

Abstract
Introduction
We investigated whether insulin resistance (IR) was associated with longitudinal age‐related change in cognition and biomarkers of Alzheimer's disease (AD) pathology and neurodegeneration in middle‐aged and older adults who were non‐demented at baseline.
Methods
IR was measured with homeostatic model assessment of insulin resistance (HOMA2‐IR). Core AD‐related cerebrospinal fluid (CSF) biomarkers and cognition were assessed, respectively, on n = 212 (1 to 5 visits) and n = 1299 (1 to 6 visits). Linear mixed models tested whether HOMA2‐IR moderated age‐related change in CSF biomarkers and cognition. Linear regressions tested whether HOMA2‐IR x apolipoprotein E ε4 allele (APOE ε4) carrier status predicted amyloid beta [Aβ] chronicity (estimated duration of amyloid positron emission tomography [PET] positivity) (n = 253).
Results
Higher HOMA2‐IR was associated with greater cognitive decline but not with changes in CSF biomarkers. HOMA2‐IR x APOE4 was not related to Aβ chronicity but was significantly associated with CSF phosphorylated tau (P‐tau)181/Aβ42 level.
Discussion
In non‐demented adults IR may not be directly associated with age‐related change in AD biomarkers. Additional research is needed to determine mechanisms linking IR to cognitive decline.
Keywords: APOE4, cognition, CSF biomarkers, insulin resistance, neurodegeneration, p‐tau181, amyloid beta
1. INTRODUCTION
Insulin resistance (IR) is a condition of reduced tissue sensitivity to the action of insulin and is often accompanied by hyperinsulinemia to control glucose levels. 1 IR increases risk for type 2 diabetes (T2D) 1 and has been related to worse episodic memory and executive function, 2 , 3 cognitive domains affected by Alzheimer's disease (AD), and aging. In samples of older adults where the majority were not diabetic, IR was associated with an increased risk for Alzheimer's clinical syndrome. 4 , 5
Although potential molecular mechanisms that link IR to AD are unclear, 6 some animal studies suggest that peripheral IR facilitates dysregulation of insulin signaling in the central nervous system, which in turn promotes tau hyperphosphorylation 7 and synaptic dysfunction. 8 , 9 Peripheral IR may also be related to amyloid beta (Aβ)42 (Aβ42) aggregation and/or accumulation through islet amyloid polypeptide (IAPP). Released by the pancreas with insulin, IAPP can be increased in IR and cross the blood‐brain barrier. 10 , 11 Misfolded forms of IAPP are hypothesized to act as a seed to activate the formation of Aβ42 fibrils or slow the clearance of Aβ42 protein from the brain. 11 , 12 , 13 , 14 In addition, hyperinsulinemia may cause substrate inhibition of insulin‐degrading enzyme, one of the enzymes responsible for Aβ degradation. 15
Results from investigations relating IR to measures of AD pathology and neurodegeneration have been mixed. Although increased risk for neuritic plaques was predicted by 10‐year antemortem IR, 16 results examining the relationship between IR and Aβ detected by Pittsburgh Compound B (PiB) positron emission tomography (PET) have been inconsistent. 17 , 18 In addition, significant relationships have not been found between IR and Aβ42 measured in cerebrospinal fluid (CSF). 2 , 19 In contrast, significant associations have been found between IR and elevated CSF phosphorylated tau181 (P‐tau181) 2 and markers of neurodegeneration, specifically elevated CSF total tau (T‐tau) 2 , 19 and decreased cerebral glucose metabolism. 20 However, 10‐year ante‐mortem IR was not related to post‐mortem tau tangle pathology, 16 and IR has not been consistently related to decreased cerebral glucose metabolism. 21 Several studies have been cross‐sectional, 2 , 18 , 19 , 20 , 21 thereby limiting the ability to investigate whether IR exacerbates aging‐related changes in biomarkers of AD pathology and neurodegeneration.
We investigated if IR moderated aging‐related change in biomarkers of AD pathology and neurodegeneration in a sample of middle‐aged and older adults who were non‐demented at baseline. We examined whether age‐related declines in CSF Aβ42/Aβ40 ratio and increases in CSF P‐tau181/Aβ42 ratio, P‐tau181, and markers of neurodegeneration (T‐tau, neurogranin, and neurofilament light chain [NfL]) were worsened in individuals with higher IR. Given prior studies indicating an increased risk for dementia due to IR, we also examined whether individuals with higher IR had greater age‐related decline in cognition.
In addition, we examined apolipoprotein E ε4 allele (APOE ε4) as a moderator of the relationship between IR and age‐related change in CSF amyloid outcome variables. APOE ε4 carriers have been found to accumulate amyloid sooner and possibly at a faster rate relative to non‐carriers, 22 and prior studies suggest a potential interaction between IR and APOE ε4. 16 Higher IR was related to increased odds of neuritic plaques in APOE ε4 carriers relative to non‐carriers. 16 We also tested whether an interaction between IR and APOE ε4 was related to Aβ chronicity, a novel measure of the estimated length of time that an individual has been PiB‐PET positive. 23
2. METHOD
2.1. Participants
Participants were enrolled in the Wisconsin Registry for Alzheimer's Prevention (WRAP), a longitudinal observational study of middle‐aged and older adults who were non‐demented at baseline. 24 The cohort is enriched for parental family history of AD; thus there is a relatively high proportion of APOE ε4 carriers. Diagnosis of mild cognitive impairment and dementia was determined through National Institute on Aging–Alzheimer's Association (NIA‐AA) criteria 25 and consensus conference. 24 Recruitment procedures have been described previously. 24 The University of Wisconsin (UW)–Madison Health Sciences Institutional Review Board approved WRAP; all participants provided written informed consent prior to enrollment.
The primary predictor in our study was IR; therefore, analyses focused on WRAP participants who had glucose and insulin values available from at least one visit (n = 1384), because both measures are required for calculating homeostatic model assessment of insulin resistance (HOMA2‐IR). 26 Participants were excluded if they did not have any useable glucose and/or insulin values whether due to: (1) failure to fast and abstain from caffeine for a minimum of 8 hours (n = 11) or (2) treatment with insulin (n = 21), a confound for the measurement of HOMA2‐IR. We excluded participants who did not have APOE ε4 data (n = 49) so that APOE ε4 carriership could be included in analyses. This resulted in a sample of n = 1303 participants. Three subsamples were selected based on availability of relevant outcomes, specifically CSF biomarkers, PiB PET for calculation of Aβ chronicity, and cognitive data (see Figure S1).
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the published literature on insulin resistance (IR) and Alzheimer's disease (AD). Although IR has been related to an increased risk of Alzheimer's clinical syndrome, results from studies associating IR with AD biomarkers have been mixed and largely limited by assessing outcomes once. We investigated the association of IR with longitudinal age‐related change in cognition and biomarkers of AD and neurodegeneration in adults non‐demented at baseline. Apolipoprotein E ε4 allele (APOE ε4) carrier status was tested as a moderator of the relationship between IR and amyloid beta (Aβ) measures.
Interpretation: Our findings suggest that IR is associated with cognitive decline but not age‐associated change in core AD‐related cerebrospinal fluid (CSF) biomarkers in non‐demented adults. APOE ε4 status moderated the relationship between IR and one Aβ measure.
Future directions: Mechanisms mediating the relationship between IR and cognitive decline, prior to dementia, require elucidation. Whether APOE ε4 and IR interact to influence AD pathology warrants further study.
In the CSF biomarker subsample (n = 211), 15 (7.1%) met cut‐off criteria for CSF amyloid and P‐tau181 positivity (see Section 2.3) at baseline; that number increased to 26 (12.3%) by study end. No participant in the CSF biomarker subsample converted to dementia by last lumbar puncture (LP). In the Aβ chronicity subsample (n = 253), 58 participants (22.9%) had a positive PiB‐PET (see Section 2.2) at the time of their most recent PiB‐PET. One participant in this subsample had dementia. In the cognitive subsample (n = 1299), four participants were diagnosed with dementia subsequent to first cognitive assessment. Sample characteristics for all three subsamples can be found in Table 1. Descriptive statistics of dependent variables and longitudinal follow‐up data for CSF biomarker and cognitive subsamples can be found in Table 2.
TABLE 1.
Descriptive statistics of demographic, health characteristics, and study variables of participants
| CSF biomarker studya (n = 212)b | Amyloid beta chronicity study (n = 253) | Cognitive studya (n = 1299) | |
|---|---|---|---|
| Age (y) | 63.1 (6.6) | 67.1 (6.1) | 58.8 (6.5) |
| Sex (female) | 138 (65.1%) | 174 (68.8%) | 915 (70.4%) |
| Race (White) | 206 (97.6%) | 242 (95.7%) | 1215 (93.5%) |
| Education (y) | 16.1 (2.1) | 16.1 (2.2) | 15.8 (2.2) |
| APOE ε4 carrier | 74 (35.1%) | 101 (39.9%) | 505 (38.9%) |
| Systolic blood pressure (mm Hg) | 126.9 (16.6) | 125.3 (12.9) | 124.7 (15.9) |
| Homeostatic model assessment of insulin resistance (HOMA2‐IR) | 1.1 (0.7) |
within‐person meanc: 1.1 (0.7) within‐person SDc: 0.3 (0.3) |
1.2 (0.9) |
| Pre‐diabetesd | 53 (25.0%) | 61 (24.1%) | 324 (24.9%) |
| Diabetesd | 10 (4.7%) | 26 (10.3%) | 78 (6.0%) |
| Mild cognitive impairment (MCI)e | 6 (2.8%) | 12 (4.7%) | 17 (1.3%) |
| Cognitive impairment (not MCI)e | 0 (0.0%) | 2 (0.8%) | 12 (0.9%) |
| Pittsburgh Compound B (PiB) PET | |||
| PiB positivitye | 58 (22.9%) | ||
| PiB chronicityg (y) |
PiB pos = 9.7 (7.9) PiB neg = −18.4 (6.3) |
||
Data presented are means (SD) or counts (%)
Data represent values collected at time of baseline HOMA2‐IR.
n = 212 had CSF neurodegeneration biomarkers; n = 211 had CSF AD pathology biomarkers.
Average of within‐person mean and SD of 3‐6 HOMA2‐IR values.
Pre‐diabetes defined as fasting glucose between 100 and 125 mg/dL (American Diabetes Association [ADA], 2010); Diabetes defined as self‐report of taking oral antidiabetic medication or, if no self‐report, fasting glucose ≥ 126mg/dL (ADA, 2010).
Diagnosed using NIA‐AA criteria (McKhann et al., 2011) and consensus conference (Johnson et al., 2018).
PiB positivity: global PiB DVR >1.19 at participant's most recent PiB‐PET.
Positive values = estimated years of PiB‐PET positivity. |Negative values| = estimated years until PiB positivity or estimated life expectancy for those with no evidence of PiB accumulation.
TABLE 2.
Descriptive statistics of dependent variables for cerebrospinal fluid (CSF) biomarker and cognitive study
| CSF biomarker study (n = 212) 1 | |||
|---|---|---|---|
| 1‐5 Lumbar Punctures (LPs) collected | |||
| Number of LPs: | Participant count (%): | ||
| 1 | 94 (44.3%) | ||
| 2 | 41 (19.3%) | ||
| 3 | 45 (21.2%) | ||
| 4 | 26 (12.3%) | ||
| 5 | 6 (2.8%) | ||
| LP collection period (years) for subsample with >1 LP: | |||
| Mean = 4.8, range 1—8.2 | |||
| CSF biomarker means (SD) at baseline and last visit | |||
| Baseline | Last visit | ||
| Aβ42/Aβ40 ratio | Raw | .06 (.02) | .06 (.02) |
| Raw | .02 (.02) | .03 (.02) | |
| P‐tau181/Aβ42 ratio | Ln | −3.9 (0.5) | −3.8 (0.6) |
| Raw | 18.1 (6.8) | 19.1 (7.4) | |
| P‐tau181 (pg/mL) | Ln | 2.8 (0.4) | 2.9 (0.4) |
| Raw | 206.7 (72.3) | 215.3 (76.5) | |
| T‐tau (pg/mL) | Ln | 5.3 (0.3) | 5.3 (0.4) |
| Raw | 829.1 (330.4) | 850.5 (336.5) | |
| Neurogranin (pg/mL) | Ln | 6.6 (0.4) | 6.7 (0.4) |
| Raw | 94.2 (50.2) | 105.5 (64.1) | |
| NfL (pg/mL) | Ln | 4.5 (0.4) | 4.6 (0.4) |
| Amyloid and P‐tau statusb: count (%) at baseline and last visit | |||
| Baseline | Last visit | ||
| Amyloid+/P‐Tau+ | 15 (7.1%) | 26 (12.3%) | |
| Amyloid+/P‐Tau‐ | 27 (12.8%) | 26 (12.3%) | |
| Amyloid‐/P‐Tau+ | 11 (5.2%) | 12 (5.7%) | |
| Amyloid‐/P‐Tau‐ | 158 (74.9%) | 147 (69.7%) | |
| Cognitive study (n = 1299) | |||
| 1‐6 PACC‐3 tests | |||
| Number of PACC‐3 tests: | Participant count (%): | ||
| 1 | 112 (8.6%) | ||
| 2 | 151 (11.6%) | ||
| 3 | 270 (20.8%) | ||
| 4 | 412 (31.7%) | ||
| 5 | 351 (27.0%) | ||
| 6 | 3 (0.2%) | ||
|
PACC‐3 collection period (years) for subsample with >1 PACC‐3 tests: | |||
|
Mean = 7.1, range 1.312.3 | |||
| Cognitive performance means (SD) at baseline and last visit | |||
|---|---|---|---|
| PACC‐3 | Baseline | Last visit | |
| .006 (0.8) | −.09 (0.8) | ||
Abbreviations: Aβ: amyloid beta; NfL: neurofilament light chain; PACC‐3: Preclinical Alzheimer's Cognitive Composite (three‐test version); P‐tau: phosphorylated tau; T‐tau: total tau.
n = 212 had CSF neurodegeneration biomarkers; n = 211 had CSF AD pathology biomarkers.
Amyloid+: CSF Aβ42/Aβ40 ratio ≤.046; P‐Tau+: CSF P‐Tau181 ≥24.8 pg/mL.
2.2. Procedures
2.2.1. Blood collection and testing
Participants fasted and abstained from caffeine for a minimum of 8 hours prior to having their blood collected during a scheduled biennial WRAP visit. The majority (70.4%) of participants had blood collected and tested within the UW Hospital and Clinics at Madison. The remaining had their blood collected and tested at the Mayo Clinic Health System in LaCrosse, WI (22.4%) or at Advocate Aurora Health in Milwaukee (7.2%). WRAP visit procedures have been described previously. 24
2.2.2. CSF collection
Participants in the CSF biomarker subsample had one to five LPs (see Table 2 for count and percentage of participants with one to five LPs). Follow‐up LPs were collected on average every 2.6 years (SD = 1.3) and were available in 55.7% of the participants. The mean follow‐up period for these participants was 4.8 years. Participants fasted for 8‐12 hours before receiving an LP in the morning for CSF collection using a Sprotte 24‐ or 25‐gauge atraumatic spinal needle. Approximately 22 mL of CSF was collected through gentle extraction using polypropylene syringes and combined in a 30 mL polypropylene tube. Samples were gently mixed, centrifuged, and aliquoted into 1.5 mL polypropylene tubes. Aliquot tubes were stored at −80°C within 30 minutes of collection.
2.2.3. PiB‐PET imaging
Participants in the Aβ chronicity subsample underwent T1‐weighted magnetic resonance imaging (MRI) for anatomical delineation and [C‐11]PiB‐PET for the quantification of cerebral Aβ. Details regarding radioligand synthesis, image acquisition, processing, and analysis of MRI and PiB‐PET images have been described previously. 27 Amyloid burden was quantified as the average cortical PiB distribution volume ratio (DVR; Logan graphical analysis, cerebellum gray matter reference region) using dynamic PiB data acquired 0‐70 minutes post‐injection with either a Siemens Biograph Horizon PET/CT or Siemens EXACT HR+ tomograph. MRI and PET image processing and quality control were performed using a pipeline that uses MATLAB (The Mathworks, Inc., Natick, MA) and SPM12 (www.fil.ion.ucl.ac.uk/spm). The cut‐point for PiB positivity was a global DVR of 1.19. 28
2.2.4. Cognitive testing
Participants completed cognitive tests during biennial WRAP visits. In the cognitive subsample, participants had one to six cognitive assessments (see Table 2 for count and percentage of participants with one to six cognitive assessments). Follow‐up cognitive testing occurred on average every 2.6 years (SD = 0.5) and was available in 91.4% of the participants. The mean follow‐up period for these participants was 7.1 years.
2.3. Measures
2.3.1. IR
IR was measured using HOMA2‐IR (i.e., the computer model), 26 which was calculated by entering fasting glucose and insulin into the HOMA calculator version 2.2.3 (University of Oxford, 2013: https://www.dtu.ox.ac.uk/homacalculator/). HOMA2‐IR provides an indication of IR in the fasting or basal state, with higher values reflecting higher IR. It has been shown to correlate strongly with clamp‐derived whole‐body insulin sensitivity. 26 Although there is no reference range, a level of 1.0 is thought to approximate normal. 26 In the study with the largest number of participants (ie, cognitive study), the average HOMA2‐IR in non‐diabetics was 1.1 (SD = 0.8) and in diabetics (defined as self‐report of taking oral antidiabetic medication or, if no self‐report, fasting glucose ≥126 mg/dL 29 ) the average level was 2.4 (SD = 1.5).
HOMA2‐IR values were calculated from insulin and glucose collected together closest in time to outcome data. For the CSF biomarker study, blood for HOMA2‐IR was collected within ± 1 year (mean = 0.25, SD = 0.39) of the baseline LP. In the cognitive study, blood for HOMA2‐IR was collected at the same visit as the earliest available cognitive assessment in the longitudinal series. In the Aβ chronicity study, a within‐person HOMA2‐IR mean and SD, calculated using three or more values out of all available HOMA2‐IR, was assessed in order to relate chronic exposure and variability in IR to Aβ chronicity. Timeframe of collection for HOMA2‐IR values used in the Aβ chronicity study was 8.2 years (SD = 2.0).
2.3.2. Apolipoprotein E ε4 (APOE ε4)
APOE ε4 was genotyped using competitive allele‐specific polymerase chain reaction (PCR)–based KASP genotyping assays for rs7412 and rs429358 (LGC Genomics, Beverly, MA). Individuals with one or two C alleles for rs429358 were coded as APOE ε4 carriers.
2.3.3. CSF biomarkers of AD pathology and neurodegeneration
CSF biomarkers of AD pathology included Aβ42/Aβ40 ratio, P‐tau181/Aβ42 ratio, and P‐tau181. CSF biomarkers of neurodegeneration included T‐tau and markers of synaptic and axonal degeneration, neurogranin, and NfL, respectively. CSF biomarker levels were measured with the Roche NeuroToolKit panel using either the Elecsys β‐Amyloid (1‐42), Total‐Tau, and Phospho‐Tau (181P) CSF immunoassays, or robust prototype assays (Roche Diagnostics International Ltd, Rotkreuz, Switzerland). T‐tau, P‐tau181, and Aβ42 were assayed on the cobas e 601; Aβ40, neurogranin, and NfL were assayed on the cobas e 411. All analyses were performed at the Clinical Neurochemistry Laboratory at the University of Gothenburg. Previously determined thresholds for amyloid (Aβ42/Aβ40) and P‐tau181 positivity were used to describe the sample. 30
2.3.4. Amyloid beta chronicity
Aβ chronicity was defined as the estimated time in years that an individual had been PiB positive at the time of last HOMA2‐IR assessment. It was calculated as the age at last HOMA2‐IR minus the estimated age at PiB positivity. To avoid missing values for individuals with no evidence of PiB accumulation, estimated age at PiB positivity was estimated as age at PET scan plus life expectancy from a sex‐specific life‐expectancy table. Information regarding the estimation of the age at PiB positivity and amyloid chronicity has been described previously. 23 Previous findings indicate that Aβ chronicity is a valid predictor of core AD‐related outcomes. Higher Aβ chronicity was significantly associated with greater odds of mild cognitive impairment/AD dementia (defined by consensus conference) and tau burden in the entorhinal cortex (assessed by MK‐6240 PET). 23
2.3.5. Preclinical Alzheimer's cognitive composite (PACC‐3)
A modified preclinical Alzheimer's cognitive composite 31 was derived from the Rey Auditory Verbal Learning (RAVLT; Trials 1‐5), 32 Logical Memory II, 33 and Digit Symbol Substitution. 34 Scores were standardized using the mean from first cognitive assessment of cognitively unimpaired participants. Details on the derivation of this metric have been described previously. 31
2.4. Statistical approach
Primary, sensitivity, and exploratory analyses for each study are discussed first prior to a discussion of secondary analyses and model fit and assumptions applicable to more than one study. Covariates included age, APOE ε4 carriership, sex, and education (a social determinant of health). Systolic blood pressure was also controlled because hypertension has been associated with IR 35 and at mid‐life increases dementia risk. 36
2.4.1. CSF biomarker study
Primary analyses: Linear mixed effects (LMEs) tested an Age x HOMA2‐IR interaction to determine whether HOMA2‐IR moderated age‐associated change in CSF biomarkers of AD pathology and neurodegeneration. Age at each LP visit was centered using average age at baseline LP. Exploratory analyses: Because APOE ε4 has been shown to influence amyloid accumulation, 22 LME tested an Age x HOMA2‐IR x APOE ε4 interaction to explore whether HOMA2‐IR interacted with APOE ε4 carrier status (ε4 carrier vs non‐carrier) to predict age‐related change in CSF Aβ42/Aβ40 and P‐tau181/Aβ42 ratios. If the three‐way interaction was not significant, the lower‐order two‐way interactions were subsequently tested.
2.4.2. Amyloid beta chronicity study
Primary analyses: Multiple linear regression was used to test whether chronic exposure and variability in IR interacted with APOE ε4 carrier status to predict Aβ chronicity at age of last HOMA2‐IR. Within‐person HOMA2‐IR means and SDs were used to measure chronic exposure and variability in IR, respectively. Age at last HOMA2‐IR was controlled. Sensitivity analyses were also conducted using PiB positivity as an outcome in two binary logistic regressions where HOMA2‐IR (within‐person ‐mean or ‐SD) x APOE ε4 was a predictor. PiB positivity was determined from the participant's most recent PiB‐PET. HOMA2‐IR within‐person mean and SD were calculated using a minimum of three HOMA2‐IR values, with the last value being collected within an average of 1.0 year (SD = 1.1) prior to the most recent PiB‐PET (n = 219). Age at most recent PiB‐PET was controlled.
2.4.3. Cognitive study
Primary analyses: LME was used to examine whether baseline HOMA2‐IR moderated age‐related decline in PACC‐3 scores. Age at each visit was centered using average age at baseline PACC‐3. Exploratory analyses. LME tested an Age x HOMA2‐IR x APOE ε4 interaction to explore whether HOMA2‐IR interacted with APOE ε4 carrier status to predict age‐related change in PACC‐3 scores. If the three‐way interaction was not significant, the lower‐order two‐way interactions were subsequently tested.
2.4.4. All studies
Secondary analyses: Simple slopes analysis was performed if an interaction was significant. If Age x HOMA2‐IR in the cognitive and CSF biomarker study or HOMA2‐IR x APOE4 in the Aβ chronicity study was not significant, the main effect of HOMA2‐IR was tested without the interaction term.
Model fit and assumptions: Natural log transformation of CSF biomarkers, except for Aβ42/Aβ40, and log10 transformation of Aβ chronicity were necessary to meet the homogeneity of variance assumption. Convergence criteria were not met for a random intercept plus age slope model when CSF Aβ42/Aβ40 and CSF ln(P‐tau181/Aβ42) were outcomes; thus a random intercept only model was used instead. Random intercept plus age slope models were used for remaining analyses in the CSF biomarker and cognitive studies. HOMA2‐IR values were transformed to z‐scores using the HOMA2‐IR mean and 1 SD unique to each study. A P < .05, uncorrected, was interpreted as significant.
3. RESULTS
3.1. CSF biomarker study
Primary analyses: When controlling for covariates and at the mean of HOMA2‐IR, age was significantly related to decreased Aβ42/Aβ40 and increased ln(P‐tau181/Aβ42), ln(P‐tau181), ln(T‐tau), ln(NfL), and ln(neurogranin) (Table 3). HOMA2‐IR did not moderate the relationship of age to CSF biomarkers (Table 3 and Figure 1). Secondary analyses: Without the Age x HOMA2‐IR interaction term, HOMA2‐IR did not explain significant variability in any CSF outcome (Table S1). Exploratory analyses: The Age x HOMA2‐IR x APOE ε4 interaction was not a significant predictor of Aβ42/Aβ40 or ln(P‐tau181/Aβ42); however, the lower order HOMA2‐IR x APOE ε4 interaction was significantly associated with ln(P‐tau181/Aβ42) in the model with the three‐way interaction term. Thus HOMA2‐IR x APOE ε4 was tested, along with Age x HOMA2‐IR and Age x APOE ε4 as a predictor of ln(P‐tau181/Aβ42) without the three‐way interaction. In the reduced model, HOMA2‐IR x APOE ε4 was significant, Age x HOMA2‐IR was not significant, and as expected, 22 Age x APOE ε4 was significant. When tested without the Age x HOMA2‐IR interaction and when controlling for Age x APOE ε4, HOMA2‐IR x APOE ε4 was a significant predictor of ln(P‐tau181/Aβ42) (Table 4). Simple slopes analysis of this interaction suggested that for every 1 SD increase in HOMA2‐IR, ln(P‐tau181/Aβ42) was .10 higher in APOE ε4 carriers (P = .06) compared with .04 lower (P = .24) in non‐carriers (Figure 2).
TABLE 3.
Results from linear mixed‐effects models investigating homeostatic model assessment of insulin resistance (HOMA2‐IR) as a moderator of the relationship between age and CSF biomarker outcomes
| (a) CSF biomarkers of AD pathology (n = 211) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Aβ42/Aβ40 ratio | Ln(P‐tau181/Aβ42 ratio) | ln(P‐tau181) | |||||||
| β | P | 95% CI | β | P | 95% CI | β | P | 95% CI | |
| Intercept | .08 | <.0001 | .06 to .10 | −4.46 | <.0001 | −5.0 to −3.9 | 2.74 | <.0001 | 2.38 to 3.10 |
| Sex (0 = female) | .0005 | .82 | −.004 to .005 | −.01 | .86 | −.15 to .13 | −.03 | .55 | −.13 to .07 |
| Education (y) | −.0009 | .09 | −.002 to .0002 | .03 | .07 | −.002 to .06 | .006 | .62 | −.02 to .03 |
| Systolic blood pressure (z‐scores) | −.0006 | .57 | −.003 to .002 | −.01 | .69 | −.08 to .05 | −.006 | .82 | −.05 to .04 |
| APOE ε4 status (0 = non‐carrier) | −.01 | <.0001 | −.02 to −.007 | .28 | <.0001 | .15 to .42 | .03 | .50 | −.06 to .13 |
| Age (y) | −.0006 | <.0001 | −.0009 to −.0004 | .02 | <.0001 | .02 to .03 | .02 | <.0001 | .01 to .02 |
| HOMA2‐IR (z‐scores) | .00008 | 0.94 | −.002 to .002 | .001 | .97 | −.06 to .06 | .03 | .14 | −.01 to .08 |
| Age x HOMA2‐IR (z‐scores) | .0001 | .31 | −.0001 to .0003 | −.001 | .67 | −.008 to .005 | −.0007 | .71 | −.005 to .003 |
| (b) CSF biomarkers of neurodegeneration (n = 212) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ln(T‐tau) | ln(NfL) | ln(neurogranin) | |||||||
| β | P | 95% CI | β | P | 95% CI | β | P | 95% CI | |
| Intercept | 5.14 | <.0001 | 4.80 to 5.49 | 4.29 | <.0001 | 3.98 to 4.61 | 6.68 | <.0001 | 6.27 to 7.09 |
| Sex (0 = female) | −.03 | .53 | −.13 to .06 | .10 | .02 | .01 to .19 | −.09 | .10 | −.20 to .02 |
| Education (y) | .008 | .47 | −.01 to .03 | .009 | .37 | −.01 to .03 | −.0008 | .95 | −.03 to .02 |
| Systolic blood pressure (z‐scores) | −.003 | .89 | −.05 to .04 | .02 | .46 | −.03 to .06 | −.002 | .93 | −.05 to .05 |
| APOE ε4 status (0 = non‐carrier) | .04 | .45 | −.06 to .13 | −.06 | .14 | −.15 to .02 | .02 | .71 | −.09 to .13 |
| Age (y) | .01 | <.0001 | .01 to .02 | .03 | <.0001 | .03 to .04 | .01 | <.0001 | .006 to .02 |
| HOMA2‐IR (z‐scores) | .03 | .12 | −.009 to .07 | .02 | .27 | −.02 to .06 | .04 | .15 | −.01 to .08 |
| Age x HOMA2‐IR (z−scores) | −.0006 | .74 | −.004 to .003 | −.004 | .13 | −.009 to .001 | −.002 | .42 | −.007 to .003 |
Notes: Random intercept model was used for CSF amyloid outcomes (Aβ42/Aβ40 ratio and Ln(P‐tau181/Aβ42 ratio); random intercept and age slope model was used for remaining CSF biomarker outcomes. Age was centered using baseline age in all models.
Abbreviations: Aβ = amyloid beta; CI = confidence interval; NfL = neurofilament light chain; P‐tau = phosphorylated tau; T‐tau = total tau.
FIGURE 1.
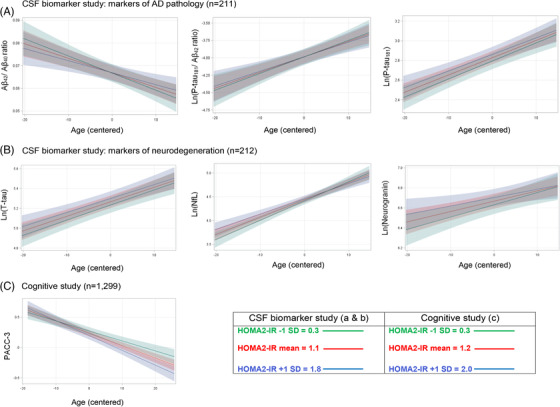
Graphs demonstrating relationship between Age x HOMA2‐IR and CSF biomarker (A and B) and cognitive (C) outcomes determined by linear mixed effects models. Age was centered at average baseline age. Notes: Random intercept model was used for CSF amyloid outcomes (Aβ42/Aβ40 ratio and ln(P‐tau181/ Aβ42 ratio); random intercept and age slope model was used for remaining CSF biomarker outcomes. The Age x HOMA2‐IR interaction was significantly related to PACC‐3 but not to any outcome in the CSF biomarker study. Aβ = amyloid beta; HOMA2‐IR = homeostatic model assessment of insulin resistance; NfL = neurofilament light chain; PACC‐3 = Preclinical Alzheimer's Cognitive Composite (version 3); P‐tau = phosphorylated tau; T‐tau = total tau
TABLE 4.
(a) Results from linear mixed effects modela (LME) testing APOE ε4 status as a moderator of the relationship between homeostatic model assessment of insulin resistance (HOMA2‐IR) and CSF ln(P‐tau181/Aβ42 ratio) (n = 211), and (b) results from LMEb testing HOMA2‐IR as a moderator of age‐related change in the preclinical Alzheimer's cognitive composite (PACC‐3) (n = 1299)
| (a) CSF biomarker study outcome: ln(P‐tau181/Aβ42 ratio) | |||
|---|---|---|---|
| β | P | 95% CI | |
| Intercept | −4.50 | <.0001 | −5.00 to −4.00 |
| Sex (0 = female) | −.003 | .97 | −.14 to .14 |
| Education (y) | .03 | .04 | .002 to .06 |
| Systolic blood pressure (z‐scores) | −.009 | .78 | −.07 to .06 |
| APOE ε4 status (0 = non‐carrier) | .25 | .0003 | .12 to .39 |
| Age (y) | .01 | .001 | .005 to .02 |
| HOMA2‐IR (z‐scores) | −.04 | .24 | −.12 to .03 |
| Age x APOE ε4 | .02 | .002 | .009 to .04 |
| HOMA2‐IR x APOE ε4 | .14 | .03 | .02 to .27 |
| (b) Cognitive study outcome: PACC‐3 | |||
|---|---|---|---|
| β | P | 95% CI | |
| Intercept | −1.23 | <.0001 | −1.49 to −.97 |
| Sex (0 = female) | −.51 | <.0001 | −.59 to −.43 |
| Education (y) | .09 | <.0001 | .08 to .11 |
| Systolic blood pressure (z‐scores) | −.02 | .39 | −.05 to .02 |
| APOE ε4 status (0 = non‐carrier) | −.09 | .02 | −.16 to −.02 |
| Age (y) | −.02 | <.0001 | −.023 to −.017 |
| HOMA2‐IR (z‐scores) | −.04 | .05 | −.07 to −.0006 |
| Age x HOMA2‐IR | −.004 | .02 | −.007 to −.0006 |
Abbreviations: CI = confidence interval.
Random intercept model when ln(P‐tau181/Aβ42 ratio) was outcome.
Random intercept and age slope model when PACC‐3 was outcome. Age was centered at average baseline age.
FIGURE 2.
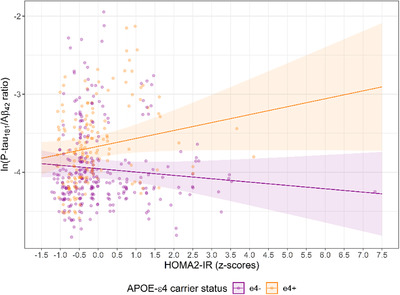
Scatterplot of HOMA2‐IR x APOE ε4 relationship with ln(P‐tau181/Aβ42 ratio) determined by a random intercept model. Notes: HOMA2‐IR mean = 1.1 (SD = 0.7); HOMA2‐IR X APOE ε4 interaction was significant and in same direction after removal of single case with extreme HOMA2‐IR z‐score (7.3), β = .15, P = .04, 95% CI = .008 to .28; Aβ = beta amyloid; HOMA2‐IR = homeostatic model assessment of insulin resistance; P‐tau = phosphorylated tau
3.2. Amyloid beta chronicity study
Primary analyses: HOMA2‐IR x APOE ε4 was not a significant predictor of Aβ chronicity (Table S2). Secondary analyses: In multiple linear regression models without the HOMA2‐IR x APOE ε4 interaction term, within‐person HOMA2‐IR mean and SD were not significant predictors of Aβ chronicity (Table S2). Sensitivity analyses: HOMA2‐IR x APOE ε4 was not a significant predictor of PiB positivity (Table S2).
3.3. Cognitive study
Primary analyses: Baseline HOMA2‐IR was a significant moderator of age‐related decline in PACC‐3 scores when controlling for sex, education, APOE ε4 carrier status, and baseline systolic blood pressure (Table 4 and Figure 1). Participants with higher baseline HOMA2‐IR experienced faster cognitive decline than participants with lower HOMA2‐IR. Simple slopes for age at 1 SD above and below the HOMA2‐IR mean were significant (P < .0001) and equaled −.024 and −.017, respectively. The Age x HOMA2‐IR interaction remained significant (β = ‐.004, P = .02) when controlling for diabetes at baseline and after removal of four participants who subsequently developed dementia. Exploratory analyses. The Age x HOMA2‐IR x APOE ε4 interaction was not significantly associated with PACC‐3 scores, indicating that the relationship between Age x HOMA2‐IR and cognition was not significantly moderated by APOE ε4 carrier status. In the model without the three‐way interaction term, the Age x HOMA2‐IR interaction was significant (β = −.004, P = .02) when controlling for HOMA2‐IR x APOE ε4 (β = .04, P = .24) and Age x APOE ε4 (β = −.009, P = .005) as well as sex, education, and baseline systolic blood pressure.
4. DISCUSSION
Using longitudinal data from predominantly healthy, non‐demented middle‐aged and older adults, we found that IR was related to age‐related change in cognition but not biomarkers of AD pathology. Specifically, higher IR was related to worse age‐associated decline in PACC‐3 scores. In the early stages of the AD continuum, IR may facilitate cognitive decline through mechanisms that are independent of AD pathology, similar to T2D, which has also been related to increased risk for Alzheimer's clinical syndrome but not post‐mortem amyloid plaque and tau tangle pathology. 37
Previous studies indicate that IR increases risk for T2D, 1 which in turn increases risk for cerebrovascular disease, cerebral infarct, and subsequent cognitive dysfunction. 37 Because our sample was generally healthy, the relationship between IR and cognitive decline observed may have been due to cerebrovascular changes occurring prior to or independent of infarct development. Indeed, in the absence of brain infarction, IR and T2D have been associated with alterations in cerebrovascular as well as neural function that potentially worsen cognitive health. For example, neurovascular coupling (ie, the regulation of blood flow in response to neural activity) was found to be altered in people with T2D, with no evidence of brain infarct or vascular lesion. 38 IR has been related to lower cerebral arterial blood flow and microvessel perfusion. 39 , 40 IR‐associated hypoperfusion has been related to cognitive deficits in patients with T2D. 39 In predominantly stroke‐free participants with T2D, IR was related to white matter hyperintensity severity and cognitive dysfunction. 41 In a mouse model of IR, increased blood‐brain barrier permeability, concurrent with neuroinflammation, was associated with subsequent cognitive decline and neurodegeneration. 42 Peripheral IR has been linked in some animal studies to central dysregulation of insulin signaling 7 , 43 and subsequent abnormalities in synaptic function. 8 , 9 In sum, multiple IR‐associated changes, such as hypoperfusion, white matter hyperintensities, increased blood‐brain barrier permeability, neuroinflammation, and dysregulation of central insulin signaling, may have contributed to cognitive decline in our participants.
IR has been associated with neurodegeneration: specifically, brain atrophy, 44 decreased cerebral glucose metabolism 20 and increased CSF T‐tau. 2 , 19 In our sample, IR did not predict age‐associated increase of the CSF concentrations of T‐tau, NfL, or neurogranin, markers of axonal and synaptic dysfunction/degeneration, respectively. Thus even though higher IR was related to worse age‐related decline in cognitive performance, it was not similarly associated with age‐associated increases in CSF biomarkers of neurodegeneration. We speculate that potential IR‐related effects on cerebrovascular and neural function may not yet have resulted in detectable neurodegeneration given that participants were non‐demented.
We did not find a significant association between IR and the average level of CSF P‐tau181 in contrast with a previous cross‐sectional study. 2 Differences in study design and sample characteristics may have contributed to the contrasting results. Our sample was younger on average and had more APOE ε4 carriers. Whether the relationship between IR and P‐tau181 is manifested in cognitively unimpaired adults older than the participants that we studied deserves further investigation. Some research suggests that peripheral IR facilitates the dysregulation of neuronal insulin signaling kinases that mediate tau hyperphosphorylation. 7 The potential interrelationship between central and peripheral IR requires further research 45 because it is unclear whether the central IR found in AD 46 precedes tau hyperphosphorylation. 46 , 47
APOE ε4 moderated the relationship between IR and average level of P‐tau181/Aβ42 ratio. Results suggested that as IR increased, APOE ε4 carriers had greater P‐tau181/Aβ42 than non‐carriers. Similar moderating effects of APOE ε4 have been found in some past research examining IR and biomarkers of AD pathology. 16 Both insulin and Aβ42 are substrates for proteolytic degradation by insulin‐degrading enzyme (IDE), 15 a protein found to be lower in AD dementia cases who were APOE ε4 carriers. 48 Elevated insulin associated with IR may reduce clearance of Aβ42 through competitive inhibition for degradation by IDE 15 to a greater extent in APOE ε4 carriers. Deficiency of IDE may also increase the IDE substrate IAPP, 49 whose misfolded forms may facilitate formation of Aβ42 fibrils or slow clearance of the protein. 11 , 13 , 14
In contrast to results for CSF P‐tau181/Aβ42, APOE ε4 was not a significant moderator of the relationship between IR and Aβ chronicity or PiB positivity. Increases in CSF P‐tau181/Aβ42 may occur prior to Aβ detectable by PiB‐PET. 50 However, because P‐tau pathology is hypothesized to follow early Aβ accumulation, 51 pathological increases in CSF P‐tau181/Aβ42 should correspond with prolonged duration of amyloid PET positivity. Indeed, Aβ chronicity was related in another study to higher entorhinal tau. 23 Further research is needed to investigate whether higher IR facilitates increased AD pathology in APOE ε4 carriers.
Our study has several strengths and limitations. Because we had longitudinal CSF core AD biomarker and cognitive data, we were able to assess age‐related change in important variables related to AD. We had multiple measures of HOMA2‐IR collected over several years, which allowed us to relate chronic exposure and variability in insulin sensitivity to a novel measure, Aβ chronicity. However, due to the correlational nature of our study, causative claims cannot be made. Although we controlled for potential confounds, we acknowledge the possibility that unmeasured factors may have influenced associations. The effect size for IR on cognition in our sample was modest. Simple slopes for age at 1 SD above and below the HOMA2‐IR mean were −.024 and −.017 respectively, demonstrating that the difference between the two slopes, although statistically significant, was small. Due to sample size constraints, we may not have had adequate power to detect a similar small effect of IR on CSF biomarkers. Furthermore, there were few participants in the CSF biomarker study who were both amyloid and P‐tau181 positive at baseline (n = 15) and by study end (n = 26), indicating that most participants did not have AD as defined biologically. Nevertheless, there were 24.6% (n = 52) who were amyloid positive, and we did demonstrate that CSF amyloid and P‐tau181 changed in the expected direction with aging. The association between IR and biomarkers of AD pathology will be examined again after more participants within our cohort develop AD.
In conclusion, higher IR was associated with worse cognitive decline but not longitudinal age‐related change in CSF biomarkers of AD pathology in non‐demented adults. IR may not be related to change in amyloid and tau in the early stages of the AD continuum; whether IR contributes to AD pathology later in the disease trajectory deserves further study. APOE ε4 moderated the relationship between IR and level of P‐tau181/Aβ42. That IR may act synergistically with APOE ε4 to influence AD pathology warrants investigation as do cerebrovascular and other mechanisms, which could be potential targets for treatment, mediating the relationship between IR and cognitive decline prior to dementia.
DECLARATION OF INTERESTS
IS is a full‐time employee and shareholder of Roche Diagnostics International Ltd. GK is a full‐time employee of Roche Diagnostics GmbH. HZ has served at scientific advisory boards for Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, and CogRx; has given lectures in symposia sponsored by Fujirebio, Alzecure and Biogen; and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. KB has served as a consultant, on advisory boards, or on data monitoring committees for Abcam, Axon, Biogen, JOMDD/Shimadzu, Julius Clinical, Lilly, MagQu, Novartis, Roche Diagnostics, and Siemens Healthineers; and is a co‐founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program. COBAS and COBAS E are registered trademarks of Roche. All remaining authors have nothing to disclose.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors would like to thank Richard Batrla for his contributions to the conception of the NeuroToolKit panel of exploratory biomarkers. This work was supported by grants from the National Institute on Aging, one of the National Institutes of Health (NIH) (P30‐AG062715; RF1‐AG027161; R01‐AG021155; R01‐AG054047), and the National Institutes of Health Office of Infrastructure Programs (S10 OD025245‐01). GE (AARF‐19‐643973) and TB (AARF‐19‐614533) are supported by the Alzheimer's Association. HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018‐02532), the European Research Council (#681712), Swedish State Support for Clinical Research (#ALFGBG‐720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809‐2016862), the AD Strategic Fund and the Alzheimer's Association (#ADSF‐21‐831376‐C, #ADSF‐21‐831381‐C, and #ADSF‐21‐831377‐C), the Olav Thon Foundation, the Erling‐Persson Family Foundation, Hjärnfonden, Sweden (#FO2019‐0228), the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No. 860197 (MIRIADE), and the UK Dementia Research Institute at UCL. KB is supported by the Swedish Research Council (#2017‐00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB‐201809‐2016615), the Swedish Alzheimer Foundation (#AF‐742881), Hjärnfonden, Sweden (#FO2017‐0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF‐agreement (#ALFGBG‐715986), and European Union Joint Program for Neurodegenerative Disorders (JPND2019‐466‐236). The Waisman Brain Imaging Laboratory is funded by the NIH National Institute of Child Health and Human Development (U54HD090256). CSF biomarkers were assayed using the exploratory Roche NeuroToolKit assays, a panel of automated robust prototype immunoassays designed to robustly evaluate biomarkers associated with key pathologic events characteristic of AD and other neurological disorders, provided by Roche Diagnostics International Ltd (Rotkreuz, Switzerland). The Roche NeuroToolKit robust prototype assays are for investigational purposes and are not approved for clinical use. Funding sources did not contribute to study design, collection and analysis of data, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Alzheimer's Association, the National Institutes of Health, or Roche Diagnostics.
Ennis GE, Koscik RL, Ma Y, et al. Insulin resistance is related to cognitive decline but not change in CSF biomarkers of Alzheimer's disease in non‐demented adults. Alzheimer's Dement. 2021;13:e12220. 10.1002/dad2.12220
REFERENCES
- 1. Martin BC, Warram JH, Krolewski AS, et al. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25‐year follow‐up study. Lancet. 1992;340(8825):925‐929. [DOI] [PubMed] [Google Scholar]
- 2. Laws SM, Gaskin S, Woodfield A, et al. Insulin resistance is associated with reductions in specific cognitive domains and increases in CSF tau in cognitively normal adults. Sci Rep. 2017;7(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tan ZS, Beiser AS, Fox CS, et al. Association of metabolic dysregulation with volumetric brain magnetic resonance imaging and cognitive markers of subclinical brain aging in middle‐aged adults. Diabetes Care. 2011;34(8):1766‐1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kuusisto J, Koivisto K, Mykkänen L, et al. Association between features of the insulin resistance syndrome and Alzheimer's disease independently of apolipoprotein E4 phenotype: cross sectional population based study. Br Med J. 1997;315(7115):1045‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schrijvers EMC, Witteman JCM, Sijbrands EJG, et al. Insulin metabolism and the risk of Alzheimer disease: the Rotterdam Study. Neurology. 2010;75(22):1982‐1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arnold SE, Arvanitakis Z, Macauley‐Rambach SL, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018;14(3):168‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kothari V, Luo Y, Tornabene T, et al. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim Biophys Acta—Mol Basis Dis. 2017;1863(2):499‐508. [DOI] [PubMed] [Google Scholar]
- 8. Arnold SE, Lucki I, Brookshire BR, et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol Dis. 2014;67:79‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu Z, Patil IY, Jiang T, et al. High‐fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PLoS One. 2015;10(5):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Enoki S, Mitsukawa T, Takemura J, et al. Plasma islet amyloid polypeptide levels in obesity, impaired glucose tolerance and non‐insulin‐dependent diabetes mellitus. Diabetes Res Clin Pract. 1992;15(1):97‐102. [DOI] [PubMed] [Google Scholar]
- 11. Jackson K, Barisone GA, Diaz E, et al. Amylin deposition in the brain: a second amyloid in Alzheimer disease?. Ann Neurol. 2013;74(4):517‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martinez‐Valbuena I, Valenti‐Azcarate R, Amat‐Villegas I, et al. Amylin as a potential link between type 2 diabetes and Alzheimer disease. Ann Neurol. 2019;86(4):539‐551. [DOI] [PubMed] [Google Scholar]
- 13. Moreno‐Gonzalez I, Edwards IIIG, Salvadores N, et al. Molecular interaction between type 2 diabetes and Alzheimer's disease through cross‐seeding of protein misfolding. Mol Psychiatry. 2017;22(9):1327‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oskarsson ME, Paulsson JF, Schultz SW, et al. In vivo seeding and cross‐seeding of localized amyloidosis. Am J Pathol. 2015;185(3):834‐846. [DOI] [PubMed] [Google Scholar]
- 15. Qiu W, Folstein M. Insulin, insulin‐degrading enzyme and amyloid‐β peptide in Alzheimer's disease: review and hypothesis. Neurobiol Aging. 2006;27(2):190‐198. [DOI] [PubMed] [Google Scholar]
- 16. Matsuzaki T, Sasaki K, Tanizaki Y, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama Study. Neurology. 2010;75(9):764‐770. [DOI] [PubMed] [Google Scholar]
- 17. Thambisetty M, Metter EJ, Yang A, et al. Glucose intolerance, insulin resistance, and pathological features of Alzheimer disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2013;70(9):1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Willette AA, Johnson SC, Birdsill AC, et al. Insulin resistance predicts brain amyloid deposition in late middle‐aged adults. Alzheimer's Dement. 2015;11(5):504‐510. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Westwood S, Liu B, Baird AL, et al. The influence of insulin resistance on cerebrospinal fluid and plasma biomarkers of Alzheimer's pathology. Alzheimers Res Ther. 2017;9(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Willette AA, Bendlin BB, Starks EJ, et al. Association of insulin resistance with cerebral glucose uptake in late middle–aged adults at risk for Alzheimer disease. JAMA Neurol. 2015;72(9):1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Byun MS, Kim HJ, Yi D, et al. Region‐specific association between basal blood insulin and cerebral glucose metabolism in older adults. NeuroImage Clin. 2019;22:101765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mishra S, Blazey TM, Holtzman DM, et al. Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE ε4 genotype. Brain. 2018;141(6):1828‐1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koscik RL, Betthauser TJ, Jonaitis EM, et al. Amyloid duration is associated with preclinical cognitive decline and tau PET. Alzheimer's Dement Diagnosis . Assess Dis Monit. 2020;12(1):e12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson SC, Koscik RL, Jonaitis EM, et al. The Wisconsin Registry for Alzheimer's Prevention: a review of findings and current directions. Alzheimer's Dement Diagnosis . Assess Dis Monit. 2018;10(1):130‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's Dement. 2011;7(3):263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27(6):1487‐1495. [DOI] [PubMed] [Google Scholar]
- 27. Johnson SC, Christian BT, Okonkwo OC, et al. Amyloid burden and neural function in people at risk for Alzheimer's disease. Neurobiol Aging. 2014;35(3):576‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Racine AM, Clark LR, Berman SE, et al. Associations between performance on an abbreviated CogState battery, other measures of cognitive function, and biomarkers in people at risk for Alzheimer's disease. J Alzheimer's Dis. 2016;54(4):1395‐1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. American Diabetes Association . Standards of medical care in diabetes–2010. Diabetes Care. 2010;33(1):S11‐S61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Hulle C, Jonaitis EM, Betthauser TJ, et al. An examination of a novel multipanel of CSF biomarkers in the Alzheimer's disease clinical and pathological continuum. Alzheimer's Dement. 2021;17(3):431‐445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jonaitis EM, Koscik RL, Clark LR, et al. Measuring longitudinal cognition: individual tests versus composites. Alzheimer's Dement Diagnosis, Assess Dis Monit. 2019;11(1):74‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schmidt M. Rey Auditory Verbal Learning Test: A Handbook. 1996. [Google Scholar]
- 33. Wechsler D. Wechsler Memory Scale ‐ Revised. The Psychological Corporation; 1987. [Google Scholar]
- 34. Wechsler D. Wechsler Adult Intelligence Scale. 3rd ed.. The Psychological Corporation; 1997. [Google Scholar]
- 35. Saad MF, Rewers M, Selby J, et al. Insulin resistance and hypertension. Hypertension. 2004;43(6):1324‐1331. [DOI] [PubMed] [Google Scholar]
- 36. Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396(10248):413‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pruzin JJ, Nelson PT, Abner EL, et al. Review: relationship of type 2 diabetes to human brain pathology. Neuropathol Appl Neurobiol. 2018;44(4):347‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duarte JV, Pereira JM, Quendera B, et al. Early disrupted neurovascular coupling and changed event level hemodynamic response function in type 2 diabetes: an fMRI study. J Cereb Blood Flow Metab. 2015;35(10):1671‐1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cui Y, Liang X, Gu H, et al. Cerebral perfusion alterations in type 2 diabetes and its relation to insulin resistance and cognitive dysfunction. Brain Imaging Behav. 2017;11(5):1248‐1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoscheidt SM, Kellawan JM, Berman SE, et al. Insulin resistance is associated with lower arterial blood flow and reduced cortical perfusion in cognitively asymptomatic middle‐aged adults. J Cereb Blood Flow Metab. 2017;37(6):2249‐2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hishikawa N, Yamashita T, Deguchi K, et al. Cognitive and affective functions in diabetic patients associated with diabetes‐related factors, white matter abnormality and aging. Eur J Neurol. 2015;22:313‐321. [DOI] [PubMed] [Google Scholar]
- 42. Takechi R, Lam V, Brook E, et al. Blood‐brain barrier dysfunction precedes cognitive decline and neurodegeneration in diabetic insulin resistant mouse model: an implication for causal link. Front Aging Neurosci. 2017;9:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu Y, Liu F, Grundke‐Iqbal I, et al. Deficient brain insulin signalling pathway in Alzheimer's disease and diabetes. J Pathol. 2011;225(1):54‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Willette AA, Xu G, Johnson SC, et al. Insulin resistance, brain atrophy, and cognitive performance in late middle‐aged adults. Diabetes Care. 2013;36(2):443‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Salas IH, De Strooper B. Diabetes and Alzheimer's disease: a link not as simple as it seems. Neurochem Res. 2019;44(6):1271‐1278. [DOI] [PubMed] [Google Scholar]
- 46. Talbot K, Wang H‐Y, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF‐1 resistance, IRS‐1 dysregulation, and cognitive decline. J Clin Invest. 2012;122(4):1316‐1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rodriguez‐Rodriguez P, Sandebring‐Matton A, Merino‐Serrais P, et al. Tau hyperphosphorylation induces oligomeric insulin accumulation and insulin resistance in neurons. Brain. 2017;140(12):3269‐3285. [DOI] [PubMed] [Google Scholar]
- 48. Cook DG, Leverenz JB, McMillan PJ, et al. Reduced hippocampal insulin‐degrading enzyme in late‐onset Alzheimer's disease is associated with the apolipoprotein E‐ε4 allele. Am J Pathol. 2003;162(1):313‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bennett RG, Hamel FG, Duckworth WC. An insulin‐degrading enzyme inhibitor decreases amylin degradation, increases amylin‐induced cytotoxicity, and increases amyloid formation in insulinoma cell cultures. Diabetes. 2003;52(9):2315‐2320. [DOI] [PubMed] [Google Scholar]
- 50. Palmqvist S, Mattsson N, Hansson O. Cerebrospinal fluid analysis detects cerebral amyloid‐β accumulation earlier than positron emission tomography. Brain. 2016;139(4):1226‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information