

Abstract

A highly chemoselective C–N bond cleavage reaction of p-methoxybenzyl- (PMB), 3,4-dimethoxybenzyl- (DMB), or cinnamyl-substituted tertiary sulfonamides in the presence of catalytic Bi(OTf)3 is presented. A wide range of sulfonamide substrates smoothly furnished the corresponding C–N bond cleavage products in good to excellent yields. Great efforts have been made to obtain insights into the reaction mechanism based on a series of control experiments and mass spectroscopy.
Introduction
The cleavage of the C–N bond of amines or amides with activated groups, such as 4-methoxybenzyl- (PMB) or dimethoxybenzyl- (DMB) including prenyl- or cinnamyl-substituents, is frequently used in organic synthesis.1 The common use of such activated groups over the other unactivated groups (alkyl- or benzyl-) was mostly privileged due to its high proclivity to undergo cleavage under mild acidic conditions. The stability of the in situ-generated carbocation and, in sequel, the use of a suitable nucleophilic scavenger in a stoichiometric amount are keys to success (Scheme 1).2 Great progress has been made in developing milder N-debenzylation conditions with substrates like PMB- or DMB-protected amines and amides.3 Nucleophilic scavengers, for example, anisole or thioanisole, and the use of excess acid are often used to increase the yield of the cleavage product. The other well-accepted methods are either Pd-C-mediated hydrogenolysis4 or oxidative cleavage mediated by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)5 or ceric ammonium nitrate (CAN).6
Scheme 1. General Approaches for C–N Bond Cleavage of Amine/Amides with π-Activated Protecting Groups.

In our recent intriguing study on Friedel–Crafts reactions,7 we unexpectedly isolated a C–N bond cleavage product 3a during the attempted Friedel–Crafts-type alkylation of tertiary sulfonamide 1d in the presence of bismuth(III)-triflate as a catalyst (Scheme 2). While the substrates 1a–b (X = CH2) underwent smoothly Friedel–Crafts-type hydroarylation and afforded the desired tetracyclic compounds 2a–b in excellent yields, under the same catalytic condition, the substrate 1d (X = NTs) gave only the C–N bond cleavage product 3a in a good yield (84%). We envisaged that this catalytic procedure might constitute a new method for the selective C–N bond cleavage of tertiary sulfonamides under the catalytic acid condition.
Scheme 2. Initial Observation of C–N Bond Cleavage of Tertiary Sulfonamide in the Presence of Catalytic Bi(OTf)3.

In prior works on C–N bond cleavage of N-alkyl sulfonamides or N,N-dialkyl sulfonamides, reagent systems with more than stoichiometric amounts such as PhI(OAc)2-I2,8 excess acid H5IO6-Cr3(OAc)7(OH)2,9 and KBr-oxone10 including the organic electron donor11 were employed. There was half-stoichiometric use of CuII(CH3CN)4PF6 (0.5 equiv)–K3PO4 (8.0 equiv) reagent systems for N-dealkylation/N-arylation of secondary sulfonamides in the presence of air.12 Solely, a single catalytic method, AgISbF6-mediated deprenylation of allylsulfonamides, has been reported at a very high temperature and under microwave-irradiated conditions.13 Furthermore, a ruthenium-catalyzed C–N bond cleavage of N-propargyl sulfonamides and amides has also been reported.14 Recently, an electrochemical method for C–N bond cleavage of secondary and tertiary N-alkyl sulfonamides has been developed.15
Nevertheless, the C–N bond cleavage of N-alkyl sulfonamides needing harsh conditions and the use of toxic metals and excess oxidants still make many of these methods inappropriate in the current scenario. Recently, Javorskis and Orentas described a highly chemoselective detosylation (N–S bond cleavage) process of N-aryl sulfonamides in the presence of either nearly or more than a stoichiometric amount of triflic acid.16 In this perspective (N–S vs C–N bond cleavage) and in view of the mechanistic standpoint (stoichiometric vs catalytic), the observation of C–N bond cleavage of tertiary N-aryl or alkyl sulfonamide using Bi(OTf)3 in a catalytic amount is still significant and needs to be further studied (Scheme 3).
Scheme 3. C–N Bond Cleavage vs N–S Bond Cleavage of Tertiary Sulfonamide.

Results and Discussion
To establish the reaction condition, N-(3,4-dimethoxy-benzyl)-4-methyl-N-phenyl-benzenesulfonamide 4a was chosen as a model substrate. The substrate 4a was reacted under various metal triflate-/halide-based Lewis acids in 1,2-dicholoroethane (ClCH2CH2Cl) at 85 °C. The key results are summarized in Table 1. Lewis acids such as Bi(OTf)3, Fe(OTf)3, Sc(OTf)3, and Al(OTf)3 afforded the desired C–N bond cleavage product 3a in good to excellent yields (78–95%; entries 1–4). Halide-based Lewis acids such as FeCl3 and AlCl3 were also tested using this catalytic method, albeit slightly lower yields were observed (entries 5 and 6). Among these Lewis acids, Bi(OTf)3 in 5 mol % was found to be the best in terms of yield (95%) and clean reaction (entry 1). However, other Bi(III)-Lewis acids with variation of counterions such as Bi(NO3)3 and BiCl3 were not found suitable for the cleavage reaction (entries 7 and 8). This observation suggests that the triflate counteranion might have played an important role in enhancing the chemical yield of the cleavage product. While the reaction was conducted at room temperature (25 °C), no C–N bond cleavage product was observed, even after 24 h. Other solvents such as 1,4-dioxane, acetonitrile, toluene, and ethanol were also studied; however, none of the solvents gave the desired product 3a as high as 1,2-dichloroethane (entries 9–12). It is noteworthy that the reaction was studied in the presence of air or N2 (balloon), and under both conditions, the cleavage product 3a was obtained in comparable yields, 95 and 92%, respectively (entries 1 and 13). Thus, the optimum conditions for this C–N bond cleavage reaction were 4a (1.0 equiv) and Bi(OTf)3 (0.05 equiv) in 1,2-dichloroethane heated at 85 °C in an open air atmosphere for 2 h, which afforded the desired cleavage product 4-methyl-N-phenyl-benzenesulfonamide 3a in a 95% yield.
Table 1. Reaction Optimizationa.

| entry | Lewis acid | solventb | time (h) | yield of 3a (%)c |
|---|---|---|---|---|
| 1 | Bi(OTf)3 | DCE | 2 | 95 |
| 2 | Fe(OTf)3 | DCE | 2 | 84 |
| 3 | Sc(OTf)3 | DCE | 3 | 81 |
| 4 | Al(OTf)3 | DCE | 3 | 78 |
| 5 | FeCl3 | DCE | 2 | 71 |
| 6 | AlCl3 | DCE | 2 | 61 |
| 7 | Bi(NO3)3 | DCE | 12 | 22 |
| 8 | BiCl3 | DCE | 12 | 18 |
| 9 | Bi(OTf)3 | 1,4-dioxane | 3 | 86 |
| 10 | Bi(OTf)3 | acetonitrile | 1.5 | 71 |
| 11 | Bi(OTf)3 | toluene | 2 | 81 |
| 12 | Bi(OTf)3 | ethanol | 2 | 43 |
| 13d | Bi(OTf)3 | DCE | 2 | 92 |
Reaction conditions: 4a (1.0 equiv, 0.25 mmol) and Lewis acid (0.05 equiv, 0.0125 mmol) were mixed together in 2 mL of solvent and heated at 85 °C.
Solvent used in the reaction without any further purification protocol.
Isolated (column purified) yield was reported.
The reaction was conducted under a N2-balloon and a degasified solvent was used (DCE = 1,2-dichloroethane).
To explore the scope of the C–N bond cleavage reactions, various DMB- or PMB-protected N-aryl sulfonamides 4 were studied under the optimized conditions. The results are summarized in Scheme 4. With the variation of the aryl moiety (Scheme 4A), the substrates bearing phenyl- (4a), p-tolyl- (4b), 2,4-dimethylphenyl- (4c), and m-chlorophenyl- (4d) substituted p-tolylsulfonamides provided the corresponding C–N bond cleavage products 3a–d in good to excellent yields (90–95%). Interestingly, the substrate with electron-rich 3,4-dimethoxyphenyl-substituted sulfonamides (4e) afforded the C–N bond cleavage product in a moderate yield (60%). In this example, additional detosylated product 3e’ (23%) through N–S bond cleavage and the intermolecular p-tolylsulfonyl-group-migrated product 3e’’ (12%) were formed. The in situ-generated triflic acid might be the plausible explanation for these observations.16 Similarly, PMB-protected arylsulfonamides (4f–i) were also examined under the stated reaction conditions, and all of the substrates afforded C–N bond cleavage products 3a, 3c, 3d, and 3f in high yields (89–95%), respectively (Scheme 4A). However, N,N-4-methylbenzyl-phenyl-substituted p-tolylsulfonamide (4j) gave the cleavage product with a little low yield (89%). However, while a 4-bromobenzyl-substituted substrate (4k) was subjected to the optimized conditions, it was found completely inactive to this catalytic method (Scheme 4B). Furthermore, we also studied the substrates with the variation of different substituents at the aryl moiety on the sulfonyl part; the results are summarized in Scheme 4C. Likewise, p-tolyl-, α-naphthyl- (4l), p- and o-trifluoromethylphenyls- (4m and 4n), and p-nitrophenyl- (4o) substitutions at the aryl moiety (Ar3) were well tolerated and gave the cleavage products in high yields (80–90%).
Scheme 4. C–N Bond Cleavage of N-Aryl Sulfonamides.

Next, we turned our focus toward the cinnamyl- and (3,4-dihydro-naphthalen-2-ylmethyl)-substituted various N-aryl sulfonamides. Successfully, all of the substrates (5a–e and 1d) well accepted this cleavage method and afforded the desired cleavage products in a range of good to high yields (Scheme 5). Likewise, electron-rich aryl-substituted sulfonamide substrates (4e) and the detosylation and tosyl-group-migrated products were also observed for the substrate 5e. Further, the effect of electronic tuning on the dibenzyl moieties (chemoselectivity) in the sulfonamides (6a–c) was examined, and expectedly, the PMB group deprotected products (3l and 3n) were obtained in good yields (Scheme 6). Interestingly, the substrate 6c with di-PMB-substituted sulfonamide afforded the fully PMB-deprotected tosylamide product 3n in 80% yield. Similarly, other N-alkyl sulfonamides (7a and 7b) also responded well to this catalytic method and afforded the desired cleavage products 3o–p in good yields (76%).
Scheme 5. C–N Bond Cleavage of N-Cinnamylsulfonamides.

Scheme 6. C–N Bond Cleavage of PMB-Protected N-Alkyl Sulfonamides (Chemoselectivity).

Several control experiments were conducted to obtain insights into the plausible reaction mechanism (Scheme 7). Irrespective of whether the Lewis acid Bi(OTf)3 or the highly acidic in situ-generated hidden Brønsted acid (triflic acid)17 plays the key role in this C–N bond cleavage reaction, we performed a reaction in anhydrous 1,2-dichoroethane in the presence of molecular sieves (MS 4A). A trace amount of the (<5%) cleavage product was formed (Scheme 7i). Another experiment, in DCE-H2O (10:1 v/v), using catalytic Bi(OTf)3 was conducted, which afforded the desired product in 93% yield (Scheme 7ii). It proves that the presence of water in the reaction system is essential for the C–N bond cleavage under optimized conditions. Further, a separate reaction using catalytic triflic acid (10 mol %) yielded 3a almost in a similar yield (89%; Scheme 7iii). With these observations, we hypothesized that the cleavage reaction undergoes in the presence of hidden in situ-generated triflic acid. Moreover, the catalytic presence of triflic acid afforded the cleavage product with very high yields, deliberately, compelling us to examine further control experiments. Consequently, a reaction was conducted in the presence of the triflic acid scavenger DTBMP (2,6-di-tertiary-butyl-4-methylpyridine (Scheme 7iv).18 The very low yield of the cleavage product obtained in this control experiment seemingly suggests that the true catalyst in the cleavage reaction could be triflic acid. The C–H oxidation at the benzylic position of the sulfonamides in the presence of oxygen (solvated oxygen/air) and followed by hemiaminal hydrolysis may give the desired product via a radical mechanism.19 However, such a possibility is unlikely as the reaction under the N2 gas atmosphere and using degasified solvent systems (DCE-H2O) yielded the desired product in a similar yield (91%; Scheme 7v). Finally, under the dark condition and also in the presence of a radical scavenger BHT20 (1.5 equiv), the same set of reaction provided the desired cleavage products 3a in high yields (Scheme 7vi,vii). Thus, it can be unambiguously concluded that the cleavage method never follows the radical steps.
Scheme 7. Control Experiments for Mechanistic Insight.

Reagent and conditions: (i) Bi(OTf)3 (5 mol %) ″dry DCE″, MS 4Α, 85 °C; (ii) Bi(OTf)3 (5 mol %), ″DCE-H2O″ (10:1), 85 °C; (iii) ″TfOH″ (10 mol %), 85 °C; (iv) Bi(OTf)3 (5 mol %), ″DCE-H2O″ (10:1), DTBMP (1.5 equiv), 85 °C; (v) Bi(OTf)3 (5 mol %), ″DCE-H2O″ (10:1), ″N2 balloon″, 85 °C; (vi) Bi(OTf)3 (5 mol %), ″DCE-H2O″ (10:1), ″dark condition″, 85 °C; (vii) Bi(OTf)3 (5 mol %), ″DCE-H2O″ (10:1), BHT (1.2 equiv), 85 °C.
Arguably, triflic acid is the true active catalyst in this cleavage method, and with this validity, a plausible reaction mechanism is proposed, which is depicted in Scheme 8. The reaction mechanism presumably starts with the protonation event. There is a dual chance of the protonation taking place on both the nitrogen atom of sulfonamide or oxygen atoms of the sulfonyl group in the tertiary sulfonamide substrate.21 To validate this, the substrates with N-aryl-N-3,4-dimethoxybenzylcarboxamides (4p–q) were reacted under the optimized conditions. These substrates completely failed to give the corresponding C–N bond cleavage products. The results from these unsuccessful substrates (with lack of a sulfonamide moiety) might be due to the higher electron-withdrawing capability of carboxamide (COR) rather than sulfonamide (SO2R). On the basis of prior reports and experimental results, it can be concluded that the initial protonation most likely is taking place on the N atom of tertiary sulfonamide (Scheme 8).21 Following the protonation, substrate 4 forms the intermediate I, which consequently releases a highly reactive benzyl carbocationic intermediate II or its resonance-stabilized intermediate III, affording the desired cleavage product 3a. The in situ formation of highly reactive intermediate II or III is also further confirmed. The cleavage reaction was executed in the presence of the external nucleophile 1,2-dimethoxybenzene (2.0 equiv) under the optimized conditions. Pleasingly, we were able to isolate the Friedel–Craft alkylated products 8a (45%) and 8b (23%) in moderate yields alongside the desired cleavage product 3a in 93% yield (Scheme 8i).
Scheme 8. Plausible Mechanism.
On the other hand, during the course of the reaction, the intermediate II or III might be trapped by water and the corresponding benzyl alcohol should be formed as a major byproduct as the reaction medium contains sufficient water. However, 3,4-dimethoxyphenylmethanol (R = OMe) was never isolated from the reaction system or traced by the TLC during the course of the reaction. Gratifyingly, a tribenzocyclononane (2,3,7,8,12,13-hexamethoxy-10,15-dihydro-5H-tribenzo[a,d,g]cyclononane) compound 10 was isolated in a 22% yield (based on sulfonamide substrate 4a). To further probe, a separate reaction was conducted with 3,4-dimethoxyphenylmethanol in the presence of a catalytic amount of Bi(OTf)3 (5 mol %) under the same optimized reaction conditions (Scheme 8ii). After 1 h, the alcohol was completely consumed, and as expected, tribenzocyclononane compound 10 was isolated in 76% yield (Scheme 8ii). The formation of compound 10 can be elucidated by acid-catalyzed trimerization reactions of 3,4,-dimethoxybenzylphenylmethanol under acidic conditions.22 It is further verified using mass spectroscopic techniques. In the mass spectrum, the intense characteristic peak exhibited at 468.2575 m/z (M+) can be attributed to compound 9, which, in turn, can be easily cyclized to afford a tribenzocyclononane product 10, which appeared at 451.2277 m/z (M + H+). Based on these control experiments and observations, we conclude that this C–N bond cleavage protocol of tertiary sulfonamides (4, 5, and 6) proceeds via the protonation event, followed by the C–N bond cleavage step (step I to step II).
Finally, the methodology was applied to the gram scale for the one-pot synthesis of N-phenyl-N-tosylacetamide 11 from 4a (Scheme 9).
Scheme 9. One-Pot Synthesis of N-Phenyl-N-tosylacetamide.

Conclusions
In conclusion, we have developed a catalytic C–N bond cleavage protocol of tertiary PMB/DMB-protected sulfonamides under a mild acidic condition. Various DMB- or PMB-protected N-aryl/alkylsulfonamides underwent smoothly and afforded highly selective C–N bond cleavage products with high yields in most of the substrates. This catalytic protocol is applicable also to the cinnamyl-substituted N-aryl sulfonamides. However, both C–N and N–S bond cleavages including tosyl migration are observed in a parallel way while the electron-rich substituted N-aryl sulfonamides are used. On the basis of several control experiments and observations, the catalytic cleavage reaction most likely is proposed, which generally comprises protonation on the nitrogen atom of N-aryl or N-alkyl tertiary sulfonamides, followed by C–N bond cleavage. The immediate stability of the in situ-generated benzylic carbocationic intermediate (II) is possibly the driving force for the highly selective C–N bond cleavage over the N–S bond cleavage (detosylation). Great efforts have been made regarding mechanism interrogation to realize a plausible reaction mechanism based on a series of control experiments and mass spectroscopy.
Experimental Section
All reactions were conducted using oven-dried glassware. Commercial AR-grade reagents were used without further purification. Solvents were dried and distilled following usual protocols. However, all of the cleavage reactions were performed in DCE directly without any further purification (bottle-grade) purchased from CDH make in India. Flash column chromatography was performed in all cases using the indicated solvent system on silica gel (230–400 mesh). Analytical thin-layer chromatography (TLC) was performed on aluminum-backed plates coated with silica gel 60 with an F254 indicator (Merck). The 1H NMR spectra were measured with 400 MHz, and 13C NMR spectra were recorded with 400 (100 MHz), using CDCl3 as the solvent. 1H NMR chemical shifts are expressed in parts per million (δ) downfield to CHCl3 (δ = 7.26), and 13C NMR chemical shifts are expressed in parts per million (δ) relative to the central CDCl3 resonance (δ = 77.0). Coupling constants in 1H NMR are in Hz. The following abbreviations classify the multiplicity: s, singlet; d, doublet; t, triple; m, multiplet or unresolved; dd, doublet of doublets. Electrospray ionization (ESI) mass spectrometry (MS) experiments were performed on Agilent Technologies 6530 Accurate-Mass Q-TOF LC/MS.
All 1,2-dihydronaphthalene-based alkenes were synthesized according to the reported literature procedure.23N-Benzyl and N-cinnamyl amines were prepared by reaction between commercially available aniline and respective aldehydes using the following standard literature procedure.24 3,4-Dihydro-naphthalen-2-ylmethyl)-aryl amines were prepared by reaction between commercially available anilines and the respective carbaldehyde using the following standard literature procedure.23 All secondary amines were further protected by following standard protocols. All Lewis acids, Brønsted acids, and other solvents and reagents used in this study were purchased from commercial suppliers.
Preparation of Substrates
General Procedure I: Synthesis of Secondary Amines
In an oven-dried round-bottom flask, aniline derivative (1.0 equiv), aldehyde (1.0 equiv), and triethyl amine (1.5 equiv) were dissolved in MeOH, and the reaction mixture was stirred at room temperature until the starting material was completely consumed and imine was formed (checked by TLC). The reaction mixture was cooled to 0 °C, and NaBH4 or Zn(BH4)2 (1.1 equiv) was added slowly and allowed to stir at room temperature until the intermediate imine was completely reduced to the corresponding amine. The reaction mixture was diluted with water and extracted with ethyl acetate. The combined organic layer was washed with brine, dried over Na2SO4, and concentrated in vaccuo, and the crude mixture was purified by flash column chromatography using petroleum ether and ethyl acetate as the eluent.
General Procedure II: Synthesis of Tertiary Sulfonamides (Protection of Secondary Amines)
All sulfonyl protected tertiary N-aryl sulfonamides were prepared from the corresponding secondary amine and sulfonyl chloride (2.0 equiv) in the presence of Et3N (3.0 equiv) and DMAP (10 mol %) in DCM at room temperature. N-Alkyl sulfonamides were synthesized via the N-alkylation reaction of monosubstituted sulfonamides with the corresponding alkyl halide following the standard literature procedure.16
General Procedure III: Friedel–Crafts Alkylation of 1,2-Dihydronaphthalene-Based Alkenes
To a stirred solution of alkene 1 (0.43 mmol, 1.0 equiv) in dichloroethane (2 mL), Bi(OTf)3 (0.05 equiv) was added, and the resultant mixture was refluxed at 85 °C for 2 h. The reaction was monitored by TLC to ensure completion. On completion, DCE was removed under reduced pressure and the crude mixture was directly subjected to purification by flash column chromatography using an EtOAc and petroleum ether mixture as the eluent, affording the desired Friedel–Crafts alkylated product 2.
General Procedure IV: C–N Bond Cleavage of Tertiary Sulfonamides
To a stirred solution of tertiary sulfonamides [4–6] (0.25 mmol, 1 equiv) in 1,2-dichloroethane (2 mL), Bi(OTf)3 (0.05 equiv) was added, and the resultant mixture was refluxed at 85 °C for 2 h. The reaction was monitored by TLC to ensure completion. On completion, DCE was removed under reduced pressure and the crude mixture was directly subjected to purification by flash column chromatography using an EtOAc and petroleum ether mixture as the eluent, affording the desired cleavage product.
Characterization data for compounds 1, 2, 3, 4, 5, 6, 7, 8, 10, and 11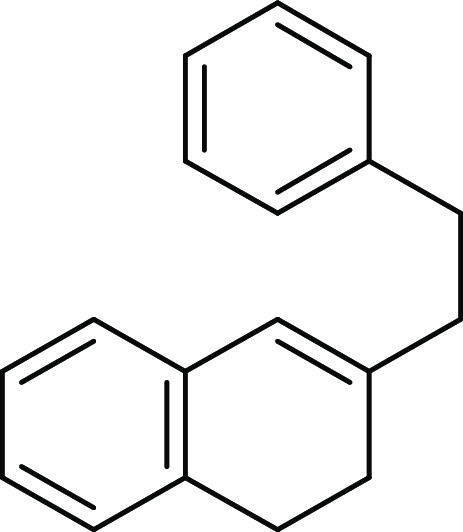
3-Phenethyl-1,2-dihydronaphthalene
(1a): Prepared
according to the reported literature procedure; 80% yield; viscous
oil. All spectroscopic data are identical to those reported in the
literature.23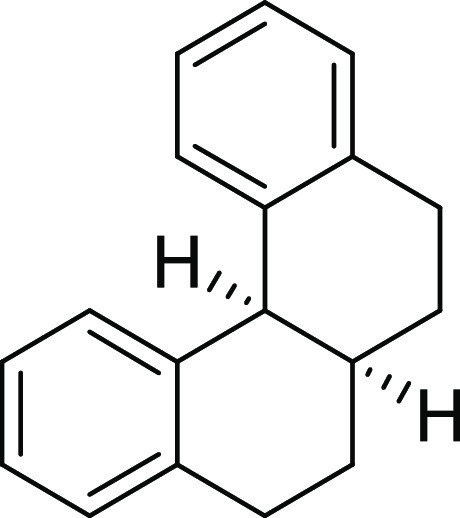
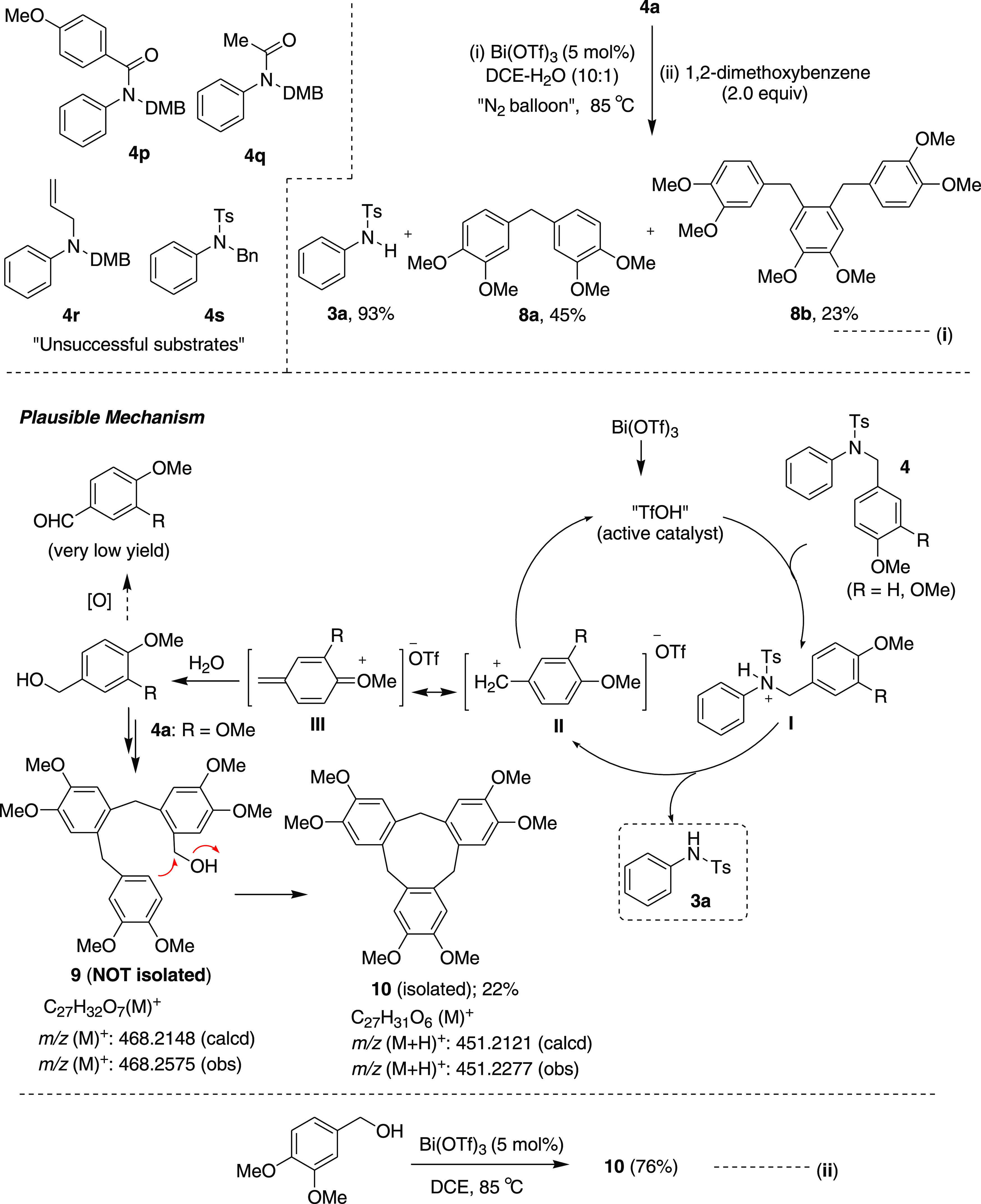
5,6,6a,7,8,12b-Hexahydrobenzo[c]phenanthrene (2a): Prepared according to general procedure III using 1a (100 mg, 0.43 mmol, 1.0 equiv); colorless
oil (94 mg, 94% yield); Rf = 0.2 (2% EA/PE); 1H NMR (CDCl3, 400 MHz): δ 7.30–7.13
(m, 8H), 4.02 (d, J = 8.8 Hz, 1H), 2.90–2.83
(m, 4H), 2.51–2.40 (m, 1H), 2.06–1.97 (m, 2H), 1.67–1.48
(m, 2H); 13C NMR (CDCl3, 100 MHz): δ 138.9,
137.5, 129.8 (2C), 128.6 (2C), 128.3, 126.0 (2C), 125.3 (3C), 42.9,
32.4, 28.1 (2C), 27.3 (2C); HRMS (ESI)+ calcd for C18H19, 235.1487 m/z (M + H)+; found, 235.1488 m/z.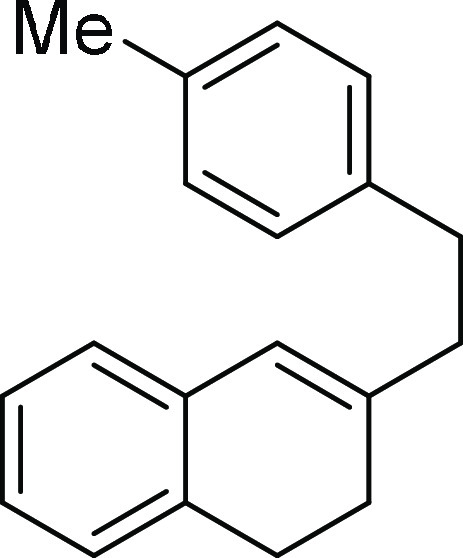
3-(4-Methylphenethyl)-1,2-dihydronaphthalene
(1b):
Prepared according to the reported literature procedure; 21% yield;
colorless oil. All spectroscopic data are identical to those reported
in the literature.23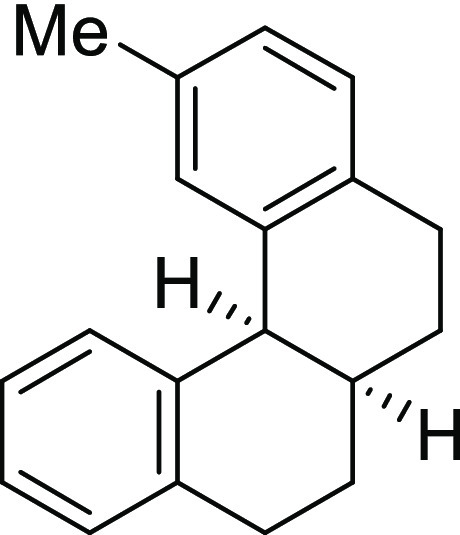
2-Methyl-5,6,6a,7,8,12b-hexahydrobenzo[c]phenanthrene (2b): Prepared according to
general procedure III using 1b (100 mg,
0.40 mmol, 1.0 equiv); colorless oil (91 mg, 91% yield); Rf = 0.2 (2% EA/PE); 1H NMR (CDCl3, 400 MHz): δ 7.34–7.19 (m, 4H), 7.17–7.06 (m,
2H), 7.00 (s, 1H), 4.04 (d, J = 10.0 Hz, 1H), 3.01–2.84
(m, 4H), 2.49–2.44 (m, 1H), 2.39 (s, 3H), 2.14–1.96
(m, 2H), 1.72–1.52 (m, 2H); 13C NMR (CDCl3, 100 MHz): δ 139.0, 138.6, 137.5, 134.6, 134.3, 130.5, 129.8,
128.5 (2C), 126.8, 125.9, 125.3, 42.8, 32.4, 27.9, 27.6, 27.3, 27.2,
21.2; HRMS (ESI)+ calcd for C19H21, 249.1643 m/z (M + H)+; found, 249.1647 m/z.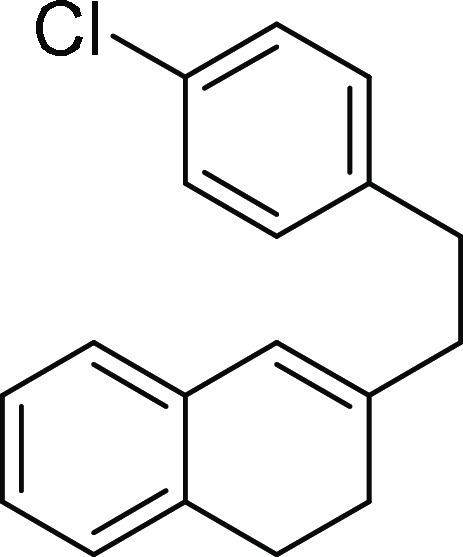
3-(4-Chlorophenethyl)-1,2-dihydronaphthalene (1c):
Prepared according to the reported literature procedure;1 40% yield; colorless oil; 1H NMR (CDCl3, 400 MHz): δ 7.46–7.24 (m, 3H), 7.19–7.12
(m, 4H), 7.06 (d, J = 12.4 Hz, 1H), 6.30 (s, 1H),
3.04–2.85 (m, 4H), 2.56 (t, J = 14.4 Hz, 2H),
2.35 (t, J = 16.0 Hz, 2H); 13C NMR (CDCl3, 100 MHz): δ 141.0, 140.5, 134.9, 134.5, 131.7, 129.9
(2C), 128.6 (2C), 127.3, 126.6, 126.4, 125.6, 123.1, 39.3, 33.7, 28.3,
27.6; HRMS (ESI)+ calcd for C18H18Cl, 269.1097 m/z (M + H)+; found, 269.1099 m/z.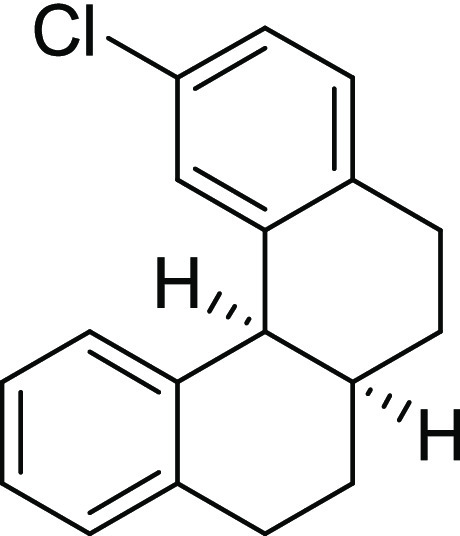
2-Chloro-5,6,6a,7,8,12b-hexahydrobenzo[c]phenanthrene (2c): The reaction was performed
according to general procedure III using 1c (100 mg, 0.37 mmol, 1.0 equiv). No cyclized product was obtained.
Yield, 0%.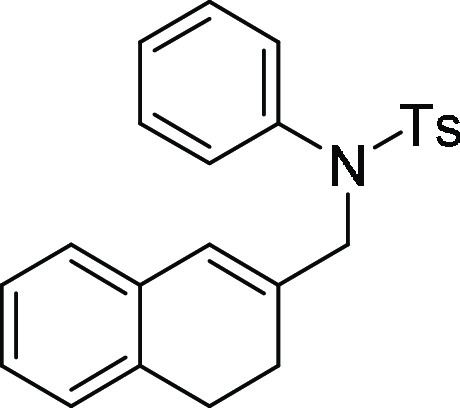
N-(3,4-Dihydronaphthalen-2-ylmethyl)-4-methyl-N-phenyl-benzenesulfonamide (1d). Prepared
according to general procedure II; 89% yield; off-white
solid; mp = 130–132 °C; 1H NMR (CDCl3, 400 MHz): δ 7.48 (d, J = 8.0 Hz, 2H), 7.24–7.21
(m, 5H), 7.05–7.02 (m, 5H), 6.84–6.83 (m, 1H), 6.10
(s, 1H), 4.27 (s, 2H), 2.67 (t, J = 8.0 Hz, 2H),
2.42 (s, 3H), 2.35 (t, J = 8.0 Hz, 2H); 13C NMR (CDCl3, 100 MHz): δ 143.3, 138.2, 135.4, 135.3,
135.2, 133.8, 129.6 (2C), 128.9 (2C), 128.8 (2C), 127.9 (2C), 127.8,
127.4, 127.2, 127.0, 126.04, 126.0, 56.4, 27.9, 24.8, 21.7; HRMS (ESI)+ calcd for C24H23NNaO2S,
412.1347 m/z (M + Na)+; found, 412.1348 m/z.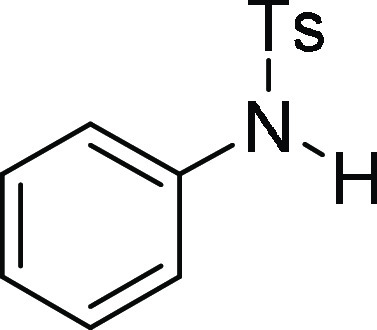
4-Methyl-N-phenyl-benzenesulfonamide (3a):25 Prepared according to general procedure III using 1d (100 mg, 0.26 mmol, 1.0 equiv);
white solid (54 mg, 84% yield). Rf = 0.25
(25% EA/PE); mp = 102–104 °C; 1H NMR (CDCl3, 400 MHz): δ 7.73 (d, J = 6.48, 2H),
7.44 (s, 1H), 7.25–7.22 (m, 4H), 7.15–7.08 (m, 3H),
2.37 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ
143.9, 136.7, 136.1, 129.7 (2C), 129.3 (2C), 127.4 (2C), 125.2, 121.5
(2C), 21.6; HRMS (ESI)+ calcd for C13H14NO2S, 248.0745 m/z (M
+ H)+; found, 248.0743 m/z.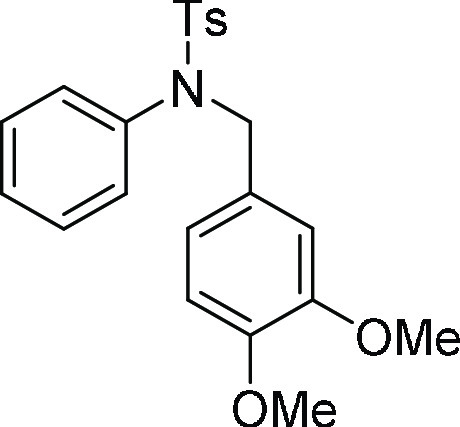
N-(3,4-Dimethoxy-benzyl)-4-methyl-N-phenyl-benzenesulfonamide (4a): Prepared
according
to general procedure II; 85% yield; off-white solid;
mp = 158–160 °C; 1H NMR (CDCl3,
400 MHz): δ 7.52 (d, J = 8.0 Hz, 2H), 7.25
(d, J = 8.0 Hz, 2H), 7.18–7.15 (m, 3H), 6.95–6.92
(m, 2H), 6.77 (s, 1H), 6.62–6.60 (m, 2H), 4.64 (s, 2H), 3.77
(s, 6H), 2.41 (s, 3H); 13C NMR (CDCl3, 100 MHz):
δ 149.0, 148.9, 143.5, 139.0, 135.8, 129.6 (2C), 129.1 (2C),
128.9 (2C), 128.4, 127.9, 127.8 (2C), 121.1, 111.7, 110.7, 55.9, 55.8,
54.6, 21.6; HRMS (ESI)+ calcd for C22H23NNaO4S, 420.1245 m/z (M + Na)+; found, 420.1245 m/z.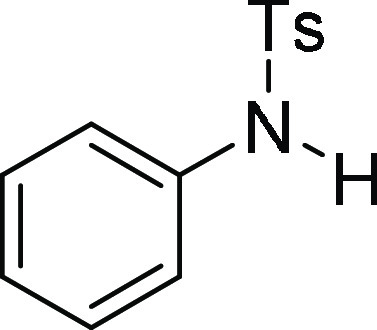
4-Methyl-N-phenyl-benzenesulfonamide
(3a):25 Prepared according
to general procedure IV using 4a (100 mg,
0.25 mmol, 1.0 equiv); white
solid (59 mg, 95% yield). All spectroscopic data are identical to 3a obtained from 1d and those reported in the
literature.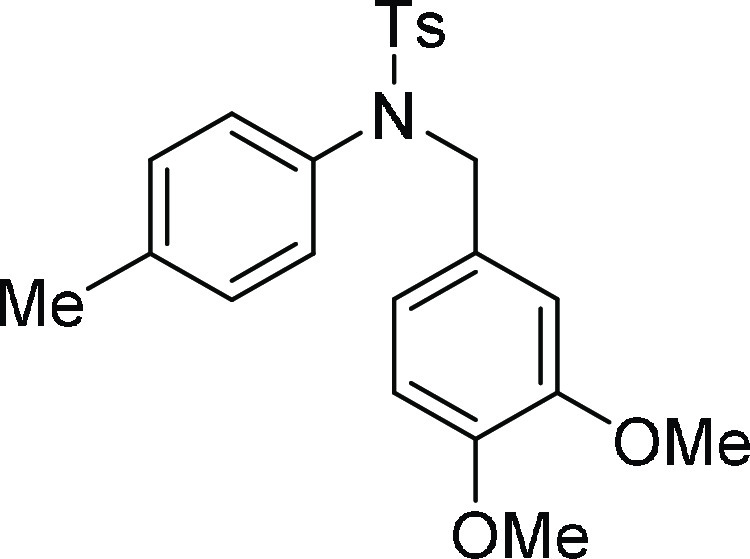
N-(3,4-Dimethoxy-benzyl)-4-methyl-N-p-tolyl-benzenesulfonamide (4b): Prepared
according to general procedure II; 83% yield; yellow
solid; mp = 130–132 °C; 1H NMR (CDCl3, 400 MHz): δ 7.54 (d, J = 8.4 Hz, 2H), 7.26
(d, J = 8.0 Hz, 2H), 6.98 (d, J =
8.4 Hz, 2H), 6.83–6.80 (m, 3H), 6.64 (s, 2H), 4.62 (s, 2H),
3.79 (s, 3H), 3.78 (s, 3H), 2.42 (s, 3H), 2.25 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 148.9, 148.4, 143.4, 137.8,
136.3, 135.9, 129.54 (3C), 129.52 (2C), 128.8 (2C), 128.5, 127.8,
121.0, 111.6, 110.7, 55.9, 55.8, 54.6, 21.6, 21.1; HRMS (ESI)+ calcd for C23H25NNaO4S,
434.1402 m/z (M + Na)+; found, 434.1401 m/z.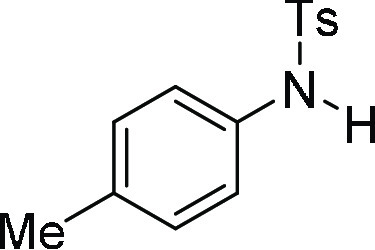
4-Methyl-N-p-tolyl-benzenesulfonamide
(3b):25 Prepared according
to general procedure IV using 4b (100 mg,
0.24 mmol, 1.0 equiv); white solid (60 mg, 95% yield); Rf = 0.2 (25% EA/PE); mp = 117–119 °C; 1H NMR (CDCl3, 400 MHz): δ 7.67 (d, J = 6.8 Hz, 2H), 7.29 (s, 1H), 7.20 (d, J = 6.4 Hz, 2H), 7.02–6.97 (m, 4H), 2.35 (s, 3H), 2.24 (s,
3H); 13C NMR (CDCl3, 100 MHz): δ 143.8,
136.0, 135.2, 133.9, 129.9 (2C), 129.7 (2C), 127.4 (2C), 122.1 (2C),
21.6, 20.9; HRMS (ESI)+ calcd for C14H16NO2S, 262.0902 m/z (M
+ H)+; found, 262.0903 m/z.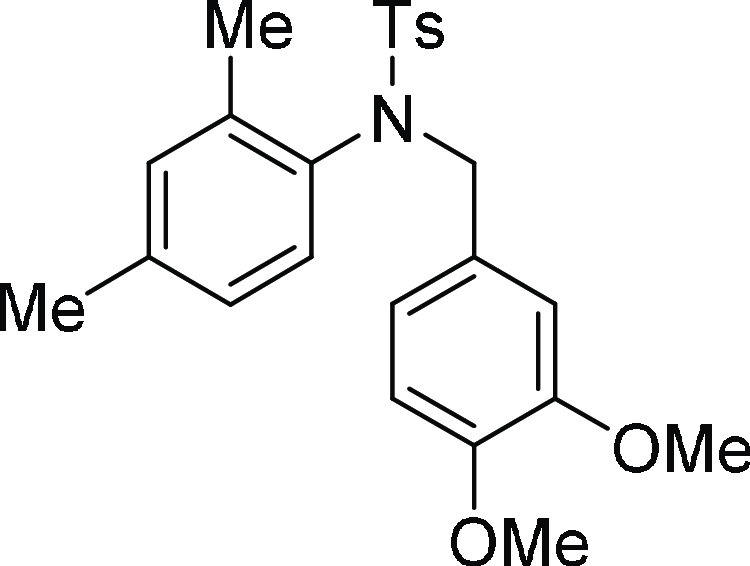
N-(3,4-Dimethoxy-benzyl)-N-(2,4-dimethyl-phenyl)-4-methyl-benzenesulfonamide
(4c): Prepared according to general procedure II; 81% yield; yellow solid; mp = 108–110 °C; 1H NMR (CDCl3, 400 MHz): δ 7.61 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 6.91 (s,
1H), 6.79 (d, J = 8.0 Hz, 1H), 6.70 (s, 1H), 6.63
(d, J = 8.0 Hz, 1H), 6.55 (d, J =
8.4 Hz, 1H), 6.45 (d, J = 8.0 Hz, 1H), 4.90 (d, J = 13.2 Hz, 1H), 4.14 (d, J = 13.2 Hz,
1H), 3.80 (s, 3H), 3.73 (s, 3H), 2.44 (s, 3H), 2.23 (s, 3H), 2.00
(s, 3H); 13C NMR (CDCl3, 100 MHz): δ 148.7,
143.4, 140.0, 138.1, 136.5, 135.3, 132.1, 129.6 (2C), 128.2, 128.0
(3C), 127.9, 126.9, 121.9, 112.5, 110.6, 55.8 (2C), 55.7, 21.7, 21.1,
18.4; HRMS (ESI)+ calcd for C24H27NNaO4S, 448.1558 m/z (M + Na)+; found, 448.1553 m/z.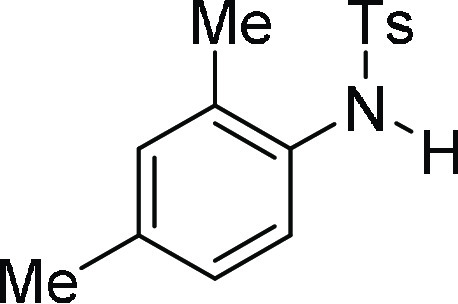
N-(2,4-Dimethyl-phenyl)-4-methyl-benzenesulfonamide
(3c):26 Prepared according
to general procedure IV using 4c (100 mg,
0.24 mmol, 1.0 equiv); white solid (62 mg, 95% yield); Rf = 0.25 (30% EA/PE); mp = 93–95 °C; 1H NMR (CDCl3, 400 MHz): δ 7.60 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H),
7.13 (d, J = 8.04 Hz, 1H), 6.93–6.88 (m, 2H),
6.50 (s, 1H), 2.38 (s, 3H), 2.25 (s, 3H), 1.95 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 143.7, 136.9, 136.4, 132.2,
131.8, 131.6, 129.7 (2C), 127.6, 127.3 (2C), 125.3, 21.7, 21.0, 17.6;
HRMS (ESI)+ calcd for C15H18NO2S, 276.1058 m/z (M + H)+; found, 276.1061 m/z.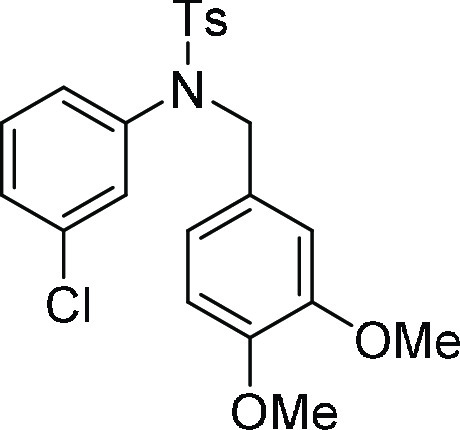
N-(3-Chloro-phenyl)-N-(3,4-dimethoxy-benzyl)-4-methyl-benzenesulfonamide
(4d): Prepared according to general procedure II; 78% yield; white solid; mp = 156–158 °C; 1H NMR (CDCl3, 400 MHz): δ 7.53 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 7.15–7.11
(m, 2H), 6.96 (s, 1H), 6.86 (d, J = 7.6 Hz, 1H),
6.76 (s, 1H), 6.63 (d, J = 7.6 Hz, 2H), 4.61 (s,
2H), 3.78 (s, 6H), 2.43 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 149.0, 148.7, 143.9, 140.2, 135.3, 134.2, 129.8,
129.7 (2C), 129.1, 128.1, 127.8 (2C), 127.7, 127.3, 121.1, 111.6,
110.8, 55.9, 55.8, 54.4, 21.6; HRMS (ESI)+ calcd for C22H22ClNNaO4S, 454.0856 m/z (M + Na)+; found, 454.0862 m/z.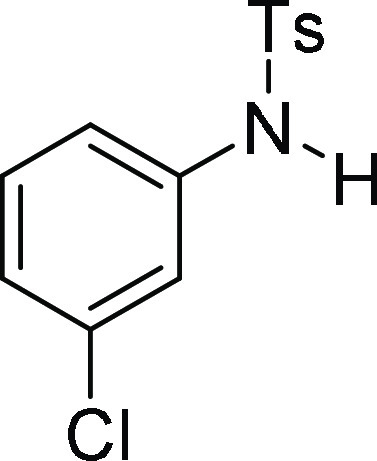
N-(3-Chloro-phenyl)-4-methyl-benzenesulfonamide
(3d):25 Prepared according
to general procedure IV using 4d (100 mg,
0.23 mmol, 1.0 equiv); white solid (59 mg, 90% yield); Rf = 0.25 (25% EA/PE); mp = 135–137 °C; 1H NMR (CDCl3, 400 MHz): δ 7.72 (d, J = 8.0 Hz, 2H), 7.56 (s, 1H), 7.24 (d, J = 8.4 Hz, 2H), 7.13 (t, J = 8.04 Hz, 2H), 7.04
(d, J = 8.0 Hz, 1H), 7.00–6.97 (m, 1H), 2.37
(s, 3H); 13C NMR (CDCl3, 100 MHz): δ 144.4,
138.0, 135.6, 134.9, 130.4, 130.0 (2C), 127.4 (2C), 125.2, 120.8,
118.9, 21.7; HRMS (ESI)+ calcd for C13H13ClNO2S, 282.0356 m/z (M + H)+; found, 282.0355 m/z.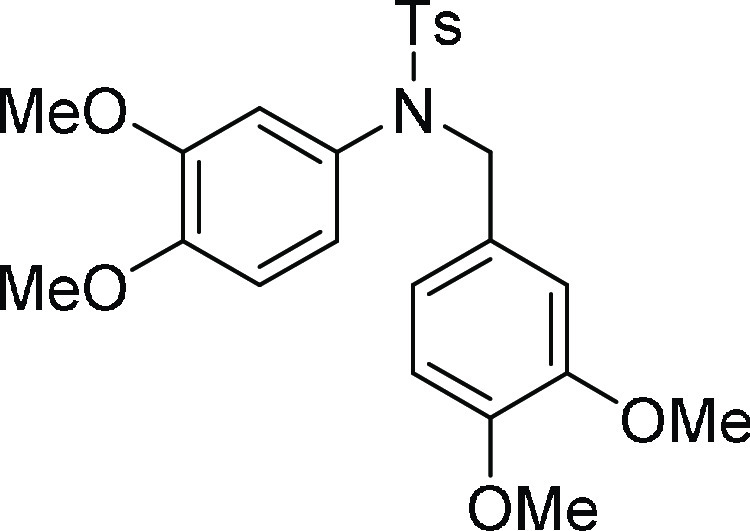
N-(3,4-Dimethoxy-benzyl)-N-(3,4-dimethoxy-phenyl)-4-methyl-benzenesulfonamide
(4e): Prepared according to general procedure II; 90% yield; off-white solid; mp = 125–127 °C; 1H NMR (CDCl3, 400 MHz): δ 7.54 (d, J = 8.04 Hz, 2H), 7.25 (d, J = 8.04 Hz, 2H), 6.79
(s, 1H), 6.64–6.58 (m, 3H), 6.43–6.36 (m, 2H), 4.58
(s, 2H), 3.77 (s, 9H), 3.61 (s, 3H), 2.40 (s, 3H); 13C
NMR (CDCl3, 100 MHz): δ 148.9, 148.7, 148.5, 148.3,
143.5, 135.9, 131.7, 129.5 (2C), 128.6, 127.9 (2C), 121.5, 121.3,
112.9, 111.8, 110.7, 110.6, 55.9 (4C), 55.1, 21.6; HRMS (ESI)+ calcd for C24H27NNaO6S,
480.1457 m/z (M + Na)+; found, 480.1463 m/z.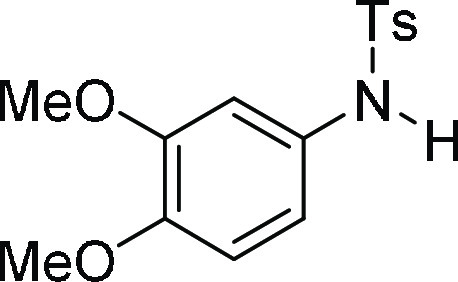
N-(3,4-Dimethoxy-phenyl)-4-methyl-benzenesulfonamide
(3e):27 Prepared according
to general procedure IV using 4e (100 mg,
0.22 mmol, 1.0 equiv); white solid (40 mg, 60% yield); Rf = 0.25 (35% EA/PE); mp = 115–117 °C; 1H NMR (CDCl3, 400 MHz): δ 7.61 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H),
6.77 (s, 1H), 6.70–6.66 (m, 2H), 6.54–6.51 (m, 1H),
3.81 (s, 3H), 3.77 (s, 3H), 2.38 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 149.3, 147.4, 143.8, 136.0, 129.6 (2C),
129.5, 127.5 (2C), 115.7, 111.3, 107.9, 56.1, 56.0, 21.6; HRMS (ESI)+ calcd for C15H18NO4S, 308.0957 m/z (M + H)+; found, 308.0958 m/z.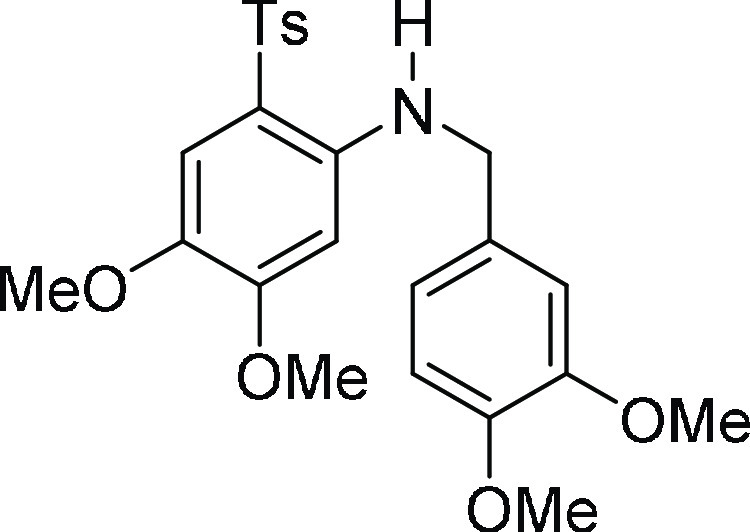
(3,4-Dimethoxy-benzyl)-[4,5-dimethoxy-2-(toluene-4-sulfonyl)-pheny]-amine
(3e′): Obtained during the course of C–N
bond cleavage of 4e according to general procedure IV; white solid, (23 mg, 23% yield); Rf = 0.2 (35% EA/PE); mp = 130–132 °C; 1H NMR (CDCl3, 400 MHz): δ 7.52 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 6.85 (s,
1H), 6.76 (d, J = 8.0 Hz, 1H), 6.56 (s, 1H), 6.49
(d, J = 8.0 Hz, 1H), 6.44 (s, 1H), 6.10 (S, 1H),
3.85 (s, 3H), 3.80 (s, 3H), 3.78 (s, 3H), 3.75 (s, 3H), 3.46 (s, 2H),
2.41 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ
149.4, 147.9, 147.8, 147.5, 143.8, 136.8, 131.6, 129.6 (2C), 127.7,
127.3, 127.2 (2C), 120.3, 113.4, 111.6, 111.5, 110.1, 56.1, 56.02,
56.01, 55.9, 37.0, 21.6; HRMS (ESI)+ calcd for C24H28NO6S, 458.1637 m/z (M + H)+; found, 458.1634 m/z.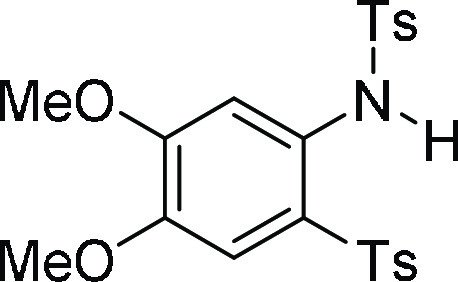
N-[4,5-Dimethoxy-2-(toluene-4-sulfonyl)-phenyl]-4-methyl-benzenesulfonamide
(3e″): Obtained during the course of C–N
bond cleavage of 4e according to general procedure IV; white solid (13 mg, 12% yield); Rf = 0.22 (35% EA/PE); mp = 123–125 °C; 1H NMR
(CDCl3, 400 MHz): δ 7.43 (d, J =
8.4 Hz, 2H), 7.38 (s, 1H), 7.31 (s, 1H), 7.02 (d, J = 8.0 Hz, 2H), 6.86 (d, J = 8.0 Hz, 2H), 6.79 (s,
1H), 6.63 (d, J = 8.4 Hz, 2H), 3.86 (s, 3H), 3.69
(s, 3H), 2.28 (s, 3H), 2.21 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 151.2, 146.4, 144.0, 136.0, 135.7, 133.5,
132.9, 130.0 (2C), 129.6 (2C), 127.4 (2C), 126.9 (2C), 118.6, 112.8,
104.8, 56.3, 56.2, 21.7, 21.1; HRMS (ESI)+ calcd for C22H24NO6S2, 462.1045 m/z (M + H)+; found, 462.1043 m/z.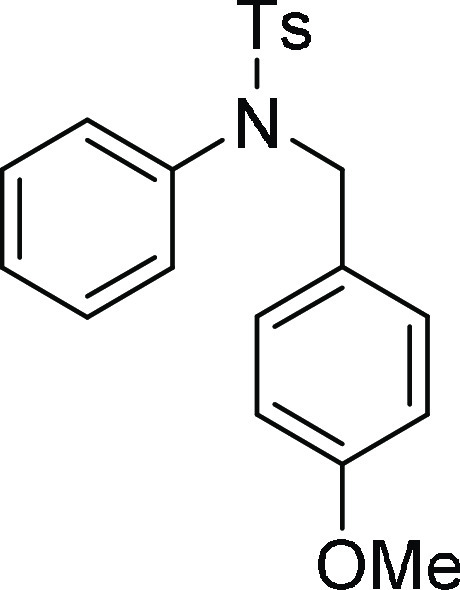
N-(4-Methoxy-benzyl)-4-methyl-N-phenyl-benzenesulfonamide (4f): Prepared
according
to general procedure II; 91% yield; off-white solid;
mp = 130–132 °C; 1H NMR (CDCl3,
400 MHz): δ 7.52 (d, J = 8.0 Hz, 2H), 7.25
(d, J = 8.0 Hz, 2H), 7.18–7.17 (m, 3H), 7.09
(d, J = 8.0 Hz, 2H), 6.95–6.92 (m, 2H), 6.71
(d, J = 8.0 Hz, 2H), 4.64 (s, 2H), 3.71 (s, 3H),
2.42 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ
159.1, 143.5, 139.0, 135.9, 130.0 (2C), 129.6 (2C), 129.2 (2C), 128.9
(2C), 128.1, 127.8 (3C), 113.8 (2C), 55.3, 54.3, 21.7; HRMS (ESI)+ calcd for C21H21NNaO3S,
390.1140 m/z (M + Na)+; found, 390.1142 m/z.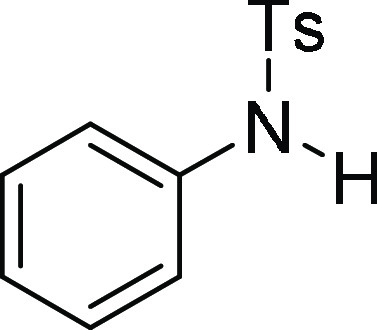
4-Methyl-N-phenyl-benzenesulfonamide (3a):25 Prepared according to general procedure IV using 4f (100 mg, 0.27 mmol, 1.0 equiv); white
solid (60 mg, 89% yield). All data are similar to the compound obtained
from 4a.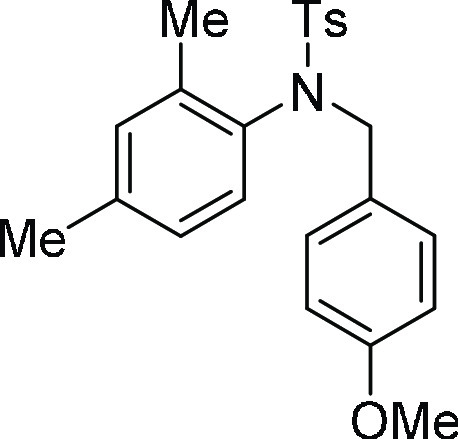
N-(2,4-Dimethyl-phenyl)-N-(4-methoxy-benzyl)-4-methyl-benzenesulfonamide
(4g): Prepared according to general procedure II; 80% yield; off-white solid; mp = 110–112 °C; 1H NMR (CDCl3, 400 MHz): δ 7.62 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 8.4 Hz, 2H), 6.91 (s, 1H), 6.80 (d, J = 8.8 Hz, 1H), 6.71 (d, J = 8.8 Hz, 2H), 6.46 (d, J = 8.0 Hz, 1H), 4.92 (d, J = 13.6 Hz,
1H), 4.14 (d, J = 13.6 Hz, 1H), 3.74 (s, 3H), 2.45
(s, 3H), 2.24 (s, 3H), 1.97 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 159.3, 143.4, 140.0, 138.1, 136.6, 135.3,
132.0, 130.8 (2C), 129.6 (2C), 128.1, 128.0 (2C), 127.9, 126.9, 113.6
(2C), 55.4, 55.3, 21.7, 21.2, 18.3; HRMS (ESI)+ calcd for
C23H25NNaO3S, 418.1453 m/z (M + Na)+; found, 418.1458 m/z.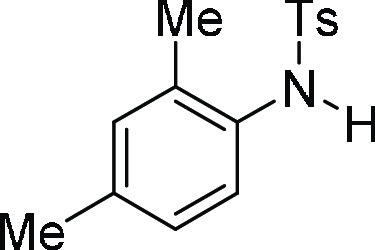
N-(2,4-Dimethyl-phenyl)-4-methyl-benzenesulfonamide
(3c):26 Prepared according
to general procedure IV using 4g (100 mg,
0.25 mmol, 1.0 equiv); white solid (66 mg, 95% yield). All data are
similar to the compound obtained from 4c.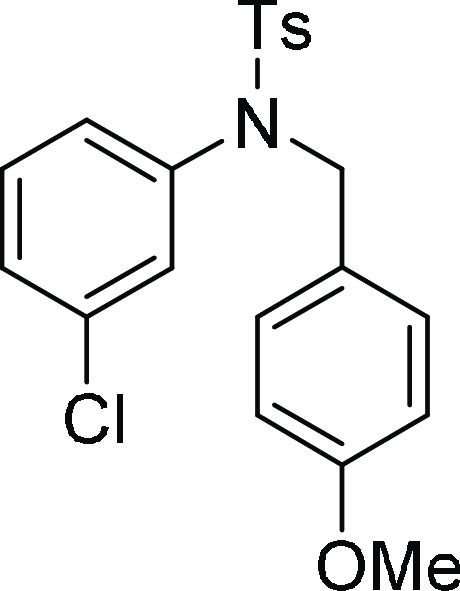
N-(3-Chloro-phenyl)-N-(4-methoxy-benzyl)-4-methyl-benzenesulfonamide
(4h): Prepared according to general procedure II; 87% yield; white solid; mp = 138–140 °C; 1H NMR (CDCl3, 400 MHz): δ 7.53 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 7.17–7.09
(m, 4H), 6.98 (s, 1H), 6.86 (d, J = 7.6 Hz, 1H),
6.74 (d, J = 8.8 Hz, 2H), 4.62 (s, 2H), 3.72 (s,
3H), 2.44 (s, 3H); 13C NMR (CDCl3, 100 MHz):
δ 159.1, 143.9, 140.2, 135.1, 134.2, 129.9 (2C), 129.7 (3C),
129.1, 128.1, 127.7 (2C), 127.4, 127.2, 113.9 (2C), 55.2, 54.0, 21.7;
HRMS (ESI)+ calcd for C21H20ClNNaO3S, 424.0750 m/z (M + Na)+; found, 424.0753 m/z.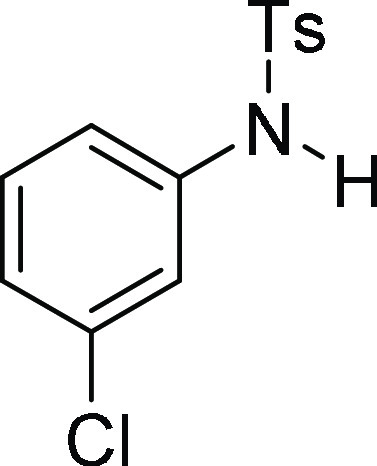
N-(3-Chloro-phenyl)-4-methyl-benzenesulfonamide
(3d):25 Prepared according
to general procedure IV using 4h (100 mg,
0.25 mmol, 1.0 equiv); white solid (67 mg, 95% yield). All data are
similar to the compound obtained from 4d.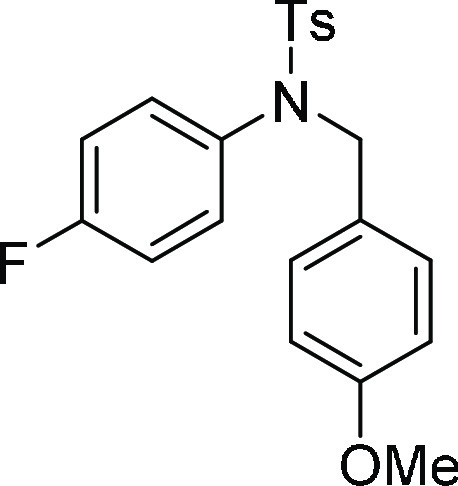
N-(4-Fluoro-phenyl)-N-(4-methoxy-benzyl)-4-methyl-benzenesulfonamide
(4i): Prepared according to general procedure II; 86% yield; white solid; mp = 142–144 °C; 1H NMR (CDCl3, 400 MHz): δ 7.53 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 7.09 (d, J = 8.4 Hz, 2H), 6.88–6.84 (m, 4H), 6.73 (d, J = 8.4 Hz, 2H), 4.61 (s, 2H), 3.71 (s, 3H), 2.43 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 161.7 (d, JC-F = 246.4 Hz), 159.1, 143.7, 135.4,
134.7 (d, JC-F = 3.04 Hz), 130.9
(d, JC-F = 8.7 Hz), 130.0 (2C),
129.7 (2C), 127.7 (3C), 127.6, 115.8 (d, JC-F = 22.5 Hz), 113.8 (3C), 55.2, 54.4, 21.6; HRMS (ESI)+ calcd for C21H20FNNaO3S, 408.1046 m/z (M + Na)+; found, 408.1043 m/z.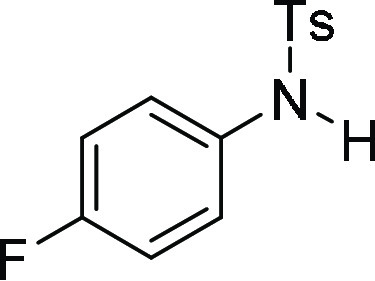
N-(4-Fluoro-phenyl)-4-methyl-benzenesulfonamide
(3f).28 Prepared according
to general procedure IV using 4i (100 mg,
0.26 mmol, 1.0 equiv); white solid (65 mg, 94% yield); Rf = 0.25 (25% EA/PE); mp = 84–86 °C; 1H NMR (CDCl3, 400 MHz): δ 7.64 (d, J = 8.0 Hz, 2H), 7.30 (s, 1H), 7.21 (d, J = 8.0 Hz, 2H), 7.07–7.04 (m, 2H), 6.90 (t, J = 8.8 Hz, 2H), 2.37 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 161.9, 159.6, 144.2, 135.7, 132.5 (d, JC-F = 2.9 Hz), 129.8 (3C), 127.4 (2C),
124.5 (d, JC-F = 8.3 Hz), 116.2
(d, JC-F = 22.6 Hz), 21.7; HRMS
(ESI)+ calcd for C13H13FNO2S, 266.0651 m/z (M + H)+; found, 266.0649 m/z.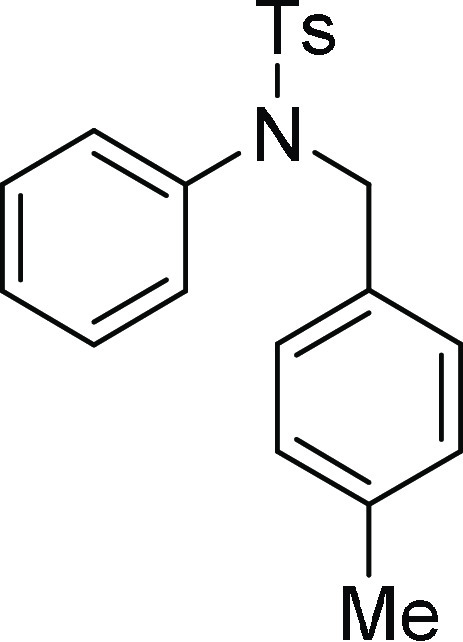
4-Methyl-N-(4-methyl-benzyl)-N-phenyl-benzenesulfonamide
(4j): Prepared according
to general procedure II; 82% yield; white solid; mp =
149–151 °C; 1H NMR (CDCl3, 400 MHz):
δ 7.52 (d, J = 8.0 Hz, 2H), 7.25 (d, J = 8.0 Hz, 2H), 7.18–7.16 (m, 3H), 7.08 (d, J = 8.0 Hz, 2H), 7.00–6.94 (m, 4H), 4.66 (s, 2H),
2.41 (s, 3H), 2.23 (s, 3H); 13C NMR (CDCl3,
100 MHz): δ 143.5, 139.1, 137.3, 135.8, 133.0, 129.6 (2C), 129.1
(2C), 129.0 (2C), 128.9 (2C), 128.6 (2C), 127.8 (3C), 54.5, 21.7,
21.2; HRMS (ESI)+ calcd for C21H21NNaO2S, 374.1191 m/z (M + Na)+; found, 374.1195 m/z.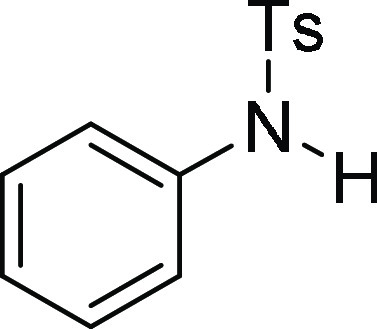
4-Methyl-N-phenyl-benzenesulfonamide
(3a):25 Prepared according
to general procedure IV using 4j (100 mg,
0.28 mmol, 1.0 equiv); white
solid (63 mg, 89% yield). All data are similar to the compound obtained
from 4a.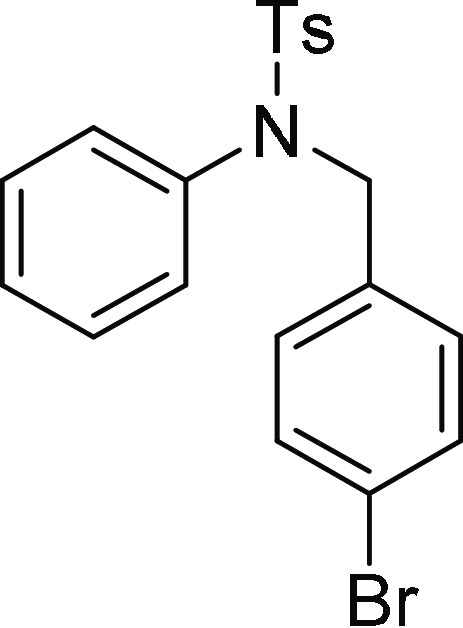
N-(4-Bromo-benzyl)-4-methyl-N-phenyl-benzenesulfonamide (4k): Prepared
according
to general procedure II; 80% yield; off-white solid;
mp = 125–127 °C; 1H NMR (CDCl3,
400 MHz): δ 7.43 (d, J = 8.4 Hz, 2H), 7.25
(d, J = 8.0 Hz, 2H), 7.18 (d, J =
8.4 Hz, 2H), 7.13–7.11 (m, 3H), 7.02 (d, J = 8.0 Hz, 2H), 6.88–6.87 (m, 2H), 4.58 (s, 2H), 2.35 (s,
3H); 13C NMR (CDCl3, 100 MHz): δ 143.8,
138.8, 135.4, 135.2, 131.6 (2C), 130.3 (2C), 129.6 (2C), 129.1 (2C),
128.9 (2C), 128.1, 127.8 (2C), 121.7, 54.2, 21.7; HRMS (ESI)+ calcd for C20H18BrNNaO2S, 440.0119 m/z (M + Na)+; found, 440.0120 m/z.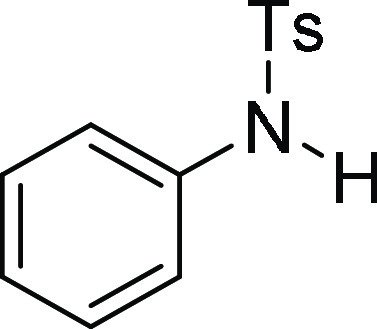
4-Methyl-N-phenyl-benzenesulfonamide (3a): The reaction was performed
according to general procedure IV using 4k (100 mg, 0.24 mmol, 1.0 equiv). No
product was obtained. Yield = 0%.
Naphthalene-1-sulfonic
Acid (3,4-dimethoxy-benzyl)-phenyl-amide
(4l): Prepared according to general procedure II; 80% yield; off-white solid; mp = 160–162 °C; 1H NMR (CDCl3, 400 MHz): δ 8.55 (d, J = 8.4 Hz, 1H), 8.16 (d, J = 7.2 Hz, 1H), 8.05 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H),
7.58–7.45 (m, 3H), 7.14–7.12 (m, 3H), 6.94–6.92
(m, 2H), 6.62–6.60 (m, 2H), 6.56–6.54 (m, 1H), 4.73
(s, 2H), 3.78 (s, 3H), 3.63 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 148.7, 148.5, 138.6, 134.5 134.4, 134.3,
131.1, 129.7 (2C), 128.9 (4C), 128.5, 128.0 (2C), 126.9, 125.6, 124.2,
121.3, 111.7, 110.6, 55.9, 55.7, 55.0; HRMS (ESI)+ calcd
for C25H23NNaO4S, 456.1245 m/z (M + Na)+; found, 456.1243 m/z.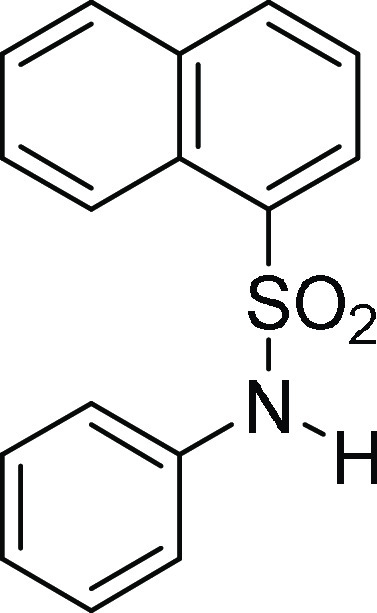
Naphthalene-1-sulfonic
Acid Phenylamide (3g):29 Prepared
according to general procedure IV using 4l (100 mg, 0.23 mmol, 1.0 equiv); white
solid (52 mg, 80% yield); Rf = 0.25 (25%
EA/PE); mp = 150–152 °C; 1H NMR (CDCl3, 400 MHz): δ 8.77 (d, J = 8.0 Hz, 1H), 8.23
(d, J = 8.0 Hz, 1H), 8.01 (d, J =
8.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.65–7.63
(m, 1H), 7.60–7.58 (m, 1H), 7.44 (t, J = 8.0
Hz, 2H), 7.11 (t, J = 8.0 Hz, 2H), 7.02–6.96
(m, 3H); 13C NMR (CDCl3, 100 MHz): δ 136.5,
134.8, 134.3, 134.1, 130.5, 129.4, 129.3 (3C), 128.7, 128.2, 127.0,
125.3, 124.3, 124.2, 121.5; HRMS (ESI)+ calcd for C16H14NO2S, 284.0745 m/z (M + H)+; found, 284.0739 m/z.
N-(3,4-Dimethoxy-benzyl)-N-phenyl-4-trifluoromethyl-benzenesulfonamide
(4m): Prepared according to general procedure II; 88% yield; off-white solid; mp = 140–142 °C; 1H NMR (CDCl3, 400 MHz): δ 7.79–7.37 (m, 4H),
7.24–7.22 (m, 3H), 6.94–6.92 (m, 2H), 6.77 (s, 1H),
6.67–6.64 (m, 2H), 4.68 (s, 2H), 3.80 (s, 3H), 3.78 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 149.6, 148.7,
142.5, 138.4, 129.2 (3C), 129.1 (2C), 128.4, 128.2 (3C), 127.8, 126.2,
126.1, 121.3, 111.7, 110.8, 55.9 (2C), 55.0; HRMS (ESI)+ calcd for C22H20F3NNaO4S, 474.0963 m/z (M + Na)+; found, 474.0961 m/z.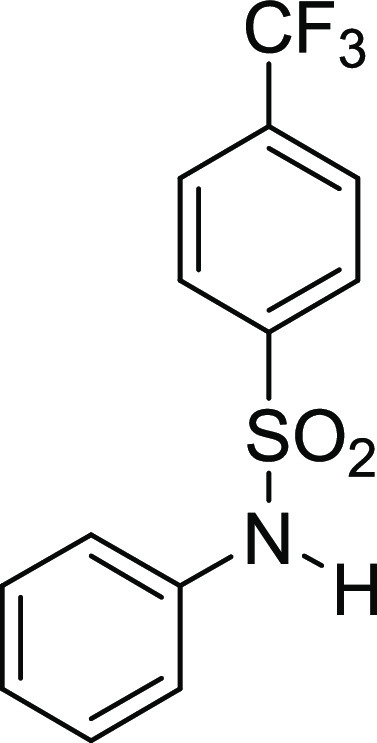
N-Phenyl-4-trifluoromethyl-benzenesulfonamide
(3h):30 Prepared according
to general procedure IV using 4m (100 mg,
0.22 mmol, 1.0 equiv); pale-yellow solid (60 mg, 90% yield), Rf = 0.25 (25% EA/PE); mp = 122–125 °C; 1H NMR (CDCl3, 400 MHz): δ 7.89 (d, J = 8.0 Hz, 2H), 7.67 (d, J = 8.0 Hz, 2H),
7.26–7.22 (m, 3H), 7.15–7.07 (m, 3H); 13C
NMR (CDCl3, 100 MHz): δ 142.5, 135.8, 134.8 (JC-F = 33.0 Hz), 129.7 (2C), 127.9 (2C),
126.4 (JC-F = 7.0, 4.0 Hz, 2C),
126.2, 123.2 (JC-F = 271.0 Hz),
122.1 (2C); HRMS (ESI)+ calcd for C13H11F3NO2S, 302.0463 m/z (M + H)+; found, 302.0462 m/z.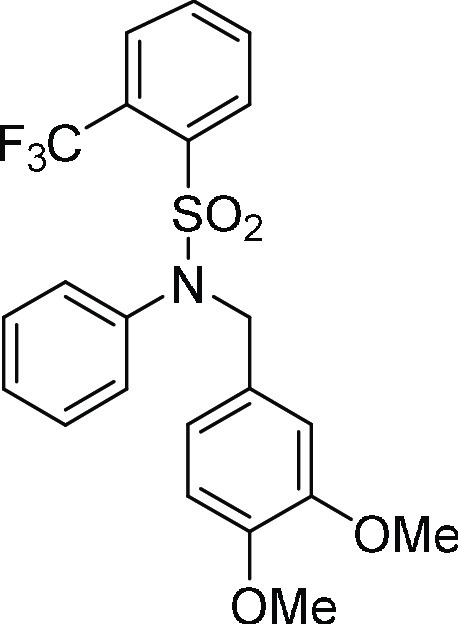
N-(3,4-Dimethoxybenzyl)-N-phenyl-2-trifluoromethyl-benzenesulfonamide
(4n): Prepared according to general procedure II; 88% yield; off-white solid; mp = 141–143 °C; 1H NMR (CDCl3, 400 MHz): δ 7.88 (d, J = 8.0 Hz, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.61 (t, J = 7.6 Hz, 1H), 7.48 (t, J = 8.0 Hz, 1H),
7.16–7.13 (m, 3H), 6.94–6.92 (m, 2H), 6.79 (s, 1H),
6.68–6.62 (m, 2H), 4.82 (s, 2H), 3.80 (s, 3H), 3.78 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 148.7 (d, JC-F = 30.1 Hz), 138.2, 137.9, 133.0 (2C),
132.7, 131.9, 129.9 (3C), 129.1 (3C), 128.6, 128.2 (2C), 121.4 (2C),
111.8, 110.6, 55.8 (2C); HRMS (ESI)+ calcd for C22H20F3NNaO4S, 474.0963 m/z (M + Na)+; found, 474.0958 m/z.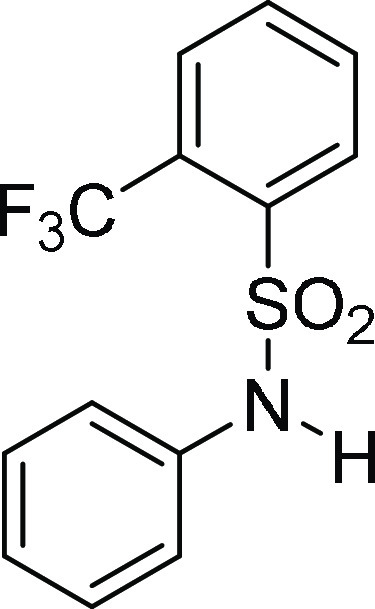
N-Phenyl-2-trifluoromethyl-benzenesulfonamide
(3i):31 Prepared according
to general procedure IV using 4n (100 mg,
0.22 mmol, 1.0 equiv); pale-yellow solid (60 mg, 90% yield); Rf = 0.25 (25% EA/PE); mp = 96–98 °C; 1H NMR (CDCl3, 400 MHz): δ 8.0 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H),
7.64 (t, J = 8.0 Hz, 1H), 7.56 (t, J = 8.0 Hz, 1H), 7.24–7.20 (m, 2H), 7.13–7.06 (m, 3H); 13C NMR (CDCl3, 100 MHz): δ 135.7, 133.2,
132.6, 132.3, 129.5 (4C), 128.6 (JC-F = 7.0 Hz), 126.1, 122.3 (3C); HRMS (ESI)+ calcd for C13H11F3NO2S, 302.0463 m/z (M + H)+; found, 302.0460 m/z.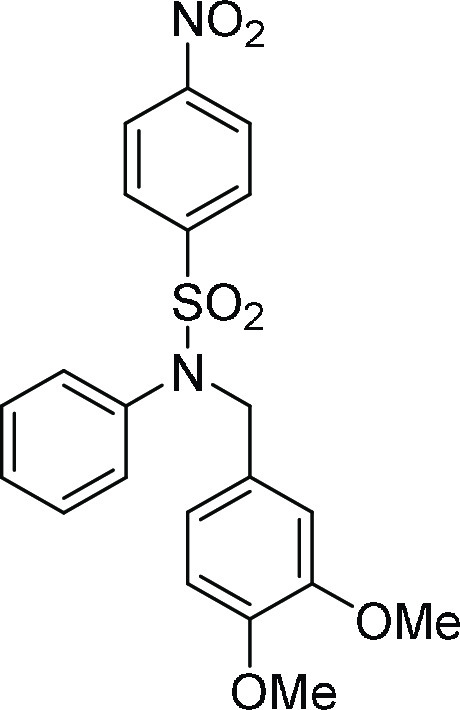
N-(3,4-Dimethoxy-benzyl)-4-nitro-N-phenyl-benzenesulfonamide (4o): Prepared
according
to general procedure II; 88% yield; pale-yellow solid;
mp = 193–195 °C; 1H NMR (CDCl3,
400 MHz): δ 8.33 (d, J = 7.6 Hz, 2H), 7.83
(d, J = 8.8 Hz, 2H), 7.26–7.23 (m, 3H), 6.93–6.91
(m, 2H), 6.77 (s, 1H), 6.69–6.63 (m, 2H), 4.70 (s, 2H), 3.81
(s, 3H), 3.80 (s, 3H); 13C NMR (CDCl3, 100 MHz):
δ 150.1, 149.0, 148.8, 144.6, 138.0, 129.4 (2C), 129.1 (2C),
128.9 (2C), 128.6, 127.6, 124.3 (2C), 121.3, 111.6, 110.7, 55.9 (2C),
55.3; HRMS (ESI)+ calcd for C21H20N2NaO6S, 451.0940 m/z (M + Na)+; found, 451.0940 m/z.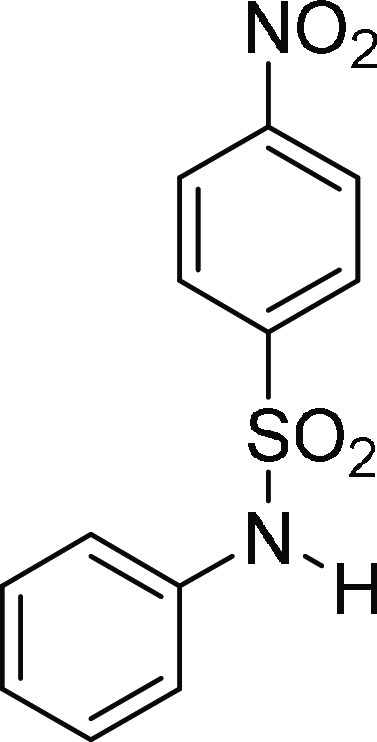
4-Nitro-N-phenyl-benzenesulfonamide
(3j):29 Prepared according
to general procedure IV using 4o (100 mg,
0.23 mmol, 1.0 equiv); pale-yellow
solid (58 mg, 90% yield); Rf = 0.25 (30%
EA/PE); mp = 168–170 °C; 1H NMR (CDCl3, 400 MHz): δ 8.28 (d, J = 8.0 Hz, 2H), 7.92
(d, J = 8.0 Hz, 2H), 7.30–7.26 (m, 2H), 7.20
(d, J = 7.2 Hz, 1H), 7.08 (d, J =
7.2 Hz, 2H), 6.79 (s, 1H); 13C NMR (CDCl3, 100
MHz): δ 150.4, 144.7, 135.4, 129.8 (2C), 128.6 (2C), 126.5,
124.4 (2C), 122.6 (2C); HRMS (ESI)+ calcd for C12H11N2O4S, 279.0440 m/z (M + H)+; found, 279.0437 m/z.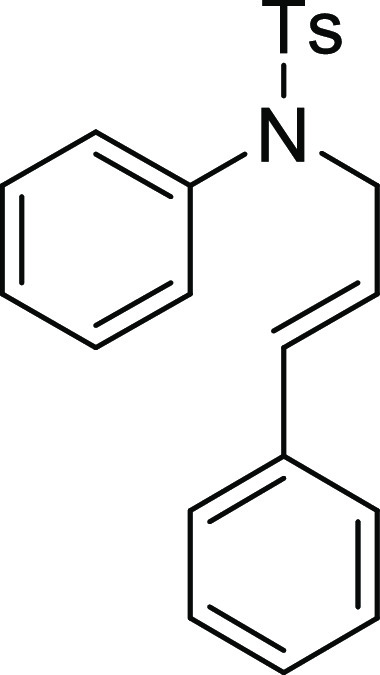
4-Methyl-N-phenyl-N-(3-phenyl-allyl)-benzenesulfonamide
(5a):32 Prepared according
to general procedure II; 80% yield; white solid; mp =
143–145 °C; 1H NMR (CDCl3, 400 MHz):
δ 7.51 (d, J = 8.4 Hz, 2H), 7.28–7.18
(m, 10H), 7.08–7.05 (m, 2H), 6.35 (d, J =
16.0 Hz, 1H), 6.12–6.04 (dt, J = 15.8, 6.5
Hz, 1H), 4.32 (dd, J = 6.8, 1.2 Hz, 2H), 2.41 (s,
3H); 13C NMR (CDCl3, 100 MHz): δ 143.6,
139.4, 136.5, 135.8, 133.8, 129.6 (2C), 129.1 (2C), 129.0 (2C), 128.6
(2C), 128.0, 127.9, 127.8 (2C), 126.6 (2C), 124.2, 53.4, 21.7; HRMS
(ESI)+ calcd for C22H21NNaO2S, 386.1191 m/z (M + Na)+; found, 386.1185 m/z.
4-Methyl-N-phenyl-benzenesulfonamide (3a):25 Prepared according to general procedure IV using 5a (100 mg, 0.28 mmol, 1.0 equiv); white
solid (55 mg, 81% yield). All data are similar to the compound obtained
from 4a.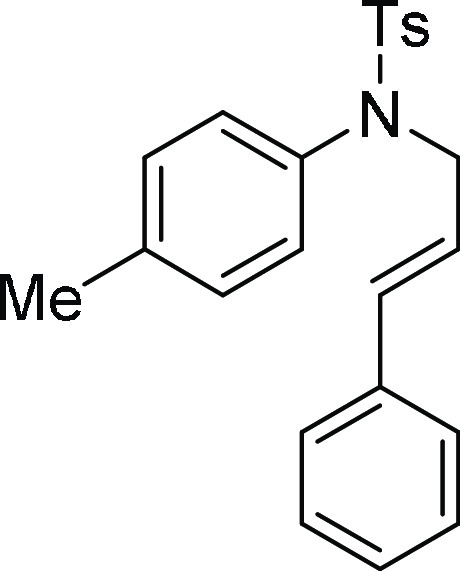
4-Methyl-N-(3-phenyl-allyl)-N-p-tolyl-benzenesulfonamide (5b):32 Prepared according to general procedure II; 80% yield; 1H NMR (CDCl3, 400 MHz):
δ 7.52 (d, J = 8.0 Hz, 2H), 7.26–7.20
(m, 7H), 7.06 (d, J = 8.0 Hz, 2H), 6.94 (d, J = 8.0 Hz, 2H), 6.36 (d, J = 15.8 Hz,
1H), 6.09 (dt, J = 15.8, 6.1 Hz, 1H), 4.30 (dd, J = 6.1 Hz, 0.8 Hz, 2H), 2.42 (s, 3H), 2.29 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 143.5, 137.9, 136.6, 136.5,
135.9, 133.7, 129.7, 129.5, 128.8, 128.6, 127.9, 126.5, 124.4, 53.5,
21.7, 21.2; HRMS (ESI)+ calcd for C23H23NNaO2S, 400.1347 m/z (M + Na)+; found, 400.1349 m/z.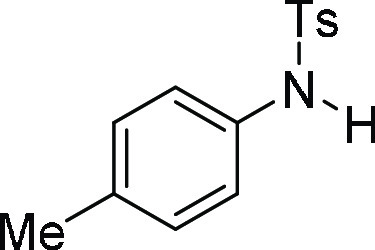
4-Methyl-N-p-tolyl-benzenesulfonamide
(3b):25 Prepared according
to general procedure IV using 5b (100 mg,
0.26 mmol, 1.0 equiv); white solid (53 mg, 78% yield). All data are
similar to the compound obtained from 4b.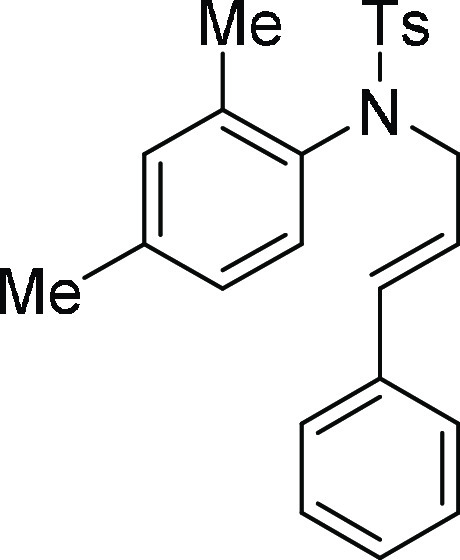
N-(2,4-Dimethyl-phenyl)-4-methyl-N-(3-phenyl-allyl)-benzenesulfonamide (5c): Prepared
according to general procedure II; 82% yield; off-white
solid; mp = 105–107 °C; 1H NMR (CDCl3, 400 MHz): δ 7.62 (d, J = 8.4 Hz, 2H), 7.28–7.20
(m, 7H), 7.04 (s, 1H), 6.84–6.82 (m, 1H), 6.52 (d, J = 7.2 Hz, 1H), 6.28 (d, J = 15.6 Hz,
1H), 6.13–6.05 (m, 1H), 4.44 (dd, J = 16.0
Hz, 5.6 Hz, 1H), 4.03 (dd, J = 14 Hz, 7.6 Hz, 1H),
2.43 (s, 3H), 2.32 (s, 3H), 2.26 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 143.4, 139.6, 138.3, 136.6, 136.5, 135.5,
134.0, 132.1, 129.5 (2C), 128.6 (2C), 128.3, 128.0 (2C), 127.9, 127.0,
126.5 (2C), 123.9, 54.4, 21.6, 21.1, 18.6; HRMS (ESI)+ calcd
for C24H25NNaO2S, 414.1504 m/z (M + Na)+; found, 414.1503 m/z.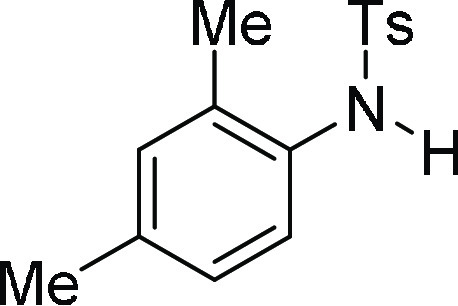
N-(2,4-Dimethyl-phenyl)-4-methyl-benzenesulfonamide
(3c):26 Prepared according
to general procedure IV using 5c (100 mg,
0.26 mmol, 1.0 equiv); white solid (53 mg, 76% yield). All data are
similar to the compound obtained from 4c.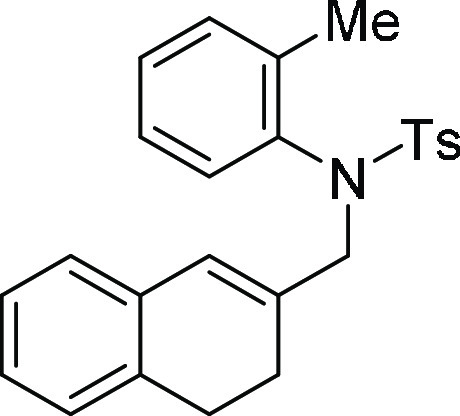
N-(3,4-Dihydro-naphthalen-2-ylmethyl)-4-methyl-N-o-tolyl-benzenesulfonamide (5d): Prepared according to general procedure II; 89% yield;
off-white solid; mp = 128–130 °C; 1H NMR (CDCl3, 400 MHz): δ 7.59 (d, J = 8.4 Hz,
2H), 7.31 (d, J = 8.0 Hz, 3H), 7.28–7.17 (m,
2H), 7.09–7.07 (m, 3H), 6.86–6.85 (m, 1H), 6.64–6.63
(m, 1H), 6.03 (s, 1H), 4.47 (d, J = 13.0 Hz, 1H),
3.93 (d, J = 13.0 Hz, 1H), 2.78–2.72 (m, 2H),
2.70 (s, 3H), 2.47 (s, 3H), 2.40–2.38 (m, 2H); 13C NMR (CDCl3, 100 MHz): δ 143.5, 139.8, 138.0, 135.0,
134.0, 132.5, 131.4, 129.4 (3C), 128.0 (3C), 127.4, 127.3, 127.1,
126.3, 126.0 (2C), 125.9, 57.3, 27.8, 25.2, 21.6, 18.6; HRMS (ESI)+ calcd for C25H26NO2S, 404.1684 m/z (M + H)+; found, 404.1678 m/z.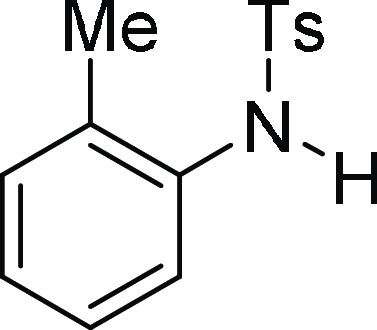
4-Methyl-N-o-tolyl-benzenesulfonamide
(3k):33 Prepared according
to general procedure IV using 5d (100 mg,
0.25 mmol, 1.0 equiv); white solid (55 mg, 84% yield); Rf = 0.25 (25% EA/PE); mp = 95–97 °C; 1H NMR (CDCl3, 400 MHz): δ 7.64 (d, J = 7.2 Hz, 2H), 7.33 (d, J = 7.6 Hz, 1H),
7.23 (d, J = 7.6 Hz, 2H), 7.15–7.09 (m, 3H),
6.57 (s, 1H), 2.40 (s, 3H), 2.03 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 143.7, 136.7, 134.5, 131.4, 130.7, 129.5
(2C), 127.1 (2C), 126.8, 126.1, 124.3, 21.5, 17.5; HRMS (ESI)+ calcd for C14H16NO2S, 262.0902 m/z (M + H)+; found, 262.0899 m/z.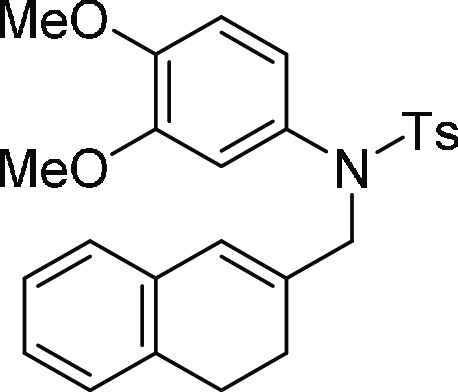
N-(3,4-Dihydro-naphthalen-2-ylmethyl-N-(3,4-dimethoxy-phenyl)-4-methyl-benzenesulfonamide (5e). Prepared according to general procedure II; 89% yield;
off-white solid; mp = 130–132 °C; 1H NMR (CDCl3, 400 MHz): δ 7.48 (d, J = 8.4 Hz,
2H), 7.24 (d, J = 8.0 Hz, 2H), 7.05–7.01 (m,
3H), 6.83–6.82 (m, 1H), 6.66 (d, J = 8.4 Hz,
1H), 6.53–6.50 (m, 2H), 6.08 (s, 1H), 4.21 (s, 2H), 3.79 (s,
3H), 3.68 (s, 3H), 2.69 (t, J = 8.0 Hz, 2H), 2.41
(s, 3H), 2.36 (t, J = 8.0 Hz, 2H); 13C
NMR (CDCl3, 100 MHz): δ 148.8, 148.6, 143.5, 135.4
(2C), 131.6, 129.5 (3C), 128.0 (3C), 127.4, 127.2, 127.0, 126.4, 126.0,
121.2, 112.6, 110.7, 56.8, 56.0, 55.9, 28.0, 24.8, 21.6; HRMS (ESI)+ calcd for C26H28NO4S, 450.1739 m/z (M + H)+; found, 450.1742 m/z.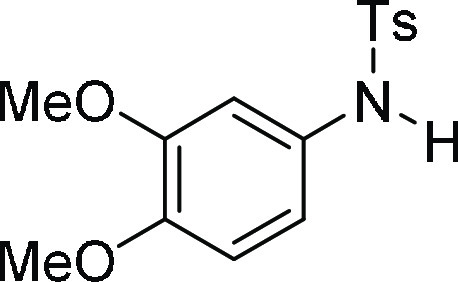
N-(3,4-Dimethoxy-phenyl)-4-methyl-benzenesulfonamide
(3e):27 Prepared according
to general procedure IV using 5e (100 mg,
0.22 mmol, 1.0 equiv); white solid (37 mg, 55% yield). All data are
similar to the compound obtained from 4e.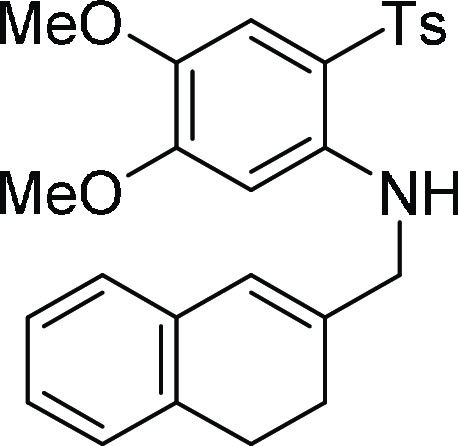
3,4-Dihydro-naphthalen-2-ylmethyl)-[4,5-dimethoxy-2-(toluene-4-sulfonyl)-phenyl
amine (Detosylated product) (5e’): Obtained during
the course of the C–N bond cleavage of 5e according
to general procedure IV; white solid (13 mg, 12% yield); Rf = 0.22 (35% EA/PE); mp = 123–125 °C; 1H NMR (CDCl3, 400 MHz): δ 7.47 (d, J = 8.4 Hz, 2H), 7.08–6.98 (m, 6H), 6.85 (s, 1H),
6.50 (s, 1H), 6.35 (s, 1H), 5.89 (s, 1H), 3.72 (s, 6H), 2.95 (s, 2H),
2.62 (t, J = 8.0 Hz, 2H), 2.26 (s, 3H), 1.89 (t, J = 8.0 Hz, 2H); 13C NMR (CDCl3, 100
MHz): δ 148.0, 147.5, 143.9, 139.3, 136.7, 134.3, 134.1, 129.7
(3C), 127.6, 127.3 (3C), 126.9, 126.7, 126.0, 124.2, 113.2, 109.8,
56.1, 56.0, 39.2, 28.0, 27.1, 21.6; HRMS (ESI)+ calcd for
C26H28NO4S, 450.1739 m/z (M + H)+; found, 450.1737 m/z.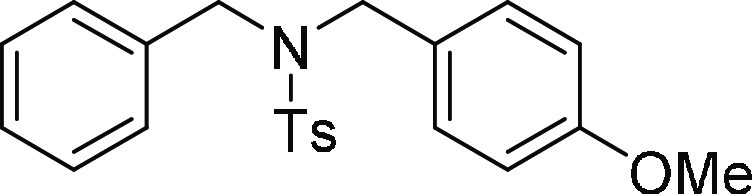
N-Benzyl-N-(4-methoxy-benzyl)-4-methyl-benzenesulfonamide
(6a): Prepared according to general procedure II; 89% yield; off-white solid; mp = 110–112 °C; 1H NMR (CDCl3, 400 MHz): δ 7.74 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 8.0 Hz, 2H), 7.23–7.22
(m, 3H), 7.08–7.07 (m, 2H), 6.98–6.95 (m, 2H), 6.75
(d, J = 8.0 Hz, 2H), 4.29 (s, 2H), 4.26 (s, 2H),
3.76 (s, 3H), 2.44 (s, 3H); 13C NMR (CDCl3,
100 MHz): δ 159.2, 143.3, 137.8, 135.9, 130.1 (3C), 129.8, 128.6
(3C), 128.6 (2C), 127.6, 127.3 (3C), 113.8, 55.3, 50.3, 49.9, 21.6;
HRMS (ESI)+ calcd for C22H24NO3S, 382.1477 m/z (M + H)+; found, 382.1473 m/z.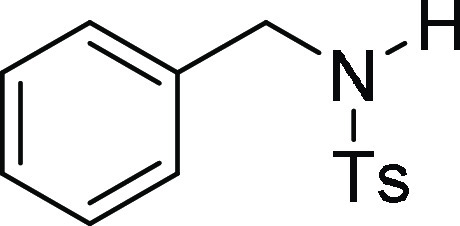
N-Benzyl-4-methyl-benzenesulfonamide (3l):34 Prepared according to general procedure IV using 6a (100 mg, 0.26 mmol, 1.0 equiv); white
solid (53 mg, 78% yield); Rf = 0.25 (25%
EA/PE); mp = 113–115 °C; 1H NMR (CDCl3, 400 MHz): δ 7.74 (d, J = 8.4 Hz, 2H), 7.29–7.23
(m, 5H), 7.19–7.17 (m, 2H), 4.97–4.95 (m, 1H), 4.09
(d, J = 6.00 Hz, 2H), 2.42 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 143.6, 136.9, 136.4, 129.8
(2C), 128.7 (2C), 128.0 (3C), 127.3 (2C), 47.3, 21.6; HRMS (ESI)+ calcd for C14H16NO2S, 262.0902 m/z (M + H)+; found, 262.0898 m/z.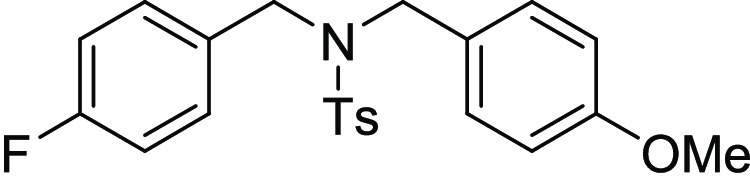
N-(4-Fluoro-benzyl)-N-(4-methoxy-benzyl)-4-methyl-benzenesulfonamide
(6b): Prepared according to general procedure II; 87% yield; off-white solid; mp = 105–107 °C; 1H NMR (CDCl3, 400 MHz): δ 7.73 (d, J = 8.4 Hz, 2H), 7.31 (d, J = 8.0 Hz, 2H), 7.04–7.01
(m, 2H), 6.94–6.86 (m, 4H), 6.73 (d, J = 8.8
Hz, 2H), 4.23 (s, 2H), 4.22 (s, 2H), 3.75 (s, 3H), 2.44 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 162.2 (d, JC-F = 244.5 Hz), 159.2, 143.4, 137.5,
131.8 (d, JC-F = 3.1 Hz), 130.2
(d, JC-F = 8.1, 2C), 130.0 (2C),
129.8 (2C), 127.4, 127.2 (2C), 115.2 (d, JC-F = 21.3, 2C), 113.8 (2C), 55.3, 50.3, 49.7, 21.6; HRMS (ESI)+ calcd for C22H23FNO3S, 400.1383 m/z (M + H)+; found, 400.1379 m/z.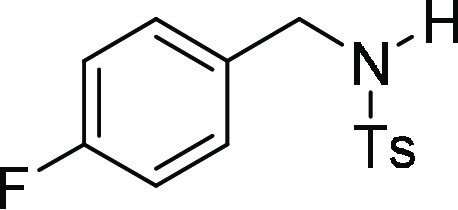
N-(4-Fluoro-benzyl)-4-methyl-benzenesulfonamide
(3m):34 Prepared according
to general procedure IV using 6b (100 mg,
0.25 mmol, 1.0 equiv); white solid (57 mg, 81% yield); Rf = 0.25 (35% EA/PE); mp = 92–94 °C; 1H NMR (CDCl3, 400 MHz): δ 7.72 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H),
7.17–7.15 (m, 2H), 6.95–6.90 (m, 2H), 5.05–5.02
(m, 1H), 4.07 (d, J = 6.4 Hz, 2H), 2.42 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 162.4 (d, JC-F = 244.8 Hz), 143.8, 136.9, 132.2,
129.9 (2C), 129.7 (d, JC-F = 8.2
Hz, 2C), 127.2 (2C), 115.6 (d, JC-F = 21.4, 2C), 46.6, 21.6; HRMS (ESI)+ calcd for C14H15FNO2S, 280.0808 m/z (M + H)+; found, 280.0809 m/z.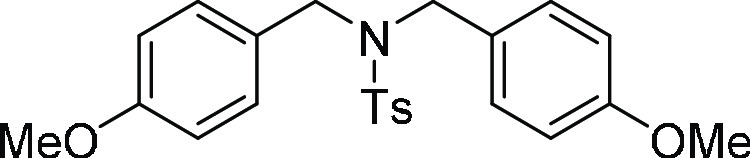
N,N-Bis-(4-methoxy-benzyl)-4-methyl-benzenesulfonamide
(6c): Prepared according to general procedure II; 89% yield; off-white solid; mp = 105–107 °C; 1H NMR (CDCl3, 400 MHz): δ 7.72 (d, J = 8.0 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H), 6.96 (d, J = 8.4 Hz, 4H), 6.75 (d, J = 8.8 Hz, 4H),
4.22 (s, 4H), 3.78 (s, 6H), 2.44 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 159.2, 143.3, 138.0, 130.0 (5C), 129.8
(2C), 127.8, 127.3 (2C), 113.8 (5C), 55.4 (2C), 49.6 (2C), 21.7; HRMS
(ESI)+ calcd for C23H26NO4S, 412.1583 m/z (M + H)+; found, 412.1583 m/z.
4-Methyl-benzenesulfonamide (3n): Prepared according
to general procedure IV using 6c (100 mg,
0.24 mmol, 1.0 equiv); white solid (33 mg, 80% yield); Rf = 0.25 (35% EA/PE); mp = 132–134 °C; 1H NMR (CDCl3, 400 MHz): δ 7.81 (d, J = 7.6 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H),
4.95 (s, 2H), 2.42 (s, 3H); 13C NMR (CDCl3,
100 MHz): δ 143.8, 139.2, 129.9 (2C), 126.6 (2C), 21.7; HRMS
(ESI)+ calcd for C7H10NO2S, 172.0432 m/z (M + H)+; found, 172.0426 m/z.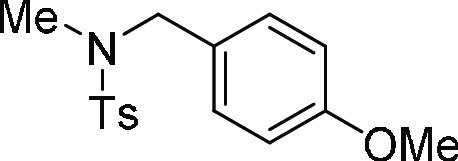
N-(4-Methoxy-benzyl)-4,N-dimethyl-benzenesulfonamide
(7a): Prepared according to general procedure II; 87% yield; white solid; mp = 69–71 °C; 1H NMR (CDCl3, 400 MHz): δ 7.71 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H),
4.04 (s, 2H), 3.79 (s, 3H), 2.54 (s, 3H), 2.45 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 159.4, 143.5, 134.3, 129.8
(3C), 127.6 (3C), 114.1 (3C), 55.4, 53.7, 34.2, 21.7; HRMS (ESI)+ calcd for C16H20NO3S, 306.1164 m/z (M + H)+; found, 306.1167 m/z.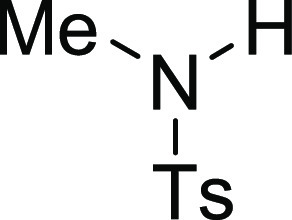
4,N-Dimethyl-benzenesulfonamide
(3o):35 Prepared according
to general procedure IV using 7a (100 mg,
0.3 mmol, 1.0 equiv); white
solid (49 mg, 76% yield); Rf = 0.2 (20%
EA/PE); mp = 70–72 °C; 1H NMR (CDCl3, 400 MHz): δ 7.74 (d, J = 8.4 Hz, 2H), 7.31
(d, J = 8.0 Hz, 2H), 4.75–4.70 (m, 1H), 2.62
(d, J = 5.6 Hz, 3H), 2.42 (s, 3H); 13C
NMR (CDCl3, 100 MHz): δ 143.6, 135.8, 129.8 (2C),
127.4 (2C), 29.4, 21.6; HRMS (ESI)+ calcd for C8H12NO2S, 186.0589 m/z (M + H)+; found, 186.0586 m/z.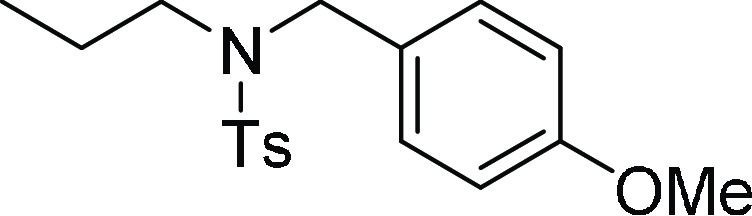
N-(4-Methoxy-benzyl)-4-methyl-N-propyl-benzenesulfonamide (7b): Prepared
according
to general procedure II; 87% yield; off-white solid;
mp = 79–81 °C; 1H NMR (CDCl3, 400
MHz): δ 7.71 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H),
6.81 (d, J = 8.4 Hz, 2H), 4.23 (s, 2H), 3.76 (s,
3H), 3.02–2.99 (m, 2H), 2.41 (s, 3H), 1.34–1.28 (m,
2H), 0.67 (t, J = 7.6 Hz, 3H); 13C NMR
(CDCl3, 100 MHz): δ 159.1, 143.1, 137.1, 129.7 (2C),
129.6 (2C), 128.5, 127.1 (2C), 113.8 (2C), 55.2, 51.3, 49.6, 21.5,
21.4, 11.2; HRMS (ESI)+ calcd for C18H24NO3S, 334.1477 m/z (M
+ H)+; found, 334.1475 m/z.
4-Methyl-N-propyl-benzenesulfonamide (3p):35 Prepared according to general procedure IV using 7b (100 mg, 0.3 mmol, 1.0 equiv); white solid (49 mg, 76% yield); Rf = 0.2 (20% EA/PE); mp = 90–92 °C; 1H NMR (CDCl3, 400 MHz): δ 7.75 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H), 4.86 (s, 1H), 2.90–2.84 (m, 2H), 2.41 (s, 3H), 1.49–1.43 (m, 2H), 0.84 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 143.4, 137.0, 129.8 (2C), 127.2 (2C), 45.0, 23.0, 21.6, 11.2; HRMS (ESI)+ calcd for C10H16NO2S, 214.0902 m/z (M + H)+; found, 214.0898 m/z.
Procedure for Control Experiments (Scheme 7)
Scheme 7i. To a stirred solution of N-(3,4-dimethoxy-benzyl)-4-methyl-N-phenyl-benzenesulfonamide 4a (0.25 mmol, 1 equiv) in superdry dichloroethane (2 mL), Bi(OTf)3 (8 mg, 0.0125 mmol, 0.05 equiv) was added along with 4Å molecular sieves, and the resultant mixture was refluxed at 85 °C for 2 h. A trace amount of the cleavage product was formed.
Scheme 7ii. To a stirred solution of N-(3,4-dimethoxy-benzyl)-4-methyl-N-phenyl-benzenesulfonamide 4a (0.25 mmol, 1 equiv) in dry dichloroethane (2 mL), water (0.2 mL) and Bi(OTf)3 (8 mg, 0.0125 mmol, 0.05 equiv) were added, and the resultant mixture was refluxed at 85 °C for 2 h. The reaction was monitored by TLC to ensure completion. On completion, DCE was removed under reduced pressure and the crude mixture was directly subjected to purification by flash column chromatography using an EtOAc and petroleum ether mixture as the eluent, affording the desired cleavage product 3a in 93% yield.
Scheme 7iii. To a stirred solution of N-(3,4-dimethoxy-benzyl)-4-methyl-N-phenyl-benzenesulfonamide 4a (0.25 mmol, 1 equiv) in dichloroethane (2 mL), TfOH (2.2 μL, 0.025 mmol, 0.1 equiv) was added, and the resultant mixture was refluxed at 85 °C for 2 h. The reaction was monitored by TLC to ensure completion. On completion, DCE was removed under reduced pressure and the crude mixture was directly subjected to purification by flash column chromatography using an EtOAc and petroleum ether mixture as the eluent, affording the desired cleavage product 3a in 89% yield.
Scheme 7iv. To a stirred solution of N-(3,4-dimethoxy-benzyl)-4-methyl-N-phenyl-benzenesulfonamide 4a (0.25 mmol, 1 equiv) in dichloroethane (2 mL), TfOH (2.2 μL, 0.025 mmol, 0.1 equiv) and the acid scavenger DTBMP (77 mg, 0.375 mmol, 1.5 equiv) were added, and the resultant mixture was refluxed at 85 °C for 2 h. A trace amount of the cleavage product was formed.
Scheme 7v. To a stirred solution of N-(3,4-dimethoxy-benzyl)-4-methyl-N-phenyl-benzenesulfonamide 4a (0.25 mmol, 1 equiv) in degassed dichloroethane (2 mL), water (0.2 mL) and Bi(OTf)3 (0.05 equiv) were added, and the resultant mixture was refluxed at 85 °C for 2 h under a nitrogen atmosphere. The reaction was monitored by TLC to ensure completion. On completion, DCE was removed under reduced pressure and the crude mixture was directly subjected to purification by flash column chromatography using an EtOAc and petroleum ether mixture as the eluent, affording the desired cleavage product 3a in 91% yield.
Scheme 7vi. To a stirred solution of N-(3,4-dimethoxy-benzyl)-4-methyl-N-phenyl-benzenesulfonamide 4a (0.25 mmol, 1 equiv) in dry dichloroethane (2 mL), water (0.2 mL) and Bi(OTf)3 (8 mg, 0.0125 mmol, 0.05 equiv) were added, and the resultant mixture was refluxed at 85 °C for 2 h under a dark condition. The reaction was monitored by TLC to ensure completion. On completion, DCE was removed under reduced pressure and the crude mixture was directly subjected to purification by flash column chromatography using an EtOAc and petroleum ether mixture as the eluent, affording the desired cleavage product 3a in 90% yield.
Scheme 7vii. To a stirred solution of N-(3,4-dimethoxy-benzyl)-4-methyl-N-phenyl-benzenesulfonamide 4a (0.25 mmol, 1 equiv) in dry dichloroethane (2 mL), water (0.2 mL), Bi(OTf)3 (8 mg, 0.0125 mmol, 0.05 equiv), and the radical scavenger BHT (66 mg, 0.3 mmol, 1.2 equiv) were added, and the resultant mixture was refluxed at 85 °C for 2 h. The reaction was monitored by TLC to ensure completion. On completion, DCE was removed under reduced pressure and the crude mixture was directly subjected to purification by flash column chromatography using an EtOAc and petroleum ether mixture as the eluent, affording the desired cleavage product 3a in 89% yield.
Preparation of Inactive Substrates 4p–4s
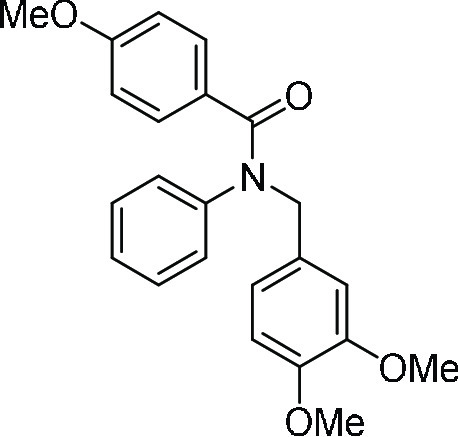
N-(3,4-Dimethoxy-benzyl)-4-methoxy-N-phenyl-benzamide (4p). To a solution of N-(3,4-dimethoxybenzyl)aniline (200 mg, 0.82 mmol) in DMF, NaH (60%
suspension, 30 mg, 1.23 mmol, 1.5 equiv) was added and stirred at
room temperature for 1 h, followed by the addition of 4-methoxybenzoyl
chloride (0.11 mL, 0.84 mmol, 1.02 equiv). The resultant mixture was
stirred at 80 °C, and after the complete consumption of reactants
(as monitored by TLC), the reaction was quenched with 1.0 M HCl aqueous
solution, extracted with EtOAc, washed with brine, dried with Na2SO4, and evaporated in vaccuo.
Purification of the crude product by flash column chromatography using
an EtOAc and petroleum ether mixture (35% EA/PE) as the eluent afforded
the desired titled compound 4p (216 mg, 70%) as a colorless
oil; 1H NMR (CDCl3, 400 MHz): δ 7.28 (d, J = 8.8 Hz, 2H), 7.17–7.08 (m, 3H), 6.89 (d, J = 7.2 Hz, 2H), 6.84 (s, 1H), 6.81–6.73 (m, 2H),
6.64 (d, J = 8.8 Hz, 2H), 5.04 (s, 2H), 3.84 (s,
3H), 3.78 (s, 3H), 3.71 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 170.1, 160.7, 148.8, 148.3, 144.0, 131.0 (2C),
130.4, 129.1 (2C), 128.1, 127.8 (2C), 126.6, 121.0, 113.0 (2C), 111.8,
110.8, 55.9 (2C), 55.3, 53.8; HRMS (ESI)+ calcd for C23H23NNaO4, 400.1525 m/z (M + Na)+; found, 400.1523 m/z.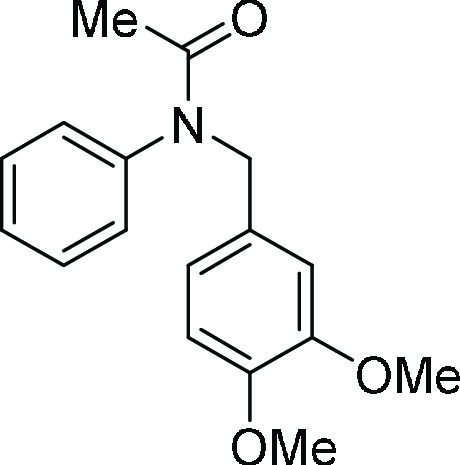
N-(3,4-Dimethoxy-benzyl)-N-phenyl-acetamide (4q): To a solution of N-(3,4-dimethoxybenzyl)aniline (200 mg, 0.82 mmol) in acetic
acid, Ac2O (0.15 mL, 1.64 mmol, 2.0 equiv) was added, and
the resultant mixture was heated to 100 °C, and after the complete
consumption of reactants (as monitored by TLC), the reaction was quenched
with saturated NaHCO3 solution, extracted with EtOAc, washed
with brine, dried with Na2SO4, and evaporated in vaccuo. Purification of the crude product by flash column
chromatography using an EtOAc and petroleum ether mixture (35% EA/PE)
as the eluent afforded the desired titled compound 4q (198 mg, 85%) as a colorless oil; 1H NMR (CDCl3, 400 MHz): δ 7.33–7.29 (m, 3H), 6.97 (d, J = 7.2 Hz, 2H), 7.64 (s, 1H), 6.72 (d, J = 8.4 Hz,
1H), 6.66 (d, J = 8.0 Hz, 1H), 4.81 (s, 2H), 3.84
(s, 3H), 3.78 (s, 3H), 1.87 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 170.4, 148.8, 148.4, 142.7, 130.1, 129.5,
128.9, 128.4, 128.0, 121.4, 119.9, 112.2, 110.8, 55.8 (2C), 52.5,
22.8; HRMS (ESI)+ calcd for C17H19NNaO3, 308.1263 m/z (M
+ Na)+; found, 308.1263 m/z.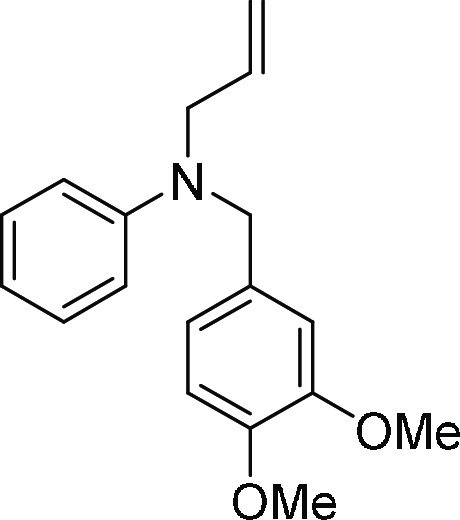
Allyl-(3,4-dimethoxy-benzyl)-phenyl-amine (4r): To
a solution of N-(3,4-dimethoxybenzyl)aniline (200
mg, 0.82 mmol) in DMF, NaH (60% suspension, 30 mg, 1.23 mmol, 1.5
equiv) was added and stirred at room temperature for 1 h, followed
by the addition of allyl bromide (0.072 mL, 0.84 mmol, 1.02 equiv).
The resultant mixture was stirred at 80 °C, and after the complete
consumption of reactants (as monitored by TLC), the reaction was quenched
with 1.0 M HCl aqueous solution, extracted with EtOAc, washed with
brine, dried with Na2SO4, and evaporated in vaccuo. Purification of the crude product by flash column
chromatography using an EtOAc and petroleum ether mixture (20% EA/PE)
as the eluent afforded the desired titled compound 4r (192 mg, 83%) as a colorless thick oil; 1H NMR (CDCl3, 400 MHz): δ 7.20 (t, J = 8.0 Hz,
2H), 6.81–6.70 (m, 6H), 5.94–5.84 (m, 1H), 5.22–5.20
(m, 1H), 5.18 (s, 1H), 4.49 (s, 2H), 4.00 (d, J =
4.8 Hz, 2H), 3.87 (s, 3H), 3.83 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 149.2 (2C), 148.0, 133.8, 131.5, 129.2
(2C), 118.7, 116.7, 116.5, 112.7 (2C), 111.2, 109.8, 56.1, 55.9, 53.9,
52.8; HRMS (ESI)+ calcd for C18H22NO2, 284.1651 m/z (M
+ H)+; found, 284.1648 m/z.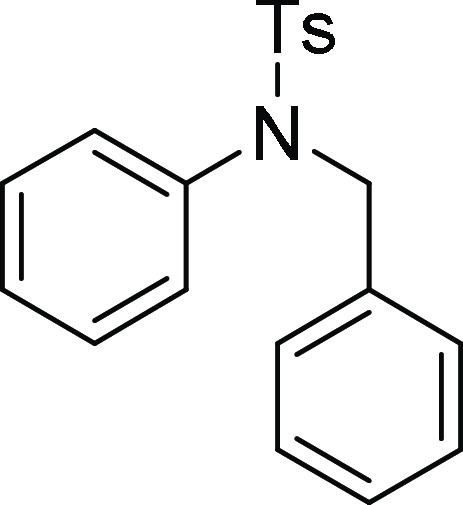
N-Benzyl-4-methyl-N-phenyl-benzenesulfonamide (4s):15 Prepared according to general procedure II; 89% yield; white solid; mp = 118–120 °C; 1H NMR (CDCl3, 400 MHz): δ 7.45 (d, J = 8.0 Hz, 2H), 7.19 (d, J = 8.4 Hz, 2H), 7.16–7.08 (m, 8H), 6.90–5.88 (m, 2H), 4.63 (s, 2H), 2.35 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 143.6, 140.0, 136.0, 135.5, 129.6 (2C), 129.0 (2C), 128.9 (2C), 128.6 (2C), 128.4 (2C), 127.9, 127.8 (2C), 127.6, 54.7, 21.7; HRMS (ESI)+ calcd for C20H19NNaO2S, 360.1034 m/z (M + Na)+; found, 360.1031 m/z.
Experimental Procedure for the Preparation of Compounds 8a and 8b
To a stirred
solution of N-(3,4-dimethoxy-benzyl)-4-methyl-N-phenyl-benzenesulfonamide 4a (0.25 mmol,
1 equiv) in dichloroethane (2 mL), Bi(OTf)3 (8 mg, 0.0125
mmol, 0.05 equiv) and 1,2-dimethoxybenzene
(0.064 mL, 0.5 mmol, 2.0 equiv) were added, and the resultant mixture
was refluxed at 85 °C for 2 h. The reaction was monitored by
TLC to ensure completion. On completion, DCE was removed under reduced
pressure and the crude mixture was directly subjected to purification
by flash column chromatography using an EtOAc and petroleum ether
mixture as the eluent, affording the desired cleavage product 3a in 93% yield along with 8a (45% yield) and 8b (23% yield).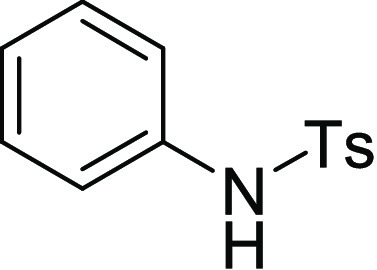
4-Methyl-N-phenyl-benzenesulfonamide
(3a): Yield = 93%; all data are consistent with the molecule
prepared according to general procedure IV.
Bis(3,4-dimethoxy-phenyl)methane (8a):36 45% yield; brown thick oil; 1H NMR
(CDCl3, 400 MHz): δ 9.84 (s, 1H), 7.45 (d, J = 7.6 Hz, 1H), 7.41(s, 1H), 6.97 (d, J = 8.0 Hz, 1H), 6.79 (d, J = 8.0 Hz, 8H), 6.70 (d, J = 10.4 Hz, 1H), 3.96 (s, 3H), 3.93 (s, 3H), 3.87 (s, 8H),
3.84 (s, 24H), 3.82 (s, 24H); 13C NMR (CDCl3, 100 MHz): δ 149.0 (2C). 147.4 (2C), 134.0 (2C), 120.8 (2C),
112.2 (2C), 111.3 (2C), 56.0 (2C), 55.9 (2C), 41.1; HRMS (ESI)+ calcd for C17H21O4, 289.1440 m/z (M + H)+; found, 289.1433 m/z. [bis(3,4-dimethoxyphenyl)methane and
3,4-dimethoxybenzaldehyde obtained in a 4:1 ratio]
1-(2,3-Dimethoxybenzyl)-5-(3,4-dimethoxybenzyl)-2,3-dimethoxybenzene (8b): 23% yield; pale-yellow oil; 1H NMR (CDCl3, 400 MHz): δ 6.75 (d, J = 8.0 Hz, 2H), 6.65 (s, 2H), 6.60–6.57 (m, 4H), 3.84 (s, 10H), 3.80 (s, 6H), 3.76 (s, 6H); 13C NMR (CDCl3, 100 MHz): δ 149.0 (2C), 147.5 (2C), 147.4 (2C), 133.6 (2C), 131.3 (2C), 120.6 (2C), 113.9 (2C), 112.0 (2C), 111.2 (2C), 56.1 (2C), 56.0 (2C), 55.9 (2C), 38.2 (2C); HRMS (ESI)+ calcd for C26H31O6, 439.2121 m/z (M + H)+; found, 439.2117 m/z.
Preparation of Tribenzocyclononane (2,3,7,8,12,13-Hexamethoxy-10,15-dihydro-5H-tribenzo[a,d,g]cyclononane) 10 (8ii)

2,3,7,8,12,13-Hexamethoxy-10,15-dihydro-5H-tribenzo[a,d,g]cyclononane (10):22 To a stirred solution of 3,4-dimethoxyphenylmethanol (0.1 g, 0.59 mmol) in dichloroethane (2 mL), Bi(OTf)3 (0.05 equiv; 0.02 g) was added, and the resultant mixture was refluxed at 85 °C for 1 h. After the complete consumption of alcohol, the reaction mixture was cooled down and next evaporated in vaccuo. Purification of the crude product by flash column chromatography using an EtOAc and petroleum ether mixture (30% EA/PE) as the eluent afforded the desired titled compound 10 (68 mg, 76% yield). 1H NMR (CDCl3, 400 MHz): δ 6.83 (s, 6H), 4.77 (d, J = 13.6 Hz, 3H), 3.84 (s, 18H), 3.55 (d, J = 13.6 H, 3H); 13C NMR (CDCl3, 100 MHz): δ 147.8, 131.9, 113.2, 56.1, 36.6; HRMS (ESI) calcd for C27H30NaO6, 473.1940 m/z (M + Na)+; found, 473.2135 m/z.
One-pot
amide synthesis in a gram scale (Scheme 9)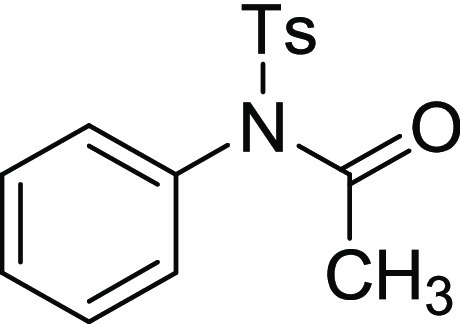
N-Acetyl-4-methyl-N-phenyl-benzenesulfonamide (11): To a stirred solution of tertiary sulfonamide 4a (1.0 g, 2.51 mmol, 1 equiv) in dichloroethane (15 mL), Bi(OTf)3 (82 mg, 0. 125 mmol, 0.05 equiv) was added, and the resultant mixture was refluxed at 85 °C for 2 h. The reaction was monitored by TLC to ensure complete consumption of 4a, and then, Ac2O (0.50 mL, 5.02 mmol, 2.0 equiv) was added to the same reaction pot. After the complete consumption of C–N bond cleavage product 3a (confirmed by TLC), the reaction was quenched with saturated NaHCO3 solution, extracted with EtOAc, washed with brine, dried with Na2SO4, and evaporated in vaccuo. Purification of the crude product by flash column chromatography using an EtOAc and petroleum ether mixture (20% EA/PE) as the eluent afforded the desired titled compound 11 (680 mg, 94% overall yield). 1H NMR (CDCl3, 400 MHz): δ 7.92 (d, J = 8.0 Hz, 2H), 7.49–7.47 (m, 3H), 7.34 (d, J = 8.0 Hz, 2H), 7.28–7.25 (m, 2H), 2.44 (s, 3H), 1.85 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 170.2, 145.1, 137.0, 136.1, 130.1 129.0 (4C), 129.5 (2C), 129.3 (2C), 25.2, 21.8; HRMS (ESI) calcd for C15H15NNaO3S, 312.0670 m/z (M + Na)+; found, 312.0670 m/z.
Acknowledgments
This work was supported by the DST, New Delhi, India [DST-INSPIRE program, IFA-CH12-79], and the Research Promotion Fund Indira Gandhi National Tribal University (IGNTU), India. A. Bhattacharya thanks the DST-INSPIRE program for his Fellowship. P. M. Shukla thanks IGNTU for his Fellowship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c02276.
Copies of 1H NMR and 13C NMR spectra for all compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Kocienski P. J.Protecting Groups; Verlag: Stuttgart, 1994. [Google Scholar]; b Wuts P. G. M.Greene’s Protective Groups in Organic Synthesis, 5th ed.; John Wiley & Sons, Inc: New Jersey, 2014; Chapter 11, pp 895–1193. [Google Scholar]
- a Wood J. L.; Stoltz B. M.; Dietrich H. J. Total Synthesis of (+)- and (-)-K252a. J. Am. Chem. Soc. 1995, 117, 10413–10414. 10.1021/ja00146a039. [DOI] [Google Scholar]; b Wood J. L.; Stoltz B. M.; Goodman S. N. Total Synthesis of (+)-RK-286c, (+)-MLR-52, (+)-Staurosporine, and (+)-K252a. J. Am. Chem. Soc. 1996, 118, 10656–10657. 10.1021/ja9626143. [DOI] [Google Scholar]
- a Murakami Y.; Watanabe T.; Kobayashi A.; Yokoyama Y. A Novel Method for the Debenzylation of Protected Indole Nitrogen. Synthesis 1984, 738–740. 10.1055/s-1984-30952. [DOI] [Google Scholar]; b Watanabe T.; Kobayashi A.; Nishiura M.; Takahashi H.; Usui T.; Kamiyama I.; Mochizuki N.; Noritake K.; Yokoyama Y.; Murakami Y. Synthetic Studies on Indoles and Related Compounds. XXVI. The Debenzylation of Protected Indole Nitrogen with Aluminum Chloride. Chem. Pharm. Bull. 1991, 39, 1152–1156. 10.1248/cpb.39.1152. [DOI] [Google Scholar]; c Paliakov E.; Strekowski L. Boron Tribromide Mediated Debenzylation of Benzylamino and Benzyloxy Groups. Tetrahedron Lett. 2004, 45, 4093–4095. 10.1016/j.tetlet.2004.03.139. [DOI] [Google Scholar]; d Okano K.; Okuyama K.; Fukuyama T.; Tokuyama H. Mild Debenzylation of Aryl Benzyl Ether with BCl3 in Presence of Pentamethylbenzene as a Non-Lewis Cation Scavenger. Synlett 2008, 13, 1977–1980. 10.1055/s-2008-1077980. [DOI] [Google Scholar]; e Nussbaumer P.; Baumann K.; Dechat T.; Harasek M. Highly Selective TFAA-Cleavage of Tertiary 2, 4-Dimethoxybenzylamines and its use in the Synthesis of Secondary Amines. Tetrahedron 1991, 47, 4591–4602. 10.1016/S0040-4020(01)86465-6. [DOI] [Google Scholar]; f Kowalski P.; Majka Z.; Kowalska T. Acid-Catalyzed N-Debenzylation of Benzylaminopyridines. Chem. Heterocycl. Compd. 1998, 34, 740–741. 10.1007/BF02252290. [DOI] [Google Scholar]; g Chern C.; Huang Y.; Kan W. M. Selective N-Debenzylation of Amides with p-TsOH. Tetrahedron Lett. 2003, 44, 1039–1041. 10.1016/S0040-4039(02)02738-7. [DOI] [Google Scholar]; h Hill B.; Liu Y.; Taylor S. D. Synthesis of α-Fluorosulfonamides by Electrophilic Fluorination. Org. Lett. 2004, 6, 4285–4288. 10.1021/ol048249z. [DOI] [PubMed] [Google Scholar]; i Trost B. M.; Fandrick D. R. DYKAT of Vinyl Aziridines: Total Synthesis of (+)-Pseudodistomin D. Org. Lett. 2005, 7, 823–826. 10.1021/ol047513l. [DOI] [PubMed] [Google Scholar]; j Trost B. M.; Krische M. J.; Radinov R.; Zanoni G. On Asymmetric Induction in Allylic Alkylation via Enantiotopic Facial Discrimination. J. Am. Chem. Soc. 1996, 118, 6297–6298. 10.1021/ja960649x. [DOI] [Google Scholar]; k Chinigo G. M.; Breder A.; Carreira E. M. Ugi-4-Component Reaction Enabling Rapid Access to the Core Fragment of Massadine. Org. Lett. 2011, 13, 78–81. 10.1021/ol102577q. [DOI] [PubMed] [Google Scholar]; l Evans P. A.; Robinson J. E. Regioselective Rh-Catalyzed Allylic Amination/Ring-Closing Metathesis Approach to Monocyclic Azacycles: Diastereospecific Construction of 2,5-Disubstituted Pyrrolines. Org. Lett. 1999, 1, 1929–1931. 10.1021/ol991064l. [DOI] [PubMed] [Google Scholar]
- a Studer M.; Blaser H. Influence of Catalyst Type, Solvent, Acid and Base on the Selectivity and Rate in the Catalytic Debenzylation of 4-Chloro-N,N-Dibenzyl Aniline with Pd/C and H2. J. Mol. Catal. A: Chem. 1996, 112, 437–445. 10.1016/1381-1169(96)00151-3. [DOI] [Google Scholar]; b Cheng C.; Sun J.; Xing L.; Xu J.; Wang X.; Hu Y. Highly Chemoselective Pd–C Catalytic Hydrodechlorination Leading to the Highly Efficient N-Debenzylation of Benzylamines. J. Org. Chem. 2009, 74, 5671–5674. 10.1021/jo9007282. [DOI] [PubMed] [Google Scholar]; c Santarem M.; Vanucci-Bacque C.; Lhommet G. Formal Total Synthesis of (+)-Gephyrotoxin. J. Org. Chem. 2008, 73, 6466–6469. 10.1021/jo801150e. [DOI] [PubMed] [Google Scholar]; d Yamamoto Y.; Shimizu E.; Ban K.; Wada Y.; Mizusaki T.; Yoshimura M.; Sajiki H.; et al. Facile Hydrogenative Deprotection of N-Benzyl Groups Using a Mixed Catalyst of Palladium and Niobic acid-on-Carbon. ACS Omega 2020, 5, 2699–2709. 10.1021/acsomega.9b03226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Baek S.; Jo H.; Kim H.; Kim H.; Kim S.; Kim D. Highly Stereoselective and Efficient Total Synthesis of (+)-Laurencin. Org Lett. 2005, 7, 75–77. 10.1021/ol047877d. [DOI] [PubMed] [Google Scholar]; b Singh S. B. Total Synthesis of Flutimide, a Novel Endonuclease Inhibitor of Influenza Virus. Tetrahedron Lett. 1995, 36, 2009–2012. 10.1016/0040-4039(95)00214-W. [DOI] [Google Scholar]
- a Alcón M.; Moyano A.; Pericàs M. A.; Riera A. Enantioselective Synthesis of Unsaturated Amino Acids Using p-Methoxybenzylamine as an Ammonia Equivalent. Tetrahedron: Asymmetry 1999, 10, 4639–4651. 10.1016/S0957-4166(99)00525-X. [DOI] [Google Scholar]; b Bull S. D.; Davis S. G.; Fenton G.; Mulvaney A. W.; Prasad R. S.; Smith A. D. Chemoselective Debenzylation of N-Benzyl Tertiary Amines with Ceric Ammonium Nitrate. J. Chem. Soc., Perkin Trans. 1 2000, 1, 3765–3774. 10.1039/b006852g. [DOI] [Google Scholar]; c Annadi K.; Wee A. G. Ceric Ammonium Nitrate Oxidation of N-(p-Methoxybenzyl) Lactams: Competing Formation of N-(Hydroxymethyl) δ-Lactams. Arkivoc 2014, 6, 108–126. 10.3998/ark.5550190.p008.840. [DOI] [Google Scholar]; d Morris J.; Wishka D. G. Vinyl Sulfonyl Esters and Amides in the Synthesis of Substituted δ-sultams and δ-sultones. J. Org. Chem. 1991, 56, 3549–3556. 10.1021/jo00011a020. [DOI] [Google Scholar]; e Fleming J. J.; McReynolds M. D.; Bois J. D. (+)-Saxitoxin: A First and Second Generation Stereoselective Synthesis. J. Am. Chem. Soc. 2007, 129, 9964–9975. 10.1021/ja071501o. [DOI] [PubMed] [Google Scholar]
- a Bhattacharya A.; Shukla P. M.; Maji B. Fe(OTf)3-Catalysed Friedel–Crafts Reaction of Benzenoid Arenes with α, β-Unsaturated Carbonyl Compounds: Easy Access to 1, 1-Diarylalkanes. R. Soc. Open Sci. 2017, 4, 170748 10.1098/rsos.170748. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Maji B.; Bhattacharya A.; Hazra S. Halogen-Induced Friedel-Crafts Alkenylation Reactions with Haloalkynes: Direct Access to gem-1,1-Dihaloalkenes. ChemistrySelect 2017, 2, 10375–10378. 10.1002/slct.201702265. [DOI] [Google Scholar]; c Maji B. Stereoselective Haliranium, Thiiranium and Seleniranium Ion-Triggered Friedel–Crafts-Type Alkylations for Polyene Cyclizations. Adv. Synth. Catal. 2019, 361, 3453–3489. 10.1002/adsc.201900028. [DOI] [Google Scholar]
- a Katohgi M.; Yokoyama M.; Togo H. Novel Sonochemical Dealkylation of N-Alkylsulfonamides in the Presence of (Diacetoxyiodo) Benzene and Iodine. Synlett 2000, 2000, 1055–1057. 10.1055/s-2000-6684. [DOI] [Google Scholar]; b Katohgi M.; Togo H. Oxidatively Sonochemical Dealkylation of Various N-Alkylsulfonamides to Free Sulfonamides and Aldehydes. Tetrahedron 2001, 57, 7481–7486. 10.1016/S0040-4020(01)00725-6. [DOI] [Google Scholar]
- Xu L.; Zhang S.; Trudell M. L. Novel N-Dealkylation of N-Alkyl Sulfonamides and N, N-Dialkyl Sulfonamides with Periodic Acid Catalyzed by Chromium (III) Acetate Hydroxide. Synlett 2004, 11, 1901–1904. 10.1055/s-2004-830851. [DOI] [Google Scholar]
- Moriyama K.; Nakamura Y.; Togo H. Oxidative Debenzylation of N-Benzyl Amides and O-Benzyl Ethers Using Alkali Metal Bromide. Org. Lett. 2014, 16, 3812–3815. 10.1021/ol501703y. [DOI] [PubMed] [Google Scholar]
- O’Sullivan S.; Doni E.; Tuttle T.; Murphy J. A. Metal-Free Reductive cleavage of C-N and S-N bonds by photoactivated electron transfer from a neutral organic donor. Angew. Chem., Int. Ed. 2014, 53, 474–478. 10.1002/anie.201306543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West M. J.; Thomson B.; Vantourout J. C.; Watson A. J. Discovery, Scope, and Limitations of an N-Dealkylation/N-Arylation of Secondary Sulfonamides under Chan-Lam Conditions. Asian J. Org. Chem. 2020, 9, 364–367. 10.1002/ajoc.201900617. [DOI] [Google Scholar]
- Inagaki F.; Hira S.; Mukai C. Silver (I)-Catalyzed Deprenylation of Allylsulfonamide Derivatives. Synlett 2017, 28, 2143–2146. 10.1055/s-0036-1589066. [DOI] [Google Scholar]
- Wang J.; Li F.; Pei W.; Yang M.; Wu Y.; Ma D.; Wang J.; et al. Selective cleavage of the N-propargyl group from sulfonamides and amides under ruthenium catalysis. Tetrahedron Lett. 2018, 59, 1902–1905. 10.1016/j.tetlet.2018.03.046. [DOI] [Google Scholar]
- Wetzel A.; Jones A. M. Electrically Driven N(sp2)–C(sp2/3) Bond Cleavage of Sulfonamides. ACS Sustainable Chem. Eng. 2020, 8, 3487–3493. 10.1021/acssuschemeng.0c00387. [DOI] [Google Scholar]
- Javorskis T.; Orentas E. Chemoselective deprotection of sulfonamides under acidic conditions: scope, sulfonyl group migration, and synthetic applications. J. Org. Chem. 2017, 82, 13423–13439. 10.1021/acs.joc.7b02507. [DOI] [PubMed] [Google Scholar]
- See the review articles:; a Ollevier T. New Trends in Bismuth-Catalyzed Synthetic Transformations. Org. Biomol. Chem. 2013, 11, 2740–2755. 10.1039/c3ob26537d. [DOI] [PubMed] [Google Scholar]; b Gaspard-Iloughmane H.; Roux C. L. Bismuth(III) Triflate in Organic Synthesis. Eur. J. Org. Chem. 2004, 2517–2532. 10.1002/ejoc.200300754. [DOI] [Google Scholar]; c Dang T. T.; Boeck F.; Hintermann L. Hidden Brønsted acid catalysis: pathways of accidental or deliberate generation of triflic acid from metal triflates. J. Org. Chem. 2011, 76, 9353–9361. 10.1021/jo201631x. [DOI] [PubMed] [Google Scholar]; d Rosenfeld D. C.; Shekhar S.; Takemiya A.; Utsunomiya M.; Hartwig J. F. Hydroamination and hydroalkoxylation catalyzed by triflic acid. Parallels to reactions initiated with metal triflates. Org. lett. 2006, 8, 4179–4182. 10.1021/ol061174+. [DOI] [PubMed] [Google Scholar]
- a Sletten E. T.; Tu Y. J.; Schlegel H. B.; Nguyen H. M. Are Brønsted Acids the True Promoter of Metal-Triflate-Catalyzed Glycosylations? A Mechanistic Probe into 1, 2-Cis-Aminoglycoside Formation by Nickel Triflate. ACS Catal. 2019, 9, 2110–2123. 10.1021/acscatal.8b04444. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dumeunier R.; Marko I. E. On the Role of Triflic Acid in the Metal Triflate-Catalysed Acylation of Alcohols. Tetrahedron Lett. 2004, 45, 825–829. 10.1016/j.tetlet.2003.11.034. [DOI] [Google Scholar]
- Yi H.; Zhang G.; Wang H.; Huang Z.; Wang J.; Singh A. K.; Lei A. Recent Advances in Radical C– H Activation/Radical Cross-Coupling. Chem. Rev. 2017, 117, 9016–9085. 10.1021/acs.chemrev.6b00620. [DOI] [PubMed] [Google Scholar]
- Fujisawa S.; Kadoma Y.; Yokoe I. Radical-Scavenging Activity of Butylated Hydroxytoluene (BHT) and its Metabolites. Chem. Phys. Lipids 2004, 130, 189–195. 10.1016/j.chemphyslip.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Chataigner I.; Panel C.; Gérard H.; Piettre S. R. Sulfonyl vs. Carbonyl Group: Which is the More Electron-Withdrawing?. Chem. Commun. 2007, 3288–3290. 10.1039/b705034h. [DOI] [PubMed] [Google Scholar]
- Wang D.; Ivanov M. V.; Mirzaei S.; Lindemana S. V.; Rathore R. An electron-transfer induced conformational transformation: from non-cofacial “sofa” to cofacial “boat” in cyclotetraveratrylene (CTTV) and formation of charge transfer complexes. Org. Biomol. Chem. 2018, 16, 5712–5717. 10.1039/C8OB01118D. [DOI] [PubMed] [Google Scholar]
- Wolstenhulme J. R.; Rosenqvist J.; Lozano O.; Ilupeju J.; Wurz N.; Engle K. M.; Gouverneur V.; et al. Asymmetric Electrophilic Fluorocyclization with Carbon Nucleophiles. Angew. Chem., Int. Ed. 2013, 125, 9978–9982. 10.1002/ange.201304845. [DOI] [PubMed] [Google Scholar]
- Everett R. K.; Wolfe J. P. Aza-Wittig Rearrangements of N-Benzyl and N-Allyl Glycine Methyl Esters. Discovery of a Surprising Cascade Aza-Wittig Rearrangement/Hydroboration Reaction. J. Org. Chem. 2015, 80, 9041–9056. 10.1021/acs.joc.5b01286. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Xie J.; Rao B.; Luo M. Iron-Catalyzed N-Arylsulfonamide Formation through Directly Using Nitroarenes as Nitrogen Sources. J. Org. Chem. 2015, 80, 3504–3511. 10.1021/acs.joc.5b00130. [DOI] [PubMed] [Google Scholar]
- Deruer E.; Coulibali S.; Boukercha S.; Canesi S. Carbon–Phosphorus Bond Formation on Anilines Mediated by a Hypervalent Iodine Reagent. J. Org. Chem. 2017, 82, 11884–11890. 10.1021/acs.joc.7b01595. [DOI] [PubMed] [Google Scholar]
- Bering L.; Vogt M.; Paulussen F. M.; Antonchick A. P. Selective, Catalytic, and Metal-free Coupling of Electron-Rich Phenols and Anilides Using Molecular oxygen as Terminal Oxidant. Org. Lett. 2018, 20, 4077–4080. 10.1021/acs.orglett.8b01631. [DOI] [PubMed] [Google Scholar]
- Sridhar R.; Srinivas B.; Kumar V. P.; Narender M.; Rao K. R. β-Cyclodextrin-Catalyzed Monosulfonylation of Amines and Amino Acids in Water. Adv. Synth. Catal. 2007, 349, 1873–1876. 10.1002/adsc.200600652. [DOI] [Google Scholar]
- Moon S. Y.; Nam J.; Rathwell K.; Kim W. S. Copper-catalyzed Chan–Lam Coupling between Sulfonyl Azides and Boronic Acids at Room Temperature. Org. Lett. 2014, 16, 338–341. 10.1021/ol403717f. [DOI] [PubMed] [Google Scholar]
- Chen K.; Chen W.; Han B.; Chen W.; Liu M.; Wu H. Sequential C–S and S–N Coupling Approach to Sulfonamides. Org. Lett. 2020, 22, 1841–1845. 10.1021/acs.orglett.0c00183. [DOI] [PubMed] [Google Scholar]
- Marset X.; Torregrosa-Crespo J.; Martínez-Espinosa R. M.; Guillena G.; Ramón D. J. Multicomponent Synthesis of Sulfonamides from Triarylbismuthines, Nitro Compounds and Sodium Metabisulfite in Deep Eutectic Solvents. Green. Chem. 2019, 21, 4127–4132. 10.1039/C9GC01541H. [DOI] [Google Scholar]
- Chan Y. C.; Yeung Y. Y. Halogen Bond Catalyzed Bromocarbocyclization. Angew. Chem., Int. Ed. 2018, 57, 3483–3487. 10.1002/anie.201800261. [DOI] [PubMed] [Google Scholar]
- Sánchez-Cantalejo F.; Priest J. D.; Davies P. W. A Gold Carbene Manifold to Prepare Fused γ-Lactams by Oxidative Cyclisation of Ynamides. Chem.–Eur. J. 2018, 24, 17215–17219. 10.1002/chem.201804378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu P.; Sun C.; Ma J.; Li F. The N-Alkylation of Sulfonamides with Alcohols in Water Catalyzed by the Water-Soluble Iridium Complex {Cp*Ir[6,6′-(OH)2bpy](H2O)}[OTf]2. Adv. Synth. Catal. 2014, 356, 447–459. 10.1002/adsc.201300711. [DOI] [Google Scholar]
- Laha J. K.; Sharma S.; Dayal N. Palladium-Catalyzed Regio-and Chemoselective Reactions of 2-Bromobenzyl Bromides: Expanding the Scope for the Synthesis of Biaryls Fused to a Seven-Membered Sultam. Eur. J. Org. Chem. 2015, 7885–7891. 10.1002/ejoc.201501032. [DOI] [Google Scholar]
- Dethe D. H.; Shukla M.; Dherange B. D. Sc(OTf)3-Catalyzed Synthesis of Symmetrical Dithioacetals and Bisarylmethanes Using Nitromethane as a Methylene Source. Org. Lett. 2020, 22, 5778–5782. 10.1021/acs.orglett.0c01831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.