

1. HTRS.P1.13
2. Successful implementation of a pediatric bleeding assessment tool in the electronic medical record in an academic clinical setting
K. Bajorek 1; C. Tarango2; S. Jones3; M. Martin4; J. Palumbo2; E. Mullins2; L. Luchtman‐Jones2
1 University of Cincinnati College of Medicine, Cincinnati, Ohio, USA; 2 Hematology, Cincinnati Children's Hospital Medical Center, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA; 3 Hematology, Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio, USA; 4 Cancer & Blood Diseases Institute, Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio, USA
Background: The Pediatric Bleeding Questionnaire (PBQ) is a validated bleeding assessment tool to standardize history‐taking and distinguish those with mild‐moderate bleeding disorders; however, use beyond the research setting has lagged. During a 4‐cycle quality improvement project, our academic pediatric hemostasis group developed and optimized the use of an electronic version among faculty and trainees. Because scores trend lower in younger children, a tool was created as a flowsheet in the electronic medical record that scores the PBQ plus 2 family history questions (HemPBQ). The questions, responses and scores are added to clinic notes as a “dot phrase.”
Objectives: For the 13 months after HemPBQ flowsheet build, we evaluated its use by 5 academic providers and their trainees in the pediatric Hemostasis group for new possible bleeding disorder clinic visits and studied patient demographic and clinical information. Hypothesis: Use of a bleeding assessment tool (HemPBQ) in the electronic medical record will be sustainable one year following its implementation.
Methods: After IRB approval, records from all new clinic visits scheduled as “possible bleeding disorder” from January 29, 2019 through February 28, 2020 were retrospectively reviewed for clinical and demographic data and for the presence of the HemPBQ in the electronic clinic note (EPIC © 2020 Epic Systems Corporation). Data are reported descriptively.
Results: Over 13 months, for 313 visits, 98.7% had a HemPBQ assessment in the clinic note. Females constituted 60% of patients, with a mean age of 4 years (4–15 IQR). The most common referral indication was for abnormal lab results (Table 1). Clinician‐initiated documentation of screening for joint hypermobility by examination or history‐taking occurred in 86 visits (27.5%).
Table 1. Demographics

Conclusions: Use of standardized bleeding history questions can provide consistency, speed documentation and serve as an educational tool for trainees. When available as a flowsheet in the electronic medical record, use of the PBQ with family history questions remained extremely high (98.7%) for new patient visits seen by 5 academic providers and their trainees, even a year after its introduction. By contrast, provider‐initiated documentation of screening for joint hypermobility was much less frequent. Future data analysis will look at the positive and negative predictive value of the HemPBQ for a bleeding disorder in new pediatric hematology clinic patients.
Other: 1. Biss TT et al. J Thromb Haemost. 2010. 2. Tosetto A et al. Semin Thromb Hemost 2016. 3. Moenen FCJI et al. Haemophilia 2018.
3. HTRS.P2.16
4. The value of immature platelet fraction of platelet apheresis products in prediction of platelet transfusion response in thrombocytopenic cancer patients
T. Bat 1; O. Maguire2; E. Turk3; J. Becker2; K. Catalfamo2; E. Wang2
1 UT Southwestern Medical Center, Dallas, Texas, USA; 2 Roswell Park Comprehensive Cancer Center, Buffalo, New York, USA; 3 Magee‐Womens Research Institute, BUffalo, New York, USA
Background: Platelet transfusion is crucial in severe thrombocytopenia to prevent potentially life‐threatening bleeding. Cancer patients are among the most abundant group receiving platelet transfusions, mainly because of increased use of myelosuppressive chemotherapy/immunotherapy regimens. The ability to precisely predict platelet response before transfusion will be a valuable tool to be used in transfusion medicine. Monitoring immature platelet fraction (IPF %) has been suggested to predict platelet response in pediatric transplant patients. This study was designed to evaluate the feasibility of using the IPF % as a predictive tool for platelet transfusion response in adult cancer patients.
Objectives: N/A.
Methods: We conducted a prospective pilot study in 47 cancer patients with mild to severe thrombocytopenia. Blood samples were obtained before and after transfusion to measure the corrected count increment (CCI). Aliquots of each aphaeresis were obtained from the blood bank services to determine IPF % and P selection expression by flow cytometry.
Results: There is no correlation between IPF % of the platelet bag and CCI (p = 0.14782632; Figure 1, Table 1). All other variables were analyzed to see if there were any other variables that may have correlated with CCI. As a result, we determined aphaeresis product platelet count/unit volume was correlated with CCI (p = 0.0361/0.0145; Table 1).
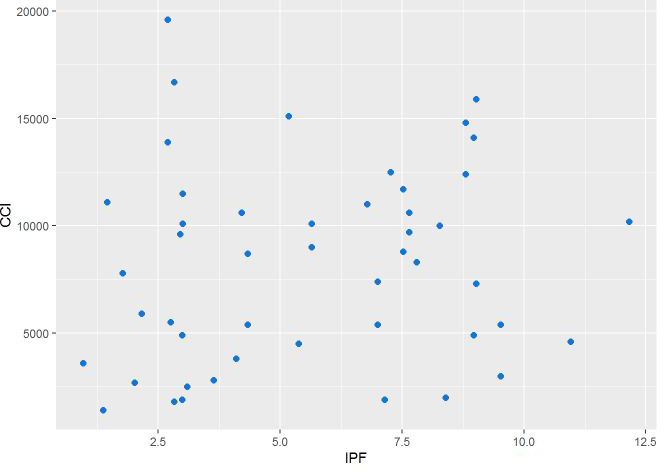
Figure 1. The scatterplot of CCI and IPF%. Spearman correlation coefficient between CCI and IPF% (rho = 0.1478, p‐value = 0.3160)
Table 1. The Spearman correlation coefficients (rho) between other variables in the study and CCI recorded. Yellow highlights statistically significant correlations between variable and CCI

Conclusions: We found platelet apheresis product’s platelet count/unit volume is correlated with the recipient’s CCI. Conversely, the measurement of IPF % and P selection lack predictive power regarding determining platelet transfusion response in thrombocytopenic cancer patients.
5. HTRS.P2.14
6. Short‐term outcome of neonatal venous thromboembolism for anticoagulated and observed patients
K. Beg; C. Toohey; C. Song; J. Journeycake; O. Khan
University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma, USA
Background: Advancements in the medical field and increased survival of premature infants has led to a rise of venous thromboembolism (VTE) in neonates. Current recommendations include careful observation, therapeutic anti‐coagulation and in some cases thrombolysis. Critically ill infants like those admitted in the neonatal intensive care unit (NICU) are more prone to bleeding complications due to disruption of the anti and pro‐coagulant balance. We hypothesize that in certain cases neonatal patients may not require anti‐coagulation and could be safely observed thus reducing their risk of bleeding complications.
Objectives: To compare the characteristics of neonates with venous thromboembolism who had expectant management vs. those treated with anticoagulation
Methods: Retrospective chart review of patients with VTE admitted to the NICU at the Oklahoma University Children’s Hospital from October 2009‐October 2019. Patients were selected if they had an ICD‐9 or ICD‐10 code specific for a venous clot. Data was collected about demographics, medical history, co‐morbidities, thrombosis characteristics, treatment and outcome.
Results: A total of 209 patient records were screened. 104 patients were eligible and included in the study. Majority of patients (83.6%) of patients had a line associated clot. Two‐third of patients (n = 66, 63.5%) received therapeutic anti‐coagulation while 34.6% were observed. The rate of complication from treatment occurred in 25% of the entire cohort, 10% of which were bleeding complications in patients receiving anti‐coagulation (Table 1). There was no difference in the odds of complete resolution between patients who were treated with therapeutic anti‐coagulation and those that were observed (OR 1.07, 95% CI 0.44–2.6, p‐value 0.88). Removal of line made no difference in outcome (OR 1.4, 95% CI 0.4–4.92, p‐value 0.27). Presence of maternal pre‐eclampsia (OR 0.17, 95% CI 0.03–0.89, p‐value 0.04) and maternal chronic hypertension (OR 0.12, 95% CI 0.02–0.6, p‐value 0.01) was associated with a reduced odds of complete resolution of the clot. Patients that were large for gestational age also tended to have reduced odds of clot resolution. (OR 0.21, 95% CI 0.04–1.15, p‐value 0.07). Conversely, patients with history of necrotizing enterocolitis had an increased odds of clot resolution (OR 7.8, 95% CI 1.0–63.2, p‐value 0.05) (Table 2).
Table 1. Line, clot and treatment characteristics of the cohort

Table 2. Effect of maternal history, comorbidities, clot characteristics on outcome

Conclusions: The short‐term outcome of neonatal VTE does not seem to differ between those that were treated with medications and those that were observed with serial imaging. Additionally, conditions that cause uteroplacental insufficiency like pre‐eclampsia and chronic hypertension may be associated with increased morbidity for neonatal VTE.
7. HTRS.P2.19
8. Clinical outcomes in patients with acute hepatic porphyria treated with givosiran who stopped hemin prophylaxis at study entry: A post‐hoc analysis from the Phase 3 envision study through month 12
H. Bonkovsky 1; J. Langendonk2; M. Balwani3; E. Sardh4; L. Gouya5; D. Rees6; P. Stein6; U. Stölzel7; A. Peiro8; D. Bissell9; S. Keel10; C. Parker11; S. Silver12; J. Windyga13; D. D'Avola14; G. Ross15; P. Steward16; B. Ritchie17; J. Oh18; P. Harper19; J. Wang20; A. Ianova21; Y. Horie22; K. Anderson23; E. Minder24; D. Vassiliou19; I. Kubisch25; E. Guillen‐Navaro26; D. Coman27; Y. Goto28
1 Wake Forest Medical Center, Winston‐Salem, North Carolina, USA; 2 Erasmus Med Coll, Rotterdam, Drenthe, Netherlands; 3 Icahn School of Medicine, Mt Sinai Medical Center, New York, New York, USA; 4 Sweden Porphyria Center, Blekinge Lan, Sweden; 5 CHU Louis Mourier Paris, Bourgogne, France; 6 Department of Clinical Biochemistry, King’s College Hospital, London, England, United Kingdom; 7 Klinikum Chemnitz gGmbH, Cheminitz, Baden‐Wurttemberg, Germany; 8 Hospital Clínic de Barcelona, Andalucia, Spain; 9 UCSF, San Francisco, California, USA; 10 U Washington, Seattle, Washington, USA; 11 Univ of Utah, Salt Lake City, Utah, USA; 12 Univ of Michigan, Ann Arbor, Michigan, USA; 13 IHIT Instytut Hematologii I Transfuzjologii, Warsaw, Dolnoslaskie, Poland; 14 Universidad de Navarra, Navarra, Spain; 15 The Royal Melbourne Hospital, Melbourne, Western Australia, Australia; 16 Royal Prince Alfred Hospital Biochemistry Dept, Ashmore and Cartier Islands, Australia; 17 University of Alberta, Alberta, Canada; 18 Gwangjingu Seoul, Seoul‐t'ukpyolsi, Republic of Korea; 19 Sweden Porphyria Center, Stockholm, Blekinge Lan, Sweden; 20 Taichung Veterans General Hospital, Taichung, Taiwan (Republic of China); 21 Triaditsa, Triaditsa, Turgovishte, Bulgaria; 22 Toyohashi University of Technology, Tokyo, Japan; 23 UTMB, Galveston, Texas, USA; 24 University Hospital of Zurich, Zurich, Switzerland; 25 University of Cologne, Cologne, Baden‐Wurttemberg, Germany; 26 Hospital Clínico Universitario Virgen de la Arrixaca. Universidad de Murcia: El Palmar, ES, Murcia, Spain; 27 Brisbane Med Ctr, Brisbane, Ashmore and Cartier Islands, Australia; 28 Taichung Veterans General Hospital, Tokyo, Japan
Background: Acute Hepatic Porphyria (AHP) is a family of rare genetic diseases due to enzyme defects in hepatic heme synthesis. Accumulation of toxic heme intermediates ALA and PBG may result in neurovisceral attacks and chronic manifestations. Intravenous hemin is approved to treat acute attacks and is sometimes used off‐label prophylactically. In the ENVISION study, givosiran, an RNAi therapeutic, reduced the composite porphyria annualized porphyria attack rate (AAR) vs. placebo (pbo). A post‐hoc analysis was conducted to evaluate outcomes in AHP patients with or without prior hemin prophylaxis prior to screening.
Objectives: ENVISION (NCT03338816) is an ongoing Phase 3 global, randomized, pbo‐controlled study, evaluating givosiran efficacy and safety in symptomatic AHP patients in a 6‐month double blind (DB) period and an open label extension (OLE) period (30 months). Patients were required to discontinue prophylactic hemin treatment at study entry, but could receive hemin for acute attacks. Outcome measures included the composite porphyria AAR ( attacks requiring hospitalization, urgent care, or IV hemin at home).
Methods: See NCT03338816.
Results: For AHP patients on prior hemin prophylaxis (median historical AAR: 9.0), a 77% reduction in mean AAR was observed with givosiran treatment vs. pbo in the DB period (Table 1). A similar reduction (63%) in mean AAR was observed in those without prior hemin prophylaxis (median historical AAR: 7.0). In both groups, further reduction in AAR was observed in patients who continued on givosiran in the OLE period (Table 1). A similar reduction in AAR was observed in both groups of pbo patients who received givosiran in the OLE (Table 1). The percentage of patients with 0 composite attacks increased in each group following 6‐months of givosiran treatment in the OLE with 55% and 67% in the patients who had continued givosiran treatment (Table 1). Additional outcomes measures will be presented.
Table 1. Clinical Outcomes in Patients with Acute Hepatic Porphyria Treated with Givosiran Who Stopped Hemin Prophylaxis at Study Entry: A Post‐hoc Analysis of Data From the Phase 3 ENVISION Study Through Month 12

Conclusions: Clinically meaningful reduction in the AAR was observed in AHP patients treated with givosiran regardless of whether they received hemin prophylaxis prior to study entry, with further AAR reduction observed in those who continued receiving givosiran during the OLE. In addition, a similar benefit was observed in pbo patients who received givosiran for 6‐months during the OLE period regardless of prior hemin prophylaxis use
Other: This submission is being presented to inform hematologists, who are the most frequent primary treater type for acute hepatic porphyia. Specifically, the study reports the results on those patients were required to stop IV hemin prophylaxis in order to enter the ENVISION trial where givosiran was evaluated. Hematologists are frequently the central provider deciding when to initiate IV hemin and how to manage side effects/toxicities during chronic administration. Thus this data is germane to the hematology community. This data was initially accepted and presented at American College of Gastroenterology in October 2020.
9. HTRS.P2.11
10. Management of recurrent disease in acquired hemophilia A
A. Boothby 1; N. Zantek2; M. Reding3; M. Mazepa2
1 University of Minnesota, Saint Paul, Minnesota, USA; 2 University of Minnesota, Minneapolis, Minnesota, USA; 3 Center for Bleeding and Clotting Disorders, University of Minnesota Medical Center, Minneapolis, Minnesota, USA
Background: Acquired hemophilia A (AHA) is a rare bleeding disorder arising from development of autoimmunity to endogenous factor VIII. Treatment centers on hemostatic therapy and immune suppressive therapy (IST) to eradicate the responsible antibody. Rituximab has shown promise as a therapeutic option for over a decade; however, there remains disagreement about the ideal setting for its use.
Objectives: Describe the experience of a subset of patients with AHA who suffered from relapsing disease and were treated with rituximab.
Methods: A single center, retrospective chart review study of cases of patients treated for relapsing AHA was conducted. Demographic, treatment, and outcomes data were collected from the electronic medical record.
Results: A cohort of 18 patients with AHA were identified. Six suffered at least one relapse. In the twelve non‐relapsing patients, only one was treated with corticosteroid monotherapy and eleven were treated with combination IST: steroid/rituximab in eight, steroid/cyclophosphamide in one patient, and more complex prolonged treatment in three. Three of six relapsing patients received corticosteroid monotherapy, two received corticosteroid/rituximab, and one was treated with corticosteroid/cyclophosphamide/therapeutic plasma exchange (TPE) at an outside center. The number of relapses ranged from one to three with follow up durations of 68 days to 23.4 years. Combination therapy with corticosteroid/rituximab was used as treatment of first relapse in five out of six patients and all three patients in second relapse. Remissions ranged from 42 days to 20 years (Figure 1). Median remission after initial treatment was 267 days while median remission after treatment of relapse was 905 days. Two patients eventually required maintenance therapy with mycophenolate mofetil. Rituximab 375 mg/m2 IV once weekly for four weeks was most frequently used; however, multiple patients were treated with additional doses in an episode, or retreated on subsequent episodes, resulting in durable remissions. Treatment related toxicities were most common with corticosteroids and included infection, hyperglycemia, cytopenia, and psychiatric syndromes.
Figure 1. Treatment course of AHA, including presentation, diagnosis, treatments, relapses, and remissions

Conclusions: This experience highlights the diverse phenotype of presentation, treatment response, and durability of remission seen in AHA. Rituximab was often used first line in combination with corticosteroids and was universally used over the course of relapsing disease. This is in contrast to current international consensus guidelines which suggest reserving first line use to those patients with poor prognosis or contraindications to corticosteroids. This data suggests use of rituximab first line as well as for relapse in previously treated patients may be appropriate. A larger database or prospective registry would allow for better understanding of prognostic factors and treatment of relapsing disease.
11. HTRS.P2.6
12. Examination and validation of a patient‐centric joint metric: “Problem Joint”; empirical evidence from the chess us dataset
T. Burke 1; I. Rodriguez Santana1; P. Chowdary2; R. Curtis3; K. Khair4; M. Laffan5; P. McLaughlin2; D. Noone1; B. O'Mahony6; J. Pasi7; M. Skinner8; J. O'Hara1
1 HCD Economics, Daresbury, Warrington, England, United Kingdom; 2 Royal Free London NHS Foundation Trust, London, England, United Kingdom; 3 Hematology Utilization Group Study (HUGS), Walnut Creek, California, USA; 4 Haemnet, London, England, United Kingdom; 5 Imperial College London, London, England, United Kingdom; 6 Irish Haemophilia Society, Dublin, Ireland; 7 Barts and The London School of Medicine and Dentistry, London, England, United Kingdom; 8 Institute for Policy Advancement Ltd., Washington DC, District of Columbia, USA
Background: Severe hemophilia is characterized by spontaneous hemarthrosis leading to progressive joint deterioration and chronic pain. Unless these recurrent hemarthroses can be prevented, patients will develop chronic synovitis, pain, and destruction of the joint. Metrics such as ‘Target joint’ and other clinical measures of joint morbidity are prevalent and widely accepted, though measures focused solely on bleeding activity, are arguably becoming less sensitive as current treatment strategies look to reduce or eradicate hemarthroses. They continue, however, to remain clinically relevant and complementary to delivery of comprehensive hemophilia care. ‘Problem Joint’ (PJ), defined as having chronic joint pain and/or limited range of movement due to chronic synovitis and/or haemophilic arthropathy, with or without persistent bleeding was derived through consensus of key opinion leaders in the haemophilia field.
Objectives: Initial research presented here was used to test the sensitivity of PJ as a patient relevant metric with respect to key outcomes for US hemophilia patients.
Methods: Data on PJs, as well as demographic, clinical and socio‐economic variables was captured within the ‘Cost of Haemophilia: A Socioeconomic Survey’ a family of datasets containing over 4,000 people with hemophilia (PwH). The US adult cohort (CHESS US+) comprised the focus of this piece. Statistical analysis explored the association of PJ count and location with respect to quality of life (EQ‐5D score), and overall work impairment, measured by the Work Productivity and Activity Impairment Questionnaire. Patients with current inhibitors were excluded from the analysis.
Results: The US cohort contained information on 345 PwH and captured adults only, with a mean age of 35 years. Approximately, 43% of PwH had one or more PJs. Nearly 42% of PwH had one or more PJs in a major joint (i.e. in the ankles, knees and/or elbows). The relationship between EQ‐5D and number of PJs showed a negative trend (Figure 1): the average EQ‐5D score was: 0.81 for those with zero PJs (N = 197);0.79 for those with one PJ (N = 24); 0.70 for two PJs (N = 29); 0.68 for three PJs (N = 24) and 0.49 for those patients with four of more PJs (N = 59). Regarding PJ location, average EQ‐5D was 0.61 (N = 145) for PwH with one or more PJ located in a major joint. For those with one or more non‐major PJs, average EQ‐5D was 0.80 (N = 200). Similarly, an increase in number of PJs meant greater work productivity impairment versus no PJs recorded: 30.08% (N = 102) vs. 19.51% (N = 137), respectively.

Figure 1. Mean EQ5D by total number of Problem Joints (N=354)
Conclusions: An increase in the number of PJs was associated with an increasing humanistic burden in PwH in the US cohort. The proposed PJ definition takes a holistic viewpoint and provides a patient relevant perspective. Further work is planned to evaluate the appropriateness of the measure, and test the sensitivity in wider cohorts.
13. HTRS.P1.12
14. Single center experience of bleeding evaluation in hemophilia carriers using ISTH‐BAT
M. Carter Febres; M. Lim
University of Utah, Salt Lake City, Utah, USA
Background: In recent years, it has been increasingly recognized that hemophilia carriers manifest a wide range of coagulation factor levels and may have bleeding tendencies even at coagulation factor levels that are within the normal range
Objectives: To evaluate bleeding symptoms in hemophilia A and B carriers using the ISTH Bleeding Assessment Tool (BAT) and to determine associations between coagulation factor levels and bleeding symptoms.
Methods: A retrospective chart review of hemophilia carriers seen at the Utah Center for Bleeding and Clotting Disorders from 2004 to 2020 was conducted. The following data were extracted: Hemophilia type, baseline factor levels, age at time of clinic visit, race/ethnicity, body mass index (BMI), family history, genetic testing (if available) and ISTH BAT scoring/characteristic symptoms. Descriptive statistics were used in the analysis.
Results: 46 hemophilia carriers were included in this study (hemophilia A, n = 27, hemophilia B n = 19). Characteristics of the cohort and the bleeding symptoms reported by the cohort are shown in Table 1. In our cohort, hemophilia carriers with high BAT score ≥6 showed a median factor level of 38 IU/dL (IQR: 25%), while hemophilia carriers with BAT score < 6 had a median factor level of 39 IU/dL (IQR: 50%).
Table 1. Demographics and bleeding characteristics

Figure 1.

ISTH BAT Score
Conclusions: Similarly, carriers with severe coagulation factor deficiencies can have serious and life‐threatening bleeding symptoms. As such, hemophilia carriers need equal attention and standard of care similar to their male counterparts. Our review of the data suggests poor discrimination of using BAT score with median factor levels, which highlights that BAT score might be more useful to predict risk of bleeding than baseline factor levels.
15. HTRS.P2.8
16. HTRS Student Research Award: Development of an immunoproteomic pipeline for the characterization of anti‐FVIII molecular and cellular repertoires in hemophilia A inhibitor patients
R. Chen 1, 3; V. Bhoj1; Valder R. Arruda2,3; B. Samelson‐Jones2,3
1 Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA; 2 Department of Pediatrics, University of Pennsylvania, Philadelphia, Pennsylvania, USA; 3 Center for Cellular and Molecular Therapeutics, The Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, USA
Background: Inhibitors, or neutralizing alloantibodies against coagulation factor VIII (FVIII), are a major cause of morbidity and mortality in inherited hemophilia A (HA). At high‐titers, inhibitors completely block the hemostatic efficacy of FVIII replacement therapy. The limited understanding of the circulating anti‐FVIII antibody and B cell compartments in patients hinders the development of preventive approaches and novel therapies for HA with inhibitors.
Objectives: To characterize both circulating anti‐FVIII antibody and the B cell receptor repertoires in HA patients with high‐titer inhibitors.
Methods: Total IgG was purified from HA patient plasma using a protein G spin column. Subsequently, anti‐FVIII antibodies were isolated from the total IgG pool using biotinylated full‐length FVIII (FVIII‐SS‐biotin) immobilized to magnetic streptavidin beads. FVIII‐SS‐biotin was produced using an optimized biotinylation procedure yielding <15 attached biotins per FVIII molecule. At this degree of biotinylation, there was only minimal reductions in FVIII specific activity, as determined by a one‐stage plasma‐based assay, and monoclonal antibody binding, as determined by ELISA.
Complete elution of FVIII‐bound IgG was achieved by reducing the disulfide biotin linkage. Both eluted IgG and IgG in the flow‐through were analyzed by LC‐MS/MS, and label‐free quantification was performed in MaxQuant using the patient’s B cell receptor library as the sequence database. Anti‐FVIII antibodies were defined as those present in both replicates of the elution with >100‐fold greater intensity than in the flow‐through.
Results: FVIII‐SS‐biotin was able to fully capture a panel of 12 monoclonal antibodies, targeting all five immunogenic domains of FVIII, spiked into normal human plasma. Monoclonal antibodies were identified in LC‐MS/MS spectra at concentrations as low as 10–100 fmol in the input. This is comparable to the sensitivity of LC‐MS/MS, highlighting that our anti‐FVIII IgG purification protocol does not limit the sensitivity of this approach.
In this study, we performed two replicate purifications from two time points of a pediatric HA patient with a high‐titer inhibitor. We achieved ≥97% capture of anti‐FVIII IgG by ELISA and complete capture of inhibitors by Bethesda assay for all replicates. Concurrently, 3–4 × 107 PBMCs from each time point were processed for high‐throughput B cell receptor sequencing. Collection and analysis of these results is ongoing.
Conclusion: We developed a novel and efficient strategy to isolate anti‐FVIII antibodies without epitope bias from HA inhibitor patients with a sensitivity sufficient for downstream immunoproteomic studies. Combining these results with ongoing paired B cell receptor sequencing will provide valuable insight into the development of the human anti‐FVIII immune response.
17. HTRS.P1.5
18. ECMO blood product utilization: The first 24 hours
A. Chidharla 1; N. Hussain2; B. Morris3; R. Jesudas4
1 University of Illinois College of Medicine, Peoria, Illinois, USA; 2 Baylor Scott & White Medical Center, Houston, Texas, USA; 3 OSF Health Care, Peoria, Illinois, USA; 4 Bleeding & Clotting Disorders Institute, Peoria, Illinois, USA
Background: Extracorporeal Membrane Oxygenation (ECMO) for cardiac support in patients with cardiac failure has increased in the recent years due to increasing clinical experience and technological advances. ECMO is invasive and associated with serious complications, including severe hemorrhage; bleeding. The initiation and maintenance of ECMO require significant blood product support. In these critically ill patients, the increased risk for hemorrhagic complications increases the need for blood product administration.
Objectives: We conducted a quality improvement (QI) project to look into the blood product utilization and anticoagulation practice within 24 hours of ECMO cannulation between pre‐intervention phase (2017‐ mid 2018) before our recommendations and post‐intervention phase (mid‐ 2018 to 2019) after the implementation of the recommendations. The aim of this QI project was to evaluate the factors that affect bleeding in patients on ECMO and reduce blood product utilization during the first 24 hours after ECMO cannulation.
Methods: A single‐center QI project 15 months pre intervention phase post intervention phase of blood product usage in the first 24 hours after ECMO cannulation and anticoagulation. Based on our evaluation, we recommended to decrease heparin dose during cannulation and to follow trauma massive transfusion protocol if there is increased bleeding. Data from the subsequent 15 months were collected and evaluated. The study comprised of 40 patients in total. The blood product utilization was compared pre and post recommendations using Wilcoxon rank‐sum test and negative binomial regression adjusting for patients’ characteristics and clinical variables.
Results: After adjusting for the patient’s characteristics and clinical variables, the FFP (p = 0.0003), PRBC (p < 0.0001), platelets (p < 0.0001), and Cryoprecipitate (p = 0.0003) usage in the post‐intervention phase are statistically significantly lower than those in pre‐intervention phase. The post‐intervention (Mean ± SD) heparin dose (in units) during the cannulation (8812.5 ± 7382.1) was lower than pre‐intervention dose of heparin (17357.1 ± 15648.1) p = 0.066. Twenty percent of patients in pre phase had Veno‐Venous extracorporeal membrane oxygenation (VV‐ ECMO), whereas 68% of patients in the post‐phase had VV‐ ECMO, p = 0.005. VV‐ ECMO patients received fewer packed red cells in (Mean ± SD) (6 ± 15.7 v 19.3 ± 22.13 Units per 24 hours p = 0.011) and platelets (3.4 ± 12.7 v 4.6 ± 5.7 Units per 24 hours p = 0.035) compared with Veno‐Arterial extracorporeal membrane oxygenation (VA‐ ECMO); patients.
Conclusions: Following our recommendations, the blood product utilization(pRBC, FFP, platelets, and cryoprecipitate) in the first 24 hours was decreased compared to the baseline, which was statistically significant. Heparin utilization during the time of canulation was lower following our recommendations. The overall usage of blood products was low in VV ECMO patients compared to VA ECMO patients.
19. HTRS.O3.2
20. Formation of prothrombinase on human endothelial cells
C. Cohen 1; N. Turner2; J. Moake2
1 Baylor College of Medicine, Texas Children's Hospital, Houston, Texas, USA; 2 Rice University, Houston, Texas, USA
Background: Thrombin is a critical protein in hemostasis. It cleaves fibrinogen to fibrin and activates other coagulation proteins. In addition, thrombin has anticoagulant functions, including binding to thrombomodulin (CD141) in order to activate protein C. The role of human endothelial cells (ECs) in hemostasis, including thrombin production, is currently of interest. We have previously demonstrated [Nature Scientific Reports (2020) 10:2005] that human ECs in culture express and produce factor (F)X and the coagulation factors necessary for FX activation, and that FX activation can occur on EC surfaces without the addition of exogenous coagulation proteins or phospholipids.
Objectives: In this study, we investigate human EC production of the components of the prothrombinase complex (FX, FV, and prothrombin [FII]), and the generation of thrombin by cleavage from prothrombin of fragment 1+2 (F1+2).
Methods: Human umbilical vein ECs (HUVECs), glomerular microvascular ECs (GMVECs), and liver sinusoidal ECs (LSECs) were grown to confluence in serum‐containing media. For protein quantification assays, ECs were washed and exposed to serum‐free media for 24 hours. Supernatants were collected and lysates were made (following additional washing) for analysis using a commercial ELISA kit for FV. For F1+2 measurements (also using a commercial ELISA kit) 5 mM CaCl2 was added to the serum‐free media. Fluorescent microscopy, combined with antibodies directed against human FII/thrombin and CD141 plus appropriate fluorescently‐tagged secondary antibodies, was used to detect these proteins on ECs grown on glass coverslips. Image analysis was utilized to determine if FII/thrombin detection overlapped with the fluorescent detection of CD141 on cell surfaces.
Results: FV protein is produced by HUVECs, GMVECs, and LSECs; and detected in the supernatant and lysates of each EC type (Figure 1a,b). Similar levels of FV were detected in the three EC supernatants (n = 3 for each cell type), although HUVEC lysates (n = 2) contained more FV than GMVECs (n = 3) and LSECs (n = 3, p < 0.0001), and LSECs contained more FV than GMVECs (p = 0.03). Fluorescent image analysis demonstrated colocalization of prothrombin (FII)/thrombin with CD141 on HUVEC surfaces (Figure 2). The prothrombin cleavage product, F1+2, was not detected in EC supernatant, although it was measured in EC lysates (Figure 1c). This may indicate surface binding of F1+2 following prothrombin cleavage. GMVECs produced more F1+2 than HUVECs (p = 0.03) and LSECs.

Figure 1. Production of factor V and prothrombin fragment 1+2 by human endothelial cells

Figure 2. Fluorescent detection of surface prothrombin plus thrombomodulin on HUVECs
Conclusions: In addition to producing the coagulation proteins necessary for FX activation, human ECs also produce FV and prothrombin. We provide evidence of active prothrombinase formation with cleavage of F1+2 fragments from prothrombin cleavage detected in EC lysates. Our data provide additional evidence that human ECs may have important roles in promoting hemostasis, in addition to their anticoagulant properties.
21. HTRS.O3.1
22. Investigating the regulation of Rap1‐integrin signaling pathway using the imMKCL megakaryocyte model
A. Dhenge 1; W. Bergmeier1; K. Eto2
1 University of North Carolina, Chapel Hill, North Carolina, USA; 2 University of Kyoto, Kyoto, Japan
Background: Blood platelets are critical for the prevention of bleeding following vascular injury. Key to their function is the activation of cell surface integrin receptors, a process that is regulated by a powerful yet tightly controlled intracellular signaling machinery. Following cellular stimulation and second messenger generation, the small GTPase Rap1 plays a central role in the assembly of the integrin activation complex. Rap1 itself cycles between an inactive, GDP‐bound and an active, GTP bound state. Rap1 activation and inactivation are mediated by CalDAG‐GEFI, a calcium‐sensitive guanine nucleotide exchange factor (GEF), and Rasa3, a GTPase activating protein (GAP), respectively. Once activated, Rap1 interacts with talin to mediate integrin activation. While the components of the Rap1‐integrin signaling module are well defined in platelets, these cells are ill‐suited for detailed studies on the spatiotemporal regulation of the pathway.
Objectives: To utilize immortalized megakaryocytic cells (imMKCLs) as a surrogate model for studying the spatiotemporal regulation of platelet Rap1‐integrin signaling.
Methods: Protein expression of CalDAG‐GEFI, Rasa3, Rap1 and talin was assessed in imMKCLs by Western blot. Integrin activation was monitored by flow cytometry (PAC1 binding) and confocal microscopy (fibrinogen binding). Lentiviral vectors are used to express wild‐type and mutant forms of CalDAG‐GEFI and Rasa3 in imMKCLs.
Results: Expression of the components of the Rap1‐talin signaling module was determined in imMKCLs during a 7‐day differentiation and in platelets derived from these cells. The expression of Rasa3 and talin did not change during imMKCL differentiation. In contrast, Rap1 expression increased over time and was highest in d7 imMKCLs. Expression of CalDAG‐GEFI was very low in d1–6 imMKCLs and increased strongly in d7 imMKCLs and imMKCL‐derived platelets. PAC‐1 binding was determined after agonist stimulation at different stages of MK differentiation. A subset of d5 imMKCLs showed robust PAC1 binding in response to protease‐activated receptor (PAR) or glycoprotein (GP)VI stimulation. Single cell confocal microscopy studies demonstrated that fibrinogen binding in activated imMKCLs is observed in cells that mount a strong second messenger (calcium flux) response. However, the kinetics of calcium flux did not correlate well with the kinetics of fibrinogen binding in stimulated d5 imMKCLs, consistent with the weak expression of CalDAG‐GEFI in these cells. In ongoing work, we are performing structure‐function studies on CalDAG‐GEFI and Rasa3 using the confocal imaging and fibrinogen binding as a readout for Rap1‐integrin signaling in these cells.
Conclusions: Our preliminary studies suggest that imMKCL cells are a useful model to investigate platelet Rap1‐talin‐integrin signaling. In ongoing work, we will express Fluorophore‐tagged proteins in imMKCLs to obtain information on the spatio‐temporal regulation of this pathway.
23. HTRS.P1.7
24. Efficacy and safety of damoctocog alfa pegol is sustained for up to ≥6 years of observation in patients aged 12–<18 years at enrollment into protect VIII
J. Ducore 1; M. Mancuso2; M. Simpson3; P. Holme4; M. Enriquez5; M. Wang6; M. Reding7
1 University of California Davis Medical Center, Sacramento, California, USA; 2 Center for Thrombosis and Haemorrhagic Disease, Humanitas Clinical and Research Center – IRCCS, Milan, Lombardia, Italy; 3 Rush University Medical Center, Chicago, Illinois, USA; 4 Department of Haematology, Oslo University Hospital and Institute of Clinical Medicine, University of Oslo, Oslo, Norway; 5 Bayer, Wuppertal, Nordrhein‐Westfalen, Germany; 6 Bayer Corporation, Whippany, New Jersey, USA; 7 Center for Bleeding and Clotting Disorders, University of Minnesota Medical Center, Minneapolis, Minnesota, USA
Background: Efficacy and safety of damoctocog alfa pegol as prophylactic and on‐demand therapy in previously treated patients with severe hemophilia A were assessed in adults/adolescents aged 12–65 years (yrs) in the phase 3 PROTECT VIII trial (NCT01580293).
Objectives: This post hoc analysis reports efficacy and safety data from the PROTECT VIII main and extension studies in a cohort of patients aged 12– < 18 yrs at enrollment.
Methods: Patients who completed the PROTECT VIII main study could enroll into the open‐label PROTECT VIII extension study. Patients receiving on‐demand treatment or prophylaxis (PPX, 30‒40 IU/kg twice weekly [2×W], 45‒60 IU/kg every 5 days [E5D], or 60 IU/kg every 7 days [E7D]) with damoctocog alfa pegol in the main study could continue their regimen or switch to any PROTECT VIII PPX regimen at any time during the extension. PPX patients who switched regimen after the first 7 days in the extension were assessed in a variable frequency group (VAR). All patients were assessed for annualized bleeding rate (ABR), joint ABR and safety outcomes. Bleeds and damoctocog alfa pegol treatments were reported in an electronic patient diary.
Results: At enrollment, 12 patients were aged 12 to less than 18 yrs and are included in this analysis; all but one were previously treated with PPX. Ten out of 12 patients had at least one investigator‐reported target joint at baseline: 5 patients had target joints in ankles, 4 in elbows and 2 in knees; 3 patients had >1 target joint. Median (range) age was 13.5 (12–17) yrs at enrollment and approximately 18 (15–23) yrs at the end of the extension. All patients completed the 36‐week main study and enrolled into the extension; total time in the full study ranged 1.28–6.17 yrs. During extension, patients were treated with damoctocog alfa pegol PPX (2×W, n = 3; E5D, n = 3; E7D, n = 2; VAR, n = 4); median (Q1; Q3) annual dose was 3885 (3243–4200) IU/kg and median (range) number of infusions/yr was 81 (54–108). Total and joint ABRs for each time period are shown in Figure 1. During the last 12 months of the extension, all target joints present at pre‐study (as reported by the investigator) or that developed during the study (3 bleeds into the same joint in 6 months), were resolved (≤2 spontaneous bleeds/yr). Drug‐related treatment‐emergent adverse events (TEAEs) were reported in 1 patient (osteoarthritis, moderate intensity). While 4 patients were reported to have had serious AEs at the end of PROTECT VIII extension (Table 1), none were study drug‐related. No patients developed FVIII inhibitors during the entire study (titer ≥0.6 Bethesda units).

Figure 1. Total and joint ABRs during PROTECT VIII in patients 12–<18 years of age at enrollment
Table 1. TEAEs during PROTECT VIII main study and extension in patients 12–<18 years of age at enrollment

Conclusions: Efficacy of damoctocog alfa pegol was maintained for up to ≥6 yrs of observation in patients who were aged 12–<18 yrs at the start of the main study. Median ABR remained low in the extension study (<2.0), all target joints present at baseline resolved and there were no discontinuations related to AEs. Thus, damoctocog alfa pegol provided continued improvement in bleed protection in this young, active population.
25. HTRS.O2.3
26. Bleeding diathesis in mice lacking JAK2 in platelets
N. Eaton 1; S. Subramaniam2; M. Schulte3; C. Drew3; D. Jakab3; S. Haberichter4; H. Weiler5; H. Falet1
1 Versiti Blood Research Institute, Milwaukee, Wisconsin; Department of Cell Biology, Neurobiology, and Anatomy, Medical College of Wisconsin, Milwaukee, Wisconsin, USA; 2 Boston University School of Medicine, Boston, Massachusetts, USA; 3 Versiti Blood Research Institute, Milwaukee, Wisconsin, USA; 4 Versiti Blood Research Institute, Milwaukee, WI; Department of Pediatrics, Medical College of Wisconsin, Milwaukee, Wisconsin; Children's Research Institute, Children's Wisconsin, Milwaukee, Wisconsin, USA; 5 Versiti Blood Research Institute, Milwaukee, Wisconsin; Department of Physiology, Medical College of Wisconsin, Milwaukee, WI, Milwaukee, Wisconsin, USA
Background: The tyrosine kinase JAK2 is a critical component of intracellular JAK/STAT cytokine signaling cascades that is prevalent in hematopoietic cells such as hematopoietic stem cells and megakaryocytes (MKs). Individuals expressing somatic JAK2 mutations such as JAK2V617F commonly develop myeloproliferative neoplasms (MPNs) associated with venous and arterial thrombosis, a leading cause of mortality.
Objectives: The role of JAK2 in hemostasis remains unclear. Here, we investigated the role of JAK2 in platelet hemostatic function using Jak2fl/fl Pf4‐Cre (Jak2Plt–/–) mice lacking JAK2 in platelets and megakaryocytes (MKs).
Methods: The hemostatic capacity of Jak2Plt–/– mice was assessed using a combination of in vivo occlusion and thrombosis models. Jak2Plt–/– platelet parameters were determined similarly and through a number of in vitro assays including adhesion under flow, static spreading, functional activation by flow cytometry, aggregation, and analysis of intracellular platelet ITAM‐based GPVI signaling.
Results: Jak2Plt–/– mice developed MK hyperplasia and splenomegaly associated with severe thrombocytosis and bleeding. This notion is supported by failure to occlude in a ferric chloride carotid artery injury model and by a cremaster muscle laser induced injury assay, where Jak2Plt–/– platelets failed to form stable thrombi. Jak2Plt–/– platelets adhered poorly to type I collagen under arterial shear rates. Jak2Plt–/– platelets also spread poorly on collagen under static conditions or on fibrinogen in response to the GPVI‐specific agonist, collagen‐related peptide (CRP). Following activation with CRP, Jak2Plt–/– platelets displayed decreased α‐granule secretion, integrin αIIbβ3 activation, and aggregation, but showed normal responses to thrombin. Jak2Plt–/– platelets had impaired intracellular signaling when activated via GPVI as assessed by phosphotyrosine expression, which was associated with abrogated phosphorylation and activation of PLC‐γ2.
Conclusions: Taken together, the results show that JAK2 deletion impairs GPVI signaling and platelet hemostatic function in mice and suggest that aberrant JAK2 signaling in patients with MPNs affects GPVI signaling leading to hemostatic platelet function.
27. HTRS.P1.14
28. Surgical procedures in patients with hemophilia: Multistakeholder perspectives from 2018 roundtables
M. Escobar 1; D. Quon2; M. Wang3; B. Warren3; A. Wufsus4; V. Ostrow4
1 University of Texas Health Science Center at Houston, Houston, Texas, USA; 2 Orthopaedic Hemophilia Treatment Center, Los Angeles, California, USA; 3 University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA; 4 Novo Nordisk Inc, Plainsboro, New Jersey, USA
Background: Owing to progress in the management of coagulation disorders over the past few decades, patients with hemophilia now have a life expectancy similar to that of the general population. As the hemophilia population is aging, a growing need exists for routine health screening and maintenance along with general surgeries or invasive procedures to help patients with comorbidities related to their age and bleeding disorder. Surgery in patients with hemophilia remains challenging and requires an experienced multidisciplinary team. The lack of procedure‐specific guidelines, especially in the context of relatively uncharted new therapies, contribute to the challenges faced when performing surgery in patients with hemophilia.
Objectives: Establishing a better understanding of the surgical process and current unmet needs may provide better awareness and support for persons with hemophilia.
Methods: Through a combination of virtual and live sessions, health care providers (HCPs), including nurses, physicians, pharmacists, physical therapists, and social workers, shared insights on the surgical process, educational needs, and future of surgery in patients with hemophilia. Transcripts from these meetings were analyzed to compile qualitative data sets.
Results: Overall, 61 HCPs from 15 states across the United States participated in 1 of 6 virtual or 3 live roundtable discussions. Most of these HCPs were associated with hemophilia treatment center–affiliated hospitals. Each center averaged 2 surgical procedures per month, most frequently dental procedures and mediport placements or removals. Most HCPs reported having a preferred referral network for surgical procedures; however, patients often elected to have surgery outside of the referral network owing to insurance or personal preference. The largest gaps identified in the surgical process included issues with rehabilitation, communication, and insurance networks (Table 1). HCPs reported that insurance providers often dictated where physical therapy occurred as well as the duration of physical therapy. HCPs also reported on the educational needs surrounding surgical procedures in patients with hemophilia. The general consensus was that a diverse group of non‐hemophilia treatment center HCPs could benefit from additional education about hemophilia and other rare bleeding disorders. This group included emergency department staff, inpatient and surgical nursing staff, obestetricians/gynecologists, pharmacists, emergency medical technicians, first responders, and intensive care unit staff.
Table 1. Largest HCP‐Identified Gaps in the Surgical Process

Conclusions: Many gaps and unmet needs remain in the surgical process for patients with hemophilia. Focusing on the gaps in communication, planning, rehabilitation, and insurance networks, and providing additional education on hemophilia to HCPs involved in the surgical process could improve the surgical experience and lead to better outcomes for patients with hemophilia.
29. HTRS.P2.15
30. The safety of eptacog beta in hemophilia patients with inhibitors in adult, pediatric, and surgical settings
M. Escobar 1; M. Callaghan2; J. Ducore3; C. Hermans4; J. Journeycake5; C. Leissinger6; J. Luck7; J. Mahlangu8; D. Quon7; M. Recht9; J. Schved10; A. Shapiro11; R. Sidonio12; M. Wang13; G. Young14; W. Alexander15; A. Al‐Sabbagh16; D. Bonzo16; T. Wilkinson17; C. Kessler18
1 University of Texas Health Science Center at Houston, Houston, Texas, USA; 2 Central Michigan University, Detroit, Michigan, USA; 3 University of California Davis Medical Center, Sacramento, California, USA; 4 Cliniques Saint‐Luc, Université Catholique de Louvain, Brussels Hoofdstedelijk Gewest, Belgium; 5 Oklahoma Center for Bleeding and Clotting Disorders, Oklahoma City, Oklahoma, USA; 6 Section of Hematology/Oncology, Tulane University School of Medicine, New Orleans, Louisiana, USA; 7 Orthopaedic Hemophilia Treatment Center, Los Angeles, California, USA; 8 Hemophilia Comprehensive Care Center, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, Gauteng, South Africa; 9 American Thrombosis & Hemostasis Network, Portland, Oregon, USA; 10 Haemophilia Treatment Centre, University Hospital Montpellier, Montpellier, Languedoc‐Roussillon, France; 11 Indiana Hemophilia & Thrombosis Center, Indianapolis, Indiana, USA; 12 Aflac Cancer and Blood Disorders Center, Emory University, Atlanta, Georgia, USA; 13 University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA; 14 Children’s Hospital Los Angeles, University of Southern California Keck School of Medicine, Los Angeles, California, USA; 15 Aoede Associates, Louisville, Kentucky, USA; 16 LFB USA, Framingham, Massachusetts, USA; 17 GLOVAL LLC, Broomfield, Colorado, USA; 18 Lombardi Comprehensive Cancer Center, Georgetown University Medical Center, Washington, District of Columbia, USA
Background: Clinical management of individuals with hemophilia is complicated by inhibitor development; bleed treatment in these patients frequently relies on the use of bypassing agents (BPAs). Eptacog beta* (EB; HEMA Biologics) is a recombinant activated human factor VII variant that provides effective treatment of bleeding in hemophilia A or B patients with inhibitors, and was approved for use in adults and adolescents (age ≥ 12) by the FDA in 2020.
Objectives: The study objective is to assess EB safety, immunogenicity, and thrombotic potential during bleed treatment and perioperative care in pediatric and adult hemophilia A or B patients with inhibitors who were enrolled in three prospective Phase 3 clinical trials (PERSEPT 1, PERSEPT 2, and PERSEPT 3).
Methods: Using a randomized crossover design, 27 subjects in PERSEPT 1 (ages 12–54) and 25 subjects in PERSEPT 2 (ages 1–11) treated bleeding episodes with an initial dose of 75 or 225 µg/kg EB followed by dosing of 75 µg/kg at prespecified intervals as determined by clinical response. Twelve PERSEPT 3 subjects (ages 2–56) received an initial pre‐operative infusion of 75 µg/kg (for minor procedures) or 200 µg/kg EB (for major surgeries) with subsequent 75 µg/kg doses of EB given intraoperatively and postoperatively as indicated. Two subjects from PERSEPT 1 and two subjects from PERSEPT 2 also participated in PERSEPT 3. Histories of thrombosis, thromboembolism, or risk factors for thrombosis were exclusionary criteria. All subjects (or their parents or legal guardians if <18 years of age) gave informed consent. Descriptive statistics were used for data analyses.
Results: Over the three trials, 60 hemophilia A or B subjects with inhibitors received 3,388 doses of EB during 1,087 exposure episodes (bleeding episodes, invasive procedures, postoperative treatments, and EB pharmacokinetic assessments). EB was well‐tolerated, and no allergic, hypersensitivity, anaphylactic, or thrombotic events occurred. No neutralizing anti‐EB antibodies were detected. Of 133 adverse events (AEs) that occurred following initial EB exposure, 10 were considered treatment‐related and 7 were serious (SAEs; Table 1). Following a major surgery that proceeded without complications, one PERSEPT 3 subject was discontinued from the trial due to post‐procedural hematoma. This patient succumbed from blood loss anemia approximately 3 days after study withdrawal; the death was found to be unrelated to EB treatment by the independent PERSEPT 3 data monitoring committee (DMC). No SAE was deemed treatment‐related by the DMC.
Table 1. The treatment‐related AEs and SAEs reported in PERSEPT 1, PERSEPT 2, and PERSEPT 3 clinical trials. The total number of treatment‐related AEs from all 3 trials is highlighted in bold

Conclusions: The results from three Phase 3 trials establish a favorable EB safety profile for treatment of bleeding and perioperative management in hemophilia patients with inhibitors. No allergic, hypersensitivity, anaphylactic, or thrombotic events were observed, and no neutralizing anti‐EB antibodies were found. EB represents a new, well‐tolerated BPA for treating hemophilia A or B patients with inhibitors.
Other: *SEVENFACT® [coagulation factor VIIa (recombinant)‐jncw].
31. HTRS.O1.3
32. Broad patient eligibility and long‐term tolerability in the first‐in‐human gene therapy study of AAVhu37.hFVIIIco in severe hemophilia A
F. Ferrante 1; S. Wiegmann2; C. Lange3; M. Braun3; L. Michaels4
1 Bayer, Basel‐Stadt, Switzerland; 2 Bayer, Wuppertal, Nordrhein‐Westfalen, Germany; 3 Bayer, Berlin, Germany; 4 Bayer, Whippany, New Jersey, USA
Background: AAVhu37.hFVIIIco comprises an adeno‐associated virus (AAV) vector with capsid serotype hu37 (AAVhu37), and a genome that directs expression of a codon‐optimized B‐domain‐deleted human factor VIII (hFVIIIco) under the control of a liver‐specific promoter/enhancer combination. Preclinical studies of AAVhu37 demonstrated efficient liver‐directed FVIII gene transfer and durable FVIII expression, however pre‐existing humoral immunity against AAV capsids may limit patient eligibility.
Objectives: Here, we assess the seroprevalence of pre‐existing neutralizing antibodies (NAbs) and anti‐drug antibodies (ADA) against AAVhu37 in humans. Updated long‐term safety data following a single dose of the gene therapy, AAVhu37.hFVIIIco, in a first‐in‐human, Phase 1/2 dose‐finding study (NCT03588299) are also reported.
Methods: Seroprevalence of NAbs and ADAs against AAVhu37 were assessed in serum derived from 100 US patients with hemophilia A. In addition, NAbs against AAV5 and AAV8 were determined. The Phase 1/2, open‐label, dose‐finding study included male patients aged ≥18 years with severe hemophilia A, receiving a single intravenous infusion of AAVhu37.hFVIIIco. Patients had no history of FVIII inhibitors, no detectable immunity to the AAVhu37 capsid above a titer of 5 and ≥150 exposure days to FVIII products. Primary endpoints were adverse events (AEs), serious AEs (SAEs) and AEs/SAEs of special interest. Secondary endpoint was change in FVIII activity from baseline. Informed patient consent and ethics committee approval were obtained.
Results: In the seroprevalence study, we observed a low seroprevalence of NAbs and ADAs with low maximum titers for NAbs (26) and ADAs (182) against AAVhu37. Based on our results, 86% of patients would be eligible for AAVhu37‐based treatment. To date, patients in Cohorts 1 (0.5 × 1013 GC/kg, n = 2), 2 (1.0 × 1013 GC/kg, n = 2) and 3 (2.0 × 1013 GC/kg, n = 2) have completed up to 21, 16 and 11 months of observation, respectively. Five (83.3%) patients from all three cohorts have shown durable and sustained FVIII levels (all ≥5%). No SAEs have been reported to date. Follow‐up data for up to 24 months will be presented.
Conclusions: AAVhu37.hFVIIIco has broad eligibility in patients with hemophilia A due to low seroprevalence of NAbs and ADA against AAVhu37, compared with other AAVs. AAVhu37.hFVIIIco has a good safety profile, and measurable and sustained expression of endogenous FVIII.
Other: All authors are employees of Bayer.
33. HTRS.O4.4
34. Clearance of fidanacogene elaparvovec vector DNA in patients with severe or moderately severe hemophilia B (HB)
P. Fogarty 1; A. Chhabra2; I. Winburn3; D. Rybin2; W. Byon4; J. Smith4; J. Marshall2; J. Rupon1
1 Pfizer Inc, Collegeville, Pennsylvania, USA; 2 Pfizer Inc, New York, New York, USA; 3 Pfizer Inc, Surrey, England, United Kingdom; 4 Pfizer Inc, Groton, Connecticut, USA
Background: Fidanacogene elaparvovec, a hepatotropic bioengineered AAV‐based vector that delivers a high‐activity factor IX (FIX) transgene, has shown durable FIX expression and low mean annualized bleeding rate/annualized infusion rate up to four years following infusion, representing the longest period of durability data for a FIX Padua‐utilizing gene therapy for HB patients to date.
Objectives: We sought to describe the kinetics of clearance of fidanacogene elaparvovec vector DNA from bodily compartments, following transduction.
Methods: Clearance of viral vector DNA was assessed in fifteen adult HB patients who had been infused with 5E+11 vg/kg of fidanacogene elaparvovec, using quantitative real‐time PCR (qPCR) analysis of peripheral blood mononuclear cells (PBMCs), saliva, urine, semen, and serum. PBMCs, saliva, urine and serum samples were collected at screening or Day 0 prior to vector infusion, and starting from Week 1 post‐vector infusion and continuing at every scheduled visit until 3 consecutive samples were negative (at or below the limit of quantification of the assay) for the given sample type. Semen samples were collected at Screening or Day 0 prior to vector infusion, and on week 1 and every 4 weeks starting from week 4 post‐vector infusion until 3 consecutive samples were negative (i.e., at or below the limit of quantification of the assay). After week 16, semen samples occurred at every scheduled visit (as opposed to every 4 weeks) if 3 consecutive negatives had not yet been obtained. Time to clearance was defined as time to reach the first negative sample of three negative samples per matrix. Descriptive statistics were used to analyze results for time to clearance and peak viral vector shedding.
Results: All subjects achieved clearance of vector DNA in all specimen types by 52 weeks following vector infusion; semen, urine, and saliva were the quickest to be cleared. All patients had cleared semen by week 12, saliva by week 8, and urine by week 7. Serum and PBMCs were the longest to clear, with serum clearing at week 22 and PBMCs at week 52 following infusion. The peak viral vector shedding in each compartment was observed during the first two weeks after infusion. Among samples with detectable viral vector load, the range was 1.63E+4 to 7.13E+5 copies/mL in saliva, 4.55E+3 to 3.61E+5 copies/mL for semen, 8.96E+2 to 3.57E3 copies/mL for urine, 6.55E+4 to 2.91E+6 copies/mL for serum, and 4.8E+3 to 4.03E+5 copies/ug for PBMCs (Table 1). There were no adverse events reported that appeared attributable to systemic vector distribution.
Table 1. Characteristics of Vector DNA Clearance in 15 subjects following transduction with fidanacogene elaparvovec1

Conclusions: Fidanacogene elaparvovec was cleared in all subjects from all compartments with 22 weeks, with the exception of PBMC which demonstrated clearance by 52 weeks. Clearance of vector DNA from bodily compartments will be assessed in a larger group of HB patients in the ongoing fidanacogene elaparvovec phase 3 clinical trial (www.clinicaltrials.gov, NCT03861273).
35. HTRS.P1.9
36. Implementation and analysis of a venous thromboembolism prophylaxis algorithm in hospitalized pediatric patients
N. Gibson, M. Federman; G. Roach
UCLA Mattel Children's Hospital, Los Angeles, California, USA
Background: Venous thromboembolism (VTE) is an under‐recognized and under‐treated hospital problem in the pediatric population. While initial data suggested low rates in children, recent analyses have shown an increase in VTE incidence (Raffini et al, Pediatrics 2009). Current evidence also demonstrates substantial morbidity and mortality in children, ranging from pulmonary embolism to post‐thrombotic syndrome to death (Biss et al, British Journal of Haematology, 2008; Goldenberg et al, Haematologica 2010). Recent pediatric studies have attempted to quantify risk factors in children to provide a framework for prophylaxis guidelines (Branchford et al, Haematologica 2012).
Objectives: This quality improvement study aims to implement a risk stratification algorithm for preventing VTE in pediatric patients and ensure appropriate VTE risk stratification and prophylaxis on hospital admission.
Methods: Based on analysis of risk factors and extrapolation of adult empiric data, an algorithm was developed to risk stratify children admitted to our institution. A “hard stop” was inserted into admission ordersets requiring risk stratification. We performed a retrospective chart review of all pediatric patients >10 years old admitted to UCLA hospitals between December 2017 and June 2018, determined whether the risk stratification was done, and compared true risk category with stratification at admission. Based on initial findings, the orderset was updated to clarify the stratification process. The pediatric history and physical template was updated to require patient ambulatory status and risk factors. A second chart review was conducted for admissions between October 2019 and March 2020.
Results: In the initial review, 552 charts were examined. 97% were risk stratified on admission and 86% were stratified correctly. While 98% of low risk patients were correctly stratified, only 41% of moderate risk patients and 25% of high risk patients were stratified correctly. 67% of incorrectly stratified moderate and high risk patients were non‐ambulatory at baseline. After making the above changes, 449 additional charts were examined. 95% were risk stratified and 86% were stratified correctly. 59% of moderate risk patients and 60% of high risk patients were stratified correctly. 41% of incorrectly stratified moderate and high risk patients were non‐ambulatory at baseline.
Conclusions: Inserting the “hard stop” into the admission orderset was effective in leading to risk stratification. The vast majority of children were stratified as low risk. Initially, while 86% overall were stratified correctly, the majority of moderate and high risk patients were not, and did not receive appropriate VTE prophylaxis. Changes made in response led to an increase in correctly stratified moderate and high risk patients, particularly in patients who were non‐ambulatory at baseline. Ongoing incorrect stratification in the moderate and high risk groups requires additional attention and will be addressed with further plan‐do‐study‐act cycles.
37. HTRS.P2.7
38. How to reduce hypercoagulability testing in the inpatient setting? A quality improvement initiative
J. Hall; C. Shaver; S. Henderson
Baylor Scott & White, Temple, Texas, USA
Background: Inpatient testing for thrombophilia should not routinely be done. It was recognized that many providers in our teaching hospital in Central Texas regularly ordered the hypercoagulability panel for patients that were hospitalized.
Objectives: The aim of this quality improvement initiative was to decrease the inpatient ordering of our hypercoagulability panel for suspected thrombophilias.
Methods: The number of hypercoagulability panels were counted pre and post intervention with data represented through use of a control chart. Target audiences were the residency programs, hospitalist division, and the hematology/oncology fellowship program. Several educational avenues were utilized to disseminate information regarding inpatient thrombophilia testing including lectures and distribution of flyers that began March 2018. The number of hypercoagulability panels were tracked pre and post intervention monthly.
Results: Hypercoagulability panel ordering initially decreased after the intervention and there was a downward trend for the remainder of 2018 with three data points lower than the pre‐intervention average and was outside the control band suggestive that there was a significant difference in the number of panels ordered. However, starting January 2019 ordering of the hypercoagulability panel increased with four data points higher than the pre‐intervention average.
Conclusions: There are challenges for sustaining a response after a quality improvement initiative as evidenced by this study. The authors are proposing that ordering of the hypercoagulability panel be limited to hematologists to reduce the number of inpatient ordering.
39. HTRS.P1.2
40. Bleeding disorder referrals to hematology clinic: A single institution experience
Z. Hudson; O. Olaiya; E. Bilynsky; H. Yeh; S. Carpenter
Children's Mercy Hospital, Kansas City, Missouri, USA
Background: Our tertiary care pediatric hematology/oncology service receives hundreds of referrals yearly for bleeding disorder evaluation. This evaluation can be challenging due to the wide variability of symptoms and the need for accurate interpretation of lab results. In 2014, Bhasin et al showed that 4% of patients referred to hematology based on a preoperative coagulation evaluation had a clinically relevant bleeding disorder. Currently there is little literature about the referral patterns to pediatric hematology and the outcomes of these referrals.
Objectives: 1. To characterize hematology referrals for bleeding disorder work up 2. To describe the diagnostic outcomes from these referrals 3. To estimate the proportion of bleeding disorders diagnosed from these referrals 4. To identify referral factors that are associated with being diagnosed with a bleeding disorder
Methods: This is a single center, retrospective chart review. Patients referred and or seen for a bleeding disorder evaluation at Children’s Mercy Hospital from 07/1/2018 until 06/30/19 were evaluated for demographics, reason for consultation, referring provider, and referral outcome. Akaike Information Criterion (AIC) was applied to logistic regression to identify factors associated with diagnosis of bleeding disorder.
Results: A total of 373 patients were included with demographics detailed in Table 1. Forty patients were diagnosed with a bleeding disorder, 78 patients were lost to follow up or had not completed the work up, and 255 patients had a bleeding disorder ruled out. Of our sample, 6% (21/373) were diagnosed with von Willebrand disease, 4% (14/373) with a platelet function disorder, and 1.3% (5/373) with a coagulation factor deficiency. Forty percent of referrals were for preoperative clearance, 36% for family history, and 57% for symptoms. Forty four percent (164/373) of the referrals were from Otolaryngology, 30% from primary care, and 27% from other specialties. The odds of a bleeding disorder diagnosis decreased by 8% for every year increment in age and was 3 times higher among patients having abnormal coagulation labs at the time of referral as compared to their counterpart. Table 2 details the logistic regression model showing referral factors that could be associated with bleeding disorder diagnosis.
Table 1. Demographics

Table 2. Logistic regression. Factors associated with bleeding disorder diagnosis.

Conclusions: This study characterizes the bleeding disorder referral patterns at our institution including the proportion of bleeding disorders diagnosed. This study highlighted certain referral factors such as age, gender, referral for preoperative clearance, previous treatment with nasal cauterization and the presence of abnormal lab values that could be predictive of the presence of a bleeding disorder. Limitations of this study are the small number of patients with confirmed diagnosis and the study being conducted at a single center. We illustrate that large‐scale studies are needed to determine referral factors associated with the diagnosis of a bleeding disorder.
41. HTRS.O2.1
42. Platelets factor 4 (PF4) binding enhances in vitro neutrophil extracellular traps (NET) capture of SARs‐CoV‐2: Therapeutic implications
M. Ishizuka; M. Kowalska; A. Sarkar; L. Rauova; M. Poncz; K. Gollomp
Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, USA
Background: SARs‐CoV‐2 infection recruits high numbers of neutrophils that extrude neutrophil extracellular traps (NETs), webs of extracellular DNA coated with citrullinated histones (cit‐His) and antimicrobial proteins. NETs have also been shown to entrap virions, concentrate antiviral proteins, and inactivate viruses. However, when NETs are degraded, they release NET degradation products (NDPs) such as cit‐His, cell‐free (cf) DNA, myeloperoxidase (MPO) and neutrophil elastase (NE) that can be toxic to the host. Our group and others have found that NETs and NDPs are highly prominent in patients with severe COVID‐19 and are associated with the development of respiratory failure (Figure 1). Platelet factor 4 (PF4) is a highly‐positively charged, platelet‐specific chemokine that aggregates polyanionic molecules like heparin and DNA. We have shown that PF4 binds to NETs, reducing the release of NDPs by preventing NET digestion by circulating nucleases. Importantly, PF4‐NET complexes markedly enhance gram‐positive and ‐negative bacterial entrapment, likely by bridging the negatively charged polyanionic phosphoribose backbone of the NET DNA scaffold to polyanionic surface molecules in the bacterial cell wall. Treatment with PF4 improved outcomes in lipopolysaccharide endotoxemia and cecal ligation and puncture models of murine sepsis.

Figure 1. Serum NDP levels among children with SARS‐CoV‐2 infection. Graphs showing a comparison of mean ± 1 SD of plasma cfDNA and citrullinated H3 level in control, minimal symptom (n = 4), severe symptom (n = 7) and multi‐systemic inflammatory syndrome in children (MIS‐C) (n = 4). (A) Plasma cfDNA level was elevated in children with severe symptoms. (B) Plasma citrullinated H3 level was elevated in children with severe symptoms and MIS‐C
Objectives: The objective of this study was to investigate whether PF4 binding to NETs is similarly protective in SARs‐CoV‐2 infection by preventing the degradation of NETs and by enhancing NET‐mediated viral capture.
Methods: We generated NET‐lined microfluidic channels. Neutrophils were isolated from healthy human donors, adhered to fibronectin‐coated channels, and incubated with phorbol myristate acetate (PMA) to induce the release of NETs. Channels were then treated with buffer alone or PF4 (100 µg/ml) to compact NETs, after which gamma‐irradiated SARS‐CoV‐2 (1 x 107 PFU) were infused at 2 dynes/cm2 for 1 hour. Viral particles were then labeled with SARS‐CoV‐2 guinea pig antiserum and visualized with a fluorescently‐labeled secondary antibody. Viral binding to NETs was quantified using confocal microscopy.
Results: Similar to that seen with bacterial attachment to NETs, we observed scant viral binding to non‐compact NETs. In contrast, there was abundant binding of SARs‐CoV‐2 aggregates to PF4 compacted NETs (Figure 2).
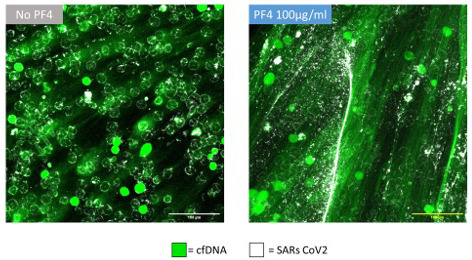
Figure 2. PF4‐NETs mediated virus entrapment. Representative confocal images of NET lined microfluidic channels infused with gamma irradiated SARS‐CoV‐2 (1 x 107 PFU) with or without PF4 (100 µg/ml). The viral particles were labeled with SARS‐CoV‐2 guinea pig antiserum and a secondary antibody
Conclusions: These findings demonstrate that PF4 plays a crucial role in NET‐mediated viral capture and suggest that PF4‐NET complexes may be part of the physiologic mechanism by which viral spread is contained in the host. Moreover, we have previously shown that an Fc‐modified version of KKO, a monoclonal antibody directed against complexes of PF4 and polyanions, markedly enhanced the protective effects of PF4 in vitro and in murine models of sepsis. Therefore, we will examine whether PF4 plus modified KKO infusions are able to limit SARS‐CoV‐2 viremia, preventing the pneumonitis and multi‐system organ dysfunction of severe COVID‐19.
43. HTRS.P1.1
44. A survey of anti‐platelet and anticoagulant use in persons with hemophilia
M. Janbain 1; M. Escobar2; B. Hardesty3; C. Kessler4; D. Nugent5; D. Quon6; M. Reding7; C. Walsh8; S. Sood9
1 Tulane University School of Medicine, New Orleans, Louisiana, USA; 2 University of Texas Health Science Center at Houston, Houston, Texas, USA; 3 Indiana Hemophilia and Thrombosis Center (IHTC), Indianapolis, Indiana, USA; 4 Lombardi Comprehensive Cancer Center, Georgetown University Medical Center, Washington, District of Columbia, USA; 5 UC Irvine School of Medicine, Irvine, California, USA; 6 Orthopaedic Hemophilia Treatment Center, Los Angeles, California, USA; 7 Center for Bleeding and Clotting Disorders, University of Minnesota Medical Center, Minneapolis, Minnesota, USA; 8 The Mount Sinai Hospital, New York, New York, USA; 9 University of Michigan, Ann Arbor, Michigan, USA
Background: With improvement in treatment, persons with hemophilia (PWH) are now living long enough to experience complications of aging, especially cardiovascular disease. For a long time, PWH were felt to have an absolute contraindication to anti‐platelet agents such as aspirin (ASA), and anticoagulants. These medications have proven efficacy in primary and secondary prevention of cardiovascular events in non‐hemophiliacs.
Objectives: There is an unmet need to determine the safety of anti‐platelet and anticoagulant medications in PWH to provide better guidance on their management.
Methods: A survey was distributed among members of the ADVANCE group, comprised of hemophilia experts from different hemophilia treatment centers (HTC) across the US interested in issues of aging with bleeding disorders, to evaluate their approach to managing PWH while on anti‐platelet and anticoagulation agents.
Results: Responses were collected from 7 different HTCs, treating a total of 3503 patients with hemophilia A (HA), B (HB) and carriers. 57 patients older than 45 yo were treated with low dose ASA (81 mg), 2 with dual anti‐platelet therapy, and 2 with anticoagulants (warfarin and rivaroxaban) (1.8% of total patients); see Table 1. Reasons for ASA use included coronary artery disease, valvular disease, peripheral vascular disease and stroke prevention. ASA dosing regimen varied between daily (70%), every other day (26%) and 3 times/week (4%). Target troughs ranged from 5%–30% and patients were started on prophylaxis or prophylaxis regimen was changed (with increased dose or frequency) by the majority of physicians to achieve higher trough. Most of the patients were treated with standard half‐life products. 2 patients with severe HB were switched to extended half‐life factor. ASA was stopped in 7 patients (12%) for GI bleed (4), GU bleed (2), and muscular bleed (1) and not resumed after bleed resolution. Bleeds occurred between a week to month after starting ASA. ASA dosing was spaced out in 4 patients due to mucocutaneous bleeds. Difficulties in achieving target was only reported in 2 patients due to adherence problems.
Table 1. Distribution of patients on antiplatelet or anticoagulation therapy by severity and type of hemophilia

Conclusions: Some observational studies in non‐hemophiliacs treated with low dose ASA suggest around 1–2 major bleeding events (GI and cerebral) / 1000 patients annually. Others suggest 1/210 would experience a serious bleeding event on low dose ASA. Our data shows that PWH on ASA experience increased risk of GI and other bleeding, with 12% of patients stopping ASA due to bleeding events. A majority of clinicians used or altered factor prophylaxis to achieve a higher trough level, albeit in a non‐standardized fashion, to ameliorate the bleeding risk. Limitation of this analysis are the small number and heterogeneity of patients. Our survey results confirm the rarity of antiplatelet and anticoagulation use in this population. Next steps are to expand survey distribution to collect more data in order to optimize the management of antiplatelet and anticoagulation agents in PWH.
45. HTRS.P2.9
46. HTRS Student Research Award: Risk factors for non‐variceal hemorrhage in patients with chronic liver disease
P. Kesavan1; A. Afzal1; L. Suhong1,2; B. Gage1; M. Schoen2; K. Sanfilippo1,2
1 Washington University School of Medicine, St. Louis, Missouri, USA; 2 St. Louis Veterans Health Administration Medical Center, St. Louis, Missouri, USA
Background: Patients with chronic liver disease (CLD) have a wide array of coagulation abnormalities, often presenting with low platelet count and an elevated international normalized ratio (INR). These laboratory abnormalities are often perceived to be risk factors for hemorrhage. However, there is a paucity of evidence to suggest that these abnormalities predict hemorrhage in patients with CLD (Segal JB, et al. Transfusion 2005). This may be due to alterations in hemostatic factors that are not measured by conventional laboratory analyses. As such, the rate of thrombin formation is not significantly different in patients with CLD compared to patients without CLD (Tripodi A, et al. Journal of Hepatology 2005). Therefore, there exists a need to investigate a variety of clinical and laboratory parameters that may predict the risk of hemorrhage among patients with CLD.
Objectives: In this study, we sought to study the association between INR and non‐variceal hemorrhage in patients with CLD. In addition, we sought to identify additional predictors of non‐variceal hemorrhage in CLD.
Methods: This is a large, retrospective cohort study using data from the Veterans Administration (VA) Informatics and Computing Infrastructure (VINCI). The study cohort consisted of VA patients diagnosed with CLD between October 1, 2002 and September 30, 2018. Patients with a history of malignancy, artificial heart valves, atrial fibrillation, and prior venous thromboembolism were excluded. Patients on anticoagulant therapy any time before or after the diagnosis of CLD were also excluded. Candidate predictor variables were selected from existing prediction models including HEMORR2HAGES, HAS‐BLED, Child‐Pugh, and MELD. When possible and appropriate, variables were entered into the model as time‐varying. Risk of non‐variceal hemorrhage within one year of CLD diagnosis was modelled using a competing risk model (Fine JP, et al. Journal of the American Statistical Association 1999).
Results: A total of 14,384 patients with CLD were included in the cohort of which there were 580 bleeding events (Table 1). In multivariate analysis (Table 2), the following predictors were found to be independent risk factors of non‐variceal hemorrhage: elevated INR (HR 1.96; 95% CI 1.60–2.39), increased bilirubin (HR 1.36; 95% CI 1.12–1.64), decreased albumin (HR 2.10; 95% CI 1.65–2.68), chronic kidney disease (HR 2.09; 95% CI 1.56–2.81), prior hemorrhage (HR 1.97; 95% CI 1.23–3.15), anemia (HR 1.68; 95% CI 1.39–2.03), alcohol abuse (HR 1.30; 95% CI 1.08–1.56), hemi‐ or paraplegia (HR 2.38; 95% CI 1.06–5.32) and dementia (HR 3.07; 95% CI 1.39–6.82).
Table 1. Demographic and clinical characteristics comparison for CLD patients diagnosed from 2002 to 2018

Table 2. Multivariate analysis of risk factors for non‐variceal hemorrhage

Conclusions: Our study of over 14,000 veterans shows that severity of liver disease at presentation, concurrent renal failure, alcohol abuse, prior hemorrhage and compromised self‐care by either dementia or paraplegia predict a higher risk of hemorrhage among patients with chronic liver disease.
47. HTRS.O1.1
48. The importance of genetic testing in diagnosing von Willebrand's disease type 2M
R. Ksayer 1; K. Zakharia1; P. Christopherson2; R. Montgomery2; M. Janbain1
1 Tulane University School of Medicine, New Orleans, Louisiana, USA; 2 Versiti Blood Research Institute, Milwaukee, Wisconsin, USA
Background: The diagnosis of von Willebrand disease (vWD) currently requires determining the antigen (Ag) and activity levels of VW protein and multimers. Frequently ordered activity testing include Ristocetin Cofactor activity (Rco), and GPIbM that evaluate VW/platelet interaction;and collagen binding test(CB) that assesses VW/collagen interaction. Most practices rely on including only one activity testing in their initial evaluation (generally Rco). Some experts recommend the upfront use of 2 different activity tests. Genetic evaluation is mainly done after several non‐conclusive tests. Several mutations have been reported with vWD type 2M. Most of them affect A1 domain, with subsequent decreased VW‐dependent platelet adhesion. A missense mutation(Ser1731Thr) in exon 30 affecting A3 domain has been described in several families with an autosomal dominant inheritance. This mutation leads to an abnormal collagen binding with normal platelet interaction. Previously described cases with this mutation had decreased VWF:CB and normal Rco activity testing.
Objectives: Studying the caveats in the laboratory diagnosis of patients with Type 2 M VWD with missense mutation (Ser1731Thr).
Methods: A review of patients with VWD Type 2M carrying the missense mutation(Ser1731Thr) within the Zimmerman cohort.
Results: We report 2 cases of VWD Type 2M. The 1st patient is a 28 years old female, with a history of heavy menstrual bleed and frequent epistaxis. Her family history was positive with a mother and identical twin sister having the same symptoms. She presented for evaluation after her niece was born with a spontaneous intracranial bleed. Her blood work showed vWFAg 67%, vWF:Rco 31% (Rco:Ag ratio 0.46), VWF:CB68% (CB:Ag ratio 1),and GP1bM71%. GP1bM did not confirm the low VWF:Rco level. Platelet aggregation studies were normal and factor VIII 100%. The 2nd patient is a 9 years old male, he had a history of easy bruising and frequent epistaxis. His blood work didn’t show evidence of vWD, with vWFAg 79%, vWF:Rco 75%, (Rco:Ag ratio 0.95), and factor VIII 78%. In both patients, genetic testing showed a missense mutation(Ser1731Thr) in exon 30. Decreased vWF:CB was the mainstay in the diagnosis of patients with vWD type 2M, especially with kits using collagen type I . Our 1st patient had normal vWF:CB(kit used type III collagen),and low Rco ratio with normal GP1bM test. The second patient had normal laboratory evaluation, with strong clinical presentation. Genetic testing was crucial to confirm the diagnosis of vWD type 2M in these patients.
Conclusions: These two cases highlight the importance of upgrading the rank of genetic testing in diagnosing vWD. Available laboratory assays used in the routine evaluation of patients with suspected vWD lack the sensitivity and specificity with high intra and inter laboratory variability.
49. HTRS.P2.18
50. Eighteen‐month interim analysis of efficacy and safety of givosiran, an RNAi therapeutic for acute hepatic porphyria, in the envision open label extension
D. Kuter 1; S. Keel2; C. Parker3; D. Rees4; U. Stoelzel5; P. Ventura6; M. Balwani7; L. Gouya8; A. Simon9; S. Liu9; J. Ko9; S. Rhyee9; S. Silver10
1 Massachusetts General Hospital, Boston, Massachusetts, USA; 2 U of Washington, Seattle, Wisconsin, USA; 3 University of Utah, Salt Lake City, Utah, USA; 4 Department of Clinical Biochemistry, King’s College Hospital, London, England, United Kingdom; 5 Saxony Porphyria Center, Department of Internal Medicine II, Klinikum, Cheminitz, Baden‐Wurttemberg, Germany; 6 University Hospital Policlinico of Modena, Modena, Lombardia, Italy; 7 Icahn School of Medicine, Mt Sinai Medical Center, New York, New York, USA; 8 Centre Français des Porphyries CRMR ‐ Porphyries, Paris, Centre, France; 9 Alnylam, Cambridge, Massachusetts, USA; 10 University of Michigan, Ann Arbor, Michigan, USA
Background: Acute hepatic porphyria (AHP) is a family of rare genetic diseases due to enzyme defects in hepatic heme biosynthesis. Induction of 5‐aminolevulinic acid synthase 1 (ALAS1), the rate‐limiting step in heme biosynthesis, leads to accumulation of heme intermediates, ALA and PBG that may result in neurovisceral attacks. ENVISION (NCT03338816) is an ongoing study, evaluating efficacy and safety of givosiran in symptomatic AHP patients in a 6‐month double blind (DB) period and a 30‐month open label extension (OLE) period.
Objectives: N/A.
Methods: ENVISION is a Phase 3 global, randomized, placebo‐controlled study. Exploratory efficacy outcome measures in the OLE included composite porphyria attacks (i.e. those requiring hospitalization, urgent care, or IV‐hemin at home), ALA/PBG levels and hemin use.
Results: As of January 10, 2020, 93/94 patients entered the OLE (placebo/givosiran = 46; givosiran/givosiran = 47). Mean exposure to givosiran at data cutoff was 12.97 [SD = 3.6] months for placebo/givosiran and 18.86 [3.6] months for givosiran/givosiran, with maximum exposure of 25.1 months. Continued treatment in givosiran/givosiran patients led to a median annualized attack rate (AAR) of 0.58 (range: 0–16.2) through Month 18. Patients in the placebo/givosiran group had an AAR of 1.62 (range: 0–11.8) after receiving givosiran for ≥12 months during the OLE period, compared with 10.65 (range: 0–51.6) whilst receiving placebo during the 6‐month DB period. The average number of attacks per patient following givosiran treatment continued to decline during the OLE period for both groups (Figure 1). Sustained ALA/PBG lowering during the OLE was accompanied by sustained reductions in hemin use, and more than half of the placebo/givosiran patients experienced 0 days of hemin use. The most common related adverse events during givosiran treatment were injection site reactions, nausea and fatigue. Hepatic and renal events were both reported in 17% of patients each during givosiran treatment. No new safety concerns occurred.

Figure 1. Eighteen‐Month Interim Analysis of Efficacy and Safety of Givosiran, an RNAi Therapeutic for Acute Hepatic Porphyria, in the ENVISION Open Label Extension
Conclusions: In the ongoing OLE period of the ENVISION study, patients receiving long‐term treatment with givosiran demonstrated a durable response in clinical efficacy, across a wide range of clinical parameters. Following the initial 6 months of givosiran treatment during the OLE, placebo/givosiran patients had a similar clinical response to that observed in givosiran/givosiran patients in the OLE period The safety profile of givosiran remained acceptable and consistent with that previously observed.
Other: This data is being submitted as an encore due to the relevance of said data in hematology. In the US and in other markets around the globe, benign and other general hematologists are very frequently the primary treating physician specialty who diagnose and manage patients with acute hepatic porphyria (AHP). This long term efficacy and safety data on a new therapeutic for AHP is therefore highly relevant to this congress' attending audience.
51. HTRS.P2.4
52. Development & testing of an asynchronous hemophilia learning module
T. Lucas
Department of Pediatrics, Division of Pediatric Hematology/Oncology, University of California San Francisco, Alameda, California, USA
Background: Hemophilia is a chronic disease; and over the pediatric lifespan of a patient, there are different management and treatment decisions required based on various disease and patient‐specific milestones. Educating trainees about this disease in the pediatric population requires many hours of patient contact so that exposure to those various milestones can be ensured. Alternatively, an interactive asynchonous online module could facilitate exposure to the most common milestones in a patient with severe hemophilia within a limited amount of time.
Objectives: This pilot project aims to produce and test an online module with new content as to its educational efficacy.
Methods: IRB approval was obtained in January 2018 and development of the module was conducted from summer 2018 thru winter 2019 in conjunction with University of California San Francisco's Office of Technology Enhanced Education using the Qualtrics platform. The module was developed based on World Hemophilia Foundation guidelines for diagnosis and treatment of hemophilia and updated to include basic information on use of emicizumab after U.S. Food and Drug Administration and European Medicines Agency approval. Data was collected via the Qualtrics platform on length of time required to complete the module, pre‐test knowledge assessment, and embedded testing within the module as well as post‐use comfort and confidence scales with qualitative comments.
Results: The module was successfully developed and administered to a total of 13 participants. Besides institutional fellows, it was also offered to young faculty and available to advance care practitioner and medical student trainees rotating thru the division. Estimated length of use of the modules was 45 minutes, providing an exposure to a full pediatric hemophilia lifespan within this limited amount of time. There was a significant increase in confidence in the topic, comfort in ordering and interpreting diagnostic testing, and discussing prophylactic therapies with those with hemophilia. There were positive ratings regarding the efficiency of using the module and how this module would inform their medical practice.
Conclusions: This pilot project demonstrated the feasibility of development of a module on an online platform, and has been shown to be easy to use, time‐limited, and increases the accuracy of medical management decisions. It can be used as a shared curricular element with various pediatric hematology/oncology programs and hemophilia treatment centers for training regarding the basics of diagnosis, treatment, and complications of pediatric hemophilia care.
53. HTRS.O3.3
54. Hematologic abnormalities and venous thromboprophylaxis in MIS‐C: Data from a multi‐disciplinary working group at a pediatric tertiary care hospital
L. Malec 1; R. Punzalan2
1 Versiti Blood Research Institute, Wauwatosa, Wisconsin, USA; 2 Versiti Blood Center of Wisconsin, Wauwatosa, Wisconsin, USA
Background: On May 15, 2020 the World Health Organization published the preliminary case definition of multisystem inflammatory syndrome (MIS‐C) in children and adolescents, a new clinical entity in the evolving COVID‐19 pandemic. COVID‐19 associated coagulopathy, with prominent elevations in d‐dimers was widely reported in adults; in contrast to the sepsis‐related disseminated intravascular coagulation (DIC), adult patients demonstrated hypercoagulability, not bleeding, with approximately 25% having overt venous thromboembolism (VTE)., There are limited data regarding coagulopathy and hypercoagulability in pediatric patients with acute COVID‐19 or MIS‐C to guide treating physicians in optimal management including VTE prevention strategies. We report the hematologic findings, VTE prophylaxis strategy, and thrombotic outcomes in a cohort of patients at a single pediatric center.
Objectives: NA.
Methods: At Children's Wisconsin we rapidly convened a multi‐disciplinary MIS‐C working group to create guidelines for evaluation and treatment of suspected/confirmed MIS‐C patients. A MIS‐C panel, including CBC, CMP, DIC panel, ESR, CRP , ferritin, troponin I, pro BNP, and COVID PCR was obtained for all patients presenting to the ER with fever for ≥4 days plus any of the following: GI symptoms, rash, bilateral non‐purulent conjunctivitis, cough, headache , and/or irritability. Specialty‐specific treatment guidelines, including modifications of an existing VTE prophylaxis guideline, were established (Table 1). As a quality improvement measure, we performed electronic query of the use of the MIS‐C panel including evaluating laboratory trends, and outcomes of cases receiving specific therapeutics, including anticoagulation. We present data regarding hematologic findings and outcomes of VTE prophylaxis in this cohort.
Table 1. Institutional algorithm for venous thromboembolism risk assessment. Patients meeting criteria for proplylactic anticoagulation would receive enoxaparin 0.5 mg twice daily unless contraindicated due to bleeding

Results: As of October 20, 2020, a total of 56 patients had an MIS‐C panel obtained in the ER, of whom 12 (21.4%) met full criteria for MIS‐C (Table 2). Of these, mild CBC abnormalities predominated as did mild coagulopathy, elevated fibrinogen levels, and d‐dimers (on average 5.5 × upper limit of normal) (Table 2). Three (25%) patients met criteria for VTE prophylaxis with enoxaparin which was continued during hospitalization in 1 patient, and for 2 weeks following hospital discharge in the remaining 2. There were no reported thromboses in any patients, including those who did not meet criteria for thromboprophylaxis. Enoxaparin was well tolerated, and no patients had bleeding events despite having mild coagulopathy.
Table 2. Demographic, laboratory and VTE prophylaxis data amongst 12 patients with MIS‐C admitted to the inpatient setting. Abnormal laboratory findings are listedin bold. CBC=complete blood count; WBC=white blood count; ALC=absolute lymphocyte count; Hb=hemoglobin; PT=prothrombin time; PTT=partial thromboplastin time; VTE=venous thromboembolism; s=seconds. * denotes Hb was normal for age

Conclusions: We report an approach to evaluation of MIS‐C, the hematologic abnormalities, and successful use of a VTE prophylaxis strategy in children. Given the drastic increase in COVID‐19 cases in Wisconsin and the US as of October 2020, and limited large‐scale pediatric studies to guide patient care, we recognize the importance of our own institutional surveillance and the need for collaboration amongst pediatric providers to gather data on outcomes of such patients.
55. HTRS.P1.17
56. Venous thromboembolic disease in children associated with COVID‐19
S. Narang; K. Morparia; P. Sharma
Newark Beth Israel Medical Center, Newark, New Jersey, USA
Background: Coronavirus disease 2019 (COVID‐19) is associated with venous thromboembolism (VTE) in adults and there are guidelines for prevention and management in adults but in children, no evidence based standard guidelines exist for prevention or management of VTE associated with COVID‐19 as little is known about VTE in children in association with COVID‐19. Antiphospholipid antibodies (lupus anticoagulant antibody, cardiolipin antibodies and beta‐2 glycoprotein 1 antibodies) in conjunction with COVID‐19 in adult patients with VTE have been noted.
Objectives: N/A
Methods: We identified 5 cases of VTE in children from April 10th 2020 till May 29th 2020. Each of those patient was tested for SARS‐CoV‐2 RNA and/or SARS‐CoV‐2 IgG and IgM. Diagnosis of VTE was made either using venous doppler or magnetic resonance (MR) imaging. Thrombophilia work up was sent on each patient, including testing for antiphospholipid antibodies.
Results: Five cases of VTE had various types of clots including pulmonary embolism (PE) and upper or lower extremity DVT (Table 1) and all had one or more antiphospholipid antibody positivity. One case was positive for SARS‐CoV‐2 RNA, two had SARS‐CoV‐2 IgM positivity and two had neither RNA nor antibody positivity. Two females had DVT with PE, and two adolescent females were diagnosed with new onset of systemic lupus erythematosus (SLE) during the time of VTE diagnosis. All patients were treated with systemic anticoagulation with heparin.
Table 1. Demographics, Type of VTE and Labs

Conclusions: Venous thromboembolic disease in children associated with anti‐phospholipid antibody is rare but we noted five cases of VTE in association with antiphospholipid antibodies in short period during pandemic, of which one case was associated with SARS‐CoV‐2 RNA and two cases suggestive of recent infection with SARS‐CoV‐2 RNA as IgM was positive. We conclude that our case series suggest VTE in children, in association with SARS‐CoV‐2, is mediated through generation of antiphospholipid antibodies. Further prospective studies can provide clarity on this supposition.
57. HTRS.P1.3
58. Catastrophic thrombotic syndrome in systemic lupus erythematosus without antiphospholipid antibodies
K. Park; N. Mirkheshti; M. Baer; H. Latif
University of Maryland, Baltimore, Maryland, USA
Background: Catastrophic thrombotic syndrome (CTS) is a rare disorder characterized by rapid onset of multi‐organ thromboembolic events over days to weeks (Kitchens et al. 2011). It is triggered by provocative factors such as infection, trauma or cancer in patients with an underlying hypercoagulable state, including antiphospholipid antibody syndrome (APS). However, no cause can be identified in some cases.
Objectives: We report a case of CTS in systemic lupus erythematosus (SLE) without identifiable cause.
Methods: Single case report.
Results: A 28‐year‐old obese woman without other comorbidities presented with menorrhagia, hemoglobin 5.7 g/dl and platelet count 6,000/mcL. She was diagnosed with immune thrombocytopenic purpura (ITP) and treated with intravenous immunoglobulin (IVIG) and corticosteroids, with improvement in platelet count to 74,000/mcL. The next day she developed a headache; her platelet count was 20,000/mcL. Imaging showed an extensive right central venous sinus thrombus (CVST). Despite thrombocytopenia, a low‐dose heparin infusion was initiated after she developed new left‐sided weakness. She had antinuclear antibodies (1:1280) in addition to antibodies to DNA and SSA. An extensive hypercoagulable work‐up was negative, including tests for lupus anticoagulant and antibodies to cardiolipin and beta‐2 glycoprotein I (Table 1). Given her ITP and these serologic markers, a diagnosis of SLE was established (2019 EULAR/ACR classification criteria). She received IVIG and high‐dose corticosteroids again, along with 4 weekly doses of rituximab, with no significant improvement in platelet count. Therefore, romiplostim was started, with a rise in platelet count to 103,000/mcL. She was discharged on enoxaparin and a steroid taper. She presented 16 days later with headache, dyspnea, right leg pain, platelet count 33,000/mcL and D‐dimer >20,000 ng/mL. Imaging showed a new left transverse and sigmoid CVST, lobar pulmonary emboli and acute thrombus of the right distal common iliac vein. Anti‐factor Xa levels were therapeutic. Antiphospholipid antibodies were again not detected. Malignancy screening was negative. Bone marrow biopsy was unrevealing. The patient was treated with plasmapheresis and high‐dose corticosteroids, per the regimen typically used for CAPS. Anticoagulation was switched to argatroban due to concern for enoxaparin failure and was then transitioned to warfarin. D‐dimer decreased and platelet count increased to 320,000/mcL. The patient will be starting immunosuppressive therapy with cyclophosphamide (Cervera et al. 2014).

Figure 1. Patient’s platelet count (blue) and d‐dimer (orange) over time with dosing of romiplostim (black) and with dosing of steroids converted to prednisone equivalents (red). Of note, patient received methylprednisolone on 9/14 which is an equivalency of 1250 mg, exceeding the max of the vertical access. Each plot point represents a dose change
Table 1. Laboratory testing performed for hypercoagulable work‐up

Conclusions: CTS can occur in SLE without markers of APS. Our patient was treated successfully with a regimen typically used for CAPS. Patients presenting with CTS in SLE should be recognized early and treated for CAPS, even in the absence of anti‐phospholipid antibodies, to prevent morbidity and mortality. (Bucciarelli et al. 2006)
59. HTRS.P2.17
60. Warfarin‐induced skin necrosis in patients with protein C and S hereditary deficiency. Systematic review
R. Pichardo‐Rodriguez 1; L. Cordova‐Cueva2; M. Saavedra‐Velasco3; O. Ruiz‐Franco1; H. Garcia‐Perdomo4
1 Servicio de Hematologia. Hospital Nacional “Dos de Mayo”, Lima, Peru; 2 Servicio de Nefrología. Hospital Nacional “Dos de Mayo”, Lima, Peru; 3 Centro de enfermedades infecciosas (CEI), Lima, Peru; 4 División de Urología. Departamento de Cirugía. Escuela de Medicina. Universidad del Valle, Cali, Valle del Cauca, Colombia
Background: Warfarin has been a fundamental pillar in the management of prothrombotic pathologies and is one of the most prescribed oral anticoagulants in North America. However, Warfarin‐induced skin necrosis has been a worrying issue for hematologists for years, since the publication of the first case by Verhagen et al in 1954. Apparently, hereditary deficiency of protein C and S, as well as other natural anticoagulants, could predispose to developing skin necrosis, however, very little is known about the behavior of this disease in this population Due to its low frequency, conduction of clinical trials is limited, and clinical trials of treatments are almost impossible. It is relevant to carry out a synthesis of the results of published clinical studies to increase the consistency of the information and better understand the clinical characteristics of this disease in patients with hereditary deficiency of protein C and S and thus make better decisions based on evidence.
Objectives: To determine the clinical characteristics and management of Warfarin‐induced skin necrosis in patients with hereditary deficiency of protein C and S.
Methods: PRISMA guidelines were followed. The search was carried out in PUBMED, EMBASE, CENTRAL. Trials were included regardless of social status or ethnic origin. Patients with hereditary deficiency of protein C and S who had skin necrosis associated with Warfarin were included. Primary outcomes: Mortality. Secondary outcomes: Time to resolution of necrosis. The CARE statement was used to assess the quality of the report. The studies were selected and evaluated by two authors who, if they did not reach a consensus, a third evaluator made the final decision. A qualitative synthesis of the results of the included studies was carried out.
Results: A total of 26 studies were identified, including 11 (reports and case series). The most frequent sex was female. The median age was 41.5 years. The most common hereditary thrombophilia was protein S deficiency (91.6%; n = 11). The median time to appearance of skin necrosis was three days with a range of 1 to 11 days. The most frequent area was the thighs and buttocks. Four patients underwent surgical debridement. Only two patients died. The shortest resolution time was six days.
Conclusions: Warfarin‐induced necrosis in patients with hereditary protein C and S deficiency can be established in less time and regardless of the dose of Warfarin. Evolution is generally favorable and pathological history has an important influence on survival. More studies are required to define the safety and efficacy of the interventions applied to treatment.
61. HTRS.O4.3
62. Efficacy and safety of etranacogene dezaparvovec in adults with severe or moderate‐severe hemophilia B: First data from the phase 3 HOPE‐B gene therapy trial
S. Pipe 1; M. Recht2; N. S. Key3; F. W. G. Leebeek4; G. Castaman5; S. U. Lattimore6; P. Van Der Valk7; K. Peerlinck8; M. Coppens9; N. O'Connell10; J. Pasi11; P. Kampmann12; K. Meijer13; A. Von Drygalski14; G. Young15; C. Hermans16; J. Astermark17; R. Klamroth18; R. S. Lemons19; N. Visweshwar20; S. Crary21; S. Kazmi22; E. Symington23; M. Escobar24; E. Gomez25; R. Kruse‐Jarres26; A. Kotowski27; D. Quon28; M. Wang29; A. P. Wheeler30
1 University of Michigan, Ann Arbor, Michigan, USA; 2 Oregon Health & Science University, Portland, Oregon, USA; 3 University of North Carolina, Chapel Hill, North Carolina, USA; 4 Erasmus University Medical Center, Rotterdam, Zeeland, Netherlands; 5 Azienda Ospedaliera Universitaria Careggi, Florence, Emilia‐Romagna, Italy; 6 The Hemophilia Center, Oregon Health and Sciences University, Portland, Oregon, USA; 7 Van Creveldkliniek, University Medical Center Utrecht, Utrecht, Utrecht, Netherlands; 8 Campus Gasthuisberg, Leuven, Belgium, Leuven, West‐Vlaanderen, Belgium; 9 Amsterdam University Medical Centers, University of Amsterdam, Amsterdam, Noord‐Holland, Netherlands; 10 National Coagulation Centre, St James' Hospital, Dublin, Dublin, Ireland; 11 Barts and The London School of Medicine and Dentistry, London, England, United Kingdom; 12 Rigshospitalet, Copenhagen, Syddanmark, Denmark; 13 University Medical Center Groningen, Groningen, Noord‐Holland, Netherlands; 14 University of California San Diego, La Jolla, California, USA; 15 Children’s Hospital Los Angeles, University of Southern California Keck School of Medicine, Los Angeles, California, USA; 16 Cliniques Univ. Saint‐Luc, Brussels, Brussels Hoofdstedelijk Gewest, Belgium; 17 Skane University Hospital, Malmo, Skane Lan, Sweden; 18 Vivantes Klinikum im Friedrichshain, Berlin, Berlin, Germany; 19 University of Utah, Salt Lake City, Utah, USA; 20 University of South Florida, Tampa, Florida, USA; 21 University of Arkansas for Medical Sciences, Little Rock, Arkansas, USA; 22 University Hospital Southampton NHS Foundation Trust, Southampton, England, United Kingdom; 23 Cambridge University, Addenbrooke's Hospital, Cambridge, England, United Kingdom; 24 University of Texas Health Science Center at Houston, Houston, Texas, USA; 25 Phoenix Children’s Hospital, Phoenix, Arizona, USA; 26 Bloodworks Northwest, Seattle, Washington, USA; 27 Hemophilia Center of Western New York, New York, New York, USA; 28 Orthopaedic Hemophilia Treatment Center, Los Angeles, California, USA; 29 School of Medicine, Hemophilia and Thrombosis Center, Anschutz Medical Campus, University of Colorado, Aurora, Colorado, USA; 30 Vanderbilt University Medical Center, Nashville, Tennessee, USA
Background: Etranacogene dezaparvovec is an investigational gene therapy for hemophilia B (HB) comprising an adeno‐associated virus serotype 5 (AAV5) vector containing a codon‐optimized Padua variant human factor IX (FIX) gene with a liver specific promoter. Although most gene therapy clinical studies exclude pts with pre‐existing neutralizing antibodies (NAb) to the capsid serotype, early clinical studies and nonhuman primate data suggest that generally prevalent titers of anti‐AAV5 NAbs may not preclude successful transduction with etranacogene dezaparvovec.
Objectives: A Phase 3, Health Outcomes with Padua gene; Evaluation in Hemophilia B (HOPE‐B; NCT03569891) was established to further assess efficacy and safety of etranacogene dezaparvovec in adults with HB with a wide range of pre‐existing NAbs to AAV5. Here we report outcomes at 26 weeks (wks).
Methods: Open‐label, single‐dose, single‐arm, multinational trial in adult males with severe or moderate‐severe HB (FIX ≤ 2%) on prior routine FIX prophylaxis. No exclusion based on pre‐existing NAbs to AAV5. Pts entered a ≥6 month lead‐in period, then received a single dose of etranacogene dezaparvovec (2 × 1013 gc/kg). Primary endpoints: FIX activity at 26 and 52 wks and 52 wk annualized bleeding rate. Secondary endpoints include adverse events (AEs), and reactive use of steroids.
Results: 54 pts were dosed and completed 26 wks of follow‐up. 23 pts had NAbs to AAV5 at baseline (BL). 38 pts had 123 bleeds during lead in despite prophylaxis. Post treatment, mean (SD; min,max) change in FIX activity from BL was +36.0% (19.7; 0,96.1, p < 0.0001) at wk26 (mean 37.2% ± 19.6). No correlation of pre‐existing NAbs with FIX activity was seen up to a titer of 678.2; n = 52, R2 = 0.078; 1 pt had a NAb titer of 3212.3 and did not respond. One other pt received a partial dose and remained on prophylaxis; 52 pts successfully discontinued routine prophylaxis. 39 pts reported 0 bleeds post‐treatment; 15 pts had a total of 21 bleeds. Treatment‐related AEs occurred in 37 pts, mostly mild (81.5%); most frequent: headache and influenza‐like illness (7 pts). No deaths occurred; no treatment‐related SAEs were reported. 7 pts had infusion‐related reactions, 1 infusion was discontinued. 9 pts received steroids for liver enzyme elevations. All discontinued steroids prior to wk26; FIX activity remained in the mild range. No inhibitors to FIX were reported. No relationship between safety and NAbs was observed.
Conclusions: The first co‐primary endpoint was met. Following a single dose of etranacogene dezaparvovec, FIX activity increased, without the need for prophylactic immunosuppression, into the mild‐to‐normal range at 26 wks in pts with severe/moderately severe HB. Importantly, this included pts with titers of pre‐existing anti‐AAV5 NAbs. Pts were able to discontinue prophylaxis and bleeding was abolished in the majority. The safety profile was consistent with early phase AAV5 studies and together these data support a favorable safety and efficacy profile for etranacogene dezaparvovec.
63. HTRS.P2.5
64. Evaluation of the hemostatic efficacy of eptacog beta (recombinant factor VIIa) for the treatment of bleeding in pediatric subjects (<12 years) with hemophilia A or B with inhibitors in persept 2
S. Pipe 1; M. Callaghan2; J. Ducore3; M. Escobar4; C. Hermans5; J. Journeycake6; J. Mahlangu7; S. Meeks8; M. Recht9; M. Wang10; G. Young11; W. Alexander12; A. Al‐Sabbagh13; D. Bonzo13; I. Mitchell14; A. Shapiro15
1 University of Michigan, Ann Arbor, Michigan, USA; 2 Central Michigan University, Detroit, Michigan, USA; 3 University of California Davis Medical Center, Sacramento, California, USA; 4 University of Texas Health Science Center at Houston, Houston, Texas, USA; 5 Cliniques Univ. Saint‐Luc, Brussels, Brussels Hoofdstedelijk Gewest, Belgium; 6 Oklahoma Center for Bleeding and Clotting Disorders, Oklahoma City, Oklahoma, USA; 7 Hemophilia Comprehensive Care Center, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, Gauteng, South Africa; 8 Aflac Cancer and Blood Disorders Center, Emory University/Children’s Healthcare of Atlanta, Atlanta, Georgia, USA; 9 American Thrombosis & Hemostasis Network, Portland, Oregon, USA; 10 School of Medicine, Hemophilia and Thrombosis Center, Anschutz Medical Campus, University of Colorado, Aurora, Colorado, USA; 11 Children’s Hospital Los Angeles, University of Southern California Keck School of Medicine, Los Angeles, California, USA; 12 Aoede Associates, Louisville, Kentucky, USA; 13 LFB, Framingham, Massachusetts, USA; 14 GLOVAL, LLC, Broomfield, Colorado, USA; 15 Indiana Hemophilia & Thrombosis Center, Indianapolis, Indiana, USA
Background: N/A.
Objectives: Eptacog beta (coagulation factor VIIa [recombinant]‐jncw, EB) is a new rFVIIa manufactured in a transgenic mammalian expression system indicated for the treatment and control of bleeding episodes (BEs) in adults and adolescents (≥12 years) with hemophilia A or B with inhibitors. A phase 3 clinical trial (PERSEPT 2, NCT02448680) in subjects <12 years of age with hemophilia A or B with inhibitors evaluated the efficacy, safety and pharmacokinetics of EB.
Methods: PERSEPT 2 was a global, multicenter, prospective, randomized crossover study of EB (75 and 225 µg/kg) initial dose regimens (IDRs). Male subjects (<12 years) with hemophilia A or B with inhibitors were enrolled. IRB approval was obtained; subjects’ legal guardians provided informed consent. Subjects were randomized to one of the IDRs for the treatment of mild/moderate BEs (Figure 1) with crossover to the alternate IDR every 3 months. Efficacy was evaluated at multiple time points for the first 24 h of each BE; the primary efficacy endpoint was at 12 h. Success at each time point was defined as good or excellent hemostasis by the subject/caretaker. In addition, to be considered successful, no additional eptacog beta, alternative hemostatic agent or blood product could be administered (and pain could not increase) following the initial good or excellent assessment and prior to 24 h.

Figure 1. The 75 µg/kg and 225 µg/kg initial dose regimen (IDR) for treatment of mild/moderate bleeding events in the PERSEPT 2 clinical trial. The initial dose regimens were selected based on PK/PD modeling from a phase 1b trial; the efficacy of these IDRs was confirmed in the PERSEPT 1 adult/adolescent trial. Additionally, the total amount of eptacog beta administered at the primary efficacy endpoint (12 h) could be the same on both IDRs
Results: Twenty‐five subjects (age range, 1–11 years; median, 5.0 years) were enrolled; 23 subjects had hemophilia A and 2 had hemophilia B. Twenty‐one subjects had high titer inhibitors (≥5 BU); the remaining had low titer inhibitors requiring the use of bypassing agents to treat BEs. A total of 546 mild/moderate BEs were treated during the trial; most (92%) were treated at home. Joint BEs accounted for 68% of all BEs; 14% were in target joints. Efficacy results at 12 and 24 h are shown in Table 1: 63% (95% CI: 51%, 74%) of mild/moderate BEs achieved hemostatic efficacy at 12 h; at 24 h this had increased to 98%. In the 225 µg/kg IDR, a median of 2 doses were required for bleed control vs a median of 3 doses in the 75 µg/kg IDR. Most BEs (98%) did not require alternative treatment and only 1.5% of BEs experienced a recurrence of bleeding at the same site within 24 hours of first treatment dose for that BE. No treatment‐related adverse events were reported. No thrombotic events or neutralizing antibodies to EB were observed. Two SAEs unrelated to EB were reported: paresis and dysentery.
Table 1. Percentage of successfully treated mild/moderate bleeding events at 12 and 24 hours in both initial dose regimens (IDRs). [Normal approximation CI taking into account the correlation among bleeding episodes for a given patient]

Conclusions: The clinically meaningful percentage of mild/moderate BEs successfully treated at 12 h (63%), high percentage of BEs resolved at 24 h (98%), low incidence of alternative hemostatic agents use (2%), low occurrence of rebleeding (1.5%), low number of doses required, and absence of treatment‐related adverse events observed in the trial demonstrate that eptacog beta provides a new and well‐tolerated at‐home hemostatic agent for the treatment and control of BEs in individuals younger than 12 years of age with hemophilia A or B with inhibitors.
65. HTRS.O2.2
66. In vitro‐grown megakaryocytes (MKS) take up the ectopic protein urokinase (UPA) and the resulting platelets (PLTS) can release the upa at sites of vascular injury
S. Zaitsev1; H. Ahn1; M. Kowalska1; K. Bdeir2; D. Cines2; D. French1; R. Camire2; V. Stepanova2; M. Poncz 1
1 The Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, USA; 2 Perelman School of Medicine at the University of Pennsylvania, Pennsylvania, USA
Background: Our goal is to develop fibrinolytic agents with a prolonged half‐life which can selectively prevent nascent thrombi while sparing established hemostatic clots. We found that single chain (sc) uPA was stored in PLT a‐granules in transgenic mice expressing uPA ectopically in PLTs during megakaryopoiesis. Infusion of such “scuPA Plts” into wild‐type mice prevented new thrombi from developing. We wish to generate in vitro‐grown MKs and released PLTs that are modified to express uPA near the point‐of‐care and then infused into patients. We have previously shown that MKs express low‐density lipoprotein (LDL) receptor‐related protein 1 (LRP1) during their maturation, whereas the PLTs that are released do not.
Objectives: N/A.
Methods: Confocal microscopy, FACS. Rose Bengal carotid artery laser injury murine model.
Results: We asked whether in vitro‐grown MKs derived from CD34+ hematopoietic cells or the induced pluripotent stem cell (iPSC)‐derived imMKCL line from Koji Eto, which were proposed for potential clinical application to make in vitro‐generated PLTs, would endocytose uPA. Both sets of MKs internalize and store scuPA and low molecular weight uPA variant modified to be thrombin‐activatable (uPA‐T). uPA uptake is blocked by LRP1 antagonist receptor‐associated protein (RAP). scuPA and uPA‐T were both endocytosed by CD34+‐MKs in the same intracellular vesicles (Figure 1A), further supporting the hypothesis that LRP1 mediates uptake of both uPA variants in MKs. Coagulation Factor (F) V is an LRP1‐dependent protein for a‐granule uptake. MKs co‐incubated with uPA‐T and FV internalized both proteins into a‐granules (Figure 1B). Co‐incubation with FV also decreased uPA‐T uptake, suggesting that both may compete for a common pathway. scuPA and uPA‐T studies with von Willebrand Factor (VWF) and with platelet factor 4 showed partial co‐localization supporting suggestion that uPA is sorted to a‐granules. We asked whether CD34+‐MK‐derived scuPA PLTs prevented nascent thrombus formation in immunodeficient NOD/SCID IL2r‐gamma null (NSG) mice that are homozygous for VWFR1326H (a mutation switching binding specificity to VWF from mouse GPIb/IX to human platelet glycoprotein (GP) Ib/IX). These mice show a mild bleeding diathesis in a Rose Bengal photochemical carotid artery injury model unless they are infused with human MKs that release functional PLTs in the pulmonary capillary bed of the recipient mouse over several hours. Thrombi developed in this model when 1 million MKs were infused with ~1%–10% of circulating platelets being human, but a similar infusion of scuPA‐human MKs did not result in occlusion (Figure 2).

Figure 1. Uptake and storage of scuPA, uPA‐T and FV by CD34+‐MKs

Figure 2. Preventing nascent thrombi by endocytosed scuPA in a Rose Bengal carotid artery murine model
Conclusions: These studies suggest that MKs internalize biologically relevant concentrations of uPA through a process likely to involve LRP1. Whether in vitro loading of MKs with scuPA or uPA‐T can be a point‐of‐care therapeutic for thromboprophylaxis in the setting of high‐risk of bleeding needs further study.
Other: R35 HL150698 (Poncz, M), R01 HL141462 (Stepanova, V), R01 HL142122 (Cines, D)
67. HTRS.P1.16
68. Unprovoked extensive deep vein thrombosis in a pediatric patient with sickle cell disease and duplicated inferior vena cava
R. Ramakrishnan; H. Imran; P. Marri; C. Ilonze
University of South Alabama, Mobile, Alabama, USA
Background: Sickle cell disease (SCD) is a known hypercoagulable state with inherent risk of thromboembolic events with attendant complications. Chronic platelet activation, increased expression of tissue factor, decreased levels of natural anticoagulants such as protein C, protein S and antithrombin III and abnormal externalization of phosphatidylserine on the surface of sickled red blood cells (RBC), all contribute to activation of coagulation cascade leading to a hypercoagulable state. Anatomical variants are infrequently associated with deep venous thrombosis (DVT) in SCD.
Objectives: Case report of an 8‐year‐old female with HbSS genotype presenting with extensive, unprovoked DVT involving the left femoral, popliteal and iliac veins and pulmonary embolism (PE) with incidental finding of duplicate inferior vena cava (IVC).
Methods: Case report and literature review.
Results: Patient initially presented with acute onset of pain around the left groin area and antalgic gait with swelling of the left lower extremity extending from the groin on physical exam with tachypnea and dyspnea. Venous Doppler ultrasound revealed the presence of an occlusive thrombus involving the left popliteal and femoral veins with extension into the IVC which was noted to be duplicated (Figure 1). Chest computed tomography revealed PE involving the right posterior basal segmental pulmonary artery. Workup revealed a grossly elevated Factor VIII level (1,249%), positive lupus anticoagulant, elevated lipoprotein A (35 mg/dL), elevated anti‐cardiolipin antibodies IgM (19 MPL), low total proteins C (37%) and functional protein S levels (34%). Treatment was initiated with unfractionated heparin drip transitioned to low molecular weight heparin for 6 months of treatment with near complete resolution. There is an anecdotal maternal history of low protein C and S, with lower extremity DVT on lifelong anticoagulation.

Figure 1. CT Abdomen and Pelvis with contrast‐ The arrow shows the thrombus
Conclusions: The presence of unprovoked, acute, extensive lower extremity venous thrombosis with PE in a patient with SCD is rare in the pediatric population compared to adolescent or adult population. Duplication of the IVC is very rare and has been associated with pulmonary embolism. This anatomical variant, in addition to perturbations in levels of natural anticoagulants and procoagulants in HbSS, may have contributed to observed thrombosis and PE. Postulated mechanisms may be related to the degree of narrowing of the vessel as it crosses the aorta. Furthermore, inherited thrombophilia workup was negative, and there was no history of pro‐thrombotic risk factors. Duplicated IVC makes this case more notable and may have led to extensive DVT at such a young age. There is a paucity of published reports of this anatomic variant leading to extensive thrombosis and PE with less than 10 cases reported. Although very rare, venous anomalies should be recognized as important causes of thrombosis which may impact outcome of thrombotic and thromboembolic events.
69. HTRS.P2.1
70. ATHN 16: Safety of coagulation factor VIIa (recombinant)‐jncw for the treatment of bleeding events in patients with hemophilia A or B with inhibitors with or without prophylactic treatment
M. Callaghan1; W. Alexander2; A. Al‐Sabbagh3; D. Bonzo3; M. Escobar4; A. Giermasz5; N. Hirsh6; J. Journeycake7; S. Nasr8; D. Quon9; T. Singleton10; M. Recht 11
1 Central Michigan University, Detroit, Michigan, USA; 2 Aoede Associates, Louisville, Kentucky, USA; 3 LFB, Framingham, Massachusetts, USA; 4 University of Texas Health Science Center at Houston, Houston, Texas, USA; 5 University of California Davis, Sacramento, California, USA; 6 American Thrombosis & Hemostasis Network, Rochester, New York, USA; 7 Oklahoma Center for Bleeding and Clotting Disorders, Oklahoma City, Oklahoma, USA; 8 GLOVAL LLC, Broomfield, Colorado, USA; 9 Orthopaedic Hemophilia Treatment Center, Los Angeles, California, USA; 10 Mississippi Center for Advanced Medicine, New Orleans, Louisiana, USA; 11 American Thrombosis & Hemostasis Network, Portland, Oregon, USA
Background: A new recombinant human activated factor VII, eptacog beta (EB; rFVIIa‐jncw), has recently received FDA approval for the treatment and control of bleeding episodes occurring in adults and adolescents (>12 years of age) with hemophilia A or B with inhibitors.
Objectives: To evaluate the safety of EB when used to treat bleeding events (BEs) in participants with hemophilia A or B with inhibitors with or without prophylactic treatment.
Methods: ATHN 16 is a phase IV multi‐center, US‐centric, open‐label study to evaluate the safety of EB when used to treat bleeding episodes in participants with hemophilia A or B with inhibitors who may be on prophylactic treatment. Participants will be followed longitudinally to a maximal study duration of 2 years from the time of enrollment. The study is anticipated to enroll approximately 50 participants in order to achieve treatment of approximately 100 BEs. Participants must have a diagnosis of hemophilia A or B with inhibitors, be 12 to 65 years of age, be willing to comply with the conditions of the protocol, be able to provide evidence of previous inhibitor levels, be willing and able to use the ATHN mobile application or a paper diary to document BEs and medication usage, and have read, understood, and documented written informed consent/assent. The following participant level data will be collected: demographics, bleeding history, medical history including diagnoses and co‐morbidities, inhibitor history, clotting factor products(s) and non‐factor product(s) treatment plan, concomitant medications, and study drug administration details. In addition, all BEs, surgical procedures and European Haemophilia Safety Surveillance (EUHASS) adverse events will be collected. Participants will be provided with nine 75 μg/kg doses of EB based on the approved initial dose recommendation. Treatment with the approved 225 μg/kg initial dose regimen will be allowable if deemed appropriate. Doses will be self‐administered or administered at a treatment facility. Duration of treatment for BEs will be at the discretion of the investigator.
Results: Reportable adverse events as defined by EUHASS, including allergic events, treatment‐emergent adverse events, transfusion‐transmitted infections, inhibitor development, thrombosis and cardiovascular events, malignancies, neurologic events, and death will be reported prospectively. Additionally, all serious adverse events will be collected. All data will be entered into electronic case report forms in ATHN’s data management system.
Conclusions: As the first interventional study sponsored by ATHN, ATHN 16 represents a crucial step forward in ATHN’s clinical research capabilities while exploring the safety of EB as therapy for BEs in participants with hemophilia A or B complicated by inhibitors with or without prophylactic treatment.
71. HTRS.P1.6
72. Eculizumab treatment for refractory thrombosis in antiphospholipid syndrome
H. Hussain1; M. Tarantino2; K. McCrae3; S. Chaturvedi4; J. Roberts 2
1 University of Illinois College of Medicine at Peoria, Peoria, Illinois, USA; 2 Bleeding and Clotting Disorders Institute, Peoria, Illinois, USA; 3 Cleveland Clinic Lerner College of Medicine, Cleveland, Ohio, USA; 4 Johns Hopkins University School of Medicine, Baltimore, Maryland, USA
Background: Antiphospholipid syndrome (APS) is characterized by arterial and/or venous thrombosis with antiphospholipid antibodies. Errors in the complement pathway has been implicated in APS pathophysiology. We present successful use of eculizumab, anti‐C5 monoclonal antibody, in controlling and preventing recurrent thrombosis in a refractory case of APS.
Objectives: To describe eculizumab use in a refractory case of APS.
Methods: Retrospective chart review.
Results: Case Description: An 18‐year‐old female was diagnosed with APS after developing extensive unprovoked deep vein thrombosis of axillary, inferior vena caval and brachiocephalic veins. Laboratory thrombophilia evaluation revealed triple positive lupus anti‐coagulant, beta‐2 glycoprotein IgM, IgA and anti‐cardiolipin antibodies (each >40 U/mL) with persistent positive titers after 12 weeks. She was refractory to multiple anticoagulants alone (enoxaparin, fondaparinux, apixaban, rivaroxaban, and warfarin), with antiplatelet (aspirin and clopidogrel) and adjunctive therapies (hydroxychloroquine, immunosuppression with steroids and rituximab, and plasmapheresis). Despite these she continued to develop recurrent thrombosis of subclavian, femoral, common femoral, iliac, popliteal and saphenous veins. She additionally developed hepatic infarction and pulmonary embolism along with failure to decrease titers after 6 weeks of plasma exchange. Following this event, eculizumab (600 mg weekly × 4 weeks followed by 900 mg every 2 weeks) was initiated in combination with fondaparinux, aspirin, clopidogrel and hydroxychloroquine. She has remained on this regimen without recurrence of thrombosis from 10/05/19 through 10/29/20 (Table 1).
Table 1. Chronology of Events: Eculizumab initiated on 9/5/2019. No evidence of venous or arterial thrombosis since 10/5/2019 to 10/29/2020

Conclusions: Recent evidence has shown complement activation in APS. This is thought to occur through C3 and C5a causing platelet and endothelial activation, leading to microvascular thrombosis. Histological evidence is supported by demonstration of anti‐beta‐2 GP1 IgG–C5b‐9 immune complexes in microvascular organ thrombi in individuals with APS leading to thrombotic microangiopathy and multiorgan failure. Evidence of complement induced thrombosis in APS led us to use eculizumab following failure of available therapies in a potentially life‐threatening event. Absence of recurrent thrombotic events over one year with eculizumab combined with anticoagulation and antiplatelet therapy highlights the potential role of complement inhibition in preventing thrombosis in APS. We have observed no decline in antiphospholipid antibody levels in a span of over one year in our patient. Importantly, there has been no recurrent thrombosis during this time. Our case suggests that eculizumab may have a role as a therapeutic option in refractory thrombosis in APS.
73. HTRS.P1.8
74. Gastrointestinal bleeding in patients with von Willebrand disease: Treatments and outcomes
J. Roberts 1; M. Escobar2; S. Acharya3; N. Hwang4; M. Wang5; S. Hale6; S. Asghar7; P. Kouides8
1 Bleeding and Clotting Disorders Institute, Peoria, Illinois, USA; 2 University of Texas Health Science Center at Houston, Houston, Texas, USA; 3 Northwell Health Hemostasis and Thrombosis Center, Cohen Children's Medical Center, Zucker School of Medicine at Hofstra/ Northwell, New Hyde Park, New York, USA; 4 Pediatric Hematology, Center for Inherited Blood Disorders, Orange, California, USA; 5 School of Medicine, Hemophilia and Thrombosis Center, Anschutz Medical Campus, University of Colorado, Aurora, Colorado, USA; 6 Takeda Pharmaceutical Company Ltd, Lexington, Massachusetts, USA; 7 HCD Economics, Daresbury, England, United Kingdom; 8 Mary M. Gooley Hemophilia Center, Rochester, New York, USA
Background: The etiology and treatment of gastrointestinal (GI) bleeds in patients with von Willebrand disease (VWD) is challenging and compounded by limited published data. Despite von Willebrand Factor (VWF) replacement therapy, GI bleeding may be refractory resulting in disparate treatment approaches and no consensus on the most efficacious therapeutic approach.
Objectives: To describe the treatment of GI bleeds in patients with congenital VWD with VWF replacement products, including recombinant VWF (rVWF) and non‐VWF therapy, and outcomes in clinical practice.
Methods: This retrospective, multicenter, observational chart review included patients with confirmed congenital VWD with ≥1 GI bleed within the last 5 years (abstraction initiated 2019 and ongoing). The target sample size is 20 patients from six US hemophilia treatment centers. Demographics and clinical information, including treatment regimens, were abstracted from patient records on all recorded GI bleeds. Clinical effectiveness was defined by treatment response, change in treatment duration or time‐to‐bleed resolution across treatment cohorts (eg, prophylaxis, on‐demand; rVWF, plasma‐derived VWF‐FVIII) at the time of a GI bleed, and any subsequent period of prophylactic treatment to prevent GI bleed recurrence. Data were analyzed descriptively.
Results: To date, data on 41 bleeds in 14 patients with Type 1 (21%), Type 2 (50%), or Type 3 (29%) VWD have been abstracted; 50% of patients were female, mean ( ± SD) age was 54.8 (21.4) years, 9 patients (64%) had a history of prior GI bleeding. 89% of patients had ≥1 recorded GI‐specific morbidity, with confirmed or suspected arterio‐venous malformations (AVM) reported as the most common GI‐specific morbidity (37%). 71% of patients were on prophylactic VWF concentrate with pdVWF‐FVIII at the initial presentation of a bleed. On‐demand treatment for GI bleeding included aminocaproic acid, tranexamic acid, pdVWF‐FVIII concentrates, rFVIII, rVWF, corticosteroids, polypectomy, and thalidomide; no patients received DDAVP. Across 41 bleeds, 49% were treated with VWF concentrates and at least one non‐VWF treatment (including a rFVIII treatment), 41% were treated with VWF concentrates, and 10% with non‐VWF treatments only. Median time to resolution of GI bleeding was 9.0 days (range, 2–40).
Conclusions: This retrospective chart review describes real‐world experience in the determination of the etiology and management of GI bleeds in patients with VWD. Of note were the breadth of treatments used and the time to bleed resolution, especially in patients with AVMs. These data highlight a persisting unmet need in the treatment of GI bleeds and underscore the opportunity to better tailor prophylaxis and target on‐demand treatment of GI bleeding in VWD. In particular, further evidence is needed to elucidate the role of AVMs in the etiology of GI bleeding in VWD patients and the effectiveness of treatments in patients with identified or suspected AVMs.
75. HTRS.O1.2
76. Von Willebrand Factor (VWF) Multiplex Activity Assay differentiation of low VWF/Type 1 von Willebrand disease (VWD) and variant VWD: Analysis from the comparative effectiveness in the diagnosis of VWD
J. Roberts 1; P. Christopherson2; M. Tarantino3; S. Gonzales3; P. Morateck2; C. Perry2; V. Flood2; T. Abshire2; R. Montgomery2; The Zimmerman Program Investigators2
1 Bleeding and Clotting Disorders Institute, Peoria, Illinois, USA; 2 Versiti Blood Research Institute, Milwaukee, Wisconsin, USA; 3 Bleeding and Clotting Disorders Institute, Peoria, Illinois, USA
Background: VWD diagnosis is challenging requiring multiple VWF activity tests using many individual assays. We have developed an ELISA‐based VWF Multiplex Activity Assay (VWF‐MAA) (Roberts JC, et al. Blood 2016)* to address this concern; however, the ability of the VWF‐MAA to discriminate between low VWF/type 1 VWD, variant VWD, and normal subjects has not been evaluated.
Objectives: To evaluate the VWF‐MAA and its ability to differentiate between low VWF/type 1 VWD, variant VWD, and normal subjects in individuals undergoing an initial laboratory evaluation for bleeding.
Methods: 177 plasma samples from the Zimmerman Program: Comparative Effectiveness in the Diagnosis of VWD were evaluated on the VWF‐MAA at the Bleeding & Clotting Disorders Institute (BCDI). Samples were obtained from 11 Centers across the US and Canada. VWF‐MAA measures the relative values of VWF:Ag (antigen), VWF:GPIbM (VWF‐platelet activity), VWF:FVIIIB (VWF‐factor VIII activity), VWF:CB3 (VWF‐collagen 3 activity), and VWFpp (VWF propeptide). Samples were compared to 30% standard control plasma by a ratio of each unique VWF activity over VWF:Ag as previously described. Data were compared to the Versiti (VBRI) research laboratory and the local Center (LC) assigned diagnosis. VWF:Ag ratio of <1.7 was considered “low VWF/type 1 VWD” as this approximates <51 U/dL when compared to 30% control plasma where control ratio is 1.
Results: 177 samples were evaluated on the VWF‐MAA and overall 129/177 (72.88%) were correctly assigned as normal (non‐VWD), low VWF/type 1, or variant VWD compared to the VBRI assigned diagnosis. When considering LC diagnosis of low VWF/type 1 in 12 samples where there was agreement with VWF‐MAA and not VBRI diagnosis, overall 141/177 (79.66%) were correctly assigned. VWF‐MAA was not able to discriminate between low VWF and type 1 VWD, thus included together in analysis to account for overall decreased VWF:Ag. Non‐VWD optical density (OD) ratio was [median(range); mean(standard deviation)], 2.22(1.73–3.97); 2.33(0.46). Low VWF/type 1 OD ratio was, 1.26(0.37–1.69); 1.24(0.30). VWF‐MAA assigned non‐VWD accurately in 29/57 (50.88%) samples, and low VWF/type 1 accurately in 93/110 (84.55%) samples. Considering LC diagnosis where there was agreement with VWF‐MAA and not VBRI diagnosis, low VWF/type 1 was accurate in 105/110 (95.45%) samples. For variant VWD, 4 type 2A VWD, 2 type 2B VWD, and 1 type 1C VWD samples were accurately assigned by the VWF‐MAA; 2 were erroneously assigned type 2A due to low VWF:CB3 OD ratio, 2 missed type 2B assignment due to lack of VWF:GPIbM enhanced OD ratio, and 2 missed type 2M/2M‐C due to normal VWF:GPIbM ratio and low VWF:Ag ratio.
Conclusions: We demonstrate that the VWF‐MAA has utility in differentiating low VWF/type 1 VWD, variant VWD, and normal subjects in individuals undergoing an initial laboratory evaluation for bleeding. Our evaluation comparing multiple hemostasis laboratory data highlight the challenges associated with current VWD diagnosis.
Other: *Blood. 2016 May 19;127(20):2472–80.
77. HTRS.P1.4
78. Drive‐through anticoagulation clinic during COVID‐19 pandemic
J. Giver; A. Dunn; B. Kerlin; A. Sankar; J. Canini; K. Monda; V. Rodriguez
Nationwide Children's Hospital, Columbus, Ohio, USA
Background: Dedicated anticoagulation clinics have demonstrated superior patient outcomes. The COVID‐19 pandemic created challenges for patient care including fear of medical appointments due to potential risk of exposure. An innovative approach to anticoagulation management was developed at our center that allows the patient to stay in their vehicle while our anticoagulation advanced practice nurse obtains blood for point‐of‐care/INR testing, education and counseling.
Objectives: 1. To assess degree of compliance to anticoagulation management pre vs. post drive‐through clinic. 2. Assess percentage and time to therapeutic range (TTR), percentage within therapeutic (TR), subtherapeutic, and supratherapeutic INR pre vs. post drive‐through clinic. 3. Evaluate bleeding and thrombosis complications during study period. 4. Assess patient/family satisfaction.
Methods: Retrospective chart review of patients on warfarin therapy who used the drive‐through clinic between April 1 and August 31, 2020. Patient demographics, underlying diagnosis, and target INR range were reviewed. Dates and values for INR, percentage of therapeutic (TR), subtherapeutic, and supratherapeutic INR and degree of compliance (≥1 INR determination per month) were collected for 6 months prior to April 1 and compared to results obtained during drive‐through clinic implementation. Data were summarized descriptively and compared using nonparametric statistical methods.
Results: A total of 17 patients were evaluated in our drive‐through clinic during the study period. Fifty‐eight percent were males (n = 10) with a median age of 15 years (range: 3–54). Primary indication for warfarin therapy included: tricuspid valve replacement (n = 1), mitral valve (n = 5), aortic valve (n = 4), Fontan (n = 5), atrial fibrillation (n = 1), and deep vein thrombosis (n = 1). Median TTR was 60.1% (range: 21.1–89.2) with a median cumulative time of anticoagulation of 3.3 years (range: 0.2–6.1 years). Overall compliance pre vs. post drive‐through clinic implementation was similar (median pre compliance 100% (95% CI: 83%–100%) vs. 100% post (95% CI: 100%–100%); p = 0.16; Figure 1). Five of six patients who had compliance less than 90% achieved an improvement with the drive‐through clinic. The median percentage of INR within TR improved significantly with the drive‐through clinic (median pre 50% (95% CI: 33%–67%) vs. median post 80% (95% CI: 57%–100%); p = 0.0103; Figure 2). No bleeding or thrombotic complications were observed. Patients and families reported 100% satisfaction with the drive‐through care.

Figure 1. Compliance with INR monitoring pre and following drive‐through anticoagulation clinic

Figure 2. Percentage INR within therapeutic range pre and post‐drive through clinic
Conclusions: A drive‐through INR clinic during COVID‐19 pandemic allowed patients to access care from their vehicle. A subgroup of patients improved compliance to INR monitoring. The median percentage of INR within TR improved significantly after implementation of the drive‐through clinic. Innovative approaches such as this clinic may improve patient compliance and adherence to anticoagulation.
79. HTRS.P2.12
80. Passive infusion logging system (PILS): A pilot
M. Santaella 1; T. Brent2; C. Nichols2; M. Rice2; M. Witkop2
1 National Hemophilia Foundation, Key Biscayne, Florida, USA; 2 National Hemophilia Foundation, New York, New York, USA
Background: According to the 2019 Sun Life Stop‐Loss Research Report, hemophilia/bleeding disorders (BD) became one of the top 20 high‐cost claim conditions in 2013 and climbed to the top 10 in 2015 where it has remained ever since. Most of BDs direct costs come directly from clotting factor (CF) (Zhou Z, et al. JMedEcon 2015). Payors have expressed concerns regarding these rising costs, warning they may have to resort to actions such as limit CF options, higher cost tiers, and dispensed drug limits. Helping payors understand how CF is dispensed, when and why it is infused, will mitigate these concerns. Logging apps exist but their use varies. While important to clinical practice, infusion logs can also improve CF assay management. A novel passive infusion logging system (PILS) may help increase the number of patients using them.
Objectives: Describe real‐time clotting factor assay management with a PILS and measure ease of use and overall satisfaction.
Methods: The National Hemophilia Foundation began a quality improvement pilot in 2017 to evaluate PILS in persons with BD (PBD) on prophylactic infusions (PI) ‐adding additional sites in 2019 and terminating in 2020. PILS had 2 advantages: it helped PBDs track infusions and bleeding episodes with minimal clicks and allowed pharmacies real‐time data of home inventory. PILS required 2 steps: (1) the pharmacy entered the prescription and dispensation information in the system and affixed an associated patient‐specific label to each vial box, and (2) during the PI, PBDs scanned the label(s) with an associated smartphone app which instantly logged the infusion’s date, time and dose, and removed it from the home inventory; breakthrough bleeds were recorded the same way plus a few extra steps describing the reason for the infusion. Surveys to assess attitudes with logging, PILS ease of use, and PBD/pharmacy satisfaction were sent at baseline, 2‐week, 3 and 6 months.
Results: Of the 36 PBD who took the baseline survey, 30 (83.3%) completed the 2‐week and 26 (72.2%) the 3 and 6‐month surveys; 14 pharmacy staff took 1 survey. Before using PILS, 18 PBD (50%) used paper logs, 15 (41.7%) an app, 5 (13.9%) did not log, 2 (5.6%) had their own spreadsheet, and 1 (2.8%) reported to the pharmacy; 41.9% were extremely/very satisfied with their system and 61.3% said they would recommend it to others. All questions were responded on a 1–5 Likert‐type scale where 1 = lowest/worse and 5 = highest/best score. PBDs’ overall satisfaction mean was > 4 and did not significantly change over time (p = 0.75); the pharmacy’s was 3.57. PBDs agreed that the app was easy to use (M = 4.5) and record an active bleed (M = 4.4). See Table 1 for PBD/pharmacy staff attitudes towards PILS. PBDs were grouped by those who had and didn’t have a logging system before PILS, attitudes towards logging were compared ‐see Table 2.
Table 1. PBD/pharmacy staff attitudes towards PILS

Table 2. PBDs’ attitudes towards logging

Conclusions: A PILS may also serve as a suitable CF assay and home inventory management system.
81. HTRS.P1.15
82. Telegenetic counseling for female My Life, Our Future participants
M. Santaella 1; C. Nichols2; S. Ryan3; M. Witkop2
1 National Hemophilia Foundation, Key Biscayne, Florida, USA; 2 National Hemophilia Foundation, New York, New York, USA; 3 Bloodworks Northwest, Eugene, Oregon, USA
Background: In the US, over 2,000 women with a family history of hemophilia with or without a formal diagnosis, were genotyped through the My Life, Our Future (MLOF) program from 2012 to 2017 (Konkle, et al. Haemophilia, 2018). Many were not established hemophilia treatment center (HTC) patients, which combined with the lack of specialized genetic counselors (GC) in many HTCs, created logistical issues for providing results and genetic counseling if needed.
Objectives: The study aimed to establish the value of specialized telegenetic counseling (TGC) in delivering sensitive genotyping results to women who had undergone MLOF testing by: describing TGC as a way to access to GCs, determining if TGC causes incremental knowledge, if it has an impact on participant's management and plan of care (POC), and its overall satisfaction.
Methods: The National Hemophilia Foundation funded the study and, with HTCs, facilitated advertisement for recruiting. It included women enrolled in MLOF who received results but no formal GC session. Using randomization, 2 groups were created. Group 1 (G1) took a baseline survey, received TGC, and completed surveys 3 and 6 months after. Group 2 (G2, control) took a baseline, 3, and 6 month surveys, and then received TGC. The women and GC completed a satisfaction survey after TGC which was done through the HIPPA compliant Zoom platform. An individualized report was sent to every woman at the end of the study.
Results: Data was collected from 2/2018 to 7/2019. All self‐referred women (N = 25) were screened: 9 were not eligible and 4 did not finish; 12 completed the study. One non‐carrier was excluded from the analysis to improve homogeneity. The sample (n = 11) included 6 women in G1 and 5 in G2. All who completed the baseline survey testing knowledge (n = 10) received perfect scores. At baseline, 1 in each group did not answer questions about a POC; 3 of the remaining 5 in G1 and 1 of the remaining 4 in G2 had a POC ‐differences were not statistically significant (p = 0.206). By the 6‐month survey, 1 in G1 and 2 in G2 acknowledged having a POC. Satisfaction with TGC was positive with 9/11 (81.8%) extremely satisfied, 1 (9.1%) neutral, and 1 (9.1%) somewhat dissatisfied. See Figure 1 for average scores. Nine (81.8%) were very and 2 (18.2%) were somewhat comfortable seeing the counselor on video. Groups differed significantly when asked if they would use TGC again (p = 0.015); all in G1 strongly agreed and, in G2, 1 (20%) strongly agreed, 1 (20%) somewhat agreed, 2 (40%) were neutral, and 1 (20%) strongly disagreed. See Figure 2 for satisfaction mean scores for telemedicine as a modality for TGC.

Figure 1. Satisfaction with TGC session

Figure 2. Satisfaction with telemedicine
Conclusions: Although a small sample, results suggest that TGC may provide a good model to increase access to specialized GCs for PWH seen in HTCs without such specialty. Because all baseline questions were answered correctly, knowledge increment could not be assessed. The impact of TGC on management and POC was not significant. However, both women and GC’s expressed great satisfaction with the experience.
83. HTRS.P1.18
84. Single‐dose pharmacokinetics, efficacy, and safety of fibrinogen concentrate for on‐demand treatment of acute bleeding and surgical prophylaxis in patients with congenital fibrinogen deficiency
C. Djambas Khayat1; S. Lohade2; F. D'Souza3; L. Gowda4; O. Zekavat5; I. Kruzhkova6; B. Schwartz 7; C. Solomon6; F. Peyvandi8
1 Hotel De Dieu de France, Beirut, Beyrouth, Lebanon; 2 Sahyadri Specialty Hospital, Pune, Maharashtra, India; 3 St.John's Medical College Hospital, Bangalore, Karnataka, India; 4 SS Institute of Medical Science and Research Center, Davangere, Karnataka, India; 5 Hematology Research Center, Nemazee Hospital, Shiraz University of Medical Sciences, Shiraz, Fars, Iran; 6 Octapharma, Lachen, Schwyz, Switzerland; 7 Octapharma, Paramus, New Jersey, USA; 8 Fondazione IRCCS Ca'Granda Ospedale Maggiore Policlinico, Angelo Bianchi Bonomi Hemophilia and Thrombosis Center and Fondazione Luigi Villa Università degli Studi di Milano, Milan, Lombardia, Italy
Background: Congenital fibrinogen deficiency (CFD) is a rare disorder characterized by lack of/low levels of functional fibrinogen. Human fibrinogen concentrate (HFC) is administered for treating bleeding episodes (BEs) and preventing blood loss during surgery in CFD patients.
Objectives: Here we compiled the total experience in clinical trials of the single‐dose pharmacokinetics (PK), efficacy and safety of a new HFC (Fibryga®, Octapharma AG) in adult/adolescent (≥12 yrs) and pediatric (<12 yrs) CFD patients.
Methods: Three studies were conducted as part of the clinical development program for this HFC. FORMA‐01/FORMA‐02 were Phase 2 PK/Phase 3 efficacy and safety studies of patients ≥12 yrs. FORMA‐04 was a Phase 3 PK/efficacy/safety study of patients <12 yrs. All PK analyses were performed examining fibrinogen levels at multiple time points after a single 70 mg/kg HFC infusion. Hemostatic efficacy was assessed for each BE and surgery by an Independent Data Adjudication Committee (IDMEAC) using objective 4‐point scales; treatment success defined as an excellent/good rating. Safety was assessed by monitoring adverse events (AEs).
Results: The median (range) age of patients was 23 years (12–53) in FORMA‐01 (n = 21), 26.5 years (12–54) in FORMA‐02 (n = 24) and 6 years (1–10) in FORMA‐04 (n = 14). PK was assessed in 21 patients ≥12 yrs and 13 patients <12 yrs and the results are shown in Table 1, displaying numerically lower values for e.g., area under concentration‐time curve (AUC), and incremental in vivo recovery (IVR) in younger patients, as described for other coagulation factor concentrates. HFC BE treatment and surgical prophylaxis doses are shown in Table 2. Hemostatic efficacy was assessed for 24 patients (89 BEs) and 9 patients (12 surgeries) in those ≥12 yrs, and 8 patients (10 BEs) and 3 patients (3 surgeries) in those <12 yrs. For BE treatment, the IDMEAC rated HFC as successful in 98.9% of patients ≥12 yrs; 100% in patients <12 yrs. For surgical prophylaxis, HFC was rated as 100% successful in both age groups. In patients ≥12 yrs, 5 AEs were reported as possibly related to HFC treatment (drug eruption (2), pyrexia, phlebitis and thrombosis). In patients <12 yrs, 2 AEs were reported as possibly related to HFC; pyrexia and portal vein thrombosis following splenectomy for spontaneous spleen rupture. No deaths or severe/serious allergic reactions were reported.
Table 1. Line, clot and treatment characteristics of the cohort

Table 2. Dose of HFC administered for the treatment of BEs and surgical prophylaxis in FORMA‐02 and FORMA‐04

Conclusions: Satisfactory PK profiles of HFC were observed for both adult/adolescent patients and pediatric patients. Numerical differences for e.g. AUC and IVR between age groups were as expected for this type of therapeutic agent. HFC efficacy was comparable between the two age groups for both BE treatment and prophylaxis of perioperative bleeding, and so was the safety data.
85. HTRS.P2.13
86. HTRS Student Research Award: Reduced health‐related quality of life is associated with depression and cognitive impairment among survivors of thrombotic thrombocytopenic purpura
S. Selvakumar 1; S. Chaturvedi2; J. Yu3; A. Moliterno3; M. Streiff3; R. Brodsky3
1 Dr. Kiran C. Patel College of Allopathic Medicine, Nova Southeastern University, Lake Mary, Florida, USA; 2 Johns Hopkins University School of Medicine, Baltimore, Maryland, USA; 3 Division of Hematology, Johns Hopkins School of Medicine, Baltimore, Maryland, USA
Background: Thrombotic thrombocytopenic purpura (TTP) is a life‐threatening blood disorder characterized by episodes of microvascular thrombosis and ischemic organ damage. TTP was previously considered an acute disorder; however, over long‐term follow‐up, TTP survivors exhibit higher rates of mortality (Deford et al., Blood 2013), cognitive impairment (Falter et al., Transfusion 2017), depression (Chaturvedi et al., Thrombosis Res. 2017), and cerebrovascular disease (Cataland et al., Am. J Hematol. 2011). The long‐term impact of TTP on psychological well‐being and health‐related quality of life (HRQOL) is understudied.
Objectives: We conducted this cross‐sectional study to determine the prevalence of reduced HRQOL and its association with depression and cognitive deficits among TTP survivors.
Methods: Participants completed two validated self‐administered surveys: Beck Depression Inventory (BDI‐II) and SF‐36 for HRQOL. Semi‐structured phone interviews were conducted to obtain patient perspectives. The NIH Toolbox Cognition Battery, an extensively validated iPad‐based cognitive assessment tool, was administered to a subset of participants.
Results: Twenty‐six adults with TTP were enrolled. Median time since last TTP episode was 21 months, median age was 48.2 (37.8–58.3), and 84.6% were female. The SF‐36 has eight domains and two summary scores (mental, MCS; physical, PCS). Compared to the reference population, TTP survivors had lower mean scores in all domains with statistical significance in six domains and summary scores: physical functioning (p < 0.01), physical role limitations (p < 0.001), general health (p = 0.0001), mental health (p < 0.01), vitality (p < 0.001), emotional role limitations (p < 0.01), MCS (p < 0.01), and PCS (p < 0.05) (Figure 1). HRQOL impairment was driven primarily by physical components. Depression (BDI‐II > 13) was present in 38%. Patients with depression had a significantly lower mean MCS (32.6 ± 7.28) compared to those without depression (51 ± 9.39, p < 0.0001). Common issues identified in semi‐structured interviews included fear of relapse (N = 15) and difficult physical recovery (N = 14). Thirteen of the participants subsequently completed cognitive testing. The associations between fluid cognition composite and crystallized cognition composite (T‐scores with normative mean, 50 ± 10) adjusted for age, sex, education, and race were compared to their corresponding MCS and PCS. The PCS was correlated with the fluid cognition composite (Pearson’s correlation r, 0.835) (Figure 2) but not the crystallized composite, and there was no association between the MCS and cognitive impairment.

Figure 1. Comparison of SF‐36 Domains and Summary Scores between TTP study participants (N = 26, blue circles) and the Reference U.S. Population (red circles)

Figure 2. Correlation between SF‐36 Physical Component Summary Scores and Fluid Cognition Composite T‐Scores Among TTP Study Participants (N = 13)
Conclusions: TTP survivors have high rates of reduced HRQOL in multiple domains, which is associated with depression. Preliminary investigation of cognitive impairment in our participants suggests a possible association between physical HRQOL and fluid cognition in TTP. Ongoing research will further explore the impact of TTP‐related cognitive deficits on depression and HRQOL.
Other: Supported by HTRS 2020 Student Research Award.
87. HTRS.P2.10
88. Magnetically powered microwheel thrombolysis of occlusive thrombi in zebrafish
C. Ku1; D. Disharoon2; D. Marr2; K. Neeves3; J. Shavit 1
1 University of Michigan, Ann Arbor, Michigan, USA; 2 Colorado School of Mines, Golden, Colorado, USA; 3 University of Colorado Denver, Denver, Colorado, USA
Background: Ischemic stroke occurs in 700,000 people yearly in the U.S., often resulting in long‐term disability. Tissue plasminogen activator (tPA) is able to effect thrombus dissolution and recanalization of occluded vessels, and is the only FDA approved treatment for ischemic stroke. However, it carries a significant risk for secondary hemorrhage. Catheter‐based thrombectomy can be effective, but cannot access small vessels and has its own risks. Therefore, additional options are needed to enhance treatment and reduce the associated risks, especially in small vessel thrombi which account for ~20% of ischemic strokes. We have previously shown that colloidal microparticles assembled into microwheels (uwheels) and functionalized with tPA, can assemble, rotate, and translate under the control of applied magnetic fields. These uwheels enable rapid thrombolysis of fibrin gels in microfluidic models of thrombosis. The zebrafish is a vertebrate that is characterized by rapid, external, and transparent development, and is essentially a living microfluidic model. The highly conserved coagulation system enables studies of hemostasis and thrombosis in the context of intact vasculature, coagulation factors, and blood cells.
Objectives: Introduce tPA functionalized uwheels into zebrafish to determine whether they can perform targeted recanalization in vivo.
Methods: We used laser‐mediated endothelial injury in the posterior cardinal vein (PCV) of 3 day old larvae, which results in rapid formation of an occlusive thrombus, followed by observation for recanalization. Infusion of tPA‐coated particles were performed without and with a magnetic field (“tPA‐particles” and “tPA‐uwheels,” respectively).
Results: The length of time for spontaneous recanalization after induced PCV thrombus formation was first established, which did not occur within 4 hours of observation, nor with infusion of saline or uncoated particles. Infusion of tPA alone resulted in lysis within 189.5 ± 70.6 minutes (n = 40), and was 232.2 ± 31.3 minutes (n = 16) for tPA‐particles without application of the magnetic field. When a magnetic field was applied to particles or tPA‐particles, only tPA‐uwheels were able to recanalize, in 11.8 ± 5.4 minutes (n = 15). tPA has been shown to have other roles in addition to plasminogen activation. In order to confirm the underlying mechanism, we evaluated tPA‐uwheels in plasminogen (plg) knockout fish. We found that using tPA‐uwheels, 90% of plg‐/‐ mutants (n = 20) were unable to be recanalized, but conversely 94% and 86% of wild‐type (n = 18) and heterozygous (n = 43) siblings, respectively, recanalized within 30 minutes.
Conclusions: Our data show that magnetically powered uwheels as a targeted tPA delivery system are dramatically more efficient at effecting plasmin‐mediated thrombolysis than systemic delivery. Further development of this system in fish and mammalian in vivo models could eventually enable a less invasive strategy for alleviating ischemia that is safer than directed thrombectomy or systemic infusion of tPA.
89. HTRS.P2.2
90. Clinical study to investigate the efficacy and safety of a von Willebrand Factor FVIII concentrate (VWF/FVIII) during prophylaxis in previously treated patients with von Willebrand disease (VWD)
R. Sidonio 1; L. Dubey2; M. Timofeeva3; K. Vilchevska4; V. Vdovin5; M. Schmid6; I. Kruzhkova6; S. Werner7
1 Aflac Cancer and Blood Disorders Center, Emory University, Atlanta, Georgia, USA; 2 Lviv National Medical University Danylo Halytskyi, Lviv, L'vivs'ka Oblast', Ukraine; 3 State Institution “National Children’s Specialized Hospital “OKHMATDYT” of the Ministry of Health of Ukraine,” Kyiv, Kyyiv, Ukraine; 4 Federal State Budgetary Institution of Science “Kirov Scientific Research Institute of Hematology and Blood Transfusion of the Federal Medical‐Biological Agency,” Kirov, Kirov, Russia; 5 Morosovskaya Children Clinical Hospital, Moscow Health Department, Department of General Hematology with the Pathology of Hemostasis, Moscow, Moskva, Russia; 6 Octapharma, Lachen, Schwyz, Switzerland; 7 Octapharma, Paramus, New Jersey, USA
Background: Inherited von Willebrand disease (VWD) is the most common inherited haemorrhagic disorder. Type 1 & 3 are characterized by quantitative von Willebrand factor (VWF) deficiency; type 2 arises from qualitative VWF deficiency, with severe bleeding reported in all subtypes. Treatment depends on VWD type/severity.
Objectives: Determine VWF/factor VIII (FVIII) concentrate (VWF/FVIII, Wilate, Octapharma AG) prophylactic treatment efficacy in previously treated patients with type 3, type 2 (except 2N), or severe type 1 VWD. Secondary objectives: determine VWF and FVIII procoagulant activity; pediatric pharmacokinetics (PK); incremental in vivo recovery (IVR); VWF/FVIII safety/tolerability and consumption data. The study also examines VWF/FVIII efficacy in bleeding episode (BE) treatment and surgical prophylaxis, quality of life (QoL) during prophylaxis, and changes in joint status and menstrual bleeding intensity.
Methods: WIL‐31 is a prospective, non‐controlled, international, Phase 3 study, enrolling 28 patients (≥6 yrs with VWD type 1, 2A, 2B, 2M, or 3). Eligible patients received frequent on‐demand treatment (ODT) with a VWF‐containing product and experienced ≥6 BEs (excluding menstrual bleeds) over 6 months, with ≥2 of these treated with a VWF‐containing product, or must have switched to prophylactic treatment with a VWF‐containing product within the past 2 years and their prior ODT bleeding history recorded. The ODT will be assessed as part of a run‐in observational study (WIL‐29) on bleeding profile prior to prophylaxis. Ten pediatric patients aged 6 to 16 years will have baseline PK profiles characterized for VWF (using ristocetin co‐factor activity [VWF:RCo]) and FVIII, based on blood samples taken pre‐ and post‐dosing. Prophylactic treatment with VWF/FVIII concentrate for 12 months will be initiated at study start or following PK. Patients will record all BEs to calculate BE frequency and annualized bleeding rate. Treatment efficacy will be assessed by the patient (and investigator in case of on‐site treatment) using a 4‐point scale. VWF/FVIII concentrate efficacy will be assessed at the end of surgery by the surgeon, post‐operatively by the hematologist, with an overall post‐operative efficacy assessment by the investigator.
Results: In July 2020, enrollment in the WIL‐29 lead ODT study was completed with 55 patients enrolled at 15 sites; Bulgaria, Croatia, Hungary, Russia, Ukraine and the USA. The first patient rolled over into the WIL‐31 prophylactic study in June 2020 and 14 patients are currently enrolled. Study end is expected Q2 2021.
Conclusions: While prophylactic treatment in other congenital bleeding disorders is widely accepted as standard of care to prevent bleeding and preserve patient QoL, there is very little data addressing long‐term VWD prophylaxis. WIL‐31 will provide data on prophylactic treatment efficacy in reducing bleeding rates, QoL impact of prophylaxis, joint status and menstrual bleed severity in VWD patients.
91. HTRS.O3.4
92. Risk of venous thromboembolism in pediatric hospitalized patients undergoing non‐cardiac surgery, a report from the Children's Hospital‐Acquired Thrombosis (CHAT) Consortium
E. Stephens 1; A. Nguyen2; J. Jaffray3; B. Branchford4; E. Amankwah5; N. Goldenberg2; E. Faustino6; N. Zakai7; A. Stillings8; E. Krava8; G. Young9; J. Fargo1
1 Akron Children's Hospital, Northeast Ohio Medical University, Akron, Ohio, USA; 2 Johns Hopkins All Children's Hospital, Saint Petersburg, Florida, USA; 3 Children’s Hospital Los Angeles, University of Southern California, Los Angeles, California, USA; 4 Versiti Medical Sciences Institute, Milwaukee, Wisconsin, USA; 5 Johns Hopkins School of Medicine, St. Petersburg, Florida, USA; 6 Yale School of Medicine, New Haven, Connecticut, USA; 7 University of Vermont Larner College of Medicine, Burlington, Vermont, USA; 8 Children’s Hospital Los Angeles, Los Angeles, California, USA; 9 Children’s Hospital Los Angeles, University of Southern California Keck School of Medicine, Los Angeles, California, USA
Background: Hospital‐acquired venous thromboembolism (HA‐VTE) is a rare complication in the pediatric population but the incidence has increased from 5.3 to 34–58 patients per 10,000. (Raffini L, et al. Pediatrics 2009) This is concerning because the mortality rate of pediatric VTE can be up to 2.2% (Monagle P., et al. Pediatric Research 2000) and up to 26% of patients can have post‐thrombotic syndrome (Goldenberg N, et al. Haematologica 2010). Surgery is a risk factor for developing HA‐VTE in pediatric patients with as many as 43% of patients developing HA‐VTE after undergoing surgery. (Jaffray J, et al. Thrombosis Research 2017) The Children’s Hospital‐Acquired Thrombosis (CHAT) Consortium maintains a large, multi‐institutional registry of HA‐VTE and non‐HA‐VTE patients. We used the CHAT Registry to identify HA‐VTE risk factors in patients who underwent non‐cardiac surgery.
Objectives: To assess whether the risk of developing HA‐VTE differs across six types of non‐cardiac surgery and identify HA‐VTE risk factors in these patients.
Methods: This analysis included 371 HA‐VTE cases and controls from the CHAT Registry who had undergone a single non‐cardiac surgery during their hospitalization. Associations between six categories of surgery and sixteen putative HA‐VTE risk factors were assessed by unadjusted and adjusted logistic regression models. Variables with p‐value ≤0.10 in unadjusted models were included in adjusted analysis for further evaluation. The final model was derived using backward selection with significance level of 0.05.
Results: From January 2012 – March 2020, 163 non‐cardiac surgery HA‐VTE cases [median age 5.7 years (IQR = 0.3–14.2)] were identified, with 208 controls [median age of 7.5 years (IQR = 3.7–12.9)]. In the final multivariable model, we found that thoracic surgery and neurosurgery had an increased risk of HA‐VTE [odds ratio (OR) = 5.73 (95% CI 1.39–23.52) and 3.41 (95% CI 1.34–8.70)] using abdominal surgery as the reference. CVC placement was associated with increased VTE risk [unadjusted OR = 31.72 (95% CI 19.23–52.33)], with temporary CVC (internal jugular, subclavian and femoral CVC) and PICC having the strongest effect [adjusted OR = 35.28 (95% CI 9.86–126.21) and 14.51 (95% CI 6.71–31.39)]. We also found that the risk of HA‐VTE increased with hospital length of stay [OR = 1.04, (95% CI 1.00–1.07), for each one‐day increase] and with prior hospitalization in the month preceding surgery [OR = 4.03 (95 % CI 1.82–8.92)] (Table 1).
Table 1. Multivariate models for VTE Case‐ Control status among participants with a single surgery (n = 371)

Conclusions: After adjustment in a multivariate model, those undergoing thoracic or neurosurgery, prior hospitalization within 1 month and the presence of a PICC line or temporary CVC were statistically significant, independent risk factors for HA‐VTE in pediatric patients who have undergone a single non‐cardiac surgery.
93. HTRS.P1.11
94. Perceptions about the use and efficacy of Extended Half‐Life (EHL) factor products in persons with hemophilia: A national survey of providers from hemophilia treatment centers in the United States
N. Swaminathan 1; V. Salinas‐Luna2; S. Acharya3; A. Sharathkumar1
1 University of Iowa Hospitals and Clinics, Iowa City, Iowa, USA; 2 Children`s Hospital of Orange County, Orange, California, USA; 3 Northwell Health Hemostasis and Thrombosis Center, Cohen Children's Medical Center, Zucker School of Medicine at Hofstra/ Northwell, New Hyde Park, New York, USA
Background: Extended Half‐Life factor products (EHLs) are an important therapeutic advancement that has reduced treatment burden in persons with hemophilia (PWH). While clinical trials evaluating the safety and efficacy of EHLs have shown promising results, reports from few centers in the United States have questioned their efficacy in treating acute bleeding diatheses in PWH. Since growing number of patients are being switched from standard half‐life (SHL) products to EHLs, it is important to gain deeper insight about their efficacy in managing acute bleeding, surgery and other practical challenges faced by clinicians.
Objectives: The objectives of this study were to examine the hemophilia treaters’ experience with the use of EHLs and to understand their perceptions about the efficacy of EHLs for the treatment of acute bleeding diatheses and surgeries in PWH.
Methods: An online multiple‐choice survey (Qualtrics®) was developed and used. The survey population included hemophilia treaters from Hemophilia Treatment Centers (HTCs) in the United States. The survey included the following domains: (i) Provider demographics, (ii) Current landscape of EHL use at the center, (iii) Clinical indications of EHLs, (iv) Laboratory monitoring of EHLs, (v) Clinical scenarios for management of bleeding diatheses and (vi) Considerations while switching from SHL to EHLs for prophylaxis and on demand treatment in PWH. Descriptive statistics was used to report categorical data.
Results: 70 providers from 149 HTCs completed the survey. Thus, the response rate was 47% (95% CI 39% to 55%) and 68% (n = 48) of respondents were pediatric hematologists. Results showed that clinicians use EHLs for a variety of indications in persons with severe and non‐severe hemophilia. About 48% of respondents had used EHLs in previously untreated patients (PUPs). Majority of respondents, ~94% (n = 66) for persons with hemophilia A (PWHA) and ~84% (n = 59) for persons with hemophilia B (PWHB), preferred to use the same EHL product for acute bleeding in PWH on prophylaxis with EHLs (Figure 1). More number of providers (~66%, n = 46) had used EHLs for acute bleeding in persons with moderate hemophilia than in mild hemophilia (~23%, n = 16). More number of providers (58.5%, n = 41) were willing to switch PWHB than PWHA (20%, n = 14) to EHLs for prophylaxis. For major surgery, > 80% of providers preferred to use SHL over EHLs in PWHA and PWHB (Figure 2).

Figure 1. What will you use to treat acute bleeding in a patient with Hemophilia on EHL product for prophylaxis?

Figure 2. What product will you use in a patient with Hemophilia requiring neurosurgery?
Conclusions: Our study informs about the perception of efficacy of EHLs in PWH among hemophilia treaters in the United States. Clinicians are comfortable with using EHLs in PWH including PUPs. The use of EHLs for acute bleeding varies based on severity of hemophilia. Most clinicians did not have concerns with the efficacy of EHLs in controlling acute bleeding in PWH. Clinicians have a higher comfort level with factor IX EHLs for prophylaxis in PWH. Most providers preferred SHLs over EHLs for major surgery. Overall, this study did not identify major concerns about the efficacy of EHLs in managing bleeding diatheses in PWH.
95. HTRS.P2.3
96. Community Voices in Research (CVR); a patient‐centric approach moving the future of inherited bleeding disorders forward
R. Vidal
The National Hemophilia Foundation, New York, New York, USA
Background: Community Voices in Research (CVR) is a community powered registry with a patient centric approach. In order to bring the community member’s voice to the forefront, the National Hemophilia Foundation (NHF) created CVR; a partnership between the bleeding disorders community and NHF, since its premiere in March 2019 over 800 participants have enrolled. Individuals and families in the bleeding disorders community and those who experience other chronic rare conditions have not typically been engaged in patient reported registries or in clinical studies during the initial study design phase, it is uncommon for patient burden to be taken into consideration as protocols are developed, however the patient’s voice is just as critical as the clinicians. CVR provides both individuals with an inherited bleeding disorder (IBD), and their family members and/or caregivers, with the opportunity to direct the future of research when they enroll and add their voices by answering surveys or participating in virtual advisory panels. Aside from providing the IBD community with the platform to amplify their voice and receive ongoing educational support via their personalized dashboard, CVR is also asking questions that have not been asked before, therefore providing the community with vital data points necessary to help move the future of IBD forward.
Objectives: Increase the understanding of the lived experience, engaging external researchers, monitor how social trends impact the population, and monitor patterns and changes longitudinally. Recognize and accept the value in the patient’s feedback, acknowledge the ability of patients/caregivers to provide input in clinical trial design before the study is developed, integrate the design of patient‐centric procedures and schedules, design patient ‐centric study designs, and overcoming internal barriers to patient engagement.
Methods: Longitudinal surveys, virtual advisory panels, embedded educational resources and rapid research cycles.
Results: On October 8th, 2020 there were a total of 795 participants enrolled in CVR; 535 (67%) reported having a bleeding disorder and 260 (33%) reported being a caregiver of a person with a bleeding disorder. When asked about their sex at birth: 416 (52.3%) females and 379 (47.7%) males, however when asked about gender identity the numbers changed slightly to 413 (51.9%) participants who identify as females and 376 (47.3%) who identify as males and 6 other; composed of 4 (0.5%) participants whom identified as transgender and 2 (0.3%) whom selected “I prefer not to answer”
Conclusions: The more participants enrolled in CVR the stronger the data and the community. CVR will establish an audience to draw in for opportunities to participate in patient‐reported outcome research and other efforts as well as providing the opportunity to educate and inform individuals and families in the bleeding disorders community (Figure 1, Figure 2).

Figure 1. What We've Learned; HTC vs Non‐HTC Care Among CVR Participants

Figure 2. What We've Learned; Women's Average Clot Size
97. HTRS.O4.2
98. Etranacogene dezaparvovec (AAV5‐padua hfix variant), an enhanced vector for gene transfer in adults with severe or moderate‐severe hemophilia B: Two year data from a phase 2B trial
A. Von Drygalski 1; A. Giermasz2; G. Castaman3; N. S. Key4; S. U. Lattimore5; F. W. G. Leebeek6; W. A. Miesbach7; M. Recht8; E. Gomez9; R. Gut10; S. Pipe11
1 University of California San Diego, La Jolla, California, USA; 2 University of California Davis, Sacramento, California, USA; 3 Azienda Ospedaliera Universitaria Careggi, Florence, Emilia‐Romagna, Italy; 4 University of North Carolina, Chapel Hill, North Carolina, USA; 5 The Hemophilia Center, Oregon Health & Science University, Portland, Oregon, USA; 6 Erasmus University Medical Center, Rotterdam, Zeeland, Netherlands; 7 University of Frankfurt, Frankfurt, Hessen, Germany; 8 Oregon Health & Science University, Portland, Oregon, USA; 9 Phoenix Children’s Hospital, Phoenix, Arizona, USA; 10 uniQure, Lexington, Massachusetts, USA; 11 University of Michigan, Ann Arbor, Michigan, USA
Background: Gene therapy for hemophilia offers the possibility to ameliorate disease severity to a mild or functionally curative state through a single administration. Etranacogene dezaparvovec (AMT‐061) is an investigational gene therapy for hemophilia B comprising an adeno associated virus serotype 5 (AAV5) vector containing a codon‐optimized Padua variant human factor IX (FIX) gene with liver specific promoter.
Objectives: We have previously demonstrated that a single dose of etranacogene dezaparvovec provides sustained FIX activity into the mild‐to normal range for up to 52 weeks post‐dose in participants with severe or moderate‐severe hemophilia B. Here, two years of follow‐up data will be presented.
Methods: A Phase 2b, open‐label, single‐dose, single‐arm, multi‐center trial (NCT03489291) in adult hemophilia B subjects who were not excluded based on neutralizing antibodies to AAV5. All subjects received a single intravenous dose of etranacogene dezaparvovec (2 × 10e13 gc/kg) and are being followed for 5‐years. The primary endpoint was FIX activity at Week 6. Secondary endpoints include e‐diary recordings of bleeds and FIX concentrate use, laboratory parameters, joint health, patient reported outcomes, and adverse events (AEs).
Results: All participants had FIX ≤1% (severe or moderately‐severe FIX deficiency), required routine FIX prophylaxis, and had neutralizing activity to AAV5 at baseline. Following AMT‐061 treatment, FIX activity increased rapidly to a mean of 31% at Week 6. At Week 52, mean FIX activity increased further to 41% with FIX activity levels of 50%, 31% and 41% in participants 1–3 respectively. There was no relationship between response to etranacogene dezaparvovec and the presence of anti‐AAV5 NAbs. As of 52 weeks, there were no bleeds post‐treatment and no requirement for FIX replacement aside from protocol specified use for perioperative management in participant 3. There were no clinically significant elevations in liver enzymes and no participants required steroids related to the treatment. One participant experienced 2 mild AEs possibly related to treatment shortly after dosing (self‐limiting headache and slightly elevated CRP). Participant 3 had hip surgery due to worsening of pre‐existing avascular necrosis (deemed unrelated to treatment by the investigator) and received FIX per protocol according to standard clinical practice. No participant developed inhibitors to FIX. Updated results to 2 years of follow‐up will be presented.
Conclusions: Patients with AAV5 NAbs are included in this Phase 2b etranacogene dezaparvovec trial and have shown sustained FIX activity into the mild‐to normal range. All participants have been able to discontinue routine prophylaxis, and there have been no bleeds post‐treatment with etranacogene dezaparvovec.
99. HTRS.O2.4
100. A randomized, pragmatic trial to assess a pediatric real‐time risk prediction model for thrombotic events
S. Walker; C. Creech; R. Moore; H. Domenico; B. French; D. Byrne; A. Wheeler
Vanderbilt University Medical Center, Nashville, Tennessee, USA
Background: The frequency of hospital acquired venous thromboembolism (HA‐VTE) is increasing in pediatric patients (Raffini et al., Pediatrics, 2009). Given that risk prediction models can identify patients at risk for venous thromboembolism better than physician judgement alone (Ellis et al., J Am Acad Orthop Surg, 2019), we developed and validated a general pediatric risk prediction model from a large, single center cohort, using 111,352 admission encounters in the derivation cohort and 44,138 encounters in a separate validation cohort. The model includes 11 variables (Table 1), easily extracted from the electronic medical record (EMR), that are available at admission and updated daily. The model has excellent discriminatory capability, with a c‐statistic of 0.907 in the derivation cohort and 0.904 in the validation cohort. To more broadly assess the utility of this model in identifying high risk pediatric inpatients, we developed the following pragmatic trial that has been approved by the Vanderbilt University Medical Center Institutional Review Board.
Table 1. Pediatric risk prediction model

Objectives: This study is designed to assess the efficacy of using a novel risk prediction model with targeted intervention to improve outcomes of hospitalized pediatric patients at high risk for HA‐VTE through a randomized, pragmatic trial.
Methods: Figure 1 displays an overview of the study scheme. All patients admitted to Monroe Carell Jr. Children’s Hospital at Vanderbilt who are 21 years of age and younger will be included in the study. The automated EMR‐based prediction model described above will be used to calculate an HA‐VTE risk prediction score on all patients. Patients randomized to the intervention arm will have their score displayed to the HA‐VTE research team, comprised of hematologists, who will review the clinical situation and make recommendations regarding prophylactic interventions, including anticoagulation, to the admitting team. The risk prediction scores of patients randomized to the control arm will not be displayed to the research team. Both groups will continue to receive current standard of care. The primary endpoint of the study is the frequency of HA‐VTE events per arm, and secondary endpoints include the number of patients in the intervention arm who receive anticoagulation and the number of bleeding‐related adverse events.

Figure 1. Overview of the study scheme
Results: The pragmatic trial is underway. We anticipate approximately 20,000 eligible patient admissions during one year of the study, with 10,000 patients expected to be in the intervention arm.
Conclusions: We report a novel study design to evaluate efficacy of a HA‐VTE predictive model in a general pediatric inpatient population. Multiple aspects of the trial occur in an automated fashion, which provides faster access to data, increased ease of use, and potential applicability across various medical centers. We anticipate that data from this trial will improve identification of pediatric patients at high risk for HA‐VTE development across the country.
Other: N/A.
101. HTRS.O4.1
102. AMT‐060 gene therapy in adults with severe or moderate‐severe hemophilia B confirm stable fix expression and sustained reductions in bleeding and factor IX use for up to 5 years
F. W. G. Leebeek 1; K. Meijer2; M. Coppens3; P. Kampmann4; R. Klamroth5; R. Schutgens6; G. Castaman7; E. Seifried8; J. Schwaeble8; H. Halvard Bönig9; E. K. Sawyer10; W. A. Miesbach11
1 Erasmus University Medical Center, Rotterdam, Zeeland, Netherlands; 2 University Medical Center Groningen, Groningen, Noord‐Holland, Netherlands; 3 Amsterdam University Medical Centers, University of Amsterdam, Amsterdam, Noord‐Holland, Netherlands; 4 Rigshospitalet, Copenhagen, Syddanmark, Denmark; 5 Vivantes Klinikum im Friedrichshain, Berlin, Germany; 6 Van Creveldkliniek University Medical Center Utrecht, Utrecht, Netherlands; 7 Azienda Ospedaliera Universitaria Careggi, Florence, Emilia‐Romagna, Italy; 8 German Red Cross Blood Service Baden‐Württemberg‐Hessen, Frankfurt, Baden‐Wurttemberg, Germany; 9 Goethe University, Frankfurt, Baden‐Wurttemberg, Germany; 10 uniQure, Lexington, Massachusetts, USA; 11 University of Frankfurt, Frankfurt, Hessen, Germany
Background: Gene therapy aims to provide long‐term therapeutic benefit from a single administration. AMT‐060 is an adeno‐associated virus serotype 5 (AAV5) vector with a codon‐optimized wildtype human factor IX (FIX) gene and liver‐specific promoter and is being evaluated in 10 participants with severe/moderate‐severe hemophilia B over 5 years (Phase 1/2 study, NCT02396342).
Objectives: To describe efficacy and safety outcomes from an analysis up to 5‐years post‐AMT‐060.
Methods: Adult males with FIX activity ≤2% and a severe bleeding phenotype received a single intravenous infusion of AMT‐060 (5 × 1012 gc/kg, Cohort 1, n = 5) or (2 × 1013 gc/kg, Cohort 2, n = 5). Assessments include FIX activity, FIX replacement use, annualized bleeding rate (ABR), treatment‐related adverse events (TRAE), immunological and inflammatory biomarkers up to 5 years (Cohort 1) and 4.5 years (Cohort 2).
Results: As of November 2019, mean FIX activity was 5.1% at 4.0 years for Cohort 1 versus 4.4% in Year 1; 6.8% in Year 2; 7.3% in Year 3; and 7.0% in Year 4. Mean FIX activity for Cohort 2 was 7.5% versus 7.1% in Year 1; 8.4% in Year 2; 7.9% in Year 3; and 7.4% in Year 4. Mean ABR during the last 12, and 6 months of observation, was 3.3 for Cohort 1 and 0.0 for Cohort 2, respectively, representing a 77% and 100% reduction in the year prior to treatment. During the same period, FIX replacement therapy consumption declined 90% (Cohort 1) and 100% (Cohort 2). Eight of 9 participants using prophylaxis at baseline were able to discontinue. No participants developed FIX inhibitors or signs of sustained AAV5 capsid‐specific T‐cell activation. As previously reported, TRAE were mainly reported in the first 3.5 months after treatment, including three participants who experienced transient mild elevations in alanine aminotransferase. One additional TRAE (joint swelling post‐exercise) was observed during the last 12 months of observation post‐treatment. Updated data, up to 5‐years of observation, will be presented.
Conclusions: Long‐term, stable endogenous FIX activity and reductions in ABR and use of FIX replacement were maintained over multiple years following a single treatment with AMT‐060. There were no additional safety concerns with longer term follow‐up. This data supports the ongoing Phase 3 study of the enhanced construct etranacogene dezaparvovec (AMT‐061), which encodes the highly active Padua FIX variant.
103. HTRS.O1.4
104. Real‐world data from a us claims database among patients with hemophilia A treated with recombinant FVIII FC fusion protein
A. Wilson 1; R. Preblick2; A. Swenson3; J. Dumont1
1 Sanofi, Cambridge, Massachusetts, USA; 2 Sanofi, Bridgewater, New Jersey, USA; 3 OM1, Inc., Boston, Massachusetts, USA
Background: Hemophilia A is characterized by spontaneous bleeds and excessive bleeding after trauma. Prophylactic factor VIII (FVIII) replacement is the standard of care to prevent life‐threatening bleeds and bleeds that cause major morbidity. Recombinant FVIII Fc fusion protein (rFVIIIFc [Sanofi, Cambridge, MA, USA]) was the first extended half‐life FVIII approved in the United States (US) and is indicated for on‐demand treatment of bleeding episodes, prophylaxis to reduce bleed frequency, and perioperative management in children and adults with hemophilia A. While there are reports of post‐market experience switching to rFVIIIFc, there is a need to further characterize real‐world use and outcomes.
Objectives: To describe patient characteristics, treatment patterns, consumption, and clinical outcomes in patients treated with rFVIIIFc in the real world.
Methods: This was a longitudinal retrospective observational cohort study of patients of all ages with hemophilia A treated with rFVIIIFc (Jan 2013–Apr 2020). Data were from the US‐based OM1® Real‐World Data Cloud (OM1, Inc., Boston, MA, USA), derived from deterministically linked, de‐identified, patient‐level health care claims from commercial insurers, Medicare and Medicaid, and patient electronic medical records (EMR). Claims and EMR data are updated quarterly and represent patients with a wide age range across the US. Patient records were included if they had ≥1 diagnosis code for hemophilia A, ≥1 record of rFVIIIFc filled or administered during the study period, and were represented in the study database for ≥12 months before the index date (the date that rFVIIIFc treatment was initiated). Study variables included demographics, hemophilia treatment patterns (analysis only of male patients with pharmacy claims during both pre‐ and post‐index periods), and clinical outcomes.
Results: During the study period, 761 patients with hemophilia A received rFVIIIFc treatment and had a mean (standard deviation [SD]) follow‐up time of 31.5 (19.2) months post‐index date. Of these, 527 patients were ≥12 years of age and 744 were male. For 219 male patients receiving rFVIIIFc and with available pharmacy claims, mean monthly factor consumption post‐index date was 50% less compared with those on FVIII prior to the index date (n = 175) (Table 1). Annualized bleed rate (ABR) was the rate of bleeding events per exposed year. Among patients with ≥6 months of exposure to rFVIIIFc, the mean (SD) ABR for those with evidence of inhibitors (n = 28) was 0.35 (1.18) versus 0.24 (1.44) for those without (n = 421). This measure is based on real‐world data, but as not all bleeds would be recorded by the OM1 data source if they were not documented in the EMR or submitted as a claim, it may underestimate ABR.
Table 1. Pharmacy claims data from 219 male patients receiving rFVIIIFc during the study period

Conclusions: Use of rFVIIIFc in patients with hemophilia A in the US was associated with a marked reduction in the mean monthly consumption of FVIII. The OM1 dataset can provide robust real‐world evidence for patients with hemophilia.
105. HTRS.P1.10
106. NHF builds a community‐driven national research agenda for inherited bleeding disorders
M. Witkop 1; M. Recht2; D. DiMichele3; L. Valentino4
1 National Hemophilia Foundation, New York, New York, USA; 2 American Thrombosis & Hemostasis Network, Portland, Oregon, USA; 3 Donna DiMichele Consulting, LLC, Washington DC, District of Columbia, USA; 4 National Hemophilia Foundation, Glenview, Illinois, USA
Background: The inherited bleeding disorders (IBD) community has witnessed significant advances in recent years thanks to novel therapeutic advances and technologies and improved diagnostic proficiency. Yet important gaps persistent, particularly for those with rare disorders and underserved populations, including women with IBD. A new initiative shaped by the voices of the patient community is underway to design and implement a national research roadmap addressing the most important needs within the community and accelerating progress through coordinated collaboration.
Objectives: To design and implement a national research agenda that embraces patient‐centric principles and holistically addresses the priorities of the IBD community, particularly among under‐represented populations. This is being informed through community listening activities and solicitation of input into the critical areas that can achieve dramatic improvements for the community today and in the future.
Methods: Virtual community listening sessions were conducted with diverse groups representing adults with IBDs, caregivers, patient organizations, chapter directors, healthcare providers, and industry representatives. These discussions will be supplemented by a survey developed by and administered through the National Hemophilia Foundation’s (NHFs) chapters as well as information gathered through NHF’s community powered registry, Community Voices in Research. These insights will be distilled into core themes, informing action by a steering committee and expert working groups as part of a State of the Science (SOS) Research Summit in 2021 culminating in a research roadmap designed and endorsed by the entire IBD community and championed by community research leaders.
Results: In total, 78 individuals participated in 9 listening sessions: 31% with an IBD, 5% caregivers, 51% healthcare professionals and 35% chapter leaders (Table 1). Important considerations were introduced to inform our research priorities such as new routes of administration and the prevention of passing a mutated gene to offspring. Patients and families cited opportunities for improvements in comprehensive care: mental health support, greater community awareness, improved tools for diagnosis and supportive care outside of the HTC settings, particularly for underserved segments of the community. Emerging trends illustrate the need for greater research progress in rare IBDs and in health disparities for under‐represented populations, including women and rare disorders, groups that often intersect. Additional insights from ongoing listening sessions will be included at the time of presentation.
Table 1. Patient listening session profiles

Conclusions: Actively soliciting the community’s views is central in our process to advance research in IBDs. At the end of the SOS process, with input from patients and families, we will have a roadmap that will positively impact and ring true to the voices heard during this planning process.
Other: Thanks to Kellie Hotz, K. Hotz Consulting Inc., for assistance with medical writing.
107. HTRS 2021 SCIENTIFIC SYMPOSIUM AUTHOR INDEX
A
Abshire T. C., HTRS.O1.2
Acharya S., HTRS.P1.8, HTRS.P1.11
Afzal A., HTRS.P2.9
Ahn H., HTRS.O2.2
Alexander W. A., HTRS.P2.1, HTRS.P2.5, HTRS.P2.15
Al‐Sabbagh A., HTRS.P2.1, HTRS.P2.5, HTRS.P2.15
Amankwah E., HTRS.O3.4
Anderson K. E., HTRS.P2.19
Arruda V. R., HTRS.P2.8
Asghar S., HTRS.P1.8
Astermark J., HTRS.O4.3
B
Baer M. R., HTRS.P1.3
Bajorek K., HTRS.P1.13
Balwani M., HTRS.P2.18, HTRS.P2.19
Bat T., HTRS.P2.16
Bdeir K., HTRS.O2.2
Becker J., HTRS.P2.16
Beg K., HTRS.P2.14
Bergmeier W., HTRS.O3.1
Bhoj V., HTRS.P2.8
Bilynsky E., HTRS.P1.2
Bissell D. M., HTRS.P2.19
Bonkovsky H., HTRS.P2.19
Bonzo D., HTRS.P2.1, HTRS.P2.5, HTRS.P2.15
Boothby A. B., HTRS.P2.11
Branchford B., HTRS.O3.4
Braun M., HTRS.O1.3
Brent T. V., HTRS.P2.12
Brodsky R. A., HTRS.P2.13
Burke T., HTRS.P2.6
Byon W., HTRS.O4.4
Byrne D. W., HTRS.O2.4
C
Callaghan M. U., HTRS.P2.1, HTRS.P2.5, HTRS.P2.15
Camire R., HTRS.O2.2
Canini J., HTRS.P1.4
Carpenter S., HTRS.P1.2
Carter Febres M., HTRS.P1.12
Castaman G., HTRS.O4.1, HTRS.O4.2, HTRS.O4.3
Catalfamo K., HTRS.P2.16
Chaturvedi S., HTRS.P1.6, HTRS.P2.13
Chen R., HTRS.P2.8
Chhabra A., HTRS.O4.4
Chidharla A., HTRS.P1.5
Christopherson P. A., HTRS.O1.1, HTRS.O1.2
Cines D., HTRS.O2.2
Cohen C. T., HTRS.O3.2
Coman D., HTRS.P2.19
Coppens M., HTRS.O4.1, HTRS.O4.3
Cordova‐Cueva L., HTRS.P2.17
Crary S., HTRS.O4.3
Creech C. B., HTRS.O2.4
Curtis R., HTRS.P2.6
D
D'avola D., HTRS.P2.19
Dhenge A., HTRS.O3.1
DiMichele D., HTRS.P1.10
Disharoon D., HTRS.P2.10
Djambas Khayat C., HTRS.P1.18
Domenico H. J., HTRS.O2.4
Drew C., HTRS.O2.3
D'Souza F., HTRS.P1.18
Dubey L., HTRS.P2.2
Ducore J., HTRS.P1.7, HTRS.P2.5, HTRS.P2.15
Dumont J., HTRS.O1.4
Dunn A., HTRS.P1.4
E
Eaton N., HTRS.O2.3
Enriquez M.M, HTRS.P1.7
Escobar M., HTRS.O4.3, HTRS.P1.1, HTRS.P1.8, HTRS.P1.14, HTRS.P2.1, HTRS.P2.15, HTRS.P2.5
Eto K., HTRS.O3.1
F
Falet H., HTRS.O2.3
Fargo J., HTRS.O3.4
Faustino E. V. S., HTRS.O3.4
Federman M., HTRS.P1.9
Ferrante F, HTRS.O1.3
Flood V. H., HTRS.O1.2
Fogarty P. F., HTRS.O4.4
French B., HTRS.O2.4
French D., HTRS.O2.2
G
Gage G., HTRS.P2.9
Garcia‐Perdomo H. A., HTRS.P2.17
Gibson N. M., HTRS.P1.9
Giermasz A., HTRS.O4.2, HTRS.P2.1
Giver J., HTRS.P1.4
Goldenberg N., HTRS.O3.4
Gollomp K., HTRS.O2.1
Gomez E., HTRS.O4.2, HTRS.O4.3
Gonzales S. E., HTRS.O1.2
Goto Y., HTRS.P2.19
Gouya L., HTRS.P2.19, HTRS.P2.18
Gowda L. S., HTRS.P1.18
Guillen‐Navaro E., HTRS.P2.19
Gut R., HTRS.O4.2
H
Haberichter S., HTRS.O2.3
Hale S. A., HTRS.P1.8
Hall J., HTRS.P2.7
Hardesty B., HTRS.P1.1
Harper P., HTRS.P2.19
Henderson S., HTRS.P2.7
Hermans C., HTRS.O4.3, HTRS.P2.5, HTRS.P2.15
Hirsh N., HTRS.P2.1
Holme P. A., HTRS.P1.7
Horie Y., HTRS.P2.19
Hudson Z., HTRS.P1.2
Hussain N., HTRS.P1.5
Hussain H., HTRS.P1.6
Hwang N. X., HTRS.P1.8
I
Ianova A., HTRS.P2.19
Ilonze C., HTRS.P1.16
Imran H., HTRS.P1.16
Ishizuka M., HTRS.O2.1
J
Jaffray J., HTRS.O3.4
Jakab D., HTRS.O2.3
Janbain M., HTRS.O1.1, HTRS.P1.1
Jesudas R., HTRS.P1.5
Journeycake J., HTRS.P2.1, HTRS.P2.5, HTRS.P2.15, HTRS.P2.14
K
Kampmann P., HTRS.O4.1, HTRS.O4.3
Kazmi S. R. S., HTRS.O4.3
Keel S., HTRS.P2.18, HTRS.P2.19
Kerlin B., HTRS.P1.4
Kesavan P., HTRS.P2.9
Kessler C., HTRS.P1.1, HTRS.P2.15
Key N. S., HTRS.O4.2, HTRS.O4.3
Khair K., HTRS.P2.6
Khan O., HTRS.P2.14
Klamroth R., HTRS.O4.1, HTRS.O4.3
Ko J., HTRS.P2.18
Kotowski A., HTRS.O4.3
Kouides P. A., HTRS.P1.8
Kowalska M. A., HTRS.O2.1, HTRS.O2.2
Krava E., HTRS.O3.4
Kruse‐Jarres R., HTRS.O4.3
Kruzhkova I., HTRS.P1.18, HTRS.P2.2
Ksayer R., HTRS.O1.1
Ku C. J., HTRS.P2.10
Kubisch I., HTRS.P2.19
Kuter D., HTRS.P2.18
L
Laffan M., HTRS.P2.6
Lange C., HTRS.O1.3
Langendonk J. G., HTRS.P2.19
Latif H., HTRS.P1.3
Lattimore S. U., HTRS.O4.2, HTRS.O4.3
Leebeek F. W. G., HTRS.O4.1, HTRS.O4.2, HTRS.O4.3
Leissinger C., HTRS.P2.15
Lemons R. S., HTRS.O4.3
Lim M., HTRS.P1.12
Liu S., HTRS.P2.18
Lohade S., HTRS.P1.18
Lucas T. L., HTRS.P2.4
Luchtman‐Jones L., HTRS.P1.13
Luck J., HTRS.P2.15
M
Maguire O., HTRS.P2.16
Mahlangu J., HTRS.P2.5, HTRS.P2.15
Malec L. M., HTRS.O3.3
Mancuso M. E., HTRS.P1.7
Marr D. W., HTRS.P2.10
Marri P., HTRS.P1.16
Marshall J. C., HTRS.O4.4
Mazepa M., HTRS.P2.11
McCrae K. R., HTRS.P1.6
McLaughlin P., HTRS.P2.6
Meeks S., HTRS.P2.5
Meijer K., HTRS.O4.1, HTRS.O4.3
Michaels L. A., HTRS.O1.3
Miesbach W. A., HTRS.O4.1, HTRS.O4.2
Minder E., HTRS.P2.19
Mirkheshti N., HTRS.P1.3
Mitchell I. S., HTRS.P2.5
Moake J. L., HTRS.O3.2
Moliterno A., HTRS.P2.13
Monda K., HTRS.P1.4
Montgomery R. R., HTRS.O1.1, HTRS.O1.2
Moore R. P., HTRS.O2.4
Morateck P. A., HTRS.O1.2
Morparia K., HTRS.P1.17
Morris B., HTRS.P1.5
Mullins E., HTRS.P1.13
N
Narang S., HTRS.P1.17
Nasr S., HTRS.P2.1
Neeves K. B., HTRS.P2.10
Nguyen A. T. H., HTRS.O3.4
Nichols C., HTRS.P1.15, HTRS.P2.12
Nugent D., HTRS.P1.1
O
O'Connell N. M., HTRS.O4.3
Oh J., HTRS.P2.19
O'Hara J., HTRS.P2.6
Olaiya O., HTRS.P1.2
O'Mahony B., HTRS.P2.6
Ostrow V., HTRS.P1.14
P
Palumbo J., HTRS.P1.13
Park K., HTRS.P1.3
Parker C., HTRS.P2.18, HTRS.P2.19
Pasi J., HTRS.O4.3, HTRS.P2.6
Peerlinck K., HTRS.O4.3
Peiro P A., HTRS.P2.19
Perry C. L., HTRS.O1.2
Peyvandi F., HTRS.P1.18
Pichardo‐Rodriguez R., HTRS.P2.17
Pipe S. W., HTRS.O4.2, HTRS.O4.3, HTRS.P2.5
Poncz M., HTRS.O2.1, HTRS.O2.2
Preblick R., HTRS.O1.4
Punzalan R., HTRS.O3.3
Q
Quon D., HTRS.P1.1, HTRS.P1.14, HTRS.P2.1, HTRS.P2.15, HTRS.O4.3
R
Ramakrishnan R., HTRS.P1.16
Rauova L., HTRS.O2.1
Recht M., HTRS.O4.2, HTRS.O4.3, HTRS.P1.10, HTRS.P2.1, HTRS.P2.5, HTRS.P2.15
Reding M. T., HTRS.P1.1, HTRS.P1.7, HTRS.P2.11
Rees D., HTRS.P2.18, HTRS.P2.19
Rhyee S., HTRS.P2.18
Rice M., HTRS.P2.12
Ritchie B., HTRS.P2.19
Roach G., HTRS.P1.9
Roberts J. C., HTRS.O1.2, HTRS.P1.6, HTRS.P1.8
Rodriguez V., HTRS.P1.4
Rodriguez Santana I., HTRS.P2.6
Ross G., HTRS.P2.19
Ruiz‐Franco O., HTRS.P2.17
Rupon J. W., HTRS.O4.4
Ryan S. E., HTRS.P1.15
S
Saavedra‐Velasco M., HTRS.P2.17
Salinas‐Luna V., HTRS.P1.11
Samelson‐Jones B., HTRS.P2.8
Sanfilippo K., HTRS.P2.9
Sankar A., HTRS.P1.4
Santaella M. E., HTRS.P2.12, HTRS.P1.15
Sardh E., HTRS.P2.19
Sarkar A., HTRS.O2.1
Sawyer E. K., HTRS.O4.1
Schmid M., HTRS.P2.2
Schoen M., HTRS.P2.9
Schulte M., HTRS.O2.3
Schutgens R., HTRS.O4.1
Schved J. C., HTRS.P2.15
Schwaeble J., HTRS.O4.1
Schwartz B., HTRS.P1.18
Seifried E., HTRS.O4.1
Selvakumar S., HTRS.P2.13
Shapiro A. D., HTRS.P2.15, HTRS.P2.5
Sharathkumar A., HTRS.P1.11
Sharma P., HTRS.P1.17
Shaver C., HTRS.P2.7
Shavit J. A., HTRS.P2.10
Sidonio R., HTRS.P2.15, HTRS.P2.2
Silver S., HTRS.P2.18, HTRS.P2.19
Simon A., HTRS.P2.18
Simpson M., HTRS.P1.7
Singleton T., HTRS.P2.1
Skinner M., HTRS.P2.6
Smith J. D., HTRS.O4.4
Solomon C., HTRS.P1.18
Song C., HTRS.P2.14
Sood S., HTRS.P1.1
Stein P., HTRS.P2.19
Stepanova V., HTRS.O2.2
Stephens E. T., HTRS.O3.4
Steward P., HTRS.P2.19
Stillings A., HTRS.O3.4
Stoelzel U., HTRS.P2.18
Stölzel U., HTRS.P2.19
Streiff M., HTRS.P2.13
Subramaniam S., HTRS.O2.3
Suhong L., HTRS.P2.9
Swaminathan N., HTRS.P1.11
Swenson A., HTRS.O1.4
Symington E., HTRS.O4.3
T
Tarango C., HTRS.P1.13
Tarantino M. D., HTRS.P1.6, HTRS.O1.2
Timofeeva M., HTRS.P2.2
Toohey C., HTRS.P2.14
Turk E., HTRS.P2.16
Turner N. A., HTRS.O3.2
V
Valentino L., HTRS.P1.10
van der Valk P., HTRS.O4.3
Vassiliou D., HTRS.P2.19
Vdovin V., HTRS.P2.2
Ventura P., HTRS.P2.18
Vidal R. P., HTRS.P2.3
Vilchevska K., HTRS.P2.2
Visweshwar N., HTRS.O4.3
von Drygalski A., HTRS.O4.2, HTRS.O4.3
W
Walker S. C., HTRS.O2.4
Walsh C., HTRS.P1.1
Wang M., HTRS.P2.15, HTRS.P1.14, HTRS.P1.8, HTRS.O4.3, HTRS.P2.5
Wang J. D., HTRS.P2.19
Wang M., HTRS.P1.7
Wang E., HTRS.P2.16
Warren B., HTRS.P1.14
Weiler H., HTRS.O2.3
Werner S., HTRS.P2.2
Wheeler A. P., HTRS.O2.4, HTRS.O4.3
Wiegmann S., HTRS.O1.3
Wilkinson T. A., HTRS.P2.15
Wilson A., HTRS.O1.4
Windyga J., HTRS.P2.19
Witkop M. L., HTRS.P2.12, HTRSP1.15, HTRSP1.10
Wufsus A., HTRS.P1.14
Y
Yeh H. W., HTRS.P1.2
Young G., HTRS.P2.5, HTRS.O3.4, HTRS.O4.3, HTRS.P2.15
Yu J., HTRS.P2.13
Z
Zaitsev S. V., HTRS.O2.2
Zakai N., HTRS.O3.4
Zakharia K., HTRS.O1.1
Zantek N., HTRS.P2.11
Zekavat O., HTRS.P1.18
Zimmerman Program Investigators, HTRS.O1.2
Disclaimer: This abstract book has been produced using author‐supplied copy. Editing has been restricted to some corrections of spelling and style where appropriate. No responsibility is assumed for any claims, instructions, methods or drugs dosages contained in the abstracts: it is recommended that these are verified independently.