

Abstract

Diastereoselective synthesis of the trans-decalin-based α-hydroxyl butanone spirocarbocycles bearing all-carbon quaternary stereogenic centers has been achieved via Norrish–Yang photocyclization of trans-decalin-substituted-2,3-butanediones using daylight. Density functional theory (DFT) calculations suggest that this diastereoselective reaction is affected by both substrate conformation and intramolecular hydrogen bonds. The developed chemistry could be applied to synthesizing the derivatives of the trans-decalin-based biologically important natural products.
Introduction
Carbocyclic four-membered ring compounds are important building blocks in organic synthesis.1 In this aspect, α-hydroxyl cyclobutanone derivatives are particularly attractive because of their usefulness as starting materials for the preparation of structurally diverse molecules.2 Because of its high ring strain,3 synthesis of cyclobutanone is challenging. Although there are various ways to prepare α-hydroxyl cyclobutanone derivatives,4 the Norrish–Yang reactions of alkyl α-diketones turns out to be one of the most effective methods of choice.5 Thanks to the effort of Inoue’s laboratory, the Norrish–Yang reaction has become an attractive method in the total syntheses of complex natural products.6
In the early 1960s,7 alkyl α-diketones were identified as useful substrates for the preparation of structurally diverse chiral 2-hydroxy-cyclobutanones. However, the diastereoselective synthesis of 2-hydroxy-cyclobutanone-based spirocarbocycle bearing an all-carbon quaternary stereogenic center8 via the Norrish–Yang reaction of alkyl α-diketones has not been reported. Here, we report our recent efforts to achieve a diastereoselective synthesis of spirocarbocycles9III via the Norrish–Yang photocyclization of α-diketones I using daylight. Our computational and experimental study indicates that the observed photocyclization proceeds diastereoselectively via a conformationally controlled hydrogen-bonding-promoted 1,5-H-atom transfer10 through the 1,4-diradical intermediate II (Figure 1a). The developed chemistry could be applied for preparing the derivatives of decalin-based biologically important natural products,11 such as IV–X (Figure 1b).
Figure 1.

(a) Application of the Norrish–Yang photocyclization of α-diketones (I) using daylight to diastereoselectively generate 2-hydroxy-cyclobutanone-based spiro-trans-decalin (II) via a conformationally controlled hydrogen-bonding-promoted 1,5-H-atom transfer through the 1,4-diradical intermediate II. (b) trans-Decalin-based natural products bearing C4, C9, and C10 stereogenic centers.
Results and Discussion
Our research began with the enantioselective synthesis of the trans-decalin-based diketone 5. Asymmetric synthesis of the Wieland–Miescher ketone 1 was achieved in 80% yield with 89% ee in the presence of a prolinamide catalyst.12 Regioselective protection of the C8 carbonyl group in diketone 1 was achieved by reaction of diketone 1 with ethane-1,2-diol in the presence of PTSA and 4 Å molecular sieves to give enone 2 in 85% yield. Ketone 3 was prepared by subjecting enone 2 to a Birch reduction by treatment with Li/NH3. The resultant enolate was then coupled with 1-bromobut-2-yne.13 Product 5 was obtained in 51% overall yield by a sequence of ketal formations and subsequent alkyne oxidation14 in the presence of RuO2/NaIO4 (Scheme 1).
Scheme 1. Enantioselective Synthesis of 1,2-Diketone 5.

With diketone 5 in hand, we then profiled its Norrish–Yang photocyclization. We systematically investigated the effects of the solvent, catalyst, and light source on the outcome of the photocyclization (Table 1). Accordingly, when diketone 5 was irradiated with a daylight lamp (65 W) at 30 °C in the solvents of CH3CN, toluene, chloroform, dichloromethane, and benzene, the annulated product 6 could be obtained in good-to-excellent yields with an excellent dr value (entries 1–5). However, when the reaction proceeded in methanol, both the yield and the dr value of the product were decreased dramatically (entry 6). We also tested the reaction under irradiation with a blue light-emitting diode (LED) lamp, but the yield was lower and the dr value was unchanged (entries 7–9). Photosensitizer addition did not significantly improve the yield or dr value of product 6 (entries 10–12), possibly because the substrate was easily excited15 and did not require initiation. We also observed that the photocyclization occurred under sunlight to afford product 6 in 64% yield and with an excellent dr without the use of additives (entry 14). This indicates that diketone 5 is a suitable substrate with sufficient stability16 for use in the desired cyclization.
Table 1. Norrish–Yang Cyclization of Diketone 5.

| entry | light source | T (°C) | solvent (0.05 M) | t (h) | yield (%)a | dr |
|---|---|---|---|---|---|---|
| 1 | daylight (65 W) | 30 | MeCN | 8 | 90 | >20:1 |
| 2 | daylight (65 W) | 30 | toluene | 10 | 80 | >20:1 |
| 3 | daylight (65 W) | 30 | CHCl3 | 10 | 96 | >20:1 |
| 4 | daylight (65 W) | 30 | CH2Cl2 | 10 | 84 | >20:1 |
| 5 | daylight (65 W) | 30 | benzene | 10 | 90 | >20:1 |
| 6b | daylight (65 W) | 30 | MeOH | 10 | 68 | >1.5:1 |
| 7 | blue LED (18 W) | 35 | MeCN | 3 | 85 | >20:1 |
| 8 | blue LED (18 W) | 35 | CHCl3 | 3 | 76 | >20:1 |
| 9 | blue LED (18 W) | 35 | CDCl3 | 5 | 77 | >20:1 |
| 10c | blue LED (18 W) | 35 | CHCl3 | 3 | 80 | >20:1 |
| 11d | blue LED (18 W) | 35 | MeCN | 3 | 85 | >20:1 |
| 12d | blue LED (18 W) | 35 | MeCN | 3 | 85 | >20:1 |
| 13 | none | 35 | CHCl3 | 12 | 0 | |
| 14e | sunlight | 37 | CHCl3 | 12 | 64 | >20:1 |
| 15f | daylight (65 W) | 30 | CHCl3 | 12 |
Reagent and conditions: the flask for the photocyclization of 5 (0.05 mmol) dissolved in a solvent was irradiated under the light source listed in the table, and the distance between the flask and the light source was ca. 4–5 cm. Product 6 was purified by flash chromatography on silica gel.
6 was obtained as a 1.5:1 mixture of diastereomers.
0.2 equiv of xanthone was added.
0.2 equiv of benzophenone was added.
6 was obtained in ca. 20% with removal of its ketal group.
3.0 equiv of (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) was added.
With the optimized cyclization conditions in hand, we evaluated the effects of the diketone motif on the annulation outcome. Four 1,2-diketone derivatives of 5 were prepared (see the Supporting Information for details), and the effects of their stereoelectronic and conformational properties on stereocontrol of the photocyclization were evaluated. The annulated products 6a–6d were formed with excellent dr values under the standard reaction conditions, but the annulated product yields decreased with increasing steric hindrance at the terminal diketone.
The effects of the substrate conformation on the annulation were evaluated by performing stereoselective reduction of the C3 carbonyl group to the corresponding hydroxyl groups and then investigating the annulations. All of the selected substrates gave the expected products (7a–7e) with excellent dr values (Table 2).
Table 2. Norrish–Yang Cyclization for the Stereoselective Synthesis of Products 6a–6d and 7a–7ea.

Reaction conditions: substrates (0.05 mmol) dissolved in CHCl3 were irradiated under a daylight lamp (65 W) at 30 °C for 10 h. The distance between the reaction flask and the light source was ca. 4–5 cm.
To account for the potential feasibility of the silyl groups in the photocyclization for the formation of 7d and 7e, we performed density functional theory (DFT) calculation to evaluate the role of intramolecular hydrogen bonding for the formation of 7d and 7e. As shown in Figure 2, in both cases, intramolecular hydrogen bondings indeed play a favorable role in the formation of products 7d and 7e.
Figure 2.
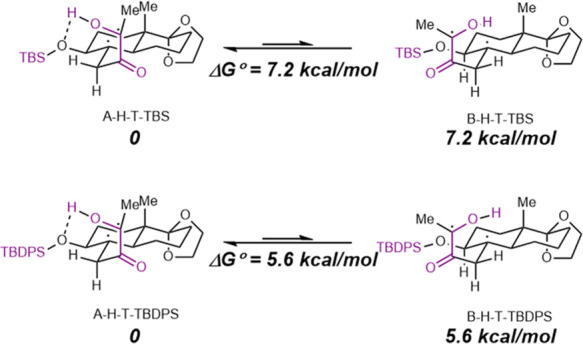
Energy profiles for the intramolecular hydrogen bonding in the syntheses of 7d and 7e.
The trans-decalin is a basic structure in many biologically important molecules such as those listed in Figure 1b. Strategies that enable the stereoselective addition of structurally diverse functional groups to this motif are therefore useful in medicinal chemistry and drug discovery. Since the α-hydroxyl butanone motif can undergo a variety of functionalization and ring expansion reactions,17 we decided to explore this synthesis feasibility using 6 as the substrate. In the event, 6 was oxidized with Pb(OAc)4 to afford ketoester 8 bearing an all-carbon quaternary chiral center at C4 in 92% yield. The Norrish–Yang photoproducts (e.g. 6a/7b/7d) could also generate corresponding ketoesters 8a/8b/8c in good yield under the same reaction condition (Scheme 2).
Scheme 2. Transformation of 2-Hydroxy-Cyclobutanone Moieties in Norrish–Yang Photoproducts.

By treatment of 6 with a catalytic amount of a boron trifluoride etherate complex, tri-substituted furan 9 could be obtained in 50% yield, which has the cafestol skeleton. By the reaction of 6 with lithium tri-tert-butoxyaluminum hydride or methyllithium, diols 10 and 11 were diastereoselectively obtained in 86% yield and 92% yield, respectively. The diastereoselective formation of diols 10 and 11 could presumably be because the bottom face of 6 is a less steric hindrance, which leads the nucleophiles to approach the carbonyl group from its bottom face, resulting in cis diol products.
To explain the diastereoselectivity at C1 of this photocyclization product 6, we performed DFT calculations (at the B3LYP-D3/def2-TZVP//B3LYP-D3/def2-SVP level) to determine the 1,5-H shift of the Norrish–Yang reactions of A-T and B-T (Figure 3).18
Figure 3.
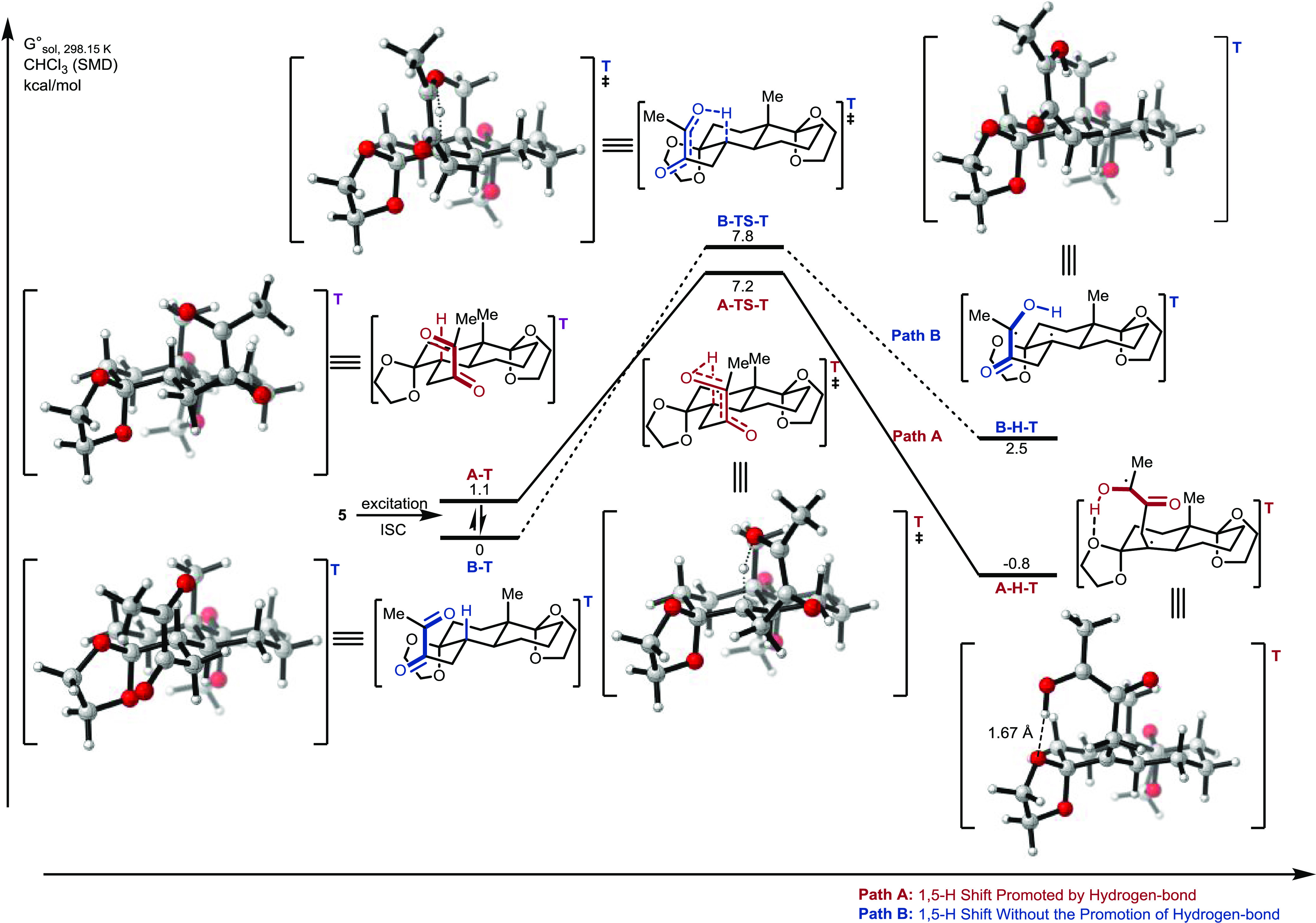
Energy profiles of the 1,5-hydrogen shift of 5 via path A and path B and solvated Gibbs free energies (SMD model) are given in kcal·mol–1 at the B3LYP-D3/def2-TZVP//B3LYP-D3/def2-SVP level.
Notably, the diketone moiety has two different conformations in the triplet state, i.e., A-T and B-T, in this photocyclization reaction. Irradiation and subsequent intersystem crossing of 5 could afford two interchangeable conformations A-T and B-T. The energy of conformation A-T is 1.1 kcal mol–1 higher than that of B-T; therefore, it is reasonable to take B-T as the reference point (0 kcal mol–1). In path A, a 1,5-H shift of A-T via A-TS-T affords the biradical species A-H-T with an energy barrier of 6.1 kcal mol–1. This process is slightly exothermic (ΔG° = −1.9 kcal mol–1). This implies that the 1,5-H shift of A-T is a reversible process. A-H-T, promoted by the stabilizing effect of the intramolecular hydrogen bonding between the O atom of the ketal and the H atom of the newly formed OH group, could generate the desired product 6 by subsequent ISC.18b On the other hand, B-T can undergo a 1,5-H shift via B-TS-T to give the biradical species B-H-T, with an energy barrier of 7.8 kcal mol–1; this is 1.7 kcal mol–1 higher than that for the transformation of A-T via A-TS-T. Without the stabilization effect of hydrogen bonding, the 1,5-H shift is endothermic (ΔG° = +2.5 kcal mol–1). The distribution of B-T is therefore much greater than that of B-H-T in this reversible process. Although the energy of conformation A-T is higher than that of B-T, path A via A-TS-T is more favorable than path B, with an activation energy difference (ΔΔG≠) of 0.6 kcal mol–1 (taking the conformation transformation into consideration, that is, B-T → A-T → A-TS-T → A-H-T). If the activation energy difference and the reversibility of the 1,5-H shift are taken into consideration in the reaction process, a conformational transformation between A-T and B-T could enable the formation of the hydrogen-bonding complex A-H-T via path A; path A would become the dominant pathway. Thus, according to our DFT investigation of the 1,5-H shift in the Norrish–Yang cyclization process, the excellent diastereoselectivity at C1 of this photocyclization product could contribute to the formation of intramolecular hydrogen bonding,18b which enables the formation of the more stable intermediate A-H-T and restrains the reverse 1,5-H shift.19,20
Conclusions
In summary, the trans-decahydronaphthalene-based spirocarbocycles bearing an all-carbon quaternary stereogenic center can be diastereoselectively prepared in good yield and high ee from their corresponding alkyl α-diketones via the Norrish–Yang photocyclization. DFT calculations suggest that the key intermediate A-H-T (Figure 2) affects this highly diastereoselective reaction via conformational restriction and formation of intramolecular hydrogen bonds.
Experimental Section
General Information
All reactions were carried out in oven-dried glassware and under an argon atmosphere with dry solvents under anhydrous conditions unless otherwise noted. Reagents were purchased at the highest commercial quality and used without further purification. Solvent purification was conducted according to Purification of Laboratory Chemicals (Peerrin, D. D.; Armarego, W. L. and Perrins, D. R., Pergamon Press: Oxford, 1980).
Yields refer to chromatographically and spectroscopically (1H nuclear magnetic resonance (NMR)) homogeneous materials. Reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm Tsingdao silica gel plates (60F-254) and visualized under UV light at 254 nm or stained with potassium permanganate (KMnO4) solution and subsequent heating. Flash column chromatography was performed using Tsingdao silica gel (60, particle size 0.040–0.063 mm).
NMR spectra were recorded on a Bruker Avance III 400 (1H: 400 MHz, 13C: 100 MHz) or Bruker Avance 500 (1H: 500 MHz, 13C: 125 MHz) spectrometer and were calibrated using a residual undeuterated solvent as an internal reference (CDCl3: 1H NMR = 7.26 ppm, 13C NMR = 77.16 ppm). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets, and td = triplet of doublets. For all of the high-resolution mass spectrometry (HRMS) measurements (ABI, QSTAR Elite), the ionization method was electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) and the mass analyzer type was TOF. IR spectra were recorded on an IR Prestige-21 FTIR spectrometer with a KBr disc. The circular dichroism (CD) spectrum was measured using the Applied Photophysics Chirascan with a 150 W Xe lamp (165–900 nm). Optical rotation values were recorded on a Rudolph Research Analytical AUTOPOL I polarimeter (Rudolph Research Co.).
Materials
2-Methyl-2-(3-oxobutyl)cyclohexane-1,3-dione (1) was synthesized by the reaction according to the literature procedures.12
Wieland–Miescher Ketone (1)
1 was synthesized according to the literature procedure:19 To a solution of 2-methyl-2-(3-oxobutyl)cyclohexane-1,3-dione (5.00 g, 25.48 mmol, 1.00 equiv) in chloroform (25 mL) was added a prolinamide catalyst (520 mg, 2.55 mmol, 0.10 equiv) at room temperature, and the resultant mixture was stirred at the same temperature for 7 days. Volatile organic materials were removed under vacuum, and the residue was purified by flash chromatography on silica gel to give 1.
(S)-8a-Methyl-3,4,8,8a-tetrahydronaphthalene-1,6(2H,7H)-dione (1)
Yellow oil, 3.63 g (20.37 mmol), 80%, ee = 89%, Rf = 0.37 (silica gel, hexane/EtOAc = 2:1, KMnO4 and UV); [α]D23 = 84.18 (c = 1.0, in CHCl3); IR νmax 2954, 1713, 1668, 1421, 1350, 1238, 1014, 940 cm–1; 1H NMR (500 MHz, CDCl3) δ 5.79 (s, 1H), 2.72–2.63 (m, 2H), 2.47–2.38 (m, 4H), 2.14–2.03 (m, 3H), 1.71–1.61 (m, 1H), 1.40 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 211.0, 198.2, 165.8, 125.8, 50.6, 37.7, 33.6, 31.8, 29.7, 23.3, 22.9; MS (ESI, m/z) calcd for C11H15O2+ [M + H]+: 179.1067, found 179.1064. High-performance liquid chromatography (HPLC) conditions: Chiralcel OD-H, hexane/i-PrOH 40/1, 1 mL/min.
Wieland–Miescher Ketal (2)21
To a flame-dried bottle containing activated 4 Å molecular sieves (6.00 g), Wieland–Miescher ketone 1 (6.00 g, 33.66 mmol, 1.0 equiv), ethane-1,2-diol (100 mL), and PTSA (6.40 g, 33.66 mmol, 1.0 equiv) were sequentially added under argon at room temperature, and the resultant mixture was then stirred at room temperature for 40 min. The reaction was worked up by addition of an ice water solution (40 mL), followed by addition of a saturated aqueous solution of NaHCO3 (80 mL) carefully. The resulting mixture was extracted with ethyl acetate (3 × 80 mL), and the combined extracts were washed with brine (2 × 15 mL) and dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by flash column chromatography on silica gel to give ketal 2.
(S)-8a-Methyl-3,4,8,8a-tetrahydro-2H-spiro[naphthalene-1,2′-[1,3]dioxolan]-6(7H)-one (2)
Yellow oil, 6.36 g (28.61 mmol), 85%, Rf = 0.40 (silica gel, hexane/EtOAc = 2:1, KMnO4 and UV); IR νmax 2950, 2882, 1620, 1332, 1238, 1173, 951, 871 cm–1; 1H NMR (500 MHz, CDCl3) δ 5.77 (d, J = 1.9 Hz, 1H), 3.97–3.90 (m, 4H), 2.43–2.35 (m, 3H), 2.34–2.21 (m, 2H), 1.91–1.83 (m, 1H), 1.78–1.73 (m, 1H), 1.72–1.62 (m, 3H), 1.32 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.3, 167.8, 125.7, 112.5, 65.4, 65.2, 45.1, 34.0, 31.5, 30.1, 27.0, 21.8, 20.6; MS (ESI, m/z) calcd for C13H18NaO3+ [M + Na]+: 245.1148, found 245.1154.
General Procedure A: Enantioselective Synthesis of 1,2-Diketones
To a solution of liquid ammonia (200 mL) was added lithium solid (683 mg, 97.62 mmol, 3.5 equiv) at −78 °C in a portionwise manner over 5 min. To this solution was added a solution of enone 2 (6.20 g, 27.89 mmol, 1.0 equiv) in tetrahydrofuran (THF) (25 mL) in a dropwise manner over 5 min, and the resultant mixture was refluxed at −33 °C for 4 h. To this mixture was added a mixed solution of water/THF (0.5 mL of H2O in 2.0 mL of THF) in a dropwise manner at the same temperature,13 and the reaction mixture was held at reflux at −33 °C for 30 min. To this solution was added a solution of 1-bromobut-2-yne (4.8 mL, 55.78 mmol, 2.0 equiv in 20 mL of THF) at −33 °C in a dropwise manner, and the reaction mixture was maintained at the same temperature for 4 h. The resultant mixture was first evaporated slowly at −33 °C, and then warmed to room temperature, followed by quenching with a saturated solution of NH4Cl (50 mL). The mixture was extracted with ethyl acetate (3 × 50 mL), and the combined extracts were washed with brine (30 mL) and dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by flash column chromatography on silica gel (EtOAc/hexane = 1:10) to give ketone 3.
To a solution of ketone 3 (4.60 g, 16.64 mmol, 1.0 equiv) and ethane-1,2-diol (60 mL) in dichloromethane (DCM) (2 mL) were added PTSA (3.16 g, 16.64 mmol, 1.0 equiv) and 4 Å MS (4.60 g) at room temperature, and the resultant mixture was stirred at the same temperature for 40 min. The reaction was worked up by addition of a mixture of ice water (20 mL) and a saturated aqueous solution of NaHCO3 (40 mL) carefully. After extraction with ethyl acetate (3 × 50 mL), the combined extracts were washed with brine (30 mL) and dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by flash column chromatography on silica gel (EtOAc/hexane = 1:10) to give 4.
To a solution of 4 (2.00 g, 6.24 mmol, 1.0 equiv) in a mixed solvent of MeCN/CCl4/H2O (8/8/12 mL) were added NaIO4 (3.33 g, 15.60 mmol, 2.5 equiv) and RuO2·H2O (19 mg, 0.12 mmol, 0.02 equiv) at 0 °C, and the resultant mixture was stirred at 0 °C for 30 min and then at room temperature for 2 h. The reaction was worked up by addition of a mixture of saturated aqueous NaHCO3 (20 mL) and saturated aqueous Na2S2O3 (15 mL) at room temperature, and the resultant mixture was extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with brine (20 mL) and dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by flash chromatography on silica gel (EtOAc/hexane = 1:10) to give 1,2-diketone 5.
Synthesis of (4aS,5S,8aS)-5-(But-2-yn-1-yl)-8a-methylhexahydro-2H-spiro[naphthalene-1,2′-[1,3]dioxolan]-6(5H)-one (3)
Yellowish oil, 4.62 g (16.82 mmol), 60%, Rf = 0.31 (silica gel, hexane/EtOAc = 5:1, KMnO4); [α]D25 = 2.69 (c = 1.0, in CHCl3); IR νmax 2950, 1710, 1448, 1185, 1107, 1067, 949, 920 cm–1; 1H NMR (500 MHz, CDCl3) δ 4.00–3.84 (m, 4H), 2.53–2.46 (m, 1H), 2.42–2.29 (m, 3H), 2.29–2.23 (m, 1H), 2.03 (td, J = 12.3, 3.3 Hz, 1H), 1.98–1.91 (m, 1H), 1.75–1.67 (m, 7H), 1.59–1.50 (m, 2H), 1.28–1.23 (m, 1H), 1.22 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 210.5, 112.6, 77.4, 76.3, 65.3, 65.1, 49.8, 45.3, 42.4, 37.7, 30.4, 30.2, 25.2, 22.8, 16.0, 14.5, 3.8; MS (ESI, m/z) calcd for C17H25O3+ [M + H]+: 277.1798, found 277.1799.
Synthesis of (4a′S,5′S,8a′S)-5′-(But-2-yn-1-yl)-8a′-methylhexahydro-2′H,5′H-dispiro[[1,3]dioxolane-2,1′-naphthalene-6′,2″-[1,3]dioxolane] (4)
Colorless oil, 3.47 g (10.84 mmol), 65%, Rf = 0.35 (silica gel, hexane/EtOAc = 5:1, KMnO4); [α]D24 = 28.68 (c = 1.0, in CHCl3); IR νmax 2954, 2878, 1439, 1264, 1189, 1127, 1042, 953 cm–1; 1H NMR (500 MHz, CDCl3) δ 4.02–3.77 (m, 8H), 2.35–2.23 (m, 1H), 2.09–2.03 (m, 1H), 1.89 (td, J = 12.4, 3.3 Hz, 1H), 1.80 (dd, J = 17.6, 3.7 Hz, 1H), 1.75 (t, J = 2.6 Hz, 4H), 1.74–1.64 (m, 4H), 1.51 (dd, J = 10.2, 3.7 Hz, 3H), 1.36–1.32 (m, 1H), 1.23–1.16 (m, 1H), 1.04 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 112.8, 110.7, 79.4, 75.1, 65.3, 65.1, 64.9, 64.8, 44.3, 44.1, 42.5, 30.6, 30.3, 27.5, 24.5, 23.2, 16.3, 14.4, 3.8; MS (ESI, m/z) calcd for C19H29O4+ [M + H]+: 321.2060, found 321.2058.
Synthesis of 1-((4a′S,5′S,8a′S)-8a′-Methylhexahydro-2′H,5′H-dispiro[[1,3]dioxolane-2,1′-naphthalene-6′,2′-[1,3]dioxolan]-5′-yl)butane-2,3-dione (5)
Yellowish oil, 1.72 g (4.88 mmol), 78%, Rf = 0.30 (silica gel, hexane/EtOAc = 5:1, KMnO4 and UV); [α]D25 = −31.39 (c = 1.0, in CHCl3); IR νmax 2930, 2354, 1702, 1362, 1288, 1151, 1043, 948 cm–1; 1H NMR (400 MHz, CDCl3) δ 3.96–3.74 (m, 7H), 3.56 (dd, J = 13.5, 6.7 Hz, 1H), 2.71 (dd, J = 13.8, 4.4 Hz, 1H), 2.49–2.31 (m, 1H), 2.26–2.15 (m, 4H), 1.79–1.58 (m, 4H), 1.53–1.36 (m, 4H), 1.35–1.09 (m, 3H), 1.02 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 198.7, 198.3, 112.6, 110.2, 65.3, 65.0, 64.3, 63.0, 43.3, 42.9, 42.6, 35.2, 29.9, 29.6, 27.2, 24.3, 23.8, 22.7, 14.2; MS (ESI, m/z) calcd for C19H28NaO6+ [M + Na]+: 375.1778, found 375.1779. Parameters for UV–vis spectroscopy: instrument model: Shimadzu UV-2450; start WL: 200.00 nm; end WL: 500.00 nm; sample concentration: 1 × 10–3 mol·L–1 FCNIrpic solution in CH3CN (HPLC grade). UV–vis spectra: (λmax: 400 nm) (Figure 4).
Figure 4.

UV–vis spectroscopy of compound 5.
General Procedure B: Optimized Norrish–Yang Cyclization
A solution of 1,2-diketone 5 (200 mg, 0.57 mmol) in chloroform (12 mL) was bubbled with Ar at 0 °C for 40 min, and the resultant mixture was then irradiated with a light lamp (Tornado High Lumen 65 W CDL 865 220V E27, Philips) for 10 h under Ar at room temperature. The solvent was removed under vacuum, and the residue was purified by flash column chromatography on silica gel (EtOAc/hexane = 1:5) to give 6 (188 mg, 96%) as a white solid.
Norrish–Yang Cyclization Product (6)
White solid, 188 mg (0.53 mmol), 96%, Rf = 0.25 (silica gel, hexane/EtOAc = 5:1, KMnO4); [α]D24 = −85.82 (c = 1.0, in CHCl3); IR νmax 3530, 2943, 1784, 1344, 1289, 1186, 1054, 950 cm–1; 1H NMR (500 MHz, CDCl3) δ 4.12–4.04 (m, 1H), 4.04–3.82 (m, 7H), 3.80–3.77 (m, 1H), 2.76 (d, J = 17.7 Hz, 1H), 2.47 (d, J = 17.7 Hz, 1H), 2.19 (dd, J = 12.6, 3.4 Hz, 1H), 1.99–1.86 (m, 2H), 1.73–1.52 (m, 6H), 1.47–1.34 (m, 5H), 1.19 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 210.6, 112.7, 112.5, 88.6, 65.3, 65.1, 64.9, 64.7, 52.6, 45.0, 42.9, 42.1, 30.5, 28.7, 27.6, 24.2, 23.7, 21.2, 16.4; MS (ESI, m/z) calcd for C19H28NaO6+ [M + Na]+: 375.1778, found 375.1779.
Norrish–Yang Cyclization Product (6a)
White solid, 186 mg (0.51 mmol), 93%, Rf = 0.29 (silica gel, hexane/EtOAc = 5:1, KMnO4); [α]D24 = −25.90 (c = 1.0, in CHCl3); IR νmax 3534, 2972, 2879, 1782, 1344, 1111, 1054, 949 cm–1; 1H NMR (500 MHz, CDCl3) δ 4.11–4.03 (m, 1H), 4.00–3.81 (m, 7H), 3.78–3.71 (m, 1H), 2.74 (d, J = 17.5 Hz, 1H), 2.41 (d, J = 17.5 Hz, 1H), 2.16 (dd, J = 12.3, 3.6 Hz, 1H), 2.05–1.86 (m, 3H), 1.79–1.74 (m, 1H), 1.72–1.51 (m, 6H), 1.43–1.32 (m, 2H), 1.17 (s, 3H), 0.96 (t, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 209.4, 112.7, 112.6, 92.0, 65.3, 65.1, 64.8, 64.7, 52.5, 45.1, 43.0, 42.1, 30.5, 28.8, 27.6, 27.1, 24.8, 23.8, 16.7, 8.2; MS (ESI, m/z) calcd for C20H30NaO6+ [M + Na]+: 389.1935, found 389.1934.
Norrish–Yang Cyclization Product (6b)
White solid, 150 mg (0.37 mmol), 75%, Rf = 0.23 (silica gel, hexane/EtOAc = 5:1, KMnO4); [α]D25 = −33.05 (c = 1.0, in CHCl3); IR νmax 3535, 2955, 2875, 1780, 1184, 1081, 1054, 949 cm–1; 1H NMR (500 MHz, CDCl3) δ 4.09–4.03 (m, 1H), 3.99–3.81 (m, 7H), 3.76 (d, J = 6.9 Hz, 1H), 2.75 (d, J = 17.5 Hz, 1H), 2.41 (d, J = 17.5 Hz, 1H), 2.17 (dd, J = 12.2, 3.7 Hz, 1H), 2.00–1.86 (m, 3H), 1.73–1.52 (m, 8H), 1.46–1.33 (m, 2H), 1.32–1.19 (m, 5H), 1.18 (s, 3H), 0.86 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 209.5, 112.7, 112.6, 92.0, 65.3, 65.1, 64.9, 64.7, 52.5, 45.2, 43.0, 42.1, 34.3, 32.5, 30.5, 28.8, 27.6, 24.9, 23.8, 23.5, 22.8, 16.8, 14.2; MS (ESI, m/z) calcd for C23H36NaO6+ [M + Na]+: 431.2404, found 431.2406.
Norrish–Yang Cyclization Product (6c)
White solid, 110 mg (0.26 mmol), 55%, Rf = 0.23 (silica gel, hexane/EtOAc = 5:1, KMnO4); [α]D25 = −37.60 (c = 1.0, in CHCl3); IR νmax 3518, 2885, 1778, 1450, 1268, 1125, 1054, 949 cm–1; 1H NMR (500 MHz, CDCl3) δ 4.13 (s, 1H), 4.08–4.01 (m, 1H), 3.99–3.89 (m, 5H), 3.89–3.82 (m, 1H), 3.79–3.70 (m, 1H), 2.69 (d, J = 17.6 Hz, 1H), 2.43 (d, J = 17.6 Hz, 1H), 2.26–2.21 (m, 2H), 2.13–2.04 (m, 1H), 2.01–1.89 (m, 2H), 1.84–1.66 (m, 6H), 1.61–1.54 (m, 4H), 1.50–1.40 (m, 1H), 1.37–1.31 (m, 1H), 1.23–1.10 (m, 8H); 13C NMR (125 MHz, CDCl3) δ 210.6, 113.1, 112.8, 94.7, 65.3, 65.1, 64.8, 64.5, 54.1, 45.2, 44.2, 43.1, 40.3, 30.4, 30.3, 28.8, 27.9, 27.9, 27.0, 27.0, 26.1, 24.0, 23.8, 17.9; MS (ESI, m/z) calcd for C24H37O6+ [M + H]+: 421.2585, found 421.2586.
Norrish–Yang Cyclization Product (6d)
White solid, 106 mg (0.27 mmol), 53%, Rf = 0.26 (silica gel, hexane/EtOAc = 5:1, KMnO4); [α]D26 = −2.40 (c = 1.0, in CHCl3); IR νmax 3522, 2959, 1774, 1475, 1396, 1122, 1054, 951 cm–1; 1H NMR (500 MHz, CDCl3) δ 4.43 (s, 1H), 4.15–4.02 (m, 1H), 4.01–3.80 (m, 6H), 3.73 (d, J = 6.9 Hz, 1H), 2.66 (d, J = 17.4 Hz, 1H), 2.40 (d, J = 17.4 Hz, 1H), 2.34–2.24 (m, 1H), 2.07–1.93 (m, 2H), 1.82–1.63 (m, 3H), 1.63–1.41 (m, 4H), 1.32 (d, J = 12.2 Hz, 1H), 1.19 (t, J = 19.5 Hz, 12H); 13C NMR (125 MHz, CDCl3) δ 210.4, 113.8, 112.8, 98.3, 65.3, 65.0, 64.7, 64.5, 57.0, 45.1, 44.0, 42.8, 38.0, 30.0, 28.8, 28.0, 27.1, 23.5, 19.2; MS (ESI, m/z) calcd for C22H34NaO6+ [M + Na]+: 417.2248, found 417.2244.
Synthesis of (1R,2R,2′S,4a′S,8a′S)-2-Hydroxy-2,4a′-dimethyl-3-oxooctahydrodispiro-[cyclobutane-1,1′-naphthalene-5′,2″-[1,3]dioxolan]-2′-yl acetate (7a)
Colorless oil, 92 mg (0.26 mmol), 46%, Rf = 0.25 (silica gel, hexane/EtOAc = 5:1, KMnO4); [α]D24 = −46.86 (c = 1.0, in CHCl3); IR νmax 3531, 2879, 1785, 1436, 1289, 1054, 883, 697 cm–1; 1H NMR (500 MHz, CDCl3) δ 5.02 (dd, J = 12.5, 4.4 Hz, 1H), 3.99–3.80 (m, 4H), 3.52 (s, 1H), 3.02 (d, J = 17.3 Hz, 1H), 2.24 (d, J = 17.3 Hz, 1H), 2.19–2.06 (m, 1H), 2.05–1.96 (m, 4H), 1.82 (td, J = 13.6, 4.1 Hz, 1H), 1.78–1.61 (m, 6H), 1.59–1.53 (m, 2H), 1.47–1.37 (m, 4H), 1.21 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 210.8, 169.6, 112.6, 89.0, 81.9, 65.3, 65.2, 46.9, 46.6, 45.9, 43.3, 30.5, 29.0, 25.4, 24.1, 23.6, 21.3, 20.9, 16.6; MS (ESI, m/z) calcd for C19H28NaO6+ [M + Na]+: 375.1778, found 375.1776.
Synthesis of (1R,2R,2′S,4a′S,8a′S)-2-Hydroxy-2′-methoxy-2,4a′-dimethyloctahydrodispiro-[cyclobutane-1,1′-naphthalene-5′,2″-[1,3]dioxolan]-3-one (7b)
White solid, 114 mg (0.35 mmol), 57%, Rf = 0.18 (silica gel, hexane/EtOAc = 5:1, KMnO4); [α]D25 = −51.33 (c = 1.0, in CHCl3); IR νmax 3495, 2875, 1788, 1462, 1234, 1115, 1017, 951 cm–1; 1H NMR (500 MHz, CDCl3) δ 4.29 (s, 1H), 3.99–3.77 (m, 4H), 3.36 (s, 3H), 3.25–3.16 (m, 1H), 2.97 (d, J = 17.0 Hz, 1H), 2.32 (d, J = 17.0 Hz, 1H), 1.91–1.85 (m, 3H), 1.71 (s, 1H), 1.70–1.60 (m, 4H), 1.56 (d, J = 13.3 Hz, 2H), 1.43–1.31 (m, 4H), 1.18 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 211.5, 112.8, 90.6, 88.5, 65.3, 65.1, 57.8, 48.1, 46.7, 46.5, 43.4, 30.6, 28.8, 24.1, 23.8, 23.6, 20.8, 16.7; MS (ESI, m/z) calcd for C18H28NaO5+ [M + Na]+: 347.1829, found 347.1828.
Synthesis of (1R,2R,2′S,4a′S,8a′S)-2′-(Benzyloxy)-2-hydroxy-2,4a′-dimethyloctahydrodispiro-[cyclobutane-1,1′-naphthalene-5′,2″-[1,3]dioxolan]-3-one (7c)
Colorless oil, 110 mg (0.27 mmol), 55%, Rf = 0.19 (silica gel, hexane/EtOAc = 5:1, KMnO4); [α]D24 = 2.31 (c = 1.0, in CHCl3); IR νmax 3495, 2874, 1787, 1398, 1233, 1115, 1017, 886 cm-1; 1H NMR (500 MHz, CDCl3) δ 7.38–7.32 (m, 2H), 7.32–7.28 (m, 1H), 7.28–7.24 (m, 2H), 4.68 (t, J = 9.5 Hz, 1H), 4.52–4.32 (m, 2H), 4.00–3.79 (m, 4H), 3.43 (dd, J = 11.3, 5.1 Hz, 1H), 2.90 (d, J = 17.0 Hz, 1H), 2.17 (d, J = 17.0 Hz, 1H), 1.96–1.94 (m, 2H), 1.86–1.78 (m, 1H), 1.76–1.52 (m, 7H), 1.42–1.30 (m, 4H), 1.20 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 211.4, 137.0, 128.6, 128.3, 128.2, 112.8, 88.7, 87.2, 71.8, 65.2, 65.1, 48.1, 46.7, 46.4, 43.3, 30.5, 28.9, 24.1, 23.9, 23.7, 20.8, 16.7; MS (ESI, m/z) calcd for C24H32NaO5+ [M + Na]+: 423.2142, found 423.2144.
Synthesis of (1R,2R,2′S,4a′S,8a′S)-2′-((tert-Butyldimethylsilyl)oxy)-2-hydroxy-2,4a′-dimethyl-octahydro-dispiro[cyclobutane-1,1′-naphthalene-5′,2″-[1,3]dioxolan]-3-one (7d)
Colorless oil, 148 mg (0.35 mmol), 74%, Rf = 0.48 (silica gel, hexane/EtOAc = 5:1, KMnO4); [α]D24 = −15.48 (c = 1.0, in CHCl3); IR νmax 3513, 2935, 2353, 1789, 1471, 1112, 895, 778 cm–1; 1H NMR (500 MHz, CDCl3) δ 4.50 (s, 1H), 3.99–3.82 (m, 4H), 3.73 (dd, J = 12.0, 4.2 Hz, 1H), 2.94 (d, J = 17.2 Hz, 1H), 2.32 (d, J = 17.2 Hz, 1H), 2.20–2.05 (m, 1H), 1.86 (d, J = 6.5 Hz, 1H), 1.78–1.61 (m, 6H), 1.56 (dd, J = 11.1, 2.5 Hz, 1H), 1.53–1.46 (m, 1H), 1.37 (s, 4H), 1.19 (s, 3H), 0.86 (s, 9H), 0.09 (d, J = 5.6 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 211.4, 112.9, 88.6, 82.7, 65.2, 65.1, 48.3, 46.6, 46.4, 43.2, 30.5, 29.1, 28.9, 25.9, 24.2, 23.8, 20.7, 17.9, 16.9, −4.4, −4.9; MS (ESI, m/z) calcd for C23H40NaO5Si+ [M + Na]+: 447.2537, found 447.2537.
Synthesis of (1R,2R,2′S,4a′S,8a′S)-2′-((tert-Butyldiphenylsilyl)oxy)-2-hydroxy-2,4a′-dimethylocta-hydrodispiro[cyclobutane-1,1′-naphthalene-5′,2″-[1,3]dioxolan]-3-one (7e)
Colorless oil, 132 mg (0.24 mmol), 66%, Rf = 0.24 (silica gel, hexane/EtOAc = 10:1, KMnO4); [α]D25 = −7.12 (c = 1.0, in CHCl3); IR νmax 3501, 2875, 1788, 1471, 1393, 1103, 966, 822 cm–1; 1H NMR (500 MHz, CDCl3) δ 7.70 (dd, J = 8.0, 1.4 Hz, 2H), 7.62 (dd, J = 8.0, 1.4 Hz, 2H), 7.49–7.32 (m, 6H), 4.70 (s, 1H), 3.89–3.63 (m, 5H), 2.94 (d, J = 17.4 Hz, 1H), 2.48 (d, J = 17.4 Hz, 1H), 2.13–2.02 (m, 1H), 1.75–1.58 (m, 5H), 1.54–1.46 (m, 1H), 1.44 (s, 3H), 1.41–1.35 (m, 1H), 1.33–1.19 (m, 3H), 1.16 (s, 3H), 1.05 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 212.1, 136.4, 136.2, 133.7, 131.9, 130.4, 130.0, 128.0, 127.7, 112.7, 88.8, 84.0, 65.1, 65.0, 48.6, 46.7, 46.4, 43.1, 30.5, 29.0, 28.5, 26.9, 24.1, 23.8, 20.9, 19.4, 17.0; MS (ESI, m/z) calcd for C33H45O5Si+ [M + H]+: 549.3031, found 549.3032.
Experimental Procedure for the Synthesis of Product 8
To a solution of 6 (200 mg, 0.57 mmol) in MeOH (20 mL) was added Pb(OAc)4 (277 mg, 0.60 mmol) at 0 °C in one portion. The mixture was stirred at 0 °C for 1 h, followed by another 4 h at 50 °C. The reaction was quenched with a saturated aqueous solution of Na2CO3 (10 mL) at room temperature and extracted with ethyl acetate (3 × 20 mL). The organic extracts were washed with brine (15 mL), dried over Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (EtOAc/hexane = 1:10) to afford 8.
Synthesis of Methyl 2-((4a′R,5′R,8a′S)-5′-acetyl-8a′-methylhexahydro-2′H,5′H-dispiro[[1,3]dioxolane-2,1′-naphthalene-6′,2″-[1,3]dioxolan]-5′-yl)acetate (8)
Colorless oil, 210 mg (0.55 mmol), 92%, Rf = 0.20 (silica gel, hexane/EtOAc = 5:1, KMnO4); 1H NMR (500 MHz, CDCl3) δ 4.01–3.76 (m, 8H), 3.56 (s, 3H), 2.84 (d, J = 13.9 Hz, 1H), 2.56 (d, J = 13.9 Hz, 1H), 2.37 (s, 3H), 2.29 (td, J = 14.0 Hz, 1H), 2.15 (dd, J = 12.8, Hz, 1H), 1.91 (td, J = 13.7 Hz, 1H), 1.79–1.73 (m, 1H), 1.72–1.63 (m, 3H), 1.61–1.39 (m, 3H), 1.37–1.30 (m, 1H), 0.92 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 209.8, 172.4, 112.7, 110.8, 65.4, 65.0, 64.6, 63.4, 61.8, 51.4, 48.3, 43.7, 38.7, 31.5, 30.3, 27.4, 27.2, 23.7, 23.5, 15.4; MS (ESI, m/z) calcd for C20H30NaO7+ [M + Na]+: 405.1884, found 405.1885.
Synthesis of Methyl 2-((4a′R,5′S,8a′S)-8a′-methyl-5′-propionylhexahydro-2′H,5′H-dispiro[[1,3]-dioxolane-2,1′-naphthalene-6′,2″-[1,3]dioxolan]-5′-yl)acetate (8a)
Colorless oil, 285 mg (0.72 mmol), 83%, Rf = 0.22 (silica gel, hexane/EtOAc = 5:1, KMnO4); 1H NMR (400 MHz, CDCl3) δ 4.05–3.75 (m, 8H), 3.54 (s, 3H), 2.91–2.72 (m, 2H), 2.69–2.50 (m, 2H), 2.30 (m, 1H), 2.16 (m, 1H), 1.92 (td, J = 13.7 Hz, 1H), 1.83–1.38 (m, 6H), 1.38–1.20 (m, 2H), 1.09 (t, J = 7.0 Hz, 3H), 0.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 212.26, 172.49, 112.77, 110.96, 65.42, 64.93, 64.61, 63.26, 61.78, 51.44, 48.34, 43.63, 38.77, 36.16, 30.34, 27.45, 27.25, 23.80, 23.48, 15.53, 8.85.; MS (ESI, m/z) calcd for C21H32NaO7+ [M + Na]+: 419.2040, found 419.2043.
Synthesis of Methyl 2-((4aR,5R,6S,8aS)-5-acetyl-6-methoxy-8a-methyloctahydro-2H-spiro[naphthalene-1,2′-[1,3]dioxolan]-5-yl)acetate (8b)
Colorless oil, 226 mg (0.64 mmol), 76%, Rf = 0.37 (silica gel, hexane/EtOAc = 4:1, KMnO4); 1H NMR (300 MHz, CDCl3) δ 3.99–3.76 (m, 4H), 3.59 (s, 3H), 3.27 (s, 3H), 3.16–3.00 (m, 2H), 2.34 (s, 3H), 2.17 (d, J = 13.5 Hz, 1H), 1.98 (d, J = 4.0 Hz, 2H), 1.82–1.28 (m, 9H), 0.88 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 210.86, 172.34, 112.80, 85.80, 65.33, 64.96, 57.54, 57.07, 51.42, 50.11, 44.15, 43.73, 31.88, 30.31, 28.66, 23.39, 23.13, 22.31, 16.05; MS (ESI, m/z) calcd for C19H30NaO6+ [M + Na]+: 377.1935, found 377.1930.
Synthesis of Methyl 2-((4aR,5R,6S,8aS)-5-acetyl-6-((tert-butyldimethylsilyl)oxy)-8a-methyloctahydro-2H-spiro[naphthalene-1,2′-[1,3]dioxolan]-5-yl)acetate (8c)
Colorless oil, 298 mg (0.66 mmol), 61%, Rf = 0.21 (silica gel, hexane/EtOAc = 4:1, KMnO4); 1H NMR (500 MHz, CDCl3) δ 4.03–3.76 (m, 5H), 3.66 (s, 3H), 2.79 (d, J = 14.4 Hz, 1H), 2.70 (d, J = 14.4 Hz, 1H), 2.36 (s, 3H), 2.07–1.94 (m, 1H), 1.86 (dd, J = 12.4, 2.6 Hz, 1H), 1.76 (dt, J = 13.1, 3.8 Hz, 2H), 1.69–1.51 (m, 5H), 1.50–1.38 (m, 2H), 1.38–1.23 (m, 3H), 0.92 (s, 3H), 0.89 (s, 9H), 0.11 (d, J = 18.3 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 211.77, 171.74, 113.05, 74.51, 65.38, 64.99, 57.94, 51.54, 46.62, 43.39, 38.79, 31.88, 30.51, 28.81, 28.54, 26.07, 23.50, 22.99, 18.20, 16.94, −4.00, −4.79; MS (ESI, m/z) calcd for C24H42NaSiO6+ [M + Na]+: 477.2643, found 477.2643.
Experimental Procedure for the Synthesis of Product 9
To a solution of 6 (200 mg, 0.57 mmol) in DCM (20 mL) was added boron trifluoride etherate (48%, 154 μL, 0.57 mmol) at −78 °C in a dropwise manner, and the resultant reaction mixture was warmed up to room temperature over 4 h. The reaction mixture was quenched by addition of an aq. HCl (1.0 M, 600 μL) at room temperature, and the mixture was extracted with ethyl acetate (3 × 20 mL). The combined extracts were washed with brine (15 mL) and dried over Na2SO4. The solvent was removed under vacuum, and the residue was purified by flash column chromatography on silica gel (EtOAc/hexane = 1:10) to give 9.
(5aS,9aS)-2-Acetyl-5a-methyl-5,5a,7,8,9,9a-hexahydronaphtho[2,1-b]furan-6(4H)-one (10)
Colorless oil, 70 mg (0.28 mmol), 50%, Rf = 0.38 (silica gel, hexane/EtOAc = 3:1, KMnO4); [α]D26 = −17.66 (c = 1.0, in CHCl3); IR νmax 2949, 2934, 1668, 1516, 1178, 1118, 1090, 914 cm–1; 1H NMR (500 MHz, CDCl3) δ 7.05 (s, 1H), 2.85–2.56 (m, 4H), 2.40 (s, 3H), 2.31 (d, J = 13.1 Hz, 1H), 2.15 (dd, J = 13.8, 6.3 Hz, 2H), 2.02 (d, J = 8.9 Hz, 1H), 1.79 (t, J = 9.2 Hz, 3H), 1.03 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 214.0, 186.2, 155.1, 152.1, 121.9, 117.2, 47.8, 42.3, 37.0, 28.9, 25.8, 25.7, 23.6, 20.4, 15.4; MS (ESI, m/z) calcd for C15H19O3+ [M + H]+: 247.1329, found 247.1329.
Experimental Procedure for the Synthesis of Product 10
To a solution of 6 (200 mg, 0.57 mmol) in THF (20 mL) was added lithium tri-tert-butoxyaluminum hydride (290 mg, 1.14 mmol) at 0 °C in a portionwise manner under Ar, and the resultant mixture was then warmed up to room temperature and stirred at room temperature for 4 h. The reaction was carefully quenched by addition of a saturated aqueous solution of potassium sodium tartrate (20 mL) at 0 °C, and the resulting mixture was stirred vigorously for another 4 h. The mixture was extracted with ethyl acetate (3 × 20 mL), and the combined extracts were washed with brine (15 mL) and dried over Na2SO4. The solvent was removed under vacuum. The residue was purified by flash column chromatography on silica gel (EtOAc/hexane = 1:5) to give 10.
Compound 10
Colorless oil, 173 mg (0.49 mmol), 86%, Rf = 0.28 (silica gel, hexane/EtOAc = 2:1, KMnO4); IR νmax 3456, 2939, 2883, 1192, 1078, 914, 732, 481 cm–1; 1H NMR (400 MHz, CDCl3) δ 4.15–3.75 (m, 9H), 3.67 (s, 1H), 3.65–3.54 (m, 1H), 2.12 (dd, J = 14.6, 7.9 Hz, 1H), 1.87 (dd, J = 10.1, 5.4 Hz, 3H), 1.70–1.54 (m, 5H), 1.54–1.45 (m, 2H), 1.39 (s, 3H), 1.24 (d, J = 24.6 Hz, 2H), 1.14 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 210.55, 113.07, 112.78, 94.68, 65.31, 65.09, 64.84, 64.50, 54.09, 45.15, 44.21, 43.11, 40.32, 30.36, 30.26, 28.77, 27.93, 27.87, 27.03, 26.98, 26.13, 24.04, 23.80, 17.86; MS (ESI, m/z) calcd for C15H19NaO3+ [M + Na]+: 377.1935, found 377.1934.
Experimental Procedure for the Synthesis of Product 11
To a solution of 6 (200 mg, 0.57 mmol) in THF (20 mL) was added methyllithium (1.79 mL, 2.85 mmol, 1.6 M) at 0 °C slowly under Ar, and the resultant mixture was then warmed up to room temperature and stirred at room temperature for 8 h. The reaction was carefully quenched by addition of a saturated aqueous solution of NH4Cl (20 mL) at 0 °C. The mixture was extracted with ethyl acetate (3 × 20 mL), and the combined extracts were washed with brine (15 mL) and dried over Na2SO4. The solvent was removed under vacuum. The residue was purified by flash column chromatography on silica gel (EtOAc/hexane = 1:5) to give 11.
Compound 11
Colorless oil, 185 mg (0.69 mmol), 97%, Rf = 0.25 (silica gel, hexane/EtOAc = 2:1, KMnO4); IR νmax 3422, 2948, 2882, 1438, 1191, 1110, 952, 733 cm–1; 1H NMR (400 MHz, CDCl3) δ 4.72 (s, 1H), 4.17–3.77 (m, 9H), 3.48 (s, 1H), 1.98–1.79 (m, 5H), 1.75 (d, J = 14.6 Hz, 1H), 1.64 (m, 5H), 1.40 (s, 3H), 1.38–1.31 (m, 1H), 1.18 (s, 3H), 1.15 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 210.55, 113.07, 112.78, 94.68, 65.31, 65.09, 64.84, 64.50, 54.09, 45.15, 44.21, 43.11, 40.32, 30.36, 30.26, 28.77, 27.93, 27.87, 27.03, 26.98, 26.13, 24.04, 23.80, 17.86; MS (ESI, m/z) calcd for C15H19NaO3+ [M + Na]+: 391.2091, found 391.2090.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (Grant No. 2018YFC0310905), the National Science Foundation of China (Grant Nos. 21632002 and 21871012), the Guangdong Provincial Key Research and Development Program (Grant No. 2020B0303070002), and the Shenzhen Outstanding Talents Training Fund.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c02054.
The authors declare no competing financial interest.
Supplementary Material
References
- For a review, see:; Leemans E.; D’hooghe M.; De Kimpe N. Ring Expansion of Cyclobutylmethylcarbenium Ions to Cyclopentane or Cyclopentene Derivatives and Metal-Promoted Analogous. Chem. Rev. 2011, 111, 3268–3333. 10.1021/cr100295j. [DOI] [PubMed] [Google Scholar]
- a Conia J. M.; Salaun J. R. Cyclobutane Ring Contractions Not Involving Carbonium Ions. Acc. Chem. Res. 1972, 5, 33–40. 10.1021/ar50049a005. [DOI] [Google Scholar]; b Carpino L. A.; Tsao J.-H. A Novel Ring Enlargement Involving Electrophilic Attack on the Dienolate Anion Derived from 7,8-Bis(trimethylsilyIoxy)-cis-bicyclo[4.2.0]octa-3,7-diene. J. Org. Chem. 1979, 44, 2564–2566. 10.1021/jo01328a049. [DOI] [Google Scholar]; c Trost B. M.; Vladuchick W. C.; Bridges A. J. Sulfur as a Regiochemical Control Element. Synthesis of 2-Alkoxy(acyloxy)-3-alkyl(aryl)thiobuta-1,3-dienes. J. Am. Chem. Soc. 1980, 102, 3548–3554. 10.1021/ja00530a040. [DOI] [Google Scholar]; d Sugimura T.; Paquette L. A. Enantiospecific Total Synthesis of the Sesquiterpene Antibiotics (-)-Punctatin A and (+)-Punctatin D. J. Am. Chem. Soc. 1987, 109, 3017–3024. 10.1021/ja00244a025. [DOI] [Google Scholar]; e Brown M. J.; Harrison T.; Herrinton P. M.; Hopkins M. H.; Hutchinson K. D.; Mishra P.; Overman L. E. Acid-Promoted Reaction of Cyclic Allylic Diols with Carbonyl Compounds. Stereoselective Ring-Enlarging Tetrahydrofuran Annulations. J. Am. Chem. Soc. 1991, 113, 5365–5378. 10.1021/ja00014a031. [DOI] [Google Scholar]; f Jenkins T. J.; Burnell D. J. Lewis Acid Catalyzed Geminal Acylation Reaction of Ketones with l,2-Bis((trimethylsilyl)oxy)cyclobutene: Direct Formation of 2,2-Disubstituted 1,3-Cyclopentanediones. J. Org. Chem. 1994, 59, 1485–1491. 10.1021/jo00085a041. [DOI] [Google Scholar]; g Lin X.-D.; Kavash R. W.; Mariano P. S. Two Interrelated Strategies for Cephalotaxine Synthesis. J. Org. Chem. 1996, 61, 7335–7347. 10.1021/jo961179s. [DOI] [PubMed] [Google Scholar]; h Gatling S. C.; Jackson J. E. Reactivity Control via Dihydrogen Bonding: Diastereoselection in Borohydride Reductions of αr-Hydroxyketones. J. Am. Chem. Soc. 1999, 121, 8655–8656. 10.1021/ja991784n. [DOI] [Google Scholar]; i Larock R. C.; Reddy C. K. 2-Alkylidenecyclopentanones via Palladium-Catalyzed Cross-Coupling of 1-(1-Alkynyl)cyclobutanols and Aryl or Vinylic Halides. Org. Lett. 2000, 2, 3325–3327. 10.1021/ol000219i. [DOI] [PubMed] [Google Scholar]; j Booker-Milburn K. I.; Baker J. R.; Bruce I. Rapid Access to Azepine-Fused Oxetanols from Alkoxy-Substituted Maleimides. Org. Lett. 2004, 6, 1481–1484. 10.1021/ol049645k. [DOI] [PubMed] [Google Scholar]; k Bray C. D.; Pattenden G. A biogenetically patterned synthetic approach to the unusual furan methylenecyclobutanol moiety in providencin. Tetrahedron Lett. 2006, 47, 3937–3939. 10.1016/j.tetlet.2006.03.158. [DOI] [Google Scholar]; l Álvarez-Dorta D.; Leon E. I.; Kennedy A. R.; Riesco-Fagundo C.; Suarez E. Sequential Norrish TypeII Photoelimination and Intramolecular Aldol Cyclization of 1,2-Diketones in Carbohydrate Systems: Stereoselective Synthesis of Cyclopentitols. Angew. Chem., Int. Ed. 2008, 47, 8917–8919. 10.1002/anie.200803696. [DOI] [PubMed] [Google Scholar]; m Renata H.; Zhou Q.; Baran P. S. Strategic Redox Relay Enables A Scalable Synthesis of Ouabagenin, A Bioactive Cardenolide. Science 2013, 339, 59–63. 10.1126/science.1230631. [DOI] [PMC free article] [PubMed] [Google Scholar]; n Ignatenko V. A.; Tochtrop G. P. Approach for Expanding Triterpenoid Complexity via Divergent Norrish-Yang Photocyclization. J. Org. Chem. 2013, 78, 3821–3831. 10.1021/jo400275p. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Ishida N.; Necas D.; Masuda Y.; Murakami M. Enantioselective Construction of 3-Hydroxypiperidine Scaffolds by Sequential Action of Light and Rhodium upon N-Allylglyoxylamides. Angew. Chem., Int. Ed. 2015, 54, 7418–7421. 10.1002/anie.201502584. [DOI] [PubMed] [Google Scholar]; p Porcu S.; Demuro S.; Luridiana A.; Cocco A.; Frongia A.; Aitken D. J.; Charnay-Pouget F.; Guillot R.; Sarais G.; Secci F. Brønsted Acid Mediated Cascade Reaction To Access 3-(2-Bromoethyl)benzofurans. Org. Lett. 2018, 20, 7699–7702. 10.1021/acs.orglett.8b03429. [DOI] [PubMed] [Google Scholar]; q Peez T.; Veronika S.; Harms K.; Koert U. Synthesis of Naphtho-cyclobutenes from α-Naphthyl Acrylates by Visible-Light Energy-Transfer Catalysis. Org. Lett. 2019, 21, 4365–4369. 10.1021/acs.orglett.9b01585. [DOI] [PubMed] [Google Scholar]; r Turnu F.; Luridiana A.; Cocco A.; Porcu S.; Frongia A.; Sarais G.; Secci F. Catalytic Tandem Friedel–Crafts Alkylation/C4–C3 Ring-Contraction Reaction: An Efficient Route for the Synthesis of Indolyl Cyclopropane-carbaldehydes and Ketones. Org. Lett. 2019, 21, 7329–7332. 10.1021/acs.orglett.9b02617. [DOI] [PubMed] [Google Scholar]; s Cuccu F.; Serusi L.; Luridiana A.; Secci F.; Caboni P.; Aitken D. J.; Frongia A. Tandem Wittig Reaction–Ring Contraction of Cyclobutanes: A Route to Functionalized Cyclopropanecarbaldehydes. Org. Lett. 2019, 21, 7755–7758. 10.1021/acs.orglett.9b02690. [DOI] [PubMed] [Google Scholar]
- Cope S. M.; Tailor D.; Nagorski R. W. Determination of the pKa of Cyclobutanone: Brønsted Correlation of the General Base-Catalyzed Enolization in Aqueous Solution. J. Org. Chem. 2011, 76, 380–390. 10.1021/jo101369w. [DOI] [PubMed] [Google Scholar]
- a Cope A. C.; Herrick E. C. Synthesis of Bicyclo [4.2.0] octane-7,8-diol, A Derivative of “Cycloöctatetraene Dichloride”. J. Am. Chem. Soc. 1950, 72, 983–987. 10.1021/ja01158a088. [DOI] [Google Scholar]; b Anderson D. R.; Koch T. H. 2,3-Bis(trimethylsilyloxy)-1,3-butadiene as a useful reactive diene in the Diels-Alder reaction. J. Org. Chem. 1978, 43, 2726–2728. 10.1021/jo00407a048. [DOI] [Google Scholar]; c Trost B. M.; Godleski S. A.; Ippen J. Stereocontrolled approach to 1,4-disubstituted 1,3-dienes. J. Org. Chem. 1978, 43, 4559–4564. 10.1021/jo00418a001. [DOI] [Google Scholar]; d Brady W. T.; Lloyd R. M. Halogenated Ketenes. 33. Cycloaddition of Ketenes and Trimethylsilyl Enol Ethers. J. Org. Chem. 1980, 45, 2025–2028. 10.1021/jo01298a064. [DOI] [Google Scholar]; e Manfrotto C.; Mella M.; Freccero M.; Fagnoni M.; Albini A. Photochemical Synthesis of 4-Oxobutanal Acetals and of 2-Hydroxycyclobutanone Ketals. J. Org. Chem. 1999, 64, 5024–5028. 10.1021/jo982053t. [DOI] [PubMed] [Google Scholar]; f Shimoda K.; Yamaoka Y.; Yoo D.; Yamada K.; Takikawa H.; et al. Total Syntheses of Allelopathic 4-Oxyprotoilludanes, Melleolides, and Echinocidins. J. Org. Chem. 2019, 84, 11014–11024. 10.1021/acs.joc.9b01589. [DOI] [PubMed] [Google Scholar]
- Yang N. C.; Yang D.-D. H. Photochemical Reactions of Ketones in Solution. J. Am. Chem. Soc. 1958, 80, 2913–2914. 10.1021/ja01544a092. [DOI] [Google Scholar]
- a Yoshioka S.; Nagatomo M. M.; Inoue M. Application of Two Direct C(sp3)–H Functionalizations for Total Synthesis of (+)-Lactacystin. Org. Lett. 2015, 17, 90–93. 10.1021/ol503291s. [DOI] [PubMed] [Google Scholar]; b Kawamata T.; Nagatomo M. M.; Inoue M. Total Synthesis of Zaragozic Acid C: Implementation of Photochemical C(sp3)–H Acylation. J. Am. Chem. Soc. 2017, 139, 1814–1817. 10.1021/jacs.6b13263. [DOI] [PubMed] [Google Scholar]
- a Urry W. H.; Trecker D. J. Photochemical reactions of 1,2-diketones. J. Am. Chem. Soc. 1962, 84, 118–120. 10.1021/ja00860a034. [DOI] [Google Scholar]; b Turro N. J.; Lee T. Molecular photochemistry. XX. Intramolecular photoreduction of alkyl α-diketones. J. Am. Chem. Soc. 1969, 91, 5651–5652. 10.1021/ja01048a045. [DOI] [Google Scholar]; c Bishop R.; Hamer N. K. 2-Hydroxycyclopentenone Derivatives from Photocyclisation of α, β-unsaturated 1, 2-Diketones: a Suggested Triplet Biradical Intermediate. J. Chem. Soc. C 1970, 1193–1197. 10.1039/J39700001193. [DOI] [Google Scholar]; d Aoyama H.; Hasegawa T.; Watabe M.; Shiraishi H.; Omote Y. Photochemical reactions of N, N-disubstituted α-oxoamides. J. Org. Chem. 1978, 43, 419–422. 10.1021/jo00397a009. [DOI] [Google Scholar]; e Hamer N. K. Photocyclisation of α-bromomethyl-1,2-diketones. Tetrahedron Lett. 1982, 23, 473–474. 10.1016/S0040-4039(00)86864-1. [DOI] [Google Scholar]; f Hamer N. K. A new route to cyclopentane-1,3-diones. Tetrahedron Lett. 1986, 27, 2167–2168. 10.1016/S0040-4039(00)84477-9. [DOI] [Google Scholar]
- a Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis; In Christoffers J.; Baro A., Eds.; Wiley-VCH: Weinheim, 2005; pp 1–335. [Google Scholar]; b Quasdorf K. W.; Overman L. E. Catalytic Enantioselective Synthesis of Quaternary Carbon Stereocentres. Nature 2014, 516, 181–191. 10.1038/nature14007. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Liu Y.; Han S.-J.; Liu W.-B.; Stoltz B. M. Catalytic Enantioselective Construction of Quaternary Stereocenters: Assembly of Key Building Blocks for the Synthesis of Biologically Active Molecules. Acc. Chem. Res. 2015, 48, 740–751. 10.1021/ar5004658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sannigrahi M. Stereocontrolled Synthesis of Spirocyclics. Tetrahedron 1999, 55, 9007–9071. 10.1016/S0040-4020(99)00482-2. [DOI] [Google Scholar]; b D’yakonov V.; Trapeznikova O. A.; de Meijere A.; Dzhemilev U. M. Metal Complex Catalysis in the Synthesis of Spirocarbocycles. Chem. Rev. 2014, 114, 5775–5814. 10.1021/cr400291c. [DOI] [PubMed] [Google Scholar]; c Xu P.-W.; Yu J.-S.; Chen C.; Cao Z.-Y.; Zhou F.; Zhou J. Catalytic Enantioselective Construction of Spiro Quaternary Carbon Stereocenters. ACS Catal. 2019, 9, 1820–1882. 10.1021/acscatal.8b03694. [DOI] [Google Scholar]
- a Natarajan A.; Wang K.; Ramamurthy V.; Scheffer J. R.; Patrick B. O. Control of Enantioselectivity in the Photochemical Conversion of α-Oxoamides into β-Lactam Derivatives. Org. Lett. 2002, 4, 1443–1446. 10.1021/ol025700i. [DOI] [PubMed] [Google Scholar]; b Griesbeck A. G.; Heckroth H. Stereoselective Synthesis of 2-Aminocyclobutanols via Photocyclization of α-Amido Alkylaryl Ketones: Mechanistic Implications for the Norrish-Yang Reaction. J. Am. Chem. Soc. 2002, 124, 396–403. 10.1021/ja0111941. [DOI] [PubMed] [Google Scholar]; c Moorthy J. N.; Samanta S.; Koner A. L.; Saha S.; Nau W. M. Intramolecular O–H···O Hydrogen-Bond-Mediated Reversal in the Partitioning of Conformationally Restricted Triplet 1,4-Biradicals and Amplification of Diastereodifferentiation in Their Lifetimes. J. Am. Chem. Soc. 2008, 130, 13608–13617. 10.1021/ja8032179. [DOI] [PubMed] [Google Scholar]
- a Rahbæk L.; Christophersen C.; Frisvad J.; Bengaard H. S.; Larsen S.; Rassing B. R. Insulicolide A: A New Nitrobenzoyloxy-Substituted Sesquiterpene from the Marine Fungus Aspergillus insulicola. J. Nat. Prod. 1997, 60, 811–813. 10.1021/np970142f. [DOI] [Google Scholar]; b Rigano D.; Aviello G.; Bruno M.; Formisano C.; Rosselli S.; Capasso R.; Senatore F.; Izzo A. A.; Borrelli F. Antispasmodic Effects and Structure–Activity Relationships of Labdane Diterpenoids from Marrubium globosum ssp. Libanoticum. J. Nat. Prod. 2009, 72, 1477–1481. 10.1021/np9002756. [DOI] [PubMed] [Google Scholar]; c Omura S.; Tomoda H.; Kim Y. K.; Nishida H. Pyripyropenes, Highly Potent Inhibitors of Acyl-CoA: Cholesterol Acyltransferase Product by Aspergillus fumigatus. J. Antibiot. 1993, 46, 1168–1169. 10.7164/antibiotics.46.1168. [DOI] [PubMed] [Google Scholar]; d Brundret K. M.; Dalziel W.; Hesp B.; Jarvis J. A. J.; Neidle S. X-Ray crystallographic determination of the structure of the antibiotic aphidicolin: a tetracyclic diterpenoid containing a new ring system. J. Chem. Soc., Chem. Commun. 1972, 1027–1028. 10.1039/c39720001027. [DOI] [Google Scholar]; e Fan Y. Y.; Zhang H.; Zhou Y.; Liu H. B.; Yue J. M.; et al. Phainanoids A–F, A New Class of Potent Immunosuppressive Triterpenoids with an Unprecedented Carbon Skeleton from Phyllanthus hainanensis. J. Am. Chem. Soc. 2015, 137, 138–141. 10.1021/ja511813g. [DOI] [PubMed] [Google Scholar]; f Li F. Z.; Li S.; Zhang P. P.; Huang Z.-H.; Zhang W. B.; Gong J.; Yang Z. A chiral pool approach for asymmetric syntheses of (−)-antrocin, (+)-asperolide C, and (−)-trans-ozic acid. Chem. Commun. 2016, 52, 12426–12429. 10.1039/C6CC06794H. [DOI] [PubMed] [Google Scholar]
- de Arriba A. L. F.; Simon L.; Raposo C.; Alcazar V.; Moran J. R. Proline imidazolidinones and enamines in Hajos–Wiechert and Wieland–Miescher ketone synthesis. Tetrahedron 2009, 65, 4841–4845. 10.1016/j.tet.2009.04.050. [DOI] [Google Scholar]
- a Mewshaw R. E.; Taylor M. D.; Smith A. B. III Indole diterpene synthetic studies. 2. First-generation total synthesis of (-)-paspaline. J. Org. Chem. 1989, 54, 3449–3462. 10.1021/jo00275a035. [DOI] [Google Scholar]; b Ali A.; Guile S. D.; Saxton J. E.; Thornton-Pett M. Synthetic Studies Towards Paspalicine: Preliminary Investigations, and the Synthesis of 3′,4′,7′,7′a,9′,10′,11′,11′a-octahydro-4′,4′,7′a-trimethylspiro[1,3-dioxolane]-2,8′(6′H)-2′H-3′,5′a-epoxynaphth[2,1-b]oxepin-2′-one. Tetrahedron 1991, 47, 6407–6426. 10.1016/S0040-4020(01)86569-8. [DOI] [Google Scholar]
- Lee-Ruff E.New Synthetic Pathways from Cyclobutanones. In Advances in Strain in Organic Chemistry; Halton B., Ed.; JAI Press: Greenwich, CT, 1991; Vol. 1. [Google Scholar]
- Sandros K.; Bäckström H. L. J.; et al. Transfer of Triplet State Energy in Fluid Solutions. II. Further Studies of the Quenching of Biacetyl Phosphorescence in Solution. Acta Chem. Scand. 1962, 16, 958–968. 10.3891/acta.chem.scand.16-0958. [DOI] [Google Scholar]
- a Bäckström H. L. J.; Sandros K. Quenching of Biacetyl Fluorescence in Solution. J. Chem. Phys. 1955, 23, 2197. 10.1063/1.1740706. [DOI] [Google Scholar]; b Turro N. J.; Engel R. Quenching of biacetyl fluorescence and phosphorescence. J. Am. Chem. Soc. 1969, 91, 7113–7121. 10.1021/ja01053a037. [DOI] [Google Scholar]
- Selected examples, see:; a Reeder L. M.; Hegedus L. S. Effect of β-Substituents on the Regioselectivity of the Diazomethane Ring Expansion of α-Methyl-α-methoxycyclobutanones to Cyclopentanones. J. Org. Chem. 1999, 64, 3306–3311. 10.1021/jo990226o. [DOI] [PubMed] [Google Scholar]; b Gadwood R. C.; Mallick I. M.; DeWinter A. J. Ring expansion of cyclobutanones to cyclopentanones via. alpha.-lithioalkyl aryl sulfoxides and selenoxides. J. Org. Chem. 1987, 52, 774–782. 10.1021/jo00381a013. [DOI] [Google Scholar]; c Fukuzawa S.; Tsuchimoto T. A facile conversion of substituted cyclobutanones to cyclopentanones by CH2I2/SmI2 system. Tetrahedron Lett. 1995, 36, 5937–5938. 10.1016/0040-4039(95)01199-R. [DOI] [Google Scholar]; d Zhou X.; Dong G. (4+1) vs (4+2): Catalytic Intramolecular Coupling between Cyclobutanones and Trisubstituted Allenes via C–C Activation. J. Am. Chem. Soc. 2015, 137, 13715–13721. 10.1021/jacs.5b09799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a He H.; Fang W.; Phillips D. L. Photochemistry of Butyrophenone: Combined Complete-Active-Space Self-Consistent Field and Density Functional Theory Study of Norrish Type I and II Reactions. J. Phys. Chem. A 2004, 108, 5386–5392. 10.1021/jp037735l. [DOI] [Google Scholar]; For the ab initio calculation of H-bonding determinded diasteroselectivity, see:; b Sinicropi A.; Barbosa F.; Basosi R.; Giese B.; Olivucci M. Mechanism of the Norrish-Yang Photocyclization Reaction of an Alanine Derivative in the Singlet State: Origin of the Chiral-Memory Effect. Angew. Chem., Int. Ed. 2005, 44, 2390–2393. 10.1002/anie.200461898. [DOI] [PubMed] [Google Scholar]
- a Wagner P. J.; Kochevar I. E.; Kemppainen A. E. Type II photoprocesses of phenyl ketones. Procedures for determining meaningful quantum yields and triplet lifetimes. J. Am. Chem. Soc. 1972, 94, 7489–7494. 10.1021/ja00776a034. [DOI] [Google Scholar]; b Scheffer J. R.; Scott C.. Crystal Structure-Solid State Reactivity Relationships: Toward a Greater Understanding of Norrish/Yang Type II Photochemistry. In Organic Photochemistry and Photobiology, 2nd ed.; CRC Press: Boca Raton, FL, 2003. [Google Scholar]
- Peerrin D. D.; Armarego W. L.; Perrins D. R.. Purification of Laboratory Chemicals; Pergamon Press: Oxford, 1980. [Google Scholar]
- Ciceri P.; Demnitz F. W. J.. An efficient, rapid and highly selective preparation of the Wieland-Miescher ketone-9-ethylene ketal. Tetrahedron Lett.; 1997; Vol. 38, pp 389–390. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.