

ABSTRACT
Microglia are highly dynamic cells crucial for developing and maintaining lifelong brain function and health through their many interactions with essentially all cellular components of the central nervous system. The frequent connection of microglia to leukodystrophies, genetic disorders of the white matter, has highlighted their involvement in the maintenance of white matter integrity. However, the mechanisms that underlie their putative roles in these processes remain largely uncharacterized. Microglia have also been gaining attention as possible therapeutic targets for many neurological conditions, increasing the demand to understand their broad spectrum of functions and the impact of their dysregulation. In this Review, we compare the pathological features of two groups of genetic leukodystrophies: those in which microglial dysfunction holds a central role, termed ‘microgliopathies’, and those in which lysosomal or peroxisomal defects are considered to be the primary driver. The latter are suspected to have notable microglia involvement, as some affected individuals benefit from microglia-replenishing therapy. Based on overlapping pathology, we discuss multiple ways through which aberrant microglia could lead to white matter defects and brain dysfunction. We propose that the study of leukodystrophies, and their extensively multicellular pathology, will benefit from complementing analyses of human patient material with the examination of cellular dynamics in vivo using animal models, such as zebrafish. Together, this will yield important insight into the cell biological mechanisms of microglial impact in the central nervous system, particularly in the development and maintenance of myelin, that will facilitate the development of new, and refinement of existing, therapeutic options for a range of brain diseases.
KEY WORDS: Microglia, Myelination, Leukodystrophy, Oligodendrocytes, Astrocytes, Genetic disease
Summary: Microglia abnormalities are increasingly linked to white matter diseases. Complementing analyses of leukodystrophy patient material with animal models yields insight into the impact and therapeutic potential of microglia across diverse brain disorders.
Introduction
Macrophages are myeloid-derived cells distributed throughout the vertebrate body that have tissue-specific subsets and functions based on their origin and tissue of residence (Varol et al., 2015; Wynn et al., 2013; Guilliams et al., 2020). Their functions extend far beyond immunological roles, contributing to processes ranging from organogenesis, tissue repair and lipid metabolism to electrical conductivity in the heart, and their organ-specific regulatory functions are thought to affect virtually every vertebrate organ (Gordon and Martinez-Pomares, 2017; Pollard, 2009). Multiple macrophage subtypes exist within the central nervous system (CNS), including microglia, as well as perivascular, meningeal and choroid plexus macrophages (Li and Barres, 2018), each possibly varying in function in accordance with its respective tissue niche (Guilliams et al., 2020). CNS macrophages exist within a complex microenvironment of neuronal and non-neuronal cells, including astrocytes and oligodendrocyte lineage cells, with which they interact extensively through a variety of direct and indirect means (Box 1, Glial cells: The Pillars of White Matter Integrity). Owing to their more clearly recognized relevance for brain function and more extensive characterization, this Review will primarily focus on microglia. Microglia originate from myeloid progenitor cells in the yolk sac that colonize the brain during early embryonic development (Herbomel et al., 1999; Ginhoux et al., 2010). Whether these yolk sac macrophages are the only source of microglia in the adult brain is still debated (Xu et al., 2015; Ferrero et al., 2018, 2020; Sheng et al., 2015; De et al., 2018). Under homeostatic conditions, microglia are long-lived and can proliferate locally, with limited peripheral contribution (Mildner et al., 2007; Sheng et al., 2015; Ajami et al., 2018). Therefore, owing to the relative isolation and enduring nature of microglia compared to many other macrophages, the effects of microglial aberrations are arguably more penetrant, or may accumulate more significantly, than might be expected in other tissues.
Box 1. Glial cells: The Pillars of White Matter Integrity.
The CNS grey matter contains mostly neuronal cell bodies that process chemo-electrical signals via axonal signaling through the white matter, enabling rapid connectivity between neurons and between brain regions. The white matter, which comprises as much as ∼60% of the brain volume in humans, is mainly composed of glial cells, capillaries and myelinated axons. Myelination is more plastic and dynamic throughout life than previously thought, and neuronal activity-dependent adaptive myelination plays a role in learning and memory (Williamson and Lyons, 2018, Mount and Monje, 2017). Neuronal activity, even that caused by epileptic seizures, can in fact induce myelination (Gibson et al., 2014, Mensch et al., 2015, Marisca et al., 2020, Hughes and Appel, 2020). Microglia and astrocytes interact with and regulate the membrane processes of oligodendrocytes, thereby shaping, maintaining and repairing myelin (reviewed by Baydyuk et al., 2020, Traiffort et al., 2020, de Waard and Bugiani, 2020). Therefore, to thoroughly understand the functions of microglia in myelination and myelin health, one should examine their dynamic interactions with oligodendrocytes and astrocytes.
Oligodendrocytes and myelin: oligodendrocytes are the myelin-producing cells of the brain, synthesizing large amounts of membrane and wrapping it around the axons in multiple compacted layers in an incredibly complex origami-like manner. Mature myelinating oligodendrocytes arise from OPCs (Bergles and Richardson, 2015). As there are many more OPCs present in the CNS throughout development and adult life than one might expect based on the number of myelinating oligodendrocytes, it has been suggested that OPCs may serve additional undiscovered purposes in the brain (Marisca et al., 2020). Myelin contains ∼40% water, whereas the remaining dry mass consists of a high proportion of lipids (∼70%) and a lower proportion of proteins (∼30%), among which myelin basic protein (MBP) accounts for 22-35% (Jahn et al., 2009). A main property of myelin is that it facilitates conduction by insulating the axon and enabling the action potential to jump from one node of Ranvier to the next, thereby greatly speeding up signal propagation. Additionally, myelin protects axons and can metabolically support them by providing carbon-based metabolites, such as lactate and pyruvate (Aggarwal et al., 2011, Philips and Rothstein, 2017).
Under homeostatic conditions, microglia modulate OPC survival and differentiation (Hagemeyer et al., 2017, Wlodarczyk et al., 2017); for example, via insulin-like growth factor (IGF1) (Ye et al., 2002, Wlodarczyk et al., 2017). Upon myelin injury, microglia can promote OPC differentiation and, consequently, remyelination (Miron et al., 2013). Additionally, microglia clear myelin debris after white matter damage, which is crucial for the remyelination process (Lampron et al., 2015, Cantuti-Castelvetri et al., 2018, Miron, 2017).
Astrocytes: Astrocytes are the most abundant cell lineage in the adult vertebrate brain, outnumbering neurons by a factor of nine in humans (Kimelberg and Nedergaard, 2010). Astrocytes are a heterogeneous cell population characterized by extended fiber-like protrusions forming a dense network. Processes surrounding the synaptic cleft are necessary for the uptake and release of neurotransmitters, including glutamate, and can regulate synapse formation and refinement (Haydon and Carmignoto, 2006, Allen and Eroglu, 2017). A subset of astrocytes forms perivascular end-feet that enwrap the capillaries and contribute to the BBB, where they can regulate blood flow and BBB integrity, and shuttle nutrients, such as glucose from the blood to neurons and other brain cells, including oligodendrocytes (Kimelberg and Nedergaard, 2010, Haydon and Carmignoto, 2006). Astrocytes in a ‘reactive’ state are associated with a broad spectrum of neurological diseases and can be either beneficial or detrimental to disease progression. This state and associated nomenclature has been recently defined by Escartin et al. (2021).
Astrocytes can secrete cytokines and growth factors that influence myelination by regulating OPC survival, oligodendrocyte differentiation and maturation (Lanciotti et al., 2013, Lundgaard et al., 2014, Baydyuk et al., 2020, Traiffort et al., 2020). Besides communication via soluble factors, direct cell-cell contacts in the form of gap junctions, formed by connexins, provide oligodendrocytes, but also neurons and axons, with lipids and glucose as both an energy supply and as substrates for myelination (Orthmann-Murphy et al., 2008, Camargo et al., 2017). As all glial cells interact, modulate and regulate one another on many levels, including signaling, metabolism and cell development, a holistic cellular view is needed to comprehend the diseased and healthy brain, especially regarding myelination and white matter integrity.
Until the early 2000s, the roles of microglia in the healthy brain were perceived almost exclusively as immune regulation and phagocytic scavenging. Mainly owing to improved real-time in vivo imaging in zebrafish and mouse models, microglia were subsequently found to be highly dynamic in ways previously not imagined. By extending and retracting their ramified processes, microglia constantly scavenge, monitor and modulate the brain environment (Nimmerjahn et al., 2005; Davalos et al., 2005; Peri and Nüsslein-Volhard, 2008; Wake et al., 2009; Tremblay et al., 2010; Hughes and Appel, 2020). For example, microglia directly shape brain development by pruning synapses (Stevens et al., 2007; Paolicelli et al., 2011; Schafer et al., 2012) and myelin sheaths (Hughes and Appel, 2020), and by clearing apoptotic neurons and myelin debris after damage, which is crucial for remyelination after injury (Neumann et al., 2009; Lampron et al., 2015; Cignarella et al., 2020). They also regulate neuronal activity by direct interactions with neuronal somata (Tremblay et al., 2010; Cserép et al., 2020; Badimon et al., 2020; Li et al., 2012), and sculpt the extracellular matrix to promote synapse formation (Nguyen et al., 2020). In fact, it is now widely agreed that microglia interact with essentially all cellular components of the CNS and have vital, yet insufficiently understood, roles in shaping brain development and maintaining normal brain function and integrity (Li and Barres, 2018). For a brief summary of the known interactions of microglia with other cells in the CNS, see Box 1. Of particular interest are their recent connections to the development and maintenance of white matter – the heavily myelinated tissue of the CNS that facilitates connectivity throughout the brain (Box 1), and is particularly damaged in genetic disorders known as leukodystrophies. However, the mechanisms that underlie the putative roles of microglia in promoting myelin health or disease remain largely unknown.
Microglia and disease
Current microglia-targeting therapies
Neuropathological, epidemiological and human genetic studies provide strong evidence that microglia play a modifying role in a wide variety of congenital and sporadic brain diseases (Graeber and Stre'rt, 1990; Mattiace et al., 1990; Perry et al., 2010; Prinz and Priller, 2014; Ulland and Colonna, 2018), and could thus represent a universal treatment target. Indeed, even though the precise roles of microglia in health and disease are incompletely understood, existing data on how hematopoietic stem cell transplantation (HSCT) therapy – a means of supplementing the endogenous myeloid cell repertoire – affects the diseased human brain strongly support this direction. HSCT provides donor myeloid cells that can migrate into the brain, likely resulting in a renewed population of healthy macrophages/microglia in the diseased CNS (Wolf et al., 2020; Bennett et al., 2018). Transplantations have been shown to promote remyelination in several leukodystrophies (Musolino et al., 2014; Krägeloh-Mann et al., 2013; Boucher et al., 2015; van Egmond et al., 2013; Cartier and Aubourg, 2010; Weinstock et al., 2020; Mochel et al., 2019; Gelfand et al., 2020), suggesting that replenishing either the abundance or function of microglia can have unprecedented benefits for a range of severe brain diseases. Inversely, transient microglial depletion by clodronate liposomes or chemical inhibitors of colony stimulating factor 1 receptor (CSF1R), a key regulator of macrophage development, chemotaxis and survival (Stanley and Chtu, 2014), can ameliorate disease progression in mouse models of neurodegenerative diseases, such as Alzheimer's disease by facilitating remyelination and alleviating symptoms of myelination defects (Casali et al., 2020; Hansen et al., 2018; Janova et al., 2018; Lloyd et al., 2019; Spangenberg and Green, 2017; Spangenberg et al., 2019).
Microglia as key players in the pathology of leukodystrophy
Leukodystrophies constitute a heterogeneous group of rare genetic disorders characterized by primary involvement of the CNS white matter and neuropathology involving glial cells (van der Knaap et al., 2019). The name ‘leukodystrophy’ is derived from Greek and literally translates to abnormal growth of the white matter. Although individual leukodystrophies are rare, the overall leukodystrophy incidence is around 1 in 7600 live births (Bonkowsky et al., 2010). The majority of leukodystrophies initiate in childhood and most affected individuals fail to reach adulthood. Cognitive and motor decline are the most common clinical symptoms, but dementia, personality changes and depression are also highly prevalent (Table 1).
Table 1.
Underlying genes, neuropathology and clinical features of five primary microgliopathies and three primary lysosomal or peroxisomal leukodystrophies

We will compare the cellular pathology of two groups of leukodystrophies to build a case for a more global involvement of microglia in neuropathology and, in doing so, highlight the broad promise of this glial cell type as a therapeutic target. First, we will describe microgliopathies, which are white matter diseases believed to be caused primarily by microglial abnormalities (Prinz and Priller, 2014), including CSF1R-related leukodystrophy, Nasu–Hakola disease (NHD) related to recessive genetic variants in TREM2 and DAP12/TYROBP, and leukodystrophies related to recessive genetic variants in the LRRC33 (also known as NRROS) or USP18 genes. Next, we will address lysosomal and peroxisomal leukodystrophies, including metachromatic leukodystrophy (MLD), X-linked adrenoleukodystrophy (X-ALD) and globoid cell leukodystrophy (GLD), also known as Krabbe disease, that all show beneficial responses to HSCT. Based on the overlapping pathological features of these two groups, we will describe possible mechanisms, supported by circumstantial evidence, through which aberrant microglia could lead to white matter abnormalities and overall disturbance of brain health. We argue that studying these diseases provides a unique opportunity to elucidate the putative roles of microglia in modulating myelin development and maintenance, while expanding our understanding of the detrimental effects caused by aberrant microglia in the human brain.
Primary microgliopathies: leukodystrophies with central microglial pathology
Glial cell pathology is a major hallmark of leukodystrophies (Garcia et al., 2020). A classification system based on the main glial contributor to the underlying pathogenesis was recently proposed (Vanderver et al., 2015; van der Knaap and Bugiani, 2017). Microgliopathies comprise disorders caused by mutations in genes predominantly expressed by microglia, ultimately leading, either directly or indirectly, to white matter defects (Prinz and Priller, 2014). Leukodystrophies related to bi-allelic loss-of-function mutations in CSF1R, TREM2, TYROBP, LRRC33/NRROS or USP18 can be categorized as primary microgliopathies (Oosterhof et al., 2019; Meuwissen et al., 2016; Schwabenland et al., 2019; Dong et al., 2020; Smith et al., 2020). To date, only CSF1R also demonstrates a dominant form of disease (Rademakers et al., 2011). These genetic disorders lead to overlapping microglial phenotypes,including disturbed distribution, gene expression and morphology (Fig. 1), which we discuss in more detail in the coming sections. Individuals affected by these genetic disorders present with severe white matter degeneration predominantly in the frontal and parietal lobes, in the corpus callosum and in periventricular regions. Axon pathology, dilated ventricles and cerebral atrophy are also highly prevalent, although the U-fibers are mostly spared. Additionally, most affected individuals present with calcifications, predominantly periventricular, although these occur specifically in the basal ganglia of NHD and USP18-related disease. An overview of clinical and pathological features can be found in Table 1.
Fig. 1.

Schematic representation of microglial phenotypes in the grey and white matter of homeostatic and leukodystrophic cortical tissue. In the homeostatic brain, most microglia are evenly distributed in the white and grey matter, and appear ramified, expressing homeostatic markers, such as IBA1, TMEM119 and P2YR12. In the leukodystrophic brain, the white matter is affected by degenerative lesions (striped pattern) and microglia are unevenly distributed, clustering in certain areas, especially within white matter lesions. Phagocytic microglia are abundant within lesions and present an amoeboid shape with large CD68+ intracellular lysosomal vacuoles. Near leukodystrophic lesions, phagocytes display lipid accumulation. Ramified microglia are localized to the grey matter, where their declining density towards the white matter lesions correlates with a gradual loss of homeostatic gene expression and gain of lysosomal CD68 expression. Grey, grey matter; yellow, white matter; stripe pattern, degenerative white matter lesions.
CSF1R-related leukodystrophy
As mentioned above, CSF1R is a key regulator of microglia and macrophage biology in vivo, regulating aspects of proliferation, migration and survival (Herbomel et al., 2001; Dai et al., 2002; Erblich et al., 2011; Oosterhof et al., 2018; Pridans et al., 2018; Rojo et al., 2019; Kuil et al., 2019b, 2020) (Fig. 2). It is a transmembrane receptor tyrosine kinase primarily expressed on mononuclear phagocytic cells (Stanley and Chitu, 2014; Hume et al., 2020). It functions as a homodimer, binding dimers of either CSF-1 or IL-34 (Stanley and Chitu, 2014). In humans, heterozygous mutations in the CSF1R gene can lead to adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP; Table 1) (Rademakers et al., 2011). Loss of CSF1R in zebrafish, mice and rats results in a severe depletion of most macrophages and microglia, though a small number of amoeboid IBA1+ cells remain (Oosterhof et al., 2018; Erblich et al., 2011; Pridans et al., 2018). Csf1r−/− mice show apparently normal myelination but have reduced oligodendrocyte numbers and rarely survive to adulthood (Erblich et al., 2011). Csf1r−/− rats have reduced myelination (Pridans et al., 2018), and csf1r-deficient zebrafish have transient myelin abnormalities, as shown in a recent preprint (Djannatian et al., 2021). Unlike mice, both Csf1r-deficient rats and zebrafish survive to adulthood.
Fig. 2.
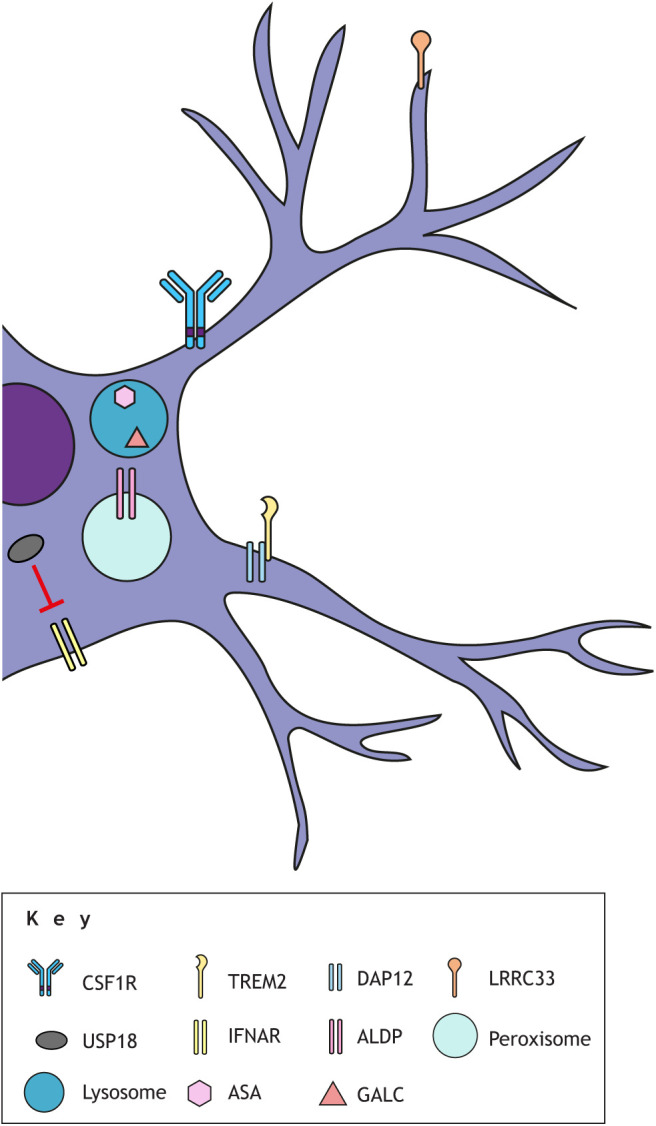
Leukodystrophy-associated proteins in microglia. Several microglial proteins, either at the cell surface or residing within organelles, are involved in leukodystrophy-related cellular processes. CSF1R is a dimeric transmembrane receptor that is a key regulator of microglia and macrophage biology, including proliferation, migration and survival, and binds ligands CSF-1 and IL-34. TREM2 is a single-pass transmembrane receptor involved in lipid metabolism and phagocytic clearance by macrophages and microglia. It binds anionic ligands, including phospholipids and bacterial components, and forms a signaling complex with DAP12, which is a dimeric transmembrane adapter required for the cell-surface expression of TREM2, forming a signaling complex. LRRC33 is a leucine-rich repeat-containing protein that anchors latent TGF-β1 at the cell surface. It is required for activation of the TGF-β1 pathway in macrophages and microglia. The USP18 isopeptidase binds to the intracellular domain of IFNAR2, thereby negatively regulating the IFN-I pathway. ALDP, a peroxisomal ABC half-transporter encoded by ABCD1, transports VLCFAs. ASA, encoded by ARSA, is a lysosomal enzyme that digests sulfatides, glycosphingolipids that are highly enriched in myelin. GALC, encoded by GALC, is a lysosomal enzyme catabolizing galactosylceramide and psychosine, both major glycosphingolipids in myelin. ASA and GALC are not known to be expressed at high levels in macrophages and microglia, but there are strong indications that their functions are required by microglia in the context of myelin.
The first records of ALSP date from 1936, when it was described as pigmentary orthochromatic leukodystrophy (POLD) and later as hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS) (Konno et al., 2018). Following the identification of their common genetic cause, they are now considered to be the same entity (Nicholson et al., 2013; Adams et al., 2018). Besides global white matter abnormalities and neuropathological features (Table 1), a major hallmark of ALSP is the presence of axonal spheroids, which contain organelles and aggregated proteins, such as neurofilament and amyloid precursor protein (APP) (Lin et al., 2010). Human postmortem histological studies showed that cells expressing IBA1 (also known as AIF1), a protein highly enriched in microglia and macrophages, and P2YR12, a microglia-specific protein, are reduced (Tada et al., 2016; Oosterhof et al., 2018) and have a decreased density with a scattered distribution in three cortical areas, both in white and grey matter (Oosterhof et al., 2018). Dense clusters of amoeboid microglia, which are associated with increased phagocytic activity, are nevertheless also observed in cortical areas, predominantly in the cerebral white matter and the corpus callosum, and these cells are strongly positive for the lysosomal protein CD68 (Oosterhof et al., 2018; Tada et al., 2016; Oyanagi et al., 2017; Kempthorne et al., 2020). CD68 is a widely used marker for visualizing phagocytes, including macrophages and microglia. Numerous lipid-laden microglia are present within degenerated white matter lesions, indicating myelin phagocytosis. Our own group recently reported that in brain tissue from a patient with bi-allelic loss-of-function CSF1R mutations, microglia are completely absent, except for sporadic perivascular clusters of phagocytic CD68+ cells (Oosterhof et al., 2019). The developmental appearance of amoeboid IBA1+ and CD68+ cells in subcortical regions, including the corpus callosum and periventricular regions (Ling and Wong, 1993), a phenomenon first described by del Rio-Hortega (1932), is likely connected to a developmental phagocytic role of these cells. Our group's experiments with zebrafish further showed that induced cell death in the brain of partial csf1r-deficient zebrafish causes a local increase in microglia numbers mainly by migration, which could further promote the local depletion of microglia in other brain areas, as observed in patients (Oosterhof et al., 2018). Altogether, these findings strongly suggest that microglial depletion or loss of homeostatic microglia, which may precede the onset of symptoms by many years, may be a key pathogenic initiating event in CSF1R-related leukodystrophy.
However, it is possible that some brain pathology may be an indirect consequence of abnormalities caused by macrophage deficiency outside of the CNS. For example, skeletal abnormalities resulting from defective osteoclasts extend to the skull bones, as seen in individuals with bi-allelic CSF1R variants and reminiscent of those observed in Csf1r-deficient zebrafish, mouse and rat models, which may additionally affect brain development (Oosterhof et al., 2019; Guo et al., 2019; Caetano-Lopes et al., 2020; Pridans et al., 2018). Furthermore, Csf1r-deficient rats exhibit a loss of liver macrophages and disrupted liver function, including dysregulation of lipid metabolism and of the GH/IGF1 system, which could be rescued by bone marrow transplantation (Keshvari et al., 2020). Meanwhile, specific depletion of microglia by knockout of a Csf1r enhancer in mice, where other brain-resident macrophages remain, results in less severe myelin and brain abnormalities than those seen in Csf1r−/− mice (Rojo et al., 2019). This suggests that macrophages beyond microglia may indirectly contribute to the severe neuropathology seen in CSF1R deficiency.
The strong effect of heterozygous mutations in CSF1R, resulting in severe progressive leukodystrophy and dramatically reduced microglia numbers, can be explained by a dominant-negative effect: all but a few causative genetic variants in ALSP patients are missense and affect one of the two tyrosine kinase domains (Konno et al., 2017). In vitro, mutant receptors can still dimerize with wild-type receptors and likely perturb their function (Hume et al., 2020; Pridans et al., 2013). Therefore, a single missense variant likely leads to a loss of function closer to 75% rather than 50%. In line with this, in several instances of reported bi-allelic loss-of-function variants, including a premature stop codon, no typical ALSP signs were reported in parents that were heterozygous for one of the variants (Oosterhof et al., 2019; Guo et al., 2019; Monies et al., 2017).
The ability to deplete microglia via CSF1R inhibition, based on mouse model studies, was recently recognized as a possible therapeutic strategy for the treatment of several brain diseases associated with inflammation, including Alzheimer's disease (Spangenberg and Green, 2017; Spangenberg et al., 2019; Casali et al., 2020; Hansen et al., 2018) and glioma (Akkari et al., 2020). In ALSP, it might be expected that providing healthy myeloid donor cells, with normal CSF1R signaling, via HSCT during an early stage of disease could provide beneficial effects, possibly by boosting the microglia pool. This is indeed the case in mice (Bennett et al., 2018). Additionally, multiple publications have reported cases in which HSCT delayed or even halted disease progression in ALSP patients (Mochel et al., 2019; Gelfand et al., 2020; Eichler et al., 2016). Importantly, whether individuals benefit from HSCT is likely highly dependent on the level of disease progression at the start of treatment and the ability of an individual to cope with the effects of myeloablative treatment.
Thus, findings in CSF1R-related leukodystrophy patients, with heterozygous and bi-allelic variants, and in CSF1R-mutant animal models strongly indicate that a loss of microglia and macrophages in development and/or in adult life strongly affects both myelin and overall brain health, and that these effects can be at least partially abrogated through HSCT.
Nasu–Hakola disease
NHD, or polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL), was described in the early 1970s by Hakola (1972) and Nasu et al. (1973). Over 200 cases have been reported in the literature since then. Unique hallmarks of this disease are bone pain and fractures caused by mild trauma and bilateral basal ganglial calcifications (Paloneva et al., 2001). Genetic analyses identified causative recessive loss-of-function mutations in one of two genes: triggering receptor expressed on myeloid cells 2 (TREM2) or DNAX activating protein of 12 kDa (DAP12), also known as TYRO protein kinase-binding protein (TYROBP) (Paloneva et al., 2002) (Fig. 2). TREM2 is a transmembrane receptor and DAP12/TYROBP a transmembrane adaptor protein that together form a signaling complex (Deczkowska et al., 2020; Ulland and Colonna, 2018). Both TREM2 and DAP12 are highly expressed in myeloid cells, including microglia. Thus, taken together with the CNS abnormalities and pathology (Table 1), NHD is considered a microgliopathy (Bianchin et al., 2004; Xing et al., 2015; Konishi and Kiyama, 2018; Bakker et al., 2000; Schmid et al., 2002). Ligands for TREM2 include lipids and bacterial components, including the Alzheimer's disease-associated ApoE and Aβ oligomers (Konishi and Kiyama, 2018). TREM2 in particular has been extensively studied in mouse and induced pluripotent stem cell (iPSC) models, mainly due to its link to microglial involvement in Alzheimer's disease pathology (reviewed by Ulland and Colonna, 2018). TREM2 is involved in lipid metabolism and TREM2+ lipid-associated macrophages in mouse adipose tissue can sense aberrations in lipid composition and respond by driving the expression of genes associated with phagocytosis, lipid catabolism and energy metabolism (Jaitin et al., 2019). Microglia in Trem2−/− mice fail to upregulate these processes in response to acute demyelination (Poliani et al., 2015; Cantoni et al., 2015). TREM2-deficient microglia can phagocytize myelin debris after demyelination but are unable to break it down, especially cholesterol, resulting in intracellular lipid accumulation (Cantoni et al., 2015; Linnartz-Gerlach et al., 2019; Nugent et al., 2020). Hence, TREM2 signaling appears to be a major pathway by which macrophages respond to changes in tissue-level lipid homeostasis, and lack of TREM2 signaling contributes to ineffective responses to myelin injury and turnover.
In genetic mouse models of NHD, microglia numbers vary depending on the age of the animal and the brain region analysed. In Trem2−/− or Dap12−/− mice, microglia numbers are typically reduced during early development but differences from wild-type mice are largely absent at later stages (Filipello et al., 2018; Poliani et al., 2015; Nataf et al., 2005; Otero et al., 2009; Kaifu et al., 2003). Loss of functional TREM2 has also been shown to impair the phagocytic capacity of microglia in a range of model systems, including mutant mice (Filipello et al., 2018; Poliani et al., 2015), iPSC-derived microglia (Garcia-Reitboeck et al., 2018; McQuade et al., 2020) and in primary mouse microglia (Takahashi et al., 2005; Hsieh et al., 2009; Kleinberger et al., 2014). Mouse models recapitulate several pathological features seen in patients (Table 1), including degeneration of the white matter (Kaifu et al., 2003; Nataf et al., 2005), bone abnormalities (Kaifu et al., 2003; Nataf et al., 2005) and axonal pathology (Cantoni et al., 2015). Importantly, a loss of microglia or macrophages is also not found in postmortem brain samples of NHD patients (Oyanagi et al., 2017). However, an increased phagocytic phenotype and intracellular waste accumulation in both ramified (Satoh et al., 2011) and CD68+ amoeboid microglia and macrophages is observed in the white matter (Paloneva et al., 2001; Oyanagi et al., 2017), which is consistent with defective processing of phagocytized material.
LRRC33/NRROS and USP18-related leukodystrophies
Two severe pediatric neurodevelopmental white matter diseases caused by bi-allelic variants in the microglia-associated genes LRRC33/NRROS or USP18 have recently been described. The pathological features of these diseases bear similarities to other microgliopathies (Table 1).
LRRC33 is a leucine-rich repeat-containing protein that can anchor latent transforming growth factor beta-1 (TGF-β1) at the cell surface and is required for the activation of TGF-β1 in macrophages and microglia (Qin et al., 2018) (Fig. 2). In addition, it is a negative regulator of reactive oxygen species (ROS) production, hence the suggested name change to NRROS (Noubade et al., 2014). TGF-β1 is a trophic factor and cytokine that is a key component of highly conserved signaling pathways with broad regulatory roles in metazoan development, including immune modulation (reviewed by Gomes et al., 2005). In brain development, TGF-β1 plays an important role in axon growth and polarity (Yi et al., 2010), regulation of astrocyte morphology, motility, proliferation and differentiation (Diniz et al., 2017), and neuron survival (Brionne et al., 2003; Gomes et al., 2005; Dong et al., 2020). In the CNS, LRRC33 is primarily expressed in microglia (Wong et al., 2017; Qin et al., 2018).
Lrrc33 knockout mice show increased CD68+ microglia numbers with an altered hypertrophic morphology, along with upregulated cell division and interferon signaling pathways (Wong et al., 2017; Qin et al., 2018). Descriptions of other neuropathological features of these mutant mice show some inconsistencies; whereas one group reported localized demyelination and noted loss of oligodendrocytes (Qin et al., 2018), another reported normal axon numbers and myelination but reactive astrocytes in the CNS (Wong et al., 2017). As conditional deletion of Lrrc33 in mice at 3 weeks of age causes no neurological effects, it is likely that LRRC33 is required for the establishment of the microglial network during development (Wong et al., 2017). So far, nine individuals with bi-allelic loss-of-function mutations in LRRC33 have been described, most of whom died between 2 and 4 years of age (Dong et al., 2020; Smith et al., 2020). Patients present with severe white matter defects and neuropathology (Table 1). Pathologically lipid-filled CD68+ macrophages lacking a homeostatic microglia gene signature were observed throughout the white matter, particularly in perivascular regions (Smith et al., 2020). Together, this suggests that microglial modulation of TGF-β signaling is key to brain development. Consistent with this, bi-allelic mutations in TGFB1 itself cause a pediatric neurodevelopmental leukoencephalopathy (Kotlarz et al., 2018).
Ubiquitin specific protease 18 (USP18) is a negative regulator of the type 1 and 3 interferon (IFN-I) pathway by binding to the intracellular domain of interferon alpha and beta receptor subunit 2 (IFNAR2) (Honke et al., 2016; Goldmann et al., 2015) (Fig. 2). Microglia that lack Usp18 exhibit elevated type I IFN signaling pathway activation (Goldmann et al., 2015). Conversely, overexpression of USP18 suppresses microglial activation by reducing the release of pro-inflammatory cytokines (Xiang et al., 2019). Microglia in Usp18-deficient mice show increased phagocytosis of myelin (Schwabenland et al., 2019) and present in clusters in the white matter, strongly resembling the microglial pathology in ALSP and NHD (Goldmann et al., 2015; Schwabenland et al., 2019). These mice exhibit diminished structural white matter integrity (Schwabenland et al., 2019) and low bone density due to osteoclast abnormalities (Yim et al., 2016). Homozygous loss-of-function variants in USP18 in humans can result in pseudo-TORCH syndrome (PTS), a type of genetic disorder termed a type I interferonopathy, which causes death within days after birth (Meuwissen et al., 2016). A striking hallmark of PTS is defective neuronal migration, partly leading to ectopic neuronal cell bodies in the white matter (Table 1). Immunohistochemistry on patient postmortem brain tissue reveals a robust presence of IBA1+ and HLA-DR+ microglia throughout the brain (Meuwissen et al., 2016). Additionally, Usp18-deficient mice exhibit an elevated number of microglia that appear to have engulfed oligodendrocytes (Schwabenland et al., 2019).
Despite differences in their underlying mechanisms, LRRC33/NRROS and USP18-related microgliopathies reveal that deficiencies in key microglial immune signaling pathways, in this instance TGF-β and interferon signaling, lead to phagocytic abnormalities in the brain and a consequent reduction in brain integrity and function, with particular involvement of the white matter.
Primary lysosome or peroxisome dysfunction in leukodystrophies: making a case for microglia
There is strong evidence indicating that microglial involvement in leukodystrophies is caused by lysosomal or peroxisomal defects. Lysosomes are key metabolic hubs for the recycling of macromolecules and metabolites (Platt et al., 2018). Impairments can lead to the accumulation or ‘storage’ of partially undigested material and the reduction of lysosomal function, as a well as a deficit of specific metabolites (Platt et al., 2018). Peroxisomes are membrane-enclosed organelles that contain enzymes involved in lipid metabolism (Titorenko and Rachubinski, 2004). Deficiencies in peroxisomal enzymes can also result in substrate accumulation. It is common that lysosomal and peroxisomal storage disorders present as severe progressive neurodegenerative diseases (Platt et al., 2018). Among these, several are also classified as leukodystrophies (Table 1): MLD, GLD and X-ALD (van der Knaap and Bugiani, 2017).
MLD patients have bi-allelic mutations in the ARSA gene that lead to deficiency of arylsulfatase A (ASA), a lysosomal enzyme that digests sulfatides – glycosphingolipids that are highly enriched in myelin sheaths (Eckhardt, 2008; Fig. 2). Accumulation of sulfatides in white matter macrophages, as well as other brain cells including neurons, astrocytes and oligodendrocytes, coincides with severe destruction of the cerebral and deep white matter (Table 1). MLD occurs in up to 2.5 cases per 100,000 individuals in the USA (Platt et al., 2018). Bi-allelic mutations in GALC, encoding the lysosomal hydrolase galactosylceramidase, cause Krabbe disease (Suzuki and Suzuki, 1970) (Fig. 2). Galactosylceramidase catabolizes galactosylceramide and galactosylsphingosine (psychosine), both of which are major glycosphingolipids in the myelin membrane (Weinstock et al., 2020). The resulting accumulation of psychosine is thought to underlie the progressive white matter disease (Suzuki, 1998). X-ALD is an X chromosome-linked disorder caused by mutations in the ABCD1 gene, encoding the ATP-binding cassette (ABC) half-transporter ALDP, resulting in the failed transport of very long-chain fatty acids (VLCFA) into the peroxisome (Kemp et al., 2001) (Fig. 2). VLCFAs aggregate predominantly in the brain and spinal cord via esterification with glycerophospholipids and cholesterol, as these lipids are abundant in the myelin membrane (Schaumburg et al., 1976). This ultimately leads to degeneration of the white matter (Table 1). The estimated incidence of X-ALD is about 1 in 14,700 live births (Bezman et al., 2001; Moser et al., 2016).
The genetic variants defining these leukodystrophies may not immediately appear directly associated with microglia, as the genes are expressed across various brain cell types. However, as microglia are the major phagocytes of the brain, defects in either lysosomes or peroxisomes are likely to substantially affect phagocytosis and waste accumulation in microglia, which could contribute to significant dysfunction. Again, support for microglial involvement in the mechanism of these pathologies is most clearly demonstrated by the beneficial effects of HSCT (reviewed by Musolino et al., 2014; van Rappard et al., 2015). In postmortem brain tissue of MLD patients who received HSCT, active ASA was clearly detectable in macrophages/microglia but not in resident oligodendrocytes and astrocytes, suggesting no cross-correction by healthy donor enzyme. Moreover, transplanted patients had increased numbers of homeostatic macrophages and oligodendrocyte lineage cells, and there was evidence of remyelination (Wolf et al., 2020). Similarly, Weinstock et al. (2020) examined enzyme cross-correction and the effects of HSCT on the peripheral nervous system (PNS) in Galc−/− mice and in postmortem spinal cord tissue of GLD patients. In mutant mice, Schwann cells, the myelinating cells of the PNS, require lysosomal Galc to maintain myelin and axonal integrity, and are unable to receive enzyme from the surrounding environment, partly due to ineffective uptake. GLD patients who received HSCT showed an improvement in axonal integrity and myelin thickness, as well as a reduction of phagocytic CD68+ microglia, fewer multinucleated phagocyte clumps (also termed globoid cells) and reduced substrate accumulation in macrophages (Weinstock et al., 2020). Although it is important to note that the effects of HSCT can vary tremendously between individuals and between leukodystrophies, and its potential effectivity is limited by the need to treat early and pre-symptomatically (Boucher et al., 2015), these data support the theory that microglia are key players in myelin repair and oligodendrocyte lineage regulation. Importantly, we consider it unlikely that the therapeutic benefit of HSCT in these examples is not linked to an initial contribution of microglial abnormalities to the myelin pathology.
Indeed, the contribution of microglia to the pathology of these leukodystrophies is supported by findings in patient postmortem brain tissue, in which CD68+ phagocytic microglia are abundant in both the affected and least-affected white matter of Krabbe disease (Del Bigio, 2018), MLD and X-ALD patient brain tissue (Bergner et al., 2019). Ramified homeostatic microglia numbers decrease continuously towards the demyelinated lesion, until there are only amoeboid phagocytic microglia left (Fig. 1; Bergner et al., 2019; Itoh et al., 2002). Importantly, amoeboid phagocytes show large intracellular phagosomes, waste accumulation and MBP inclusions, indicating disrupted (myelin) phagocytosis (Bergner et al., 2019; Percy et al., 1994). Galc+/− mice do no exhibit a neuropathological phenotype in normal circumstances, but upon induced demyelination, microglia show impaired myelin phagocytosis and reduced Trem2 expression, which is necessary for clearing myelin (Scott-Hewitt et al., 2017). In accordance, patient-derived PNS macrophages show defective myelin degradation, resulting in waste material accumulation (Weinstock et al., 2020). Together, these studies indicate that lysosomal or peroxisomal dysfunction results in waste accumulation in microglia, presumably due to improper degradation, which worsens in the proximity of white matter lesions. Although these diseases likely also have major non-microglial components, the effects seen in microglia appear to be cell-autonomous and not simply responses to pathological processes in other cells of the CNS. This is reminiscent of lysosomal storage disorder phenotypes in zebrafish models (Box 2: The Zebrafish – In Vivo Modelling of Glial Cell Dynamics and Myelination in Genetic Brain Disorders), in which abnormal microglia are typically seen in very early embryogenesis, right at the stages at which they seem to shape brain development (Berg et al., 2016; Kuil et al., 2019a; Sanderson et al., 2021).
Box 2. The Zebrafish – In Vivo Modelling of Glial Cell Dynamics and Myelination in Genetic Brain Disorders.
Zebrafish (Danio rerio) are especially suitable for studying early brain development and pediatric disease: the embryos develop rapidly, ex utero, are transparent, and it is now generally accepted that all the major glial cells known in mammals (Box 1) are also present in zebrafish. Importantly, they share extensive conservation of function, morphology and gene expression (Lyons and Talbot, 2014; Marisca et al., 2020; Hughes and Appel, 2019; Oosterhof et al., 2017; Mu et al., 2019; Chen et al., 2020). Cellular dynamics and interactions, including processes such as myelination and myelin maintenance, can be studied non-invasively with high spatiotemporal precision. With over 70% of genes shared with humans and carrying orthologs of ∼82% of human disease-associated genes, and continuously improving genetic tools, the zebrafish is an increasingly popular model for investigating genetic disorders (Howe et al., 2013). For more information, please see extensive reviews by Duncan et al. (2011), Ackerman and Monk (2016) and Rutherford and Hamilton (2019) on zebrafish and rodent models of leukodystrophies and myelination.
Transgenic zebrafish lines expressing fluorescent proteins under the control of specific promoters can be used to visualize cell populations in real time (Preston and Macklin, 2015). This allows cell type-specific in vivo monitoring of, for example, the calcium signaling of neurons and astrocytes (Akerboom et al., 2012; Mu et al., 2019), cell-cell interactions, cell dynamics and intracellular processes, such as phagocytosis (van Ham et al., 2014). Zebrafish allow real-time non-invasive imaging of cells in their natural environment in a whole organism. By imaging fish on consecutive days, researchers can easily follow individual cells longitudinally. This also allows for an unbiased approach to the discovery of novel cell biological processes. Recent examples of this are the discovery of microglial regulation of neuronal activity (Li et al., 2012), pruning of myelin sheaths during development (Hughes and Appel, 2020), regulation of myelination by neuronal activity via synaptic vesicle release (Mensch et al., 2015) and the existence of functionally distinct subsets of oligodendrocytes in the spinal cord (Marisca et al., 2020).
Zebrafish have a tremendous regenerative capacity after injury, which can be exploited to identify molecules and pathways that regulate myelin and axonal regeneration, which could, for example, aid in treatment strategies for spinal cord injury (Cigliola et al., 2020). Drug screening can be performed rapidly, on a large scale and in vivo, which is a major advantage compared to murine models (MacRae and Peterson, 2015). Drug screens in zebrafish have another unexpected benefit: as drugs are screened in a complete organism, any leads discovered in zebrafish are more likely to be effective in additional in vivo systems. Indeed, several drugs discovered only recently in zebrafish have made it to phase 2 clinical trials [e.g. Dravet syndrome (Baraban et al., 2013)]. Another drug screen in an abcd1−/− zebrafish model shows the potential for leukodystrophies, revealing metabolic rerouting of saturated to mono-unsaturated VLCFAs following pharmacologically increased expression of the enzyme SCD1 (Raas et al., 2021). This led to fewer behavioral abnormalities and reduced lipid toxicity, making it a promising therapeutic strategy for X-ALD and other peroxisomal diseases.
Overlapping pathologies indicating possible mechanisms: a perfect storm?
Although differences among leukodystrophies do remain profound, we argue that the overlapping pathological features described in this Review are both significant and revealing (Table 1). A detailed examination of these similarities, combined with recent insights from genetic disease models, will aid in understanding the shared underlying mechanisms and cell types involved, which in turn will facilitate the identification of novel targets for treatment. These analyses will also yield important clues about the function of microglia and macrophages in myelin homeostasis and repair in particular, and in maintaining brain health overall. Here, we suggest a variety of possible ways, supported by circumstantial evidence from rare genetic disease, in which aberrant microglia may lead to white matter abnormalities (Fig. 3).
Fig. 3.

Loss of microglial functions could lead to white matter degeneration by affecting multiple intercellular connections. Routes whereby microglia, directly or indirectly, affect the white matter (WM) have been supported by data from experimental studies as described here; however, their relevance to disease remains to be fully explored. Astrocytes compensate for a lack of microglial phagocytosis by becoming more phagocytic, although they are less efficient than microglia and this response may result in the neglect of critical astrocytic functions. Together with the increased astrocytic reactivity observed in leukodystrophies, this can result in disturbed lipid and metabolic supply to oligodendrocytes (OL) and an unsupportive ECM environment for OPCs. Additionally, altered interactions between the BBB and astrocytic end-feet can perturb metabolic supply to brain cells. Aberrant microglia can lead to insufficient trophic support for OPCs and oligodendrocytes, and a diminished oligodendrocyte lineage. Aberrant microglia may also exhibit perturbed clearance and pruning capacity and contribute to impaired remyelination. Both aberrant microglia and affected astrocytes can cause neuronal stress due to neurotoxicity, ineffective phagocytosis and/or dysregulation of neuroactivity. Axonal pathology and abnormal neuronal activation can affect myelination and, in turn, the degeneration of myelin results in a loss of metabolic support for axons. In sum, white matter degeneration in leukodystrophies is likely preceded by distinct effects of aberrant microglia, possibly forming a ‘perfect storm’ of parallel effects particularly detrimental for the myelinated white matter tracts. Solid arrows indicate established interaction/consequence. Dashed arrows indicate hypothesized interaction/consequence in leukodystrophic brain.
Axonal pathology
One overlapping hallmark of leukodystrophies is axonal pathology, which may precede white matter degeneration. In fact, a brain biopsy of a patient with ALSP showed abundant spheroids, which are pathological swellings of neuronal processes, 2.5 years before a postmortem examination that revealed fewer spheroids (Marotti et al., 2004). Additionally, the occipital lobe in ALSP patient brains does not show obvious myelin changes but does present axonal spheroids (Jin et al., 2015). Axonal spheroids can be caused by impaired ATP-dependent axonal transport of proteins, lipids, organelles and autophagy-lysosomal degradation, processes that are implicated in several neurological diseases (Sleigh et al., 2019). Other axon pathologies, such as the neuritic beading regularly found in neurodegenerative diseases, are linked to microglial glutamate release leading to inhibition of mitochondrial activity and a rapid drop in neuronal ATP levels (Takeuchi et al., 2005). This energy loss leads to defective axonal transport and potential subsequent excitotoxic neuronal death. Importantly, as myelin provides protection and metabolic support to axons (Aggarwal et al., 2011), myelin degeneration also likely results in axonal defects, making it difficult to pinpoint the initial culprit (Fig. 3).
Axon loss could also occur due to failed axon guidance and growth during development. Defective neuronal migration is observed in USP18-related leukodystrophy patients (Meuwissen et al., 2016). In the complete absence of microglia, as in bi-allelic CSF1R mutations, there is almost no development of the corpus callosum. This is supported by studies in mouse models showing that microglia are crucial for dorsal axon guidance in the corpus callosum (Pont-Lezica et al., 2014), and for axon guidance and growth in other parts of the brain (Rigby et al., 2020; Squarzoni et al., 2014). However, Csf1r-deficient rats have only a slight thinning of the corpus callosum (Keshvari et al., 2020), and there were no apparent commissural abnormalities in zebrafish deficient for csf1r (Oosterhof et al., 2018).
Microglia have been shown in mouse studies to prune synapses and axons, which is important for normal brain development (Paolicelli et al., 2011; Schafer et al., 2012). Another intriguing function has been identified in zebrafish, in which neuronal activity steers microglial processes and facilitates direct contact with highly active neurons to reduce stimulus-evoked neuronal excitation (Li et al., 2012). In mice, microglia can also suppress neuronal activity by an adenosine-mediated mechanism (Badimon et al., 2020). Microglia can sense and catabolize the extracellular ATP released by neurons and astrocytes via the purinergic P2YR12 (Badimon et al., 2020). Microglial ablation, or P2RY12 blockade, can induce amplification and synchronization of neuronal excitation, leading to seizures (Badimon et al., 2020). P2RY12-dependent somatic microglia-neuron junctions are found in mouse and human brains, and, upon injury, trigger P2RY12-dependent neuronal protection and regulation of neuronal calcium load (Cserép et al., 2020). Thus, microglial processes can sense, monitor and inhibit neuronal activity and protect neuronal functions. The absence of normally functioning microglia can thereby lead to overexcitation and seizures (Badimon et al., 2020). An as-yet unstudied potential implication of this process is the possibility of aberrant neuronal activity patterns and neuroplasticity in patients with abnormal microglia (Fig. 3). As neuronal activity can regulate and drive myelination, numerous studies, including a recent preprint (Knowles et al., 2020), indicate that these altered activity patterns could subsequently lead to abnormal myelination patterns (Mensch et al., 2015; Marisca et al., 2020; Hughes and Appel, 2020).
Phagocytic clearance and lipid metabolism
Both NHD and storage-related leukodystrophies show that defective myelin and lipid processing by microglia could be an initiating event for white matter defects. Myelin membranes are incredibly lipid rich, as lipids are required for myelin stability (Schmitt et al., 2015). Hence, disrupted lipid metabolism could cause abnormal membrane lipid composition or the accumulation of toxic lipid species in the brain environment (Schmitt et al., 2015).
Lysosomal and peroxisomal storage-related leukodystrophies have shown that insufficient waste degradation leads to the accumulation of myelin debris and other phagocytic cargo in microglia. During normal aging, the degradative capacity of microglia is known to decrease, leading to lysosomal storage (Safaiyan et al., 2016). Recently, abnormal microglia have been identified in aging white matter in mice. These are characterized by the upregulation of phagocytic activity and lipid metabolism, are found in clusters in the white matter and engage in clearing degenerated myelin (Safaiyan et al., 2021). Dysfunctional myelin recycling by microglia presumably results in excessive myelin debris and (lipid) toxins in the brain environment, which could impede the availability of lipids for myelin formation, and proper clearance of debris is crucial for remyelination (Lampron et al., 2015; Cantuti-Castelvetri et al., 2018). Owing to the high plasticity of developing myelin, which in humans continues into adulthood and is linked to learning and memory (Mount and Monje, 2017; Williamson and Lyons, 2018; Bonetto et al., 2020), white matter could be specifically and devastatingly affected by progressive microglial storage dysfunction. The adult-onset cases of microgliopathies described here could involve an accumulation of detrimental microglial dysfunction throughout life, leading to a progressive inability to maintain white matter integrity. Impairment of myelin degradation, as occurs during normal aging, could be the final blow from where pathology starts to manifest in clinical symptoms.
In addition to clearing myelin debris from the brain environment, microglia also actively phagocytize myelin sheaths during development, a process regulated by neuronal activity. Interestingly, when microglia are ablated, oligodendrocytes maintain excessive myelin sheaths (Hughes and Appel, 2020) and show ultrastructural myelin pathologies, as described in a recent preprint (Djannatian et al., 2021). This suggests that in the absence of healthy functioning microglia, myelin sheaths are not properly pruned during myelin development. One can speculate that improper pruning during development can lead to an unstable myelin structure and integrity (Fig. 3). How and whether this could affect myelin health is not yet fully understood.
Aberrant astrocytes
Astrocytes characterized by elevated GFAP expression and hypertrophic processes, often termed reactive astrocytes (Box 1; Escartin et al., 2021), are detected in virtually all neurodegenerative diseases, including the leukodystrophies described in this article. Astrogliotic scars are particularly abundant in the most affected white matter lesions in leukodystrophy. Reactive astrocytes can be both pro-inflammatory and neurotoxic, leading to the death of neurons and oligodendrocytes, by losing the ability to promote neuronal survival, synaptogenesis and phagocytosis (Bi et al., 2013; Liddelow and Barres, 2017; Liddelow et al., 2017). However, reactive astrocytes can also have beneficial effects, including limiting tissue damage after injury and modulating the immune response (Faulkner et al., 2004; Myer et al., 2006; Escartin et al., 2021). Hence, it is still unclear whether reactive astrocytes are detrimental to the progression of leukodystrophies or are serving a protective purpose. Similarly, it is unclear what relationship they have to the observed white matter defects.
Two studies have shown that both microglial dysfunction (Konishi et al., 2020) and microglia depletion (Damisah et al., 2020) lead to elevated astrocytic phagocytosis. An enhanced phagocytic phenotype is also observed in reactive astrocytes after brain ischemia (Morizawa et al., 2017). Although astrocytes can phagocytize apoptotic cell bodies and debris, they are much less efficient than microglia (Damisah et al., 2020). When taking over the role of the major phagocyte in the brain in the absence of (functioning) microglia, astrocytes may fail to execute critical astrocytic functions important for maintaining brain health (Fig. 3). For example, lipid and nutrient supply by astrocytes to oligodendrocytes via connexin-coupled gap junctions is essential for proper myelination (Camargo et al., 2017; Orthmann-Murphy et al., 2008). Abnormalities in astrocytic end-feet and blood-brain barrier (BBB) defects have also been noted in neurodegenerative diseases (Oksanen et al., 2019). The surrounding of vessels by astrocytic end-feet is vital for BBB integrity and overall brain health (Box 1). Compromised BBB integrity could contribute to the calcifications observed in most microglia-related leukodystrophy patients (Table 1). Notably, the BBB appears to be intact in Csf1r−/− rats (Pridans et al., 2018), and vascular barriers may even be less permissive. Further research is needed to establish whether and how astrocytes lose certain functions vital for white matter and overall brain health in the absence of functioning microglia.
Important clues on the impact of aberrant astrocytes on white matter integrity come from astrocyte-related leukodystrophies. Gain-of-function mutations in GFAP lead to Alexander disease (AxD), which is characterized by a severe reactive astrocyte phenotype, including hypertrophic GFAP+ processes and astrocytic cytoplasmic inclusions of GFAP called Rosenthal fibers (Sosunov et al., 2018). AxD mouse models have revealed reduced glutamate transporter expression on astrocytes, indicating decreased glutamate uptake by astrocytes (Tian et al., 2010; Hagemann et al., 2009), which is supported by an observed glutamate toxicity-induced neuron loss in a Drosophila AxD model (Wang et al., 2011). Astrocytes remove ∼90% of all released glutamate in the brain, thereby maintaining glutamate homeostasis by regulating the balance between glutamate uptake and release in the synaptic cleft (Mahmoud et al., 2019). Excessive glutamate toxicity is hypothesized to be one of the mechanisms driving myelin loss in AxD (Sosunov et al., 2018). As elevated GFAP+ astrocytes are also abundantly present in microglia-related leukodystrophies, and glutamate toxicity is a known cause of axon pathology, a similar astrocyte phenotype could partly underlie the white matter defects in microglia-related leukodystrophies (Fig. 3). Deposition of the glycosaminoglycan hyaluronan in the ECM, which occurs in the astrogliosis seen in various diseases and in AxD mice, has also been suggested to contribute to myelin defects (Sosunov et al., 2018). Hyaluronan inhibits oligodendrocyte precursor cell (OPC) maturation (Box 1, Fig. 3) and is thought to be important to the pathology of vanishing white matter disease, another astrocyte-related leukodystrophy (Sosunov et al., 2018). Microglia engulf the ECM in an IL-33-dependent manner to promote synapse plasticity and deficiency of this process leads to an accumulation of ECM proteins around synapses and dendrites (Nguyen et al., 2020). Hence, disturbed white matter integrity due to abnormalities in ECM composition, resulting from the inability of microglia to properly prune and maintain it, is a promising future line of investigation.
The zebrafish: a promising leukodystrophy model
Looking forward, it will be important to combine and compare data from diverse models, human material and clinical observations of disease to improve insight into the cellular brain. Rare brain diseases can give unique and precious clues into human genetics, physiology and pathology. Nevertheless, high quality postmortem brain tissue is very rare, and studies often allow only limited quantitative analysis. So far, the majority of available leukodystrophy animal models have been mice. Hence, their neuropathological and clinical features are thoroughly characterized. Mouse models allow thorough investigation by highly standardized techniques and reagents in a well-characterized system. Yet, despite the substantial advantages of mouse models, direct in vivo cell biological observations are still challenging, and developmental processes in particular are relatively obscured. Cerebral organoids derived from iPSCs are improving but are not yet able to effectively model myelination and the native interactions of multiple glial cell types. The zebrafish has repeatedly proven its value as a system for unparalleled developmental observations of in vivo cellular mechanisms, and its many unique advantages readily complement data obtained through other model systems (Box 2). Especially for understanding the highly interactive cellular processes of myelination and glial cell development in leukodystrophy, the ability to perform real-time, unbiased and non-invasive imaging of dynamic cellular interactions during early embryonic development in a whole organism makes the zebrafish a promising model that can take the field far beyond the current knowledge (Rutherford and Hamilton, 2019; Keefe et al., 2020). The major players in leukodystrophies have been well-characterized in zebrafish (Herbomel et al., 1999; Peri and Nüsslein-Volhard, 2008; Oosterhof et al., 2017; Marisca et al., 2020; Park et al., 2002; Mu et al., 2019; Chen et al., 2020) and have led to major discoveries in the functions of glial cells and myelination (Mensch et al., 2015; Almeida et al., 2018; Djannatian et al., 2019; Hughes and Appel, 2019, 2020; Marisca et al., 2020; Mu et al., 2019; Li et al., 2012). Indeed, a number of genetic zebrafish models have provided important data on the disease mechanisms of several leukodystrophies (Pant et al., 2019; Strachan et al., 2017), including the involvement of microglia in the early disease pathology of CSF1R-related leukodystrophy (Oosterhof et al., 2018, 2019) and RNASET2-related leukodystrophy (Haud et al., 2011; Hamilton et al., 2020; Weber et al., 2020), and the identification of possible therapeutic targets for the treatment of X-ALD (Raas et al., 2021).
Conclusions
Leukodystrophies involving microglia raise intriguing hypotheses regarding the impact of macrophages and microglia on the developing and adult brain, in particular on myelination. In addition, they may help provide insight regarding the issues of optimal macrophage and microglia enhancement or depletion strategies that, although avoiding detrimental side effects related to macrophage function, could treat human disease. It is now clear that depleting microglia, at least temporarily via CSF1R inhibitors, can be beneficial in mouse models of Alzheimer's disease, and therefore has clinical potential. Conversely, studies of rare human disease complemented by zebrafish and rodent disease models has demonstrated that there are undoubtedly negative effects associated with microglial depletion. Therefore, understanding the breadth of roles microglia have in normal human brain development and in the maintenance of adult brain health would be very helpful to further fine-tune methods of modulating microglia in disease to mitigate disease progression and benefit recovery. Insight from CSF1R-related leukodystrophies and CSF1R-deficient animal models could assist in deducing the consequences of the absence or local depletion of macrophages and microglia on the human brain.
It is clear that several important questions remain to be answered regarding how microglia, as well as peripheral macrophages, affect myelin development and the maintenance of white matter and brain health in both development and adult life. For example, defective osteoclasts and other macrophage populations can lead to skeletal abnormalities, as observed in some of the leukodystrophies described here. Skeletal abnormalities are present in several microglia-related leukodystrophies and are likely caused by abnormal osteoclast populations, which originate from similar hematopoietic progenitors as other macrophages and microglia (Bar-Shavit, 2007). In the case of bi-allelic loss of CSF1R function, skeletal abnormalities are identified across vertebrate model systems. It is unclear whether this bone phenotype could contribute to the neuropathology, opening up intriguing opportunities for future research.
Integrating the application of novel methods on postmortem brain tissue of patients, including single-cell and high-throughput genomic, proteomic and metabolomic approaches with genetic model systems, in particular the zebrafish, will aid in unraveling how both microglia and other brain cell types modulate white matter integrity and overall brain health. Importantly, rare genetic brain disorders provide a unique window into human brain physiology and continue to provide clues to the inner workings of the brain and to potential avenues of therapeutic modulation that may be pursued for a broad range of neurological diseases.
Acknowledgements
We thank Dr Stefan Barakat for valuable feedback on the manuscript and all of the members of the van Ham lab for constructive discussion.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Funding
L.E.S. is supported by a LEaDing fellowship from the European Union Horizon 2020 research and innovation programme, under the Marie Skłodowska-Curie grant agreement number 707404. T.J.v.H. is supported by Erasmus Universiteit Rotterdam fellowships.
References
- Ackerman, S. D. and Monk, K. R. (2016). The scales and tales of myelination: using zebrafish and mouse to study myelinating glia. Brain Res. 1641, 79-91. 10.1016/j.brainres.2015.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, S. J., Kirk, A. and Auer, R. N. (2018). Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP): Integrating the literature on hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD). J. Clin. Neurosci. 48, 42-49. 10.1016/j.jocn.2017.10.060 [DOI] [PubMed] [Google Scholar]
- Aggarwal, S., Yurlova, L. and Simons, M. (2011). Central nervous system myelin: structure, synthesis and assembly. Trends Cell Biol. 21, 585-593. 10.1016/j.tcb.2011.06.004 [DOI] [PubMed] [Google Scholar]
- Ajami, B., Samusik, N., Wieghofer, P., Ho, P. P., Crotti, A., Bjornson, Z., Prinz, M., Fantl, W. J., Nolan, G. P. and Steinman, L. (2018). Single-cell mass cytometry reveals distinct populations of brain myeloid cells in mouse neuroinflammation and neurodegeneration models. Nat. Neurosci. 21, 541-551. 10.1038/s41593-018-0100-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerboom, J., Chen, T.-W., Wardill, T. J., Tian, L., Marvin, J. S., Mutlu, S., Calderon, N. C., Esposti, F., Borghuis, B. G., Sun, X. R.et al. (2012). Optimization of a GCaMP calcium indicator for neural activity imaging. J. Neurosci. 32, 13819-13840. 10.1523/JNEUROSCI.2601-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkari, L., Bowman, R. L., Tessier, J., Klemm, F., Handgraaf, S. M., de Groot, M., Quail, D. F., Tillard, L., Gadiot, J., Huse, J. T.et al. (2020). Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 12, eaaw7843. 10.1126/scitranslmed.aaw7843 [DOI] [PubMed] [Google Scholar]
- Allen, N. J. and Eroglu, C. (2017). Cell biology of astrocyte-synapse interactions. Neuron 96, 697-708. 10.1016/j.neuron.2017.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allewelt, H., Taskindoust, M., Troy, J., Page, K., Wood, S., Parikh, S., Prasad, V. K. and Kurtzberg, J. (2018). Long-term functional outcomes after hematopoietic stem cell transplant for early infantile Krabbe disease. Biol. Blood Marrow Transplant. 24, 2233-2238. 10.1016/j.bbmt.2018.06.020 [DOI] [PubMed] [Google Scholar]
- Almeida, R. G., Pan, S., Cole, K. L. H., Williamson, J. M., Early, J. J., Czopka, T., Klingseisen, A., Chan, J. R. and Lyons, D. A. (2018). Myelination of neuronal cell bodies when myelin supply exceeds axonal demand. Curr. Biol. 28, 1296-1305.e5. 10.1016/j.cub.2018.02.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badimon, A., Strasburger, H. J., Ayata, P., Chen, X., Nair, A., Ikegami, A., Hwang, P., Chan, A. T., Graves, S. M., Uweru, J. O.et al. (2020). Negative feedback control of neuronal activity by microglia. Nature 586, 417-423. 10.1038/s41586-020-2777-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker, A. B. H., Hoek, R. M., Cerwenka, A., Blom, B., Lucian, L., Mcneil, T., Murray, R., Phillips, J. H., Sedgwick, J. D. and Lanier, L. L. (2000). DAP12-deficient mice fail to develop autoimmunity due to impaired antigen priming. Immunity 13, 345-353. 10.1016/S1074-7613(00)00034-0 [DOI] [PubMed] [Google Scholar]
- Bar-Shavit, Z. (2007). The osteoclast: a multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J. Cell. Biochem. 102, 1130-1139. 10.1002/jcb.21553 [DOI] [PubMed] [Google Scholar]
- Baraban, S. C., Dinday, M. T. and Hortopan, G. A. (2013). Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 4, 2410. 10.1038/ncomms3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baydyuk, M., Morrison, V. E., Gross, P. S. and Huang, J. K. (2020). Extrinsic factors driving oligodendrocyte lineage cell progression in CNS development and injury. Neurochem. Res. 45, 630-642. 10.1007/s11064-020-02967-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, F. C., Bennett, M. L., Yaqoob, F., Mulinyawe, S. B., Grant, G. A., Hayden Gephart, M., Plowey, E. D. and Barres, B. A. (2018). A combination of ontogeny and CNS environment establishes microglial identity. Neuron 98, 1170-1183.e8. 10.1016/j.neuron.2018.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, R. D., Levitte, S., O'sullivan, M. P., O'leary, S. M., Cambier, C. J., Cameron, J., Takaki, K. K., Moens, C. B., Tobin, D. M., Keane, J.et al. (2016). Lysosomal disorders drive susceptibility to tuberculosis by compromising macrophage migration. Cell 165, 139-152. 10.1016/j.cell.2016.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles, D. E. and Richardson, W. D. (2015). Oligodendrocyte development and plasticity. Cold Spring Harb. Perspect. Biol. 8, a020453. 10.1101/cshperspect.a020453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergner, C. G., van der Meer, F., Winkler, A., Wrzos, C., Turkmen, M., Valizada, E., Fitzner, D., Hametner, S., Hartmann, C., Pfeifenbring, S.et al. (2019). Microglia damage precedes major myelin breakdown in X-linked adrenoleukodystrophy and metachromatic leukodystrophy. Glia 67, 1196-1209. 10.1002/glia.23598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezman, L., Moser, A. B., Raymond, G. V., Rinaldo, P., Watkins, P. A., Smith, K. D., Kass, N. E. and Moser, H. W. (2001). Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann. Neurol. 49, 512-517. 10.1002/ana.101 [DOI] [PubMed] [Google Scholar]
- Bi, F., Huang, C., Tong, J., Qiu, G., Huang, B., Wu, Q., Li, F., Xu, Z., Bowser, R., Xia, X.-G.et al. (2013). Reactive astrocytes secrete lcn2 to promote neuron death. Proc. Natl. Acad. Sci. USA 110, 4069-4074. 10.1073/pnas.1218497110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchin, M. M., Capella, H. M., Chaves, D. L., Steindel, M., Grisard, E. C., Ganev, G. G., da Silva, J. P., Jr, Neto Evaldo, S., Poffo, M. A., Walz, R.et al. (2004). Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy--PLOSL): a dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell. Mol. Neurobiol. 24, 1-24. 10.1023/B:CEMN.0000012721.08168.ee [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto, G., Kamen, Y., Evans, K. A. and Káradóttir, R. T. (2020). Unraveling Myelin Plasticity. Front Cell Neurosci 14, 156. 10.3389/fncel.2020.00156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkowsky, J. L., Nelson, C., Kingston, J. L., Filloux, F. M., Mundorff, M. B. and Srivastava, R. (2010). The burden of inherited leukodystrophies in children. Neurology 75, 718-725. 10.1212/WNL.0b013e3181eee46b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher, A. A., Miller, W., Shanley, R., Ziegler, R., Lund, T., Raymond, G. and Orchard, P. J. (2015). Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: the largest single-institution cohort report. Orphanet J. Rare Dis. 10, 94. 10.1186/s13023-015-0313-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brionne, T. C., Tesseur, I., Masliah, E. and Wyss-Coray, T. (2003). Loss of TGF-β1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron 40, 1133-1145. 10.1016/S0896-6273(03)00766-9 [DOI] [PubMed] [Google Scholar]
- Caetano-Lopes, J., Henke, K., Urso, K., Duryea, J., Charles, J. F., Warman, M. L. and Harris, M. P. (2020). Unique and non-redundant function of csf1r paralogues in regulation and evolution of post-embryonic development of the zebrafish. Development 147, dev192211. 10.1242/dev.192211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo, N., Goudriaan, A., van Deijk, A.-L. F., Otte, W. M., Brouwers, J. F., Lodder, H., Gutmann, D. H., Nave, K.-A., Dijkhuizen, R. M., Mansvelder, H. D.et al. (2017). Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Biol. 15, e1002605. 10.1371/journal.pbio.1002605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantoni, C., Bollman, B., Licastro, D., Xie, M., Mikesell, R., Schmidt, R., Yuede, C. M., Galimberti, D., Olivecrona, G., Klein, R. S.et al. (2015). TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 129, 429-447. 10.1007/s00401-015-1388-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri, L., Fitzner, D., Bosch-Queralt, M., Weil, M.-T., Su, M., Sen, P., Ruhwedel, T., Mitkovski, M., Trendelenburg, G., Lütjohann, D.et al. (2018). Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 359, 684-688. 10.1126/science.aan4183 [DOI] [PubMed] [Google Scholar]
- Cartier, N. and Aubourg, P. (2010). Hematopoietic stem cell transplantation and hematopoietic stem cell gene therapy in X-linked adrenoleukodystrophy. Brain Pathol. 20, 857-862. 10.1111/j.1750-3639.2010.00394.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casali, B. T., Macpherson, K. P., Reed-Geaghan, E. G. and Landreth, G. E. (2020). Microglia depletion rapidly and reversibly alters amyloid pathology by modification of plaque compaction and morphologies. Neurobiol. Dis. 142, 104956. 10.1016/j.nbd.2020.104956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J., Poskanzer, K. E., Freeman, M. R. and Monk, K. R. (2020). Live-imaging of astrocyte morphogenesis and function in zebrafish neural circuits. Nat. Neurosci. 23, 1297-1306. 10.1038/s41593-020-0703-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cigliola, V., Becker, C. J. and Poss, K. D. (2020). Building bridges, not walls: spinal cord regeneration in zebrafish. Dis. Model. Mech. 13, dmm044131. 10.1242/dmm.044131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cignarella, F., Filipello, F., Bollman, B., Cantoni, C., Locca, A., Mikesell, R., Manis, M., Ibrahim, A., Deng, L., Benitez, B. A.et al. (2020). TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 140, 513-534. 10.1007/s00401-020-02193-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserép, C., Pósfai, B., Lénárt, N., Fekete, R., László, Z. I., Lele, Z., Orsolits, B., Molnár, G., Heindl, S., Schwarcz, A. D.et al. (2020). Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 367, 528-537. 10.1126/science.aax6752 [DOI] [PubMed] [Google Scholar]
- Dai, X.-M., Ryan, G. R., Hapel, A. J., Dominguez, M. G., Russell, R. G., Kapp, S., Sylvestre, V. and Stanley, E. R. (2002). Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 99, 111-120. 10.1182/blood.V99.1.111 [DOI] [PubMed] [Google Scholar]
- Damisah, E. C., Hill, R. A., Rai, A., Chen, F., Rothlin, C. V., Ghosh, S. and Grutzendler, J. (2020). Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci. Adv. 6, eaba3239. 10.1126/sciadv.aba3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., Littman, D. R., Dustin, M. L. and Gan, W.-B. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752-758. 10.1038/nn1472 [DOI] [PubMed] [Google Scholar]
- De, S., van Deren, D., Peden, E., Hockin, M., Boulet, A., Titen, S. and Capecchi, M. R. (2018). Two distinct ontogenies confer heterogeneity to mouse brain microglia. Development 145, dev152306. 10.1242/dev.152306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waard, D. M. and Bugiani, M. (2020). Astrocyte-Oligodendrocyte-Microglia Crosstalk in Astrocytopathies. Front Cell Neurosci 14, 608073. 10.3389/fncel.2020.608073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska, A., Weiner, A. and Amit, I. (2020). The physiology, pathology, and potential therapeutic applications of the TREM2 signaling pathway. Cell 181, 1207-1217. 10.1016/j.cell.2020.05.003 [DOI] [PubMed] [Google Scholar]
- del Bigio, M. R. (2018). Autopsy neuropathology findings in a child with chronic infantile krabbe leukoencephalopathy. Pediatr. Neurol. 82, 51-52. 10.1016/j.pediatrneurol.2018.03.002 [DOI] [PubMed] [Google Scholar]
- del Rio-Hortega, P. (1932). Microglia. In Cytology and Cellular Pathology of the Nervous System, Vol. 2 (ed. Hoeber W.), pp. 483-534. New York: Penfield. [Google Scholar]
- Diniz, L. P., Tortelli, V., Matias, I., Morgado, J., Bérgamo Araujo, A. P., Melo, H. M., Seixas da Silva, G. S., Alves-Leon, S. V., de Souza, J. M., Ferreira, S. T.et al. (2017). Astrocyte transforming growth factor Beta 1 protects synapses against Aβ oligomers in Alzheimer's disease model. J. Neurosci. 37, 6797-6809. 10.1523/JNEUROSCI.3351-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djannatian, M., Timmler, S., Arends, M., Luckner, M., Weil, M.-T., Alexopoulos, I., Snaidero, N., Schmid, B., Misgeld, T., Möbius, W.et al. (2019). Two adhesive systems cooperatively regulate axon ensheathment and myelin growth in the CNS. Nat. Commun. 10, 4794. 10.1038/s41467-019-12789-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djannatian, M., Weikert, U., Safaiyan, S., Wrede, C., Deichsel, C., Kislinger, G., Ruhwedel, T., Campbell, D. S., VAN Ham, T., Schmid, B.et al. (2021). Myelin biogenesis is associated with pathological ultrastructure that is resolved by microglia during development. bioRxive 2021.02.02.429485. [DOI] [PMC free article] [PubMed]
- Dong, X., Tan, N. B., Howell, K. B., Barresi, S., Freeman, J. L., Vecchio, D., Piccione, M., Radio, F. C., Calame, D., Zong, S.et al. (2020). Bi-allelic LoF NRROS variants impairing active TGF-β1 delivery cause a severe infantile-onset neurodegenerative condition with intracranial calcification. Am. J. Hum. Genet. 106, 559-569. 10.1016/j.ajhg.2020.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan, I. D., Kondo, Y. and Zhang, S.-C. (2011). The myelin mutants as models to study myelin repair in the leukodystrophies. Neurotherapeutics 8, 607-624. 10.1007/s13311-011-0080-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt, M. (2008). The role and metabolism of sulfatide in the nervous system. Mol. Neurobiol. 37, 93-103. 10.1007/s12035-008-8022-3 [DOI] [PubMed] [Google Scholar]
- Eichler, F. S., Li, J., Guo, Y., Caruso, P. A., Bjonnes, A. C., Pan, J., Booker, J. K., Lane, J. M., Tare, A., Vlasac, I.et al. (2016). CSF1R mosaicism in a family with hereditary diffuse leukoencephalopathy with spheroids. Brain 139, 1666-1672. 10.1093/brain/aww066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erblich, B., Zhu, L., Etgen, A. M., Dobrenis, K. and Pollard, J. W. (2011). Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS ONE 6, e26317. 10.1371/journal.pone.0026317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin, C., Galea, E., Lakatos, A., O'callaghan, J. P., Petzold, G. C., Serrano-Pozo, A., Steinhäuser, C., Volterra, A., Carmignoto, G., Agarwal, A.et al. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24, 312-325. 10.1038/s41593-020-00783-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner, J. R., Herrmann, J. E., Woo, M. J., Tansey, K. E., Doan, N. B. and Sofroniew, M. V. (2004). Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 24, 2143-2155. 10.1523/JNEUROSCI.3547-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero, G., Mahony, C. B., Dupuis, E., Yvernogeau, L., DI Ruggiero, E., Miserocchi, M., Caron, M., Robin, C., Traver, D., Bertrand, J. Y.et al. (2018). Embryonic microglia derive from primitive macrophages and are replaced by cmyb-dependent definitive microglia in zebrafish. Cell Rep. 24, 130-141. 10.1016/j.celrep.2018.05.066 [DOI] [PubMed] [Google Scholar]
- Ferrero, G., Miserocchi, M., Di Ruggiero, E. and Wittamer, V. (2020). A csf1rb mutation uncouples two waves of microglia development in zebrafish. Development 148, dev194241. 10.1242/dev.194241 [DOI] [PubMed] [Google Scholar]
- Filipello, F., Morini, R., Corradini, I., Zerbi, V., Canzi, A., Michalski, B., Erreni, M., Markicevic, M., Starvaggi-Cucuzza, C., Otero, K.et al. (2018). The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 48, 979-991.e8. 10.1016/j.immuni.2018.04.016 [DOI] [PubMed] [Google Scholar]
- Garcia, L. M., Hacker, J. L., Sase, S., Adang, L. and Almad, A. (2020). Glial cells in the driver seat of leukodystrophy pathogenesis. Neurobiol. Dis. 146, 105087. 10.1016/j.nbd.2020.105087 [DOI] [PubMed] [Google Scholar]
- Garcia-Reitboeck, P., Phillips, A., Piers, T. M., Villegas-Llerena, C., Butler, M., Mallach, A., Rodrigues, C., Arber, C. E., Heslegrave, A., Zetterberg, H.et al. (2018). Human induced pluripotent stem cell-derived microglia-like cells harboring TREM2 missense mutations show specific deficits in phagocytosis. Cell Rep. 24, 2300-2311. 10.1016/j.celrep.2018.07.094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand, J. M., Greenfield, A. L., Barkovich, M., Mendelsohn, B. A., VAN Haren, K., Hess, C. P. and Mannis, G. N. (2020). Allogeneic HSCT for adult-onset leukoencephalopathy with spheroids and pigmented glia. Brain 143, 503-511. 10.1093/brain/awz390 [DOI] [PubMed] [Google Scholar]
- Gibson, E. M., Purger, D., Mount, C. W., Goldstein, A. K., Lin, G. L., Wood, L. S., Inema, I., Miller, S. E., Bieri, G., Zuchero, J. B.et al. (2014). Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344, 1252304. 10.1126/science.1252304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., Mehler, M. F., Conway, S. J., Ng, L. G., Stanley, E. R.et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841-845. 10.1126/science.1194637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann, T., Zeller, N., Raasch, J., Kierdorf, K., Frenzel, K., Ketscher, L., Basters, A., Staszewski, O., Brendecke, S. M., Spiess, A.et al. (2015). USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J. 34, 1612-1629. 10.15252/embj.201490791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes, F. C. A., Sousa, V. O. and Romão, L. (2005). Emerging roles for TGF-β1 in nervous system development. Int. J. Dev. Neurosci. 23, 413-424. 10.1016/j.ijdevneu.2005.04.001 [DOI] [PubMed] [Google Scholar]
- Gordon, S. and Martinez-Pomares, L. (2017). Physiological roles of macrophages. Pflugers Arch. 469, 365-374. 10.1007/s00424-017-1945-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber, M. B. and Stre'rt, W. J. (1990). Microglia: immune network in the CNS. Brain Pathol. 1, 2-5. 10.1111/j.1750-3639.1990.tb00630.x [DOI] [PubMed] [Google Scholar]
- Guilliams, M., Thierry, G. R., Bonnardel, J. and Bajenoff, M. (2020). Establishment and Maintenance of the Macrophage Niche. Immunity 52, 434-451. 10.1016/j.immuni.2020.02.015 [DOI] [PubMed] [Google Scholar]
- Guo, L., Bertola, D. R., Takanohashi, A., Saito, A., Segawa, Y., Yokota, T., Ishibashi, S., Nishida, Y., Yamamoto, G. L., Franco, J. F. S.et al. (2019). Bi-allelic CSF1R mutations cause skeletal dysplasia of dysosteosclerosis-Pyle disease spectrum and degenerative encephalopathy with brain malformation. Am. J. Hum. Genet. 104, 925-935. 10.1016/j.ajhg.2019.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann, T. L., Boelens, W. C., Wawrousek, E. F. and Messing, A. (2009). Suppression of GFAP toxicity by αB-crystallin in mouse models of Alexander disease. Hum. Mol. Genet. 18, 1190-1199. 10.1093/hmg/ddp013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeyer, N., Hanft, K.-M., Akriditou, M., Unger, N., Park, E. S., Stanley, E. R., Staszewski, O., Dimou, L. and Prinz, M. (2017). Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 134, 441-458. 10.1007/s00401-017-1747-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakola, H. P. (1972). Neuropsychiatric and genetic aspects of a new hereditary disease characterized by progressive dementia and lipomembranous polycystic osteodysplasia. Acta Psychiatr. Scand. Suppl. 232, 1-173. [PubMed] [Google Scholar]
- Hamilton, N., Rutherford, H. A., Petts, J. J., Isles, H. M., Weber, T., Henneke, M., Gärtner, J., Dunning, M. J. and Renshaw, S. A. (2020). The failure of microglia to digest developmental apoptotic cells contributes to the pathology of RNASET2-deficient leukoencephalopathy. Glia 68, 1531-1545. 10.1002/glia.23829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, D. V., Hanson, J. E. and Sheng, M. (2018). Microglia in Alzheimer's disease. J. Cell Biol. 217, 459-472. 10.1083/jcb.201709069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haud, N., Kara, F., Diekmann, S., Henneke, M., Willer, J. R., Hillwig, M. S., Gregg, R. G., Macintosh, G. C., Gartner, J., Alia, A.et al. (2011). rnaset2 mutant zebrafish model familial cystic leukoencephalopathy and reveal a role for RNase T2 in degrading ribosomal RNA. Proc. Natl. Acad. Sci. USA 108, 1099-1103. 10.1073/pnas.1009811107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon, P. G. and Carmignoto, G. (2006). Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 86, 1009-1031. 10.1152/physrev.00049.2005 [DOI] [PubMed] [Google Scholar]
- Herbomel, P., Thisse, B. and Thisse, C. (1999). Ontogeny and behaviour of early macrophages in the zebrafish embryo. Development 126, 3735-3745. 10.1242/dev.126.17.3735 [DOI] [PubMed] [Google Scholar]
- Herbomel, P., Thisse, B. and Thisse, C. (2001). Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process. Dev. Biol. 238, 274-288. 10.1006/dbio.2001.0393 [DOI] [PubMed] [Google Scholar]
- Honke, N., Shaabani, N., Zhang, D.-E., Hardt, C. and Lang, K. S. (2016). Multiple functions of USP18. Cell Death Dis. 7, e2444. 10.1038/cddis.2016.326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe, K., Clark, M. D., Torroja, C. F., Torrance, J., Berthelot, C., Muffato, M., Collins, J. E., Humphray, S., Mclaren, K., Matthews, L., et al. (2013). The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498-503. 10.1038/nature12111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, C. L., Koike, M., Spusta, S. C., Niemi, E. C., Yenari, M., Nakamura, M. C. and Seaman, W. E. (2009). A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 109, 1144-1156. 10.1111/j.1471-4159.2009.06042.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, A. N. and Appel, B. (2019). Oligodendrocytes express synaptic proteins that modulate myelin sheath formation. Nat. Commun. 10, 4125. 10.1038/s41467-019-12059-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, A. N. and Appel, B. (2020). Microglia phagocytose myelin sheaths to modify developmental myelination. Nat. Neurosci. 23, 1055-1066. 10.1038/s41593-020-0654-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume, D. A., Caruso, M., Ferrari-Cestari, M., Summers, K. M., Pridans, C. and Irvine, K. M. (2020). Phenotypic impacts of CSF1R deficiencies in humans and model organisms. J. Leukoc. Biol. 107, 205-219. 10.1002/JLB.MR0519-143R [DOI] [PubMed] [Google Scholar]
- Itoh, M., Hayashi, M., Fujioka, Y., Nagashima, K., Morimatsu, Y. and Matsuyama, H. (2002). Immunohistological study of globoid cell leukodystrophy. Brain Dev. 24, 284-290. 10.1016/S0387-7604(02)00057-8 [DOI] [PubMed] [Google Scholar]
- Jahn, O., Tenzer, S. and Werner, H. B. (2009). Myelin proteomics: molecular anatomy of an insulating sheath. Mol. Neurobiol. 40, 55-72. 10.1007/s12035-009-8071-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin, D. A., Adlung, L., Thaiss, C. A., Weiner, A., Li, B., Descamps, H., Lundgren, P., Bleriot, C., Liu, Z., Deczkowska, A.et al. (2019). Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell 178, 686-698.e14. 10.1016/j.cell.2019.05.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janova, H., Arinrad, S., Balmuth, E., Mitjans, M., Hertel, J., Habes, M., Bittner, R. A., Pan, H., Goebbels, S., Begemann, M.et al. (2018). Microglia ablation alleviates myelin-associated catatonic signs in mice. J. Clin. Invest. 128, 734-745. 10.1172/JCI97032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, C., Washimi, Y., Yoshida, K., Hashizume, Y. and Yazawa, I. (2015). Characterization of spheroids in hereditary diffuse leukoencephalopathy with axonal spheroids. J. Neurol. Sci. 352, 74-78. 10.1016/j.jns.2015.03.033 [DOI] [PubMed] [Google Scholar]
- Kaifu, T., Nakahara, J., Inui, M., Mishima, K., Momiyama, T., Kaji, M., Sugahara, A., Koito, H., Ujike-Asai, A., Nakamura, A.et al. (2003). Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J. Clin. Invest. 111, 323-332. 10.1172/JCI16923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe, M. D., Soderholm, H. E., Shih, H.-Y., Stevenson, T. J., Glaittli, K. A., Bowles, D. M., Scholl, E., Colby, S., Merchant, S., Hsu, E. W.et al. (2020). Vanishing white matter disease expression of truncated EIF2B5 activates induced stress response. eLife 9, e56319. 10.7554/eLife.56319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp, S., Pujol, A., Waterham, H. R., van Geel, B. M., Boehm, C. D., Raymond, G. V., Cutting, G. R., Wanders, R. J. A. and Moser, H. W. (2001). ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: role in diagnosis and clinical correlations. Hum. Mutat. 18, 499-515. 10.1002/humu.1227 [DOI] [PubMed] [Google Scholar]
- Kempthorne, L., Yoon, H., Madore, C., Smith, S., Wszolek, Z. K., Rademakers, R., Kim, J., Butovsky, O. and Dickson, D. W. (2020). Loss of homeostatic microglial phenotype in CSF1R-related Leukoencephalopathy. Acta Neuropathol. Commun. 8, 72. 10.1186/s40478-020-00947-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshvari, S., Caruso, M., Teakle, N., Batoon, L., Sehgal, A., Patkar, O. L., Ferrari-Cestari, M., Snell, C. E., Chen, C., Stevenson, A.et al. (2021). CSF1R-dependent macrophages control postnatal somatic growth and organ maturation. PLoS Genet. 17, 6. 10.1371/journal.pgen.1009605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg, H. K. and Nedergaard, M. (2010). Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics 7, 338-353. 10.1016/j.nurt.2010.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger, G., Yamanishi, Y., Suarez-Calvet, M., Czirr, E., Lohmann, E., Cuyvers, E., Struyfs, H., Pettkus, N., Wenninger-Weinzierl, A., Mazaheri, F.et al. (2014). TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6, 243ra86. 10.1126/scitranslmed.3009093 [DOI] [PubMed] [Google Scholar]
- Knowles, J. K., Soane, C., Frost, E., Tam, L. T., Fraga, D., Xu, H., Batra, A., Ni, L., Villar, K., Saucedo, T.et al. (2020). Maladaptive myelination promotes epileptogenesis in absence epilepsy. bioRxive 2020.08.20.260083. [DOI] [PMC free article] [PubMed]
- Konishi, H. and Kiyama, H. (2018). Microglial TREM2/DAP12 signaling: a double-edged sword in neural diseases. Front. Cell Neurosci. 12, 206. 10.3389/fncel.2018.00206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi, H., Okamoto, T., Hara, Y., Komine, O., Tamada, H., Maeda, M., Osako, F., Kobayashi, M., Nishiyama, A., Kataoka, Y.et al. (2020). Astrocytic phagocytosis is a compensatory mechanism for microglial dysfunction. EMBO J. 39, e104464. 10.15252/embj.2020104464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno, T., Yoshida, K., Mizuno, T., Kawarai, T., Tada, M., Nozaki, H., Ikeda, S.-I., Nishizawa, M., Onodera, O., Wszolek, Z. K.et al. (2017). Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur. J. Neurol. 24, 37-45. 10.1111/ene.13125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno, T., Kasanuki, K., Ikeuchi, T., Dickson, D. W. and Wszolek, Z. K. (2018). CSF1R-related leukoencephalopathy: a major player in primary microgliopathies. Neurology 91, 1092-1104. 10.1212/WNL.0000000000006642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlarz, D., Marquardt, B., Barøy, T., Lee, W. S., Konnikova, L., Hollizeck, S., Magg, T., Lehle, A. S., Walz, C., Borggraefe, I.et al. (2018). Human TGF-β1 deficiency causes severe inflammatory bowel disease and encephalopathy. Nat. Genet. 50, 344-348. 10.1038/s41588-018-0063-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krägeloh-Mann, I., Groeschel, S., Kehrer, C., Opherk, K., Nägele, T., Handgretinger, R. and Müller, I. (2013). Juvenile metachromatic leukodystrophy 10 years post transplant compared with a non-transplanted cohort. Bone Marrow. Transplant. 48, 369-375. 10.1038/bmt.2012.155 [DOI] [PubMed] [Google Scholar]
- Kuil, L. E., López Martí, A., Carreras Mascaro, A., van den Bosch, J. C., van den Berg, P., van der Linde, H. C., Schoonderwoerd, K., Ruijter, G. J. G. and VAN Ham, T. J. (2019a). Hexb enzyme deficiency leads to lysosomal abnormalities in radial glia and microglia in zebrafish brain development. Glia 67, 1705-1718. 10.1002/glia.23641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuil, L. E., Oosterhof, N., Geurts, S. N., van der Linde, H. C., Meijering, E. and van Ham, T. J. (2019b). Reverse genetic screen reveals that Il34 facilitates yolk sac macrophage distribution and seeding of the brain. Dis. Model. Mech. 12, dmm037762. 10.1242/dmm.037762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuil, L. E., Oosterhof, N., Ferrero, G., Mikulášová, T., Hason, M., Dekker, J., Rovira, M., van der Linde, H. C., van Strien, P. M. H., de Pater, E.et al. (2020). Zebrafish macrophage developmental arrest underlies depletion of microglia and reveals Csf1r-independent metaphocytes. eLife 9, e53403. 10.7554/eLife.53403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampron, A., Larochelle, A., Laflamme, N., Préfontaine, P., Plante, M.-M., Sánchez, M. G., Yong, V. W., Stys, P. K., Tremblay, M.-E. and Rivest, S. (2015). Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 212, 481-495. 10.1084/jem.20141656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciotti, A., Brignone, M. S., Bertini, E., Petrucci, T. C., Aloisi, F. and Ambrosini, E. (2013). Astrocytes: emerging stars in leukodystrophy pathogenesis. Transl. Neurosci. 4, 144-164. 10.2478/s13380-013-0118-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. and Barres, B. A. (2018). Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225-242. 10.1038/nri.2017.125 [DOI] [PubMed] [Google Scholar]
- Li, Y., Du, X.-F., Liu, C.-S., Wen, Z.-L. and Du, J.-L. (2012). Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev. Cell 23, 1189-1202. 10.1016/j.devcel.2012.10.027 [DOI] [PubMed] [Google Scholar]
- Liddelow, S. A. and Barres, B. A. (2017). Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957-967. 10.1016/j.immuni.2017.06.006 [DOI] [PubMed] [Google Scholar]
- Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., Bennett, M. L., Münch, A. E., Chung, W.-S., Peterson, T. C.et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481-487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, W. L., Wszolek, Z. K. and Dickson, D. W. (2010). Hereditary diffuse leukoencephalopathy with spheroids: ultrastructural and immunoelectron microscopic studies. Int. J. Clin. Exp. Pathol. 3, 665-674. [PMC free article] [PubMed] [Google Scholar]
- Ling, E.-A. and Wong, W.-C. (1993). The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia 7, 9-18. 10.1002/glia.440070105 [DOI] [PubMed] [Google Scholar]
- Linnartz-Gerlach, B., Bodea, L. G., Klaus, C., Ginolhac, A., Halder, R., Sinkkonen, L., Walter, J., Colonna, M. and Neumann, H. (2019). TREM2 triggers microglial density and age-related neuronal loss. Glia 67, 539-550. 10.1002/glia.23563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd, A. F., Davies, C. L., Holloway, R. K., Labrak, Y., Ireland, G., Carradori, D., Dillenburg, A., Borger, E., Soong, D., Richardson, J. C.et al. (2019). Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat. Neurosci. 22, 1046-1052. 10.1038/s41593-019-0418-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgaard, I., Osório, M. J., Kress, B. T., Sanggaard, S. and Nedergaard, M. (2014). White matter astrocytes in health and disease. Neuroscience 276, 161-173. 10.1016/j.neuroscience.2013.10.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons, D. A. and Talbot, W. S. (2014). Glial cell development and function in zebrafish. Cold Spring Harb. Perspect. Biol. 7, a020586. 10.1101/cshperspect.a020586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae, C. A. and Peterson, R. T. (2015). Zebrafish as tools for drug discovery. Nat. Rev. Drug Discov. 14, 721-731. 10.1038/nrd4627 [DOI] [PubMed] [Google Scholar]
- Mahmoud, S., Gharagozloo, M., Simard, C. and Gris, D. (2019). Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells 8, 184. 10.3390/cells8020184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marisca, R., Hoche, T., Agirre, E., Hoodless, L. J., Barkey, W., Auer, F., Castelo-Branco, G. and Czopka, T. (2020). Functionally distinct subgroups of oligodendrocyte precursor cells integrate neural activity and execute myelin formation. Nat. Neurosci. 23, 363-374. 10.1038/s41593-019-0581-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotti, J. D., Tobias, S., Fratkin, J. D., Powers, J. M. and Rhodes, C. H. (2004). Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: report of a family, historical perspective, and review of the literature. Acta Neuropathol. 107, 481-488. 10.1007/s00401-004-0847-x [DOI] [PubMed] [Google Scholar]
- Mattiace, L. A., Davies, P., Yen, S.-H. and Dickson, D. W. (1990). Microglia in cerebellar plaques in Alzheimer's disease. Acta Neuropathol. 80, 493-498. 10.1007/BF00294609 [DOI] [PubMed] [Google Scholar]
- Mcquade, A., Kang, Y. J., Hasselmann, J., Jairaman, A., Sotelo, A., Coburn, M., Shabestari, S. K., Chadarevian, J. P., Fote, G., Tu, C. H.et al. (2020). Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer's disease. Nat. Commun. 11, 5370. 10.1038/s41467-020-19227-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensch, S., Baraban, M., Almeida, R., Czopka, T., Ausborn, J., EL Manira, A. and Lyons, D. A. (2015). Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat. Neurosci. 18, 628-630. 10.1038/nn.3991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, M. E. C., Schot, R., Buta, S., Oudesluijs, G., Tinschert, S., Speer, S. D., Li, Z., van Unen, L., Heijsman, D., Goldmann, T.et al. (2016). Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 213, 1163-1174. 10.1084/jem.20151529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner, A., Schmidt, H., Nitsche, M., Merkler, D., Hanisch, U.-K., Mack, M., Heikenwalder, M., Brück, W., Priller, J. and Prinz, M. (2007). Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 10, 1544-1553. 10.1038/nn2015 [DOI] [PubMed] [Google Scholar]
- Miron, V. E. (2017). Microglia-driven regulation of oligodendrocyte lineage cells, myelination, and remyelination. J. Leukoc. Biol. 101, 1103-1108. 10.1189/jlb.3RI1116-494R [DOI] [PubMed] [Google Scholar]
- Miron, V. E., Boyd, A., Zhao, J.-W., Yuen, T. J., Ruckh, J. M., Shadrach, J. L., van Wijngaarden, P., Wagers, A. J., Williams, A., Franklin, R. J. M.et al. (2013). M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 16, 1211-1218. 10.1038/nn.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochel, F., Delorme, C., Czernecki, V., Froger, J., Cormier, F., Ellie, E., Fegueux, N., Lehericy, S., Lumbroso, S., Schiffmann, R.et al. (2019). Haematopoietic stem cell transplantation in CSF1R-related adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. J. Neurol. Neurosurg. Psychiatry 90, 1375-1376. 10.1136/jnnp-2019-320701 [DOI] [PubMed] [Google Scholar]
- Monies, D., Maddirevula, S., Kurdi, W., Alanazy, M. H., Alkhalidi, H., Al-Owain, M., Sulaiman, R. A., Faqeih, E., Goljan, E., Ibrahim, N.et al. (2017). Autozygosity reveals recessive mutations and novel mechanisms in dominant genes: implications in variant interpretation. Genet. Med. 19, 1144-1150. 10.1038/gim.2017.22 [DOI] [PubMed] [Google Scholar]
- Morizawa, Y. M., Hirayama, Y., Ohno, N., Shibata, S., Shigetomi, E., Sui, Y., Nabekura, J., Sato, K., Okajima, F., Takebayashi, H.et al. (2017). Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 8, 28. 10.1038/s41467-017-00037-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser, H. W., Mahmood, A. and Raymond, G. V. (2007). X-linked adrenoleukodystrophy. Nat. Clin. Pract. Neurol. 3, 140-151. 10.1038/ncpneuro0421 [DOI] [PubMed] [Google Scholar]
- Moser, A. B., Jones, R. O., Hubbard, W. C., Tortorelli, S., Orsini, J. J., Caggana, M., Vogel, B. H. and Raymond, G. V. (2016). Newborn screening for X-Linked Adrenoleukodystrophy. Int J Neonatal Screen 2, 15. 10.3390/ijns2040015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount, C. W. and Monje, M. (2017). Wrapped to adapt: experience-dependent myelination. Neuron 95, 743-756. 10.1016/j.neuron.2017.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu, Y., Bennett, D. V., Rubinov, M., Narayan, S., Yang, C.-T., Tanimoto, M., Mensh, B. D., Looger, L. L. and Ahrens, M. B. (2019). Glia accumulate evidence that actions are futile and suppress unsuccessful behavior. Cell 178, 27-43.e19. 10.1016/j.cell.2019.05.050 [DOI] [PubMed] [Google Scholar]
- Musolino, P. L., Lund, T. C., Pan, J., Escolar, M. L., Paker, A. M., Duncan, C. N. and Eichler, F. S. (2014). Hematopoietic stem cell transplantation in the leukodystrophies: a systematic review of the literature. Neuropediatrics 45, 169-174. 10.1055/s-0033-1364179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myer, D. J., Gurkoff, G. G., Lee, S. M., Hovda, D. A. and Sofroniew, M. V. (2006). Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 129, 2761-2772. 10.1093/brain/awl165 [DOI] [PubMed] [Google Scholar]
- Nasu, T., Tsukahara, Y. and Terayama, K. (1973). A lipid metabolic disease- “membranous lipodystrophy”-an autopsy case demonstrating numerous peculiar membrane-structures composed of compound lipid in bone and bone marrow and various adipose tissues. Acta Pathol. Jpn. 23, 539-558. 10.1111/j.1440-1827.1973.tb01223.x [DOI] [PubMed] [Google Scholar]
- Nataf, S., Anginot, A., Vuaillat, C., Malaval, L., Fodil, N., Chereul, E., Langlois, J.-B., Dumontel, C., Cavillon, G., Confavreux, C.et al. (2005). Brain and bone damage in KARAP/DAP12 loss-of-function mice correlate with alterations in microglia and osteoclast lineages. Am. J. Pathol. 166, 275-286. 10.1016/S0002-9440(10)62251-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann, H., Kotter, M. R. and Franklin, R. J. M. (2009). Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132, 288-295. 10.1093/brain/awn109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, P. T., Dorman, L. C., Pan, S., Vainchtein, I. D., Han, R. T., Nakao-Inoue, H., Taloma, S. E., Barron, J. J., Molofsky, A. B., Kheirbek, M. A.et al. (2020). Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell 182, 388-403.e15. 10.1016/j.cell.2020.05.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson, A. M., Baker, M. C., Finch, N. A., Rutherford, N. J., Wider, C., Graff-Radford, N. R., Nelson, P. T., Clark, H. B., Wszolek, Z. K., Dickson, D. W.et al. (2013). CSF1R mutations link POLD and HDLS as a single disease entity. Neurology 80, 1033-1040. 10.1212/WNL.0b013e31828726a7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn, A., Kirchhoff, F. and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314-1318. 10.1126/science.1110647 [DOI] [PubMed] [Google Scholar]
- Noubade, R., Wong, K., Ota, N., Rutz, S., Eidenschenk, C., Valdez, P. A., Ding, J., Peng, I., Sebrell, A., Caplazi, P.et al. (2014). NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature 509, 235-239. 10.1038/nature13152 [DOI] [PubMed] [Google Scholar]
- Nugent, A. A., Lin, K., van Lengerich, B., Lianoglou, S., Przybyla, L., Davis, S. S., Llapashtica, C., Wang, J., Kim, D. J., Xia, D.et al. (2020). TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105, 837-854.e9. 10.1016/j.neuron.2019.12.007 [DOI] [PubMed] [Google Scholar]
- Oksanen, M., Lehtonen, S., Jaronen, M., Goldsteins, G., Hämäläinen, R. H. and Koistinaho, J. (2019). Astrocyte alterations in neurodegenerative pathologies and their modeling in human induced pluripotent stem cell platforms. Cell. Mol. Life Sci. 76, 2739-2760. 10.1007/s00018-019-03111-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterhof, N., Holtman, I. R., Kuil, L. E., van der Linde, H. C., Boddeke, E. W. G. M., Eggen, B. J. L. and van Ham, T. J. (2017). Identification of a conserved and acute neurodegeneration-specific microglial transcriptome in the zebrafish. Glia 65, 138-149. 10.1002/glia.23083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterhof, N., Kuil, L. E., van der Linde, H. C., Burm, S. M., Berdowski, W., van Ijcken, W. F. J., van Swieten, J. C., Hol, E. M., Verheijen, M. H. G. and van Ham, T. J. (2018). Colony-Stimulating Factor 1 Receptor (CSF1R) regulates microglia density and distribution, but not microglia differentiation in vivo. Cell Rep. 24, 1203-1217 e6. 10.1016/j.celrep.2018.06.113 [DOI] [PubMed] [Google Scholar]
- Oosterhof, N., Chang, I. J., Karimiani, E. G., Kuil, L. E., Jensen, D. M., Daza, R., Young, E., Astle, L., van der Linde, H. C., Shivaram, G. M.et al. (2019). Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am. J. Hum. Genet. 104, 936-947. 10.1016/j.ajhg.2019.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthmann-Murphy, J. L., Abrams, C. K. and Scherer, S. S. (2008). Gap junctions couple astrocytes and oligodendrocytes. J. Mol. Neurosci. 35, 101-116. 10.1007/s12031-007-9027-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero, K., Turnbull, I. R., Poliani, P. L., Vermi, W., Cerutti, E., Aoshi, T., Tassi, I., Takai, T., Stanley, S. L., Miller, M.et al. (2009). Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat. Immunol. 10, 734-743. 10.1038/ni.1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyanagi, K., Kinoshita, M., Suzuki-Kouyama, E., Inoue, T., Nakahara, A., Tokiwai, M., Arai, N., Satoh, J.-I., Aoki, N., Jinnai, K.et al. (2017). Adult onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and Nasu-Hakola disease: lesion staging and dynamic changes of axons and microglial subsets. Brain Pathol. 27, 748-769. 10.1111/bpa.12443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paloneva, J., Autti, T., Raininko, R., Partanen, J., Salonen, O., Puranen, M., Hakola, P. and Haltia, M. (2001). CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology 56, 1552-1558. 10.1212/WNL.56.11.1552 [DOI] [PubMed] [Google Scholar]
- Paloneva, J., Manninen, T., Christman, G., Hovanes, K., Mandelin, J., Adolfsson, R., Bianchin, M., Bird, T., Miranda, R., Salmaggi, A.et al. (2002). Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656-662. 10.1086/342259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant, D. C., Dorboz, I., Schluter, A., Fourcade, S., Launay, N., Joya, J., Aguilera-Albesa, S., Yoldi, M. E., Casasnovas, C., Willis, M. J.et al. (2019). Loss of the sphingolipid desaturase DEGS1 causes hypomyelinating leukodystrophy. J. Clin. Invest. 129, 1240-1256. 10.1172/JCI123959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., Giustetto, M., Ferreira, T. A., Guiducci, E., Dumas, L.et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456-1458. 10.1126/science.1202529 [DOI] [PubMed] [Google Scholar]
- Park, H.-C., Mehta, A., Richardson, J. S. and Appel, B. (2002). olig2 is required for zebrafish primary motor neuron and oligodendrocyte development. Dev. Biol. 248, 356-368. 10.1006/dbio.2002.0738 [DOI] [PubMed] [Google Scholar]
- Percy, A. K., Odrezin, G. T., Knowles, P. D., Rouah, E. and Armstrong, D. D. (1994). Globoid cell leukodystrophy: comparison of neuropathology with magnetic resonance imaging. Acta Neuropathol. 88, 26-32. 10.1007/BF00294356 [DOI] [PubMed] [Google Scholar]
- Peri, F. and Nüsslein-Volhard, C. (2008). Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 133, 916-927. 10.1016/j.cell.2008.04.037 [DOI] [PubMed] [Google Scholar]
- Perry, V. H., Nicoll, J. A. R. and Holmes, C. (2010). Microglia in neurodegenerative disease. Nat. Rev. Neurol. 6, 193-201. 10.1038/nrneurol.2010.17 [DOI] [PubMed] [Google Scholar]
- Philips, T. and Rothstein, J. D. (2017). Oligodendroglia: metabolic supporters of neurons. J. Clin. Invest. 127, 3271-3280. 10.1172/JCI90610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt, F. M., D'azzo, A., Davidson, B. L., Neufeld, E. F. and Tifft, C. J. (2018). Lysosomal storage diseases. Nat. Rev. Dis. Primers 4, 27. 10.1038/s41572-018-0025-4 [DOI] [PubMed] [Google Scholar]
- Poliani, P. L., Wang, Y., Fontana, E., Robinette, M. L., Yamanishi, Y., Gilfillan, S. and Colonna, M. (2015). TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Invest. 125, 2161-2170. 10.1172/JCI77983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard, J. W. (2009). Trophic macrophages in development and disease. Nat. Rev. Immunol. 9, 259-270. 10.1038/nri2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pont-Lezica, L., Beumer, W., Colasse, S., Drexhage, H., Versnel, M. and Bessis, A. (2014). Microglia shape corpus callosum axon tract fasciculation: functional impact of prenatal inflammation. Eur. J. Neurosci. 39, 1551-1557. 10.1111/ejn.12508 [DOI] [PubMed] [Google Scholar]
- Preston, M. A. and Macklin, W. B. (2015). Zebrafish as a model to investigate CNS myelination. Glia 63, 177-193. 10.1002/glia.22755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridans, C., Sauter, K. A., Baer, K., Kissel, H. and Hume, D. A. (2013). CSF1R mutations in hereditary diffuse leukoencephalopathy with spheroids are loss of function. Sci. Rep. 3, 3013. 10.1038/srep03013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridans, C., Raper, A., Davis, G. M., Alves, J., Sauter, K. A., Lefevre, L., Regan, T., Meek, S., Sutherland, L., Thomson, A. J.et al. (2018). Pleiotropic impacts of macrophage and microglial deficiency on development in rats with targeted mutation of the Csf1r locus. J. Immunol. 201, 2683-2699. 10.4049/jimmunol.1701783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz, M. and Priller, J. (2014). Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 15, 300-312. 10.1038/nrn3722 [DOI] [PubMed] [Google Scholar]
- Qin, Y., Garrison, B. S., Ma, W., Wang, R., Jiang, A., Li, J., Mistry, M., Bronson, R. T., Santoro, D., Franco, C.et al. (2018). A milieu molecule for TGF-beta required for microglia function in the nervous system. Cell 174, 156-171.e16. 10.1016/j.cell.2018.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raas, Q., van de Beek, M.-C., Forss-Petter, S., Dijkstra, I. M. E., Deschiffart, A., Freshner, B. C., Stevenson, T. J., Jaspers, Y. R. J., Nagtzaam, L. M., Wanders, R. J. A.et al. (2021). Metabolic rerouting via SCD1 induction impacts X-linked adrenoleukodystrophy. J. Clin. Invest. 131. 10.1172/JCI142500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers, R., Baker, M., Nicholson, A. M., Rutherford, N. J., Finch, N. C., Soto-Ortolaza, A., Lash, J., Wider, C., Wojtas, A., Dejesus-Hernandez, M.et al. (2011). Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 44, 200-205. 10.1038/ng.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby, M. J., Gomez, T. M. and Puglielli, L. (2020). Glial cell-axonal growth cone interactions in neurodevelopment and regeneration. Front. Neurosci. 14, 203. 10.3389/fnins.2020.00203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo, R., Raper, A., Ozdemir, D. D., Lefevre, L., Grabert, K., Wollscheid-Lengeling, E., Bradford, B., Caruso, M., Gazova, I., Sánchez, A.et al. (2019). Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat. Commun. 10, 3215. 10.1038/s41467-019-11053-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford, H. A. and Hamilton, N. (2019). Animal models of leukodystrophy: a new perspective for the development of therapies. FEBS J. 286, 4176-4191. 10.1111/febs.15060 [DOI] [PubMed] [Google Scholar]
- Safaiyan, S., Kannaiyan, N., Snaidero, N., Brioschi, S., Biber, K., Yona, S., Edinger, A. L., Jung, S., Rossner, M. J. and Simons, M. (2016). Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 19, 995-998. 10.1038/nn.4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safaiyan, S., Besson-Girard, S., Kaya, T., Cantuti-Castelvetri, L., Liu, L., Ji, H., Schifferer, M., Gouna, G., Usifo, F., Kannaiyan, N.et al. (2021). White matter aging drives microglial diversity. Neuron 109, 1100-1117. 10.1016/j.neuron.2021.01.027 [DOI] [PubMed] [Google Scholar]
- Sanderson, L. E., Lanko, K., Alsagob, M., Almass, R., Al-Ahmadi, N., Najafi, M., Al-Muhaizea, M. A., Alzaidan, H., Aldhalaan, H., Perenthaler, E.et al. (2021). Bi-allelic variants in HOPS complex subunit VPS41 cause cerebellar ataxia and abnormal membrane trafficking. Brain 144, 769-780. 10.1093/brain/awaa459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh, J.-I., Tabunoki, H., Ishida, T., Yagishita, S., Jinnai, K., Futamura, N., Kobayashi, M., Toyoshima, I., Yoshioka, T., Enomoto, K.et al. (2011). Immunohistochemical characterization of microglia in Nasu-Hakola disease brains. Neuropathology 31, 363-375. 10.1111/j.1440-1789.2010.01174.x [DOI] [PubMed] [Google Scholar]
- Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., Ransohoff, R. M., Greenberg, M. E., Barres, B. A. and Stevens, B. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691-705. 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaumburg, H. H., Powers, J. M., Raine, C. S., Johnson, A. B., Kolodny, E. H., Kishimoto, Y., Igarashi, M. and Suzuki, K. (1976). Adrenoleukodystrophy: a clinical, pathological and biochemical study. Adv. Exp. Med. Biol. 68, 379-387. 10.1007/978-1-4684-7735-1_25 [DOI] [PubMed] [Google Scholar]
- Schmid, C. D., Sautkulis, L. N., Danielson, P. E., Cooper, J., Hasel, K. W., Hilbush, B. S., Sutcliffe, J. G. and Carson, M. J. (2002). Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 83, 1309-1320. 10.1046/j.1471-4159.2002.01243.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, S., Castelvetri, L. C. and Simons, M. (2015). Metabolism and functions of lipids in myelin. Biochim. Biophys. Acta 1851, 999-1005. 10.1016/j.bbalip.2014.12.016 [DOI] [PubMed] [Google Scholar]
- Schwabenland, M., Mossad, O., Peres, A. G., Kessler, F., Maron, F. J. M., Harsan, L.-A., Bienert, T., von Elverfeldt, D., Knobeloch, K.-P., Staszewski, O.et al. (2019). Loss of USP18 in microglia induces white matter pathology. Acta Neuropathol. Commun. 7, 106. 10.1186/s40478-019-0757-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Hewitt, N. J., Folts, C. J., Hogestyn, J. M., Piester, G., Mayer-Pröschel, M. and Noble, M. D. (2017). Heterozygote galactocerebrosidase (GALC) mutants have reduced remyelination and impaired myelin debris clearance following demyelinating injury. Hum. Mol. Genet. 26, 2825-2837. 10.1093/hmg/ddx153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, J., Ruedl, C. and Karjalainen, K. (2015). Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity 43, 382-393. 10.1016/j.immuni.2015.07.016 [DOI] [PubMed] [Google Scholar]
- Sleigh, J. N., Rossor, A. M., Fellows, A. D., Tosolini, A. P. and Schiavo, G. (2019). Axonal transport and neurological disease. Nat. Rev. Neurol. 15, 691-703. 10.1038/s41582-019-0257-2 [DOI] [PubMed] [Google Scholar]
- Smith, C., Mccoll, B. W., Patir, A., Barrington, J., Armishaw, J., Clarke, A., Eaton, J., Hobbs, V., Mansour, S., Nolan, M.et al. (2020). Biallelic mutations in NRROS cause an early onset lethal microgliopathy. Acta Neuropathol. 139, 947-951. 10.1007/s00401-020-02137-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosunov, A., Olabarria, M. and Goldman, J. E. (2018). Alexander disease: an astrocytopathy that produces a leukodystrophy. Brain Pathol. 28, 388-398. 10.1111/bpa.12601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg, E. E. and Green, K. N. (2017). Inflammation in Alzheimer's disease: Lessons learned from microglia-depletion models. Brain Behav. Immun. 61, 1-11. 10.1016/j.bbi.2016.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg, E., Severson, P. L., Hohsfield, L. A., Crapser, J., Zhang, J., Burton, E. A., Zhang, Y., Spevak, W., Lin, J., Phan, N. Y.et al. (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat. Commun. 10, 3758. 10.1038/s41467-019-11674-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squarzoni, P., Oller, G., Hoeffel, G., Pont-Lezica, L., Rostaing, P., Low, D., Bessis, A., Ginhoux, F. and Garel, S. (2014). Microglia modulate wiring of the embryonic forebrain. Cell Rep 8, 1271-1279. 10.1016/j.celrep.2014.07.042 [DOI] [PubMed] [Google Scholar]
- Stanley, E. R. and Chitu, V. (2014). CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 6, a021857. 10.1101/cshperspect.a021857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., Micheva, K. D., Mehalow, A. K., Huberman, A. D., Stafford, B.et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164-1178. 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- Strachan, L. R., Stevenson, T. J., Freshner, B., Keefe, M. D., MIRANDA Bowles, D. and Bonkowsky, J. L. (2017). A zebrafish model of X-linked adrenoleukodystrophy recapitulates key disease features and demonstrates a developmental requirement for abcd1 in oligodendrocyte patterning and myelination. Hum. Mol. Genet. 26, 3600-3614. 10.1093/hmg/ddx249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, K. (1998). Twenty five years of the “psychosine hypothesis": a personal perspective of its history and present status. Neurochem. Res. 23, 251-259. 10.1023/A:1022436928925 [DOI] [PubMed] [Google Scholar]
- Suzuki, K. and Suzuki, Y. (1970). Globoid cell leucodystrophy (Krabbe's disease): deficiency of galactocerebroside beta-galactosidase. Proc. Natl. Acad. Sci. USA 66, 302-309. 10.1073/pnas.66.2.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada, M., Konno, T., Tada, M., Tezuka, T., Miura, T., Mezaki, N., Okazaki, K.-I., Arakawa, M., Itoh, K., Yamamoto, T.et al. (2016). Characteristic microglial features in patients with hereditary diffuse leukoencephalopathy with spheroids. Ann. Neurol. 80, 554-565. 10.1002/ana.24754 [DOI] [PubMed] [Google Scholar]
- Takahashi, K., Rochford, C. D. P. and Neumann, H. (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647-657. 10.1084/jem.20041611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi, H., Mizuno, T., Zhang, G., Wang, J., Kawanokuchi, J., Kuno, R. and Suzumura, A. (2005). Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J. Biol. Chem. 280, 10444-10454. 10.1074/jbc.M413863200 [DOI] [PubMed] [Google Scholar]
- Tian, R., Wu, X., Hagemann, T. L., Sosunov, A. A., Messing, A., Mckhann, G. M. and Goldman, J. E. (2010). Alexander disease mutant glial fibrillary acidic protein compromises glutamate transport in astrocytes. J. Neuropathol. Exp. Neurol. 69, 335-345. 10.1097/NEN.0b013e3181d3cb52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titorenko, V. I. and Rachubinski, R. A. (2004). The peroxisome: orchestrating important developmental decisions from inside the cell. J. Cell Biol. 164, 641-645. 10.1083/jcb.200312081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traiffort, E., Kassoussi, A., Zahaf, A. and Laouarem, Y. (2020). Astrocytes and microglia as major players of myelin production in normal and pathological conditions. Front. Cell Neurosci. 14, 79. 10.3389/fncel.2020.00079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay, M.-E., Lowery, R. L. and Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8, e1000527. 10.1371/journal.pbio.1000527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulland, T. K. and Colonna, M. (2018). TREM2 - a key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 14, 667-675. 10.1038/s41582-018-0072-1 [DOI] [PubMed] [Google Scholar]
- van der Knaap, M. S. and Bugiani, M. (2017). Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 134, 351-382. 10.1007/s00401-017-1739-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap, M. S., Schiffmann, R., Mochel, F. and Wolf, N. I. (2019). Diagnosis, prognosis, and treatment of leukodystrophies. Lancet Neurol. 18, 962-972. 10.1016/S1474-4422(19)30143-7 [DOI] [PubMed] [Google Scholar]
- van Egmond, M. E., Pouwels, P. J. W., Boelens, J.-J., Lindemans, C. A., Barkhof, F., Steenwijk, M. D., van Hasselt, P. M., van der Knaap, M. S. and Wolf, N. I. (2013). Improvement of white matter changes on neuroimaging modalities after stem cell transplant in metachromatic leukodystrophy. JAMA Neurol 70, 779-782. 10.1001/jamaneurol.2013.629 [DOI] [PubMed] [Google Scholar]
- van Ham, T. J., Brady, C. A., Kalicharan, R. D., Oosterhof, N., Kuipers, J., Veenstra-Algra, A., Sjollema, K. A., Peterson, R. T., Kampinga, H. H. and Giepmans, B. N. G. (2014). Intravital correlated microscopy reveals differential macrophage and microglial dynamics during resolution of neuroinflammation. Dis. Model. Mech. 7, 857-869. 10.1242/dmm.014886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rappard, D. F., Boelens, J. J. and Wolf, N. I. (2015). Metachromatic leukodystrophy: Disease spectrum and approaches for treatment. Best Pract. Res. Clin. Endocrinol. Metab. 29, 261-273. 10.1016/j.beem.2014.10.001 [DOI] [PubMed] [Google Scholar]
- Vanderver, A., Prust, M., Tonduti, D., Mochel, F., Hussey, H. M., Helman, G., Garbern, J., Eichler, F., Labauge, P., Aubourg, P.et al. (2015). Case definition and classification of leukodystrophies and leukoencephalopathies. Mol. Genet. Metab. 114, 494-500. 10.1016/j.ymgme.2015.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varol, C., Mildner, A. and Jung, S. (2015). Macrophages: development and tissue specialization. Annu. Rev. Immunol. 33, 643-675. 10.1146/annurev-immunol-032414-112220 [DOI] [PubMed] [Google Scholar]
- Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S. and Nabekura, J. (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974-3980. 10.1523/JNEUROSCI.4363-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L., Colodner, K. J. and Feany, M. B. (2011). Protein misfolding and oxidative stress promote glial-mediated neurodegeneration in an Alexander disease model. J. Neurosci. 31, 2868-2877. 10.1523/JNEUROSCI.3410-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, T., Schlotawa, L., Dosch, R., Hamilton, N., Kaiser, J., Schiller, S., Wenske, B., Gartner, J. and Henneke, M. (2020). Zebrafish disease model of human RNASET2-deficient cystic leukoencephalopathy displays abnormalities in early microglia. Biol Open 9, bio049239. 10.1242/bio.049239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock, N. I., Shin, D., Dhimal, N., Hong, X., Irons, E. E., Silvestri, N. J., Reed, C. B., Nguyen, D., Sampson, O., Cheng, Y.-C.et al. (2020). Macrophages Expressing GALC Improve Peripheral Krabbe Disease by a Mechanism Independent of Cross-Correction. Neuron 107, 65-81.e9. 10.1016/j.neuron.2020.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, J. M. and Lyons, D. A. (2018). Myelin dynamics throughout life: an ever-changing landscape? Front. Cell Neurosci. 12, 424. 10.3389/fncel.2018.00424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarczyk, A., Holtman, I. R., Krueger, M., Yogev, N., Bruttger, J., Khorooshi, R., Benmamar-Badel, A., De Boer-Bergsma, J. J., Martin, N. A., Karram, K.et al. (2017). A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 36, 3292-3308. 10.15252/embj.201696056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, N. I., Breur, M., Plug, B., Beerepoot, S., Westerveld, A. S. R., van Rappard, D. F., de Vries, S. I., Kole, M. H. P., Vanderver, A., van der Knaap, M. S.et al. (2020). Metachromatic leukodystrophy and transplantation: remyelination, no cross-correction. Ann. Clin. Transl. Neurol. 7, 169-180. 10.1002/acn3.50975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, K., Noubade, R., Manzanillo, P., Ota, N., Foreman, O., Hackney, J. A., Friedman, B. A., Pappu, R., Scearce-Levie, K. and Ouyang, W. (2017). Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nat. Immunol. 18, 633-641. 10.1038/ni.3743 [DOI] [PubMed] [Google Scholar]
- Wynn, T. A., Chawla, A. and Pollard, J. W. (2013). Macrophage biology in development, homeostasis and disease. Nature 496, 445-455. 10.1038/nature12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, J., Zhang, X., Fu, J., Wang, H. and Zhao, Y. (2019). USP18 overexpression protects against focal cerebral Ischemia injury in mice by suppressing microglial activation. Neuroscience 419, 121-128. 10.1016/j.neuroscience.2019.09.001 [DOI] [PubMed] [Google Scholar]
- Xing, J., Titus, A. R. and Humphrey, M. B. (2015). The TREM2-DAP12 signaling pathway in Nasu-Hakola disease: a molecular genetics perspective. Res. Rep. Biochem. 5, 89-100. 10.2147/RRBC.S58057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J., Zhu, L., He, S., Wu, Y., Jin, W., Yu, T., Qu, J. Y. and Wen, Z. (2015). Temporal-spatial resolution fate mapping reveals distinct origins for embryonic and adult microglia in zebrafish. Dev. Cell 34, 632-641. 10.1016/j.devcel.2015.08.018 [DOI] [PubMed] [Google Scholar]
- Ye, P., Li, L., Richards, R. G., Diaugustine, R. P. and D'ercole, A. J. (2002). Myelination is altered in insulin-like growth factor-I null mutant mice. J. Neurosci. 22, 6041-6051. 10.1523/JNEUROSCI.22-14-06041.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, J. J., Barnes, A. P., Hand, R., Polleux, F. and Ehlers, M. D. (2010). TGF-beta signaling specifies axons during brain development. Cell 142, 144-157. 10.1016/j.cell.2010.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim, H. Y., Park, C., Lee, Y. D., Arimoto, K.-I., Jeon, R., Baek, S. H., Zhang, D.-E., Kim, H.-H. and Kim, K. I. (2016). Elevated response to type I IFN enhances RANKL-mediated osteoclastogenesis in Usp18-knockout mice. J. Immunol. 196, 3887-3895. 10.4049/jimmunol.1501496 [DOI] [PubMed] [Google Scholar]