

Abstract
α-Synuclein aggregation at the synapse is an early event in Parkinson’s disease and is associated with impaired striatal synaptic function and dopaminergic neuronal death. The cysteine string protein (CSPα) and α-synuclein have partially overlapping roles in maintaining synaptic function and mutations in each cause neurodegenerative diseases. CSPα is a member of the DNAJ/HSP40 family of co-chaperones and like α-synuclein, chaperones the SNARE complex assembly and controls neurotransmitter release. α-Synuclein can rescue neurodegeneration in CSPαKO mice. However, whether α-synuclein aggregation alters CSPα expression and function is unknown. Here we show that α-synuclein aggregation at the synapse is associated with a decrease in synaptic CSPα and a reduction in the complexes that CSPα forms with HSC70 and STGa. We further show that viral delivery of CSPα rescues in vitro the impaired vesicle recycling in PC12 cells with α-synuclein aggregates and in vivo reduces synaptic α-synuclein aggregates increasing monomeric α-synuclein and restoring normal dopamine release in 1-120hαSyn mice. These novel findings reveal a mechanism by which α-synuclein aggregation alters CSPα at the synapse, and show that CSPα rescues α-synuclein aggregation-related phenotype in 1-120hαSyn mice similar to the effect of α-synuclein in CSPαKO mice. These results implicate CSPα as a potential therapeutic target for the treatment of early-stage Parkinson’s disease.
Keywords: α-synuclein, CSPα, Parkinson’s disease, synapse, DNAJ chaperone family
CSPα and αsynuclein (αsyn) are involved in synaptic function and neurotransmitter release via parallel pathways. Caló et al. show that αsyn aggregation at the synapse is associated with a reduction in CSPα. An increase in CSPα reduces synaptic αsyn aggregates, rescuing impaired striatal dopamine release in αsyn transgenic mice.
Introduction
Alpha-synuclein (α-syn) is a synaptic protein involved in vesicle clustering, assembly of the soluble N-ethylmaleimide sensitive fusion attachment protein receptor (SNARE) complex and neurotransmitter release. Point mutations and duplication/triplication of the α-syn gene (SNCA) cause Parkinson’s disease1 and α-syn aggregates form the Lewy bodies characteristic of Parkinson’s disease.2,3 C-terminal truncation of α-syn, found in Lewy bodies, promotes its aggregation.4,5 We previously described 1-120 truncated human α-synuclein (1-120hαSyn) transgenic mice expressing C-terminally truncated α-syn under the control of the tyrosine hydroxylase (TH) promoter in the absence of the endogenous protein,6 where α-syn aggregation in the striatal terminals is associated with redistribution of SNARE proteins and impairment in dopamine (DA) release, features present in Parkinson’s disease patients.7 Growing evidence points to presynaptic terminals as the initial site of neurodegeneration in Parkinson’s disease,7-10 as shown in our transgenic MI2 mice, where synaptic dysfunction with α-syn aggregation preceded DA cell death, both rescued by an oligomer modifier.10
The cysteine string protein α (CSPα/DNAJC5) is a vesicle-associated protein that regulates neurotransmitter release, exocytosis/endocytosis coupling and SNARE complex assembly through a pathway parallel to that of α-syn11–13 and DNAJC proteins have been linked to parkinsonism.14 CSPα function is mediated by the DNAJ domain, which activates the ATPase activity of the heat shock cognate 70 kDa protein (HSC70).15,16 The CSPα mechanism of action is functionally associated with α-syn in that overexpression of α-syn abolishes lethal neurodegeneration in CSPα-KO (knockout) mice and ablation of all three (α,β,γ)-syn genes results in SNARE complex assembly deficit with an increase in CSPα.17,18 However, whether CSPα levels and activity change with α-syn aggregation and SNARE protein redistribution or if CSPα can rescue the synaptic pathology associated with α-syn aggregation is not known.
In this study we show that CSPα expression and function are impaired in the presynaptic terminals of 1-120hαSyn mice concomitantly with the presence of α-syn aggregates and a reduction in evoked DA release. We further show that expression of CSPα rescues both α-syn aggregation-dependent deficit in vesicle cycle in vitro and impaired DA release in vivo. This effect is associated in vivo with reduction in the number of striatal synaptic α-syn aggregates as shown by immunoblotting and dSTORM super resolution analysis of tissue sections.
Materials and methods
Mice
Transgenic 1-120hαSyn and control mice without endogenous α-syn (C57BL/6OlaHsd / C57Bl/6S, background strain for the transgenic mice)6,7 and control C57Bl/6J mice with endogenous α-syn were used in this study.
Regulated animal procedures were carried out under the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body (AWERB), under project license no. 7008383.
Immunostaining
Brains from paraformaldehyde-perfused mice were sectioned and 30 µm free-floating sections were incubated overnight at 4°C with primary antibodies (anti-α-syn, BD Transduction Laboratories 610787, 1:700), anti-DNAJC5 (Millipore AB1576, 1:500), anti-TH (Millipore LNC1-MAB318, 1:500), anti-NeuN (Novus, 77686AF405, 1:500) as previously described.7 Staining was visualized using the ABC Elite Kit (Vector Laboratories) and 3,3′-diaminobenzidine (DAB) or Alexa-labelled secondary antibodies (Invitrogen A11001, A21247) and imaged using a Leitz DMRB microscope or a Leica TCS SPE confocal microscope.
Co-immunoprecipitation and immunoblotting
Total proteins were extracted from mouse striata in PBS containing 0.1% Tween 20 and Protease Inhibitor cocktail (Roche) with or without 1 mM non-hydrolysable ADP (Sigma). Immunoprecipitation followed previous protocols.7 Briefly, proteins (0.8–1 mg) were rotated overnight at 4°C with 5 µg of mouse anti-SGTa antibody (Abcam AB116439, 1:500) or control mouse IgG and protein G Dynabeads (Invitrogen). Immunocomplexes were eluted by denaturation in NuPAGE LDS sample buffer (Invitrogen). Synaptosomal fractions were extracted using Syn-PERTM synaptic protein extraction reagent (Thermo Scientific). Proteins were resolved on a 4–12% gradient SDS-PAGE gel (Invitrogen), transferred onto nitrocellulose membranes (Bio-Rad), incubated with peroxidase-conjugated secondary antibodies (GE Healthcare) and visualized with chemiluminescent substrates (Thermo Fisher Scientific), as previously described.10 Antibodies were: mouse anti-DNAJC5 (Millipore1:500), anti-VAMP2 (Abcam AB181869 1:500), anti-ATPase HSC70 (Synaptic systems, 149011, 1:500), anti-SGTa (Abcam, AB116439 1:500) and rabbit anti-β-actin (Abcam, AB8226 1:10 000). For α-syn immunoblotting, total proteins from mouse striata were extracted in PBS/0.3% TritonTM X-100 (Sigma) with Protease inhibitor cocktail (Roche); synaptosomal fractions were prepared and processed as described above and immunoblotting performed using anti-α-syn antibody (BD Transduction Laboratories, 610787 1:500) or MJFR 14-6-4-2 (Abcam, 209538 1:10 000). For reprobing, blots were stripped using RestoreTM western blot stripping buffer following the manufacturer’s instructions (Thermo Fisher Scientific, 21059).
AAV vector injections
Human full-length CSPα/DNAJC5 cDNA, a gift from Prof. R. D. Burgoyne (Liverpool University, UK), was subcloned into a pAAV vector under the PGK promoter and packaged in serotype 6 AAV particles as described.19 Vector suspension was diluted to 1 × 1013 viral genome containing particles (vg)/ml and an AAV6 empty vector (EV) used as control. For vector injections, animals were anaesthetized with 2% isoflurane, placed in a stereotaxic frame (David Kopf Instruments) and injected bilaterally with 2 μl of the virus (0.2 μl/min flow rate) in the substantia nigra (SN) at the following coordinates: AP = −3.1, L = +1.7, DV = −3.9 below dural surface relative to the Bregma according to the Mouse Brain Atlas.20 This area of the SN was selected to avoid possible injection-related damage of the substantia nigra pars compacta (SNc) from where the striatal terminal and synapse investigated derive.
Retention of FM1-43
PC12 cells stably expressing 1-120hαSyn were plated onto coverslips in 12-well plates at 7 × 105 cells/ml.7 Cells were infected with 1 μl 1 × 1013 vg/ml AAV6CSPα or control AAV6EV for 24 h, then grown for 4 days in fresh medium. To stimulate vesicle endocytosis of the FM1-43 dye (Invitrogen), PC12 cells were depolarized with KCl (Hank’s balanced salts medium with Ca2+ and Mg2+, 90 mM KCl, 63 mM NaCl) then incubated with 15 µM FM1-43 for 90 s at room temperature and unbound dye removed by 10 min wash in PBS/1 mM scavenger dye ADVASEP-7 (Sigma). Cells were re-incubated with depolarizing solution for 90 s at room temperature.7,21 Only vesicles with impaired release retained the dye. Cells were then washed, fixed with 4% paraformaldehyde and stained with anti-α-syn antibody (BD Transduction Laboratories, 610787 1:500) overnight at 4°C. Signal was detected using a Leica SPE 4 confocal microscope. FM1-43 positive puncta above the threshold fluorescence set by the AAV6EV transduced cells were counted using ImageJ analysis software. Between 700 and 1000 cells were counted for each experimental condition.
Optical densitometry analysis of striatal TH+ fibres
Fixed brains from 12-month-old control and 1-120hαSyn mice sectioned as previously described were immunostained with anti-TH antibody (Abcam, AB112 1:500) and ABC Elite Kit at three coronal levels: +0.74, +0.14, and −0.70 relative to Bregma. The 8-bit greyscale images were captured using a Nikon Eclipse Ni-U microscope, and the optical density of dopaminergic, TH-positive fibre innervation was measured in the striatal areas using ImageJ 1.52p. The obtained values were corrected by subtraction of the optical density of the background measured in the cortex, where TH-positive innervation is negligible.
Quantification of TH+ neurons in substantia nigra pars compacta
Fixed AAVEV, AAVCSPα or non-injected mouse brains were processed for double immunofluorescence by incubation with an anti-TH antibody (Millipore, LNC1-MAB318 1:500) and Alexa Fluor® 405 conjugated anti-NeuN (Novus, 77686AF405, 1:500) in PBS/0.3% TritonTM X-100/1% donkey serum overnight. TH staining was visualized with Alexa Fluor® 488 conjugated secondary antibody as well as DAB. Images were acquired using a Leica DFG 3000g camera mounted on a Leica DMI 4000B inverted microscope at ×20. Digitalized images in six 30-µm thick coronal sections per mouse, evenly spaced at 180 µm within the range covering the SNc between −2.8 mm and −3.85 mm AP from Bregma, were used to count the number of TH+ and NeuN+ cells in the SNc using the cell count plugin in ImageJ. In some sections, TH staining was also developed using 3,3′-DAB. Four mice per group were used for the quantification.
In vivo microdialysis
In vivo microdialysis was performed as previously reported.7,10 A microdialysis cannula (CMA Microdialysis) was placed in anaesthetized mice in the right medial striatum (AP = +0.7, L = +1.7, from bregma, DV = −2.1 from the skull surface20). The following day, a CMA/7 microdialysis probe was inserted into the guide cannula and microdialysis was performed at a constant flow rate (2 µl/min) with artificial CSF (ACSF: 140 mM NaCl, 7.4 mM glucose, 3 mM KCl, 0.5 mM MgCl2, 1.2 mM CaCl2, 1.2 mM Na2HPO4, 0.3 mM NaH2PO4, pH 7.4). Dialysates were collected every 20 min in tubes containing 5 µl of 0.2 M perchloric acid to prevent dopamine oxidation and assayed for dopamine, homovanillic acid and 3,4-dihydroxyphenylacetic acid. Two fractions (20–40 min) were collected to evaluate baseline release, ACSF was then replaced by ACSF containing 50 mM KCl and three more fractions collected (60–100 min). High KCl ACSF was then replaced by basal ACSF and two more fractions (120–140 min) were collected after which mice were sacrificed, and brains used for immunohistochemistry or immunoblotting. DA and homovanillic acid levels in the dialysate were measured by high-performance liquid chromatography.7,10
dSTORM
Thirty-micrometre free-floating striatal sections were stained for anti-α-syn (BD Transduction Laboratories, 610787 1:300) and Alexa FluorTM Plus 647 secondary antibody (Invitrogen, A21247 1:2000) in the presence of TetraSpeckTM microspheres (Invitrogen) to correct for drift during imaging.
Images were acquired with Photometrics EMCCD camera on a Nikon Ti-2E inverted microscope in near-TIRF mode. Image stacks consisted of 10 000 frames (50 ms/frame) on the field of view. Four to five fields of view per section and four mice per group were tested.
The image stacks were drift corrected and analysed using PeakFit in the open source ImageJ plugin GDSC SMLM, followed by a custom script. Briefly, this grouped the fluorescence signals into clusters (monomers or aggregates) based on their spatiotemporal distribution to determine their area. The aggregate size reported is the square root of the area.22
Experimental design and statistical analysis
Immunoblotting
Relative band intensity (RI) was calculated using ImageJ. β-Actin-normalized CSPα, HSC70, STGa, αSyn and VAMP2 levels were analysed with two-tailed Student’s t-test.
Co-immunoprecipitation
RI of CSPα, HSC70, and SGTa alongside corresponding input proteins were normalized to β-actin and analysed using one-way ANOVA with Bonferroni’s multiple comparisons test.
FM1-43 dye retention
Data were evaluated using two-way ANOVA with Bonferroni’s multiple comparison test.
Microdialysis
DA release was normalized to the baseline fraction (0 min) and expressed as fold difference relative to the average DA release directly following K+ stimulation (60 min fraction) in the control group. DA release in 1-120hαSyn or control mice treated with CSPα or EV was analysed using two-way ANOVA with Bonferroni’s multiple comparison test.
dSTORM
The species observed by dSTORM were divided into monomer/aggregates based on their size with recombinant monomers having a median size < 36.5 nm [20.65 ± 2 × 7.9, mean ± 2 × standard deviation (SD)10]. Data were analysed using the software https://github.com/Eric-Kobayashi/SR_toolkit.
Other analyses
Median size was analysed using a two-tailed Student’s t-test; size distribution differences between AAVCSPα and AAVEV-treated groups (monomers and aggregates combined) was calculated from a cumulative histogram using a Kolmogorov-Smirnov test.
Visualization of α-syn monomers by dSTORM was done by immunohistochemistry and the presence of the antibodies needs to be considered when evaluating the size of the monomeric protein. The smallest 25% of the objects in the AAVEV treated mouse images were assumed to be monomers, the mean size of which was determined as described above. The threshold to distinguish monomer and aggregates with 95% confidence was therefore 36.5 nm (mean + 2 × SD), similar to the threshold value previously reported.10
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material.
Results
CSPα expression levels are altered in the striatum of 1-120hαSyn mice
α-Syn aggregates were present in the striatum of 1-120hαSyn 12-month-old mice, whereas no α-syn staining was present in background control mice lacking the endogenous protein, as shown previously.6,7 CSPα staining in the striatum of control mice revealed a punctate pattern that was less intense in 1-120hαSyn mice at 12 months of age (Fig. 1A). By immunoblotting no significant difference was present in CSPα levels between controls and 1-120hαSyn mice in total tissue homogenates. However, a significant reduction (66.5%) in CSPα amount was present in synaptosomal protein extracts at 12 months of age in 1-120hαSyn mice compared to controls (homogenates RI: control 1.27 ± 0.18, 1-120hαSyn 1.07 ± 0.17; synaptic fraction: control 1.76 ± 0.11, 1-120hαSyn 0.59 ± 0.14) (Fig. 1B). This CSPα reduction was age-dependent in that no changes in CSPα levels were observed in the synaptosomal fractions of 1-120hαSyn transgenic mice compared to C57Bl6S controls at 3 months of age when transgenic 1-120hαSyn is expressed but not yet aggregated (synaptic fraction RI: control 0.7 ± 0.09, 1-120hαSyn 0.7 ± 0.04) (Supplementary Fig. 1A). CSPα in synaptic terminals forms a trimeric complex with HSC70 and SGTa. To test whether this function was perturbed in the striatum of 1-120hαSyn mice, we used immunoprecipitation in the presence of ADP. We found a reduction in the amount of CSPα and HSC70 complexes in 1-120hαSyn mice compared to controls (RI: HSC70: control 93 031 ± 3690, 1-120hαSyn 53 078 ± 1726; CSPα: control 69 117 ± 3181, 1-120hαSyn 18 097 ± 2762; SGTa: control 87 661 ± 5166, 1-120hαSyn 81 385 ± 4499). No changes in SGTa, HSC70 and CSPα expression levels were found in the total homogenate input (Fig. 1C). HSC70 and SGTa, that form a complex with CSPα, were not changed in the synaptic fraction of 12-month-old mice (HSC70, control 0.29 ± 0.05, 1-120hαSyn 0.42 ± 0.1; SGTa, control 1.32 ± 0.06, 1-120hαSyn 1.28 ± 0.08) (Supplementary Fig. 1B and C). The level of the synaptic protein VAMP2 was also not changed in either the total homogenate (control 0.25 ± 0.06, 1-120hαSyn 0.23 ± 0.03) or the synaptic fraction (control 0.72 ± 0.07, 1-120hαSyn 0.65 ± 0.03), as previously reported7 (Supplementary Fig. 1D) suggesting no deficit in synapse number.
Figure 1.

CSPα expression is altered in the striatum of 12-month-old 1-120hαSyn mice. (A) CSPα staining in striatal sections from controls and 1-120hαSyn mice. Note the loss of CSPα puncta intensity in 1-120hαSyn mice compared to controls (arrowheads indicate typical CSPα puncta). Scale bar = 20 µm. (B) Left: Immunoblot for anti-CSPα and β-actin (βAct) in total homogenates and synaptic fractions from striata of control and 1-120hαSyn mice. Right: RI of CSPα levels normalized to β-actin in total homogenates and synaptic fractions. Data are presented as mean ± standard error of the mean (SEM) of n = 5–6 mice, ***P < 0.0001 (Student’s t-test). (C) Left: immunoblots of HSC70, CSPα, SGTa after co-immunoprecipitation with anti-SGTa antibody with non-hydrolysable ADP. CSPα and HSC70 complexes with SGTa are reduced in 1-120hαSyn mice compared to controls whereas input levels do not change (bottom, CSPα, HSC70 and SGTa and correspondent β-actin). Right: RI of HSC70, CSPα, SGTa relative to their input levels (total homogenates prior to co-immunoprecipitation). Values in graph represent n = 4–5 mice in three independent experiments; ***P < 0.001 (one-way ANOVA with Bonferroni’s multiple comparison test).
Taken together, these findings suggest that the presence of aggregated α-syn in 1-120hαSyn mice is selectively associated with a reduction of CSPα levels in striatal synaptic terminals hampering its chaperone activity.
CSPα rescues vesicle cycling impairment in PC12 cells expressing 1-120hαSyn
As previously reported, expression of 1-120hαSyn in PC12 cells alters vesicle endo/exocytosis.7 To determine whether CSPα could rescue the α-syn-related vesicle turnover impairment, PC12 cells stably expressing 1-120hαSyn were transduced with either an AAV6 vector encoding human CSPα (AAVCSPα), or an empty AAV6 control vector (AAVEV) that does not express a protein. In a control experiment, non-transfected PC12 cells were also treated with AAVCSPα or EV (Fig. 2).
Figure 2.

CSPα rescues the vesicle cycle impairment in PC12 cells expressing 1-120hαSyn. (A) FM1-43 dye fluorescence (left, green) and α-syn staining (right, red) in cells treated with empty vector (EV) or CSPα. Note the reduction in FM1-43 dye retention in cells stably expressing 1-120hαSyn treated with CSPα compared to their EV-treated counterpart (arrows). Treatment with CSPα or EV had no effect in non-transfected PC12 cells (endo). (B) Quantification of the number of cells that retained FM1-43 dye above basal levels in every treatment group. Values are mean ± SEM of n = 12 independent experiments. ***P < 0.001, two-way ANOVA with Bonferroni’s multiple comparison test. Scale bar = 10 µm.
A cycle of vesicle uptake and release was induced by K+ in the presence of FM1-43 dye and the number of cells that internalized and retained the dye above a threshold value of fluorescence was counted. Fluorescence, due to vesicle retention, was increased in cells stably expressing 1-120hαSyn following K+ treatment, compared to untransfected cells (endo) indicating a release deficit as also previously shown7 (Fig. 2). Expression of CSPα in 1-120hαSyn expressing cells reduced the number of cells that retained FM1-43 while the deficit was maintained in cells treated with EV. Furthermore, AAVCSPα or AAVEV infections did not alter the release of FM1-43 from wild-type cells expressing endogenous α-syn (Fig. 2B) (cell number: 1-120hα-syn: EV = 55 ± 4.9, CSPα = 16.6 ± 4.6; endo: EV 16.9 ± 3.6, CSPα 17.9 ± 3). Thus, CSPα overexpression selectively rescues the synaptic vesicle cycle impairment caused by the expression/aggregation of human truncated α-syn.
CSPα delivery in vivo restores dopamine release
To investigate whether the effect of CSPα observed in vitro was present in vivo, we injected AAVCSPα and AAVEV into the SN of transgenic 1-120hαSyn mice that display a progressive decrease in striatal DA release. This impairment is not due to significant loss of striatal dopaminergic innervation, (quantification of TH+ fibres in the striatum at 12 months of age: control 100 ± 12.8, 1-120hαSyn 80.7 ± 22.4; Supplementary Fig. 2) or loss of TH neurons in the SNc following AAV injection (nigral TH+ neurons number injected to non-injected ratio: AAVEV 1.1 ± 0.008, AAVCSPα 1.01 ± 0.03; Supplementary Fig. 3). No change in total neuron number between injected and non-injected mice SNc was also confirmed by NeuN staining (Supplementary Fig. 3B). We first investigated the time course of CSPα expression after injection of AAVCSPα and AAVEV in the SN of control mice, and CSPα levels were measured by immunoblotting in striatal extracts at 4, 6 and 8 weeks post-injection. A 2-fold increase in CSPα expression in the striatum was present up to 8 weeks post-injection (4 weeks: CSPα = 1.37 ± 0.09, EV = 0.71 ± 0.07, 6 weeks: CSPα = 1.22 ± 0.04, EV = 0.48 ± 0.04; 8 weeks: CSPα = 1.21 ± 0.03, EV = 0.58 ± 0.06) (Supplementary Fig. 4). The increase of CSPα expression following AAVCSPα injection was confirmed in the striatum of 1-120hαSyn as shown by immunohistochemistry (Supplementary Fig. 4C). The expression of CSPα in striatum of AAVCSPα injected 1-120hαSyn mice was similar to that in control mice without (C57Bl/6S) or with (C57Bl/6J) endogenous α-syn (Supplementary Fig. 5), showing that the lack of α-syn does not alter CSPα expression per se. An increase in CSPα was also present in neuronal somata in the SNc in 1-120hαSyn mice following AAVCSPα injection (Supplementary Fig. 6).
Therefore, AAVCSPα or AAVEV were injected in the SN of 10-month-old 1-120hαSyn and control mice and the striatal DA release was measured at 8 weeks post-injection using in vivo microdialysis.
As previously shown, K+-evoked DA release was significantly reduced in untreated 1-120hαSyn mice compared to untreated controls7 [fractions: 60, 80, 100 min normalized to peak control mice value (60 min); 0.47 ± 0.045 versus 1 ± 0.03 (60 min), 0.21 ± 0.03 versus 0.63 ± 0.18 (80 min), 0.12 ± 0.03 versus 0.42 ± 0.18 (100 min)]. Notably, expression of CSPα restored DA release levels in 1-120hαSyn mice back to the control values (0.98 ± 0.06 fraction 60 min, 0.77 ± 0.08 fraction 80 min, 0.74 ± 0.08 fraction 100 min) whereas the EV vector was ineffective (0.46 ± 0.05 fraction 60 min, 0.38 ± 0.05 fraction 80 min, 0.29 ± 0.03 fraction 100 min) (Fig. 3).
Figure 3.
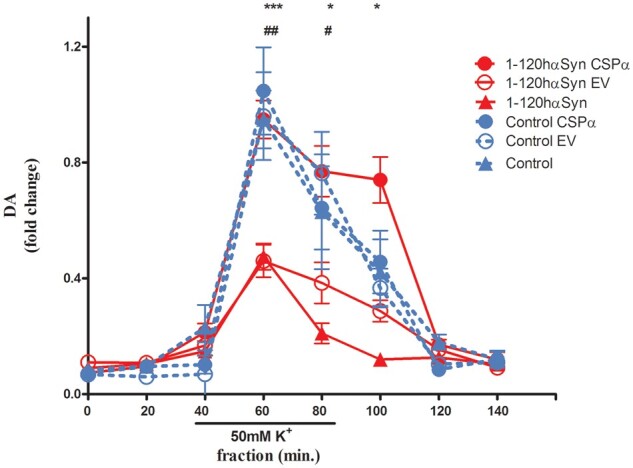
CSPα restores dopamine release impairment in 1-120hαSyn mice. Striatal DA release after infusion of 50mM KCl for 60 min during in vivo microdialysis. Twelve-month-old 1-120hαSyn mice showed a significant reduction in DA release following KCl stimulation compared with controls (***P < 0.001, *P < 0.05). Values are expressed as fold change normalized to peak control mice value. Increase of CSPα following AAVCSPα injection restored DA release to control levels in 1-120hαSyn mice whereas treatment with AAVEV was ineffective (fractions: 60 min ##P < 0.01, 80 min #P < 0.05). No significant difference was observed in the 60 and 80 min fractions between CSPα and EV-treated or untreated control mice. Values are mean ± SEM of n = 5–7 1-120hαSyn and n = 5–6 control mice per treatment group, two-way ANOVA, Bonferroni’s multiple comparison test.
The effect of CSPα was associated with α-syn aggregation, because no difference was observed in control mice after treatment with CSPα or EV, (CSPα: 1.05 ± 0.15 versus EV: 0.98 ± 0.15 fraction 60 min, CSPα: 0.64 ± 0.14 versus EV: 0.76 ± 0.1 fraction 80 min, CSPα: 0.46 ± 0.1 versus EV: 0.37 ± 0.07 fraction 100 min) (Fig. 3).
These results show that increased expression of CSPα in vivo specifically rescues impaired DA release associated with 1-120 α-syn aggregation.
CSPα reduces α-syn aggregates and increases α-syn monomers in the striatum of 1-120hαSyn mice
To investigate whether CSPα increase had an effect on aggregation of α-syn in 1-120hαSyn mice, immunostaining for α-syn was performed in striata of 12-month-old mice after microdialysis. Striata from EV-treated mice showed intensely stained α-syn aggregates in the neuropil, while the staining was reduced in CSPα-treated mice (Fig. 4A). To characterize the α-syn species in CSPα animals, we used dSTORM and compared the number of aggregated versus monomeric α-syn species at the nanoscopic level in both CSPα and EV-injected mice. In CSPα-injected mice the number of α-syn aggregates was reduced compared to EV-treated mice [aggregate number relative units (RU) = % of all species that are aggregated: CSPα = 66 ± 2%, EV = 78 ± 2%] (Fig. 4B).
Figure 4.

CSPα reduces α-syn aggregates and increases α-syn monomers in the striatum of 1-120hαSyn mice. (A) α-Syn immunostaining in the striata of CSPα and EV treated 1-120hαSyn mice. Scale bar = 10 µm. (B) Aggregate number (RU) versus monomeric α-syn in AAVEV (EV) and AAVCSPα (CSPα)-treated mice based on dSTORM analysis using anti-α-syn Syn1 antibody. A significant reduction in the number of aggregates versus monomeric α-syn is present after AAVCSPα injection. Two-tailed Student’s t-test **P⩽ ≤ 0.01; n = 4 mice per group. (C) Median size of α-syn aggregates showing no significant difference between EV-and CSPα-injected mice. Two-tailed Student’s t-test P > 0.01, n = 4 mice. (D) Cumulative histograms for α-syn species distribution in EV and CSPα-treated mice (left) and difference between the two distributions (right). Frequency values of α-syn species at the 36.5 nm intercept; <36.5 nm, EV 0.245, CSPα 0.411; >36.5 nm EV 0.754, CSPα 0.588. Kolmogorov-Smirnov test **P < 0.01, number of measured species EV: 4583, CSPα: 2574. (E) Representative dSTORM images of α-syn staining in CSPα or EV injected 1-120hαSyn mouse striatum. Note the difference in size of α-syn aggregates (single α-syn aggregates enlarged in boxed areas; scale bar insets = 200 nm).
We then analysed the difference in aggregate size distribution between EV- and CSPα-injected mice. No difference in the median size of α-syn aggregates between the CSPα-injected and EV injected group was found; however, CSPα-injected mice exhibited a significant 16.6% reduction in α-syn aggregates (>36.5 nm) and conversely, an increase in monomeric species (<36.5 nm) (Fig. 4C and D) indicating that CSPα reduced the number of α-syn aggregates and increased the number of the α-syn monomeric species as presented in Fig. 4E. The increase in monomeric species and reduction of α-syn aggregates was also observed by immunoblotting of extract from isolated striatal synaptosomal fractions (RI: α-syn monomeric levels, AAVCSPα 1.1 ± 0.008, AAVEV 0.38 ± 0.11; high molecular weight α-syn bands (aggregated α-syn) AAVCSPα 0.34 ± 0.07, AAVEV 0.68 ± 0.09; Supplementary Fig. 7).
Discussion
α-Syn is predominantly localized at the synapse where it is involved in SNARE complex assembly and synaptic vesicle turnover.17,23–28 CSPα is also present at the synapse where it assists in folding of client proteins involved in SNARE complex formation, neurotransmitter release and exo/endocytosis.11–13 α-Syn is functionally interconnected with CSPα as shown by its ability to rescue the neurodegeneration characteristic of CSPα-KO mice11 but whether CSPα is affected by α-syn aggregation is not clear. Here we show that CSPα immunoreactivity is reduced in the striatum of 12-month-old 1-120hαSyn mice compared to controls. This decrease was found specifically in the synaptosomal fraction indicating that CSPα reduction is spatially co-localizing with α-syn aggregation. CSPα decrease appears to be associated with α-syn aggregation in that synaptosomal CSPα levels in 1-120hαSyn mice are similar to control mice at 3 months of age when no synaptic α-syn aggregates are present.7 As a synaptic co-chaperone, CSPα binds to HSC70 and the adapter protein SGTa to regulate vesicle fusion. By co-immunoprecipitation we found that CSPα/HSC70 complexes with SGTa are reduced in the striatum, an effect that may be attributed to CSPα reduction. These findings indicate that α-syn synaptic aggregation affects CSPα levels and function. To determine whether an increase of CSPα could be beneficial and restore vesicle cycling altered by 1-120hα-syn expression, we first tested this in PC12 cells stably expressing 1-120hα-syn. We found that indeed their abnormal vesicle cycle, indicated by an increase in FM1-43 retention, was restored by CSPα overexpression. This prompted us to investigate whether an increase of CSPα in vivo in the striatum of 1-120hαSyn mice could restore DA release. Using microdialysis we found that CSPα expression restored the striatal DA release reduced by synaptic α-syn aggregation, dSTORM super-resolution microscopy performed in the tissue after microdialysis indicated a reduction in the number of α-syn aggregates and a concomitant increase in monomeric species confirmed then by immunoblotting of synaptosomal striatal fraction of 1-120hαSyn mice. It is unclear whether CSPα prevents monomeric α-syn aggregation or contributes to dissociation of the α-syn aggregates. Since both HSC70-family small chaperones and other DNAJs have been shown to affect α-syn aggregation,29,30 CSPα in our model could contribute to facilitating HSC70 ATPase activity as the burden of misfolded α-syn increases. Although it cannot be excluded that the increase in CSPα per se could improve dopamine release, the fact that no change was observed in control mice would support the hypothesis that CSPα acts by affecting α-syn-related alterations. The increase in monomeric α-syn is similar to what we observed in MI2 transgenic mice after anle138b treatment,10 whether the CSPα beneficial effect is due to the reduction of aggregates or the increase of monomeric α-syn remains to be determined.
In conclusion, our data reveal for the first time that α-syn aggregation impairs expression of synaptic CSPα and formation of its functional complexes with HSC70 and that an increase of CSPα reduces synaptic α-syn aggregates and increases monomeric α-syn while rescuing striatal DA release impairment. These results point to CSPα and related chaperones as therapeutic avenues for restoring normal synaptic function in early Parkinson’s disease.
Supplementary Material
Acknowledgements
We wish to thank Prof RD Burgoyne for the CSPα cDNA, Dr E. Dimou and Dr J. McColl for advice on dSTORM imaging and Dr MP Fransen and Dr G Vivacqua for critically reading the manuscript.
Funding
This work was supported by funding from the MJ Fox Foundation, the Cure PD Trust, Parkinson’s UK, the Royal Society and the UK Dementia ResearchInstitute, which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer's Society and Alzheimer's Research UK.
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
Glossary
- 1-120hαSyn
1-120 truncated human α-synuclein
- CSPα
cysteine string protein
- DA
dopamine
- EV
AAV6 empty vector
- SNARE
soluble N-ethylmaleimide sensitive fusion attachment protein receptor
References
- 1. Lunati A, Lesage S, Brice A.. The genetic landscape of Parkinson's disease. Rev Neurol. 2018;174(9):628–643. [DOI] [PubMed] [Google Scholar]
- 2. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M.. Alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M.. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. [DOI] [PubMed] [Google Scholar]
- 4. Baba M, Nakajo S, Tu PH, et al. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 5. Crowther RA, Jakes R, Spillantini MG, Goedert M.. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 1998;436:309–312. [DOI] [PubMed] [Google Scholar]
- 6. Tofaris GK, Garcia Reitböck P, Humby T, et al. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): implications for Lewy body disorders. J Neurosci. 2006;26:3942–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garcia-Reitbock P, Anichtchik O, Bellucci A, et al. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson's disease. Brain. 2010;133:2032–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Janezic S, Threlfell S, Dodson PD, et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A. 2013;110:E4016–E4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakata Y, Yasuda T, Fukaya M, et al. Accumulation of alpha-synuclein triggered by presynaptic dysfunction. J Neurosci. 2012;32:17186–17196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wegrzynowicz M, Bar-On D, Calo L, et al. Depopulation of dense α-synuclein aggregates is associated with rescue of dopamine neuron dysfunction and death in a new Parkinson's disease model. Acta Neuropathol. 2019;138:575–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC.. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. [DOI] [PubMed] [Google Scholar]
- 12. Tobaben S, Thakur P, Fernández-Chacón R, Südhof TC, Rettig J, Stahl B.. A trimeric protein complex functions as a synaptic chaperone machine. Neuron. 2001;31:987–999. [DOI] [PubMed] [Google Scholar]
- 13. Zhang YQ, Henderson MX, Colangelo CM, et al. Identification of CSPalpha clients reveals a role in dynamin 1 regulation. Neuron. 2012;74:136–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roosen DA, Blauwendraat C, Cookson MR, Lewis PA.. DNAJC proteins and pathways to parkinsonism. FEBS J. 2019;286:3080–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Braun JE, Wilbanks SM, Scheller RH.. The cysteine string secretory vesicle protein activates Hsc70 ATPase. J Biol Chem. 1996;271:25989–25993. [DOI] [PubMed] [Google Scholar]
- 16. Chamberlain LH, Burgoyne RD.. Activation of the ATPase activity of heat shock proteins Hsc70/Hsp70 by cysteine-string protein. Biochem J. 1997;322:853–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC.. Alpha-ynuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gorenberg EL, Chandra SS.. The role of co-chaperones in synaptic proteostasis and neurodegenerative disease. Front Neurosci. 2017;11:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Löw K, Aebischer P, Schneider BL.. Direct and retrograde transduction of nigral neurons with AAV6, 8, and 9 and intraneuronal persistence of viral particles. Hum Gene Ther. 2013;24:613–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paxinos G, Franklin KBJ.. The mouse brain in stereotaxic coordinates. Amsterdam: Elsevier Academic Press; 2004. [Google Scholar]
- 21. Gaffield MA, Betz WJ.. Imaging synaptic vesicle exocytosis and endocytosis with FM dyes. Nat Protoc. 2006;1:2916–2921. [DOI] [PubMed] [Google Scholar]
- 22. Whiten DR, Zuo Y, Calo L, et al. Nanoscopic characterisation of individual endogenous protein aggregates in human neuronal cells. Chembiochem. 2018;19:2033–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iwai A, Masliah E, Yoshimoto M, et al. The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. [DOI] [PubMed] [Google Scholar]
- 24. Nemani VM, Lu W, Berge V, et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burré J, Sharma M, Südhof TC.. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A. 2014;111:E4274–E4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Diao J, Burre J, Vivona S, et al. Native alpha-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. Elife. 2013;2:e00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vargas KJ, Schrod N, Davis T, et al. Synucleins have multiple effects on presynaptic architecture. Cell Rep. 2017;18:161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Longhena F, Faustini G, Spillantini MG, Bellucci A.. Living in promiscuity: The multiple partners of alpha-synuclein at the synapse in physiology and pathology. Int J Mol Sci. 2019;20:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pemberton S, Madiona K, Pieri L, Kabani M, Bousset L, Melki R.. Hsc70 protein interaction with soluble and fibrillar alpha-synuclein. J Biol Chem. 2011;286:34690–34699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Whiten DR, Cox D, Horrocks MH, et al. Single-molecule characterization of the interactions between extracellular chaperones and toxic α-synuclein oligomers. Cell Rep. 2018;23(12):3492–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material.