

SUMMARY
In ribosomopathies, perturbed expression of ribosome components leads to tissue-specific phenotypes. What accounts for such tissue-selective manifestations as a result of mutations in the ribosome, a ubiquitous cellular machine, has remained a mystery. Combining mouse genetics and in vivo ribosome profiling, we observe limb patterning phenotypes in ribosomal protein (RP) haploinsufficient embryos and uncover selective translational changes of transcripts controlling limb development. Surprisingly, both loss of p53, which is activated by RP haploinsufficiency, and augmented protein synthesis rescue these phenotypes. These findings are explained by the identification that p53 functions as a master regulator of protein synthesis, at least in part, through transcriptional activation of 4E-BP1. 4E-BP1, a key translational regulator, in turn, facilitates selective changes in the translatome downstream of p53, and thereby explains how RP haploinsufficiency may elicit specificity to gene expression. These results provide an integrative model to understand how in vivo tissue-specific phenotypes emerge in ribosomopathies.
Keywords: ribosomopathy, translational control, p53, ribosome profiling, ribosome protein haploinsufficiency
Graphical Abstract
eTOC
Tiu et al. show that ribosomal protein (RP) haploinsufficiency in the developing mammalian limb leads to patterning defects driven in part by p53-mediated translational regulation through induction of 4E-BP1, a translational repressor. This finding integrates p53 and translational dysregulation into a cohesive in vivo model of RP haploinsufficiency.
INTRODUCTION
Ribosomopathies are diseases characterized by mutations in ribosome components or key ribosome biogenesis factors that produce unexpected tissue-specific phenotypes. For example, Diamond Blackfan Anemia (DBA), which arises from haploinsufficiency of ~20 different essential ribosomal proteins (RPs), converges upon a common set of poorly understood congenital birth defects such as craniofacial, digit, and limb abnormalities, as well as selective impairment of erythroid differentiation (Clinton and Gazda, 1993; Yelick and Trainor, 2015). While the disease phenotypes in DBA have been the topic of intense study, the mechanisms underlying them, particularly congenital birth defects, are still poorly understood.
Many studies suggest that in vivo phenotypes underlying ribosomopathies can arise from the activation of the stress-induced transcription factor p53. In particular, mutations in RPs are thought to lead to p53 stabilization and activation through a mechanism in which nucleolar stress and defective ribosome biogenesis result in inhibition of the major p53 negative regulator, Mdm2 (Deisenroth and Zhang, 2010; Dutt et al., 2011; Morgado-Palacin et al., 2015; Narla and Ebert, 2010). p53 stabilization is thought to contribute to ribosomopathies through transcriptional activation of genes canonically involved in regulating cell proliferation and apoptosis (Deisenroth and Zhang, 2010). The role of p53 in generating these phenotypes is underscored when examining RP haploinsufficiency in vivo. Studies in both mice and zebrafish models of DBA have demonstrated that RP haploinsufficiency can lead to tissue-specific defects, which can be rescued by loss of p53 (Barlow et al., 2010; Danilova et al., 2008; McGowan and Mason, 2011; McGowan et al., 2008; Sulic et al., 2005). While these findings highlight the role of p53 in the development of disease phenotypes, how p53-mediated global effects on cells, such as cell cycle arrest and apoptosis, can lead to tissue-specific developmental phenotypes remains a paradox.
Alternatively, it has been suggested that mutations in RPs may lead to translational dysregulation independent of p53 (Dalla Venezia et al., 2019; Khajuria et al., 2018). Despite this, changes to translation have been largely unexplored within in vivo developmental contexts associated with RP haploinsufficiency. A recent ex vivo cell culture study identified translational defects in hematopoietic cells upon knockdown of RPs mutated in DBA, and suggests that ribosome levels may play a role in this dysfunction (Khajuria et al., 2018). However, it is perplexing how this hypothesis accounts for complex in vivo phenotypes and how it can be reconciled with the known role of p53 in the etiology of ribosomopathy phenotypes.
Taken together, it remains a mystery what roles p53-dependent and - independent mechanisms play in the development of in vivo tissue-specific phenotypes associated with RP haploinsufficiency. Here, we genetically inactivate one allele of the core RP, Rps6 (eS6), in the developing mouse limb bud as a model system for investigating the complex phenotypes linked to ribosomopathies. Strikingly, we find that Rps6 haploinsufficiency leads to selective limb phenotypes reminiscent of those observed in DBA. By combining comprehensive mouse genetics and in vivo ribosome profiling, we demonstrate that translational dysfunction plays a critical role in development of tissue-specific phenotypes upon Rps6 haploinsufficiency. Moreover, our ribosome profiling uncovered hundreds of transcripts which undergo differential translational regulation upon Rps6 haploinsufficiency. The majority of these transcript-specific translational changes are unexpectedly rescued by loss of p53, indicating that translational control and p53 activation are in fact coupled. To this end, we demonstrate that p53 in addition to its bona fide role in transcriptional regulation, is also a master regulator of protein synthesis. This function is, at least in part, mediated through p53-dependent induction of a key translational regulator, 4E-BP1 (eukaryotic initiation factor 4E-binding protein 1), which is known to have a role in translational control of selective mRNAs (Thoreen et al., 2012; Truitt et al., 2015). Together this work provides an integrative model wherein mutations in core components of the ribosome activate p53 to directly lead to transcript-specific changes in cap-dependent translation by modulating 4E-BP1 expression.
RESULTS
RP haploinsufficiency leads to selective developmental phenotypes
To delineate how haploinsufficiency of an essential RP leads to tissue-specific phenotypes, we perturbed expression of Rps6 (Panić et al., 2006; Volarevic et al., 2000) in the developing mouse embryo. Rps6 loss of function has served as a model to study ribosomopathies, as conditional Rps6 hemizygosity within the hematopoietic compartment leads to erythroid phenotypes similar to that of other RPs mutated in ribosomopathies, such as DBA (McGowan et al., 2011). However, constitutive Rps6 haploinsufficiency is embryonic lethal (Panić et al., 2006). Therefore, to investigate how congenital defects arise in an in vivo context due to RP haploinsufficiency without affecting organismal viability, we turned to the developing limb bud. Developing limbs are a well-established model system for studying tissue patterning and are where defects emerge in ribosomopathies. Importantly, patients diagnosed with DBA have been characterized with limb defects including hypoplastic digit 1 and radial abnormalities (Gazda et al., 2008; Hurst et al., 1991).
During development, limbs arise from a small bud of mesenchymal cells that includes chondrocyte progenitors, which serve as precursors to all the skeletal elements of the limb covered by a surface ectoderm. The apical ectodermal ridge (AER) is a morphologically distinct ectoderm surrounding the distal tip of the limb bud that serves as an important signaling center that promotes limb outgrowth and survival of the underlying mesenchyme (Zeller et al., 2009) (Figure 1A). The basic vertebrate limb skeleton derives from a cartilaginous model: a single proximal long bone within the stylopod segment (humerus; femur), followed by two long bones within the zeugopod segment (radius, ulna; tibia, fibula) and then the distal autopod segment comprising of wrist or ankle, and digits. Using two distinct and well-established Cre lines driven by the Prx1 promoter/enhancer (Logan et al., 2002) or the Msx2 promoter (Sun et al., 2000), we conditionally deleted one allele of Rps6 in either the limb mesenchyme or AER, reflecting the two major cell lineages that either give rise to the limb cartilage template or control limb outgrowth and skeletal patterning, respectively. Within the forelimb of Prx1Cre;Rps6lox/+ embryos, a significant decrease of Rps6 mRNA and protein was detected as early as day 10.5 of embryonic development (E10.5) when Cre recombinase is broadly activated throughout the limb bud mesenchyme (Figure 1B; S1A; S4A–B) (Logan et al., 2002). At E17.5, Prx1Cre;Rps6lox/+ embryos demonstrated striking selective underdevelopment (hypoplasia) or loss (aplasia) of the radius compared to the ulna in addition to scapular hypoplasia; hypoplasia/absence of the proximal humerus; and selective hypoplasia/absence of digits 1, 2, 4, and 5 marked by reduced phalangeal numbers, with digits 1 and 5 exhibiting the most severe phenotypes (Figure 1D–E; S1B–D; Table S1). Interestingly, the Prx1Cre;Rps6lox/+ phenotype is reminiscent of those found in DBA patients, as mentioned above (Gazda et al., 2008; Hurst et al., 1991). In contrast, the hindlimbs displayed a much milder phenotype consisting of a hypoplastic digit 5 and a smaller, medially displaced patella (Figure 1F–G; S1E–J). Surprisingly, we did not observe any phenotype upon Rps6 haploinsufficiency in the AER both within the forelimb and hindlimb (see Msx2Cre;Rps6lox/+; Figure 1H–I; S1K–N). In comparison to the selective skeletal phenotypes observed in the Prx1Cre;Rps6lox/+ embryos, the lack of phenotype upon Rps6 haploinsufficiency in the AER in the Msx2Cre;Rps6lox/+ embryos further suggests tissue and context specific effects of RP haploinsufficiency.
Figure 1. Rps6 haploinsufficiency in the developing limb bud mesenchyme leads to selective patterning defects marked by p53 activation and reduced global protein synthesis.
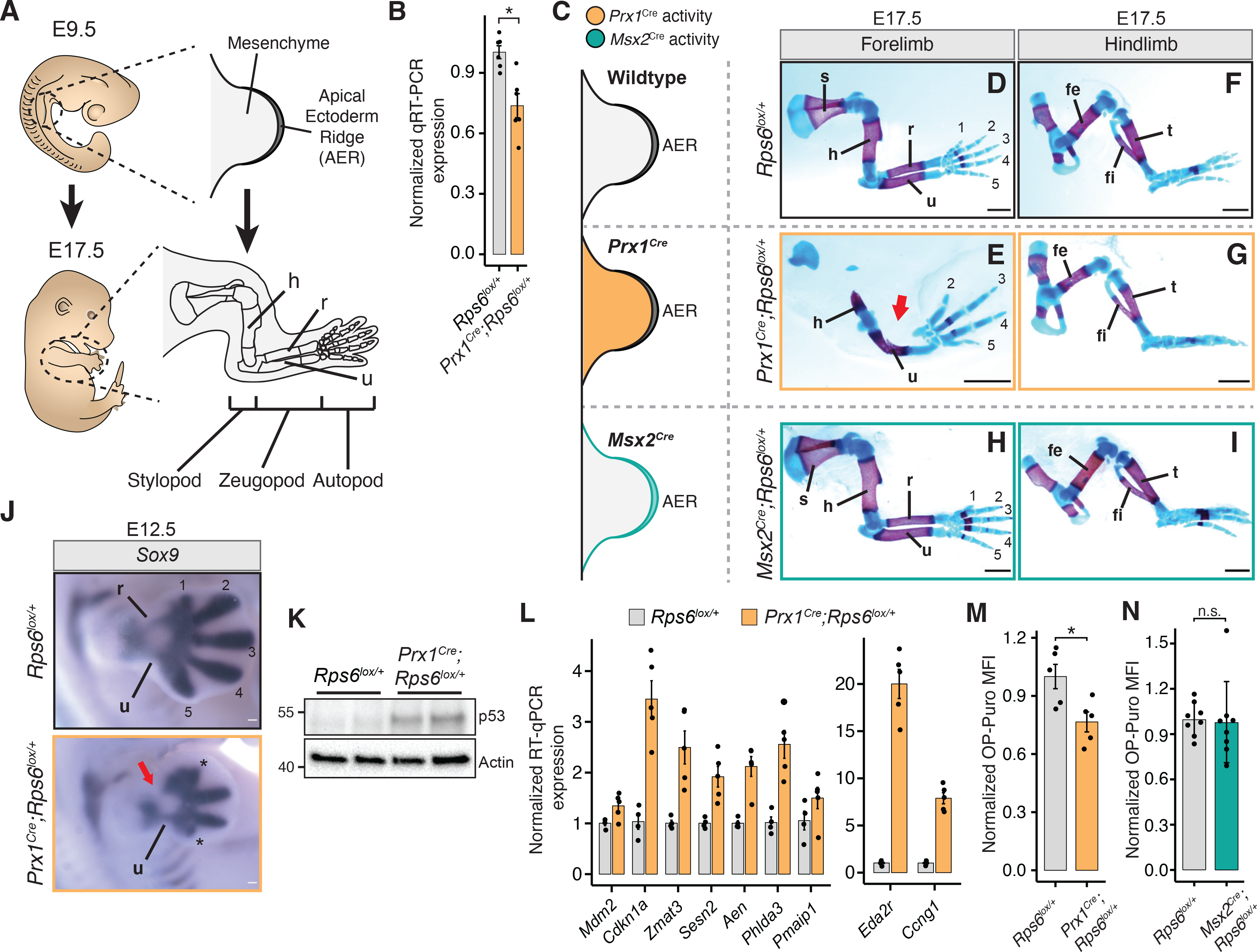
(A) Overview of forelimb development. AER, apical ectodermal ridge; h, humerus; r, radius; u, ulna.
(B) RT-qPCR of Rps6 mRNA from whole E10.5 forelimbs.
(C) Cre recombinase activity distribution in limb bud mesenchyme (Prx1Cre; orange), or in the overlying AER (Msx2Cre; green).
(D-I), E17.5 forelimbs and hindlimbs from WT (top row), Prx1Cre;Rps6lox/+ (middle row), and Msx2Cre;Rps6lox/+ (bottom row). Bone, red; Cartilage, blue. Numbers indicate digits. Arrow indicates absence of radius. Scale bars, 1 mm.
(J) Sox9 in situ hybridization of E12.5 forelimbs. Numbers indicate mesenchymal digit condensations. *Impaired digit development. Arrow indicates absence of radius. Scale bars, 0.1 mm.
(K) Representative p53 Western blot in whole E10.5 forelimbs. n = 2 embryos each.
(L) RT-qPCR of p53 target genes in whole E10.5 forelimbs. n = 4 embryos (Rps6lox/+), n = 5 embryos (Prx1Cre;Rps6lox/+).
(M) OPP MFI of cells dissociated from whole E10.5 Prx1Cre;Rps6lox/+ forelimbs normalized to WT. n = 5 embryos; MFI = median fluorescence intensity.
(N) OPP MFI of E11.5 Msx2Cre;Rps6lox/+ ectodermal cells normalized to WT. n = 8 embryos.
p53 activation and decreased protein synthesis coincide with limb patterning defects
The selective radial and digit phenotypes in Prx1Cre;Rps6lox/+ embryos were observed as early as E12.5 by in situ hybridization of Sox9, the earliest marker of mesenchymal condensations, which prefigure cartilage elements, suggesting that these phenotypes result from defects in patterning prior to cartilage formation (Figure 1J). We hypothesized that the observed phenotypes may arise from either p53 activation or perturbed protein synthesis. Thus, we first asked if p53 is activated in E10.5 Prx1Cre;Rps6lox/+ limb buds, approximately one day after Cre-mediated Rps6 recombination in the limb mesenchyme. Indeed, we observed spatially homogeneous p53 activation and transcriptional upregulation of known p53 target genes (Figure 1K–L; Figure S1O) (Bowen et al., 2019; Brady et al., 2011). This was accompanied by increased apoptosis and mildly decreased cell proliferation that were not spatially restricted within the limb bud mesenchyme (Figure S1P–S). We also observed p53 activation in the ectoderm of Msx2Cre;Rps6lox/+ embryos (Figure S2A–B). Next, we measured changes in global translation in limb cells by quantifying incorporation of O-propargyl-puromycin (OPP) into nascent peptides of freshly dissociated E10.5 limb mesenchymal cells (Figure S2C) (Signer et al., 2014). We observed reduced OPP incorporation in Prx1Cre;Rps6lox/+ versus control, indicating that protein synthesis is impaired upon Rps6 reduction (Figure 1M). Conversely, we did not observe a decrease in OPP incorporation in cells derived from the separated ectoderm layer of Msx2Cre;Rps6lox/+ limb buds (Figure 1N), despite p53 activation and decreased Rps6 mRNA levels in this tissue (Figure S2A–B). Taken together, these data suggest that both p53 activation and defects in global protein synthesis may be associated with limb patterning phenotypes upon Rps6 haploinsufficiency. Moreover, as Rps6 haploinsufficiency in the mesenchyme leads to a patterning phenotype and decreased protein synthesis whereas haploinsufficiency in the AER does not, this suggests that diminished protein synthesis may be an important driver of tissue-selective phenotypes upon RP haploinsufficiency.
mTORC1 activation and loss of p53 rescue limb patterning defects
As both p53 activation and translational dysfunction accompany the Prx1Cre;Rps6lox/+ limb phenotypes, we turned to in vivo genetics to determine the contribution of decreased protein synthesis and/or p53 activation to phenotype (Figure 2A). First, we genetically modulated global protein synthesis by manipulating the mTOR pathway (Saxton and Sabatini, 2017). mTOR signaling functions are distributed between at least two distinct mTOR protein complexes: mTORC1 and mTORC2. One of the major functions of mTORC1 signaling is controlling protein synthesis levels by phosphorylating key translational regulators, which subsequently leads to upregulation of cap-dependent protein synthesis (Saxton and Sabatini, 2017; Signer et al., 2014; Zhang et al., 2014) (Figure 2B). To address whether translation is implicated in phenotype upon Rps6 haploinsufficiency, we activated mTORC1 in the limb by conditionally inactivating the tuberous sclerosis complex (TSC), which specifically represses mTORC1 (Garami et al., 2003; Zhang et al., 2014), via genetic deletion of Tsc2. Remarkably, deletion of Tsc2 in developing limbs rescued the Rps6 haploinsufficiency radius patterning phenotype (Prx1Cre;Rps6lox/+;Tsc2lox/lox vs. Prx1Cre;Rps6lox/+;Tsc2+/+; Figure 2C–F; S3A–B). This phenotypic rescue is accompanied by a concurrent increase in protein synthesis (Figure 2G). While we only observed a partial rescue, this may be explained by the incomplete restoration of protein synthesis back to wildtype (WT) levels upon loss of Tsc2 (Figure 2G). In addition, loss of Tsc2 did not restore Rps6 levels nor did it abrogate the p53 activation induced by Rps6 haploinsufficiency (Figure 2H; S3A,C–D). Taken together, these results suggest that perturbed translation may contribute to the developmental patterning phenotypes observed upon Rps6 reduction.
Figure 2. mTORC1 activation with corresponding augmented protein synthesis rescues Rps6 haploinsufficiency phenotypes.
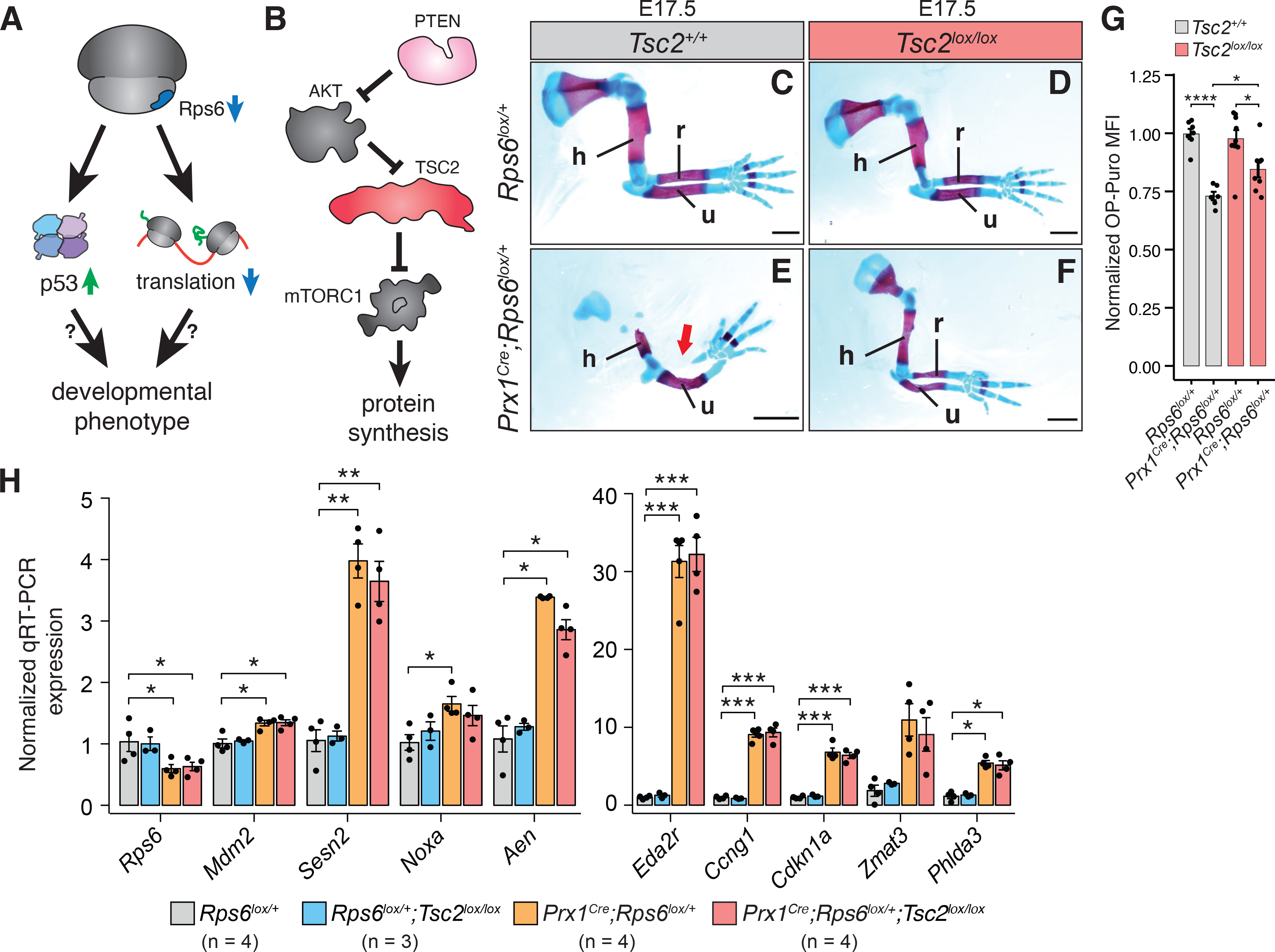
(A) Overview of potential pathways leading to developmental phenotypes upon Rps6 haploinsufficiency, specifically p53 activation and translation dysregulation.
(B) Schematic of mTORC1 regulation and downstream effects.
(C-F) Representative E17.5 forelimbs of WT (Rps6lox/+) and Prx1Cre;Rps6lox/+ embryos in Tsc2 WT (Tsc2+/+) or Tsc2 conditional loss (Tsc2lox/lox) backgrounds. Arrow indicates absence of radius. Scale bars, 1 mm.
(G) OPP MFI of cells dissociated from whole E10.5 forelimbs normalized to WT. n = 7 embryos, (Rps6lox/+;Tsc2+/+); n = 6 embryos, (Prx1Cre;Rps6lox/+;Tsc2+/+); n = 9 embryos, (Rps6lox/+;Tsc2lox/lox); n = 8 embryos, (Prx1Cre;Rps6lox/+;Tsc2lox/lox).
(H) RT-qPCR of p53 target genes from whole E10.5 forelimbs. n = 4 (Rps6lox/+, Prx1Cre;Rps6lox/+, Prx1Cre;Rps6lox/+;Tsc2lox/lox), n = 3 (Rps6lox/+;Tsc2lox/lox).
To formally address the potential contribution of p53 activation to limb phenotypes upon Rps6 haploinsufficiency, we next genetically ablated p53. Surprisingly, deletion of p53 in the Prx1Cre;Rps6lox/+ background also resulted in the rescue of the limb phenotype (Figure 3A–D; S3E–H). Together these findings show that either activation of the mTORC1 pathway with associated restoration of global protein synthesis or inactivation of p53 results in a genetic rescue of Rps6 haploinsufficient patterning phenotypes.
Figure 3. p53 loss rescues Rps6 haploinsufficiency phenotypes, and p53 activation mediates translational changes upon Rps6 reduction.
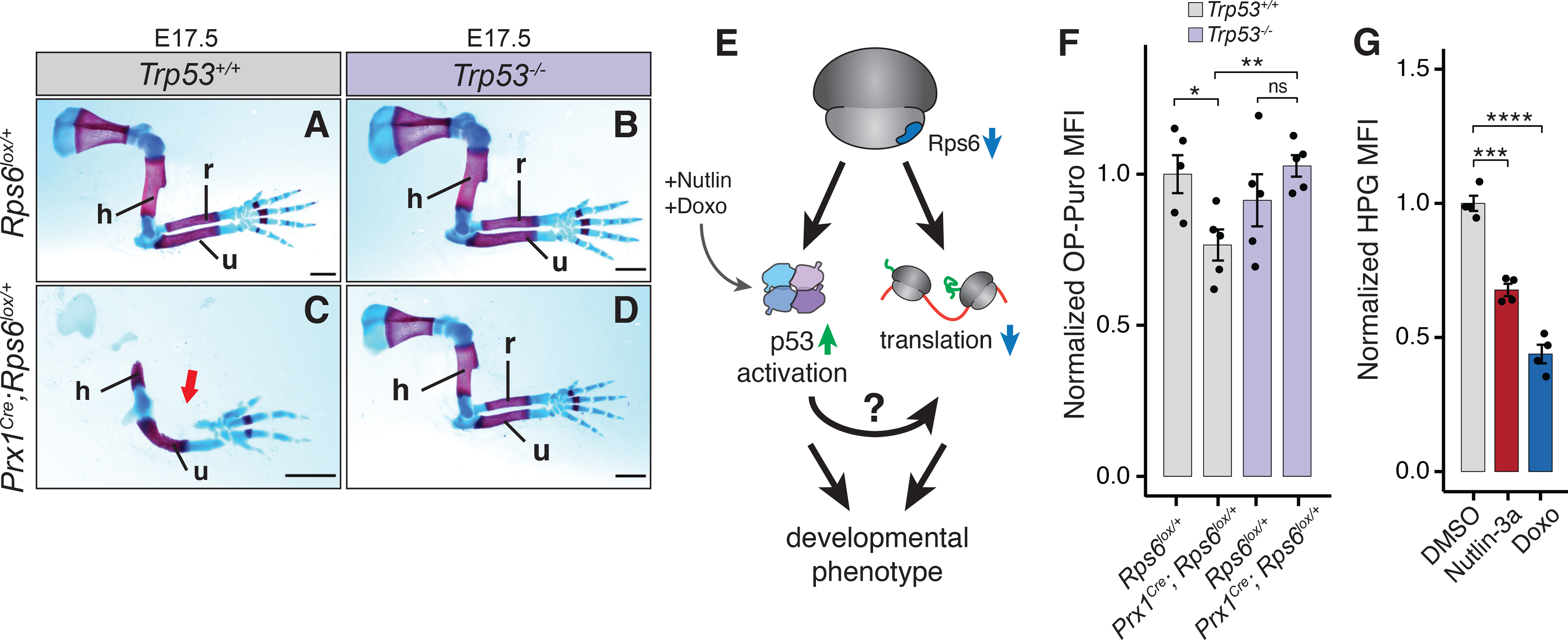
(A-D) E17.5 forelimbs of WT and Prx1Cre;Rps6lox/+ embryos in Trp53 WT (Trp53+/+) and Trp53 null (Trp53−/−) backgrounds. Arrow indicates absence of radius. Scale bars, 1 mm.
(E) Potential pathways for p53-dependent translational control upon Rps6 haploinsufficiency.
(F) OPP MFI of cells dissociated from whole E10.5 forelimbs normalized to WT (Rps6lox/+). n = 5 embryos.
(G) HPG MFI of mouse embryonic fibroblasts treated with Nutlin-3a or Doxo normalized to DMSO treated control. 8 h treatment, n = 4.
These findings raise the possibility that p53 activity and translational control may be unexpectedly coupled in the context of in vivo RP haploinsufficiency. In support of this, past studies in cultured cells have suggested that p53 may regulate translation (Kasteri et al., 2018), for example by suppressing mTORC1 activity by activating Sestrin, a negative regulator of mTORC1 (Budanov and Karin, 2008; Loayza-Puch et al., 2013). However, in the context of Rps6 haploinsufficiency in vivo, we do not observe a significant difference in mTORC1 activity in Prx1Cre;Rps6lox/+ vs. WT E10.5 limb buds (Figure S4A–B). Therefore, we explored whether p53 regulates protein synthesis through a yet unknown mechanism (Figure 3E). Strikingly, loss of p53 rescues the protein synthesis defect observed upon reduction of Rps6 (Figure 3F). To determine whether p53 activation alone is sufficient to repress global protein synthesis, we assayed the incorporation of the methionine analog L-Homopropargylglycine (HPG) into nascent peptides in primary mouse embryonic fibroblasts (MEFs) treated with two different p53 small molecule activators. Nutlin-3a specifically blocks Mdm2-mediated p53 destabilization, while doxorubicin (Doxo) induces genotoxic stress (Vassilev et al., 2004; Wang et al., 2004). Nutlin-3a and Doxo treatment led to a decrease in global protein synthesis within 8 h of treatment (Figure 3G). Importantly, we did not observe a significant difference in apoptosis and only a modest decrease in cell proliferation after 8 h of Nutlin-3a treatment (Figure S4C–D). Collectively, these findings demonstrate that activation of p53 is required to repress protein synthesis upon Rps6 haploinsufficiency and that p53-dependent control of translation occurs through an unknown mechanism independent of mTORC1 pathway activation.
p53 drives translational changes upon Rps6 haploinsufficiency
We next asked what transcripts may be selectively perturbed upon RP haploinsufficiency in a p53-dependent and -independent manner. To address this question, we performed ribosome profiling directly on E10.5 Rps6 haploinsufficient limb buds in a Trp53+/+ or Trp53−/− background (Figure 4A–B; S5) (Ingolia et al., 2011). We observed significant translational change for many transcripts upon Rps6 reduction (Figure 4A, Table S3). Most of these changes are rescued by loss of p53 (Figure 4B; S5E; Table S3–4). Interestingly, the translation efficiency (TE) changes of a smaller subset of differentially translated mRNAs, which included p53 regulators and cytoplasmic RPs (see below), were not dependent on p53 (Figure 4B; S5F; Table S4).
Figure 4. Ribosome profiling reveals p53-dependent and -independent translation changes upon Rps6 haploinsufficiency.
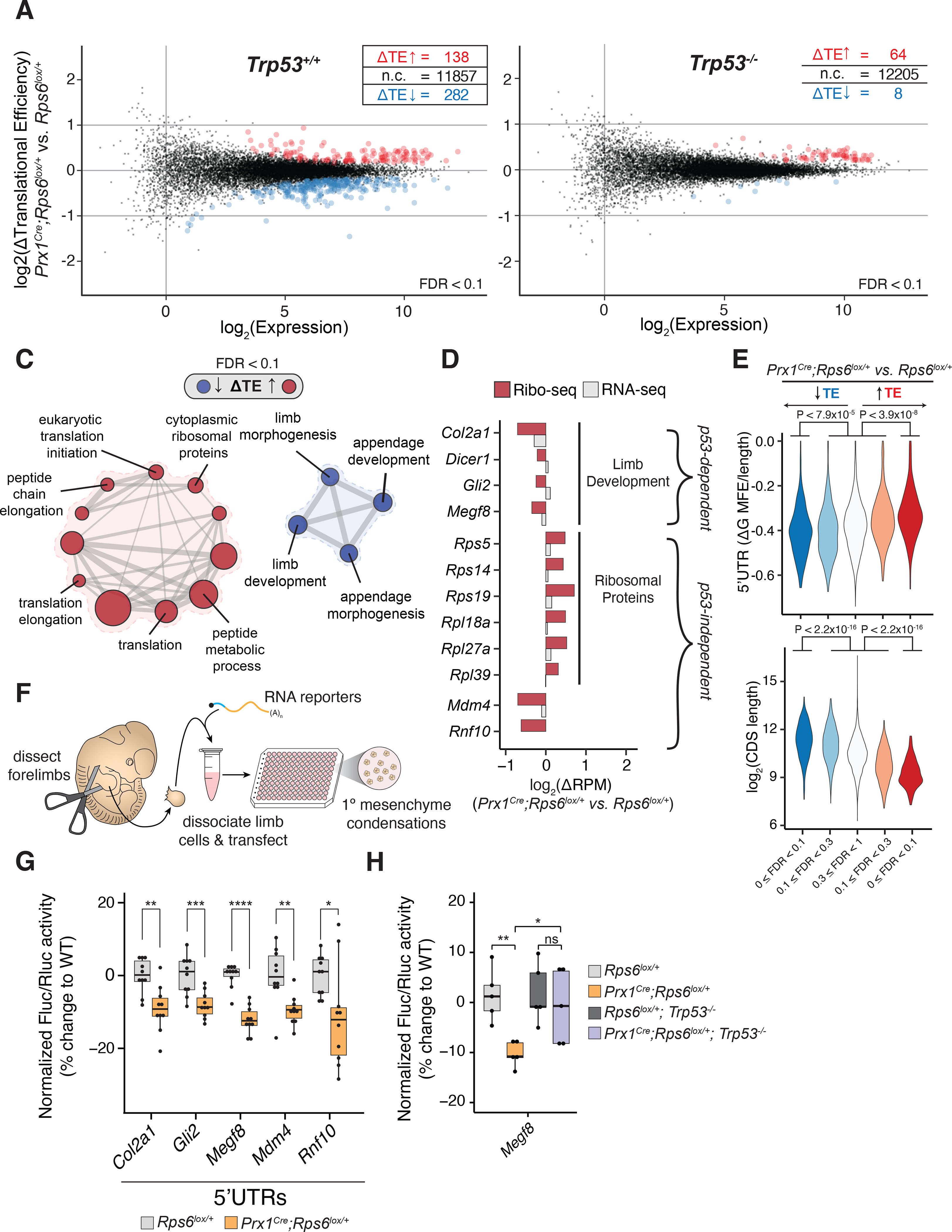
(A-B) MA plot of change in translational efficiency (ΔTE) in whole E10.5 Prx1Cre;Rps6lox/+ vs. Rps6lox/+ forelimbs in Trp53+/+ (A) and Trp53−/− (B) backgrounds. red, ΔTE > 0; blue, ΔTE < 0; n = 3; FDR < 0.1. (C) Gene set enrichment analysis for transcripts changing in TE in E10.5 Prx1Cre;Rps6lox/+ vs. Rps6lox/+ forelimbs. node size, gene set size; edge size, gene set overlap; FDR < 0.1.
(D) Relative change in ribosome footprints (red) and mRNA expression (gray) for select high-confidence transcripts (see Methods).
(E) Violin plots quantifying ΔTE relative to computationally predicted 5’UTR structuredness normalized to UTR length (ΔG MFE / 5’UTR length; see Methods) and relative to CDS length. Transcripts are stratified by direction of ΔTE (blue, down; red, up) and FDR; Mann-Whitney U test.
(F) Workflow of the primary limb micromass assay.
(G) Fluc/Rluc activity from RNA reporter transfection in whole E11.5 forelimb micromass cultures normalized to the geometric mean of control Pkm and Cnot10 5’UTR activities and WT (Rps6lox/+). Pkm and Cnot10 were chosen as controls given that they did not change in TE upon Rps6 haploinsufficiency (Table S3).
(H) Fluc/Rluc activity of Megf8 5’UTR RNA reporter transfected into whole E11.5 forelimb micromass cultures normalized to control Pkm 5’UTR activity and mean of WT (Rps6lox/+) or Trp53-null (Rps6lox/+;Trp53−/−) background.
Gene set enrichment analysis revealed that translationally repressed gene sets include those involved in limb development (Figure 4C; Table S5), which correlates with the observed phenotypes. Hence, the selective changes in translational control may account for the specificity in the limb patterning defects observed. For example, high confidence transcripts in these gene sets include Col2a1, Dicer1, Gli2, and Megf8 (Figure 4D; see Method Details for selection criteria). Col2a1 encodes for collagen type II alpha 1 chain, a cartilage component that lays the framework for skeletal development of most bones (Lee et al., 1989). Dicer1, which encodes an RNase essential for miRNA maturation, is essential for proper limb maturation and survival of mesenchymal cells (Harfe et al., 2005). Furthermore, loss of Gli2, a component of the Sonic Hedgehog pathway, leads to shortened limbs with particular shortening of the radial bone (Mo et al., 1997), while mutations in Megf8 lead to limb developmental defects in humans (Twigg et al., 2012). In addition, translational repression of these select transcripts involved in limb development is p53-dependent, consistent with the requirement of p53 for the development of limb phenotypes upon Rps6 haploinsufficiency (Figure S5E–F).
Translationally upregulated mRNAs were enriched in gene sets that included those containing cytoplasmic RPs (Figure 4C–D; Figure S5F; Table S5). In contrast to the p53-dependent translationally repressed limb development transcripts, RP translational upregulation is p53-independent (50 RPs out of 64 p53-independent upregulated transcripts; Figure S5F; Table S4). Our in vivo results may thus represent a possible translational feedback mechanism to restore RP homeostasis when core RP expression is perturbed. We also observed p53-independent translational downregulation of the p53 negative regulator Mdm4 and of the RING finger-containing protein, Rnf10 (Figure 4D; S5F; see Discussion). Ribosome profiling results were validated using sucrose gradient polysome fractionation and RT-qPCR (Figure S6A–C). Taken together, these data demonstrate that complex translational remodeling occurs upon Rps6 haploinsufficiency that consists of a p53-dependent program associated with transcripts that correlate with phenotypic changes and a smaller p53-independent program that may mediate homeostatic responses to RP perturbations.
Next, we investigated what features may regulate differential translational sensitivity of transcripts to Rps6 haploinsufficiency. Previous studies have demonstrated that cis-features such as ORF length and 5’UTR structuredness contribute to translational control (Weinberg et al., 2016). Indeed, we find that translationally repressed transcripts upon Rps6 reduction have longer ORFs whereas transcripts that are upregulated in translation have shorter ORFs (Figure 4E; S6D). Additionally, repressed transcripts have more structured 5’UTRs whereas upregulated mRNAs tend to have less structured 5’UTRs (Figure 4E). Notably, the p53-dependent, translationally repressed limb developmental genes (i.e. Col2a1, Dicer1, Gli2, and Megf8) have long ORF lengths and are predicted to have highly structured 5’UTRs (Table S6).
Given these observations, we asked whether the 5’UTRs of differentially translated mRNAs contribute to their translational regulation upon Rps6 haploinsufficiency. To this end, we in vitro transcribed and transfected Firefly luciferase reporter mRNAs harboring candidate 5’UTRs alongside control Renilla luciferase mRNA into primary limb micromass cultures derived from Rps6lox/+ or Prx1Cre;Rps6lox/+ limb buds (Figure 4F). Primary limb micromass cultures are a well-established model system to study limb mesenchyme and cartilage formation that mimics many of the in vivo steps of cellular differentiation (Barna and Niswander, 2007). In comparison to two control 5’UTRs taken from transcripts whose translation was unaffected by Rps6 haploinsufficiency (Pkm and Cnot10), we observed reduced reporter activity driven by the 5’UTRs of translationally repressed transcripts upon Rps6 haploinsufficiency (Figure 4G). Importantly, the reporter activity from the Megf8 5’UTR was rescued upon loss of p53 in the context of Rps6 haploinsufficiency, suggesting that the 5’UTR contributes to p53-dependent translational repression (Figure 4H). Overall, these findings demonstrate that 5’UTRs are important cis-regulatory elements that may contribute to differential translational sensitivity to RP haploinsufficiency.
p53 modulates translational control by upregulating 4E-BP1 expression
We next sought to determine how RP haploinsufficiency leads to p53-dependent translational remodeling (Figure 5A). Thus, we first investigated whether these translational changes depend on the transcriptional transactivation activity of p53. Indeed, in MEFs expressing the p53 transactivation dead mutant Trp53QM/QM (Brady et al., 2011), we did not observe a reduction in protein synthesis (Figure 5B; S7A) after treatment with Nutlin-3a or Doxo, suggesting that p53-dependent translational repression requires p53 transactivation activity. We therefore asked what p53 transcriptional targets may be responsible for these translational changes. Strikingly, we found that transcription of Eif4ebp1 (4E-BP1), which encodes a master regulator of cap-dependent translation (Lin et al., 1994; Sonenberg and Hinnebusch, 2009), is induced upon Rps6 reduction in embryonic limb buds in a p53-dependent manner (Figure 5C–D). This upregulation was also reflected at the protein level (Figure 5E–F; S7B). This suggests that Eif4ebp1 may be a transcriptional target of p53. Notably, we did not observe transcriptional induction of the closely related homolog, Eif4ebp2 (Figure 5D), suggesting that this response was specific to Eif4ebp1. This increase in 4E-BP1 RNA and protein levels was mirrored in NIH3T3 cells treated with Nutlin-3a (Figure 5G–I). Interestingly, we did not detect Eif4ebp1 upregulation despite p53 activation in the AER-containing ectoderm of Msx2Cre;Rps6lox/+ limb buds, which also did not demonstrate a phenotype or a reduction in protein synthesis (Figure 1H–I,N; S2A–B; S7C). This suggests that there may be additional tissue-specific regulatory mechanisms governing Eif4ebp1 expression.
Figure 5. p53 activation leads to transcriptional upregulation of Eif4ebp1.
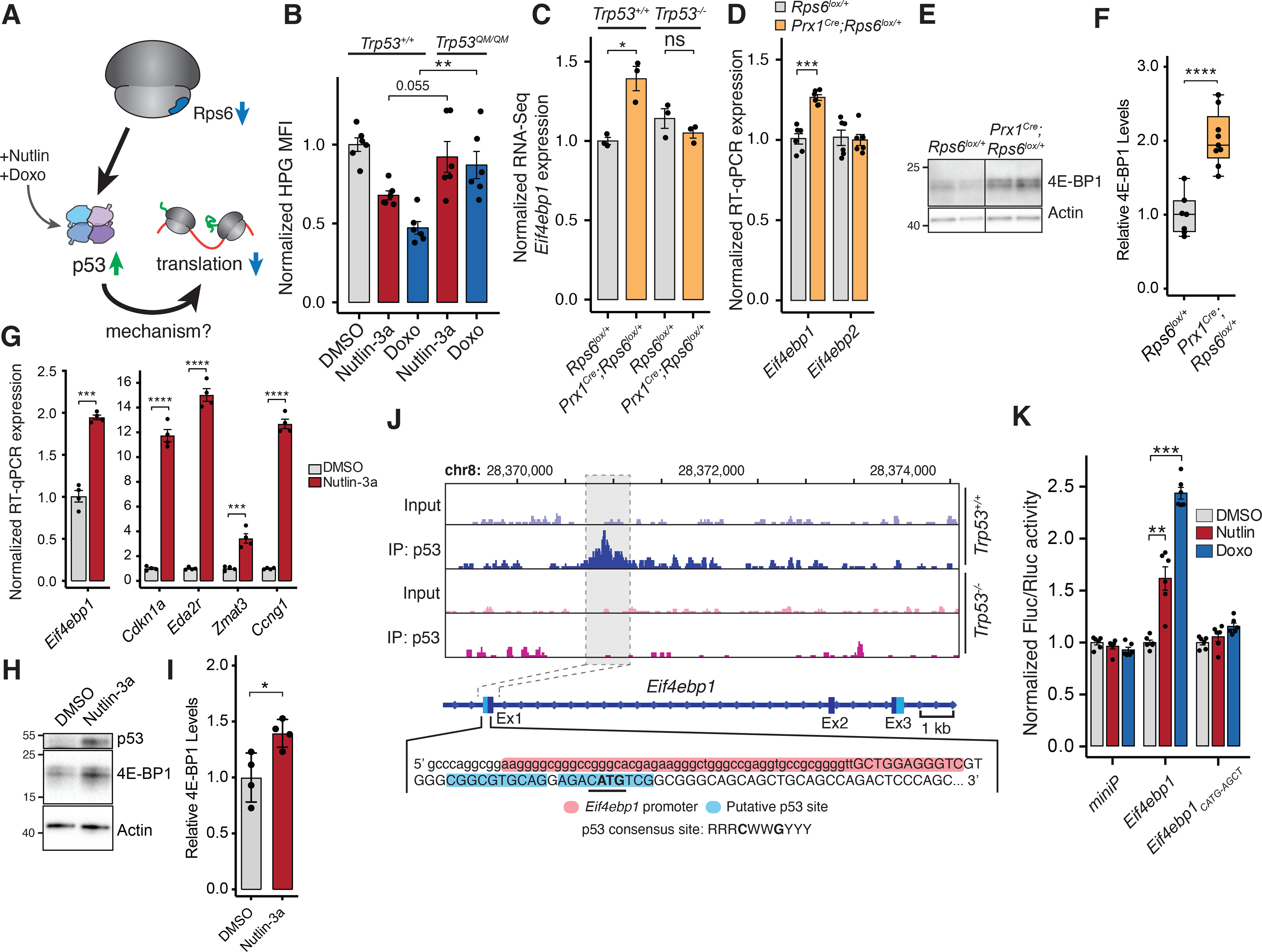
(A) Potential model of p53-dependent translational control upon Rps6 haploinsufficiency.
(B) HPG MFI of MEFs expressing WT or transactivation dead p53 (Trp53QM) treated with Nutlin-3a or Doxo normalized to mean of DMSO control expressing WT p53. 8 h treatment, n = 6.
(C) Eif4ebp1 expression from RNA-Seq of whole E10.5 forelimbs normalized to WT (Rps6lox/+;Trp53+/+), n = 3.
(D) Expression of Eif4ebp1 and Eif4ebp2 mRNA from whole E10.5 forelimbs.
(E-F) Representative 4E-BP1 Western blot (E) and quantification (F) from Figure S7B of E10.5 Rps6lox/+ and Prx1Cre;Rps6lox/+ forelimb mesenchyme cells after ectoderm removal with values normalized to Actin and Rps6lox/+. n = 7, Rps6lox/+; n = 9, Prx1Cre;Rps6lox/+.
(G) Expression of Eif4ebp1 and p53 target genes by RT-qPCR from NIH3T3 cells treated with DMSO or Nutlin-3a for 8 h.
(H-I) Western blot of 4E-BP1 from NIH3T3 cells treated with Nutlin-3a for 8 h. Shown is a representative blot (H) with quantification (I) from 4 independent replicates.
(J) p53 ChIP-Seq gene track of Eif4ebp1 locus (Kenzelmann Broz et al., 2013) from Doxo-treated Trp53+/+ and Trp53−/− MEFs. Capital letters = transcription state site; red highlight = Eif4ebp1 promoter; blue highlight = putative p53-binding region; bold = Eif4ebp1 start codon; underline = p53 core binding sequence.
(K) Fluc/Rluc activity normalized to DMSO control of each construct. Plasmid containing a minimal promoter, the Eif4ebp1 region, or mutated Eif4ebp1 region (Figure S7F) were transfected into NIH3T3 cells and treated with DMSO, Nutlin-3a, or Doxo for 8 h; n = 6.
To determine whether Eif4ebp1 is a direct p53 transcriptional target gene, we re-analyzed our previously generated genome-wide p53 ChIP-Seq data (Kenzelmann Broz et al., 2013). This data revealed a ~0.4 kb region in the first exon of Eif4ebp1 containing a putative p53 binding site, which was validated by ChIP-qPCR (Figure 5J; S7D). This finding was specific to Eif4ebp1, and no candidate p53 binding site was found in the Eif4ebp2 locus (Figure S7E). When cloned into a luciferase reporter construct, the ~0.4 kb Eif4ebp1 fragment was sufficient to drive expression of luciferase upon p53 activation (Figure 5K; S7F). Mutation of the core p53 binding sequence (Eif4ebp1CATG-AGCT) abolished p53-dependent luciferase activity, revealing a genuine cis-regulatory element responsible for p53-dependent induction of Eif4ebp1 expression (Figure 5K).
The primary function of 4E-BP1 is to repress the major cap-binding protein, eukaryotic initiation factor 4E (eIF4E) by occluding eIF4G binding (Sonenberg and Hinnebusch, 2009). eIF4E is rate limiting for the translation of select mRNAs, including those with specific sequences or structures within their 5’UTRs. For example, highly structured 5’UTRs are more sensitive to eIF4E dosage as it enhances the unwinding of RNA secondary structures during ribosome scanning by eIF4A (Pelletier and Sonenberg, 1985; Pelletier et al., 2015). In addition, distinct 5’UTR sequences such as the cytosine rich motif (CERT) also confer specificity to eIF4E activity (Truitt et al., 2015). Thereby, increased expression of 4E-BP1 inhibits the formation of the eIF4E translation initiation complex and alters the translation of eIF4E-sensitive transcripts (Figure 6A). To test whether p53 activation leads to 4E-BP1-mediated translational repression, we first performed cap-binding assays to determine if there is increased binding of 4E-BP1 to eIF4E upon p53-activation. Indeed, we observed increased 4E-BP1-eIF4E binding in Nutlin-3a treated cells (Figure 6B) accompanied by a concurrent decrease in eIF4G binding, an initiation factor that promotes cap-dependent translation when bound to eIF4E (Figure 6B). Next, we measured whether p53-dependent repression of global protein synthesis requires 4E-BP (Figure 6C; S7G). As 4E-BP1 and 4E-BP2 are functionally redundant, we first knocked-down both proteins to prevent compensation of 4E-BP1 by 4E-BP2 (Bi et al., 2017; Ding et al., 2018). 4E-BP1/2 knockdown rescues to a large extent the global protein synthesis repression observed upon Rps6 reduction in Prx1Cre;Rps6lox/+ primary micromass cultures and in Nutlin-3a treated cells (Figure 6C–D). This effect was mirrored when 4E-BP1 was knocked-down alone in Nutlin-3a treated cells (Figure S7H).
Figure 6. p53 controls translation in part through 4E-BP1.
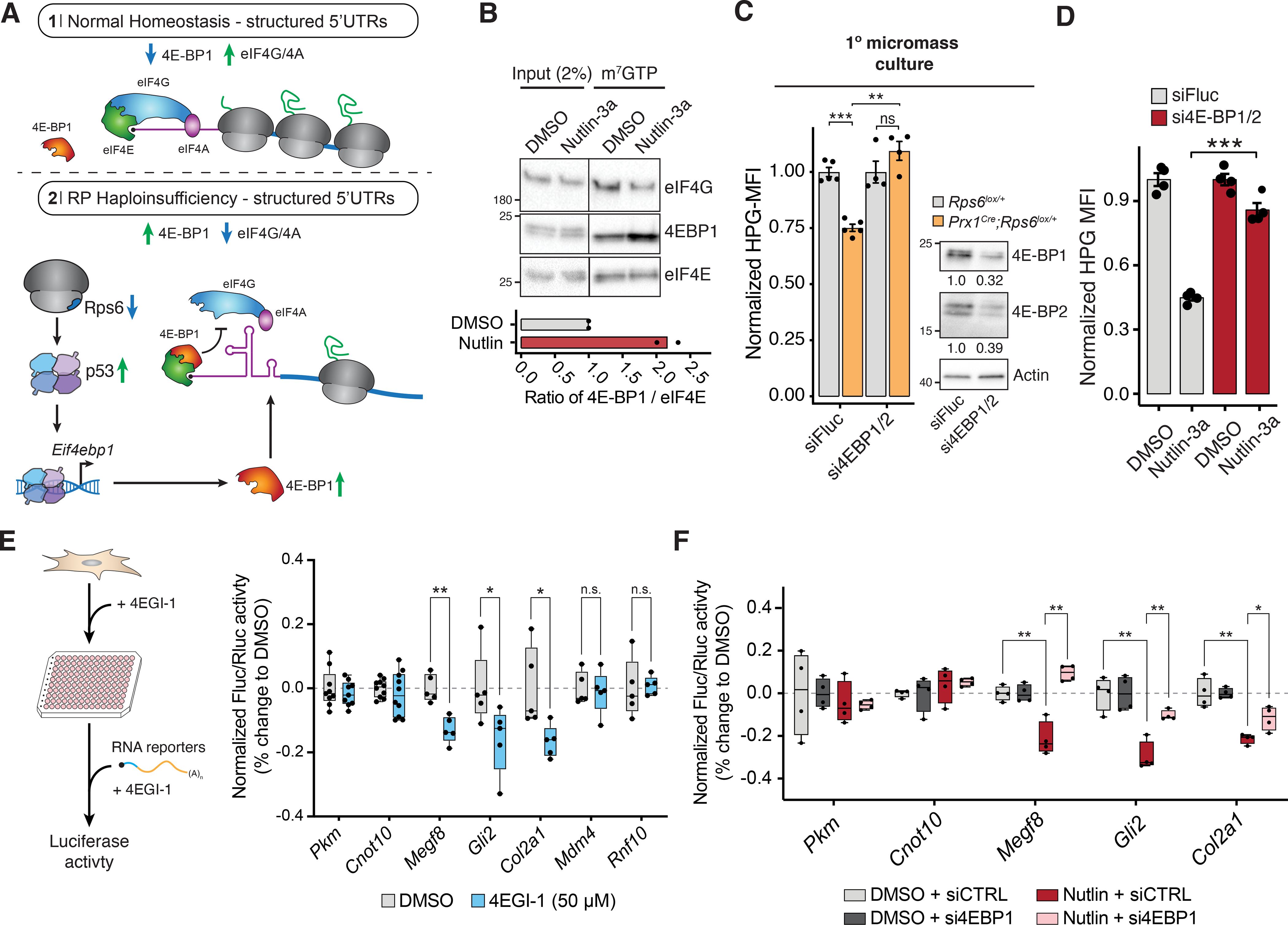
(A) mRNAs with highly structured 5’UTRs are particularly sensitive to eIF4E/4E-BP1-mediated regulation (Pelletier and Sonenberg, 1985; Pelletier et al., 2015). In normal homeostasis (1), eIF4E recruits eIF4G/4A, which helps unwind structured 5’UTRs and promote translation. Upon RP haploinsufficiency (2), p53-mediated 4E-BP1 induction may lead to selective translational repression of structured mRNAs by blocking eIF4E-eIF4G/4A binding.
(B) Cap-binding assay of NIH3T3 cells treated with Nutlin-3a for 8 h. Bottom: ratio of 4E-BP1 to eIF4E in cap-binding assays; n = 2.
(C) Left: HPG MFI upon 4E-BP1/2 siRNA treatment in primary Rps6lox/+ and Prx1Cre;Rps6lox/+ limb micromass cultures from whole E10.5 forelimbs. Values normalized to respective wildtype of each siRNA condition; n = 4. Right: Western blot of 4E-BP1 and 4E-BP2 levels in primary Rps6lox/+ limb micromass cultures after siRNA treatment for 16 h. Numbers indicate quantification of proteins normalized to siFluc.
(D) HPG MFI upon 4E-BP1/2 siRNA treatment in NIH3T3 cells normalized to mean of respective DMSO control for each knockdown condition; n = 4.
(E) Left: Schematic of 4EGI-1 treatment and luciferase RNA reporter assay in C3H/10T1/2 mesenchymal cells. Right: Fluc/Rluc activity of RNA reporters transfected into C3H/10T1/2 cells after 4 h 4EGI-1 treatment. Activity was normalized to the geometric mean of Pkm and Cnot10 5’UTR reporters and mean of DMSO control.
(F) Fluc/Rluc activity from transfection of RNA reporters in NIH3T3 cells treated with either DMSO or Nutlin-3a for 8 h after siRNA knockdown for 16 h with control or 4E-BP1 siRNA. Activity was normalized to the geometric mean of control Pkm and Cnot10 5’UTR activities and mean of respective DMSO control of each knockdown condition.
Our previous results demonstrated that key limb development transcripts are translationally down-regulated in accordance with their 5’UTR features (Figure 4G). As a result, we asked if the 5’UTRs of translationally down-regulated transcripts upon Rps6 haploinsufficiency contribute to sensitivity of 4E-BP1-mediated translational regulation. To this end, we treated C3H/10T1/2 mesenchymal cells with 4EGI-1 (eIF4E/eIF4G interaction inhibitor 1), which functionally acts a small molecule mimic of 4E-BP1 (see Method Details) (Sekiyama et al., 2015), prior to transfection with RNA reporter constructs harboring 5’UTRs shown to be sensitive to RP haploinsufficiency (Figure 4D,G; 6E). We found that reporter constructs containing 5’UTRs whose translational regulation was p53-dependent (Megf8, Gli2, Col2a1) to be more sensitive to 4EGI-1 treatment compared to p53-independent 5’UTRs (Mdm4, Rnf10) or control 5’UTRs that do not change in TE in our ribosome profiling experiment (Pkm, Cnot10) (Figure 6E). Additionally, we asked whether 4E-BP1 is required for translational repression of RNA reporter constructs driven by 5’UTRs of p53-dependent transcripts upon p53 activation. Compared to control 5’UTRs (Pkm and Cnot10), we observed reduced reporter activity driven by the 5’UTRs of p53-dependent translationally repressed transcripts (Megf8, Gli2, and Col2a1) (Figure 6F). This repression is rescued upon knockdown of 4E-BP1, demonstrating that 4E-BP1 is required for p53-dependent translational repression of these mRNAs via their 5’UTRs. Together, these data further show that 4E-BP1-mediated translation regulation contributes to the selective p53-dependent translational changes observed upon Rps6 haploinsufficiency.
To further understand if 4E-BP1 upregulation is a general phenomenon associated with depletion of core RPs, we knocked down RPs commonly mutated in ribosomopathies such as DBA and 5q- myelodysplastic syndrome, Rps19 and Rps14, in primary limb micromass cultures and assessed p53 activation and Eif4ebp1 expression. As with Rps6 haploinsufficiency, we found that p53 is activated and Eif4ebp1 expression is induced upon depletion of these RPs (Figure 7). Together, these results suggest a common pathway via the p53–4EBP1-eIF4E axis by which selective changes in translation occur upon ribosome perturbation, including in the context of RPs directly mutated in ribosomopathies.
Figure 7. Depletion of RPs commonly mutated in ribosomopathies induces Eif4ebp1 expression.
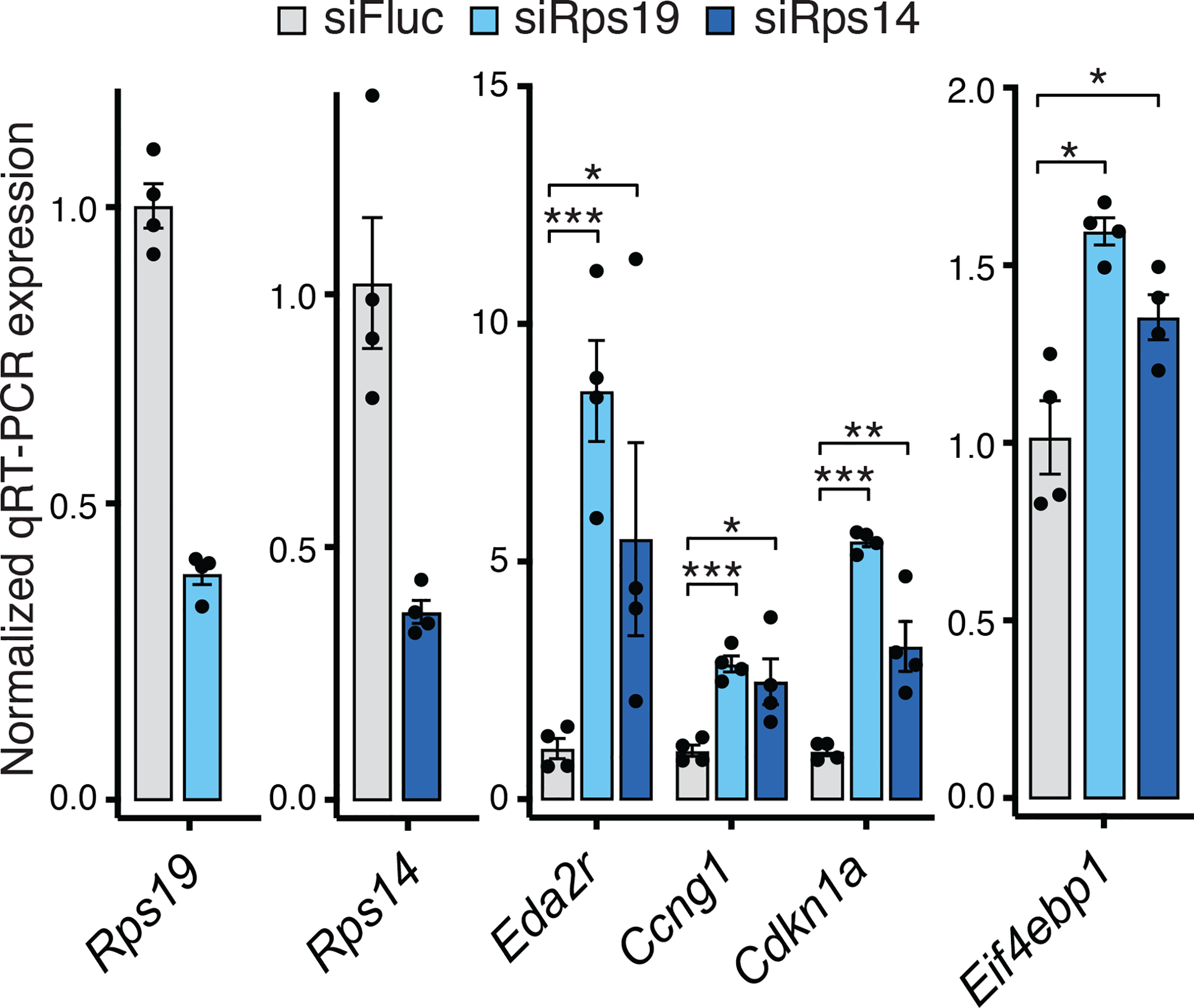
RT-qPCR of p53 target genes (Eda2r, Ccng1, Cdkn1a) and Eif4ebp1 in primary forelimb micromass cultures after Rps19 and Rps14 siRNA knockdown. n = 4.
DISCUSSION
Ribosomopathies caused by haploinsufficiency of essential RPs present with a variety of phenotypes including craniofacial, digit, and limb abnormalities, as well as selective impairment of erythroid differentiation. How such phenotypes, and in particular congenital birth defects, are produced from mutations in essential RPs has been an outstanding mystery. Studies investigating the role of p53 in ribosomopathies have found that p53 activation contributes to phenotypes in many model systems although it is unclear how p53 leads to such complex phenotypes (Danilova et al., 2008; Dutt et al., 2011; Jones et al., 2008; McGowan et al., 2008, 2011; Sulic et al., 2005). The few studies that have suggested that translation dysregulation plays a role in ribosomopathies typically hypothesized that these translational changes occur directly from impaired ribosome function independently of p53 (Horos et al., 2012; Khajuria et al., 2018; Ludwig et al., 2014). Such studies knocked down RPs in ex vivo models to levels that are likely much lower than what would be physiologically expected in an RP haploinsufficient disease. It has therefore been difficult to capture a physiologically relevant change in translation underlying in vivo tissue phenotypes. Thus, while p53-dependent or -independent mechanisms have typically been considered to be mutually exclusive processes that underlie ribosomopathies, our work demonstrates that they are interconnected. In particular, we demonstrate an unexpected role of p53 in mediating much of the translation program downstream of RP haploinsufficiency. We show that p53 induces the transcription of 4E-BP1, which is the main negative regulator of eIF4E-mediated cap-dependent translation (Lin et al., 1994; Sonenberg and Hinnebusch, 2009). Thereby, one way which translational specificity can arise upon RP haploinsufficiency is through an intermediary pathway, the p53–4E-BP1-eIF4E axis, which links RP haploinsufficiency to selective changes in cap-dependent translation.
Our discovery that p53-mediated induction of 4E-BP1 underlies many of the translational changes upon RP haploinsufficiency also suggests mechanisms by which tissue specificity can emerge. Tissue specific phenotypes could emerge either through 4E-BP1-mediated selective translation of key developmental transcripts within tissues or through differential expression and activity of 4E-BP1 itself among different tissues. The 4E-BP1-eIF4E axis is a rate-limiting component of translation initiation with inherent selectivity (Duncan et al., 1987; Pelletier et al., 2015; Truitt et al., 2015). As a result, 4E-BP1 induction may lead to a translational program in which perturbed translation of select transcripts, which are differentially sensitive to eIF4E activity, contribute to tissue-specific phenotypes. For example, upon Rps6 haploinsufficiency, we observe p53-dependent translational repression of key limb developmental transcripts that correlate with the observed limb phenotypes. In addition, we also observed that differential 4E-BP1 expression and response among different tissues may also contribute to cell and tissue-specific phenotypes upon RP haploinsufficiency. We demonstrate that although conditional Rps6 haploinsufficiency led to p53 activation in both the limb mesenchyme and AER, only the limb mesenchyme exhibited 4E-BP1 upregulation, translation repression, and a phenotype. As a result, future studies characterizing tissue-specific 4E-BP1 expression and investigating mechanisms underlying such selectivity would provide further insight into tissue specificity in ribosomopathies.
Our findings demonstrating a role for p53-mediated induction of 4E-BP1 in translational control upon RP haploinsufficiency also suggest possible therapeutic strategies to treat ribosomopathies. Treatment with l-leucine, which upregulates translation via mTORC1 activation, was shown to rescue developmental phenotypes in ribosomopathy models (Jaako et al., 2012; Payne et al., 2012). Given that our work demonstrates that in vivo phenotypes upon RP haploinsufficiency are driven in part by translational perturbation via the p53–4E-BP1-eIF4E axis, more potent activators of cap-dependent translation and/or eIF4E activity may serve as enticing candidates to treat ribosomopathies.
Although most of the translational changes observed upon Rps6 haploinsufficiency were p53-dependent, we also observed a small number of p53-independent translational changes that included translational upregulation of many ribosomal proteins and translational repression of the p53 negative regulator Mdm4. We hypothesize that these changes represent possible post-transcriptional quality control mechanisms that allow cells to quickly respond to ribosome dysfunction. For example, translational upregulation of RPs upon haploinsufficiency of a specific RP would allow for increased RP production that may mitigate the effects of RP depletion. In fact, translation of Rps6 itself was upregulated (Table S3–4), suggesting that the phenotype may have been more severe with a further decrease in Rps6 protein levels had it not been for this feedback mechanism. Upregulation of RP translation was particularly unexpected given that ex vivo studies demonstrated a marked decrease in RP translation after RP depletion (Khajuria et al., 2018), suggesting that results from in vivo RP haploinsufficiency models may differ from RP knockdown in cultured cells. Furthermore, translational repression of Mdm4, a negative regulator of p53 that cooperates with Mdm2 to suppress p53 activity (Marine et al., 2007), may serve as a rapid post-transcriptional quality control sensor to activate p53 in the context of RP haploinsufficiency.
Ribosome biogenesis is one of the most energetically expensive programs in the cell. By coupling alterations in essential RP expression directly to cap-dependent translation, p53 may ensure that a modified cellular translatome continues to support cellular growth and survival under suboptimal conditions. Interestingly, recent findings suggest that tissue selective RP haploinsufficiency phenotypes in Drosophila also largely occur indirectly via a regulatory response mediated by the bZip-domain protein Xrp1 rather than by a direct decrease in ribosome levels (Kongsuwan et al., 1985; Lee et al., 2018). As opposed to Xrp1 in Drosophila, we show that p53 itself drives the majority of the translational changes in an in vivo mammalian model of RP haploinsufficiency. Whereas we demonstrate in our work that p53-dependent transcriptional control of Eif4ebp1 regulates at least a part of this translational response, it is likely that other yet-to-be defined p53 targets mediate other aspects of this translational program. Therefore, characterization of p53-dependent remodeling of the translational landscape is likely to yield greater insights and novel therapeutic strategies to target ribosomopathies.
LIMITATIONS OF THE STUDY
Our work demonstrates translational downregulation of mRNAs including those critical for proper limb development upon Rps6 haploinsufficiency. Although translationally perturbed limb development transcripts such as Col2a1, Dicer1, Gli2, and Megf8 could be associated with the specific limb phenotypes observed, it is unclear which of these transcripts actually contribute to phenotype. In fact, it is likely that translational perturbation of several transcripts combinatorially act to perturb proper limb development. Further combinatorial in vivo genetic interaction experiments would be required to elucidate which transcripts contribute to phenotype upon Rps6 haploinsufficiency.
Murine Rps6 haploinsufficiency recapitulates many of the cardinal phenotypes of ribosomopathies, including limb defects as demonstrated in this work, and blood developmental defects (McGowan et al., 2008, 2011). We selected this model given the strong phenotype that it produces (Panić et al., 2006). However, heterozygous loss of function mutations in Rps6 have not been identified in ribosomopathies, likely because of the severity of constitutive Rps6 haploinsufficiency in the developing embryo. Nevertheless, we do observe a conserved mechanism whereby reduction in RPs associated with ribosomopathies (Rps19 and Rps14) triggers p53 activation and subsequent upregulation of Eif4ebp1, suggesting that this is a more general mechanism that is not specifically linked to any single RP. Overall, this common pathway could explain how mutations in multiple RP genes converge upon similar phenotypes. Given that we focused on perturbation of one particular RP in the context of one particular tissue in mice, further work examining the in vivo role of 4E-BP1 in the manifestation of ribosomopathy phenotypes in other disease relevant tissues such as the hematopoietic compartment and in other RP haploinsufficient states in both human tissue samples and mice is an important future direction.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Maria Barna (mbarna@stanford.edu).
Materials availability
All plasmids and DNA constructs generated in this study are available upon request. All antibodies, chemicals, cell lines, and most mouse lines used in this study are commercially available. All other unique materials are also available upon request.
Data and code availability
All custom codes are available upon request from the corresponding author. Data that support the findings of this study have been deposited in the Gene Expression Omnibus under accession number GSE135722.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse husbandry.
All animal work was reviewed and approved by the Stanford Administrative Panel on Laboratory Animal Care (APLAC). The Stanford APLAC is accredited by the American Association for the Accreditation of Laboratory Animal Care (AAALAC). All mice used in the study were housed at Stanford University. Mice were maintained on mixed backgrounds and the following previously described alleles were used: Rps6lox (Volarevic et al., 2000), Prx1Cre (Logan et al., 2002), Msx2Cre (Sun et al., 2000), Tsc2lox (Hernandez et al., 2007), Trp53null (Jacks et al., 1994), and RosamTmG/+ (Muzumdar et al., 2007). Rps6lox mice were generously provided by George Thomas (IDIBELL). Prx1Cre, Msx2Cre, and Tsc2lox mice were obtained from the Jackson Laboratories. Genotyping was performed using standard PCR protocols using primers described in aforementioned publications or on the Jackson Laboratories website. Common Cre primers were used to genotype Cre lines (http://mgc.wustl.edu/protocols/pcr_genotyping_primer_pairs). The following primers were used for Rps6lox genotyping: Rps6lox_1 GCTTCTACTTCTAAGTCTGAGTCCAGTC, Rps6lox_2 TCCTGCCGAGAAAGTATCCATCATG, and Rps6lox_3 CTGCAGCCTTTTCTTTTAGCATACCTG. To generate embryos, a male mouse was housed with 1–2 females overnight. Female mice were considered pregnant at E0.5 on the day a vaginal plug was observed. Embryos were harvested from pregnant females that had been euthanized via CO2 exposure, according to APLAC-approved methods. Mice used for a given experiment were colony-matched. Mouse embryos for a given experiment were somite matched. Mouse embryos were harvested at E9.5, E10.5, E11.5, E12.5, E13.5, and E17.5. Biological sex was determined for embryos used in ribosome profiling experiments via PCR using primers targeting the Sry locus: Sry F - TTG TCT AGA GAG CAT GGA GGG CCA TGT CAA, Sry R - CCA CTC CTC TGT GAC ACT TTA GCC CTC CGA (https://mgc.wustl.edu/protocols/pcr_genotyping_primer_pairs), with sex incorporated as batch variables in the analysis (see below in “Ribosome profiling analysis”). For all other experiments and phenotypic analyses, biological sex was not determined and data from male and female mice were pooled because no obvious difference was found between sexes.
Cell Lines.
Primary MEFs were derived from E13.5 mouse embryos using standard protocols and cultured under standard conditions in DMEM supplemented with 2 mM L-glutamine and 10% fetal bovine serum (FBS). Cells were used for experiments at passages 2–5. NIH3T3 and C3H/10T1/2 cells were purchased from the American Type Culture Collection and grown under standard conditions in DMEM supplemented with 10% FBS and 2 mM L-glutamine. Cells were passaged 1:6 roughly every 2–3 days. All cell lines used in this study were mycoplasma-free. All cell lines were grown in humidified CO2 incubators at 37°C and 5% CO2. See below for limb micromass culture conditions.
METHOD DETAILS
Skeleton and cartilage staining.
Bone and cartilage staining of E17.5 embryos were performed as per standard methods. Skeletal images were acquired using a Zeiss SteREO Discovery.V8 microscope and a Zeiss AxioCam MRc5 camera with an external standard as scale. Skeletal elements were measured using the ImageJ Fiji plug-in with genotypes blinded during measurement.
Separation of limb ectoderm and mesenchyme.
Limb bud experiments were performed either on whole forelimbs consisting of both mesenchyme and ectoderm layers or on limbs enriched for mesenchyme with the ectoderm removed. Because mesenchyme isolation requires trypsinization and time to microdissect, we used whole limbs rather than isolated mesenchyme for several of our experiments, especially those that measured temporally sensitive processes such as translation (polysome profiles, ribosome profiling, etc.). For most cases, the main limitation of including un-recombined tissue might be the potential to decrease the sensitivity of our assays. As a result, hits we do find in whole limb experiments, such as those from the ribosome profiling experiment, likely represent strongly differentially translated targets.
For experiments in which the ectoderm was removed, the separation of the limb ectodermal jacket from the mesenchymal layer was done as previously described (Zeller et al., 1989). Briefly, forelimb buds of E10.5 or E11.5 embryos were dissected in 1×PBS. Dissected limb buds were then digested in 3% trypsin in 1×PBS for 25 min at 4°C with gentle shaking. Trypsin digestion was quenched by moving the limb buds to 10% FBS in PBS. The limb buds were then gently vortexed for 15 seconds to dislodge the mesenchymal and ectodermal layers. The ectodermal jackets were further removed from the individual limb buds under a dissecting microscope in 1×PBS using forceps. Separated samples were inspected visually before being moved to separate tubes. Samples were either flash frozen and stored at −80°C or used immediately.
For OPP labeling, dissected ectoderm was dissociated from the mesenchyme layer of E11.5 forelimb buds by incubating in 1% trypsin at room temperature for 20 min. The ectoderm was then separated from the mesenchyme layer and resuspended in DMEM/F12, no phenol red, 10% FBS and labeled with OPP (20 μM) as described below.
In situ hybridization.
In situ hybridization for Sox9 at E12.5 was performed as per standard protocols (Lufkin, 2007) with modifications to achieve good probe penetration and signal. Digoxigenin (DIG) labeled RNA probes were generated from XhoI linearized T3 promoter Sox9 plasmid (gift from Chi-Chung Hui, University of Toronto). Fixed and rehydrated embryos were treated with 20 μg/mL proteinase K in PBS+0.1% Tween-20 (PBST) for 35 minutes at room temperature. After post-fixation and subsequent washes, embryos were incubated overnight at 70°C with hybridization mix containing 0.5 μg/mL denatured DIG-labeled probe. After washes and pre-blocking, embryos were incubated overnight at 4°C with 1:2000 alkaline phosphatase-conjugated anti-DIG antibodies (Sigma/Roche, 11093274910). After washes and prior to development, embryos were washed 2×20 min with 100 mM NaCl, 100 mM Tris-HCl pH 9.5, 50 mM MgCl2, 0.1% Tween-20, 2 mM levamisole. Embryos were then developed with BM Purple AP substrate (Sigma/Roche, 11442074001) at 4°C for >1 day until adequate signal to noise ratio was achieved. The reaction was then subsequently inactivated, post-fixed, and stored. Embryos were then imaged with a Leica MZ16 FA microscope with a Leica DFC480 camera with an external standard used for scale.
Vectors and in vitro transcription.
For generation of 5’UTR reporter constructs, sequences were obtained from ENSEMBL and isoforms were chosen based on alignment to the RNA sequencing results presented herein (Table S2). 5’UTR gene fragments were synthesized (Twist Biosciences) and cloned immediately upstream of a Firefly luciferase gene encoded in pGL3-FLB (Leppek et al., 2020). Each 5’UTR-FLuc construct was PCR amplified using primers flanking the 5’UTR and FLuc gene and incorporating a T7 RNA polymerase promoter at the 5’ end. PCR amplicons were column purified (NEB, T1030) and used as a template for in vitro transcription reactions with a mMESSAGE mMACHINE® T7 Transcription Kit (Ambion, AM1344) followed by the addition of a poly(A) tail using a Poly(A) Polymerase Tailing Kit (Lucigen, PAP5104H). In vitro synthesized capped and poly(A) tailed RNA was purified using PureLink RNA Mini Columns (Thermo Fisher, 12183020). RNA integrity was confirmed via denaturing agarose gel electrophoresis.
For testing p53 transcriptional activity, a 423 bp gene fragment of Eif4ebp1 (Table S2) was amplified from genomic DNA of primary mouse embryonic fibroblasts and cloned in lieu of the minimal promoter sequence within pGL4.23[luc2/miniP]. All constructs were sequence verified.
siRNA-mediated knockdowns.
NIH3T3 cells were plated at a density of 1.0×105 cells/well in a 6-well plate and allowed to attach for at least 6 h at 37°C. Cells were washed with 1×PBS before being transfected with 25 nM of non-targeting control siRNA #2 (siFluc; Dharmacon, D-001210–02-05) or siRNA target against 4E-BP1 and 4E-BP2 (Dharmacon) using Dharmafect Reagent I (Dharmacon, T-2001) as per manufacturer’s protocol. Cells were incubated for 16 h at 37°C in antibiotic-free media. Following incubation, the siRNA-containing media was replaced with complete DMEM media (as described above) and cells were treated with either DMSO, Nutlin-3a, or Doxorubicin.
For siRNA treatment in micromass limb cultures, limbs from E11.5 embryos were dissected and cells dissociated as described. Subsequently, 1.5×106 cells were reverse transfected with 25 nM siRNA as per manufacturer’s protocol using Dharmafect I (Dharmacon, T-2001) in Limb micromass media and plated in a 96-well plate. Cells were incubated for 20 h before use in subsequent experiments. Note that siRNA knockdowns of Rps19 and Rps14 were performed in micromass cultures derived from wildtype C57BL/6J mice.
For si4E-BP1 knockdown and mRNA reporter experiments, NIH3T3 cells (5.0×104 cells/well in a 12-well plate) were transfected with 25 nM of non-targeting control siRNA #3 (Dharmacon, D-001210–03-05) or siRNA target against 4E-BP1 (Dharmacon) using Dharmafect I (Dharmacon, T-2001) as per manufacturer’s protocol. Cells were incubated for 16 h at 37°C in antibiotic-free media. The next day, media was removed and cells were treated with either DMSO or Nutlin-3a (10 μM) in DMEM supplemented with 10% FBS for 4h before being transfected with Firefly Luciferase reporter RNA (50 ng) and Renilla Luciferase control RNA (10 ng) as described below. Cells were incubated for a further 4 h in the presence of DMSO or Nutlin-3a at 37°C before being harvested for luciferase assays.
RT-qPCR quantification of RNA expression.
RNA from embryonic limbs or cell culture was extracted with TRIzol (Thermo Fisher, 15596026) as per manufacturer’s protocol followed by DNase I treatment for 30 min at 37°C. RNA was then purified via Zymo RNA Clean & Concentrator columns (Zymo Research). Subsequently, 100 ng of RNA was used for first strand cDNA synthesis using the iScript Supermix (Bio-rad, 1708841). cDNA was diluted 18-fold and 4 μL was used in a reaction for SYBR green detection with SsoAdvanced SYBR Green supermix (Bio-Rad, catalog no. 1725270) on a CFX384 machine (Bio-rad). All primer sequences are available in Table S2. In general, Ct values were normalized to those of housekeeping genes and then to wildtype, DMSO, or siRNA controls. For Figure 1B: Expression was normalized to the geometric mean of housekeeping genes Actb, Tbp, Ubb, Exo5, Pkm2, and Nadk2 then to the mean of wildtype values. For Figure 1L: Expression was normalized to Actb and to the mean of wildtype values. For Figure 2H: Expression was normalized to the geometric mean of Actb and Tbp then to the mean of wildtype. For Figure 5D: Expression was normalized to the geometric mean of housekeeping genes Actb, Tbp, Ubb, Exo5, Pkm2, Nadk2 and mean of wildtype. Figure 5G: Expression was normalized to the geometric mean Actb and NupL1 and then to DMSO. For Figure 7: Expression was normalized to the geometric mean of TBP and NupL1 and then to siRNA control (siFluc).
Western blots.
Equal amounts of protein were resolved on either a 15% or a 4–20% Tris-glycine gradient SDS-PAGE gel and transferred to a polyvinylidene difluoride Immobilon-FL membrane (PVDF; Bio-rad). Membranes were blocked for 30 min at room temperature with 5% BSA in TBST (50 mM Tris, 150 mM NaCl, 1% Tween-20, pH 7.4). Blots were incubated for 24 h at 4°C with the following primary antibodies: mouse anti-GAPDH (Cell Signaling, 97166), mouse anti-actin (Cell Signaling, 3700S), rabbit anti-Rps6 (Cell Signaling, 2217L), rabbit anti-4E-BP1 (Cell Signaling, 9644S), rabbit anti-4E-BP2 (Cell Signaling, 2845S), rabbit anti-phospho-p70 S6 Kinase (Cell Signaling, 9205S), rabbit anti-p70 S6 Kinase (Cell Signaling, 9202S), rabbit anti-phopsho-ULK1(Ser757) (Cell Signaling, 6888T), rabbit anti-ULK1 (Cell Signaling, 8054T), rabbit anti-p53 (Leica Biosystems, CM5), rabbit anti-TSC2 (Cell Signaling, 4308T), rabbit anti-eIF4G (Cell Signaling, 2498S), mouse anti-eIF4E (BD Transduction, 610269). All antibodies were used at a dilution of 1:1000 in 5% BSA-TBST, unless stated otherwise. Membranes were washed 3 times for 5 min in TBST before incubation for 1 h at room temperature with secondary antibodies: donkey anti-mouse (1:5000; GE Healthcare, NA931–1ML), or donkey anti-rabbit (1:5000; GE Healthcare, NA934–1ML) coupled to Horseradish Peroxidase. Membranes were then washed 3 times for 5 min in TBST before detection using Clarity Western ECL Substrate (Bio-rad, 170–5061) and imaging on a ChemiDoc MP (Bio-rad, 17001402). All blots were quantified using ImageJ v2.0.0.
Micromass cultures and RNA transfection luciferase assays.
E11.5 mouse limbs were harvested in DMEM/F12, no phenol red, 10% FBS, pen/strep. Cells were then dissociated in Dissociation Buffer (1% trypsin in HBSS without Ca+2 or Mg+2) and incubated for 30 min at 37°C. Single-cell suspensions were obtained by quenching the dissociation with Limb Media (DMEM/F12 HEPES, 10% FBS) and passing through a cell strainer. Cells were then pelleted and resuspended in Limb Media. Next, 1.0×105 cells were co-transfected with capped and poly(A)-tailed FLuc (100 ng) and RLuc reporter RNAs (10 ng) using Lipofectamine 2000 (Thermo Fisher, 11668–019) before seeding into a single well of a 96-well Nunclon Delta Microplate (Thermo Fisher, 167008). Cells were incubated for 6 h at 37°C before being harvested in 1× Passive Lysis Buffer and subjected to a Dual-Luciferase Reporter Assay System as per manufacturer’s instructions (Promega, E1980) on a GloMax-Multi plate reader (Promega).
DNA transfection luciferase assays.
NIH3T3 cells were seeded at a density of 1.0×105 cells/well in a 6-well plate and incubated at 37°C overnight. The following day cells were co-transfected with 2 μg of FLuc plasmid DNA and 20 ng of RLuc transfection control plasmid (pRL) per well using Lipofectamine 2000 (Thermo Fisher, 116688–019) and incubated for 16 h at 37°C. Cells were then treated with either DMSO, Nutlin-3a (10 μM), or Doxorubicin (0.2 μg/mL) for 8 h. Cells were harvested and lysed in 50 μL of 1×Passive Lysis Buffer. Lysate was cleared of debris by centrifugation at 10,000 RCF for 5 min, and 20 μL of resulting supernatant was used to measure luciferase activity with the Dual-Luciferase Reporter Assay System (Promega, E1980) on a GloMax-Multi plate reader (Promega).
4EGI-1 treatment of 10T1/2 cells and luciferase assays.
Mechanistically, 4EGI-1 binds with eIF4E to specifically disrupt association of eIF4G, a large scaffold protein that recruits the 40S ribosomal subunit, while promoting and stabilizing the binding of 4E-BP1 (Sekiyama et al., 2015). 10T1/2 cells were seeded at a density of 4.0×105 cells/well in a 96-well plate and incubated at 37°C overnight. The next day, cells were treated with 50 μM 4EGI-1 (Sigma, 324517) or DMSO in DMEM supplemented with 10% FBS and 2 mM L-glutamine and incubated for 4 h at 37°C. Following treatment, cells were co-transfected with capped and poly(A)-tailed FLuc (100 ng) and RLuc reporter RNAs (10 ng) using Lipofectamine 2000 (Thermo Fisher, 11668–019). Cells were then re-treated with 50 μM 4EGI-1 or DMSO and incubated for a further 4 h at 37°C. After a total of 8 h of treatment (4 h prior to transfection and 4 h after transfection), cells were harvested in 1× Passive Lysis Buffer and luciferase activities were measured using a Dual-Luciferase Reporter Assay System as per manufacturer’s instructions (Promega, E1980) on a GloMax-Multi plate reader (Promega).
P53 transactivation domain mutated MEFs.
MEFs were derived from previously generated knock-in mice expressing p53 wild-type (Trp53LSL-WT/LSL-WT) or Trp53QM/QM (Trp53LSL−25,26,53,54/LSL−25,26,53,54) mutant (Brady et al., 2011). In these MEFs, expression of both Trp53 alleles is silenced through upstream transcriptional stop elements flanked by loxP recombination sites. To reactivate p53 expression, MEFs were infected with adenoviruses expressing Cre recombinase (Ad5 CMV-Cre) or empty virus (Ad5-CMV-Empty) as control at an approximate multiplicity of infection of 100. Viruses were purchased from University of Iowa Viral Vector Core Facility. Infected cells were cultured for 48 hours at 5% CO2, 37°C before initiating drug treatments and HPG assay. Cells were treated with 0.2 μg/ml doxorubicin or 10 μM Nutlin-3a for indicated times (see below). High efficiency of recombination (>90% cells) was confirmed by immunostaining of p53. P53 stabilization upon doxorubicin and Nutlin-3a treatment was confirmed by Western blotting using p53 CM5 antibody (Leica Biosystems).
Chromatin immunoprecipitation qPCR.
MEFs were grown in DMEM containing 10% FCS and seeded at 7 × 106 cells per 10 cm dish one day prior to the ChIP experiment. After treatment with 0.2 μg/ml doxorubicin for 6h, cells were harvested to prepare chromatin for immunoprecipitation using p53 polyclonal antibodies (NCL-L-p53-CM5p; Leica Biosystems). ChIPs were performed essentially as described previously (Kenzelmann Broz et al., 2013). Chromatin-immunoprecipitated DNA was quantified by qPCR using SYBR Green and a 7900HT Fast Real-Time PCR machine (Applied Biosystems) and primers specific for Eif4ebp1 (Table S2). The signals obtained from the ChIP were analyzed by the percent input method.
Cap-binding assays.
NIH3T3 cells were seeded at a density of 1.0×106 in 10 cm tissue culture plates and incubated overnight at 37°C. Cells were then treated with DMSO or Nutlin-3a (10 μM) for 8 h. Subsequently, cells were harvested and washed twice with ice cold 1×PBS before being lysed for 30 min on ice with occasional agitation in Cap Lysis Buffer (10 mM Tris-HCl pH 7.5. 140 mM KCl, 4 mM MgCl2, 1 mM DTT, 1 mM EDTA, 1% Nonidet P-40) supplemented with protease and phosphatase inhibitor for 30 min on ice with occasional agitation. Lysates were cleared via centrifugation at 10,000 RCF for 15 min at 4°C. Resulting supernatants were collected and protein concentration was determined using a BCA assay (Thermo Fisher). 250 μg of protein was pre-cleared using 50 μL of agarose beads (Jena Biosciences, AC-100S) in Cap Lysis Buffer for 30 min at 4°C with rocking. Next, the beads were pelleted at 1000 RCF and the supernatant was added to 50 μL of γ-aminophenyl-m7GTP (C10-spacer)-agarose beads (Jena Biosciences, AC-155S) in Cap Lysis Buffer overnight at 4°C with rocking. Supernatant was removed and the beads were washed twice with Cap Lysis Buffer followed by 1×PBS. Beads were then resuspended in 100 μL of 1× SDS loading buffer and boiled for 5 min. 25 μL of each sample was used for SDS-PAGE and Western blot analysis for eIF4E, eIF4G, and 4E-BP1.
Proliferation and apoptosis assays.
Cell proliferation and apoptosis levels were measured using EdU (5-ethynyl-2´-deoxyuridine) incorporation and Annexin V staining, respectively. Briefly, 1.0×105 NIH3T3 cells were seeded in a 6-well plate and incubated overnight at 37°C. The following day cells were treated with DMSO or Nutlin-3a (10 μM) for 4 or 8 h. For proliferation measurements, one hour before incubation was complete, cells were treated with 10 μM EdU using a Click-iT EdU Alexa Fluor 488 flow Cytometry Assay Kit (Thermo Fisher, C10425) and processed as per manufacturer’s instructions. For apoptosis measurements, cells were harvested, washed twice with Cell Staining Buffer, and labeled using a FITC Annexin V and propidium iodide staining kit as per manufacturer’s protocol (BioLegend, 640914). Both EdU- and Annexin V-labeled samples were analyzed on a LSRII flow cytometer (BD Biosciences) using software packages CellQuest and FlowJo v10.
For whole mount cell death assays, dissected embryos were incubated with 1 mL of 5 μM LysoTracker Red DND-99 (Invitrogen) in HBSS for 45 min at 37°C. Embryos were washed 4X with HBSS and then fixed in 4% paraformaldehyde overnight at 4°C. Embryos were washed with PBS + 0.1% Tween-20 and then dehydrated stepwise into 100% methanol in which they were stored at −20°C. Prior to imaging, they were rehydrated stepwise into PBS + 0.1% Tween-20 and imaged on a fluorescence microscope.
Immunofluorescence on cryosections.
Immunofluorescence on cryosections was performed per standard protocols. Mouse embryos were dissected and fixed in ice-cold 4% PFA for 1 hour, washed four times with PBS + 0.1% Tween-20, equilibrated in 30% sucrose 0.1M potassium phosphate buffer, pH 7.4, embedded in O.C.T., and stored at −80°C. Embryos were sectioned transversely into 12 um thick sections using a Leica cryostat, and frozen on slides at −80°C until use. For immunofluorescence staining, all washes and incubations were done using blocking buffer (PBS with 1% goat serum and 0.1% Triton X-100). Sections were blocked for 1 hr in blocking buffer, incubated in primary antibody overnight at 4C, washed three times, incubated for 1 hr at room temperature with secondary antibody, and washed three more times before mounting. Primary antibodies were used at the following concentrations: 1:200 Leica anti-p53 (CM5P-L), 1:500 Sigma anti-phospho-Histone H3 (06–570), 1:600 Cell Signaling Technology Cleaved Caspase-3 (9661S). Goat anti-rabbit AF568 secondary from Life Technologies (A11036) was used for all slides at 1:500. Slides were imaged using a Zeiss EC 10x Plan-Neofluar Ph1 objective (NA = 0.3), and acquired using a CSU-X1 UltraVIEW Spinning disc with a Hamamatsu EM-CCD camera and Volocity software. Slides comparing the same antibodies were imaged using consistent imaging settings and processing. For phospho-Histone H3 quantification, total cell count in the limb bud from multiple embryos was quantified from DAPI staining with Spot Counter in ImageJ, and phospho-Histone H3 positive cells were counted manually. Images are representative of average percentages of phospho-Histone H3 cells in Prx1Cre;Rps6lox/+ and wildtype sections.
Measurement of global protein synthesis.
Briefly, O-propargyl-puromycin (OPP) labeling of embryonic limbs was done as follows. E10.5 embryos were dissected in filming media (DMEM/F12, no phenol red, 10% FBS). Forelimbs were removed and dissociated in Dissociation Buffer (1% trypsin in HBSS without Ca+2 or Mg+2) for 15 min at 37°C. Resuspended forelimb cells were used for downstream labeling and analysis. For OPP or L-homopropargylglycine (HPG) labeling of cultured cells, primary MEFs or NIH3T3 cells were seeded at a density of 1.0×105 cells/well in a 6-well plate and allowed to attach overnight. The following day, cells were treated with DMSO, Nutlin-3a (10 μM), or Doxorubicin (0.2 μg/mL) before metabolic labeling. For OPP incorporation, cells were labeled with 20 μM of OPP in DMEM plus drug for 30 min at 37°C. For HPG labeling, two hours before each timepoint, cells were methionine starved in Met dropout media (DMEM supplemented with 10% dialyzed FBS, 25 mM L-cysteine, 2 mM L-glutamine, no methionine) for 45 min at 37°C. Cells were then labeled with 50 μM HPG (Thermo Fisher, C10186) or 50 μM L-Met (control) for 1 h at 37°C. Following metabolic labeling, cells were harvested and washed with twice 1×PBS. Cell pellets were resuspended in Zombie Violet Live-Dead Stain (1:500 in PBS; BioLegend, 423113) and incubated for 15 min in the dark. Cells were then washed with Cell Staining Buffer (0.1% NaN3, 2% FBS in HBSS) before being fixed in 1% PFA for 15 min on ice. Subsequently, cells were permeabilized overnight at 4°C in Perm Buffer (0.1% Saponin, 0.1% NaN3, 3% FBS in PBS). The next day, cells were washed twice with Cell Staining Buffer (without 0.1% NaN3), labeled with an Alexa Fluor 555 Picolyl Azide dye (Thermo Fisher, C10642) and incubated for 30 min at room temperature in the dark. Labeled cells were washed and resuspended in Cell Staining Buffer before being analyzed on either a LSRII flow cytometer (BD Biosciences) or a NovoCyte Quanteon (Agilent Technologies) using software packages CellQuest and FlowJo v10.
Sucrose density gradients and RT-qPCR.
E10.5 mouse embryonic limbs were harvested in cold HBSS (Thermo Fisher, 14025–076) containing 100 μg/ml cycloheximide (CHX). Limb pairs from 3–4 embryos of a given genotype were pooled and lysed via vigorous pipetting and 30 minute incubation at 4°C in 175 μL lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 15 mM MgCl2, 1 mM DTT, 8% glycerol, 1% Triton X-100, 100 μg/ml CHX (Sigma-Aldrich, C7698–1G), 200 U/mL SUPERase RNase Inhibitor (Thermo Fisher, AM2696), 20 U/ml Turbo DNAse (Thermo Fisher, AM2238), and 1X Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher, 78442). Samples were centrifuged at 1,300xg, 5 min and 10,000xg, 5 min to remove cell debris. Samples were then layered onto a 15–45% sucrose gradient or 25–50% sucrose gradient containing 20 mM Tris-HCl pH 7.5, 100 mM NaCl, 15 mM MgCl2, and 100 μg/ml CHX. 15–45% gradient was made on a Biocomp Model 108 Gradient Master. 25–50% gradient was made by sequentially freezing at −80°C a 5-step gradient (50%, 43,75%, 37.5%, 31.25%, 25% sucrose). Samples layered on gradients were spun on a Beckman SW-60 rotor at 35,000 RPM, 2.5 h, 4°C. After centrifugation, gradients were fractionated using a Density Gradient Fraction System (Brandel, BR-188). To normalize for fraction volume in subsequent RT-qPCR experiments, 100 pg of in vitro transcribed RNA containing Renilla and Firefly luciferase (in vitro transcribed from the pRF-HCV vector) were added to each fraction. Equal volumes of fractions were then pooled as described in the text. RNA was then extracted by mixing samples with Acid-Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1) (Thermo Fisher, AM9722), incubating at 65°C, 5 min, and subsequent centrifugation at 21,000xg, 10 min, RT. The aqueous phase was obtained, mixed 1:1 with 100% ethanol, and further purified using the RNA Clean & Concentrator-5 kit (Zymo Research, R1013). Samples were treated 30 minutes with TURBO DNase per manufacturer protocol (Thermo Fisher, AM2238), and subsequently purified again using the RNA Clean & Concentrator-5 kit. Next, 20 ng of RNA was used for first strand cDNA synthesis using the iScript Supermix (Bio-rad, 1708841). cDNA was diluted 20-fold and 4 μL was used in a reaction for SYBR green detection with SsoAdvanced SYBR Green supermix (Bio-Rad, catalog no. 1725270) on a CFX384 machine (Bio-rad). All primer sequences are available in Table S2.
For analysis, the candidate transcript qPCR Ct value in each pooled fraction was normalized to that of the spike-in in vitro transcribed Renilla luciferase RNA. Normalized Ct values were converted from log to linear space, multiplied by the number of fractions combined per pool, and then normalized to the sum total across all pooled fractions for a given sample. These values were compared between genotypes for each fraction using a paired, two-tailed t-tests with samples paired based on day of collection and somite count.
Ribosome profiling of embryonic limb buds.
Ribosome profiling was performed as described before (Ingolia et al., 2012) with modifications. Details are described below. E10.5 mouse embryonic limbs were harvested in cold HBSS (Thermo Fisher, 14025–076) containing 100 μg/ml cycloheximide (CHX). Choice of RNase was particularly important given that low input samples from a pair of E10.5 embryonic limbs were used. Although RNase I offers better codon resolution than RNase A/T1, RNase I has been shown to degrade ribosomes when used in high concentrations relative to input RNA whereas RNase A and T1 better maintain ribosome integrity at a greater range of concentrations despite only cutting single stranded RNA at C/U and G, respectively (Cenik et al., 2015). Given that E10.5 embryonic limbs yield low amounts of RNA, RNase A and T1 were used to prevent excess ribosome degradation.
Embryonic heads were collected for subsequent genotyping. Limb pairs from each embryo were lysed via vigorous pipetting and 30 minute incubation at 4°C in 215 μL buffer A (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 15 mM MgCl2, 1 mM DTT, 8% glycerol, 1% Triton X-100, 100 μg/ml CHX (Sigma-Aldrich, C7698–1G), 20 U/ml Turbo DNAse (Thermo Fisher, AM2238), and Complete Protease Inhibitor EDTA-free (Sigma-Aldrich, 11836170001). Lysates were cleared via sequential centrifugation at 1,300g, 5 min and 10,000g, 10 min at 4°C. For RNA input for RNA-Seq, 70 μL of lysate was diluted in 55 μL of water and stored at −80°C in 375 μL of TRIzol LS (Thermo Fisher, 10296010). For ribosome profiling, 120 μL of cleared lysate was treated with 0.5 μg RNase A (Thermo Fisher, AM2271) and 300 U RNase T1 (Thermo Fisher, EN0541) for 30 min, RT with gentle rocking. The reaction was stopped by the addition of 100 U SUPERase RNase Inhibitor (Thermo Fisher, AM2694). Ribosomes were enriched by adding 110 μL lysate onto 900 μL sucrose cushion buffer (1 M sucrose in buffer A containing 20 U/mL SUPERase RNase Inhibitor), and centrifuging in a TLA 120.2 rotor (Beckman) 70,000 rpm, 4 h, 4 °C. The ribosome pellet containing the ribosome footprinted Ribo-Seq library was resuspended in 500 μL TRIzol.
Library preparation was adapted from previous protocols(Flynn et al., 2015; Ingolia et al., 2012) and the ARTseq Ribosome Profiling Kit manual (Epicentre, Illumina). In summary, total RNA and ribosome footprints were extracted using the Direct-zol Micro Kit (Zymo, R2060) with in-column DNase I treatment. To have an adequate amount of RNA for subsequent steps, pairs of samples of the same genotype were pooled to generate a single replicate sample, brought up to 90 μL with water, and precipitated with 150 μL isopropanol overnight −80°C after addition of 10 μL 3 M NaOAc pH 5.5 and 1.5 μL 15 mg/mL GlycoBlue Coprecipitant (Thermo Fisher, AM9515). The samples were then centrifuged at 21,000g, 30 min, 4°C, supernatant was removed, and the RNA pellet was washed 500 μL cold 75% ethanol. Pellets were dried for 10 min, RT and resuspended in 15 μL nuclease free water. After extraction and precipitation, both ribosome footprinting and total RNA samples were depleted of rRNA using the Ribo-Zero Gold rRNA Removal Kit (H/M/R) (Illumina, catalog no. MRZG126) with the modification that the 50°C incubation was not performed for the ribosome footprinting samples. The samples were then column purified (RNA Clean & Concentrator 5, Zymo Research, catalog no. R1016) with the modifications that 2 volumes of RNA Binding Buffer and 4.5 volumes of ethanol were added to ribosome footprinting samples to purify small RNAs and 1 volume of RNA Binding Buffer and 1 volume of ethanol was added to total RNA samples to isolate RNA > 200 nt. Total RNA samples were then fragmented by partial alkaline hydrolysis. The samples were diluted to 100 μL with 5 mM Tris-HCl, pH 7.5 and incubated with 100 μL 2x alkaline fragmentation buffer (100 mM Na2CO3 pH 9.2, 2 mM EDTA) for 20 minutes at 95°C. The reaction was neutralized with 440 μL STOP Buffer (70 μL 3 M NaOAc pH 5.5, 2 μL Glycoblue, and 370 μL nuclease free water) and isopropanol precipitated overnight at −80°C.
Ribosome protected fragments and total RNA samples were then size selected by running the samples out on a 15% TBE-Urea polyacrylamide gel. Ribosome protected fragments were size selected between 28-nt and 34-nt as marked by RNA oligonucleotides oNTI199 and oNTI265, respectively (Ingolia et al., 2011). Total RNA samples were size selected between 34–70 nt as marked by a 10 bp DNA ladder (Invitrogen, catalog no. 10821015). Gel slices were crushed and extracted at room temperature overnight in 400 μL RNA extraction buffer (300 mM NaOAc pH 5.5, 1 mM EDTA, 0.25% SDS) and isopropanol precipitated. Samples were then 3’dephosphorylated by denaturing at 65°C for 5 min and incubating with T4 PNK (NEB, catalog no. M0201S) in a 10 μL reaction (7 μL precipitated RNA, 1 μL 10x T4 PNK Buffer, 1 μL SUPERase Inhibitor, 1 μL 10 U/μL T4 PNK) at 37°C for 1 hour. The reaction was stopped through heat inactivation at 65°C for 20 min. To ligate adaptor, samples were then incubated with 0.5 μL of 50 μM Universal miRNA Cloning Linker (NEB, catalog no. S1315S) and denatured at 65°C for 5 min. The denatured sample was then incubated with 1 μL T4 RNA Ligase 2, truncated KQ (NEB, M0373S), 1 μL 10x buffer, 6 μL 50% PEG 8000, and 1.5 μL water for 4.5 h at 25°C. Free adaptor was then removed by addition of 1 μL 10 U/μL 5’-Deadenylase (NEB, M0331S). 1 μL 10 U/μL RecJ Exonuclease (Lucigen/Epicentre, RJ411250). and 1 μL 20 U/μL SUPERase Inhibitor. Samples were then purified using Zymo RNA Clean & Concentrator-5 columns using the protocol above to preserve small RNAs (100 μL sample, 200 μL RNA binding buffer, 450 μL 100% ethanol).
To perform reverse transcription, 10 μL sample was incubated with 2 μL of 1.25 μM RT primer (see Table S2) and denatured at 65°C for 5 min. Reverse transcription (RT) was performed with primers containing sample barcodes and unique molecular identifiers. RT was performed with SuperScript III (Thermo Fisher, 18080–044) in a 20 μL reaction (48°C, 40 min). RNA was then hydrolyzed by adding 2.2 μL of 1N NaOH and incubating for 20 min at 98°C. Samples were then purified using Zymo RNA Clean & Concentrator-5 columns (100 μL sample, 200 μL RNA binding buffer, 450 μL 100% ethanol). RT products were size selected and gel extracted from 10% TBE Urea polyacrylamide gels as described above instead that DNA extraction buffer was used for overnight extraction (300 mM NaCl, 10 mM Tris-HCl pH 8, 1 mM EDTA, 0.1% SDS). Eluate was then isopropanol precipitated overnight at −80°C overnight. Samples were then circularized with CircLigase (Illumina, CL4115K) in a 20 μL reaction (15 μL cDNA, 2 μL 10x CircLigase Buffer, 1 μL 1 mM ATP, 1 μL MnCl2, 1 μL CircLigase) for 12 hours at 60°C and subsequently purified by Zymo RNA Clean & Concentrator 5 columns (100 μL sample, 200 μL RNA binding buffer, 450 μL 100% ethanol) and eluted with 12 μL 10 mM Tris-HCl pH 8. 1 μL of library was used for PCR amplification with Phusion High-Fidelity DNA Polymerase (Thermo Fisher, catalog no. F530S) (98°C 30s, 98°C 10s, 65°C 10s, 72°C 5s) for 9–10 cycles (see Table S2 for primers). PCR product was PAGE purified from native 8% TBE polyacrylamide gels, extracted overnight using DNA extraction buffer, and isopropanol precipitated for > 2 h at −80°C. DNA was measured and quality controlled on the Agilent 2100 Bioanalyzer (High-Sensitivity DNA) by the Stanford Protein and Nucleic Acid (PAN) Facility. Libraries were sequenced by the Stanford Functional Genomics Facility (SFGF) on the Illumina NextSeq 500 (1×75nt).
Ribosome profiling analysis.
For analysis pre-processing, UMI-tools(Smith et al., 2017) was used to extract sample barcodes and UMIs. Reads were demultiplexed, sample barcodes were removed, and reads were quality filtered (fastq_quality_filter -Q33 -q 20 -p 70) using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). The 3’ adapter sequence CTGTAGGCACCATCAAT was removed using cutadapt (Martin, 2011), and the 5’ end of each read was then removed using fastx_trimmer from FASTX-Toolkit. To remove reads that aligned to rRNA, tRNA, and snRNAs, reads were first aligned to these sequences using bowtie2(Langmead and Salzberg, 2012) (bowtie2 parameters: -L 18) and subsequently discarded. Filtered reads that did not align to rRNA/tRNA/snRNAs were then aligned using bowtie2 (bowtie2 parameters: --norc -L 18) to an GRCm38/mm10 transcriptome reference derived from UCSC/GENCODE VM20 knownCanonical annotations filtered for high confidence transcripts that contain at least one of the following: a Uniprot ID, a RefSeq ID, or an Entrez ID. PCR duplicates were then removed using UMI-tools. Ribosome protected fragments (RPFs) were then parsed for uniquely aligned reads, separated into read length groups, and ribosome A site positions were determined by offsetting the distance of the 5’ end of each read to canonical start sites in each length group and adding 4 nucleotides(Ingolia et al., 2011). RNA-Seq reads were also parsed for uniquely aligned reads and were assigned to the center of each read. RPFs and RNA-Seq reads were then counted using the transcriptome annotations detailed above. Reads aligning to the CDS (with the first 15 codons and last 5 codons removed) were used for RPF libraries, and reads aligning to the entire transcript were used for RNA-Seq libraries. Transcripts with counts per million (cpm) > 0.75 for at least 3 ribosome profiling libraries were retained for downstream analysis. RPF and RNA-Seq libraries were then normalized separately by the method of trimmed mean of M-values (TMM) using edgeR(Robinson et al., 2010).
Differential translation analysis was performed using voom(Law et al., 2014) and limma(Ritchie et al., 2015). The following design matrix was constructed (~0 + genotype and type of library (RPF vs. RNA) + replicate batch + sex). The corresponding contrast matrix comparing translational efficiency (RPF - RNA) for each of the pairs of genotypes of interest (e.g., Prx1Cre;Rps6lox/+;Trp53+/+ - Prx1Cre;Rps6lox/+;Trp53+/+, Prx1Cre;Rps6lox/+;Trp53−/− - Prx1Cre;Rps6lox/+;Trp53−/−, etc.) was also constructed. The data was then voom transformed to remove mean-variance count heteroscedasticity. Using the design and contrast matrices mentioned above, linear models were fit to the data using limma with empirical Bayes moderation. Significant differentially translated genes were defined as those with FDR < 0.1 obtained using the Benjamini-Hochberg method. Note that although standard ribosome profiling cannot detect alterations in global protein synthesis, this technique can pinpoint relative changes in the translation efficiency (TE) of specific transcripts (McGlincy and Ingolia, 2017).
Gene set enrichment analysis was performed using Camera (Wu and Smyth, 2012) with mouse GO Biological Processes gene sets obtained from http://download.baderlab.org/EM_Genesets/current_release/Mouse/Entrezgene/. Gene sets were filtered such that all genes in gene sets have expression values in the dataset. Gene sets were also filtered to only include those with > 10 and < 500 genes. Enriched gene sets were visualized using Enrichment Map (Merico et al., 2010) and Cytoscape. For feature analyses, UTR RNA folding energies were obtained from UCSC Genome Browser (foldUtr5 and foldUtr3). Normalized predicted structuredness is reflected by the change in free energy (ΔG) of the minimum free energy (MFE) structure normalized by feature length.
To select for high-confidence translationally repressed transcripts involved in limb development, transcripts were selected such that 1) the TE was significantly decreased in Prx1Cre;Rps6lox/+ vs. Rps6lox/+ embryos, and 2) the transcripts also had a significant decrease in RPF, which filters for transcripts whose changes in TE are driven primarily by ribosome footprints and not by changes in RNA transcript levels. The transcripts that then intersected with the significantly enriched limb-related GO categories (appendage development, appendage morphogenesis, limb development, and limb morphogenesis) were selected. This set of criteria would select for high confidence transcripts in these gene sets whose decrease in TE is primarily due to reduction in ribosome footprints and not to changes in transcript levels.
QUANTIFICATION AND STATISTICAL ANALYSIS
All measurements were sampled from individual biological replicates. Replicates for mouse embryo experiments consisted of individual limbs, individual embryos, or pools of embryos if material was limiting. Experiments consisted of multiple embryos from multiple litters. Unless otherwise stated, when groups of continuous data are compared, two-tailed t-tests with unequal variance were performed; * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001; for bar plots, error bars = SEM. For box-plots, center line, median; box limits, first and third quartiles; whiskers, max or min value no further than 1.5x interquartile range. Otherwise, statistical tests and definitions of significance are described in respective figure legends and methods for each experiment. No statistical methods were used to predetermine sample size.
Supplementary Material
Table S2. Primers and oligos. Related to STAR Methods.
Table S3. Ribosome profiling results from Prx1Cre;Rps6lox/+ vs. Rps6lox/+ E10.5 limbs. Related to Figures 4 and S5.
Table S4. Ribosome profiling results from Prx1Cre;Rps6lox/+;Trp53−/− vs. Rps6lox/+;Trp53−/− E10.5 limbs. Related to Figures 4 and S5.
Table S5. Gene set enrichment analysis of translational efficiency changes in Prx1Cre;Rps6lox/+ vs. Rps6lox/+ E10.5 limbs. Related to Figure 4.
Table S6. RNA folding energies and lengths of transcripts. Related to Figure 4.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-GAPDH | Cell signaling | Cat# 97166 |
| mouse anti-actin | Cell signaling | Cat# 3700S |
| rabbit anti-Rps6 | Cell signaling | Cat# 2217L |
| rabbit anti-4E-BP1 | Cell signaling | Cat# 9644S |
| rabbit anti-4E-BP2 | Cell signaling | Cat# 2845S |
| rabbit anti-phospho-p70 S6 Kinase(T389) | Cell signaling | Cat# 9205S |
| rabbit anti-p70 S6 Kinase | Cell signaling | Cat# 9202S |
| rabbit anti-phopsho-ULK1(Ser757) | Cell signaling | Cat# 6888T |
| rabbit anti-ULK1 | Cell signaling | Cat# 8054T |
| rabbit anti-TSC2 | Cell signaling | Cat# 4308T |
| rabbit anti-eIF4G | Cell signaling | Cat# 2498S |
| rabbit anti-p53 | Leica Biosystems | Cat# CM5P-L |
| mouse anti-eIF4E | BD Biotransduction | Cat# 610269 |
| anti-phospho-Histone H3 | Sigma | Cat# 06-570 |
| Cleaved Caspase-3 | Cell signaling | Cat# 9661S |
| Goat anti-rabbit AF568 | Life Technologies | Cat# A11036 |
| donkey anti-mouse HRP | GE Healthcare | Cat# NA931-1ML |
| donkey anti-rabbit HRP | GE Healthcare | Cat# NA934-1ML |
| alkaline phosphatase-conjugated anti-DIG | Sigma | Cat# 11093274910 |
| Bacterial and virus strains | ||
| Ad5 CMV-Cre | University of Iowa Viral Vector Core Facility | Cat# Ad5CMVCre |
| Ad5 CMV-empty | University of Iowa Viral Vector Core Facility | Cat# Ad5CMVempty |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| O-propargyl-puromycin | Medchem Source LLP | Cat# JA-1024 |
| L-homopropargylglycine | Thermo Fisher | Cat# C10186 |
| 4EGI-1 (eIF4E/eIF4G interaction inhibitor) | Millipore Sigma | Cat# 324517 |
| Dimethyl sulfoxide (DMSO, cell culture grade) | Sigma | Cat# D2650 |
| Nutlin-3a | Cayman Chemicals | Cat#18585 |
| Doxorubicin hydrochloride | Sigma | Cat# D1515 |
| TURBO DNase | Ambion | Cat# AM2238 |
| SUPERase In RNase Inhibitor | Ambion | Cat# AM2696 |
| TRIzol | Invitrogen | Cat# 15596 |
| iScript Supermix | Bio-Rad | Cat# 1708840 |
| SsoAdvanced SYBR Green supermix | Bio-Rad | Cat# 1725270 |
| SDS-PAGE gels | Bio-Rad | Cat# 567-1095, 456-1096 |
| Semi-dry Trans-Blot Turbo system | Bio-Rad | Cat# 170-4273 |
| Clarity Western ECL Substrate | Bio-Rad | Cat# 170-5061 |
| Dulbecco’s Modified Eagle’s Medium | GIBCO | Cat# 11965–118 |
| Fetal calf serum | EMD Millipore | Cat# TMS-013-B |
| DMEM/F-12, HEPES, with phenol red | Thermo Fisher | Cat# 11330032 |
| Penicillin-Streptomycin Solution 100X | Thermo Fisher | Cat# 15140163 |
| Opti-MEM | GIBCO | Cat# 11058-021 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668-019 |
| Dharmafect I | Dharmacon | Cat# T-2001-02 |
| siGENOME smart pool siRNA 4EBP1 | Dharmacon | Cat# M-058681-01-0005 |
| siGENOME smart pool siRNA 4EBP2 | Dharmacon | Cat# M-044972-01-0005 |
| siGENOME control siRNA #3 | Dharmacon | Cat# D-001210-03-05 |
| siGENOME control siRNA #2 (siFluc) | Dharmacon | Cat# D-001210-02-05 |
| Zombie Violet Live-Dead Stain | BioLegend | Cat# 423113 |
| BM Purple AP substrate | Sigma | Cat# 11442074001 |
| LysoTracker Red DND-99 | Invitrogen | Cat# L7528 |
| Alizarin Red S | Sigma | A5533 |
| Alcian Blue 8GX | Sigma | A5268 |
| HBSS | Thermo Fisher | 14025-076 |
| Cycloheximide | Sigma-Aldrich | C7698 |
| Halt Protease and Phosphatase Inhibitor Cocktail | Thermo Fisher | 78442 |
| Acid-Phenol: Chloroform, pH 4.5 (with IAA, 125:24:1) | Thermo Fisher | AM9722 |
| Complete Protease Inhibitor EDTA-free | Sigma-Aldrich | 11836170001 |
| TRIzol LS | Thermo Fisher | 10296010 |
| RNase A | Thermo Fisher | AM2271 |
| RNase T1 | Thermo Fisher | EN0541 |
| GlycoBlue Coprecipitant | Thermo Fisher | AM9515 |
| 10 bp DNA ladder | Invitrogen | 10821015 |
| T4 PNK | NEB | M0201S |
| Universal miRNA Cloning Linker | NEB | S1315S |
| T4 RNA Ligase 2, truncated KQ | NEB | M0373S |
| 5’-Deadenylase | NEB | M0331S |
| RecJ Exonuclease | Lucigen/Epicentre | RJ411250 |
| Critical commercial assays | ||
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1980 |
| Monarch Gel Extraction Kit | NEB | Cat# T1020S |
| NEBuilder HiFi DNA Assembly Master Mix | NEB | Cat# E2621S |
| RNA Clean and Concentrator-5 columns | Zymo Research | Cat# R1016 |
| RNA PureLink columns | Ambion | Cat# 12183018 |
| mMESSAGE mMACHINE® T7 Transcription Kit | Ambion | Cat# AM 1344 |
| Poly(A) Polymerase Tailing Kit | Lucigen | Cat# PAP5104H |
| Agarose beads | Jena Biosciences | Cat# AC-100S |
| γ-aminophenyl-m7GTP (C10-spacer)-agarose beads | Jena Biosciences | Cat# AC-155S |
| Click-iT EdU Alexa Fluor 488 flow Cytometry Assay Kit | Thermo Fisher | Cat# C10425 |
| FITC Annexin V and propidium iodide staining kit | BioLegend | Cat# 640914 |
| Click-iT™ Plus Alexa Fluor™ 555 Picolyl Azide Toolkit | Thermo Fisher | Cat# C10642 |
| Direct-zol Micro Kit | Zymo | R2060 |
| Ribo-Zero Gold rRNA Removal Kit (H/M/R) | Illumina | MRZG126 |
| SuperScript III | Thermo Fisher | 18080-044 |
| CircLigase | Illumina | CL4115K |
| Phusion High-Fidelity DNA Polymerase | Thermo Fisher | F530S |
| Deposited data | ||
| Ribosome profiling (ribosome footprints and RNA-Seq) data from E10.5 Rps6lox/+;Trp53+/+; Prx1Cre;Rps6lox/+;Trp53+/+; Rps6lox/+;Trp53−/−; Prx1Cre;Rps6lox/+;Trp53−/− embryonic limb buds | This paper | GEO: GSE135722 |
| p53 ChIP-seq in primary MEFs | (Kenzelmann Broz et al., 2013) | GEO: GSE46240 |
| Experimental models: cell lines | ||
| NIH3T3 | ATCC | CAT#CRL-1658 |
| C3H10T1/2 | ATCC | CAT#CCL-226 |
| Mouse embryonic fibroblasts (Trp53LSL-WT/LSL-WT) | (Brady et al., 2011) | N/A |
| Mouse embryonic fibroblasts (Trp53LSL-25,26,53,54/LSL-25,26,53,54) | (Brady et al., 2011) | N/A |
| Experimental Models: organisms/strains | ||
| Mouse: Rps6lox | (Volarevic et al., 2000) | N/A |
| Mouse: Prx1Cre | (Logan et al., 2002); Jackson Laboratories | JAX: 005584 |
| Mouse: Msx2Cre | (Sun et al., 2000); Jackson Laboratories | JAX: 027892 |
| Mouse: Tsc2lox | (Hernandez et al., 2007); Jackson laboratories | JAX: 027458 |
| Mouse: Trp53null | (Jacks et al., 1994) | N/A |
| Mouse: RosamTmG | (Muzumdar et al., 2007) | N/A |
| Oligonucleotides | ||
| Oligonucleotides for ribosome profiling, RT-qPCR, ChIP-PCR, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| pGL3-Pkm-FLB-stop | This Paper | N/A |
| pGL3-Cnot10-FLB-stop | This Paper | N/A |
| pGL3-Mdm4-FLB-stop | This Paper | N/A |
| pGL3-Rnf10-FLB-stop | This Paper | N/A |
| pGL3-Megf8-FLB-stop | This Paper | N/A |
| pGL3-Col2a1-FLB-stop | This Paper | N/A |
| pGL3-Gli2-FLB-stop | This Paper | N/A |
| pGL4.23 | Promega | CAT#E8411 |
| pGL4.23-p53-4EBP1_delMiniP | This Paper | N/A |
| pGL4.23-Delp53-4EBP1_delMiniP | This Paper | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism | Graphpad Software Inc. | Version 8.0 |
| FlowJo | BD Biosciences | Version 10.7.1 |
| Volocity | Quroum Technologies | Version 6.3.1 |
| UMI-tools | (Smith et al., 2017) | https://github.com/CGATOxford/UMI-tools |
| FASTX-Toolkit | http://hannonlab.cshl.edu/fastx_toolkit/ | http://hannonlab.cshl.edu/fastx_toolkit/ |
| cutadapt | (Martin, 2011) | https://cutadapt.readthedocs.io/en/stable/ |
| bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/ |
| voom | (Law et al., 2014) | https://www.rdocumentation.org/packages/limma/versions/3.28.14/topics/voom |
| limma | (Ritchie et al., 2015) | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Camera | (Wu and Smyth, 2012) | https://www.rdocumentation.org/packages/limma/versions/3.28.14/topics/camera |
| Enrichment Map | (Merico et al., 2010) | http://apps.cytoscape.org/apps/enrichmentmap |
| Cytoscape | https://cytoscape.org/ | https://cytoscape.org/ |
| RStudio | https://rstudio.com/ | https://www.rstudio.com/ |
| samtools | (Li et al., 2009) | http://www.htslib.org/ |
| Mouse GRCm38/mm10 UCSC/GENCODE VM20 knownCanonical annotation | https://genome.ucsc.edu/ | https://genome.ucsc.edu/ |
| Enrichment Map Mouse Gene Sets | http://download.baderlab.org/EM_Genesets/current_release/Mouse/entrezgene/ | http://download.baderlab.org/EM_Genesets/current_release/Mouse/entrezgene/ |
| ggplot2 | (Wickham, 2016) | https://ggplot2.tidyverse.org/ |
| Other | ||
| Density Gradient Fraction System | Brandel | Cat# BR-188 |
| CFX384 Touch qPCR machine | Bio-Rad | Cat# 1855485 |
| ChemiDoc MP | Bio-Rad | Cat# 17001402 |
| Biocomp Model 108 Gradient Master | BioComp | N/A |
| LSRII Flow Cytometer | BD Biosciences | N/A |
| NovoCyte Quanteon Flow Cytometer | Agilent Technologies | N/A |
| CFX384 Touch Real-Time PCR Detection System | Bio-Rad | Cat# 1855485 |
| GloMax-Multi Plate Reader | Promega | Cat# E7081 |
| SW-60 rotor | Beckman | Cat# 335649 |
| TLA 120.2 rotor | Beckman | Cat# 357656 |
Highlights.
Ribosomal protein (RP) haploinsufficiency in mouse limb causes patterning defects
A p53-dependent translational regulatory program contributes to this phenotype
p53 regulates translation through induction of 4E-BP1, a translation repressor
Ribosome profiling reveals p53-dependent and -independent translational changes
ACKNOWLEDGMENTS
The authors thank Kotaro Fujii, Katrina Cabaltera, Jing Huang, Naomi Genuth, and David Mahoney for their experimental contributions as well as the Stanford FACS facility and SFGF; George Thomas (IDIBELL) for generously providing the Rps6lox mice; and Barna lab members for critical feedback and discussion.
Funding:
Supported by New York Stem Cell Foundation grant NYSCF-R-I36 (M.B.), New York Stem Cell Robertson Investigator (M.B.), NIH grant 1R01HD086634 (M.B.), Alfred P. Sloan Research Fellowship (M.B.), Pew Scholars Award (M.B.), NIH grant R35CA242986 (D.R.), American Cancer Society RP-19-181-01-RMC (D.R.), March of Dimes Foundation grant 6-FY15-189 (L.D.A.), NIH R35 grant CA197591 (L.D.A.), Paul and Daisy Soros Fellowships for New Americans (G.C.T), Stanford Medical Scientist Training Program (G.C.T.), Canadian Institutes for Health Research Fellowship (C.H.K. and O.Z.), Stanford Dean’s Fellowship (C.H.K.), Helen Hay Whitney Foundation Fellowship (O.Z.), NIH/NIDDK grant K08DK119561 (C.M.F.), National Institutes of Health Developmental Biology Training Grant 5T32GM007790-41 (H.D.R.), Campini Foundation and the University of California, San Francisco, Department of Pediatrics K12 (Award 5K12HDO72222-05) (C.M.F.), and the Jane Coffin Childs Fund Postdoctoral Fellowship (M.E.B.). D.R is an American Cancer Society Research Professor.
Footnotes
DECLARATION OF INTERESTS
D.R. is a shareholder of eFFECTOR Therapeutics, Inc. and a member of its scientific advisory board.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Barlow JL, Drynan LF, Hewett DR, Holmes LR, Lorenzo-Abalde S, Lane AL, Jolin HE, Pannell R, Middleton AJ, Wong SH, et al. (2010). A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat. Med 16, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna M, and Niswander L (2007). Visualization of cartilage formation: insight into cellular properties of skeletal progenitors and chondrodysplasia syndromes. Dev. Cell 12, 931–941. [DOI] [PubMed] [Google Scholar]
- Bi C, Zhang X, Lu T, Zhang X, Wang X, Meng B, Zhang H, Wang P, Vose JM, Chan WC, et al. (2017). Inhibition of 4EBP phosphorylation mediates the cytotoxic effect of mechanistic target of rapamycin kinase inhibitors in aggressive B-cell lymphomas. Haematologica 102, 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen ME, McClendon J, Long HK, Sorayya A, Van Nostrand JL, Wysocka J, and Attardi LD (2019). The Spatiotemporal Pattern and Intensity of p53 Activation Dictates Phenotypic Diversity in p53-Driven Developmental Syndromes. Dev. Cell 50, 212–228.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. (2011). Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, and Karin M (2008). p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik C, Cenik ES, Byeon GW, Grubert F, Candille SI, Spacek D, Alsallakh B, Tilgner H, Araya CL, Tang H, et al. (2015). Integrative analysis of RNA, translation, and protein levels reveals distinct regulatory variation across humans. Genome Res. 25, 1610–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton C, and Gazda HT (1993). Diamond-Blackfan Anemia. In GeneReviews(®), Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, Bird TD, Fong C-T, Mefford HC, Smith RJ, et al. , eds. (Seattle (WA: ): University of Washington, Seattle; ), p. [Google Scholar]
- Dalla Venezia N, Vincent A, Marcel V, Catez F, and Diaz J-J (2019). Emerging role of eukaryote ribosomes in translational control. Int. J. Mol. Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilova N, Sakamoto KM, and Lin S (2008). Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood 112, 5228–5237. [DOI] [PubMed] [Google Scholar]
- Deisenroth C, and Zhang Y (2010). Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene 29, 4253–4260. [DOI] [PubMed] [Google Scholar]
- Ding M, Van der Kwast TH, Vellanki RN, Foltz WD, McKee TD, Sonenberg N, Pandolfi PP, Koritzinsky M, and Wouters BG (2018). The mTOR Targets 4E-BP1/2 Restrain Tumor Growth and Promote Hypoxia Tolerance in PTEN-driven Prostate Cancer. Mol. Cancer Res 16, 682–695. [DOI] [PubMed] [Google Scholar]
- Duncan R, Milburn SC, and Hershey JW (1987). Regulated phosphorylation and low abundance of HeLa cell initiation factor eIF-4F suggest a role in translational control. Heat shock effects on eIF-4F. J. Biol. Chem 262, 380–388. [PubMed] [Google Scholar]
- Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, Wilson FH, Currie T, Khanna-Gupta A, Berliner N, et al. (2011). Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 117, 2567–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RA, Martin L, Spitale RC, Do BT, Sagan SM, Zarnegar B, Qu K, Khavari PA, Quake SR, Sarnow P, et al. (2015). Dissecting noncoding and pathogen RNA-protein interactomes. RNA 21, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garami A, Zwartkruis FJT, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, and Thomas G (2003). Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466. [DOI] [PubMed] [Google Scholar]
- Gazda HT, Sheen MR, Vlachos A, Choesmel V, O’Donohue M-F, Schneider H, Darras N, Hasman C, Sieff CA, Newburger PE, et al. (2008). Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am. J. Hum. Genet 83, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, McManus MT, Mansfield JH, Hornstein E, and Tabin CJ (2005). The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci USA 102, 10898–10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez O, Way S, McKenna J, and Gambello MJ (2007). Generation of a conditional disruption of the Tsc2 gene. Genesis 45, 101–106. [DOI] [PubMed] [Google Scholar]
- Horos R, Ijspeert H, Pospisilova D, Sendtner R, Andrieu-Soler C, Taskesen E, Nieradka A, Cmejla R, Sendtner M, Touw IP, et al. (2012). Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood 119, 262–272. [DOI] [PubMed] [Google Scholar]
- Hurst JA, Baraitser M, and Wonke B (1991). Autosomal dominant transmission of congenital erythroid hypoplastic anemia with radial abnormalities. Am. J. Med. Genet 40, 482–484. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, and Weissman JS (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, and Weissman JS (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc 7, 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaako P, Debnath S, Olsson K, Bryder D, Flygare J, and Karlsson S (2012). Dietary L-leucine improves the anemia in a mouse model for Diamond-Blackfan anemia. Blood 120, 2225–2228. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, and Weinberg RA (1994). Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4, 1–7. [DOI] [PubMed] [Google Scholar]
- Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey J-P, Glynn EF, Ellington L, Du C, Dixon J, et al. (2008). Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 14, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasteri J, Das D, Zhong X, Persaud L, Francis A, Muharam H, and Sauane M (2018). Translation Control by p53. Cancers (Basel) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, and Attardi LD (2013). Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 27, 1016–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajuria RK, Munschauer M, Ulirsch JC, Fiorini C, Ludwig LS, McFarland SK, Abdulhay NJ, Specht H, Keshishian H, Mani DR, et al. (2018). Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell 173, 90–103.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongsuwan K, Yu Q, Vincent A, Frisardi MC, Rosbash M, Lengyel JA, and Merriam J (1985). A Drosophila Minute gene encodes a ribosomal protein. Nature 317, 555–558. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Vissing H, Ramirez F, Rogers D, and Rimoin D (1989). Identification of the molecular defect in a family with spondyloepiphyseal dysplasia. Science 244, 978–980. [DOI] [PubMed] [Google Scholar]
- Lee C-H, Kiparaki M, Blanco J, Folgado V, Ji Z, Kumar A, Rimesso G, and Baker NE (2018). A regulatory response to ribosomal protein mutations controls translation, growth, and cell competition. Dev. Cell 46, 456–469.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppek K, Fujii K, Quade N, Susanto TT, Boehringer D, Lenarčič T, Xue S, Genuth NR, Ban N, and Barna M (2020). Gene- and Species-Specific Hox mRNA Translation by Ribosome Expansion Segments. Mol. Cell 80, 980–995.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TA, Kong X, Haystead TA, Pause A, Belsham G, Sonenberg N, and Lawrence JC (1994). PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science 266, 653–656. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza-Puch F, Drost J, Rooijers K, Lopes R, Elkon R, and Agami R (2013). p53 induces transcriptional and translational programs to suppress cell proliferation and growth. Genome Biol. 14, R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan M, Martin JF, Nagy A, Lobe C, Olson EN, and Tabin CJ (2002). Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80. [DOI] [PubMed] [Google Scholar]
- Ludwig LS, Gazda HT, Eng JC, Eichhorn SW, Thiru P, Ghazvinian R, George TI, Gotlib JR, Beggs AH, Sieff CA, et al. (2014). Altered translation of GATA1 in Diamond-Blackfan anemia. Nat. Med. 20, 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lufkin T (2007). In situ hybridization of whole-mount mouse embryos with RNA probes: hybridization, washes, and histochemistry. CSH Protoc. 2007, pdb.prot4823. [DOI] [PubMed] [Google Scholar]
- Marine J-CW, Dyer MA, and Jochemsen AG (2007). MDMX: from bench to bedside. J. Cell Sci 120, 371–378. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j. 17, 10. [Google Scholar]
- McGlincy NJ, and Ingolia NT (2017). Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan KA, and Mason PJ (2011). Animal models of Diamond Blackfan anemia. Semin. Hematol 48, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan KA, Li JZ, Park CY, Beaudry V, Tabor HK, Sabnis AJ, Zhang W, Fuchs H, de Angelis MH, Myers RM, et al. (2008). Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat. Genet 40, 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan KA, Pang WW, Bhardwaj R, Perez MG, Pluvinage JV, Glader BE, Malek R, Mendrysa SM, Weissman IL, Park CY, et al. (2011). Reduced ribosomal protein gene dosage and p53 activation in low-risk myelodysplastic syndrome. Blood 118, 3622–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, and Bader GD (2010). Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgado-Palacin L, Varetti G, Llanos S, Gómez-López G, Martinez D, and Serrano M (2015). Partial Loss of Rpl11 in Adult Mice Recapitulates Diamond-Blackfan Anemia and Promotes Lymphomagenesis. Cell Rep. 13, 712–722. [DOI] [PubMed] [Google Scholar]
- Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, Chik KW, Shi XM, Tsui LC, Cheng SH, et al. (1997). Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 124, 113–123. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, and Luo L (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. [DOI] [PubMed] [Google Scholar]
- Narla A, and Ebert BL (2010). Ribosomopathies: human disorders of ribosome dysfunction. Blood 115, 3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panić L, Tamarut S, Sticker-Jantscheff M, Barkić M, Solter D, Uzelac M, Grabusić K, and Volarević S (2006). Ribosomal protein S6 gene haploinsufficiency is associated with activation of a p53-dependent checkpoint during gastrulation. Mol. Cell. Biol 26, 8880–8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne EM, Virgilio M, Narla A, Sun H, Levine M, Paw BH, Berliner N, Look AT, Ebert BL, and Khanna-Gupta A (2012). L-Leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood 120, 2214–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, and Sonenberg N (1985). Insertion mutagenesis to increase secondary structure within the 5’ noncoding region of a eukaryotic mRNA reduces translational efficiency. Cell 40, 515–526. [DOI] [PubMed] [Google Scholar]
- Pelletier J, Graff J, Ruggero D, and Sonenberg N (2015). Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res. 75, 250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiyama N, Arthanari H, Papadopoulos E, Rodriguez-Mias RA, Wagner G, and Léger-Abraham M (2015). Molecular mechanism of the dual activity of 4EGI-1: Dissociating eIF4G from eIF4E but stabilizing the binding of unphosphorylated 4E-BP1. Proc Natl Acad Sci USA 112, E4036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signer RAJ, Magee JA, Salic A, and Morrison SJ (2014). Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, and Hinnebusch AG (2009). Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulic S, Panic L, Barkic M, Mercep M, Uzelac M, and Volarevic S (2005). Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53-dependent checkpoint response. Genes Dev. 19, 3070–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Lewandoski M, Meyers EN, Liu YH, Maxson RE, and Martin GR (2000). Conditional inactivation of Fgf4 reveals complexity of signalling during limb bud development. Nat. Genet. 25, 83–86. [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, and Sabatini DM (2012). A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, Seo Y, Barna M, and Ruggero D (2015). Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell 162, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SRF, Lloyd D, Jenkins D, Elçioglu NE, Cooper CDO, Al-Sannaa N, Annagür A, Gillessen-Kaesbach G, Hüning I, Knight SJL, et al. (2012). Mutations in multidomain protein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization. Am. J. Hum. Genet 91, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. [DOI] [PubMed] [Google Scholar]
- Volarevic S, Stewart MJ, Ledermann B, Zilberman F, Terracciano L, Montini E, Grompe M, Kozma SC, and Thomas G (2000). Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 288, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Wang S, Konorev EA, Kotamraju S, Joseph J, Kalivendi S, and Kalyanaraman B (2004). Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. intermediacy of H(2)O(2)- and p53-dependent pathways. J. Biol. Chem 279, 25535–25543. [DOI] [PubMed] [Google Scholar]
- Weinberg DE, Shah P, Eichhorn SW, Hussmann JA, Plotkin JB, and Bartel DP (2016). Improved Ribosome-Footprint and mRNA Measurements Provide Insights into Dynamics and Regulation of Yeast Translation. Cell Rep. 14, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2 - Elegant Graphics for Data Analysis (New York, NY: Springer-Verlag New York; ). [Google Scholar]
- Wu D, and Smyth GK (2012). Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 40, e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelick PC, and Trainor PA (2015). Ribosomopathies: Global process, tissue specific defects. Rare Dis. 3, e1025185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller R, Jackson-Grusby L, and Leder P (1989). The limb deformity gene is required for apical ectodermal ridge differentiation and anteroposterior limb pattern formation. Genes Dev. 3, 1481–1492. [DOI] [PubMed] [Google Scholar]
- Zeller R, López-Ríos J, and Zuniga A (2009). Vertebrate limb bud development: moving towards integrative analysis of organogenesis. Nat. Rev. Genet 10, 845–858. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Nicholatos J, Dreier JR, Ricoult SJH, Widenmaier SB, Hotamisligil GS, Kwiatkowski DJ, and Manning BD (2014). Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 513, 440–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Primers and oligos. Related to STAR Methods.
Table S3. Ribosome profiling results from Prx1Cre;Rps6lox/+ vs. Rps6lox/+ E10.5 limbs. Related to Figures 4 and S5.
Table S4. Ribosome profiling results from Prx1Cre;Rps6lox/+;Trp53−/− vs. Rps6lox/+;Trp53−/− E10.5 limbs. Related to Figures 4 and S5.
Table S5. Gene set enrichment analysis of translational efficiency changes in Prx1Cre;Rps6lox/+ vs. Rps6lox/+ E10.5 limbs. Related to Figure 4.
Table S6. RNA folding energies and lengths of transcripts. Related to Figure 4.
Data Availability Statement
All custom codes are available upon request from the corresponding author. Data that support the findings of this study have been deposited in the Gene Expression Omnibus under accession number GSE135722.