

Abstract
Rationale:
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a rare disease, manifested by syncope or sudden death in children or young adults under stress conditions. Mutations in the Ca2+ release channel/ryanodine receptor (RyR2) gene account for about 60% of the identified mutations. Recently, we found and described a mutation in RyR2 N-terminal domain, RyR2R420Q.
Objective:
to determine the arrhythmogenic mechanisms of this mutation.
Methods and Results:
Ventricular tachycardias under stress conditions were observed in both CPVT patients and KI mice. During action potential recording (by patch-clamp in KI mouse cardiomyocytes and by microelectrodes in mutant hiPSC-CM) we observed an increased occurrence of delayed after-depolarizations (DADs) under isoproterenol stimulation, associated with increased Ca2+ waves during confocal Ca2+ recording in both mouse and human RyR2R420Q cardiomyocytes. In addition, Ca2+-induced Ca2+-release, as well as a rough indicator of fractional Ca2+ release, were higher and Ca2+ sparks longer in the RyR2R420Q expressing cells. At the ultrastructural nanodomain level, we observed smaller RyR2 clusters and widened junctional sarcoplasmic reticulum (jSR) measured by g-STED super-resolution and electronic microscopy, respectively. The increase in jSR width might be due to the impairment of RyR2R420Q binding to junctophilin-2, as there were less junctophilin-2 co-immunoprecipitated with RyR2R420Q. At the single current level, the RyR2R420Q channel dwells longer in the open state at low [Ca2+]i, but there is predominance of a subconductance state. The latter might be correlated with an enhanced interaction between the N-terminus and the core solenoid, a RyR2 inter-domain association that has not been previously implicated in the pathogenesis of arrhythmias and sudden cardiac death.
Conclusions:
the RyR2R420Q CPVT mutation modifies the interdomain interaction of the channel and weaken its association with junctophillin-2. These defects may underlie both nanoscale disarrangement of the dyad and channel dysfunction.
Keywords: CPVT, RyR2, cardiomyocyte calcium handling, calcium induced calcium release, ryanodine receptor, calcium sparks, cardiac myocyte, arrhythmia (mechanisms)
Subject Terms: Animal Models of Human Disease, Calcium Cycling/Excitation-Contraction Coupling, Ion Channels/Membrane Transport
Graphical Abstract
INTRODUCTION
Sudden Cardiac Death (SCD) represents about 15% of all deaths in developed countries1. It may occur in a structurally diseased heart, but also in apparently normal hearts where a channelopathy, i.e., a genetic disease affecting ion channel activity, is at the origin of the lethal tachyarrhythmia. Catecholaminergic polymorphic ventricular tachycardia (CPVT)2 is an inherited disease characterized by exercise- or stress-induced ventricular polymorphic tachycardia episodes in the absence of apparent structural heart disease. The disease is highly malignant, often manifesting for the first time in childhood or adolescence as syncopal events and/or SCD. CPVT patients have a mortality rate of ~30-50% by the age of 35 years when untreated. Mutations in the type 2 ryanodine receptor (RyR2) gene account for about 60% of the identified mutations and they are inherited following an autosomal dominant trait. Other CPVT-related genes encode for proteins which may bind to and modulate the RyR2 channel3. Thus CPVT is clearly an arrhythmogenic disorder affecting directly or indirectly RyR2 function, pointing to intracellular Ca2+ mishandling as the origin of the disease.
The RyR2 is a homotetramer with hydrophobic segments of the four identical subunits forming a central pore3. Usually pathogenic RyR2 CPVT mutations are arbitrarily clustered into three discrete protein regions or “hot spots”, including the N-terminus (32%), central (30%) and C-terminus domain (38%)4, 5. While a loss-of-function RyR2 mutation has been described6, 7, the vast majority of the mutations studied so far induce gain-of-function changes in RyR2. Different molecular mechanisms underlying defective RyR2 channel regulation have been proposed, including increased sensitivity to cytosolic8, 9 and sarcoplasmic reticulum (SR) luminal10 Ca2+, abnormal regulation by FKBP12.611 or calmodulin12 and defective intra- and inter-subunit 13, 14.
Recently, we described a new mutation in the RyR2 N-terminal domain (NTD), RyR2R420Q, in a Spanish family diagnosed for CPVT15. Remarkably, this mutation has also been found in other CPVT families16-19, and other pathogenic mutations at the same amino acid have also been identified18-20 suggesting that Arg420 forms part of a critical domain that controls RyR2 function. The molecular and cellular mechanisms underlying pathogenicity of this domain are still elusive despite several attempts to shed light on the subject21-23.
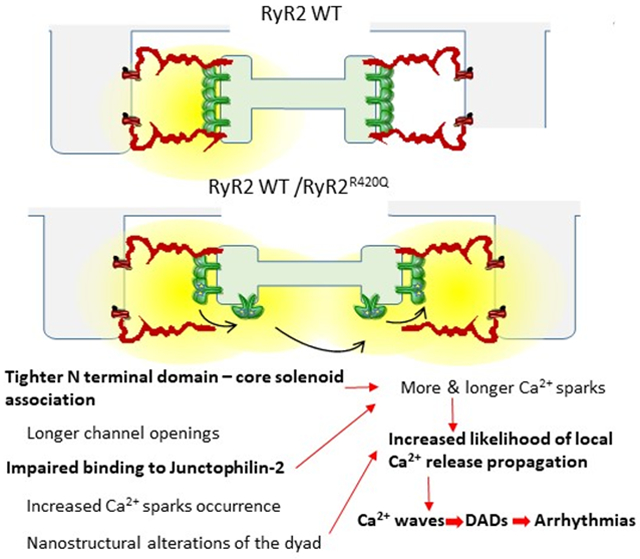
Herein we analyzed the arrhythmogenic mechanism of the N-terminal mutation RyR2R420Q using cardiac myocytes from mice harboring this mutation as well as induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CM) from a RyR2R420Q CPVT patient. Using multiple state-of-the-art approaches, we show that the RyR2R420Q mutation induces: a) alterations of channel intrinsic properties, with increased interaction between the NTD and the core solenoid, which may underlie enhanced subconductance state open probability at low Ca2+; and b) impairs the channel association with junctophilin-2, which affects both the ultrastructural organization of the dyads, and the distribution and size of the RyR2 clusters.
METHODS
Data Availability.
The authors declare that all supporting data are available within the article and its supplementary files.
Data was obtained from human, mouse, and heterologous system (HEK293 cell culture) expressing the RyR2R420Q CPVT mutation, using the methods detailed in the Online Supplement.
The Spanish family diagnosed with CPVT due to RyR2R420Q mutation15 underwent familial evaluation with a protocol conforming to the ethical guidelines of the 1975 Declaration of Helsinki previously approved by the local research ethics committee. Informed consent was obtained from each individual. Human induced pluripotent stem cells derived cardiomyocytes (hiPSC-CM) were obtained from patients’ blood samples (after personal consent and ethical approval) in Nantes iPSC core facility, (https://sfrsante.univ-nantes.fr/plates-formes/modeles-cellulaires-et-geniques/plate-forme-ipsc-nantes-cellules-souches-pluripotentes-induites-1101640.kjsp) and differentiated into cardiac myocytes in the laboratory.
Murine experiments were performed in male and female mice heterozygous RyR2R420Q knock in (KI) mice and wild-type (WT) littermates (4- 6 months old) in accordance with the ethics principles laid down by the French Ministry of Agriculture and ECC directive 86/609/ECC under protocol agreement n° B9201901.
Please see the Major Resources Table in the Supplemental Materials.
Statistical Analysis.
Statistical analysis were conducted using OriginPro and R statistical computing software. First we performed normality tests (which include the Kolmogorov-Smirnov, the Kolmogorov-Smirnov-Lilliefors, the Anderson-Darling, D'Agostino K-Squared, the Chen-Shapiro and the Shapiro-Wilk tests providing “can’t reject normality” or “reject normality” output) to determine whether each group constitutes a normally distributed population. When every tests gave “Can’t reject normality” output, we pursued with Levene's Test to check the homogeneity of variance between groups. If both criteria were met, we used either Student’s t-test or two way ANOVA or a conditional hierarchical linear mixed effect model used (lme4 R package24) to compare continuous variables between groups to take into account the multiple observations per animal. The group with or without treatment was the fixed effects, and animals were a random effect nested in the group; in the case of repeated measurements on cells, we added a random effect for cell in animal. When conditions of parametric tests were not met, we used either a Mann-Whitney test or the data were submitted to an Aligned Rank Transform procedure in ARTool R package (https://cran.r-project.org/web/packages/ARTool/readme/README.html,25;26), which allowed a non-parametric factorial ANOVA to be performed taking into account the multiple observations per animal and repeated measurements, when appropriated. Post hoc pairwise multiple comparisons were adjusted with Benjamini and Hochberg method. For proportions we used either Fisher’s Exact Test for Count Data or Generalized linear mixed model fit by maximum likelihood in case of repeated measures. Specific procedure analysis and output for each figure panels and tables are given in online Table III and no outliers were eliminated. Data are presented either as mean ± se or as box (with median and 25-75% range) and whisker (Min-Max). Differences were considered statistically significant if p<0.05 and presented in figures as *for p<0.05, **for p<0.005 and ***for p<0.0005. When p was found not statistically significant (p> 0.05), the exact p value is also provided in online Table III, and no sign is shown in the Figures.
The representative examples are chosen the closer to mean or median.
RESULTS
Alteration of Ca2+ homeostasis in RyR2R420Q induces ventricular arrhythmias.
Knock-in mice carrying the RyR2R420Q mutation found in CPVT patients15 presented a CPVT phenotype under stress challenges27. Compared to WT littermates, KI mice showed normal ventricular cardiac function, as assessed by M-Mode echocardiography (Online Table I) in line with the findings in RyR2R420Q patients15. Under basal conditions, telemetry-obtained electrocardiograms in freely moving KI and their WT littermates did not reveal significant changes, except lower heart rate (HR) in female KI mice, as previously shown27 (Online Table II). However, under emotional stress induced by short episodes (15s) of warm air blowing with a hairdryer, the occurrence of evoked ventricular tachycardia (VT) was higher in KI compared to WT (p<0.05 for genotype x hairdryer stress interaction, Fig. 1A). Likewise, Isoproterenol (ISO) injection elicited more episodes of VT in KI than in WT mice (p<0.05 for genotype x Iso treatment interaction, Fig. 1B). Thus, the KI mice recapitulate the main phenotype of human RyR2R420Q mutation carriers, namely, a normal cardiac structure and function, and stress-triggered ventricular arrhythmias. Consistent with this in vivo phenotype, delayed afterdepolarizations (DADs) and/or triggered activity (TA) were present in almost all patch-clamped ISO-stimulated KI ventricular cardiomyocytes, whereas less than half WT cardiomyocytes showed those pro-arrhythmic events (Fig. 1C). In the presence of Ca2+ chelator BAPTA (5 mM) into the patch pipette, ISO-induced DADs and TA were completely prevented in all myocytes (Fig. 1C, right), assigning a critical role to intracellular Ca2+ handling in the generation of this proarrhythmic cellular behavior. Of translational importance, we observed similar electrical abnormalities upon ISO perfusion (1 μM) during action potential (AP) recording (by a microelectrode technique) in cell monolayers of human hiPSC-CM from a carrier patient (III:7 from15) whose stress ECG is presented in Fig.1D. Compared to the hiPSC-CMs from his genotype- and phenotype-negative brother (Ctl) (III:4 from15), hiPSC-CMs from the RyR2R420Q patient (Mu) presented higher incidence of DADs after ISO treatment (Fig. 1E&F).
Figure 1. RyR2R420Q induces arrhythmias.

A. Left. Example of an ECG recording of KI mice showing ventricular tachycardia (VT) (arrows) after emotional stress (hairdryer blowing protocol). Right. VT episodes during 10 minutes recording in mice before and under emotional stress in 6 WT and 7 KI mice. B. Left. VT occurrence in KI mouse after 1 mg/Kg ISO injection. Right. Incidence of VT after ISO injection in 14 WT and 15 KI mice. C. Left. Representative AP recordings from WT and KI myocytes with no Ca2+ chelator (left) or with 5 mM BAPTA (right) in the intracellular solution during 100 nM ISO perfusion. The extra AP, consistent with triggered activity (TA) is marked by an arrow. The other arrow points to a DAD. Right. The occurrence of DADs / Triggered Activity (TA) measured in WT and KI myocytes, either without Ca2+ chelator in the intracellular solution (left panels) or in the presence of 5 mM BAPTA (right) expressed as % of cells displaying at least one event during 1 Hz stimulation. Numbers on the bars indicates n of positive cells/total. D. Exercise-induced ventricular arrhythmias during a Bruce protocol exercise testing (third and fourth stage) in a patient carrying the mutation RyR2R420Q (speed recording at 25mm/s) showing ventricular ectopic beats, bidirectional couplets and non-sustained ventricular tachycardia. E. AP recorded in an hiPSC-CM from the same patient (in D) showing DADs (marked by red arrows) during ISO perfusion (1 μM). F. DADs incidence in human hiPSC-CMs in basal conditions and during ISO perfusion in 19 control cells (Ctl) and 30 RyR2R420Q carrier cells (Mu). *p<0.05; **p<0.05; ***p<0.005. Statistical tests and exact p values are provided in Online Table III.
To gain mechanistic insights, we next analyzed RyR2R420Q function in situ by recording spontaneous Ca2+ sparks in intact quiescent cardiomyocytes. Figure 2A shows examples of line scan confocal images displaying Ca2+ sparks in ventricular cells from WT and KI littermates under basal conditions and during 100 nM ISO perfusion. The spontaneous Ca2+ sparks frequency was slightly (~1.5 fold) but significantly higher in KI than in WT cells (Fig. 2B), with ISO enhancing this parameter in both cell types (Fig. 2C). This overactivity of RyR2R420Q function was accompanied by Ca2+ sparks with higher peak amplitude that lasted significantly longer in cardiomyocytes from KI mice (full duration at half maximum amplitude), albeit no significant difference was observed in width (full width at half maximum amplitude) (Online Fig. I). These alterations in Ca2+ spark characteristics during diastolic periods in KI cells resulted in enhanced Ca2+ release per Ca2+ spark (Ca2+ spark mass, Online Fig. I) and in enhanced Ca2+ spark-mediated Ca2+ leak from the SR as evaluated in each cell through averaged Ca2+ spark mass multiplied by Ca2+ spark frequency (Fig. 2D). Of note, ISO significantly augmented those Ca2+ sparks parameters in both cell groups in a similar manner (Fig 2 C&E, Online Fig. I, no significant difference).
Figure 2. Mice and human cardiomyocytes expressing RyR2R420Q present more diastolic Ca2+ release.

A. Line scan confocal images showing Ca2+ sparks in ventricular cardiomyocytes isolated from a WT and a KI mouse in basal conditions and during 100 nM ISO perfusion. B. Ca2+ sparks frequency measured as number of events recorded per second in a 100 μm line of scan in 31 WT and 31 KI cells. C. Ca2+ spark frequency increase by 100nM ISO in 9 WT and 18 KI cells. D. Estimation of total Ca2+ release as Ca2+ sparks, calculated by multiplying the Ca2+ spark mass by the Ca2+ spark frequency in 27 WT and 28 KI cells. E. Percentage of increase in Ca2+ release through Ca2+ sparks by ISO in 8 WT and 18 KI cells. F. Line scan images of mouse ventricular cardiomyocytes from WT and KI cells without and with ISO. G. Percentage of arrhythmic cells during protocol such as in F. H. Example of RyR2 immunolabelling of an h-iPS-CM. I. Left. Line scan images of a h-iPS-CM from the CPVT patient electrically stimulated at 1 HZ in absence and presence of 1 μM ISO. Spontaneous diastolic Ca2+ release as Ca2+ waves are marked by the red arrows. Right. Percentage of arrhythmic cells in the presence of ISO in hiPS-CM control (Ctl) and mutant carrier (Mu). *p<0.05; **p<0.05; ***p<0.005. Statistical tests and exact p values are provided in Online Table III..
The exacerbated RyR2R420Q activity at rest might underlie the increased occurrence of arrhythmogenic events observed in AP recordings during patch-clamp experiments, which were completely blunted by intracellular Ca2+ chelation (Fig. 1C). Indeed, KI cardiomyocytes exhibited a strikingly higher incidence of Ca2+ waves following 4-Hz burst pacing, after 100 nM ISO exposure (Fig. 2F & G). Similarly, in 1-Hz electrically paced hiPSC-CMs from a RyR2R420Q carrier patient, which expresses RyR2s in a striated pattern (Fig. 2H), ISO elicited aberrant diastolic Ca2+ release during the diastolic periods (Fig. 2I, left), consistent with the DAD occurrence (Fig. 1E). Pooled data (Fig. 2I, right) showed a higher incidence of these aberrant diastolic Ca2+ releases in mutant RyR2R420Q hiPSC-CMs compared to control cells.
The above data shows that RyR2R420Q increases diastolic SR Ca2+ leak under resting conditions and increases propensity for Ca2+-dependent triggered arrhythmic activities, which are exacerbated by ISO, pointing out a pivotal role for SR Ca2+ mishandling. We next analyzed global intracellular Ca2+ ([Ca2+]i) handling in intact mouse cells by confocal microscopy. Examples of [Ca2+]i transients evoked at 2 Hz and then exposed to rapid 10 mM caffeine (as an index of the SR Ca2+ load) in WT or KI cardiomyocytes before and under ISO application are presented in Figure 3A. Consistent with the normal heart function detected with echocardiography in mice (Online Table I) and patients15, cardiomyocytes from KI mice showed normal [Ca2+]i transient amplitude and decay kinetics compared to WT, both at 2 and 4 Hz (Fig. 3B,C; no significant difference was observed between genotypes). β-adrenergic stimulation with ISO increased the [Ca2+]i transient amplitude and accelerated its decay time in both cell types (p<0.05 in each case assessed using paired t-test, Fig. 3D). While the ISO effect in accelerating [Ca2+]i transient decay time was similar between WT and KI, the increase in its peak was significantly smaller in KI mice (Fig. 3D). In regard to the SR Ca2+ load, KI cardiomyocytes had lower SR Ca2+ content than WT cells, but β-adrenergic stimulation blurred that difference (p<0.05 for genotype x Iso treatment interaction, Fig. 3A&E). Time of decay of the caffeine evoked [Ca2+]i transient was not different between the two groups (Fig. 3F), suggesting similar Na+/Ca2+ exchanger (NCX) function in the forward mode. Consistent with the kinetics of electrically and caffeine-evoked [Ca2+]i transients that suggest normal SERCA and NCX function, the protein levels of these proteins were similar in WT and KI hearts (Online Fig. II A,B). Likewise, the protein levels of the SERCA regulator phospholamban, total and phosphorylated, were not statistically different between WT and KI hearts (Online Fig. II D-F). The enhanced Ca2+ leak in KI cells correlated with decreased Post-rest Potentiation (defined as the first [Ca2+]i transient amplitude after a period of 1 minute rest normalized by the [Ca2+]i at steady state during electrical stimulation at 4 Hz: 1.41±0.067 in 10 WT cells, vs 1.19±0.06 in 11 KI cells, p<0.05). Taking into consideration the limitation of the non ratiometric fluorescence Ca2+ dye, these results suggest lower SR Ca2+ load with normal [Ca2+]i transients resulting in enhanced “fractional release” (roughly calculated by dividing the amplitudes of caffeine-evoked [Ca2+]i transient by the electrically-evoked [Ca2+]i transient) in KI cardiomyocytes (Fig. 3G), i.e., for a given amount of Ca2+ stored in the SR, there was greater Ca2+ release during each twitch. Similar findings occurred in hiPSC-CMs from a RyR2R420Q carrier compared to hiPSC-CMs from his genotype-negative brother (Fig 3H & I).
Figure 3. Mouse and human cardiomyocytes expressing RyR2R420Q show enhanced release per SR Ca2+ load.

A Line scan images of a cardiomyocyte isolated from a WT and a KI mouse before and during 100nM ISO perfusion. The thick line over the images represent 10 mM caffeine perfusion. B. [Ca2+]i transient amplitude represented as the maximum value of F/F0, where F is the fluorescence signal and F0 the diastolic fluorescence during field stimulation at 2 Hz in 39 cells WT (black open circles) and 40 KI cells (red open circles); and at 4 Hz (triangles) in 16 WT and 12 KI cells. C. Decay time constant of the [Ca2+]i transients from the same cell groups than in B obtained by fitting the descending portion of the fluorescence trace to a single exponential function. D. ISO effect on [Ca2+]i transient amplitude (left bars) and on decay time (right bars) obtained by normalizing in each cell the values in the presence of ISO to the values before ISO application in 16 WT and 18 KI cells during stimulation at 2 Hz. Independently, each group showed a significant ISO effect . E. Ca2+ transient evoked by rapid caffeine application (10 mM) provided as peak fluorescence evoked by caffeine (F) during electrical stimulation at 2 Hz, normalized to the resting fluorescence (F0) in cardiomyocytes from both groups in absence and presence of ISO (20 WT cells, 16 WT cells after ISO, 19 KI cells, red hatched bar, 14 KI cells in the presence of ISO). F. Decay time constant obtained as in C but during caffeine application in the same cells than in E. G. “Fractional release”, calculated as the peak F/F0 evoked by electrical stimulations at 4 Hz, normalized by the caffeine-evoked peak F/F0 in absence (open circles) or presence (filled circles) of 100nM ISO. H. Left, line scan of h-iPS-CM from a control (CTL) and mutant carrier (Mu) during filed stimulation at 1 Hz. Right, line scan images from h-iPS-CM during caffeine application (time of caffeine presence is noted by the thick line). I. “Fractional release” (obtained as in G) for 37 control (black) and 40 RyR2R420Q carriers (red) hiPS-CMs. J. Representative line-scans and traces of cytosolic (upper traces) and luminal (SR) Ca2+ signal (bottom traces) in permeabilized cardiomyocytes from WT and KI mice. 10 mM caffeine was used to determine the minimal fluorescence signal when the SR is empty. K. Quantitative analysis of SR Ca2+ wave threshold in permeabilized cardiomyocytes with a [Ca2+]cyt of 100 nM in 36 WT and 34 KI cells. *p<0.05; **p<0.05; ***p<0.005 Statistical tests and exact p values are provided in Online Table III.
Taken together, these data indicate that in a normal twitch, the amount of Ca2+ release as a function of the SR Ca2+ stored is higher in RyR2R420Q expressing cardiomyocytes. We then wondered whether arrhythmogenic Ca2+ waves also occur at a lower SR Ca2+ load. We monitored SR Ca2+ in permeabilized myocytes using SR-entrapped Fluo-5N, while bathed in an internal solution that induces Ca2+ waves. As shown in Figure 3 J&K, the SR [Ca2+] threshold (SR Ca2+ baseline) that evokes Ca2+ waves was significantly lower in RyR2R420Q cardiomyocytes than in control cells.
As the presence of the RyR2R420Q mutation increases Ca2+ release at a given SR Ca2+ load (“fractional release”), we analyzed whether the RyR2R420Q mutation also alters Ca2+ influx-triggered Ca2+ release. Figure 4A shows simultaneous recordings of Ca2+ current (ICa) and its evoked [Ca2+]i transient at 0 mV in a WT cell (top) and a KI cell (bottom, red traces). Summary data (Fig. 4B) reveal that the RyR2R420Q mutation had no effect on the ICa density-voltage relationship relative to WT cardiomyocytes (the maximal ICa conductance was similar between both groups (in pS): 205.7±25.1 in 10 WT cells, vs. 219.7±14.8 in 15 KI cells), while the [Ca2+]i transient amplitude in KI cells was significantly increased throughout the entire voltage range. As a result, the Ca2+-induced Ca2+ release (CICR) gain, calculated by normalizing the [Ca2+]i transient amplitude by the integral of ICa, was higher in KI cells, as exemplified at −20 mV in Figure 4C. Hence, there was a greater Ca2+ release flux through RyR2R420Q than WT channels over a given Ca2+ influx. The higher Ca2+ release could induce faster Ca2+-dependent inactivation of the ICa. Indeed, the Ca2+-dependent fast time constant of inactivation was accelerated in KI cardiomyocytes (Fig. 4D), while there were not significant differences in the slow time constant of inactivation (not shown). Of note, ICa voltage dependent activation and inactivation properties (Fig. 4E) showed no significant differences among groups (activation: V0.5= −13.5±1.7 mV and K=5.0±0.4 in 10 WT cells, and V0.5= −14.6±1.1 mV and K= 5.6±0.4 in 15 KI cells; inactivation: V0.5= −25.1±0.8 mV and K= 4.3±0.1 in 7 WT cells, and V0.5= −26.8±0.9 mV and K= 4.2±0.3 in 11 KI cells). Consequently, the “window” current, determined by the voltage dependence of activation and inactivation overlap, is not altered in KI mice. The latter is important because the “window” current produces a sustained Ca2+ influx during the AP, contributes to SR load, and may also underlie arrhythmogenic events28. Upon β-adrenergic challenge by 100 nM ISO, no difference neither on genoype nor in ISO effect was observed for ICa (p>0.05 for genotype x Iso treatment interaction, Fig. 4F). However, the elicited [Ca2+]i transient amplitude, which was higher in KI cells at basal conditions, showed less ISO effect in this group (p<0.05 for genotype x Iso treatment interaction, Fig. 4G).
Figure 4. ICa in KI mice is normal but triggers more Ca2+ release.

A Representative examples of ICa traces and their evoked intracellular [Ca2+]i transients simultaneously recorded in WT and KI myocytes. B Average Ca2+ transient amplitude (Top panel) and ICa density (lower panel) voltage dependence relationships in 8 WT and 16 KI myocytes. Peak fluorescence signal at each depolarizing step (F) was normalized by basal fluorescence measured at −80 mV (F−80). C Average Ca2+ induced Ca2+ release gain, measured in each cell as peak of F/F−80 normalized to the ICa integral during depolarization at −20 mV. D Fast constant time of inactivation, obtained by fitting the inactivation of the ICa trace to a double exponential. E. Average ICaL activation/inactivation curves. Activation in 8 WT and 16 KI cells. Inactivation in 7 WT and 11 KI cells. F. ICa density at −20 mV before (open circles) and during (closed circles) 100 nM ISO perfusion in 5 cells from each group. G. [Ca2+]i transient amplitude (F/F−80) elicited by the ICa from the same cell groups than in F. Color code: WT myocytes= black; KI myocytes= red. * p<0.05; **p<0.05; ***p<0.005. Statistical tests and exact p values are provided in Online Table III.
RyR2 clustering and ultrastructure of the dyad.
CICR is initiated at the dyads, where clusters of RyR2 channels located at the junctional SR (jSR) are in close proximity to the L-type Ca2+ channels (LTCC), located at the transverse tubules. In order to test whether the RyR2R420Q affects clustering, we acquired super-resolution images of immuno-labelled RyR2 channels. Figure 5A shows deconvoluted images of RyR2 Ca2+ release units (CRUs) after threshold analyses on a WT (left) and a KI (right) samples. We found that the distribution of cluster size was different between groups (Fig. 5B) with higher relative frequency of smaller clusters in KI cells, resulting in clusters mean size significantly smaller in the KI cardiomyocytes (in μm2: 0.034±1.76E-4, n=46393 CRU in WT, vs 0.032±1.32E-4 in 84553 CRU in KI). The reduction in the RyR2 cluster size could reflect a decreased number of expressed channels, if the mutation would interfere with protein expression. However, the measured total RyR2 expression level by Western Blots (Fig. 5C) showed that both WT and KI similarly express RyR2, ruling out this hypothesis. In addition to the smaller cluster size, CRUs distribution in KI cells showed greater dispersion along the Z line, with lower relative frequency of clusters closer to the line, and higher relative frequency of clusters less aligned (Fig. 5D).
Figure 5. RyR2R420Q induces nanodomain structural alterations.

A. Deconvoluted super-resolution image of RyR2 in a cardiomyocyte from a WT mouse and a KI mouse. B. Cluster size relative frequency distribution in WT (open bars) and KI cells (red bars) (9516 clusters in 49 cells from 3 WT, 14165 clusters in 147 cells from 5 KI). C. Representative examples of Western Blot of RyR2 and GAPDH in WT and KI mice, as indicated. Right panel shows the quantification of the RyR2 bands, normalized to GAPDH in the two experimental groups (18 WT hearts, and 20 KI mice hearts). D. Relative frequency distribution of the distance of each cluster to the line connecting most, measured in WT cells (white wider bars), and KI cells (red narrower bars) from the same cell groups than in B. E. CRUs, specialized intracellular junctions between the jSR (labeled in yellow in a) and T-tubules (labelled in green in a). The cytoplasmic domains of RyR2, i.e. the feet, are visible as evenly spaced densities (pointed by a series of arrows in panel b) spanning the gap between jSR and T-tubule. The jSR is wrapped around the T-tubule to form a CRU (panel a), or associated to form a CRU with multiple (two or more) couplons (panel b). In WT, couplons are usually quite extended (panels a & b), while in KI cardiomyocytes we may find couplons, which appear fairly normal in length (panels c & e), and other ones in which the jSR is either shorter or apparently fragmented (panels d, f, g & h). The jSR contains a classic chain-like electron dense polymer (single arrows in panels b and d), representing CSQ2. In KI myocytes, the chain-like polymer of condensed CSQ2 may be sometimes missing in some portions of the jSR. F. Left: Western blot of immunoprecipitated proteins with JPH2 antibody from WT and KI hearts and then exposed successively (after stripping) to RyR2, Cav1.2 and JPH2 antibodies. Middle: Quantitative analysis of RyR2 co-IP with JPH2 in hearts from 5 WT mice and 12 KI mice. Right: Quantitative analysis of LTCC (Cav1.2) co-IP with the RyR2 in 5 WT and 8 KI hearts. G. Western blot of immunoprecipitated proteins with JPH2 antibody from control and RyR2R420Q (Mu) h-iPS-CM and then exposed successively (after stripping) to RyR2, and JPH2 antibodies. On the right, quantitative analyses from 6 different differentiations each. H. Western Blot of JPH2 immunoprecipitated CSQ2 in 6 WT and 11 KI hearts. **p<0.05 and ***p<0.0005 Statistical tests and exact p values are provided in Online Table III.
Data presented in Figure 4 shows alteration in CICR gain in RyR2R420Q KI cardiomyocytes. We previously found reduced CICR gain in heart failure29 and proposed changes in the structure of dyads as a mechanism to explain the decreased CICR gain30. To provide an additional and independent test to robustly support our super-resolution approach, we used electron microscopy (EM) to analyze the structure of CRUs, which are specialized intracellular junctions located between the SR (labeled in yellow in Fig. 5 Ea) and the T-tubules (labeled in green in Fig. 5 Ea). CRUs are the structures containing clusters of RyR2 channels and constitute the elemental functional units for Ca2+ release at the nanoscopic level during EC coupling. The cytoplasmic domain of RyR2 channels, i.e. the “feet”, were visible as evenly spaced densities spanning the gap between jSR and the T-tubules in EM images at high magnification (pointed by the arrows in Fig. 5Eb and 5Ed). In WT cardiomyocytes, CRUs were almost exclusively positioned in proximity to the Z-lines and the T-tubule presents often a wide profile (Fig. 5 Ea & c) with the jSR being wrapped around it (Fig 5 Ea), or associated to it forming multiple (two or more) couplons (Fig. 5Eb showing 1 CRU with 2 couplons). We also evaluated the (number/area) of CRUs (a T-tubule associated to jSR, one or more) and of couplons, (consisting of individual jSR profiles associated to T-tubules) in both WT and KI cardiac cells. CRUs and couplons were significantly reduced in KI cells (by ~30%; Table 1A, columns A and B), indicating that about 1/3 of release sites were missing. As we saw differences in the SR membrane, we assessed caveolin content, to see if it could be an unnoticed defect on caveolae, but both WT and KI had similar caveolin content (Online Fig. II G)
Table 1: EM data.
A. Quantitative analysis of frequency and size of calcium release units (CRUs). Column A, the number of CRUs per surface unit; Column B, the number of contacts between SR and T-tubules or couplons; Column C, the average length of each couplon; Columns D and E, the estimated the average area of each couplon and the approximate number of RyR2-feet that could be contained in each couplon, respectively. In parenthesis, the number of fibers analyzed (columns A and B), and total number of couplons analyzed (column C). B. jSR width and total SR volumes in CRUs. Column A, the width of junctional SR (nm) and Column B, the fraction of free SR to the total SR volume. In parenthesis, the number of CRUs (A) and number of fibers analyzed (B). All data were collected from 4 hearts in RYRWT/WT and 4 hearts in RYRR420Q/WT. Data are shown as mean ± SE.
| A | |||||
|---|---|---|---|---|---|
| A | B | C | D | E | |
| No. CRUs /20 μm2 |
No. of couplons /20 μm2 |
Average length of each couplon (nm) |
Estimated average area of each couplon |
Estimated No. of RYR2- feet /couplon |
|
| WT | 2.94 ± 0.11 (136) |
2.60 ±0.17 (66) |
344.4 ± 22.2 (104) |
0.151 μm2 | 179 |
| KI | 1.94 ± 0.15*** (135) |
3.76 ±0.20*** (214) |
285.5 ± 12.0* (252) |
0.104 μm2 | 123 |
| B | |||
|---|---|---|---|
| A | B | ||
| jSR width | Free SR Volume/Total Volume, % |
||
| RYRWT/WT | 28.7 ± 0.3 (244) |
2.13 ± 0.06 (114) |
|
| RYRR420Q/WT | 33.6 ± 0.4* (290) |
1.34 ± 0.08** (118) |
|
p<0.05
p<0.005. See Online table III for statistical test and exact p value.
We then analyzed the morphology of the CRUs. In WT cardiomyocytes, couplons were usually quite extended (one in Fig. 5Ea and two in Fig. 5Eb). However, in KI cardiomyocytes the shape of couplons ranged from fairly normal to shorter in length (Fig. 5Ec & e) and/or apparently fragmented jSR (Fig. 5Ed, f, g & h). Thus, we measured the length of the jSR/T-tubule contacts, i.e. length of couplons, and found that the average size of individual couplons is shorter in KI cardiac cells (Table 1A, column C). Based on these data we then estimated: a) the average area of contact between jSR and T-tubule in each couplon (Table 1, column D) by assuming that their shape is approximately round and that the EM sections cut a random cord of this circle (see Methods in Online Supplement for more detail); and b) the approximate number of RyR2-feet which would be contained in a couplon of such a size (Table 1A, column E), assuming that the whole area of a couplon is filled with RyR231. These estimates indicated that couplons in KI cardiomyocytes were significantly smaller and therefore probably contain a significantly lower number of RyR2-feet (Table 1A, columns D and E respectively), in agreement with our super-resolution data. Since RyR2 expression levels were not different in the two groups (Fig. 5C), this could imply that there might be more orphaned RyR2 in the KI model, which may produce SR Ca2+ leak not visible as Ca2+ sparks. As matter of fact, the SR Ca2+ leak assessed by the tetracaine protocol32 (Online Fig. III), was higher in KI cardiomyocytes than in WT.
We also analyzed jSR volume. Whereas the jSR cisternae were narrow and flat in WT (Fig. 5E a & b), they had a more variable/irregular profile in KI (Fig. 5Ec-f), with slightly wider sections. To confirm this visual observation and to verify possible changes in total SR volume, we measured the mean width of the jSR and quantified the percentage of fiber volume occupied by the free SR (i.e. longitudinal SR). This analysis indicated that the mean jSR lumen is on average wider in KI than in WT cardiomyocytes but free-SR volume is smaller in KI mice (Table 1B).
We next explored potential mechanisms by which a RyR2 mutation could alter CRU clustering. Junctophilin-2 (JPH2) has recently emerged as a junctional protein that directly affects the CRU clustering of RyR2, as well as the Ca2+ release function33. We thus hypothesized that the RyR2R420Q mutation could hamper RyR2-JPH2 interaction and hence disturb RyR2 clustering. Co-immunoprecipitation (co-IP) of JPH2 and RyR2R420Q was strongly reduced while co-IP with the L-type Ca2+ channel (Cav1.2) was not significantly modified (Fig. 5F). The disturbed interaction of RyR2R420Q with JPH2 could partly explain the ultrastructural changes observed in KI cardiomyocytes. Likewise, JPH2 co-IP of RyR2 was weaker in the mutant than in the non-mutant control hiPSC-CM (Fig. 5G). Expression of other important RyR2 binding proteins such as calsequestrin, calmodulin and FKBP12/12.6, were not altered in KI mice (Online Fig. II C,H,I). Co-IP analyses showed that their binding to RyR2R420Q was not significantly different than to WT RyR2 (Online Fig. IV A-E). As FKBP12/12.6 has been shown to bear weaker binding to other CPVT RyR2 mutations34, we repeated the co-IP experiments in human context. Our data from 4 different differentiations of each control and mutant hIPSC-CM showed similar FKBP12/12.6 binding in both groups (Online Fig. IV F)
We also analyzed the appearance of the jSR content. The jSR cisternae in WT contained the classic chain-like electron dense polymer that runs parallel to the SR membrane (Fig. 5Eb, single arrows), representing Calsequestrin-2 (CSQ2). In KI myocytes, the chain-like polymer of condensed CSQ2 was present in some CRUs (Fig. 5 Ec & d), and missing in others, while the jSR lumen was either empty or containing some electron-dense material not well organized/clustered as in normal jSR/T-tubule junctions (Fig. 5 Ee-h). This observation suggested an inadequate assembly of CSQ2 inside the jSR lumen. Indeed, a similar reduction in the size of junctional domains was also observed in other mouse models carrying mutations or ablations of excitation-contraction (EC) coupling proteins35, 36 such as CSQ2R33Q/R33Q 37. However, evaluation of CSQ2 protein expression in both groups did not shown significant difference (Online Fig. II C).
RyR2R420Q function.
The alterations in the dyad ultrastructure outlined above for KI cardiomyocytes are supposed to result in conspicuous changes in Ca2+ spark duration (Online Fig. I). This finding may be explained by the theory of Ca2+ spark termination by local depletion of Ca2+ in the jSR 38. In this scenario, local depletion of Ca2+ in KI cells during a Ca2+ spark would take longer, since their jSR is wider and hence, its local volume bigger. We addressed this hypothesis analyzing the effect of intracellular hypo-osmolarity (assuming a subsequent SR swelling) on Ca2+ sparks characteristics in permeabilized rat ventricular cardiomyocytes (Online Fig. V). Changing the internal solution from normal to hypo-osmotic internal solution didn’t significantly modify Ca2+ spark frequency (Online Fig. V A) nor Ca2+ spark duration (although showed a tendency to prolong it: p=0.09, Online table III, Online Fig. V B,) but reduced Ca2+ spark amplitude (Online Fig. V C) without affecting Ca2+ spark width (Online Fig V D). Thus, increased jSR width alone does not completely explain all the RyR2R420Q-related functional alterations, although it may contribute to Ca2+ spark prolongation. Furthermore, in permeabilized KI mouse cells under hypoosmotic solution, the Ca2+ spark duration was not prolonged but reduced compared to WT myocytes (Online Fig. V F), supporting the hypothesis that alteration of this property may be the result of SR swelling. However, KI permeabilized cells still presented higher Ca2+ sparks frequency than WT (Online Fig. V E) and decreased in all characteristics (Online Fig. V F-H), which may be the reflect of higher RyR2 open probability, and smaller clusters, respectively. Thus, our data shows that increased jSR width alone does not completely explain all the RyR2R420Q-related functional alterations, although suggest that it may contribute. A complementary and/or alternative mechanism that might account for the increased Ca2+ spark frequency could be an enhanced Ca2+ sensitivity of RyR2R420Q, analogous to that of some C-terminal RyR2 mutations7, 8. We used [3H]ryanodine binding to compare the Ca2+-dependent activation of RyR2 channels from WT and KI SR microsomes, an index of RyR2 open probability, as [3H]ryanodine binds only to the open state of RyR2 channels. The Ca2+-dependent [3H]ryanodine binding curve was not different among groups, normalized to either the total amount of RyR2 present in each sample (Fig. 6A) or to the maximum binding at pCa5 (Fig. 6B). Likewise, similar Ca2+ spark frequency was observed at different [Ca2+]i in permeabilized ventricular WT and KI cardiomyocytes (Fig. 6C)
Figure 6. RyR2R420Q unitary current recordings show alterations.

A and B [3H]Ryanodine binding to SR microsomes from 4 WT (black open circles) and from 4 KI hearts (red open circles) normalized to the protein content in A and the binding of each sample at pCa5 in B. C. Ca2+ spark frequency (measured as in Fig. 2B) recorded in permeabilized cells (46 WT and 41 KI cells from 4 animals in each group) plotted as a function of the [Ca2+]i . Symbols as in A. D. WT RyR2 single-channel conducting Ca2+. Traces obtained at 0 mV of membrane potential, in the cytosolic presence of 500 nM free Ca2+ (top) and 1 μM free Ca2+ (bottom). E Amplitude histograms obtained from 3-min single-channel recordings and fitted with multi-Gaussian functions (ordinate shown as square root of the bin counts). F Single-channel recordings of RyR2R420Q conducting Ca2+, as in D. In all traces c denotes the closed state. Filtering, 800 Hz. G. As in E but for RyR2R420Q. H. Single-channel recordings of RyR2R420Q recorded at +30 mV of membrane potential, in symmetrical 250 mM Cs-methanesulfonate, 20 mM HEPES, 1 mM BAPTA, and at the indicated cytosolic free [Ca2+], Channel openings shown as positive deflections with dashed lines indicating the sub-conductance states, and the solid lines the current levels for the closed and open states. I. Open probability of the full conductance (black squares) and added sub-conductance (blue circles) states plotted as a function of the cytosolic free [Ca2+]. Full conductance open probability fitted with a sigmoidal function. Statistical tests outcome are provided in Online Table III.
RyR2R420Q show intrinsic functional alteration
Taken together, our results suggest that the Ca2+ sensitivity of the RyR2R420Q is not drastically different to that of the WT RyR2, and open the possibility that other molecular mechanisms contribute to the pathogenicity of this mutation. To directly assess RyR2R420Q function, we reconstituted cardiac SR microsomes from WT and homozygous KI mice (to ensure that all RyR2 channels bear the mutation) in planar lipid bilayers and recorded single channel activity. Figure 6D&F shows examples of unitary current activity from WT and RyR2R420Q channels at two different cytosolic Ca2+ concentrations. Channel transitions to sub-conductance states were rare for WT RyR2 when Ca2+ is used as ion carrier (Fig. 6E). In contrast, RyR2R420Q openings to sub-conductance states of variable dwell times were common (Fig. 6F&G). We detected transitions to sub-conductance states of ~one- or two-thirds of the main conductance, which were evident components of the amplitude distribution histograms (Fig. 6 E&G, n=4). Furthermore, the probability of occurrence of sub-conductance states was inversely proportional to the cytosolic [Ca2+], i.e., the sub-conductance events were more frequent at lower cytosolic [Ca2+], and less frequent at higher [Ca2+] (Fig. 6 H&I, Online VI). Of note, the phosphorylation state of RyR2-Ser2814 (phosphorylated by CaMKII) and RyR2-Ser2808 (phosphorylated by both CaMKII and PKA), which could purportedly generate sub-conductance states39, did not differ in ventricular protein extracts from WT and KI mice (Online Fig.VII A-C). Likewise, RyR2 phosphorylation at S2367 and SPEG content40 was similar in WT and KI hearts (Online Fig. VII D&E).
The altered biophysical properties of RyR2R420Q may originate from defective intermolecular interactions caused by the mutation. Indeed, the NTD regulates the activity of the homotetrameric RyR2 channel via two distinct inter-domain interactions: it forms an interface with a neighboring NTD and a separate interface with the core solenoid3, 41. Having previously demonstrated that the arrhythmogenic mutations R176Q and L433P disrupt RyR2 N-terminus self-association but not NTD interaction with the core solenoid,14, 41 we hypothesize that a similar scheme might be at work for the RyR2R420Q mutation. To ascertain the involvement of R420Q in RyR2 NTD tetramerization14, 41, we expressed the cMyc-tagged RyR2 N-terminus (NT, residues 1-906) in HEK293 cells, subjected it to chemical cross-linking with glutaraldehyde, and analyzed oligomer formation by immunoblotting. Surprisingly, the R420Q mutation did not disrupt N-terminus tetramer formation, as indicated by quantification of the anti-cMyc immunoreactive bands corresponding to monomer and tetramer using densitometry analysis (p>0.05 for genotype x time interaction, Fig. 7A). To assess the effect of R420Q in RyR2 N-terminus interaction with the core solenoid41, we co-expressed NTWT/NTR420Q with HA-tagged RyR2-CT (residues 3529-4967). HA tagged-RyR2-CT was then immunoprecipitated with AbHA and the presence of NTWT or NTR420Q was analyzed by immunoblotting using AbcMyc. Immunoprecipitation of RyR2-CT with anti-HA resulted in successful co-precipitation of both NTWT and NTR420Q. Notably, the R420Q mutation resulted in statistically significant 2-fold increase in NT interaction with RyR2-CT (Fig. 7B). It is possible that the tighter interaction between the NTD and the core solenoid slightly changes the conformation of the channel, altering its binding to accessory proteins, as for JPH2 (Fig. 5F&G). In addition, we observed that the RyR2R420Q co-IP with FKBP12/12.6 was not significanlty different than WT RyR2, and normal for calmodulin (Online Fig. VII).
Figure 7. A. RyR2R420Q N terminal domain shows tighter interaction with the core solenoid.

Chemical cross-linking assays of HEK293 cell homogenates expressing cMyc-tagged NTWT (RyR2 residues 1-906) or NTR420Q. Cell homogenates were incubated with glutaraldehyde for the indicated time points under reducing (10 mM DTT) conditions and analyzed by immunoblotting using AbcMyc; monomer (M) and tetramer (T) are indicated with the arrows. Densitometry analysis (n = 10 in each group) performed on the bands corresponding to tetramer and monomer moieties was used to calculate tetramer formation. Data are normalized for WT and given as mean value ± SEM; B. Co-immunoprecipitation assays from HEK293 cells co-expressing NTWT (n=8) or NTR420Q (n=10) together with HA-tagged RyR2-CT (residues 3529-4967). HA-RyR2-CT was immunoprecipitated with AbHA from CHAPS-solubilized cell lysates and the presence of co-precipitated NTWT/NTR420Q was analyzed by immunoblotting using AbcMyc (top). To detect immuno-isolated HA-RyR2-CT, 1/10th of IP samples was analyzed by immunoblotting using AbHA (bottom). Non-immune rabbit IgG served as negative control. An aliquot of HEK293 cell lysate corresponding to 1% of the amount processed in the co-IP assay was included in the gels to assess protein expression. Data summary (n ≥ 8) for NT specific binding (non-immune IgG IP signal subtracted from anti-HA IP signal) following densitometry analysis and normalization to each construct’s respective lysate (taken as 100%). * p<0.05 Statistical tests and exact p values are provided in Online Table III.
DISCUSSION
This integrated study combining human and mouse cardiomyocyte data on the RyR2R420Q CPVT mutation is the first comprehensive description of an ultrastructural defect underlying ventricular arrhythmias in type 1 CPVT. This disease is regarded as a type of channelopathy, for which no cardiac structural abnormality is supposed to be present. Our results directly challenge that definition at a nanostructural level. Specifically, we find that RyR2R420Q channel binding to JPH2 is disturbed and that this defect can underlie both the ultrastructural remodeling of the dyad, displaying smaller CRUs/couplons and wandering edges of the junctional SR, and functionally the enhanced Ca2+ release through Ca2+ sparks in RyR2R420Q compared to the WT RyR2. Moreover, we identify a molecular defect underlying RyR2R420Q dysregulation, namely a tighter interaction between the N-terminus and core solenoid domains, which may underlie channel openings to subconductance states.
We created a mouse model bearing the RyR2R420Q mutation we previously identified in CPVT patients15, 27, as well as hiPS cells differentiated into cardiomyocytes obtained from volunteer family members, and experimented with channel domains directly affected by the mutation. This multi-level approach yielded remarkable mechanistic insight and allowed us to correlate data obtained in both human and mouse cardiac cells. We found in both mouse and human cardiomyocytes that RyR2R420Q induced VT under emotional stress, which might be the result of DADs. These electrical proarrhythmic alterations were dependent on spontaneous Ca2+ release during diastolic periods both in mouse and human cardiomyocytes. Overall, these are consistent findings in CPVT models, but the molecular mechanism underlying RyR2R420Q channel dysfunction is unprecedented.
We previously found enhanced Ca2+ sensitivity of the RyR2R4496C CPVT mutation8, 9, which is located at the C terminal portion of the channel. Hence, we tested this possible mechanism in RyR2R420Q, which is located at the N-terminal portion. However, our analysis by two different approaches failed to unequivocally point to a difference in Ca2+ sensitivity of this mutation. A surprising finding was the longer lasting Ca2+ sparks, a characteristic of the mutation that we hypothesized could be due to enlarged jSR cisterna, since local SR Ca2+ depletion at the CRU is a potential mechanism for Ca2+ spark termination38. The enlarged jSR width in KI may thus induce a delay in local Ca2+ depletion. We mimicked this jSR enlargement with experimentally-induced SR swelling and, Ca2+ sparks tend to lasted significantly longer (p=0.09 online Table III) in permeabilized cardiomyocytes bathed in hypo-osmotic solution compared to normo-osmotic solution. It is possible that the experimental settings of permeabilized cells, where Ca2+ spreads faster away from local sources due to unlimited diffusional barriers, makes that the small increase in Ca2+ spark duration by lowering osmolarity didn’t reach statistical significance. Enlargement of SR was shown in a model of CSQ2 KO mice, and proposed to be a compensatory mechanism to the lack ot this intraluminal protein, where the expression of triadine was also almost absent35. However, our CPVT model does not show any significant alteration in the expression of these intraluminal proteins, so the origin of the enlargement should differ, and could be due to less tight interaction between TT and jSR membranes by JPH2 weaker interaction to the RyR2R420Q. Bathing permeabilized cells from WT and KI mouse in hyposomotic solution reversed the prolongation in Ca2+ spark duration, suggesting that this ultrastructural reorganization may contribute to this alteration. However, Ca2+ spark frequency was still higher, and all characteristics were smaller in KI cardiomyocytes (the latter could be the reflect of smaller CRUs), showing that other mechanisms, as intrinsic altered function, or binding to accessory proteins, which modulate RyR2 activity, need to be considered.
In fact, a salient question from the ultrastructural alteration of the dyad is, how can a point mutation in RyR2 alter the jSR shape? We examined the role of JPH2, a protein associated with both SR and plasmalemmal membranes that maintains the integrity of the dyads42. The amount of JPH2 that could be co-immunoprecipitated with RyR2 was severely decreased in RyR2R420Q. This is consistent with the reduced size of CRU/couplons and CRU cluster size. In fact, JPH2 overexpression induces opposite effects to those we found here, namely, an enlargement of the couplon size33. Further, JPH2 has been reported to affect RyR2 function, since downregulation of JPH2 enhances Ca2+ spark frequency, whereas its overexpression induces an opposite effect33, 43. Thus, the decreased binding of RyR2R420Q to JPH2 could not only explain the ultrastructural alteration, but also the functional alteration (enhanced Ca2+ spark occurrence) of the RyR2R420Q. At present, it still remains unclear how the R420Q mutation impairs the RyR2 interaction with JPH2 but it is unlikely that the R420 residue directly participates in the interaction with JPH2 because it is not surface-exposed. The structure of the RyR2 NTD carrying the R420Q mutation has been resolved by X-ray crystallography and shown to induce local conformational alterations44. Our data thus suggest that the conformational disturbance of the RyR2R420Q, results in an increased NTD interaction with the core solenoid, favors unbinding of JPH2 and finally produces nano-disturbances in CRUs with uneven and on average wider jSR containing less polymerized CSQ2, which by itself has also been implied to contribute to RyR2 closure and Ca2+ spark termination45. Loss of JPH2 binding to the RyR2 is also known to increase Ca2+ spark occurrence, and has been related to atrial fibrillation46. Thus, the altered binding of RyR2R420Q to JPH2 produces both ultrastructural and functional alterations in this CPVT mutation. Functionally, the Ca2+ spark probability is enhanced on one hand, and the Ca2+ spark duration is additionally prolonged on the other, favoring propagation of Ca2+ release and initiation of arrhythmogenic Ca2+ waves.
In our clinical description of this mutation, we first performed an in vitro analysis of the mutant protein15. RyR2R420Q expressed in HEK293 cells showed enhanced activity at low [Ca2+]i and lower activity at high [Ca2+]i suggesting gain of function at low [Ca2+]i and loss of function at high [Ca2+]i. The enhanced activity at low [Ca2+]i can manifest in cardiac myocytes as an increase in the Ca2+ spark and wave frequency. HEK293 cells are not known to express JPH2 and thus the dysfunction found in those heterologous systems cannot be attributed to dyadic alterations and may not be extrapolated to human pathophysiology. In fact, the RyR2R420Q channel also exhibits functional alterations as detected in single channel analyses. At low [Ca2+]i, the RyR2R420Q channel is preferentially open in sub-conductance states with higher open probability than the WT RyR2. The RyR2R420Q induced subconductance state, although of smaller amplitude than full openings, may induce greater Ca2+ flux per unit time, thus enhancing Ca2+ spark frequency, since the opening of one RyR2R420Q, even at lower conductance, may trigger the opening of other RyRs in the same cluster (mutated or not) and produce a full Ca2+ spark. Further, the intrinsic alteration in RyR2R420Q openings, which favors sub-conductance states at low [Ca2+]i, suggests that the ultrastructural alteration in the RyR2R420Q channel44 alters its gating properties even though it still remains unclear how a mutation in the NTD can alter the channel’s conductance.
The R420 site appears as an important residue for RyR2 function. While most of the CPVT RyR2 mutations have been found in a single family, the RyR2R420Q mutation has been reported, as far as we know, in four apparently unrelated families15-17, 27, and the RyR2R420W in two families18, 19. Unlike other RyR2 NTD mutations (e.g. R176Q, L433P)14, 41, the R420Q does not perturb the N-terminus self-association. Instead, R420Q strengthens the interaction of the N-terminus with the core solenoid within the C-terminal pore-forming region. Residue 420, which is buried within the NTD structure, does not form part of the NTD-core solenoid (nor NTD-NTD) interface(s)3. Thus, substitution of Arg420 by glutamine does not directly impact inter-domain interactions but rather induces local conformational changes enabling the NTD to make more stable contacts with the core solenoid, which probably reduces the channel conductance. These alterations may also render a weaker association to JPH2, which destabilizes the channel in the closed state, increasing its activity. However, this enhanced Ca2+ leak does not induce macroscopic heart remodeling in RyR2R420Q KI mice as in CPVT patients and other CPVT models, neither at cardiac imaging (echocardiography) nor at pathological examination even at 1 year of age.
In conclusion, our data show that the CPVT mutation RyR2R420Q, induces a tighter interaction between the NTD and the core solenoid domains of the channel, which yields both a dysfunction at the single channel level and an impaired association with JPH2 responsible for nanostructural alterations of the dyad and functional channel alterations, representing a new mechanism for the genesis of CPVT.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Catecholaminergic Polymorphic Ventricular Tachycardia, CPVT, is a rare disease causative of syncope and sudden cardiac death in apparently healthy children and young adults.
Most of identified mutations affect the calcium (Ca2+) release channel, the ryanodine receptor type 2 (RyR), and are categorized as CPVT1.
Proposed mechanisms in CPVT1 mostly point to enhanced RyR2 sensitivity to Ca2+ either cytosolic or luminal.
We identified a family with a mutation in the Ca2+ release channel, the RyR2R420Q.
What New Information Does This Article Contribute?
Our data show that the CPVT mutation RyR2R420Q, which induces a tighter interaction between the N terminal and core solenoid domains of the channel, produces both dysfunction at the single channel level and impaired association with the junctophilin-2 protein, responsible for nano structural alterations of the dyad, representing a new mechanism for the genesis of CPVT.
Thus, our multi-level approach allowed us to correlate data obtained in both human and mouse cardiac cells, being the first identification of an ultrastructural defect underlying ventricular arrhythmias in type 1 CPVT.
The molecular mechanism underlying RyR2R420Q channel dysfunction opens new areas to better understand mechanisms of cardiac sudden death not only in CPVT1 patients, but in other acquired diseases where RyR2 function is altered such as in heart failure, or diseases affecting organs where RyR2 is expressed, such as brain and pancreas.
Catecholminergic Polymorphic Ventricular Tachycardia (CPVT) type 1 is caused by mutations in the ryanodine receptor (RyR2). Using integrated methods ranging from the molecule to the whole living being, we elucidated new arrhythmogenic mechanisms in the RyR2R420Q CPVT mutation, both in a RyR2R420Q KI mouse model and in induced pluripotent stem cell-derived cardiomyocytes from a RyR2R420Q CPVT carrier patient. Our molecular analyses identified a tighter association between the N terminal domain and the core solenoid of the RyR2R420Q channel, which produces longer openings in subconductances states. Moreover, we revealed of a lesser affinity for Junctophilin 2, producing a nano structural disarrangement at the dyad with wider junctional SR and disarranged Ca2+ release units. These molecular and subcellular defects produce more and longer Ca2+ sparks, with higher propensity of arrhythmogenic Ca2+ waves associated with delayed after depolarizations under β–adrenergic stimulation both in human and murine studies. These findings are the first comprehensive description of an ultrastructural defect underlying ventricular arrhythmias in CPVT type 1.
ACKNOWLEDGEMENTS
We thank Patrick Lechêne for writing a program to analyze AP performed by microelectrodes in hiPSC-CM and help with figures formatting, Florence Lefebvre for help with cardiomyocyte cell isolation, and Gladys René-Corail for administrative assistance. We also thanks Xander Wehrens for generously sharing anti RyR2 phospho-S2367 antibody. We thank the patients who readily made themselves available to take part in this project, and the iPSc core facility of Nantes where Laurent David and Quentin Francheteau did the hiPSC from blood patients.
SOURCES OF FUNDING
This work was funded by Inserm and University Paris Sud, and grants from ANR (ANR-19-CE14-0031-01 to AMG), LabEx LERMIT (ANR-10-LABX-33), “Instituto de Salud Carlos III”; FEDER “Union Europea, Una forma de hacer Europa” [PI18/01582], La Fe Biobank [PT17/0015/0043] and Memorial Nacho Barberá to EZ; Swiss National Science Foundation (SNSF grants no. 31003A 179325 and 310030 156375 to EN), British Heart Foundation (FS/15/30/31494) to SZ; Italian Telethon ONLUS Foundation (GGP19231) and Italian MIUR (PRIN #2015ZZR4W3) to FP; NIH grants to JRF (R01GM111397) and HHV-AMG (2R01HL055438-22); and European Union H2020 (MSCA-RISE AMD-734931-6) to AMG.
HHV was recipient of a Fullbright-Tockeville chair to work on AMG laboratory. JLA and CRV were recipient of a visiting program Alambert from University Paris-Sud to work in AMG laboratory. AZJr was recipient of a post-doctoral position from University Paris-Sud. R.R was recipient of a postdoctoral fellowship from CORDDIM (Région Ile de France). LY was recipient of the CSC (Chinese Scholarship council).
Nonstandard Abbreviations and Acronyms:
- ANOVA
Analysis of Variance
- AP
action potential
- ART
Aligned Rank Transformed
- BCA
bicinchoninic acid
- BW
Body weight
- CaMKII
Ca2+ /Calmodulin Kinase II
- CICR
Ca2+-induced Ca2+ release
- Cm
Membrane capacitance
- co-IP
Co-immunoprecipitation
- CPVT
Catecholaminergic polymorphic ventricular tachycardia
- CRUs
Ca2+ release units
- CSQ
calsequestrin
- CSQ2
Calsequestrin-2
- DADs
Delayed After-Depolarizations
- EC
excitation-contraction
- ECG
Electrocardiogram
- EF
Left ventricular ejection fraction
- EM
electron microscopy
- F
fluorescence
- F0
basal fluorescence
- FKBP
FK506 binding protein
- FS
Left ventricular fractional shortening
- GLM
Generalized linear mixed model
- gSTED
gated Stimulated Emission Depletion
- hiPSC-CM
human Induced Pluripotent Stem Cell-derived Cardiomyocytes
- HR
heart rate
- HW
Heart weight
- ICa
Ca2+ current
- ISO
Isoproterenol
- JPH2
Junctophilin-2
- jSR
junctional Sarcoplasmic Reticulum
- KI
Knock In
- LTCC
L-type Ca2+ channels
- LV
Left ventricular
- ME
Mixed Effects
- NCX
sodium calcium exchanger
- NTD
N-terminal domain
- PKA
Protein Kinase A
- PLB
phospholamban
- RM
Repeated Measures
- RyR2
Type 2 Ryanodine Receptor
- SCD
Sudden Cardiac Death
- SR
sarcoplasmic reticulum
- STED
Stimulated Emission Depletion
- TA
triggered activity
- VT
ventricular tachycardia
- WT
wild-type
Footnotes
DISCLOSURES
No conflict of interest
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.Mehra R Global public health problem of sudden cardiac death. Journal of electrocardiology. 2007;40:S118–122 [DOI] [PubMed] [Google Scholar]
- 2.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512–1519 [DOI] [PubMed] [Google Scholar]
- 3.Peng W, Shen H, Wu J, Guo W, Pan X, Wang R, Chen SR, Yan N. Structural basis for the gating mechanism of the type 2 ryanodine receptor ryr2. Science. 2016;354. [DOI] [PubMed] [Google Scholar]
- 4.George CH JH, Thomas NL, Fry DL, Lai FA. Ryanodine receptors and ventricular arrhythmias: Emerging trends in mutations, mechanisms and therapies. J Mol and Cell Cardiol. 2007;42:34–50 [DOI] [PubMed] [Google Scholar]
- 5.Yano M, Yamamoto T, Ikeda Y, Matsuzaki M. Mechanisms of disease: Ryanodine receptor defects in heart failure and fatal arrhythmia. Nat Clin Pract Cardiovasc Med. 2006;3:43–52 [DOI] [PubMed] [Google Scholar]
- 6.Jiang D CW, Wang R, Zhang L, Chen SR. . Loss of luminal ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proc Natl Acad Sci U S A. . 2007;104:18309–18314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao YT, Valdivia CR, Gurrola GB, Powers PP, Willis BC, Moss RL, Jalife J, Valdivia HH. Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function. Proc Natl Acad Sci U S A. 2015;112:E1669–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fernandez-Velasco M, Rueda A, Rizzi N, Benitah J-P, Colombi B, Napolitano C, Priori SG, Richard S, Maria Gomez A. Increased ca2+ sensitivity of the ryanodine receptor mutant ryr2(r4496c) underlies catecholaminergic polymorphic ventricular tachycardia. Circulation Research. 2009;104:201–U116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neco P, Torrente AG, Mesirca P, Zorio E, Liu N, Priori SG, Napolitano C, Richard S, Benitah J-P, Mangoni ME, Gomez AM. Paradoxical effect of increased diastolic ca2+ release and decreased sinoatrial node activity in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circulation. 2012;126:392-+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang D XB, Zhang L, Chen SR. Enhanced basal activity of a cardiac ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ Res. . 2005;97:1173–1181 [DOI] [PubMed] [Google Scholar]
- 11.Lehnart SE, Wehrens XH, Laitinen PJ, Reiken SR, Deng SX, Cheng Z, Landry DW, Kontula K, Swan H, Marks AR. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation. 2004;109:3208–3214 [DOI] [PubMed] [Google Scholar]
- 12.Hwang HS, Nitu FR, Yang Y, Walweel K, Pereira L, Johnson CN, Faggioni M, Chazin WJ, Laver D, George AL Jr., Cornea RL, Bers DM, Knollmann BC. Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ Res. 2014;114:1114–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uchinoumi H, Yano M, Suetomi T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, Doi M, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ Res. 2010;106:1413–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seidel M, Thomas NL, Williams AJ, Lai FA, Zissimopoulos S. Dantrolene rescues aberrant n-terminus intersubunit interactions in mutant pro-arrhythmic cardiac ryanodine receptors. Cardiovasc Res. 2015;105:118–128 [DOI] [PubMed] [Google Scholar]
- 15.Domingo D, Neco P, Fernandez-Pons E, Zissimopoulos S, Molina P, Olague J, Suarez-Mier MP, Lai FA, Gomez AM, Zorio E. Non-ventricular, clinical, and functional features of the ryr2(r420q) mutation causing catecholaminergic polymorphic ventricular tachycardia. Rev Esp Cardiol (Engl Ed). 2015;68:398–407 [DOI] [PubMed] [Google Scholar]
- 16.Medeiros-Domingo A BZ, Tester DJ, et al. . The ryr2-encoded ryanodine receptor/calcium release channel in patients diagnosed previouslywith either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long qt syndrome: A comprehensive open reading frame mutational analysis. . J Am Coll Cardiol. 2009;54:2065–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arad M, Glikson M, El-Ani D, Monserrat-Inglesias L. A family with recurrent sudden death and no clinical clue. Annals of noninvasive electrocardiology : the official journal of the International Society for Holter and Noninvasive Electrocardiology, Inc. 2012;17:387–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishio H, Iwata M, Suzuki K. Postmortem molecular screening for cardiac ryanodine receptor type 2 mutations in sudden unexplained death: R420w mutated case with characteristics of status thymico-lymphatics. Circ J. 2006;70:1402–1406 [DOI] [PubMed] [Google Scholar]
- 19.Bauce B, Rampazzo A, Basso C, Bagattin A, Daliento L, Tiso N, Turrini P, Thiene G, Danieli GA, Nava A. Screening for ryanodine receptor type 2 mutations in families with effort-induced polymorphic ventricular arrhythmias and sudden death: Early diagnosis of asymptomatic carriers. J Am Coll Cardiol. 2002;40:341–349 [DOI] [PubMed] [Google Scholar]
- 20.Tester DJ, Spoon DB, Valdivia HH, Makielski JC, Ackerman MJ. Targeted mutational analysis of the ryr2-encoded cardiac ryanodine receptor in sudden unexplained death: A molecular autopsy of 49 medical examiner/coroner's cases. Mayo Clinic proceedings. 2004;79:1380–1384 [DOI] [PubMed] [Google Scholar]
- 21.Okudaira N, Kuwahara M, Hirata Y, Oku Y, Nishio H. A knock-in mouse model of n-terminal r420w mutation of cardiac ryanodine receptor exhibits arrhythmogenesis with abnormal calcium dynamics in cardiomyocytes. Biochem Biophys Res Commun. 2014;452:665–668 [DOI] [PubMed] [Google Scholar]
- 22.Borko L, Bauerova-Hlinkova V, Hostinova E, Gasperik J, Beck K, Lai FA, Zahradnikova A, Sevcik J. Structural insights into the human ryr2 n-terminal region involved in cardiac arrhythmias. Acta crystallographica. Section D, Biological crystallography. 2014;70:2897–2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novak A, Barad L, Lorber A, Gherghiceanu M, Reiter I, Eisen B, Eldor L, Itskovitz-Eldor J, Eldar M, Arad M, Binah O. Functional abnormalities in ipsc-derived cardiomyocytes generated from cpvt1 and cpvt2 patients carrying ryanodine or calsequestrin mutations. Journal of cellular and molecular medicine. 2015;19:2006–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. 2015. 2015;67:48 [Google Scholar]
- 25.Wobbrock JO, Findlater L, Gergle D, Higgins JJ. The aligned rank transform for nonparametric factorial analyses using only anova procedures. Proceedings of the SIGCHI Conference on Human Factors in Computing Systems. 2011:143–146 [Google Scholar]
- 26.Elkin LA, Kay M, Higgins JJ, Wobbrock JO. An aligned rank transform procedure for multifactor contrast tests. arXiv preprint arXiv:2102.11824. 2021 [Google Scholar]
- 27.Wang YY, Mesirca P, Marques-Sule E, Zahradnikova A Jr., Villejoubert O, D'Ocon P, Ruiz C, Domingo D, Zorio E, Mangoni ME, Benitah JP, Gomez AM. Ryr2r420q catecholaminergic polymorphic ventricular tachycardia mutation induces bradycardia by disturbing the coupled clock pacemaker mechanism. JCI Insight. 2017;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirano Y, Moscucci A, January CT. Direct measurement of l-type ca2+ window current in heart cells. Circ Res. 1992;70:445–455 [DOI] [PubMed] [Google Scholar]
- 29.Gómez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. [DOI] [PubMed] [Google Scholar]
- 30.Gomez AM, Guatimosim S, Dilly KW, Vassort G, Lederer WJ. Heart failure after myocardial infarction: Altered excitation-contraction coupling. Circulation. 2001;104:688–693 [DOI] [PubMed] [Google Scholar]
- 31.Protasi F, Sun XH, Franzini-Armstrong C. Formation and maturation of the calcium release apparatus in developing and adult avian myocardium. Dev Biol. 1996;173:265–278 [DOI] [PubMed] [Google Scholar]
- 32.Pereira L, Bare DJ, Galice S, Shannon TR, Bers DM. 0beta-adrenergic induced sr ca2+ leak is mediated by an epac-nos pathway. J Mol Cell Cardiol. 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Munro ML, Jayasinghe ID, Wang Q, Quick A, Wang W, Baddeley D, Wehrens XH, Soeller C. Junctophilin-2 in the nanoscale organisation and functional signalling of ryanodine receptor clusters in cardiomyocytes. Journal of cell science. 2016;129:4388–4398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D'Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. Fkbp12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840 [DOI] [PubMed] [Google Scholar]
- 35.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chopra N, Knollmann BC. Triadin regulates cardiac muscle couplon structure and microdomain ca(2+) signalling: A path towards ventricular arrhythmias. Cardiovasc Res. 2013;98:187–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qin J, Valle G, Nani A, Nori A, Rizzi N, Priori SG, Volpe P, Fill M. Luminal ca2+ regulation of single cardiac ryanodine receptors: Insights provided by calsequestrin and its mutants. J Gen Physiol. 2008;131:325–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cannell MB, Kong CH, Imtiaz MS, Laver DR. Control of sarcoplasmic reticulum ca2+ release by stochastic ryr gating within a 3d model of the cardiac dyad and importance of induction decay for cicr termination. Biophys J. 2013;104:2149–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. Pka phosphorylation dissociates fkbp12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell. 2000;101:365–376 [DOI] [PubMed] [Google Scholar]
- 40.Campbell HM, Quick AP, Abu-Taha I, Chiang DY, Kramm CF, Word TA, Brandenburg S, Hulsurkar M, Alsina KM, Liu HB, Martin B, Uhlenkamp D, Moore OM, Lahiri SK, Corradini E, Kamler M, Heck AJR, Lehnart SE, Dobrev D, Wehrens XHT. Loss of speg inhibitory phosphorylation of ryanodine receptor type-2 promotes atrial fibrillation. Circulation. 2020;142:1159–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seidel M, de Meritens CR, Johnson L, Parthimos D, Bannister M, Thomas NL, Ozekhome-Mike E, Lai FA, Zissimopoulos S. Identification of an amino-terminus determinant critical for ryanodine receptor/ca2+ release channel function. Cardiovasc Res. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: A novel family of junctional membrane complex proteins. Molecular cell. 2000;6:11–22 [DOI] [PubMed] [Google Scholar]
- 43.Reynolds JO, Quick AP, Wang Q, Beavers DL, Philippen LE, Showell J, Barreto-Torres G, Thuerauf DJ, Doroudgar S, Glembotski CC, Wehrens XH. Junctophilin-2 gene therapy rescues heart failure by normalizing ryr2-mediated ca2+ release. International journal of cardiology. 2016;225:371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimlicka L, Tung CC, Carlsson AC, Lobo PA, Yuchi Z, Van Petegem F. The cardiac ryanodine receptor n-terminal region contains an anion binding site that is targeted by disease mutations. Structure. 2013;21:1440–1449 [DOI] [PubMed] [Google Scholar]
- 45.Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beavers DL, Wang W, Ather S, Voigt N, Garbino A, Dixit SS, Landstrom AP, Li N, Wang Q, Olivotto I, Dobrev D, Ackerman MJ, Wehrens XHT. Mutation e169k in junctophilin-2 causes atrial fibrillation due to impaired ryr2 stabilization. J Am Coll Cardiol. 2013;62:2010–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Llach A, Mazevet M, Mateo P, Villejouvert O, Ridoux A, Rucker-Martin C, Ribeiro M, Fischmeister R, Crozatier B, Benitah JP, Morel E, Gómez AM. Progression of excitation-contraction coupling defects in doxorubicin cardiotoxicity. J Mol Cell Cardiol. 2019;126:129–139 [DOI] [PubMed] [Google Scholar]
- 48.Pidoux G, Gerbaud P, Dompierre J, Lygren B, Solstad T, Evain-Brion D, Tasken K. A pka-ezrin-cx43 signaling complex controls gap junction communication and thereby trophoblast cell fusion. Journal of cell science. 2014;127:4172–4185 [DOI] [PubMed] [Google Scholar]
- 49.Loaiza R, Benkusky NA, Powers PP, Hacker T, Noujaim S, Ackerman MJ, Jalife J, Valdivia HH. Heterogeneity of ryanodine receptor dysfunction in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2013;112:298–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chamberlain BK, Levitsky DO, Fleischer S. Isolation and characterization of canine cardiac sarcoplasmic reticulum with improved ca2+ transport properties. J Biol Chem. 1983;258:6602–6609 [PubMed] [Google Scholar]
- 51.Ramos-Franco J, Gomez AM, Nani A, Liu Y, Copello JA, Fill M. Ryanodol action on calcium sparks in ventricular myocytes. Pflugers Arch. 2010;460:767–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of ca(2+) release units and couplons in skeletal and cardiac muscles. Biophys J. 1999;77:1528–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Franzini-Armstrong C, Nunzi G. Junctional feet and particles in the triads of a fast-twitch muscle fibre. J Muscle Res Cell Motil. 1983;4:233–252 [DOI] [PubMed] [Google Scholar]
- 54.Block BA, Imagawa T, Campbell KP, Franzini-Armstrong C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol. 1988;107:2587–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mobley BA, Eisenberg BR. Sizes of components in frog skeletal muscle measured by methods of stereology. J Gen Physiol. 1975;66:31–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Loud AV. A method for the quantitative estimation of cytoplasmic structures. J Cell Biol. 1962;15:481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruiz-Hurtado G, Li L, Fernandez-Velasco M, Rueda A, Lefebvre F, Wang Y, Mateo P, Cassan C, Gellen B, Benitah JP, Gomez AM. Reconciling depressed ca2+ sparks occurrence with enhanced ryr2 activity in failing mice cardiomyocytes. Journal of General Physiology. 2015;146:295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fernandez-Velasco M, Ruiz-Hurtado G, Rueda A, Neco P, Mercado-Morales M, Delgado C, Napolitano C, Priori SG, Richard S, MariaGomez A, Benitah JP. Ryrca(2+) leak limits cardiac ca2+ window current overcoming the tonic effect of calmodulinin mice. Plos One. 2011;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benitah JP, Gomez AM, Bailly P, Daponte JP, Berson G, Delgado C, Lorente P. Heterogeneity of the early outward current in ventricular cells isolated from normal and hypertrophied rat hearts. Journal of Physiology-London. 1993;469:111–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fernandez-Tenorio M, Niggli E. Real-time intra-store confocal ca(2+) imaging in isolated mouse cardiomyocytes. Cell Calcium. 2016;60:331–340 [DOI] [PubMed] [Google Scholar]
- 61.Macquaide N, Tuan HT, Hotta J, Sempels W, Lenaerts I, Holemans P, Hofkens J, Jafri MS, Willems R, Sipido KR. Ryanodine receptor cluster fragmentation and redistribution in persistent atrial fibrillation enhance calcium release. Cardiovasc Res. 2015;108:387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, Palecek SP. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating wnt/beta-catenin signaling under fully defined conditions. Nat Protoc. 2013;8:162–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burridge PW, Holmstrom A, Wu JC. Chemically defined culture and cardiomyocyte differentiation of human pluripotent stem cells. Curr Protoc Hum Genet. 2015;87:21 23 21–21 23 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all supporting data are available within the article and its supplementary files.
Data was obtained from human, mouse, and heterologous system (HEK293 cell culture) expressing the RyR2R420Q CPVT mutation, using the methods detailed in the Online Supplement.
The Spanish family diagnosed with CPVT due to RyR2R420Q mutation15 underwent familial evaluation with a protocol conforming to the ethical guidelines of the 1975 Declaration of Helsinki previously approved by the local research ethics committee. Informed consent was obtained from each individual. Human induced pluripotent stem cells derived cardiomyocytes (hiPSC-CM) were obtained from patients’ blood samples (after personal consent and ethical approval) in Nantes iPSC core facility, (https://sfrsante.univ-nantes.fr/plates-formes/modeles-cellulaires-et-geniques/plate-forme-ipsc-nantes-cellules-souches-pluripotentes-induites-1101640.kjsp) and differentiated into cardiac myocytes in the laboratory.
Murine experiments were performed in male and female mice heterozygous RyR2R420Q knock in (KI) mice and wild-type (WT) littermates (4- 6 months old) in accordance with the ethics principles laid down by the French Ministry of Agriculture and ECC directive 86/609/ECC under protocol agreement n° B9201901.
Please see the Major Resources Table in the Supplemental Materials.