC-seq libraries were prepared following the omni-ATAC protocol with minor modification52. 50,000 cells were collected by sorting and washed twice with cold-PBS at 600 × g for 5 minutes. Cell pellets were resuspended in 50 μl ATAC-lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP40, 0.1% Tween 20, 0.01% Digitonin ), and incubated on ice for 3 min, after which 1 ml washing buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween 20) was added, and the cells were spun down at 1000 × g for 10 min at 4 °C. The supernatant was removed carefully, and the nuclei were resuspended in 50 μl of transposition mix (25 μl of TD buffer [20 mM Tris-HCl pH 7.6, 10 mM MgCl2, 20% dimethylformamide], 2.5 μl of 2 μM transposase, 16.5 μl PBS, 0.5 μl 1% digitonin, 0.5 μl 10% Tween-20, 5 μl water) and incubated at 37 °C for 30 min. DNA was purified with a Qiagen MinElute Kit (Qiagen). Libraries were amplified with KAPA HiFi HS Real-time PCR master mix, and sequenced on an Illumina Novaseq 6000 sequencer (paired-end 50-bp reads). RNA-seq libraries were prepared following the SMARTseq2 protocol53 modification. Total RNA was extracted from 10,000 sorted cells by using the RNeasy Micro plus kit (Qiagen) and following the SMARTseq2 protocol as described. Libraries were prepared using the Nextera XT LibraryPrep kit (Illumina), and sequenced on an Illumina Novaseq 6000 sequencer (paired-end 50-bp reads).
ATAC seq analysis: Genome browser tracks
Paired raw reads were aligned to the Mus musculus genome (mm10) using bowtie (version 1.0.0 and -X 2000 -m 1 --best --strata -tryhard -S –fr)54. Unmapped reads were trimmed to remove adapter sequences and clipped by 1 base pair with Trim_galore (version 0.4.3)55,56 before being aligned again (-X 2500 -m 1 --best --strata -tryhard -S -- fr -v 3 -e 100). Sorted alignments from the first and second alignments were merged together with samtools (version 1.8)57, followed by removal of reads aligned to the mitochondrial genome using a custom perl script (version v5.18.1). Duplicated reads were removed with Picard tools’ Mark Duplicates (version 1.94)58. Reads aligning to the blacklisted regions (generated by Alan Boyle and Anshul Kundaje as part of the ENCODE and modENCODE’s projects)59 were removed by using bedtools intersect (version v2.27.1)60. Subnucleosomal fragments were defined as mapped pair reads with insertion distance smaller than 100 base pairs, obtained from merged mapping results. Tn5 footprint was obtained by adapting Jiang Li’s preShift.pl script, to take the strand orientation of a given read to take 9 base pairs around the start or end of the forward and reverse reads ([−4,5] and [−5,4] respectively); preShift.pl script is available in “https://github.com/riverlee/ATAC/blob/master/code/preShift.pl” and adaptation found in “https://github.com/Edahi/NGSDataAnalysis/blob/master/ATAC-Seq/Tn5_bed9bp_full.pl”. For quality control purposes, we used Xi Chen’s Fragment_length_density_plot.py python script. The Script is available in “https://github.com/Edahi/NGSDataAnalysis/blob/master/ATAC-Seq/Fragment_length_density_plot.py”. This program plots the histogram of the distances among the mapped usable reads. Final mapping results were processed using HOMER’s makeTagDirectory followed by makeMultiWigHub.pl programs (version v4.10.4)61 to produce normalized bigwig genome browser tracks for the whole mapping results, the Tn5 footprint and the subnucleosomal reads separately.
ATAC seq analysis: Differentially accessible regions
We used the complete fragments for peak calling using MACS2 callpeak function (version 2.1.1.20160309 and -q 0.0001 --keep-dup all --nomodel –call-summits)62. The narrowpeak files from all samples and replicates for in vivo (or in vitro) experiments were merged with bedtools merge (version v2.27.1)60 to generate a universe of peaks, used to obtain the Tn5 footprint signal from each sample. After limma-voom normalization63 performed in the Tn5 signal, and a linear model fitter to each region, computation of significance statistics for differential enrichment (accessibility) was done by empirical Bayes moderation of the standard errors, with [−1,1] (lfc) as the interval for null hypothesis. A region was considered differentially accessible (DARs) if a Log2FC ≥ 1 and Adj p-value ≤0.05 threshold was met. The Tn5 signals from in vivo and in vitro experiments were analyzed independently from one another. The MA plots used the merged signal from replicates. Used R64 packages: IRdisplay65, limma66, edgeR67, Glimma68, Mus.musculus69, RColorBrewer70, ggplot271, GenomicRanges& GenomicAlignments72, and pheatmap73.
ATAC seq analysis: Venn diagrams
DARs from TIL’s were intersected with bedtools intersect (version v2.27.1)60 with default parameters (1bp overlapped considered an overlap) against the exhaustion- or the activation-related regions from Mognol et al.36 (GSE88987). The overlaps were used to plot the Venn diagrams for both BATF and pMIG TILs. One-tailed Fisher test (Fisher’s exact test on 2×2 contingency tables in MATLAB)74 was done to calculate the significance of the overlaps.
ATAC seq analysis: Heatmaps
The z-score from the limma-voom63 normalized signal from TIL and CD8+ T cell samples in the regions of interest (pMIG or BATF DARs from either TILs or CD8+ T cells) was clustered by the region’s signal (cluster_rows = T) and plotted using the R library pheatmap73.
ATAC seq analysis: Quartiles boxplots from ChIP regions
The raw Tn5 signal72 from the 2504 ChIP-seq regions meeting the criterion log2(Tn5 signal in BATF-overexpressing cells/ Tn5 signal in pMIG control cells) ≥ 3 was RPM-normalized for both BATF and pMIG CD8+ T cells, with the RPM per replicate averaged. The regions were subdivided in quartiles with respect to the pMIG Tn5 RPM signal and the signal for both ATAC- and ChIP-seq data were then plotted71 altogether.
ATAC seq analysis: Known motifs analysis
A region was called differentially accessible when it had a two-fold difference and an adjusted p-value (false discovery rate, FDR) lower than 0.05, and was repeated for in vitro experiments. The Differentially Accessible Regions per condition and per experiment (BATF and pMIG, in vivo and in vitro) were used as input for HOMER’s findMotifsGenome.pl (version v4.10.4)57.
RNA-seq analysis: Genome browser tracks
Paired reads were mapped to STAR75 using the parameters (--outFilterMultimapNmax 30 --outReadsUnmapped Fastx --outSAMattributes All --outSAMprimaryFlag OneBestScore --outSAMstrandField intronMotif --outSAMtype BAM SortedByCoordinate --quantMode GeneCounts). Mapping results were processed using HOMER’s makeTagDirectory61 twice, once for the individual replicates and a subsequent one merging them (for a less crowded genome browser session), followed by makeMultiWigHub.pl programs (version v4.10.4) to produce normalized bigwig genome browser tracks.
RNA-seq analysis: MA plots of differential gene expression (TILs)
Counts per gene were obtained from STAR’s “STAR_gene_counts” (version subread-2.0.0-source)75 Differential Gene Expression was done with R (version 3.5.2) and these packages: IRdisplay65, limma66, edgeR67, Glimma68, Mus.musculus69, RColorBrewer70, gplots76. In brief, count reads from STAR were read and voom-normalized after both CPM conversion and removal of genes whose CPM was lower than 1 across less than a third of total samples. After limma-voom normalization performed in the gene’s signal, and a linear model fitter to each gene, computation of significance statistics for differential gene expression (DGE) was done by empirical Bayes moderation of the standard errors, without intervals for the null hypothesis ([0,0] lfc). A gene was considered DGE if the adjusted p-value (FDR) ≤0.1 threshold was met. Colors in the MA plots for these genes indicate these parameters (red indicates genes more expressed in BATF-transduced compared to control pMIG-transduced cells; blue, vice versa; gray indicates genes that are not differentially expressed).
RNA-seq analysis: MA plots of differential gene expression (in vitro)
Similarly processed as in the previous section, now using an interval for the null hypothesis of [-log2(1.2), log2(1.2)] lfc. A gene was considered DGE if both the absolute Log2FC was ≥ 2 and the adjusted p-value (FDR) ≤0.05 threshold was met.
RNA-seq analysis: Gene signal heatmaps
The heatmaps are composed of the top 100 most significant (adjusted p-value) differentially expressed genes in pMIG control cells between 0 h and 6 h after restimulation. The limma-voom normalized signal for all of the pMIG-, BATF- and HKE- transduced samples was Z-score transformed gene-wise. The Z-score normalized data were then used to plot the heatmaps with the heatmap.2 function from gplots76 R package.
ChIP-seq analysis: Genome browser tracks
Paired raw reads were aligned to the Mus musculus genome (version mm10) using bwa77 mem (version 0.7.15-r1144- dirty). Unmapped reads were trimmed to remove adapter sequences and clipped by 1 base pair with Trim_galore (version 0.4.3)55,56 before being aligned again. Sorted alignments from the first and second alignments were merged together with samtools (version 1.8), followed by removal of reads aligned to the mitochondrial genome using a custom perl script (version v5.18.1). Duplicated reads were removed with Picard tools’ Mark Duplicates (version 1.94)58. Reads aligning to the blacklisted regions (generated by Alan Boyle and Anshul Kundaje as part of the ENCODE and modENCODE’s projects) were removed by using bedtools60 intersect (version v2.27.1). Final mapping results were processed using the HOMER61 makeTagDirectory followed by makeMultiWigHub.pl programs (version v4.10.4) to produce normalized bigwig genome browser tracks.
ChIP-seq analysis: Venn diagram
For each sample, peaks were called using MACS262 (version 2.1.1.20160309) callpeak function, using the sample’s respective input dataset, qvalue of 0.05 --keep-dup all and --nomodel parameters. The narrowpeak files among replicates were merged using bedtools merge60 (version v2.27.1). To identify overlapping genes by the merged narrowpeak files per condition, we used the UCSC Mus musculus mm10 annotation genes. Called peaks were assigned to a gene if they overlapped with a window containing the body of the gene (the longest transcription unit for the gene locus definition) plus the 20-kb region upstream of the TSS and the 5-kb region downstream of the 3’ end of the gene. Each gene was considered only once and the whole gene set was used to find shared genes among the samples being compared. The overlap was conducted with the bedtools60 intersect function (version v2.27.1). Venn diagrams of shared overlapping genes were produced using R (version 3.5.2) as well as the libraries VennDiagram78 (doi.org/10.1186/1471-2105-12-35) and “viridis”79.
ChIP-seq analysis: Probability per base pair BATF binding site
Peaks from BATF-transduced CD8+ T cells subjected to ChIP-Seq with anti-BATF antibodies were functionally annotated to the mm10 using HOMER61 annotatePeak.pl program. Distance to nearest TSS and gene name were filtered from the annotation results. A sublist of the genes differentially expressed between BATF- and pMIG-transduced CD8+ T cells, identified by RNA-seq analysis, was used to subset separately the peak annotation results for genes upregulated and downregulated in BATF-transduced cells. The genomic histograms were generated using R (3.5.2)64 and ggplot271 with all the peak results, whereas the upregulated and downregulated histograms used the subset of genes generated above. The percentage of genes closer than 20 kb was obtaining by taking the absolute value to the closest TSS that was lower than or equal to 20 kb. The distances were numerically sorted and an empirical cumulative distribution function was generated based on the data.
ChIP-seq analysis: Removal of spurious peaks
All the peaks from all the different conditions and replicates were merged into a singularity table keeping track of which condition belonged to what region. For the superset of peaks belonging to the αBATF IP, we kept peaks whose average RPM input signal across the pMIG-, BATF-, and HKE-transduced INPUT samples was lower than 0.75 times the αBATF IP RPM signal from BATF-overexpressing cells. Similarly, for the aIRF4 IP superset, we kept peaks where said INPUT signal was lower than 0.75 times that of the αIRF4 IP RPM signal from pMIG control cells. These filtered supersets were used for all subsequent analysis.
ChIP-seq analysis: Normalized αIRF4 ChIP-seq reads report accurately on IRF4 binding
It cannot be taken for granted that a difference in the normalized αIRF4 signal (in RPM) between pMIG and BATF-overexpressing cells reports on a change in IRF4 binding at the peak in question. The general issue is that normalization of the IRF4 signal at a particular peak to total mapped reads introduces a second independent variable into the measurement. If, for example, there were free IRF4 in the nucleus of pMIG cells, and if overexpressed BATF recruited this additional IRF4 to sites in DNA, then a greater total amount of IRF4-bound DNA would be precipitated from BATF-overexpressing cells. For any individual site where exactly the same amount of IRF4-bound DNA was precipitated as from pMIG cells, the normalization would result in an artifactually lower RPM value.
To address this issue, we utilized a subset of nonspecific background DNA regions that are equally represented in the input samples and in immunoprecipitated samples from the same cells. The reads mapping to these regions in immunoprecipitated samples— which seem to represent a low fraction of input DNA carried along by nonspecific binding to the protein A/protein G beads— can serve as an internal standard. Specifically, we selected the twenty spurious peak regions with the largest ATAC-seq signal in pMIG cells (see preceding section), since the high total signal ensured that any fractional contribution to the signal from actual IRF4 binding would be negligible. The spurious ‘αIRF4’ ChIP-seq signal from these regions was consistently the same in BATF-overexpressing and pMIG cells (Extended Data Fig. 9), which implies that normalization does not distort the comparison between BATF-overexpressing and pMIG samples, and that a decrease in the normalized αIRF4 signal for an individual specific αIRF4 peak means that there was an actual decrease in IRF4 binding at that peak.
ChIP-seq analysis: Scatter and contour plots
Each scatterplot is based on the log2 of the RPM IP signal of a subset of regions representing those of interest (for example, αBATF IP signal from BATF-overexpressing cells versus αBATF IP signal from pMIG control cells). We took the union of peaks for the illustrated samples and fetched the αIRF4 and/or the αBATF average RPM IP signal (as indicated in the graphs) followed by a log2 transformation. These normalized signals were then processed in R using ggplot’s’ function geom_bin2d(bins = 300) for the scatterplots (density, i.e. occurrences of points per region) and geom_density_2d(bins=30) for the contour plots71.
ChIP-seq analysis: Overlap measurement as reads-in-peaks percentage (RiP%)
For the Venn diagrams of Fig. 6a and Extended Data Fig. 8b, we took the union of the peaks in the two conditions considered (for example, the union of αBATF ChIP-seq peaks from BATF-overexpressing and from pMIG control cells), conserving the information of whether an individual peak was unique to one condition or shared between the two conditions. For each condition, we divided the number of reads that mapped to peaks shared by both conditions by the total reads mapped to peaks in that condition, obtaining the RiP% in shared peaks and— by complementation— the RiP% in unique peaks.
ChIP-seq analysis: Histograms of signal distribution among subsets of peaks
For the histograms of Fig. 6a and Extended Data Fig. 8b, the identities of peaks that were unique to a specified condition, or shared with a second condition, were used to fetch the RPM-normalized ChIP-seq signals for peaks in each subset. The values were then log2 transformed, the median value calculated from the log2-transformed data, and its distribution was plotted as a histogram using R’s ggplot2 function “geom_histogram”71.
ChIP-seq analysis: Heatmaps
We used the deepTools80 computeMatrix function (with parameters --referencePoint center -a 1000 -b 1000 --binSize 50 --averageTypeBins mean --missingDataAsZero -p 4) to compute the signal matrices across all the conditions. The regions that were used are the input-corrected peaks, one peakset per condition. The bigwig datasets used to fetch the signal were the HOMER-normalized bigwigs (same ones as used in the genome browser track). We then proceeded to give this program’s output as input to the deepTools’ plotHeatmap function (with parameters --averageType mean --plotType se --averageTypeSummaryPlot mean --sortRegions descend --sortUsing mean --sortUsingSamples 6 --refPointLabel Center --missingDataColor yellow).
Statistical Analysis
No statistical method was used to predetermine sample size. No data were excluded from the analyses. Tumor-bearing mice were randomly assigned to adoptive-transfer treatment groups. In most cases, tumor sizes were measured in a blinded manner by DLAC staff, except during the holiday season or when the institute was under restricted access due to the COVID-19 shut-down. Investigators were not blinded to sample identity when analyzing T cells recovered from the tumors. Details of the sample sizes, replicates, and statistical tests used are provided in the individual figure legends.
Data availability
The RNA-seq, ATAC-seq, and ChIP-seq data have been deposited at the Gene Expression Omnibus (GSE154747). Other data that support the findings of this study are available from the corresponding authors upon request.
Code availability
The base code used to preprocess ATAC-seq, RNA-seq, and ChIP-seq data, and the code used for analysis of differential gene expression, have been deposited in Github: https://github.com/Edahi/NGSDataAnalysis.
Extended Data
Extended Data Fig. 1. Identification of bZIP transcription factors capable of increasing NFAT:AP-1 reporter activity.
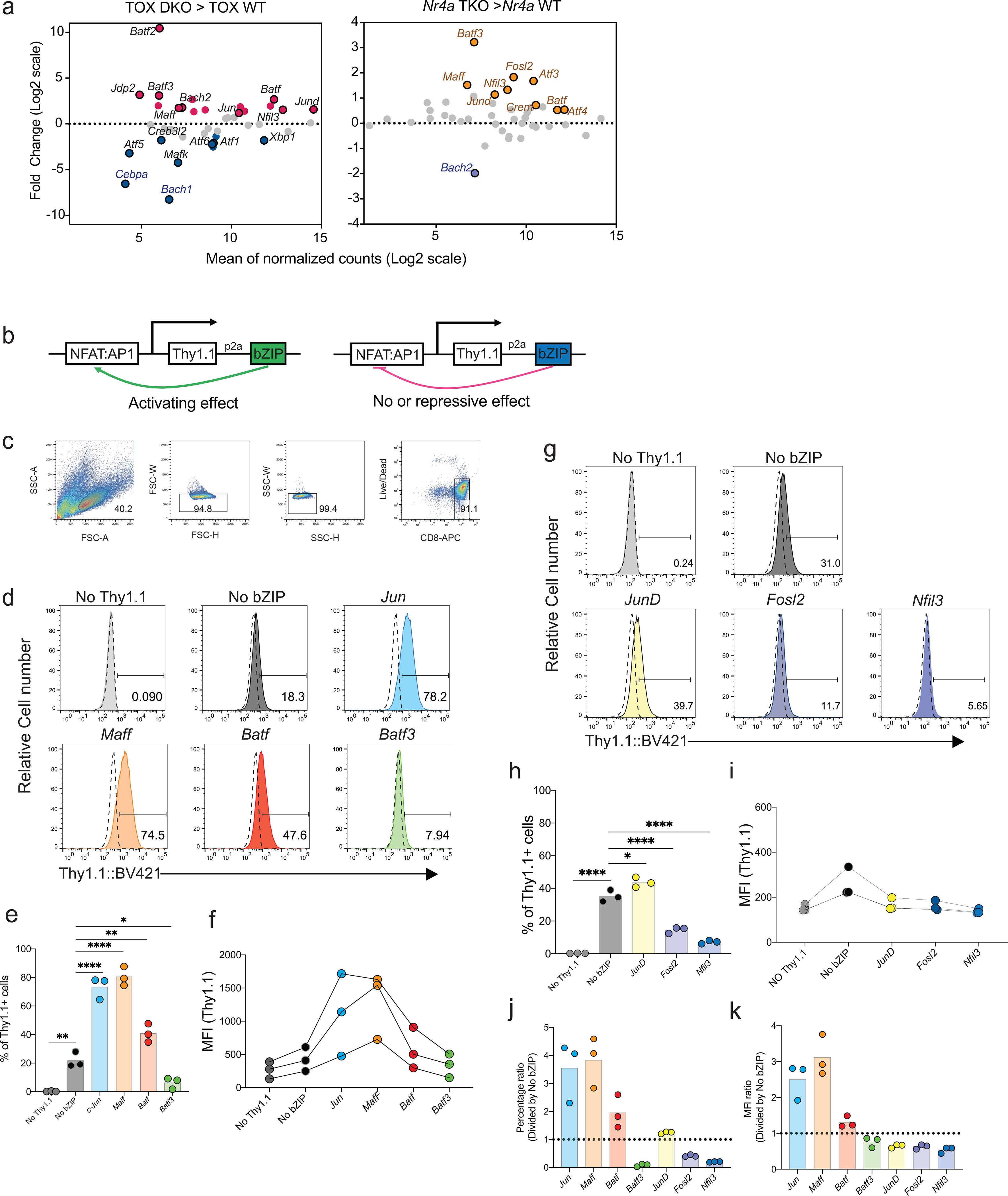
a, MA plots of basic region-leucine zipper (bZIP) transcription factor gene expression in TOX-depleted (TOX DKO, left) or NR4A-depleted (Nr4a TKO, right) CAR TILs5,6— which mount increased anti-tumor responses— relative to control CAR TILs. Differentially expressed genes (adjusted p-value < 0.1, log2 fold-change ≥ 0.5 or ≤ −0.5) are highlighted; selected genes are labeled.
b, Basis of the experiment to identify bZIP transcription factors that activate an NFAT:AP-1 reporter through a positive feedback loop, either directly by binding adjacent to NFAT on the NFAT:AP-1 composite site or indirectly by increasing the expression or activity of NFAT or AP-1. Mouse CD8+ T cells were transduced with retroviral expression vectors encoding a Thy1.1 reporter, separated by a P2A sequence from a co-expressed bZIP transcription factor, and under the control of six tandem NFAT:AP1 sites upstream of a minimal IL-2 promoter. Transcription factors for testing were chosen based on the data analysis in a.
c, Gating strategy for the experiments.
d, g, Histograms of Thy1.1 expression after CD8+ T cell transduction. No Thy1.1, transduced with empty retrovirus without Thy1.1 or bZIP transcription factor; No bZIP, transduced with retroviral vector encoding Thy1.1 but no bZIP transcription factor, a condition that assesses the background induction of Thy1.1 by endogenous NFAT and AP-1; Jun, Maff, Batf, and Batf3 (d) and JunD, Fosl2, and Nfil3 (g), transduced with vector encoding the indicated bZIP transcription factor. e, h, Percentage of Thy1.1+ cells in three replicate experiments.
f, i, MFI of Thy1.1 expression in these experiments.
j, k, Results for each sample, normalized to those of the No bZIP control from the same experiment.
Each circle in e, f, h, I, j and k represents one mouse. Data are representative of (c, d, g) or obtained from (e, f, h-k) three biological replicate experiments. Data in e and h were analyzed by one-way ANOVA. *p≤0.05; **P≤0.01; ***P≤0.001; ****P≤0.0001.
Extended Data Fig. 2. Phenotypic analyses of pMIG- and BATF-transduced CAR TILs.
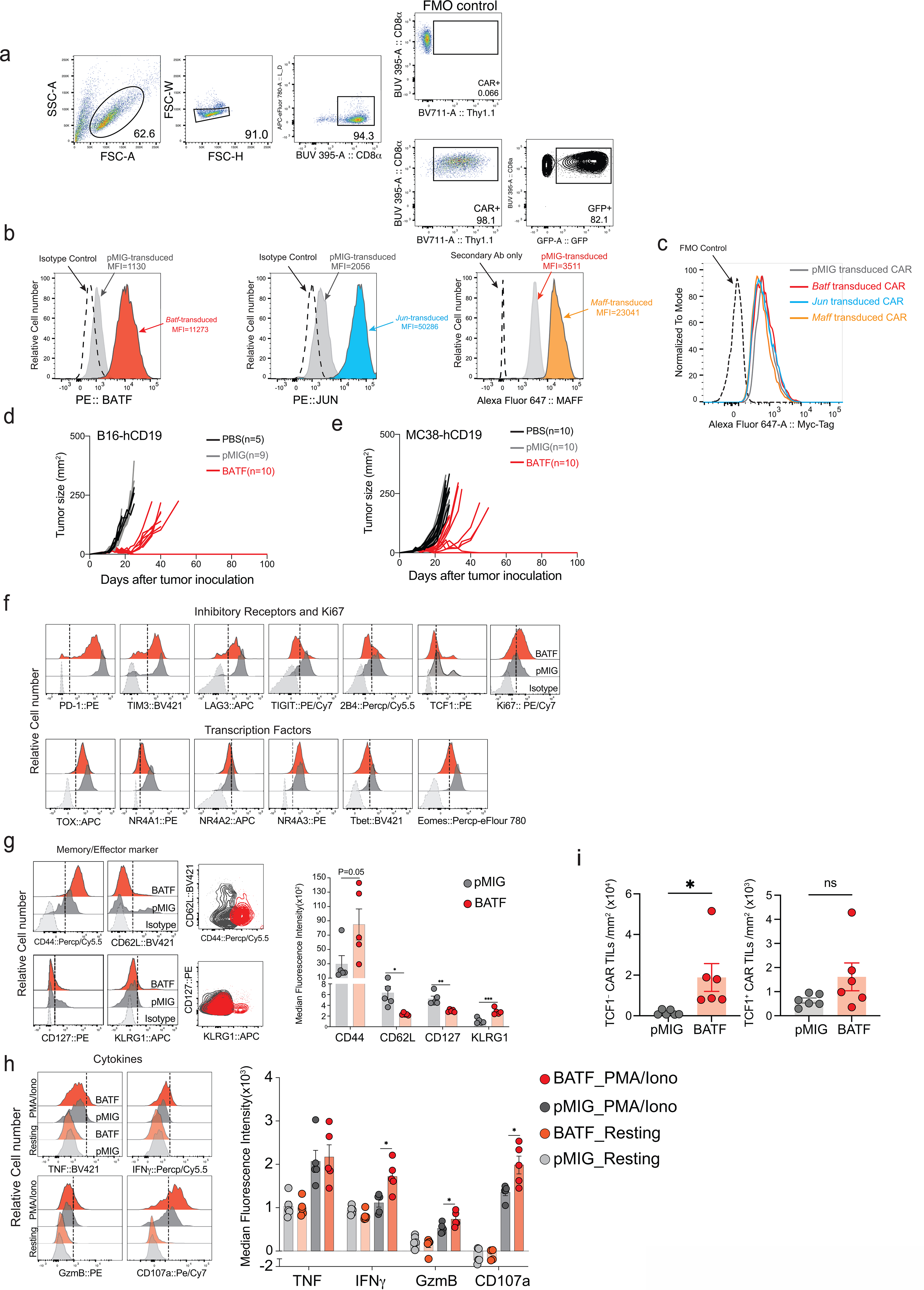
a, Retroviral transduction efficiencies for CAR and MSCV-IRES-eGFP retroviral expression plasmids, assessed as expression of Thy1.1 and GFP respectively. FMO, fluorescence-minus-one control.
b, Histograms showing BATF (left), JUN (middle), and MAFF (right) expression after retroviral transduction of CD8+ T cells with the corresponding retroviral expression plasmids or pMIG empty-vector control, assessed by flow cytometry with antibodies to the endogenous proteins.
c, Histograms showing CAR expression (assessed by staining for the Myc tag) in pMIG- (grey), BATF- (red), JUN- (sky blue) and MAFF- (orange) transduced CAR T cells.
d-e, Replicate tumor growth experiments using B16F0-hCD19 (d) and MC38-hCD19 (e) tumor cells. 1×105 tumor cells were injected subcutaneously into the left flank of C57BL/6 mice at day 0 (D0) in 100 μl phosphate-buffered saline (PBS); 3×106 control pMIG-, JUN-, MAFF-, or BATF-transduced CAR T cells were adoptively transferred by retro-orbital injection at day 7. Tumor sizes were measured by caliper.
f, Histograms showing expression of the indicated markers by each group of CAR TILs, assessed by flow cytometry.
g, Left panels, Histograms showing expression of CD44, CD62L, CD127 and KLRG1. Middle panels, Overlaid contour plots of CD44 and CD62L (top) and CD127 and KLRG1 (bottom) in pMIG- (grey) and BATF- (red) transduced CAR TILs. Right panel, expression of the markers quantitated as MFI.
h, Left panels, Histograms showing expression of TNF, IFNγ, granzyme B, and CD107a after resting in T cell media or after stimulation with PMA and ionomycin for 4 h. Right panel, expression of the markers quantitated as MFI.
i, pMIG (n=6) and BATF (n=6). Quantitation of TCF1+ and TCF1– CAR TILs.
Each circle in g, h, and i represents one mouse, and the bar graphs represent the mean ± standard error of mean (s.e.m.). Data in d-i were obtained from two independent biological experiments. Data in g, h, and i were analyzed by two-tailed unpaired Student’s t-test.
Extended Data Fig. 3. Mass cytometric analyses.
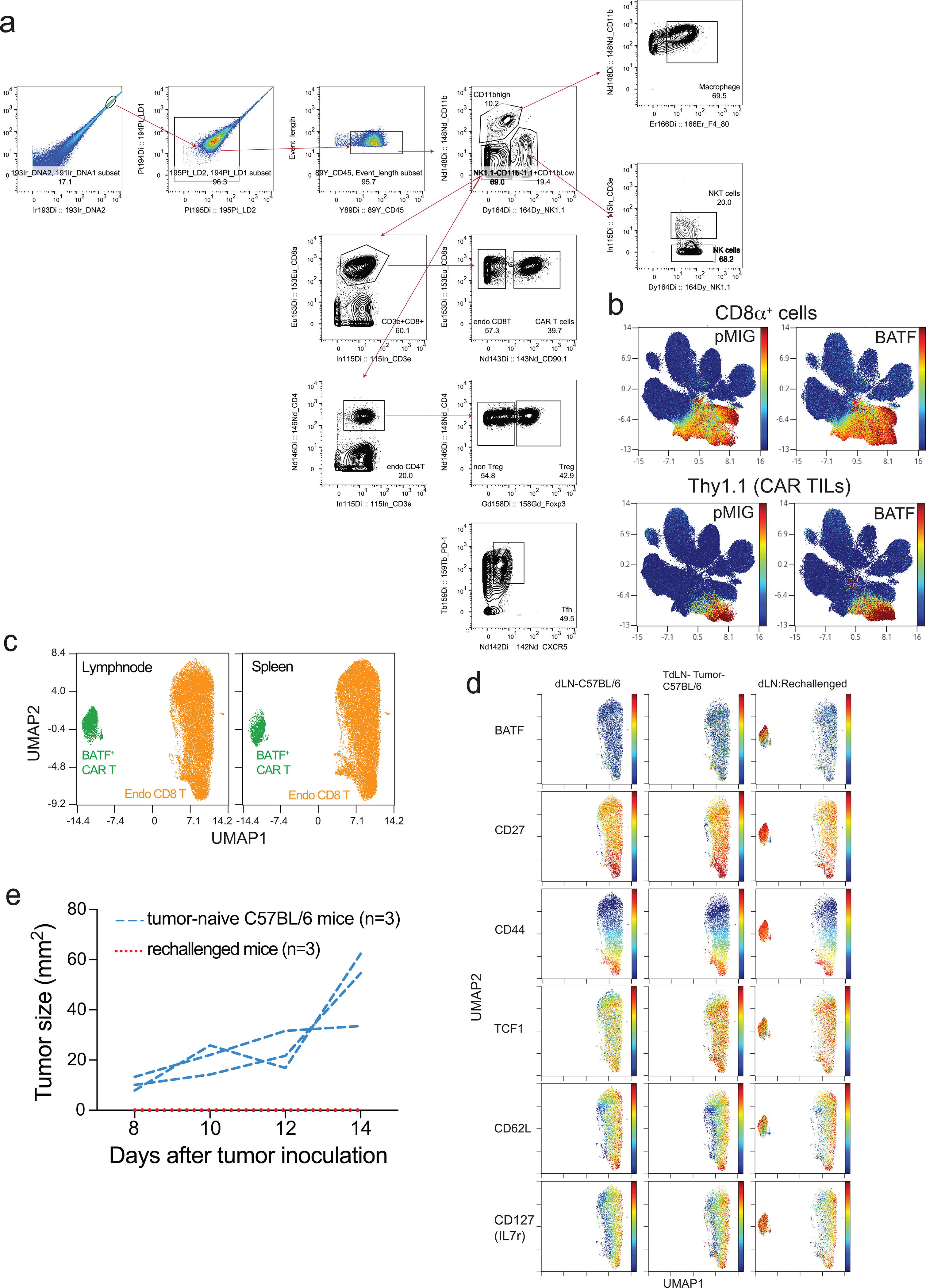
a, Gating strategy for mass cytometry of tumor-infiltrating lymphocytes (TILs).
b, UMAP plots of data from TILs of tumour-bearing mice, showing expression of CD8α (top) as a marker for all CD8+ T cells (including endogenous T cells) and Thy1.1 (bottom) as a marker for adoptively transferred pMIG- or BATF-transduced CAR T cells.
c, Mass cytometric analysis of CD8+ T cells from draining lymph nodes (left) and spleens (right) of mice in the tumor rechallenge experiments (Fig. 3). Endogenous CD8+ T cells and BATF-transduced CAR T cells are clearly distinguishable in the UMAP views.
d, Expression of selected surface markers on lymphocytes obtained from three groups of age-matched C57BL/6 mice: inguinal lymph nodes of completely unmanipulated, non-tumor-bearing mice (left panels); draining (inguinal) lymph nodes of mice inoculated with tumors 14 days previously (middle panels), and draining (inguinal) lymph nodes of rechallenged mice. The UMAP plots show that each marker is expressed by all or by a large fraction of lymph node CAR T cells of the rechallenged mice. Each group of samples in c and d was pooled from 5 mice. The data are representative of two independent biological experiments.
e, Replicate of the experiment in Fig. 3e. 1×105 B16F0-hCD19 tumor cells were injected subcutaneously into the right flank of age-matched C57BL/6 mice (n=3) to yield the “tumor-naïve” control group, or into the right flank of the surviving tumor-free mice (n=3) from the experiment of Extended Data Fig. 2d. In this replicate B16-hCD19 rechallenge experiment, CAR T cells accounted for ~10% of CD8+ T cells in spleen and LN of the mice, and exhibited a memory phenotype (CD44high, CD62Lhigh, TCF1high) as in the first experiment.
Extended Data Fig. 4. BATF overexpression improves effector function in human CAR T cells.
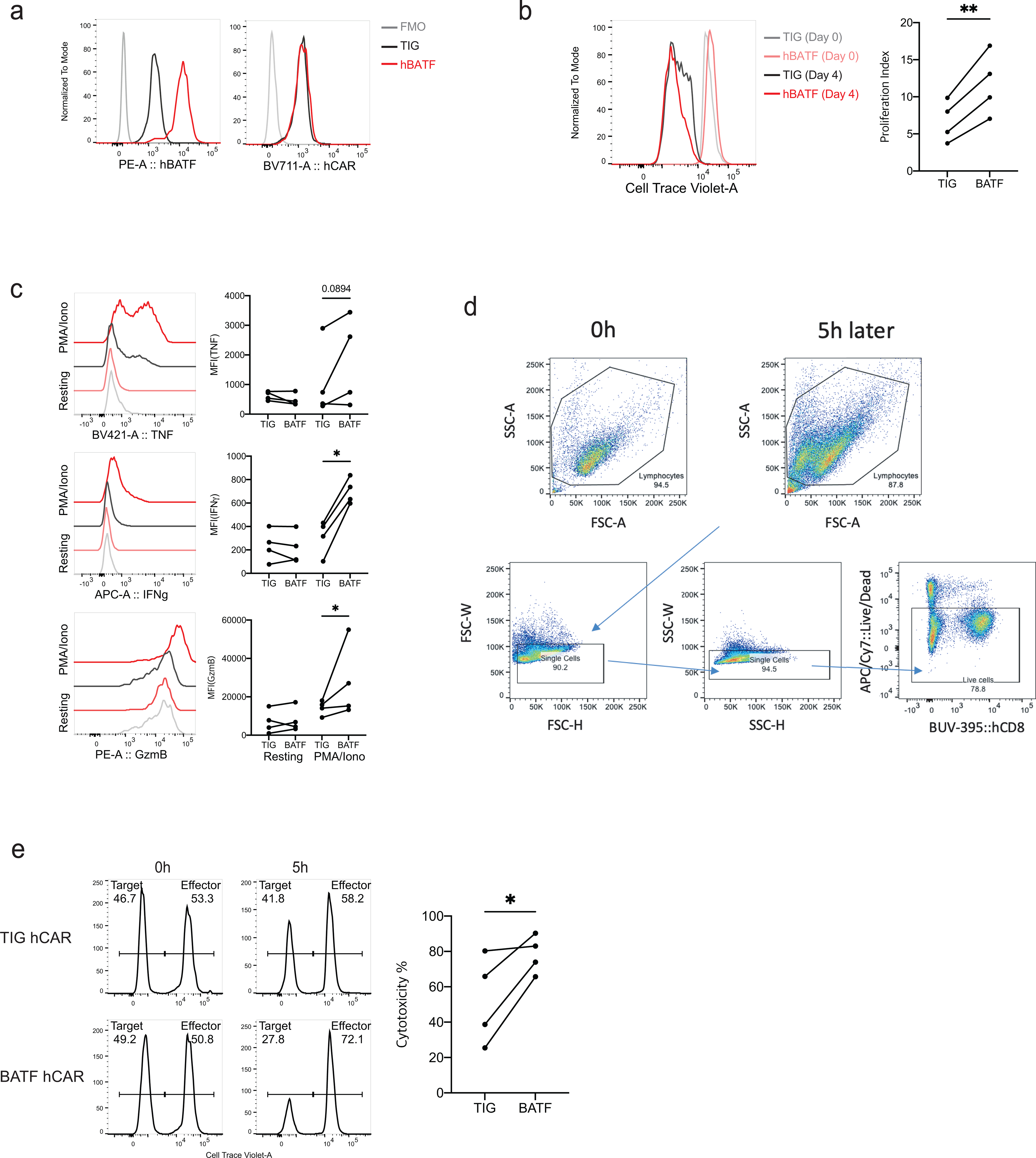
a, Histograms showing expression of human BATF (hBATF, left panel) and human CAR (hCAR [stained by Goat Anti-Armenian Hamster IgG (H+L)], right panel) in the corresponding lentivirally transduced human CD8+ T cells. FMO, fluorescence-minus-one control; TIG, control cells transduced with the empty vector.
b, Left, Histograms showing CellTrace Violet (CTV) dilution in lentivirally transduced human CD8+ CAR T cells. Right, Proliferation index calculated as (Day 0 CTV MFI) / (Day 4 CTV MFI).
c, Histograms showing expression of the indicated cytokines after resting in X-Vivo media or after stimulation with PMA and ionomycin for 4 h.
d-e, Human CAR T cells were labeled with CellTrace Violet and co-cultured with NALM6 cells for 5 h.
d, Gating strategy for in vitro cytotoxicity assay of control (TIG) or hBATF-transduced human CAR T cells.
e, Histograms showing ratio between target cells (NALM6) and effector cells (human CAR T cells). Percent cytotoxicity was calculated as (1 − (R5/R0)) × 100, where R5 = (target cells as % of total at 5 h) / (effector cells as % of total at 5h), R0 = (target cells as % of total at 0 h) / (effector cells as % of total at 0 h).
Each dot in b-e represents an individual donor. Data were obtained from four biological experiments and analyzed by one-tailed unpaired Student’s t-test
Extended Data Fig. 5. Tumor growth and TIL expansion/survival in mice receiving BATF-transduced, BATF-HKE-transduced, or BATF-deficient CD8+ T cells.

a, Tumor growth curves for the individual mice from Fig. 4b–c (PBS (n=12), pMIG (n=16), BATF (n=25) and HKE (n=12)).
b-f, 1×105 B16F0-hCD19 tumor cells were subcutaneously injected into the left flank of C57BL/6 mice at day 0 (D0). 100 μl of PBS, without cells or containing 1.5×106 CAR T cells transduced with retroviral expression plasmids encoding either pMIG control, BATF, or BATF HKE-mutant, were adoptively transferred into C57BL/6 recipient mice by retro-orbital injection on day 12. TILs were isolated on days 13, 16, 19, and 22.
c, Expression of CAR T cell marker Thy1.1 on CD8+ TILs on the indicated days.
d-f, Frequencies and MFIs of the indicated PD-1- and TIM3-expressing populations from Fig. 4j,k.
g-m, 1×105 B16F0-hCD19 tumor cells were injected subcutaneously into the left flank of C57BL/6 mice at day 0 (D0). 1.5×106 wild-type (WT, n=4) or BATF-deficient (BATF KO, n=4) CAR T cells were adoptively transferred at day 12. Tumor-infiltrating lymphocytes were isolated at day 20.
h, Tumor growth curves for individual mice (dashed lines) and average (bold lines) of all tumor growth curves in a group.
i-k, Contour plots of Thy1.1 expression in CD8+ TILs (i), percentages of Thy1.1+ CAR TILs (j) and numbers of Thy1.1+ CAR TILs normalized to tumor size (k) in tumor-bearing BATF WT or BATF KO mice.
l, Contour plots of PD-1 and TIM3 expression (left) and percentage of PD-1hiTIM3+ cells (right) in WT or BATF KO CAR TILs.
m, Contour plots of PD-1 and TOX expression (top left), TIM3 and TCF1 expression (bottom left), and percentages of the indicated populations (right) in WT or BATF KO CAR TILs.
Data in a were obtained from three independent experiments. Each circle in d-f and j-m represents one mouse, and the bar graphs represent the mean ± standard error of mean (s.e.m.). Data in d-m are representative of two independent experiments. Data in j-m were analyzed by two-tailed unpaired Student’s t-test.
Extended Data Fig. 6. Tumor growth rates, survival curves, and phenotypic analysis of CAR TILs.
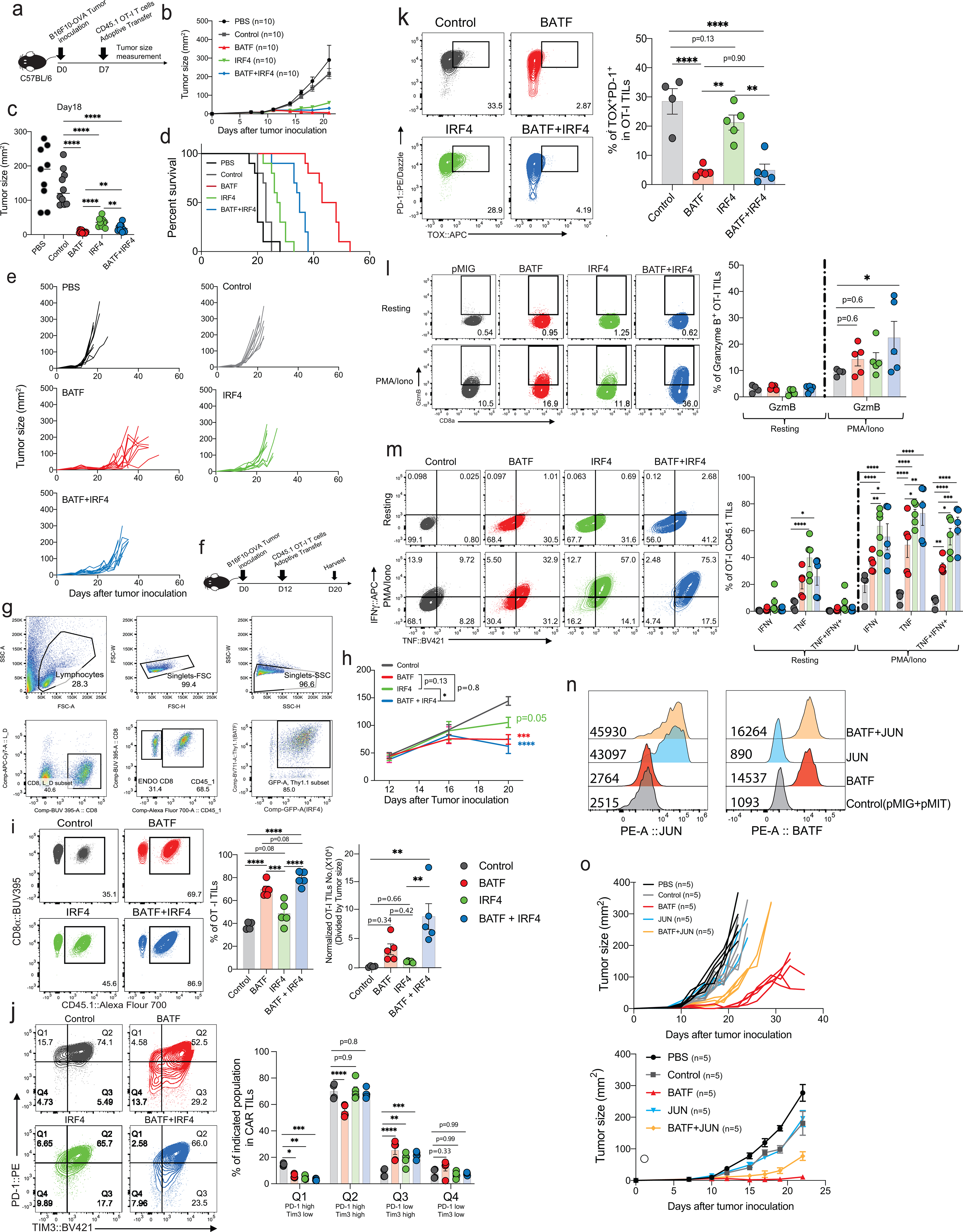
a-e, 2.5×105 B16F10-OVA tumor cells were injected subcutaneously into the left flank of C57BL/6 mice at day 0 (D0). 100 μl of PBS (n=10), without cells or containing 3×106 OT-I T cells transduced with retroviral expression plasmids encoding pMIG control (n=10), BATF(n=10), IRF4(n=10), or BATF+IRF4(n=10), were adoptively transferred by retro-orbital injection at day 7.
b, Averaged tumor growth curves for all mice in the indicated groups.
c, Tumor sizes measured in individual mice at day 18.
d, Mouse survival curves.
e, Tumor growth curves in individual mice.
f-m, 2.5×105 B16F10-OVA tumor cells were injected subcutaneously into the left flank of C57BL/6 mice at day 0. 1×106 pMIG control (n=4)-, BATF(n=5)-, IRF4(n=5)-, or BATF+IRF4(n=5)-transduced OT-I cells were adoptively transferred at day 12. TILs were isolated at day 20.
g, Gating strategy for flow cytometric analysis of OT-I TILs.
h, Averaged tumor growth curves for all mice in the indicated groups.
i, Left, Contour plots of CD8α and CD45.1 expression in OT-I TILs. Middle, Percentage of OT-I TILs in CD8+ TILs. Right, Number of OT-I TILs normalized to tumor size.
j, Left, Contour plots of PD-1 and TIM3 expression in each group of OT-I TILs. Right, Percentages of the indicated PD-1- and TIM3-expressing cell populations.
k, Left, Contour plots of PD-1 and TOX expression in the indicated OT-I TILs. Right, Percentage of PD-1+TOX+ cells in OT-I TILs.
l, m, Left, Contour plots of expression of granzyme B (l) and the indicated cytokines (m) under resting conditions or after PMA/ionomycin stimulation for 4 h. Right, Percentages of OT-I TILs expressing granzyme B (l) or the indicated cytokines (m) under resting conditions or after PMA/ionomycin stimulation for 4 h. Data obtained from two biological experiments
n, Histograms showing JUN and BATF expression in the indicated groups of transduced OT-I T cells.
o, Tumor growth curves for individual mice given pMIG control-, BATF-, JUN-, or BATF+JUN-transduced OT-1 cells (top) and averaged tumor growth curves for all mice in each group (bottom). Experimental scheme as in a.
Each circle in i, j, k, l, and m represents one mouse, and the bar graphs represent the mean ± standard error of mean (s.e.m.). Data were obtained from two independent biological experiments. Data in h, j, l, and m were analyzed by two-way ANOVA test; data from i and k, by one-way ANOVA test. *p≤0.05; **p≤0.01; ***p≤0.001; ****p≤0.0001.
Extended Data Fig. 7. Differentially accessible regions in BATF compared to pMIG-transduced CAR TILs.
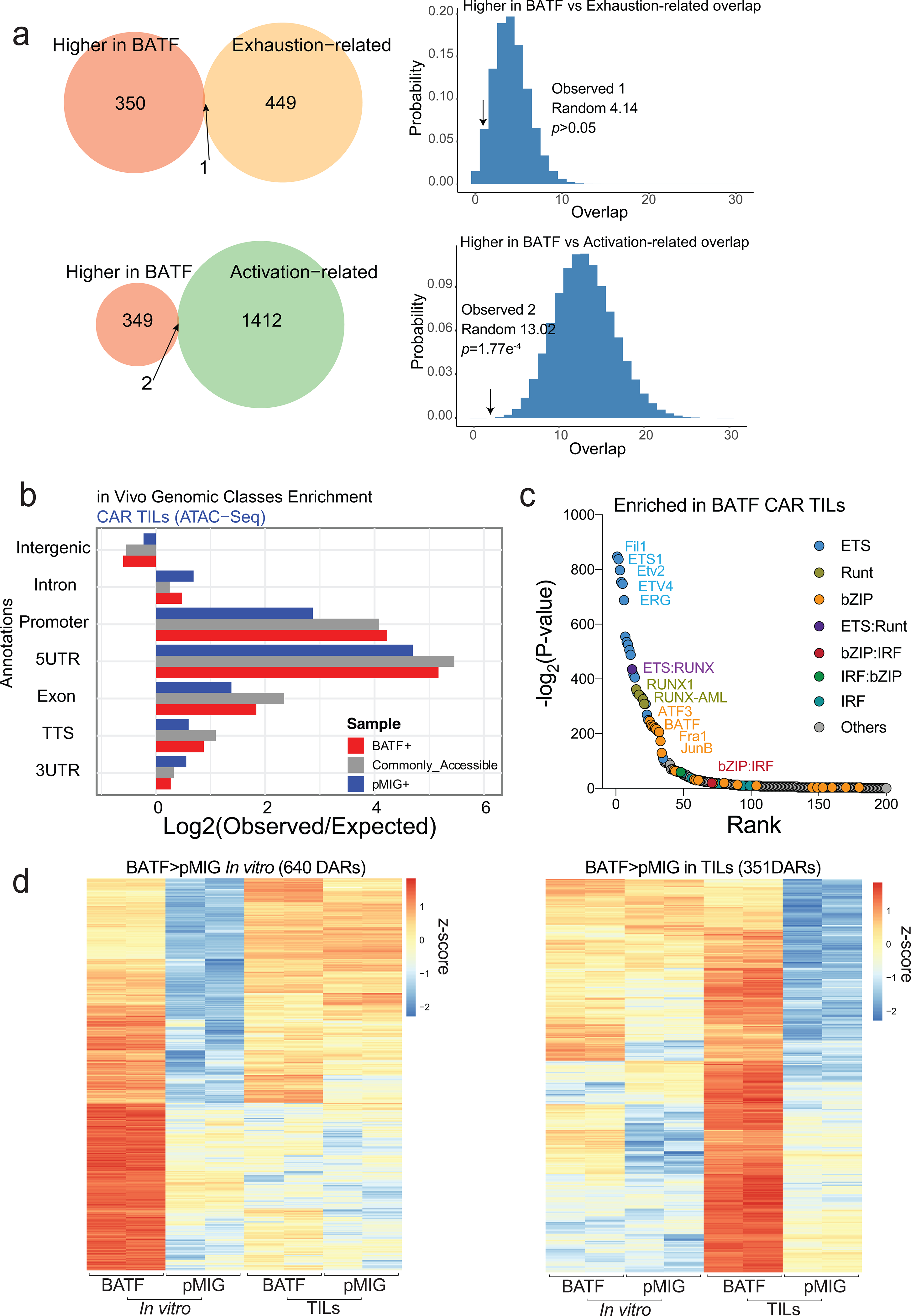
a, Left, Venn diagrams showing the overlap of the 351 regions more accessible in BATF- versus pMIG-transduced TILs (Fig. 5c) with the exhaustion-related (top) or activation-related (bottom) regions from Mognol et al36. Right, Histograms illustrate the significance calculation by one-tailed Fisher’s exact test.
b, Genomic annotation of the commonly and differentially accessible regions in CAR TILs.
c, Enrichment for transcription factor binding motifs in regions differentially accessible in BATF CAR TILs.
d, Heatmap of ATAC-seq signal (z-score) from BATF- and pMIG-transduced CD8+ T cells or CAR TILs, in the 640 regions more accessible in BATF-transduced compared to pMIG-transduced CD8+ T cells (left; see Fig. 5b) and in the 351 regions more accessible in BATF-transduced compared to pMIG CAR TILs (right; see Fig. 5c). Each column represents a biological replicate. Data obtained from two biological experiments
Extended Data Fig. 8. BATF and IRF4 binding and gene expression changes in pMIG-, HKE-, and BATF-transduced cells.
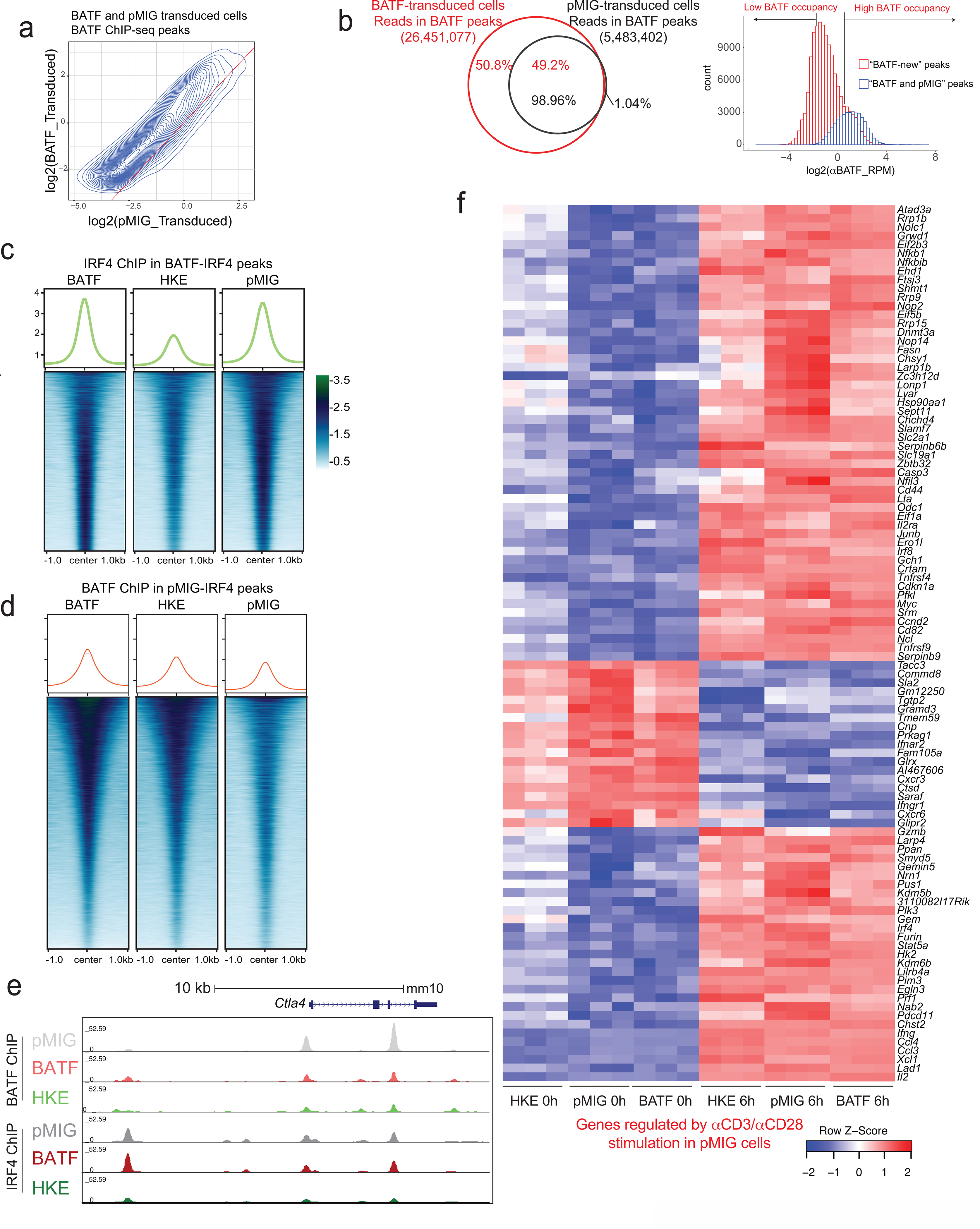
a, Contour plot relating BATF ChIP-seq signals (log2(CPM)) in BATF-transduced CD8+ T cells to signals from the corresponding peaks in pMIG-transduced cells.
b, Left, Distribution of BATF ChIP-seq reads in peaks in BATF-transduced cells (red) versus pMIG-transduced cells (black). Right, BATF ChIP-seq signal distribution in the “BATF and pMIG” peaks common to the two conditions (blue) and in “BATF-new” peaks observed only in BATF-transduced cells (red). The BATF-new peaks defined here are largely the same as the BATF-only peaks in Fig. 6a.
c, Heatmaps of the IRF4 ChIP-seq signal in BATF-transduced, BATF-HKE-transduced, and pMIG-transduced cells, at IRF4 peak locations called in BATF-transduced cells. Curves at the top show the average signal taken over all peaks in the respective heatmap.
d, Heatmaps of the BATF ChIP-seq signal in BATF-transduced, BATF-HKE-transduced and pMIG-transduced cells, at IRF4 peak locations called in pMIG-transduced cells. Curves at the top show the average signal taken over all peaks in the respective heatmap.
e, Genome browser view of Ctla4 gene locus showing BATF and IRF4 ChIP-seq signals from pMIG-, BATF-, and HKE-transduced CD8+ T cells.
f, Heatmap of normalized RNA-seq reads (as z-scores) under the indicated conditions, for the top 100 genes differentially expressed after αCD3/αCD28 stimulation of pMIG-transduced cells. Data obtained from two or three biological experiments.
Extended Data Fig. 9. Redistribution of IRF4 binding in BATF-overexpressing cells.
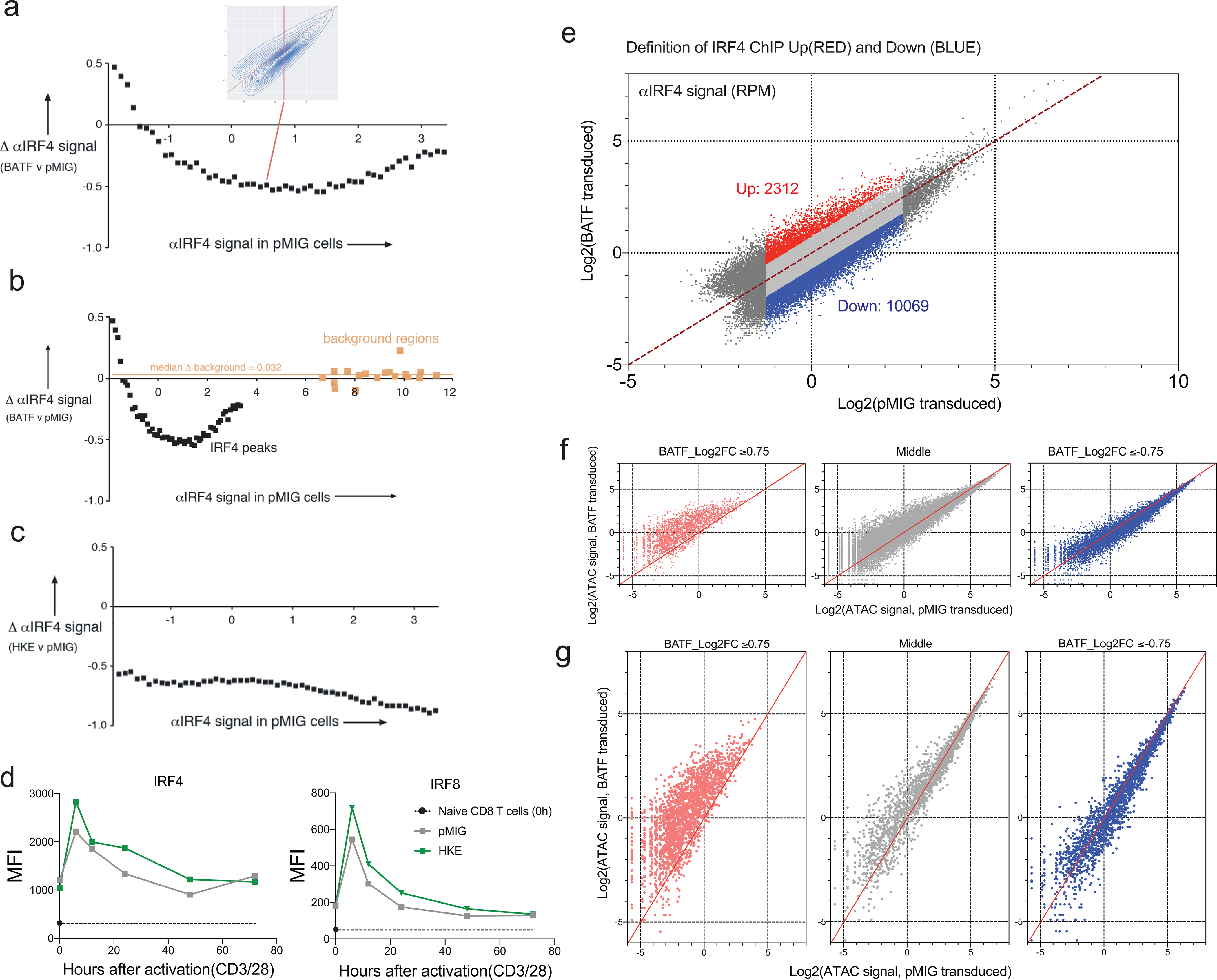
a, The redistribution of the normalized IRF4 ChIP-seq signal in BATF-overexpressing cells is most evident when the median deviation of the y-coordinate (αIRF4 ChIP-seq signal in BATF-overexpressing cells) from the diagonal in Fig. 7c, left, is plotted as a function of position on the x-axis (αIRF4 ChIP-seq signal in control cells). The median for all peaks in each slice of 0.1 log2(CPM) units on the x-axis was determined. The inset replicates Fig. 7c, left, with a red rectangle indicating the slice between log2(CPM)=0.5 and log2(CPM)=0.6 on the x-axis.
b, Spurious “αIRF4” ChIP-seq regions (as defined in Methods), incorporated into the graph of panel a.
c, IRF4 does not redistribute in BATF-HKE-overexpressing cells. The median deviation of the y-coordinate (αIRF4 ChIP-seq signal in HKE-overexpressing cells) from the diagonal in Fig. 7c, right, is plotted as a function of position on the x-axis (αIRF4 ChIP-seq signal in control cells), as in a.
d, IRF4 (left) and IRF8 (right) expression detected by flow cytometry (MFI) in pMIG- and HKE-transduced CD8+ T cells that had been expanded in vitro, at the indicated times after restimulation with αCD3/αCD28. The black symbol on the y-axis shows expression in naïve CD8+ T cells. Values for pMIG-transduced and naïve CD8+ T cells were obtained in the same experiments and are replotted from Fig. 6f. Overexpression of BATF-HKE did not attenuate IRF4 or IRF8 induction.
e, Similar to Fig. 8a. Dot plot highlighting regions of the IRF4 ChIP-seq data from Fig. 7c where IRF4 binding increases (log2FC ≥ 0.75, red dots), does not change substantially (light grey dots), or decreases (log2FC ≤ −0.75, blue dots) in BATF-overexpressing relative to pMIG-transduced cells. Peaks with very low (log2(signal) < −1.25) or high (log2(signal) > 2.5) IRF4 binding in pMIG cells were judged unlikely to be informative and were omitted from the analysis.
f, ATAC-seq signal (CPM) in peaks in each of the three categories defined in e.
g, Similar to b, but downsampling the subsets to match the 2312 regions with increased IRF4 binding in BATF-transfected cells, for clearer visualization.
Data were obtained from (a-c, e-g), or are representative of (d), two independent biological experiments.
Supplementary Material
Acknowledgements
We would like to thank C. Kim, D. Hinz, and C. Dillingham of the LJI flow cytometry core facility for CyTOF experiments; D.Hinz, C. Dillingham, S. Ellis, M. Haynes, S. Sehic, and C. Kim for cell sorting, J. Day, C Kim, and K. Tanguay of the LJI Next Generation Sequencing Facility for sequencing; and the LJI Department of Laboratory Animal Care (DLAC) and the animal facility for excellent support; and R. Nowak for excellent laboratory management. This work was funded in part by NIH R01 grants AI109842 and AI040127 (to P.H. and A.R.) and U01 grant DE028227 (to A.R.); NIH S10 instrumentation grants RR027366 and OD018499 (to the La Jolla Institute); AACR-Genentech Immuno-oncology Research Fellowship 18–40-18-SEO and Donald J. Gogel Cancer Research Institute Irvington Fellowship (to H.S.); University of California Institute for Mexico and the United States (UC-MEXUS) and El Consejo Nacional de Ciencia y Tecnología (UCMEXUS/CONACYT) predoctoral fellowship (to E.G.-A); NIH T32 predoctoral training grant in the UCSD Cardiovascular Bioengineering Training Program (to W.Z.); Cancer Research Institute Irvington Fellowship (to C.Y.); an Independent Investigator Fund (La Jolla Institute/Kyowa Kirin) and a Career Transition Award from the National Cancer Institute (K22CA241290) (to C.-W. J. L.).
Competing interests
A.R. and P.G.H. are recipients of a grant from Lyell Immunopharma for a separate research project related to cancer immunotherapy. None of the other authors has any competing interests.
References
- 1.Blank CU et al. Defining ‘T cell exhaustion.’ Nat Rev Immunol 19, 665–674 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McLane LM, Abdel-Hakeem MS & Wherry EJ CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol 37, 457–495 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Pereira RM, Hogan PG, Rao A & Martinez GJ Transcriptional and epigenetic regulation of T cell hyporesponsiveness. J Leukocyte Biol 102, 601–615 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu X et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seo H et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc National Acad Sci 116, 12410–12415 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott AC et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 1–5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X et al. TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J Hepatol 71, 731–741 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Alfei F et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Khan O et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Macián F et al. Transcriptional Mechanisms Underlying Lymphocyte Tolerance. Cell 109, 719–731 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Martinez GJ et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8+ T Cells. Immunity 42, 265–278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hogan PG, Chen L, Nardone J & Rao A Transcriptional regulation by calcium, calcineurin, and NFAT. Gene Dev 17, 2205–2232 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Karin M, Liu Z & Zandi E AP-1 function and regulation. Curr Opin Cell Biol 9, 240–246 (1997). [DOI] [PubMed] [Google Scholar]
- 15.Oh-hora M et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol 9, 432–443 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott-Browne JP et al. Dynamic Changes in Chromatin Accessibility Occur in CD8+ T Cells Responding to Viral Infection. Immunity 45, 1327–1340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hogan PG Calcium–NFAT transcriptional signalling in T cell activation and T cell exhaustion. Cell Calcium 63, 66–69 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lynn RC et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurachi M et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol 15, 373–383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy TL, Tussiwand R & Murphy KM Specificity through cooperation: BATF–IRF interactions control immune-regulatory networks. Nat Rev Immunol 13, 499–509 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Li P et al. BATF–JUN is critical for IRF4-mediated transcription in T cells. Nature 490, 543–546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ciofani M et al. A Validated Regulatory Network for Th17 Cell Specification. Cell 151, 289–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glasmacher E et al. A Genomic Regulatory Element That Directs Assembly and Function of Immune-Specific AP-1–IRF Complexes. Science 338, 975–980 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tussiwand R et al. Compensatory dendritic cell development mediated by BATF–IRF interactions. Nature 490, 502–507 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Man K et al. Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 47, 1129–1141.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Quigley M et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med 16, 1147–1151 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Im SJ et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurtulus S et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1−CD8+ Tumor-Infiltrating T Cells. Immunity 50, 181–194.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siddiqui I et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Miller BC et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Utzschneider DT et al. T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Brogdon J, June CH., Loew A, Maus M & Scholler J Treatment of cancer using humanized anti-CD19 chimeric antigen receptor. WO patent WO2014153270A1 (2014).
- 33.Iwata A et al. Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF–IRF4 transcription factor complex. Nat Immunol 18, 563–572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao Z, Casey KA, Jameson SC, Curtsinger JM & Mescher MF Programming for CD8 T Cell Memory Development Requires IL-12 or Type I IFN. J Immunol 182, 2786–2794 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huber JP & Farrar JD Regulation of effector and memory T‐cell functions by type I interferon. Immunology 132, 466–474 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mognol GP et al. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc National Acad Sci 114, E2776–E2785 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jadhav RR et al. Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc National Acad Sci 116, 14113–14118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muthusamy N, Barton K & Leiden JM Defective activation and survival of T cells lacking the Ets-1 transcription factor. Nature 377, 639–642 (1995). [DOI] [PubMed] [Google Scholar]
- 39.Grenningloh R et al. Ets-1 Maintains IL-7 Receptor Expression in Peripheral T Cells. J Immunol 186, 969–976 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wherry EJ T cell exhaustion. Nat Immunol 12, 492–499 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Wherry EJ & Kurachi M Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, He Y, Hao J, Ni L & Dong C High Levels of Eomes Promote Exhaustion of Anti-tumor CD8+ T Cells. Front Immunol 9, 2981 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li P & Leonard WJ Chromatin Accessibility and Interactions in the Transcriptional Regulation of T Cells. Front Immunol 9, 2738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pham D et al. Batf Pioneers the Reorganization of Chromatin in Developing Effector T Cells via Ets1-Dependent Recruitment of Ctcf. Cell Reports 29, 1203–1220.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang YK, Zuo Z & Stormo GD Quantitative profiling of BATF family proteins/JUNB/IRF hetero-trimers using Spec-seq. Bmc Mol Biol 19, 5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Man K et al. The transcription factor IRF4 is essential for TCR affinity–mediated metabolic programming and clonal expansion of T cells. Nat Immunol 14, 1155–1165 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Wei J et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 576, 471–476 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xin G et al. A Critical Role of IL-21-Induced BATF in Sustaining CD8-T-Cell-Mediated Chronic Viral Control. Cell Reports 13, 1118–1124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nicholson IC et al. Construction and characterisation of a functional CD19 specific single chain Fv fragment for immunotherapy of B lineage leukaemia and lymphoma. Mol Immunol 34, 1157–1165 (1997). [DOI] [PubMed] [Google Scholar]
- 50.Roybal KT et al. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hooijberg E, Bakker AQ, Ruizendaal JJ & Spits H NFAT-controlled expression of GFP permits visualization and isolation of antigen-stimulated primary human T cells. Blood 96, 459–466 (2000). [PubMed] [Google Scholar]
- 52.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Picelli S et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9, 171–81 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krueger Felix. “Trim galore.” A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files 516: 517. (2015). [Google Scholar]
- 56.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17, 10–12 (2011). [Google Scholar]
- 57.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.“Picard Toolkit.” Broad Institute, GitHub Repository. http://broadinstitute.github.io/picard/; Broad Institute. (2018). [Google Scholar]
- 59.Amemiya HM, Kundaje A & Boyle AP The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep-uk 9, 9354 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heinz S et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Law CW, Chen Y, Shi W & Smyth GK voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15, R29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/. (2018). [Google Scholar]
- 65.Kluyver T, Angerer P & Schulz J IRdisplay: “Jupyter” Display Machinery. R package version 0.7.0. https://CRAN.R-project.org/package=IRdisplay. [Google Scholar]
- 66.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47–e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Su S et al. Glimma: interactive graphics for gene expression analysis. Bioinformatics 33, btx094 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bioconductor Core Team. Mus.musculus: Annotation package for the Mus.musculus object. R package version 1.3.1. (2015). [Google Scholar]
- 70.Neuwirth Erich. RColorBrewer: ColorBrewer Palettes. R package version 1.1–2. https://CRAN.R-project.org/package=RColorBrewer. (2014). [Google Scholar]
- 71.Wickham H ggplot2, Elegant Graphics for Data Analysis. R 147–168, doi: 10.1007/978-3-319-24277-4_7. (2016). [DOI] [Google Scholar]
- 72.Lawrence M et al. Software for Computing and Annotating Genomic Ranges. Plos Comput Biol 9, e1003118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kolde Raivo. pheatmap: Pretty Heatmaps. R package version 1.0.12. https://CRAN.R-project.org/package=pheatmap. (2019). [Google Scholar]
- 74.MATLAB. version 7.10.0 (R2010a). Natick, Massachusetts: The MathWorks Inc. (2010). [Google Scholar]
- 75.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Warnes GR et al. gplots: Various R Programming Tools for Plotting Data. R package version 3.0.1.1. https://CRAN.R-project.org/package=gplots. [Google Scholar]
- 77.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen Hanbo. VennDiagram: Generate High-Resolution Venn and Euler Plots. R package version 1.6.20. https://CRAN.R-project.org/package=VennDiagram. (2018). [Google Scholar]
- 79.Garnier Simon. viridis: Default Color Maps from “matplotlib”. R package version 0.5.1. https://CRAN.R-project.org/package=viridis. (2018). [Google Scholar]
- 80.Ramírez F, Dündar F, Diehl S, Grüning BA & Manke T deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq, ATAC-seq, and ChIP-seq data have been deposited at the Gene Expression Omnibus (GSE154747). Other data that support the findings of this study are available from the corresponding authors upon request.