

Abstract
Genome-wide association studies (GWASs) have identified dozens of loci associated with risk of chronic obstructive pulmonary disease (COPD). However, identifying the causal variants and their functional role in the appropriate cell type remains a major challenge. We aimed to identify putative causal variants in 82 GWAS loci associated with COPD susceptibility and predict the regulatory impact of these variants in lung-cell types. We used an integrated approach featuring statistical fine mapping, open chromatin profiling, and machine learning to identify functional variants. We generated chromatin accessibility data using the Assay for Transposase-Accessible Chromatin with High-Throughput Sequencing (ATAC-seq) for human primary lung-cell types implicated in COPD pathobiology. We then evaluated the enrichment of COPD risk variants in lung-specific open chromatin regions and generated cell type–specific regulatory predictions for >6,500 variants corresponding to 82 COPD GWAS loci. Integration of the fine-mapped variants with lung open chromatin regions helped prioritize 22 variants in putative regulatory elements with potential functional effects. Comparison with functional predictions from 222 Encyclopedia of DNA Elements (ENCODE) cell samples revealed cell type–specific regulatory effects of COPD variants in the lung epithelium, endothelium, and immune cells. We identified potential causal variants for COPD risk by integrating fine mapping in GWAS loci with cell-specific regulatory profiling, highlighting the importance of leveraging the chromatin status in relevant cell types to predict the molecular effects of risk variants in lung disease.
Keywords: functional genomics, chromatin accessibility, machine learning, COPD, genome-wide association study
Chronic obstructive pulmonary disease (COPD) is a complex and heterogeneous disorder characterized by irreversible airflow obstruction. COPD susceptibility is determined by environmental factors, mainly cigarette smoke exposure, and genetic factors, as evidenced by family and population studies. The largest genome-wide association study (GWAS) for COPD to date, examining over 250,000 individuals, identified 82 genetic loci significantly associated with disease susceptibility (1). Furthermore, some of these COPD susceptibility loci overlap with associations for lung function, pulmonary fibrosis, and asthma (2). Genetic perturbation of COPD susceptibility loci in mouse and cellular models have elucidated the contributions of HHIP (hedgehog interacting protein) and FAM13A (family with sequence similarity 13 member A) to cigarette smoke–induced emphysema (3–6). However, identification of causal genes and functional variants at the majority of COPD GWAS loci remains elusive.
Statistical fine-mapping methods can help determine the likely causal variants from highly correlated “tag SNPs” due to linkage disequilibrium (LD) (7–11). Still, the majority of GWAS-associated variants are noncoding, making it difficult to ascribe function or even putative target genes. This challenge is compounded by tissue- and cell type–specific gene expression. As common genetic trait associations are enriched in functional DNA elements (12, 13), statistical fine mapping with integration of genomic regulatory annotations at GWAS loci may assist in implicating functional variants.
For many cell types and tissues, profiling epigenomic marks has revealed robust regulatory landscapes (12, 14–17). However, currently available lung-specific epigenomic profiling and chromatin accessibility data are limited to a few cell lines and cell types. The Assay for Transposase-Accessible Chromatin with High-Throughput Sequencing (ATAC-seq) has emerged as a robust and sensitive method to profile accessible chromatin (18–21). ATAC-seq identifies accessible chromatin regions by using a hyperactive Tn5 transposase to simultaneously fragment and tag genomic DNA with sequencing adaptors. Accessible chromatin regions are therefore preferentially sequenced, providing genome-wide information on regulatory regions (18). We applied ATAC-seq in four human lung primary cell types implicated in COPD pathology (large airway epithelial cells and small airway epithelial cells [SAECs], alveolar type II [AT-II] pneumocytes, and lung fibroblasts [LFs]) to 1) determine cell type–specific regulatory chromatin regions, 2) prioritize the potential causal variants at COPD-associated loci, and 3) predict the possible molecular effects of risk variants in disease. We hypothesized that variants associated with COPD risk are enriched in accessible regulatory regions of chromatin in disease-relevant lung-cell types and that cell type–specific, sequence-based analysis would identify candidate functional regulatory variants for COPD.
Methods
Overview of Study Design
In this study, by using ATAC-seq, we profiled open chromatin regions (OCRs) in five COPD-relevant lung-cell types, including four types of human lung primary cells. We also performed statistical fine mapping using Probabilistic Identification of Causal SNPs (PICS) and generated a credible set of functional variants for 82 COPD GWAS loci. We also applied stratified LD score regression (LDSC), a method to examine the enrichment of genetic signal in different genomic regions. Lastly, we performed a machine-learning method using either OCRs in five lung-cell types or in 222 Encyclopedia of DNA Elements (ENCODE) lines with DNase I hypersensitivity data set lines to train the model. We then applied the model and predicted functional variants in 82 COPD GWAS loci.
ATAC-seq of Human Primary Lung-Cell Types and Cell Lines
Human primary lung-cell types (AT-II cells, LFs, SAECs, and normal human bronchial epithelial [NHBE] cells) were obtained from either donor tissue or commercial sources. SAECs, NHBE cells, and LFs were cultured in appropriate media before ATAC-seq transposition and library preparation. Omni–ATAC-seq library preparation was performed as previously described (21). For a detailed description of lung-cell isolation and culture, ATAC-seq library preparation, and PCR quantification, see the data supplement.
ATAC-seq Sequencing, Alignment, and Peak Calling
ATAC-seq libraries were sequenced using pair-end sequencing on the Illumina NextSeq 500 system to an average depth of 16.9 million, with 2 × 79 bp reads per library. After quality assessment and adapter trimming, reads were aligned to the Genome Reference Consortium Human Build 37 reference genome with Bowtie (version 1.0.1.) (22). Mitochondrial reads and PCR duplicates were filtered. ATAC-seq signal peaks were called using Model-based Analysis of Chromatin Immunoprecipitation Sequencing 2 (MACS2), and blacklisted repetitive artifacts were filtered out. The genomic distribution of peaks, fragment lengths, frequency and enrichment of peaks/reads in known enhancers and transcription start sites were evaluated as quality assessment of the libraries. We compared the individual replicate libraries by using Pearson correlation and hierarchical clustering of the samples. For downstream analysis of cell-type peak sets, the replicates were concatenated before peak calling. For a detailed description of ATAC-seq sequencing and bioinformatic analysis, see the data supplement.
Fine Mapping of COPD GWAS Loci and Enrichment in Lung-Accessible Chromatin
To generate a credible set of putative causal variants from the COPD GWAS summary statistics, we used the PICS algorithm. This method uses a model based on Bayesian statistics to infer the probability of each SNP in linkage, with the lead SNP being the causal variant using the association signal of the lead SNP and LD information from 1000 Genomes reference populations (8). We used the association signals of the 82 lead SNPs identified by Sakornsakolpat and colleagues (1) in the overall GWAS meta-analysis for COPD. We conducted the fine-mapping analysis via the PICS web portal using the 1000 Genomes European reference population (https://pubs.broadinstitute.org/pubs/finemapping/pics.php). Direct overlap of variants in the COPD credible set with lung OCRs was assessed by using the Fisher’s exact test, and variants in OCRs were identified. We computed partitioned heritability using LDSC to estimate the overall enrichment of COPD heritability in lung OCRs compared with the baseline model of 24 genomic functional annotations (23). For a detailed description of COPD fine mapping, enrichment, and LDSC analysis, see the data supplement.
Lung-Specific Regulatory Sequence Analysis and Prediction
We applied the machine-learning algorithm “gkm-SVM” (gapped k-mers [gkm]–support vector machine [SVM]) (24) to the combined ATAC-seq peak sets to evaluate the cell type–specific regulatory models. Positive training sets for each cell type included all significant peaks defined by MACS2 (q-value < 0.05), and randomly sampled negative training sets were generated by matching length, guanine-cytosine content, and repeat fractions of the positive sets. We trained a gkm-SVM using large-scale gkm-SVM (https://github.com/Dongwon-Lee/lsgkm) and measured the classification performance using receiver operating characteristic curves with fivefold cross-validation (25). Weight files for change in gkm-SVM score (deltaSVM) analysis were generated by scoring all possible nonredundant k-mers (k = 10) for each cell type–specific model. To compute deltaSVM scores for the ENCODE samples, precomputed weight files were downloaded from the same website. For a detailed description of gkm-SVM training and deltaSVM analysis, see the data supplement.
Availability of Sequencing Data and Visualization Tools for Results
All raw sequencing data and processed ATAC-seq data sets discussed in this publication have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus and are accessible through accession number GSE152779. We created an Integrative Genomics Viewer session and an application using R (R Foundation for Statistical Computing) and Shiny (RStudio) to facilitate the search for the overlap of COPD credible sets in lung OCRs and integration with other relevant data sources. For a full description and access to the data, see the data supplement.
Results
ATAC-seq Identifies Open Chromatin in Primary Lung-Cell Subtypes
To identify OCRs, we performed ATAC-seq on human bronchial epithelial (HBE) cell line 16HBE cells, primary NHBE cells, SAECs, AT-II pneumocytes, and LFs (Figure 1A). All primary cells were isolated from healthy donors consisting of both males and females and smokers and nonsmokers, with ages ranging from 19 to 50 (see Table E1 in the data supplement).
Figure 1.
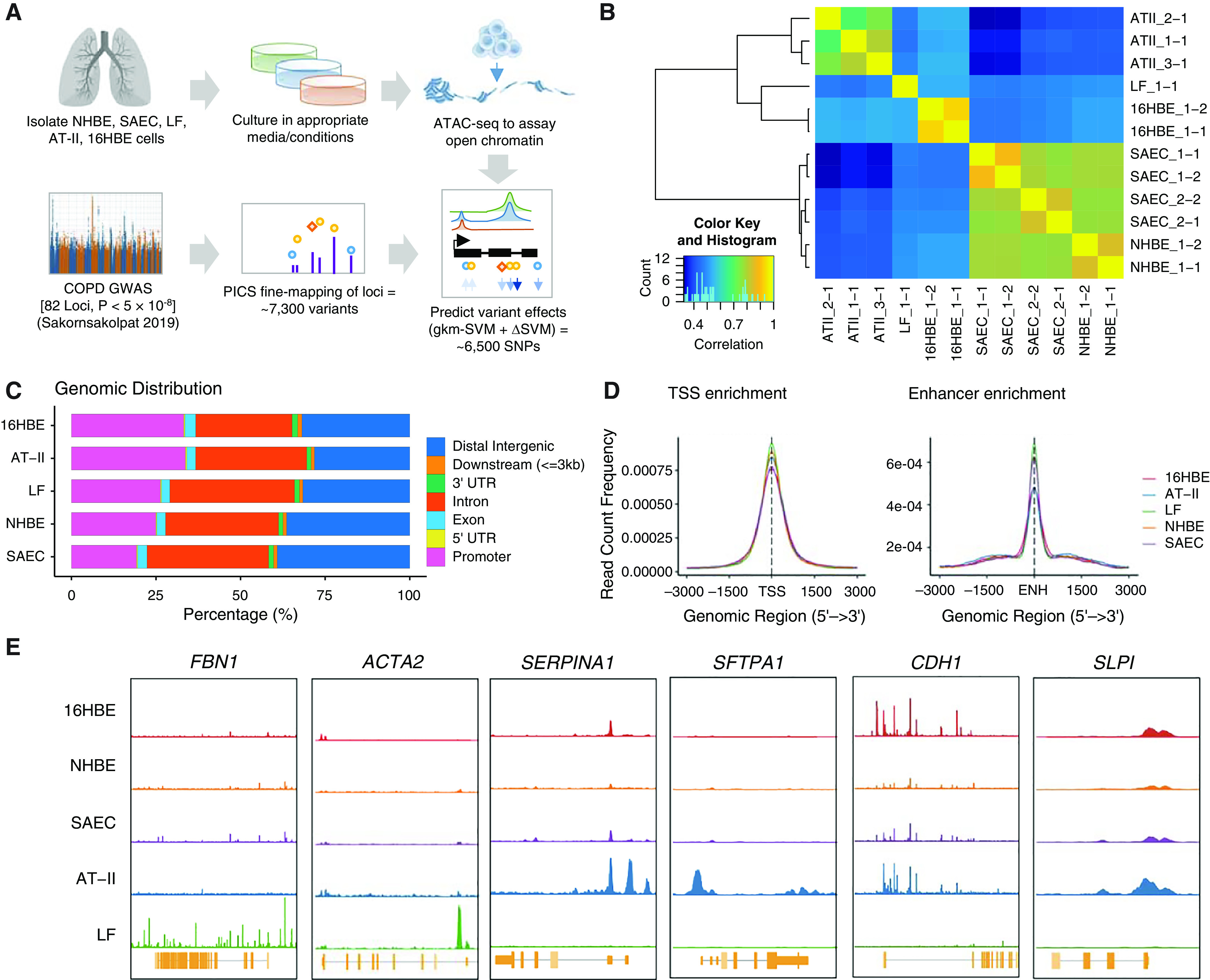
Assays of open chromatin in lung-cell subtypes. (A) Schematic outline of study design. Briefly, lung-cell types were obtained or isolated from healthy donor tissue and cultured in appropriate conditions before lysis and transposition according to the Assay for Transposase-Accessible Chromatin with High-Throughput Sequencing (ATAC-seq) protocol. Open chromatin regions (OCRs) were then used to evaluate the enrichment of chronic obstructive pulmonary disease (COPD) risk variants in cell-specific regulatory regions and construct predictive models of functional variant effects. (B) Correlation of peak sets from individual ATAC-seq libraries by Pearson correlation. The color scale indicates the degree of correlation (r2). (C) Genomic distribution of OCRs (percentages) for each cell type–specific peak set after combining replicates. (D) Profile of OCRs enriched at TSSs and genome-wide enhancers in all five cell types. (E) Cell type–specific signal of chromatin accessibility of nearby genes with known cell type–specific expression in lungs. ΔSVM = change in gkm-SVM score; AT-II = alveolar type II; ENH = enhancer midpoint; gkm-SVM = gapped k-mers support vector machine; GWAS = genome-wide association study; HBE = human bronchial epithelial; LF = lung fibroblast; NHBE = normal HBE; SAEC = small airway epithelial cell; TSS = transcription start site; UTR = untranslated region.
After sequencing, we observed a high correlation between technical replicates and observed that the samples clustered according to cell type, with peaks in the NHBE cells and SAECs sharing the highest degree of similarity (mean r = 0.78; Figure 1B), whereas the 16HBE cell line was less correlated with the primary epithelial cells (mean r = 0.49). For further quality-control assessment, we evaluated the distribution of fragment lengths and the fractions of reads in peaks for individual libraries (see Figure E1).
In total, we identified 131,845 OCRs in SAECs, 79,503 OCRs in NHBE cells, 71,556 OCRs in LFs, 52,777 OCRs in AT-II cells, and 53,564 OCRs in 16HBE cells (Tables E2–E6). Analysis of the genomic distribution of the OCRs suggested enrichment in promoters, introns, and distal intergenic regions, as previously reported (26–29) (Figure 1C). To further assess the quality of the data, we confirmed that peaks were enriched at transcription start sites and permissive enhancer regions genome-wide (Figure 1D and Table E7). We also confirmed the specificity of OCRs at well-characterized cell type–specific genes (30) (Figure 1E).
Overlap and Enrichment of COPD GWAS Variants in Lung Open Chromatin
To identify candidate COPD causal variants, we first performed fine mapping on our recent GWAS (1) using PICS, an algorithm that seeks to reconcile the LD relationships in causal fine mapping (8). The resulting PICS set included 7,285 variants corresponding to 82 GWAS loci (Table E8). Based on PICS, 20 of the 82 (24.4%) index SNPs scored an estimated probability greater than 50% for being the causal SNP (Table E8), whereas the remaining majority of COPD loci contained many candidate causal SNPs unresolved by LD.
We then evaluated the overlap and enrichment of COPD GWAS variants in the five lung cell–specific sets of OCRs by using Fisher’s exact tests for each lung cell type. Overlap of COPD-associated variants with OCRs was nominally significant (P < 0.05) in LFs, AT-II cells, SAECs, and 16HBE cells but not in NHBE cells (P = 0.12) (Figure 2A). Overlap in LFs, AT-II cells, SAECs, and 16HBE cells remained significant after Holm-Bonferroni correction for multiple tests. Across all cell types, 250 SNPs from the PICS fine-mapped set overlapped at least one OCR (Figure E2). Six GWAS lead SNPs directly overlapped an OCR in a single cell type (Figure 2B), suggesting that cell type–specific open chromatin was linked to COPD risk. In addition, the GWAS index SNP at 5q13.2, reference SNP 34651 (rs34651), overlapped an OCR at the promoter and exon 1 of TNPO1 (transportin-1). This finding is consistent with our prior report identifying TNPO1 as the causal gene using regulatory fine mapping (1) and suggests that rs34651 is the causal variant at this locus that regulates TNPO1 expression in multiple lung-cell types.
Figure 2.

Overlap and enrichment of COPD heritability in lung-specific open chromatin. (A) Results of Fisher’s exact test for overlap between COPD credible sets and lung-cell OCRs. The significance of the overlap [−log10(P-val)], number of SNPs overlapping OCRs, and proportion of total credible set overlapping OCRs is indicated in each cell type. The color of significance bars indicates the ratio of obs/exp overlaps. (B) Matrix of top index SNPs from the credible sets that directly overlap OCRs in lung cells. The blue boxes indicate overlap of the SNP with an OCR in each cell type. The posterior probability (PICS) of the index SNP being the causal SNP is indicated. (C) COPD heritability enrichment (standard error) and significance level [−log10(P-val)] of different functional genomic annotations estimated in various cell types using partitioned linkage disequilibrium score regression. Enrichment in lung-cell OCRs is highlighted in green. Baseline annotations are indicated in gray bars. DHS = DNaseI HS; ENCODE = Encyclopedia of DNA Elements; HS = hypersensitivity; logEnrichP = logarithmic enrichment P-val; obs/exp = observed/expected; PICS = Probabilistic Identification of Causal SNPs; P-val = P value; rs = reference SNP; TNPO1 = transportin-1; UCSC = University of California Santa Cruz.
To further evaluate the relevance of these lung OCRs to COPD, we used stratified LDSC, a method that allowed us to examine the enrichment of the genetic signal in different genomic regions and cell types (23). We found enrichment across cell types, with the strongest enrichment of chromatin accessibility in AT-II epithelial cells (enrichment = 15.1-fold; P = 3.55 × 10−5; Figure 2C); AT-II enrichment remained nominally significant after conditioning on other functional annotations (Figure E3).
gkm-SVM Predicts Cell-Specific Functional Impact of COPD Risk Variants
To determine whether specific variants in identified regulatory regions have functional consequences, we used a machine-learning method, deltaSVM (24, 31–34), to train cell type–specific models of open chromatin and predict the effects of regulatory variants within the COPD GWAS locus credible sets. The deltaSVM method uses an SVM classification algorithm to identify predictive oligomers of length k that do not require a precise sequence match (gkm) and computes the relative impact of single-nucleotide variants on their predicted functional effect.
We first trained gkm-SVM models using the ATAC-seq peaks for each lung-cell type and determined that all five lung-cell type–specific regulatory models had high predictive power as determined by fivefold cross-validation (Figure E4). To predict the impact of COPD risk variants, we calculated the deltaSVM score for each SNP in the fine-mapped COPD variants (credible set) by summing the change in weight between alleles for each of the ten 10-bp sequences (10-mers) encompassing the SNP. We computed deltaSVM scores for 6,509 SNPs from the initial credible set (excluding insertion–deletion mutations and variants that could not be mapped to the Database of Single Nucleotide Polymorphisms [dbSNP]). Across all SNPs in the COPD credible set and in each of the five cell types, deltaSVM scores followed a normal distribution (Figure 3A). As the values of deltaSVM scores (negative or positive) have been shown to correlate well with effect sizes of DNase I sensitivity quantitative trait loci, we expect variants with negative deltaSVM scores to indicate decreased chromatin accessibility, relative to the reference allele, and vice versa (31). To account for differences in scores due to variable sizes of training sets, we scaled each set of scores for subsequent analyses.
Figure 3.
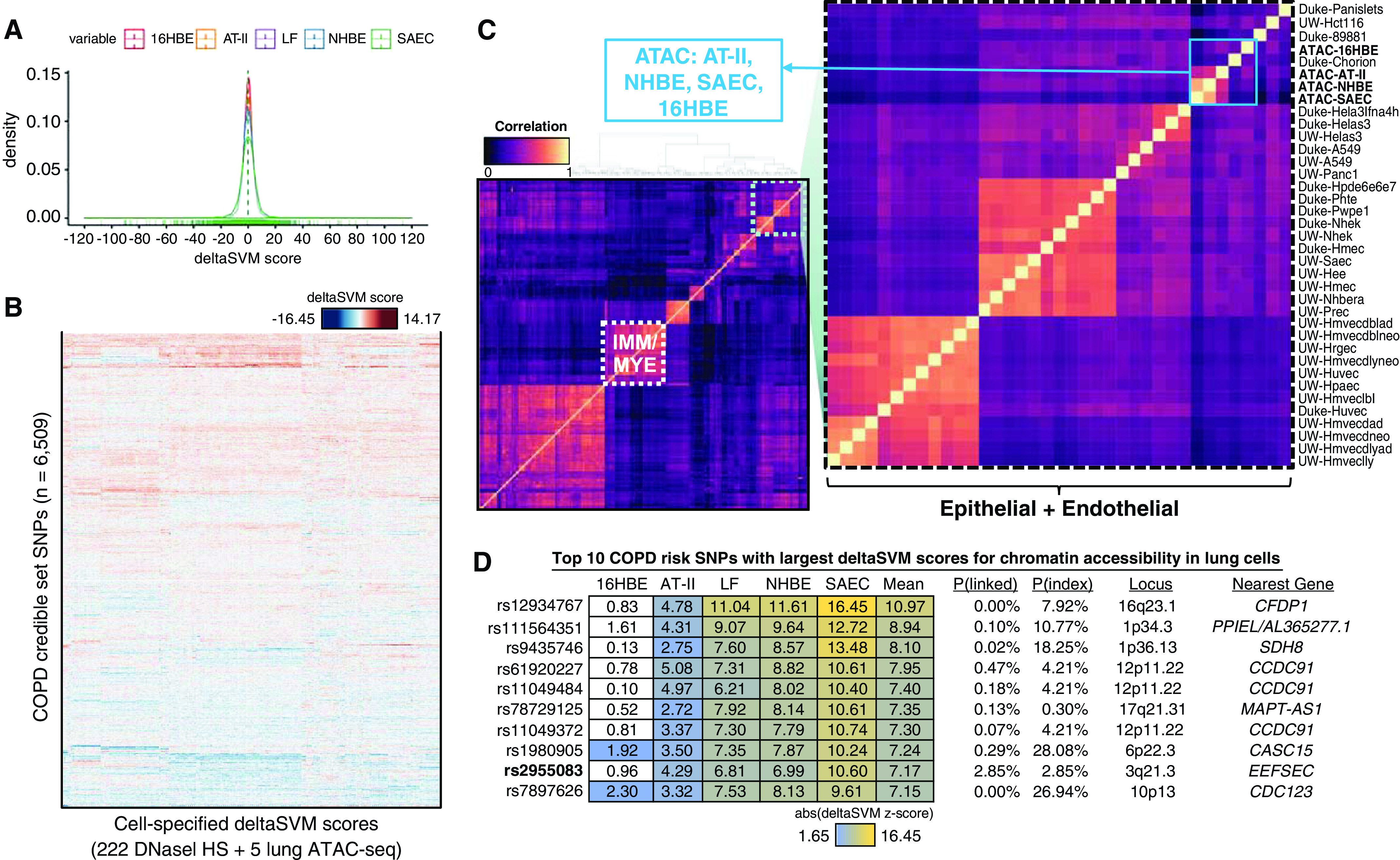
deltaSVM predicts cell-specific functional impact of COPD risk variants. (A) Distribution of raw deltaSVM scores for 6,509 COPD SNPs in the PICS credible set for ATAC-seq open chromatin models of SAECs, NHBE cells, AT-II pneumocytes, LFs, and 16HBE cells. (B) Heatmap of deltaSVM z-scores for COPD risk SNPs in five lung ATAC-seq samples compared with scores from 222 ENCODE DHS samples. (C) Correlation heatmap of deltaSVM scores for COPD risk SNPs across all 227 samples. (D) Table depicting the top 10 COPD risk SNPs by mean absolute deltaSVM z-score in the five lung ATAC-seq models. The PICS probability (of being the causal variant at a given locus) for each SNP [P(linked)] and the probability of the index SNP [P(index)] are indicated. In bold, rs2955083 is the index SNP at the 3q21.3 locus. IMM/MYE = immune and myeloid cells; UW = University of Washington.
In addition, we also calculated deltaSVM scores for the credible set of SNPs using weights previously calculated for 222 ENCODE DNase I hypersensitivity data sets (31), encompassing a large diversity of tissues and cell types. Some loci had large deltaSVM scores that were unique to lung-cell types (Figure 3B), in contrast to other cell type–ubiquitous loci. By correlating the deltaSVM scores across all cell types, we observed distinct profiles of predicted effects by COPD risk variants on clusters of cell types, including in immune/myeloid cells and endothelial/epithelial cell types (Figure 3C). Scores from our SAEC, NHBE, and AT-II ATAC-seq models were clustered together with other public DNase data in epithelial cells from the bronchus or small airways (Figure 3D). Scores from our LF ATAC-seq model, although still strongly correlated with the lung ATAC-seq models, clustered with fibroblasts from other tissues.
To identify the variants with the greatest likelihood of functional regulatory effects, we looked for SNPs in the credible set that both overlapped an experimentally identified OCR and resulted in a significant deltaSVM score. From the initial credible set of 6,509 COPD risk SNPs (Table E9), only 22 met both criteria, including 9 in AT-II cells (Table 1). We further identified the cell type for each SNP with the largest positive and negative predicted scores (Table E10).
Table 1.
List of 22 COPD Fine-Mapped SNPs with Significant deltaSVM Scores Overlapping Lung OCRs
| rsID | Nearest Gene | r2 (to Index) | deltaSVM Score (z-Score) |
||||
|---|---|---|---|---|---|---|---|
| 16HBE | AT-II | LF | NHBE | SAEC | |||
| rs6662037 | SLC30A10 | 0.763 | 0.50 | −1.08 | −1.43 | −1.42 | −1.88* |
| rs9861425 | ADCY5 | 0.937 | −4.69 | −3.22 | −3.66* | −3.13 | −2.81 |
| rs6440001 | ZBTB38 | 0.532 | 1.76* | 0.60 | 0.19 | −0.23 | 0.17 |
| rs2013701 | FAM13A | 0.775 | 2.40 | 1.39 | 2.43* | 1.31 | 1.52 |
| rs2904259 | FAM13A | 0.774 | −2.86* | −3.58 | −5.07* | −4.84* | −3.52* |
| rs262121 | ADGRG6 | 0.788 | −2.15* | 0.48 | 0.96 | 0.08 | −0.13 |
| rs36062557 | CDC123 | 0.761 | 0.70 | 2.06* | 0.31 | 0.75 | 0.53 |
| rs4319455 | SFTPD | 0.530 | −0.81 | −1.70* | −0.87 | −0.13 | 0.00 |
| rs11191829 | STN1 | 0.870 | −1.94* | 0.21 | −0.66 | 0.39 | −0.21 |
| rs4918067 | STN1 | 0.516 | −0.32 | −1.77* | −1.91* | −1.80 | −0.98 |
| rs11191847 | STN1 | 0.524 | 0.50 | 1.75* | 0.56 | 0.64 | 0.92 |
| rs1372212 | ARNTL | 0.567 | 1.68 | 2.24 | 2.00 | 1.85* | 1.68* |
| rs11049415 | CCDC91 | 0.837 | −1.66* | −0.62 | −0.69 | −0.90 | −0.97 |
| rs4941489 | SERP2 | 0.527 | 2.61 | 1.66* | 1.67 | 2.07 | 1.07 |
| rs55781567 | CHRNA3 | 0.890 | 2.28* | 2.08* | 2.12* | 2.13* | 1.20 |
| rs8056080 | CFDP1 | 0.932 | −1.54 | 0.56 | −1.64 | −1.75* | −0.79 |
| rs4888403 | CFDP1 | 0.836 | 1.05 | 1.34 | 1.25 | 1.87* | 0.90 |
| rs2227322 | THRA | 0.533 | 2.46 | 1.80* | 1.50 | 2.23 | 1.46 |
| rs80233201 | SPPL2C | 0.978 | −0.27 | −1.67* | −0.30 | −1.20 | −0.59 |
| rs17564493 | SPPL2C | 0.878 | 0.47 | 2.01* | 0.79 | 0.75 | 0.69 |
| rs4438347 | SOX9 | 0.598 | −0.29 | 2.24 | 3.50* | 4.15 | 4.23 |
| rs12158631 | SYN3 | 0.503 | 3.28 | 2.32 | 1.83 | 1.93* | 0.91 |
Definition of abbreviations: AT-II = alveolar type II; COPD = chronic obstructive pulmonary disease; deltaSVM = change in gkm-SVM score; HBE = human bronchial epithelial; LF = lung fibroblast; NHBE = normal HBE; OCR = open chromatin region; rs = reference SNP; rsID = rs identifier; SAEC = small airway epithelial cell; SOX9 = SRY-box transcription factor 9; SYN3 = synapsin III.
Nine SNPs with significant deltaSVM scores overlaps with OCRs in AT-II cells are indicated in bold.
Indicates overlap with OCR in this cell type and a significant deltaSVM z-score.
Examination of SNP Predictions at Previously Studied Risk Loci
To assess our prediction model, we first examined the lung-specific deltaSVM scores at two well-defined COPD loci for which we have multiple types of supporting evidence on causal genes and causal variants in COPD-relevant cell types. At the top COPD GWAS locus at 4q31 upstream of HHIP, we identified 78 SNPs in the fine-mapped set (Figure 4A). The lead SNP (rs13140176) in the COPD GWAS analysis (PICS probability ≥90%) was also the lead SNP in an analysis of lung function (35) and had large deltaSVM scores in AT-II and 16HBE cells, with the G allele (COPD risk allele) being predicted to cause decreased chromatin accessibility in both cell types. Five additional variants in 16HBE cells (rs11938745, rs12509311, rs11100860, rs1828591, and rs1489759), one additional variant in NHBE cells (rs36023701), and one additional variant in AT-II cells (rs34265962) also had predicted functional impact. Two SNPs had significant deltaSVM scores in multiple cell types (rs13142776 and rs1489763), although these were accompanied by low PICS probabilities. However, neither of the previously identified functional variants, rs1542725 and rs6537296 (36), nor the previous COPD GWAS index SNP rs13141641 (2) at this locus were predicted to have large effects by deltaSVM, possibly due to different cell types used in the previous functional assays. Together, fine mapping of GWAS signals and functional predictions of variant effects on chromatin accessibility by deltaSVM provide the most support for rs13140176 as one possible causal SNP in the HHIP COPD GWAS region.
Figure 4.
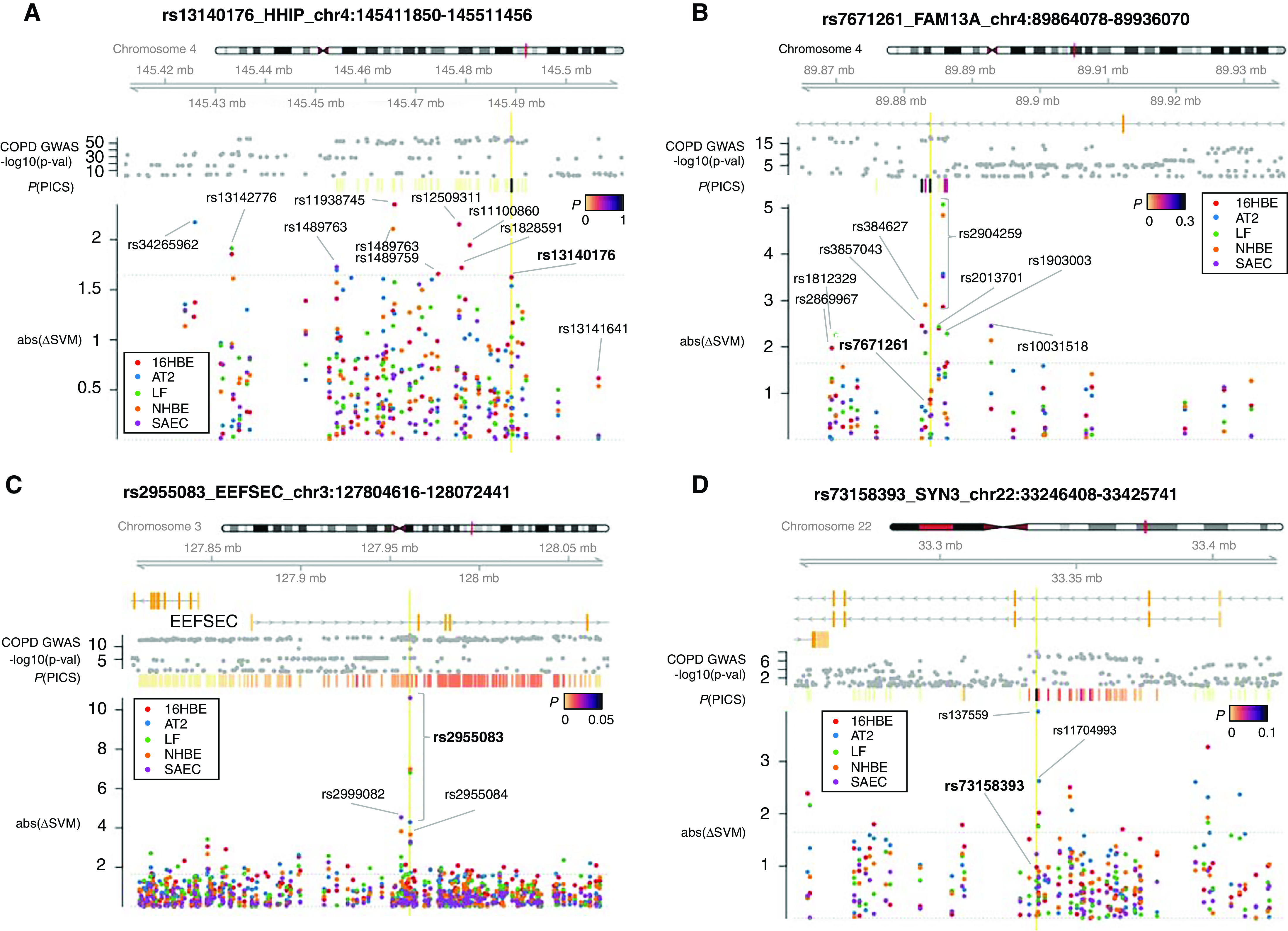
deltaSVM of chromatin accessibility predicts causal SNPs at COPD risk loci. Plots of abs(ΔSVM) z-scores for SNPs in the PICS credible set at COPD risk loci near (A) HHIP, (B) FAM13A, (C) EEFSEC, and (D) TIMP3 (TIMP metallopeptidase inhibitor 3)/SYN3 (synapsin III) are shown. deltaSVM scores were calculated from gkm-SVM models trained on ATAC-seq data from SAECs, NHBE cells, AT-II pneumocytes, LFs, and 16HBE cells. For each plot, the COPD genome-wide association significance [−log10(P-val)] from the Sakornsakolpat and colleagues (1) 2019 study and calculated PICS probabilities indicated in colored bars [P(PICS)] are shown. The location of the index SNP at each locus is depicted by a vertical yellow line. abs(ΔSVM) = absolute deltaSVM; AT2 = AT-II.
At the 4q22.1 locus (within FAM13A), the functional variant rs2013701, previously identified by using a massively parallel reporter assay (MPRA) followed by validation with reporter assays and endogenous genome-editing assays (6), had significant deltaSVM scores in 16HBE and LF cells, further supporting this SNP as a putative causal SNP in this locus. We also examined lung deltaSVM scores of 24 SNPs at the FAM13A locus within the credible set (Figure 4B). Seven other SNPs were predicted to impact lung open chromatin (rs286997, rs1812329, rs3857043, rs384627, rs2904259, rs1903003, and rs10031518). The SNP rs2904259, which is <700 bp from rs2013701, was predicted to have the greatest impact on chromatin accessibility in all five lung-cell types. In addition, both rs2013701 and rs2904259 overlap an OCR in all five lung-cell types. The lead COPD risk variant, rs7671261 (P value = 1.4 × 10−18) (1), showed no evidence of allele-specific activity in the MPRA study, which is consistent with the lack of evidence of functional impact in lung cells indicated by deltaSVM.
Because statistical fine mapping alone cannot adequately resolve the association signals at most COPD GWAS loci, we examined whether deltaSVM scores could help identify putative causal variants at especially ambiguous loci. At 3q21.3 (near EEFSEC), from 216 credible set SNPs scored by deltaSVM (Figure 4C), we identified 46 SNPs (21.3%) with significant impact on open chromatin in any of the five lung-cell types. Notably, rs2955083, the index variant at the locus, has the largest predicted effect observed (in AT-II cells, SAECs, NHBE cells, and LFs), despite a PICS probability of only 2.9%. Thus, at this locus where the genetic association signal is ambiguous and for which the fine-mapping approach was unable to narrow down the functional variants, the deltaSVM functional predictions provide evidence supporting a functional role for the lead SNP rs2955083.
The deltaSVM can rather accurately predict the tissue and cell type–specific impact of variants on regulatory elements (31). Given enrichment of COPD heritability in AT-II cells (Figure 2C), we examined whether any SNPs were predicted to preferentially impact regulatory sequences in AT-II cells on the basis of deltaSVM scores. At the 22q12.3 locus (in the intronic region of SYN3 [synapsin III] and downstream of TIMP3 [TIMP metallopeptidase inhibitor 3]), six SNPs were predicted to impact regulatory elements in AT-II cells, including two (rs137559 and rs11704993) in high LD with the lead GWAS variant, rs73158393 (Figure 4D). In addition, we also observed AT-II–specific functional predictions at the 5q15 locus (in between SPATA9 [spermatogenesis associated 9] and RHOBTB3 [rho-related BTB domain–containing 3]; Figures E5–E2), 1p36.13 locus (near MFAP2 [microfibril-associated protein 2]; Figures E5–E46), 22q11.21 locus (intronic region of MICAL3 [microtubule-associated monooxygenase, calponin, and LIM domain–containing 3]; Figures E5–E48), 17q24.3 locus (near CASC17 [cancer susceptibility 17] and SOX9 [SRY-box transcription factor 9]; Figures E5–E58), and 5q35.1 locus (near FGF18 [fibroblast growth factor 18]; Figures E5–E61).
To further visualize these results in the context of other data types including gene expression, in addition to the GWAS, and to make the results accessible, we created a companion R/Shiny application and an Integrative Genomics Viewer (see Figures E6–E9). These are available for download at http://www.copdconsortium.org/.
Discussion
Using genetic data, statistical fine-mapping algorithms, open chromatin profiling, and machine-learning models, we evaluated the genetic landscape of COPD and sought to identify potential key cell types and causal variants for COPD. Using these approaches, we identified 250 SNPs directly overlapping accessible chromatin, including 22 high-confidence SNPs with putative impacts on regulatory elements specific to lung-cell types.
By profiling open chromatin in five human lung-cell types using ATAC-seq, we were able to identify OCRs with high resolution and generate predictive models of the functional impact of noncoding sequence variation. We further demonstrated the enrichment of COPD heritability in the accessible chromatin of these cell types by using LDSC. Previous examination of partitioned heritability in COPD by Sakornsakolpat and colleagues (1) revealed enrichment in epigenomic marks (H3K4Me1, H3K27ac) and DNase-hypersensitivity sites in fetal lung and gastrointestinal smooth muscle. Analysis of single-cell chromatin accessibility profiles in mouse lung demonstrated cell-specific enrichment in endothelial cells, B cells, and type 1 and type 2 alveolar epithelial cells (1, 37–39). Although our analysis underscored the importance of using relevant cell types to investigate the functional impact of sequence variation in complex disease, it also suggests the limitations of routinely used immortalized cell lines for experimental validation. In our study, chromatin accessibility and deltaSVM scores in the 16HBE cell line only modestly correlated with the primary HBE cells.
Among the 82 COPD GWAS loci, we evaluated putative functional SNPs at well-defined loci that have extensive replication and functional validation studies (loci near HHIP and FAM13A) as well as the recently identified loci (EEFSEC and others) for which there are no experimental data yet in the literature. At the FAM13A locus, the deltaSVM analysis supported rs2013701 as the most likely functional variant, consistent with our previous MPRA analysis and subsequent series of functional evaluations (6). At the HHIP locus (4q31.21), we observed significant deltaSVM scores for the lead SNP from the most recent GWAS for COPD susceptibility (1), rs13140176, suggesting putative functional effects of this SNP in AT-II cells and 16HBE cells. The risk allele rs13140176-G leads to decreased chromatin accessibility in AT-II cells as predicted by deltaSVM, providing evidence of a possible mechanism to explain that this risk allele is also associated with lower HHIP expression (1, 40). Future validation of regulatory roles for rs13140176 on HHIP expression in human AT-II cells may help confirm its function. Although we did not find any evidence of a putative function for previously reported functional SNPs rs1542725 and rs6537296 (36) on the basis of deltaSVM scores, it is notable that the previously identified 2.4-kb enhancer region (36) spanning these two SNPs lies directly adjacent to rs13140176 (<250 bp upstream). Moreover, the previously reported SNP affecting SP3 binding (rs1542725) is located <800 bp from rs13140176. Additional functional studies within this genomic region are warranted because there may be multiple functional variants regulating transcription of HHIP.
At the FAM13A locus (4q22.1), we compared deltaSVM predictions with an experimental MPRA study in which our group had previously identified 45 variants with regulatory activity in the Beas-2B HBE cell line (6). Five variants that were analyzed in this study as part of the PICS credible set demonstrated allele-specific activity in the MPRA experiments (rs11722033, rs1964516, rs2013701, rs7671167, and rs7674369). Only rs2013701, previously identified as a functional variant in this locus by using reporter assays and endogenous genome-editing assays, scored significant deltaSVM values in our study (in 16HBE and LF cells). Scores for rs2013701 in the other lung-cell types (AT-II, NHBE, and SAEC) were increased but did not reach statistical significance. Thus, our analysis confirmed that rs2013701 has regulatory capacity and that it may in fact be at least one of the causal variants at this locus.
We further demonstrated the usefulness of this approach to help identify putative functional variants in loci where the LD pattern cannot be resolved by statistical fine mapping. At the EEFSEC locus (3q21.3), we found strong evidence supporting a functional role for the lead SNP, rs2955083, in all four primary lung-cell types, despite low causal probability calculated by PICS due to extensive LD. Of interest, rs2955083, located in the intron between exons 1 and 2 of the EEFSEC gene, has previously been associated with COPD and lung function (1, 2, 35, 41, 42). Based on HaploReg version 4.1 annotations, this variant intersects promoter and enhancer histone marks in ENCODE lung tissues and may alter 25 different regulatory motifs, including members of the Fox (forkhead box) transcription factor family, which are critical regulators of lung morphogenesis and maintain gene expression in differentiated epithelial cells (2, 43–46). Future experimental validation may help to determine the transcription factor bound to this variant, tissue-specific function of the variant, and target gene(s) of the variant.
To our knowledge, our study presents the first bulk profiling of open chromatin and modeling of sequence-based regulatory impact in multiple lung-cell types, including human AT-II cells, a critical cell type for COPD, with little prior epigenetic data. This is especially pertinent for COPD because, using stratified LDSC, we demonstrated that disease heritability is enriched specifically in AT-II OCRs compared with OCRs of other lung-cell types. This evidence supports previous findings that COPD heritability is enriched in AT-II cells (based on human single-cell gene expression in lungs and murine single-cell ATAC-seq) (1). It has been shown that deltaSVM predictions are highly cell type dependent and that training on the appropriate cell type is crucial for accurately identifying disease-associated SNPs (31). Therefore, we hypothesized that training the gkm-SVM on AT-II OCRs and calculating deltaSVM scores would improve identification of functional COPD variants. Our discovery of several loci with strong putative effects in AT-II cells compared with the other lung-cell types supports this hypothesis but will require further experimental validation. This was especially true at the 22q11.21 (intronic region of MICAL3) locus, where the lead SNP rs9617650 (PICS probability = 96.7%) scored the largest absolute deltaSVM score in AT-II cells compared with all other cell types (including ENCODE DNase hypersensitivity). AT-II cells also play a central role in other lung diseases, such as idiopathic pulmonary fibrosis, and their role in producing surfactant is critical to normal lung function (47, 48). Thus, these chromatin accessibility profiles and functional predictions may further elucidate the role of genetic variants in other lung traits.
We recognize several limitations of this present study. First, a small sample size of cell types and technical limitations precluded the analysis of replicates for some cell types. Future profiling of additional lung-cell types (such as cells from fetal lung, type I alveolar cells, smooth muscle cells, and pulmonary microvascular endothelial cells) may more comprehensively inform the functional impact of COPD risk variants. Furthermore, these investigations will benefit by including open chromatin data from specific contexts, including different stages of lung development and lung samples from patients with COPD. In addition, because of feasibility, the AT-II epithelial cells in our studies were collected immediately after flow-sorting enrichment, whereas other cell types were cultured for a small number of passages (<4) before collection for ATAC-seq; this technical difference could have influenced our results. Second, the optimal method for identifying putative causal SNPs in complex diseases is evolving. Fine mapping using additional statistical methods (such as those that allow multiple causal variants at a locus and those that jointly model functional annotations) may result in different SNP sets to evaluate for functional impact. Third, our approach focuses on causal SNPs that may alter the chromatin state and ultimately gene expression. Other molecular mechanisms such as alternative splicing (49) or deleterious coding variants (1) are beyond our current identification approach. Interestingly, at the 10q22.3 locus, the lead SNP rs721917, with a PICS probability of 97.2%, is a missense variant, resulting in a methionine-to-threonine mutation in surfactant protein D. We also observed a large deltaSVM score for rs721917 in AT-II cells, suggesting that it may influence gene transcriptional regulation as well as protein structure. Overall, confirmation of these prioritized functional variants in this study requires future functional variations by combined molecular biology approaches such as reporter assays and CRISPR-mediated genome editing targeting the variants in relevant lung-cell types. Despite the limitations we have outlined, our results demonstrate that integrating ATAC-seq information with genetic-association and statistical fine-mapping evidence can assist in the challenging task of identifying functional variants for complex diseases like COPD.
Footnotes
Supported by National Institutes of Health grants R33 HL120794, R01 HL137927, R01 HL147148, P01 HL114501, and P01 HL132825. Human lung specimens were provided by the University of North Carolina Tissue Procurement and Cell Culture Core that is supported by National Institutes of Health grant P30DK065988 and Cystic Fibrosis Foundation Grant BOUCHE19RO.
Author Contributions: C.J.B., M.H.C., E.K.S., and X.Z. designed and supervised the study. C.J.B. designed, conducted and analyzed the Assay for Transposase-Accessible Chromatin with High-Throughput Sequencing experiments. M.H.C. and X.Z. supervised the experimental work and computational/statistical analysis. C.J.B. and F.D. optimized the Assay for Transposase-Accessible Chromatin with High-Throughput Sequencing protocol and collected sequencing data. J.L. provided bioinformatic support, conducted linkage disequilibrium score regression analysis, and generated the R/Shiny application. F.G. isolated human alveolar type II epithelial cells and lung fibroblasts. S.H.R. provided human lung tissue samples. C.J.B., J.L., M.H.C., E.K.S., and X.Z. participated in the manuscript writing. All authors reviewed and approved of the final version to be published.
This article has a data supplement, which is accessible from this issue's table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0475OC on March 30, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Sakornsakolpat P, Prokopenko D, Lamontagne M, Reeve NF, Guyatt AL, Jackson VE, et al. SpiroMeta Consortium; International COPD Genetics Consortium. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat Genet. 2019;51:494–505. doi: 10.1038/s41588-018-0342-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hobbs BD, de Jong K, Lamontagne M, Bossé Y, Shrine N, Artigas MS, et al. COPDGene Investigators; ECLIPSE Investigators; LifeLines Investigators; SPIROMICS Research Group; International COPD Genetics Network Investigators; UK BiLEVE Investigators; International COPD Genetics Consortium. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet. 2017;49:426–432. doi: 10.1038/ng.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lao T, Jiang Z, Yun J, Qiu W, Guo F, Huang C, et al. Hhip haploinsufficiency sensitizes mice to age-related emphysema. Proc Natl Acad Sci USA. 2016;113:E4681–E4687. doi: 10.1073/pnas.1602342113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yun JH, Morrow J, Owen CA, Qiu W, Glass K, Lao T, et al. Transcriptomic analysis of lung tissue from cigarette smoke-induced emphysema murine models and human chronic obstructive pulmonary disease show shared and distinct pathways. Am J Respir Cell Mol Biol. 2017;57:47–58. doi: 10.1165/rcmb.2016-0328OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang Z, Knudsen NH, Wang G, Qiu W, Naing ZZC, Bai Y, et al. Genetic control of fatty acid β-oxidation in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2017;56:738–748. doi: 10.1165/rcmb.2016-0282OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castaldi PJ, Guo F, Qiao D, Du F, Naing ZZC, Li Y, et al. Identification of functional variants in the FAM13A chronic obstructive pulmonary disease genome-wide association study locus by massively parallel reporter assays. Am J Respir Crit Care Med. 2019;199:52–61. doi: 10.1164/rccm.201802-0337OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaid DJ, Chen W, Larson NB. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet. 2018;19:491–504. doi: 10.1038/s41576-018-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hormozdiari F, Kostem E, Kang EY, Pasaniuc B, Eskin E. Identifying causal variants at loci with multiple signals of association. Genetics. 2014;198:497–508. doi: 10.1534/genetics.114.167908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hormozdiari F, van de Bunt M, Segrè AV, Li X, Joo JWJ, Bilow M, et al. Colocalization of GWAS and eQTL signals detects target genes. Am J Hum Genet. 2016;99:1245–1260. doi: 10.1016/j.ajhg.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W, McDonnell SK, Thibodeau SN, Tillmans LS, Schaid DJ. Incorporating functional annotations for fine-mapping causal variants in a Bayesian framework using summary statistics. Genetics. 2016;204:933–958. doi: 10.1534/genetics.116.188953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trynka G, Raychaudhuri S. Using chromatin marks to interpret and localize genetic associations to complex human traits and diseases. Curr Opin Genet Dev. 2013;23:635–641. doi: 10.1016/j.gde.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-Seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenleaf WJ. Assaying the epigenome in limited numbers of cells. Methods. 2015;72:51–56. doi: 10.1016/j.ymeth.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, et al. An improved ATAC-Seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P-R, et al. ReproGen Consortium; Schizophrenia Working Group of the Psychiatric Genomics Consortium; RACI Consortium. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghandi M, Lee D, Mohammad-Noori M, Beer MA. Enhanced regulatory sequence prediction using gapped k-mer features. PLOS Comput Biol. 2014;10:e1003711. doi: 10.1371/journal.pcbi.1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee D. LS-GKM: a new gkm-SVM for large-scale datasets. Bioinformatics. 2016;32:2196–2198. doi: 10.1093/bioinformatics/btw142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McClymont SA, Hook PW, Soto AI, Reed X, Law WD, Kerans SJ, et al. Parkinson-associated SNCA enhancer variants revealed by open chromatin in mouse dopamine neurons. Am J Hum Genet. 2018;103:874–892. doi: 10.1016/j.ajhg.2018.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fullard JF, Giambartolomei C, Hauberg ME, Xu K, Voloudakis G, Shao Z, et al. Open chromatin profiling of human postmortem brain infers functional roles for non-coding schizophrenia loci. Hum Mol Genet. 2018;29:2812. doi: 10.1093/hmg/ddy229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y, Zheng D, Cvekl A. Profiling of chromatin accessibility and identification of general cis-regulatory mechanisms that control two ocular lens differentiation pathways. Epigenetics Chromatin. 2019;12:27. doi: 10.1186/s13072-019-0272-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pastor WA, Liu W, Chen D, Ho J, Kim R, Hunt TJ, et al. TFAP2C regulates transcription in human naive pluripotency by opening enhancers. Nat Cell Biol. 2018;20:553–564. doi: 10.1038/s41556-018-0089-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Natarajan A, Yardimci GG, Sheffield NC, Crawford GE, Ohler U. Predicting cell-type-specific gene expression from regions of open chromatin. Genome Res. 2012;22:1711–1722. doi: 10.1101/gr.135129.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee D, Gorkin DU, Baker M, Strober BJ, Asoni AL, McCallion AS, et al. A method to predict the impact of regulatory variants from DNA sequence. Nat Genet. 2015;47:955–961. doi: 10.1038/ng.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee D, Kapoor A, Safi A, Song L, Halushka MK, Crawford GE, et al. Human cardiac cis-regulatory elements, their cognate transcription factors, and regulatory DNA sequence variants. Genome Res. 2018;28:1577–1588. doi: 10.1101/gr.234633.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beer MA. Predicting enhancer activity and variant impact using gkm-SVM. Hum Mutat. 2017;38:1251–1258. doi: 10.1002/humu.23185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kapoor A, Lee D, Zhu L, Soliman EZ, Grove ML, Boerwinkle E, et al. Multiple SCN5A variant enhancers modulate its cardiac gene expression and the QT interval. Proc Natl Acad Sci USA. 2019;116:10636–10645. doi: 10.1073/pnas.1808734116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lutz SM, Cho MH, Young K, Hersh CP, Castaldi PJ, McDonald M-L, et al. ECLIPSE Investigators; COPDGene Investigators. A genome-wide association study identifies risk loci for spirometric measures among smokers of European and African ancestry. BMC Genet. 2015;16:138. doi: 10.1186/s12863-015-0299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou X, Baron RM, Hardin M, Cho MH, Zielinski J, Hawrylkiewicz I, et al. Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Hum Mol Genet. 2012;21:1325–1335. doi: 10.1093/hmg/ddr569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;1:e90558. doi: 10.1172/jci.insight.90558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ardini-Poleske ME, Clark RF, Ansong C, Carson JP, Corley RA, Deutsch GH, et al. LungMAP Consortium. LungMAP: the molecular atlas of lung development program. Am J Physiol Lung Cell Mol Physiol. 2017;313:L733–L740. doi: 10.1152/ajplung.00139.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell. 2018;174:1309–1324, e18. doi: 10.1016/j.cell.2018.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wain LV, Shrine N, Artigas MS, Erzurumluoglu AM, Noyvert B, Bossini-Castillo L, et al. Understanding Society Scientific Group; Geisinger-Regeneron DiscovEHR Collaboration. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet. 2017;49:416–425. doi: 10.1038/ng.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shrine N, Guyatt AL, Erzurumluoglu AM, Jackson VE, Hobbs BD, Melbourne CA, et al. Understanding Society Scientific Group. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet. 2019;51:481–493. doi: 10.1038/s41588-018-0321-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costa RH, Kalinichenko VV, Lim L. Transcription factors in mouse lung development and function. Am J Physiol Lung Cell Mol Physiol. 2001;280:L823–L838. doi: 10.1152/ajplung.2001.280.5.L823. [DOI] [PubMed] [Google Scholar]
- 45.Li S, Wang Y, Zhang Y, Lu MM, DeMayo FJ, Dekker JD, et al. Foxp1/4 control epithelial cell fate during lung development and regeneration through regulation of anterior gradient 2. Development. 2012;139:2500–2509. doi: 10.1242/dev.079699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Golson ML, Kaestner KH. Fox transcription factors: from development to disease. Development. 2016;143:4558–4570. doi: 10.1242/dev.112672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitsett JA, Weaver TE. Hydrophobic surfactant proteins in lung function and disease. N Engl J Med. 2002;347: 2141–2148. doi: 10.1056/NEJMra022387. [DOI] [PubMed] [Google Scholar]
- 48.Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–1952. doi: 10.1016/S0140-6736(17)30866-8. [DOI] [PubMed] [Google Scholar]
- 49.Saferali A, Yun JH, Parker MM, Sakornsakolpat P, Chase RP, Lamb A, et al. COPDGene Investigators; International COPD Genetics Consortium Investigators. Analysis of genetically driven alternative splicing identifies FBXO38 as a novel COPD susceptibility gene. PLoS Genet. 2019;15:e1008229. doi: 10.1371/journal.pgen.1008229. [DOI] [PMC free article] [PubMed] [Google Scholar]