

Abstract
A major limitation to understanding the associations of human leukocyte antigen (HLA) and CD8+ and CD4+ T cell receptor (TCR) genes with disease pathophysiology is the technological barrier of identifying which HLA molecules, epitopes, and TCRs form functional complexes. Here we present a high-throughput epitope identification system that combines capture of T cell-secreted cytokines by barcoded antigen-presenting cells (APCs); cell sorting; and next-generation sequencing to identify class I- and II-restricted epitopes starting from highly complex peptide-encoding oligonucleotide pools. We engineered APCs to express anti-cytokine antibodies, a library of DNA-encoded peptides, and multiple HLA class I or II molecules. We demonstrate that these engineered APCs link T cell activation-dependent cytokines with the DNA that encodes the presented peptide. We validated this technology by showing that we could select known targets of viral epitope-, neoepitope-, and autoimmune epitope-specific TCRs, starting from mixtures of peptide-encoding oligonucleotides. Then, starting from ten TCRβ sequences that are found commonly in humans but lack known targets, we identified seven CD8+ or CD4+ TCR-targeted epitopes encoded by the human cytomegalovirus (CMV) genome. These included known epitopes, as well as a class I and a class II CMV epitope that have not been previously described. Thus, our cytokine capture-based assay makes use of a signal secreted by both CD8+ and CD4+ T cells, and allows pooled screening of thousands of encoded peptides to enable epitope discovery for orphan TCRs. Our technology may enable identification of HLA-epitope-TCR complexes relevant to disease control, etiology, or treatment.
One Sentence Summary:
Cytokine-capturing antigen-presenting cells allow screening of complex DNA libraries for CD8+ and CD4+ T cell-targeted epitopes.
INTRODUCTION
Variation in human leukocyte antigen (HLA) and T cell receptor (TCR) genes is associated with risk of infection and autoimmunity (1, 2), and can influence patient survival to cancer immunotherapy (3, 4). Identification of the specific complexes between HLA molecules, epitopes, and TCRs – resulting in T cell activation – can thus provide fundamental information about disease pathogenesis and lead to the generation of T cell-based treatments, such as TCR-based cellular therapies (5–7). However, there remain technological barriers to identifying which HLA, epitopes, and TCRs productively lead to T cell activation. These barriers in part emerge from the significant inter- and intra-individual variation in HLA (8) and TCR genes (9, 10), as well as the vast potential space of peptide epitopes.
Traditional epitope identification technologies – including functional assays such as ELISPOT (enzyme-linked immunospot) (11) and ICS (intracellular cytokine staining) (12) – are in widespread use to detect HLA-epitope-TCR complexes. ELISPOT and ICS rely on capture of T cell activation-dependent cytokines – endogenous signals with high signal-to-noise ratios (11, 12). These methods have had broad and important applications, e.g. to identify epitopes targeted by CD8+ or CD4+ T cells in disease contexts such as cancer and natural/vaccine-elicited immunity against pathogens (13, 14). However, traditional assays are particularly limited in assessing large epitope candidate sets due to the high costs of peptide synthesis (15, 16).
One approach to scale peptide space is to use highly complex oligonucleotide pools (17, 18) to encode peptide libraries (15). Many thousands of peptide-encoding oligonucleotides can be synthesized on an array at a cost lower by orders of magnitude (18) relative to the synthesis of peptides. Methods exploiting libraries of DNA-encoded peptides presented on defined HLA molecules have recently been described, in conjunction with various mechanisms to generate signal to detect the presence of HLA-epitope-TCR interactions (Fig. 1A) (19–22). However, applications of these methods have largely relied on expressing single HLA class I molecules (e.g. HLA-A*02:01). The ability of these methods to screen both class I- as well as class II-presented peptides remains unclear (20–22), and/or their ability to concurrently test multiple HLA alleles is limited, secondary to the need for extensive optimization of each recombinant HLA (19, 23, 24) or to fusion of peptide libraries to an HLA molecule (19, 21, 25). Development of a high-throughput epitope identification technology that combines both HLA and peptide diversity may enable identification of HLA-epitope-TCR complexes whose identification is intractable using existing assays.
Fig. 1. Establishing a system for epitope identification by APC cytokine capture.
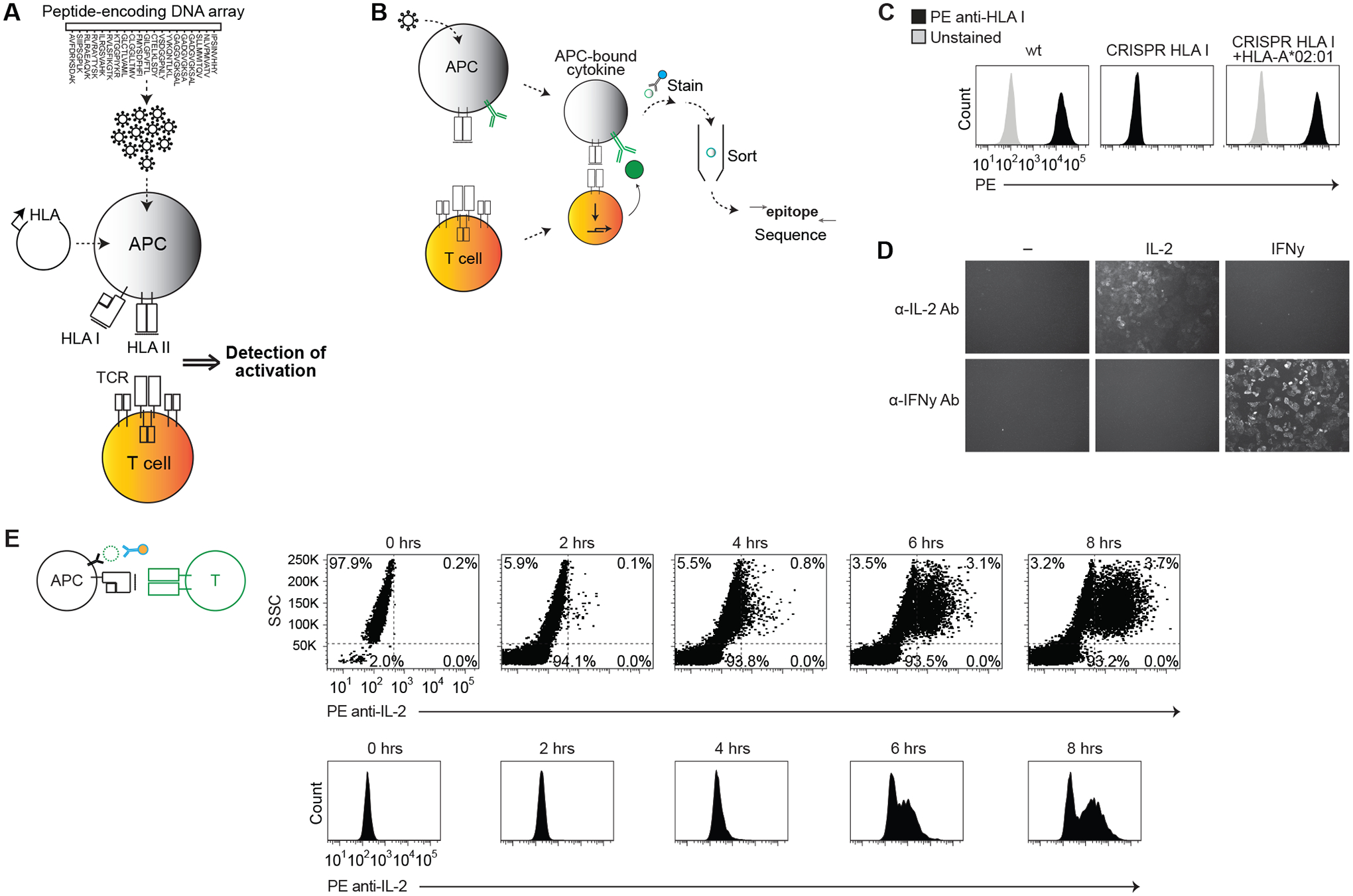
(A and B) Schematic representation of system to express a library of peptide-encoding genes on specified HLA proteins (A), and to identify APCs that present epitopes by capturing T cell activation-dependent cytokines on the APC surface (B). (C) Flow cytometric analysis of HLA class I expression on wild-type HeLa cells, class I KO cells (CRISPR HLA I), or class I KO cells stably expressing HLA-A*02:01. (D) Fluorescent microscopy of HeLa cells expressing membrane-bound anti-IL-2 or anti-IFN-γ antibody, and incubated for 2 hours with 100 ng/mL recombinant IL-2, 100 ng/mL recombinant IFN-γ, or with no cytokine. Cells were stained with respective PE anti-cytokine antibodies. (E) Flow cytometric analysis showing SSC versus PE anti-IL-2 antibody staining (top) or PE histogram (bottom) for indicated time intervals after co-culture of C25 TCR-expressing T cells with APCs expressing HLA-A*02:01 and a CMV epitope-encoding gene. Data are representative of two or three independent experiments.
Here we develop a strategy in which signal is generated by capturing cytokines – similar to traditional functional assays (11, 12) – but in which the anti-cytokine antibodies are directly expressed by the APCs along with an encoded peptide and multiple HLA class I or II genes. After APC/T cell co-culture, APCs that become coated with cytokine are sorted, and the encoded epitopes are read out by next-generation sequencing (NGS). We demonstrate that our method has the sensitivity to detect a single epitope from a pool of thousands of peptide-encoding oligonucleotides, and can identify epitopes for orphan CD8+ and CD4+ TCRs.
RESULTS
Establishing a system for epitope identification using APC-bound anti-cytokine antibodies
We sought to develop a method (fig. S1A) that accepts highly complex peptide-encoding oligonucleotide pools as an input (Fig. 1A), and uses a cytokine readout to pinpoint T cell-targeted epitopes (Fig. 1B). In cytokine detection assays such as the ELISPOT, antibodies linked to solid phase capture cytokines secreted by activated T cells (fig. S1B). We reasoned that (i) stably encoding the peptide library within APCs and (ii) expressing an anti-cytokine antibody on the APC surface instead of on solid phase (fig. S1B) gives the assay the ability to sort epitopes using activation-dependent cytokines.
First, we generated HLA class I knockout (KO) APC lines. Following transfection of CRISPR/Cas9 cassettes targeting class I loci (table S1), class I expression was eliminated from HeLa and HEK293T cells, which were used as model APCs (Fig. 1C and fig. S1C). We were then able to stably express defined HLA genes in these cells (Fig. 1C).
For class I antigen presentation, we cloned peptide-encoding minigenes 3’ of the human IL2 signal sequence, in a lentiviral vector backbone, to stably express defined peptides (fig. S1D) that are predicted to cleave off the signal sequence (26). Alternatively, longer peptide-encoding genes were synthesized that undergo further antigen processing prior to presentation (table S2).
To capture cytokines on the APC surface, we expressed in the APCs antibody-encoding heavy and light chain genes – here, encoding antibodies to IL-2 or IFN-γ (Fig. 1B and table S3). Addition of IL-2 or IFN-γ to these “cytokine-capturing” APCs affixed cytokine to the APC surface in a cytokine-specific manner (Fig. 1D).
We then assayed for cytokine capture during culture of the APCs with T cells. We co-cultured cytokine-capturing APCs expressing HLA-A*02:01 and a cytomegalovirus (CMV) epitope-encoding minigene, with TCR knockout Jurkat T cells (fig. S1E) expressing the C25 TCR (27) (table S4), which recognizes the CMV epitope. After staining the APCs with fluorescently-labeled anti-IL-2 antibody, negligible staining was seen with APCs alone, while cytokine staining on the APC surface increased during co-culture (Fig. 1E and fig. S1H).
These results establish development of an engineered APC that can present integrated epitopes on defined HLA molecules, and can capture cytokines from activated T cells.
Engineered APCs capture cytokine only in the context of functional HLA-epitope-TCR complexes
To test the specificity of the assay, we first used a KRAS p.G12D-reactive TCR that was previously identified in a tumor-infiltrating lymphocyte (TIL) (6). We transduced cytokine-capturing APCs with HLA-C*08:02 alone, or with KRAS peptide-encoding minigenes (Fig. 2, A and B). The TCR was specific for the p.G12D mutant (Fig. 2B, fig. S2A, and table S5). In addition, we transduced cytokine-capturing APCs with HLA-A*02:01 alone, or in combination with influenza, CMV, or NY-ESO-1 epitope-encoding minigenes (table S2). We co-cultured these APCs with T cells expressing JM22 TCR (influenza-reactive) (28), C25 TCR, or 1G4 TCR (NY-ESO-1-reactive) (29) (table S4). Surface-bound IL-2 was detectable only when the APCs expressed HLA and epitope pairs corresponding to the expected TCR specificities (fig. S2B).
Fig. 2. Engineered APCs capture cytokine in an HLA class I or II epitope-specific manner.
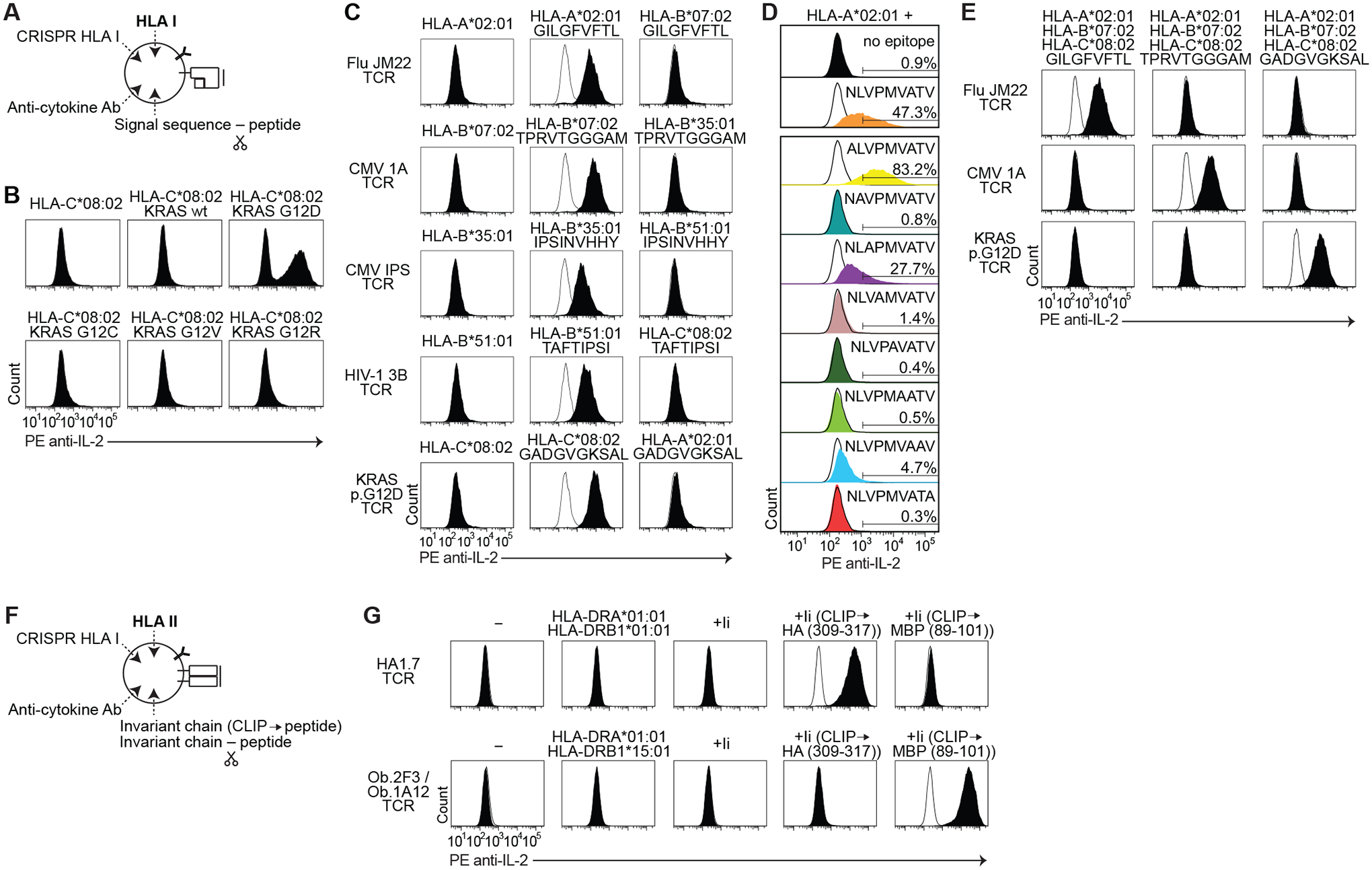
(A) Schematic of genetic alterations to engineer cytokine-capturing APCs. Each class I minigene is fused to a signal sequence. (B) Flow cytometric analysis of IL-2 cell surface capture after co-culture of KRAS p.G12D-reactive TCR-expressing T cells with APCs expressing HLA-C*08:02 with or without encoded KRAS peptides. (C to E) Histogram of IL-2 cell surface capture after co-culture of indicated TCR-expressing T cells with APCs expressing indicated HLA and encoded peptides, including encoded peptides that bind to different HLA (C); alanine substitution variants of NLVPMVATV (D); or encoded peptides co-expressed with multiple HLA genes (E). (F) Schematic of genetic alterations to engineer cytokine-capturing APCs for class II presentation. Class II encoded peptides are fused to the invariant chain (Ii) either replacing CLIP or with an intervening cathepsin-cleavage sequence. (G) Histogram of IL-2 cell surface capture after co-culture of indicated TCR-expressing T cells with APCs expressing indicated HLA and peptide-encoding genes. Peptides were encoded in place of CLIP. Wild-type Ii was used as a control. For each TCR, histogram of HLA alone is overlaid in white. Data are representative of two or three independent experiments.
Next, we tested recognition of epitopes presented by different HLA-A, -B, or -C proteins. We co-transduced cytokine-capturing APCs with defined HLA and epitope-encoding minigenes, and co-cultured these APCs with T cells expressing corresponding HLA/epitope-reactive TCRs (30–32) (table S2 and S4). Surface-bound IL-2 was detected after co-culture with each known HLA-epitope-TCR complex (Fig. 2C). We then permuted the HLA genes. Co-culture with these alternative HLA genes did not result in substantial surface-bound IL-2 relative to the cognate HLA alone (Fig. 2C and table S5).
To demonstrate the quantitative nature of the signal, which may allow separation of epitopes that activate T cells with differential strengths (fig. S2C), we created an alanine scan library of the NLVPMVATV epitope (table S2), individually transduced each construct into cytokine-capturing APCs with the HLA-A*02:01 gene, and co-cultured these APCs with C25 TCR-expressing T cells. Expression of the p.N495A (ALVPMVATV) mutant consistently elicited brighter signal (83.2% of APCs gated as PE anti-IL2 antibody-positive in a representative experiment) than expression of the wild-type epitope (47.3% gated); the p.V497A (NLAPMVATV) mutant elicited moderate signal (27.7% gated); and the p.T502A (NLVPMVAAV) mutant elicited dim signal (4.7% gated) (Fig. 2D). Expression of the other mutants (p.L496A, p.P498A, p.M499A, p.V500A, and p.V503A) did not elicit substantial signal (0.8%, 1.4%, 0.4%, and 0.3% gated, respectively) relative to expression of HLA-A*02:01 alone (0.9% gated) (Fig. 2D). These results are consistent with a crystal structure (27) that showed that C25 TCR makes extensive interactions with positions 498–500. Mutation of positions 496 and 503 are predicted to weaken binding to HLA-A*02:01 (table S5).
Finally, we reasoned that we could increase the number of HLA genes that could be tested concurrently. To demonstrate this, we co-transduced cytokine-capturing APCs with HLA-A*02:01, HLA-B*07:02, and HLA-C*08:02; along with GILGFVFTL, TPRVTGGGAM, or GADGVGKSAL epitope-encoding genes. We co-cultured these APCs with T cells expressing JM22, 1A, or KRAS p.G12D-reactive TCRs. Surface-bound IL-2 was detected when the APCs expressed epitopes corresponding to expected TCR specificities (Fig. 2E).
Taken together, these results suggest that our engineered APCs could express epitopes presented by HLA-A, HLA-B, or HLA-C; could be co-transduced with multiple HLA; could be labeled in a T cell activation-dependent manner; and could distinguish epitopes eliciting strong cytokine release from those eliciting weak cytokine release.
Adapting the system to identify HLA class II epitopes
We designed an analogous system to label class II epitope-expressing APCs (Fig. 2F). While the class I KO APCs do not express class II on their surface at baseline (fig. S2D), non-professional APCs can support class II expression, either after transduction with CIITA (33), or after expression of defined class II genes (34). As CIITA induces endogenous class II expression (fig. S2D), transient expression of CRISPR/Cas9 cassettes are needed to knockout endogenous class II genes (HLA I/II KO APCs) (fig. S2, E and F) so that the APCs express only desired HLA genes (fig. S2, G and H). To direct encoded peptides into endosomal compartments for class II presentation, encoded peptides can be fused to CD74. In one version, we fused the peptide-encoding minigene to the 3’ of CD74 with an intervening cathepsin S cleavage sequence (35); in the second version, we encoded the peptide sequence in place of the CLIP-encoding region in CD74 (34) (Fig. 2F and fig. S2I).
To test these systems, first, we co-transduced HLA I/II KO APCs with HLA-DRA*01:01, HLA-DRB1*01:01, and a class II epitope-encoding gene from influenza A hemagglutinin (HA) fused to the 3’ of CD74. In parallel, we transduced TCR KO T cells with an HA-reactive TCR, HA1.7 (36). Co-culture of these APCs and T cells led to surface-bound IL-2 on the APCs (fig. S2J). Second, we transduced the HLA I KO APCs (in the absence of CIITA) with HLA-DRA*01:01 and HLA-DRB1*01:01, or with HLA-DRA*01:01 and HLA-DRB1*15:01. We co-transduced these cells with the influenza epitope-encoding gene or with a class II autoimmune epitope-encoding gene from myelin basic protein (MBP) (table S2) (37). In parallel, we transduced TCR KO T cells with HA1.7 TCR or with the MBP-reactive TCRs, Ob.1A12 or Ob.2F3 (38). After co-culture of the APCs and T cells, expression of the epitope-encoding sequences either fused to the invariant chain or replacing CLIP, but not fused to a signal sequence, led to cytokine capture on the APCs in an epitope-specific manner (Fig. 2G and fig. S2K).
Taken together, these results demonstrated that our engineered APCs can capture cytokine in the context of both HLA class I and II epitopes, and their respective TCRs.
Identification of a T cell epitope from a pooled oligonucleotide library
Using these APCs, we developed a method to locate a specific epitope amongst a pool of peptide-encoding oligonucleotides (fig. S3A). First, we created a library consisting of 32 CMV, Epstein-Barr virus (EBV), or influenza (Flu) (CEF) epitope-encoding minigenes (Fig. 3A) (39). Sequencing of the library showed representation of each epitope (fig. S3B). We transduced the library into HLA-A*02:01-expressing APCs at a multiplicity of infection (m.o.i.) <1 (approximately 0.2–0.5) to create a library of epitope-expressing APCs (Fig. 3B and fig. S3A).
Fig. 3. Identification of a target epitope from a pooled oligonucleotide library.
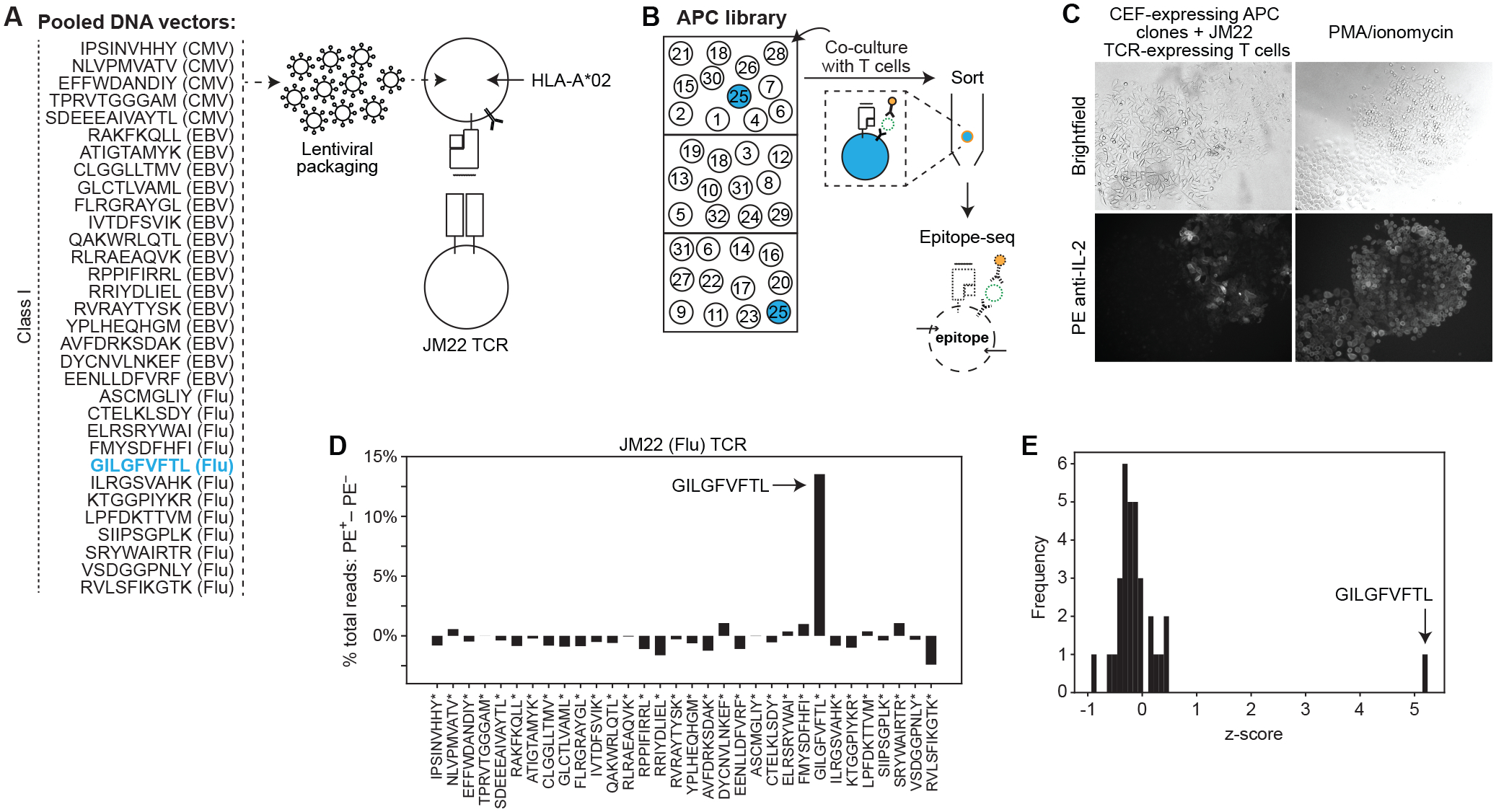
(A) Schematic showing the pooled set of 32 CEF epitope-encoding genes. HLA-A*02:01-expressing APCs were transduced with the pooled peptide-encoding genes at m.o.i. <1, and co-cultured with T cells expressing JM22 TCR. (B) Schematic of strategy to identify the target epitope. The APC library is seeded into wells in numbers less than the diversity of the encoded peptide library. T cells are added to all wells. APCs expressing the target epitope-encoding gene become labeled with cytokine and can be selected. (C) Brightfield (top) and fluorescent (bottom) microscopy of HLA-A*02:01- and CEF epitope-expressing (m.o.i. <1) APC clones that were clonally expanded, co-cultured with JM22 TCR-expressing T cells, and then stained with PE anti-IL-2 antibody. PMA (80 ng/mL) and ionomycin (1000 ng/mL) were added to the T cells and APCs as a positive control (right). (D) Difference between sorted and flow-through cells in percentage of total read counts for each encoded peptide. (E) Frequency of z-score measurements from screening data. Data are representative of two independent experiments.
We seeded the APC library at low density relative to library diversity (i.e. 3.5–10 APCs/well relative to 32 possible epitope-expressing APCs), aiming for <1 positive clone per well to limit the possibility of signal leakage (Fig. 3B and fig. S3A). In total, we seeded ≥1000 cells (i.e. 1000–8000) so that each epitope in the library was represented multiple times (Fig. 3B). To each well, we added T cells expressing JM22 TCR (28). After co-culture, we stained APCs with fluorescent molecule-conjugated anti-IL-2 antibody (Fig. 3C and fig. S3C), separated PE+ and unlabeled APCs, and sequenced the epitope-encoding genes by NGS.
In a representative experiment (table S6A), in the pulldown of PE+ cells, 17.4% of total peptide-encoding reads encoded the JM22 target, GILGFVFTL (fig. S3D). In comparison, GILGFVFTL was represented by 3.9% of peptide-encoding reads in the unlabeled population (fig. S3D). This difference of 13.5% (Fig. 3D) was the most significant outlier among the 32 encoded peptides, with a z-score of 5.2 (Fig. 3E). All 31 of the other encoded peptides had a z-score <1 (Fig. 3E). Replication showed similar results (table S6A and fig. S3, E and F). These results demonstrated that our assay can identify a targeted epitope starting from a mixed pool of peptide-encoding oligonucleotides.
Identification of an HLA class II epitope from a pooled oligonucleotide library
We performed an analogous screen with HLA class II epitopes, using a pooled DNA library consisting of 19 CMV, EBV, Flu, or Clostridium tetani (CEFT) class II epitope-encoding genes (fig. S3G) (40, 41). We transduced this library into HLA-DRA*01:01 and HLA-DRB*01:01 co-expressing APCs at m.o.i. <1 (approximately 0.2–0.5), and co-cultured the APC library with T cells expressing HA1.7 TCR.
In a representative experiment (table S6B), in the pulldown of PE+ cells, 11.4% of total peptide-encoding reads encoded the HA1.7 target, PKYVKQNTLKLAT (fig. S3H). In comparison, PKYVKQNTLKLAT was represented by 2.4% of peptide-encoding reads in the unlabeled population (fig. S3H). This difference of 9.0% (fig. S3, I and J) was the most significant outlier among the 19 encoded peptides, with a z-score of 3.7 (fig. S3K). Replication showed similar results (table S6B and fig. S3, I and J). These results demonstrated that our assay can be used to identify class II epitopes starting from mixed pools of peptide-encoding oligonucleotides.
Scale-up of the assay to identify a TIL-targeted neoepitope from an oligonucleotide array
We then determined whether we could identify the neoepitope target of a tumor-infiltrating T cell amongst a pool of thousands of putative epitopes synthesized on an oligonucleotide array. As proof-of-principle, we used the KRAS p.G12D-reactive TCR (6). We created a pooled library of 2,100 oligonucleotides encoding all 8–12 amino acid (aa) peptides that contain one of 42 common driver mutations (Fig. 4A and table S6C). 1,967 peptide-encoding sequences (93.6% of expected library diversity) were identified in the input DNA library (Fig. 4B) at the depth sequenced (fig. S4A). We transduced the library into HLA-C*08:02-expressing APCs at m.o.i. <1 (approximately 0.8), and co-cultured the APC library with KRAS p.G12D-reactive TCR-expressing T cells.
Fig. 4. Scale-up to identify a targeted neoepitope from an oligonucleotide array.
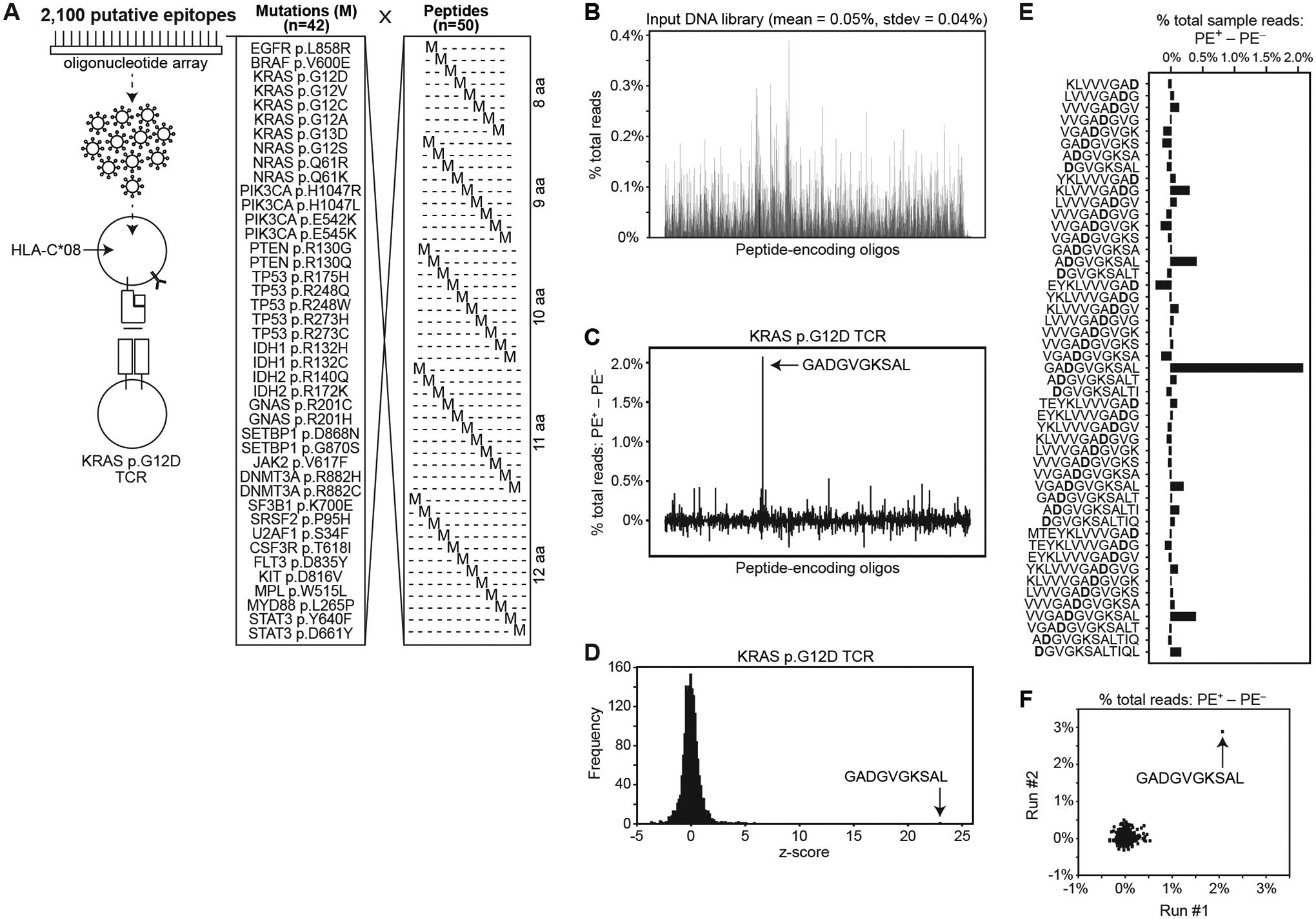
(A) Schematic showing the set of 2,100 peptide-encoding minigenes encoding each of 42 common somatic mutations in all possible positions in 8–12 aa peptides. HLA-C*08:02-expressing APCs were transduced with the pooled peptide-encoding genes at m.o.i. <1, and co-cultured with T cells expressing KRAS p.G12D-reactive TCR. (B) Read count of amplified peptide-encoding sequences from the input DNA library. GADGVGKSAL epitope is shown in blue. (C) Difference between sorted and flow-through cells in percent total read count for each encoded peptide. (D) Frequency of z-score measurements from screening data. (E) Difference between sorted and flow-through cells in percent total read count for each encoded peptide; only epitopes containing the KRAS p.G12D mutation are shown. (F) Comparison between run #1 and run #2 of differences in percent total read count between sorted and flow-through cells for each encoded peptide. Data are representative of two independent experiments.
In a representative experiment (table S6C), in the pulldown of PE+ cells, 2.2% of total peptide-encoding reads encoded the KRAS p.G12D TCR target, GADGVGKSAL (fig. S4B). In comparison, GADGVGKSAL was represented by 0.09% of peptide-encoding reads in the unlabeled population (fig. S4B). This difference of 2.1% (Fig. 4C) was the most significant outlier among the library, with a z-score of 23.0 (Fig. 4D). Other sequences that encompass the p.G12D mutation but at a shifted position and/or different length were not significantly enriched (Fig. 4E). Replication showed similar results (Fig. 4F, fig. S4C, and table S6C).
We also created a pooled library of 3,005 oligonucleotides (table S6D) encoding all 8–12 aa peptides that contain one of 61 somatic mutations identified in the patient (4095) in whom the KRAS p.G12D-reactive TIL was found (6). We transduced the library into APCs that were co-transduced with all six of patient 4095’s class I genes (fig. S4D). After co-culture of the APC library with T cells expressing the KRAS p.G12D-reactive TCR, we again identified GADGVGKSAL as the most significant outlier (fig. S4E and table S6D), with a z-score of 25.6 (fig. S4F and table S6D).
Taken together, these results demonstrated that our assay can identify a TCR-targeted epitope – in this case a tumor neoepitope – amongst a library of thousands of peptide-encoding oligonucleotides synthesized on a DNA array.
Fine mapping epitope sequences using tiled encoded peptides
We reasoned that our method can be used to fine map epitopes targeted by a reactive T cell: e.g. the minimal epitope targeted within a gene or genome. As proof of principle, we used the Ob.1A12 TCR, a class II-restricted TCR identified in a patient with multiple sclerosis (38). ENPVVHFFKNIVTPR was previously identified as an optimal 15 aa epitope within the MBP protein (37).
We created a pooled DNA library that encoded all 15 aa peptides in MBP (Fig. 5, A and B, fig. S5A, and table S6E), and created fusions with CD74 (fig. S2I, middle panel). We transduced this library into HLA-DRA*01:01 and HLA-DRB1*15:01 co-expressing APCs at m.o.i. <1 (approximately 0.25–0.75), and co-cultured the APCs with T cells expressing the Ob.1A12 TCR.
Fig. 5. Fine mapping epitope sequences using tiled encoded peptides.
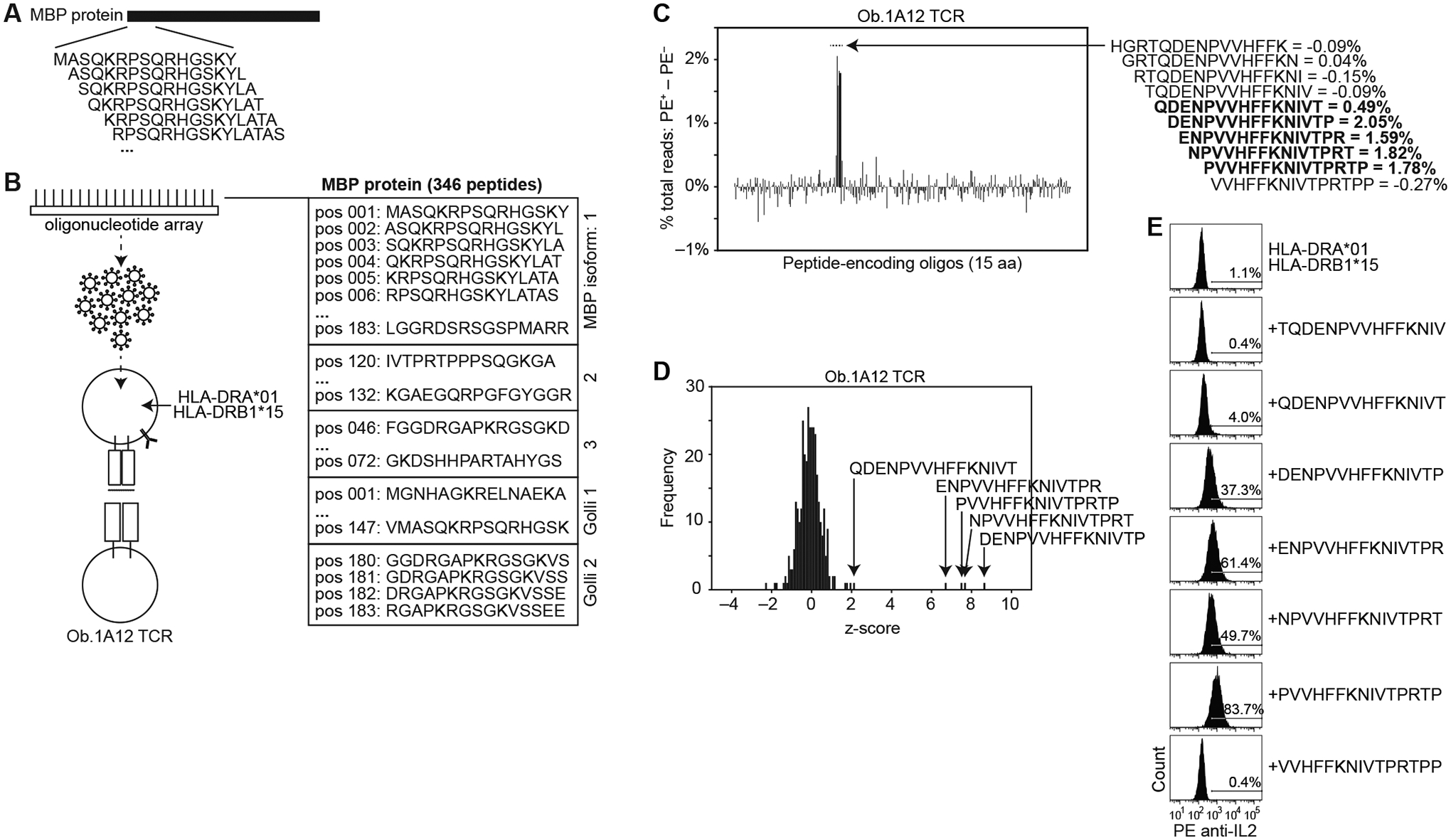
(A) Schematic showing in silico construction of an encoded peptide library consisting of all 15 aa peptides in the MBP protein. (B) Schematic showing the positions (pos) of the 346 unique encoded peptides from indicated MBP isoforms. Peptide-encoding genes were cloned 3’ of CD74 with an intervening cathepsin-cleavage sequence. HLA-DRA*01:01- and HLA-DRB1*15:01-expressing APCs were transduced with the pooled peptide-encoding genes at m.o.i. <1, and co-cultured with T cells expressing Ob.1A12 TCR. (C) Difference between sorted and flow-through cells in percent total read count for each encoded peptide. Values of selected peptide sequences are shown on the right. (D) Frequency of z-score measurements from screening data (left). (E) Histogram of IL-2 cell surface capture after co-culture of Ob.1A12 TCR-expressing T cells with APCs expressing HLA-DRA*01 and HLA-DRB1*15:01 alone, or with indicated peptide-encoding genes. Screening data are representative of one independent experiment run in triplicate. Flow cytometry data are representative of three independent experiments.
In a representative experiment (table S6E), in the pulldown of PE+ cells, five overlapping peptides had z-scores >2 (Fig. 5, C and D, and fig. S5B). Replication showed similar results (fig. S5, C to F, and table S6E). We confirmed our screening results using individual encoded peptides, showing that each of the five epitopes elicit T cell activation, while the flanking peptides did not elicit substantial signal relative to expression of HLA alone (Fig. 5E). Expression of QDENPVVHFFKNIVT consistently resulted in dimmer signal (4.0% of APCs gated as PE+ in a representative experiment) relative to expression of the other four epitopes (37.3%, 61.4%, 49.7%, and 83.7% gated) (Fig. 5E and fig. S5G), consistent with its weaker enrichment in the screen (Fig. 5, C and D). Consistent with our functional screen, the four strongest T cell-activating peptides (Fig. 5E) were the top four predicted binders to HLA-DRB1*15:01 (fig. S5H).
These results show that multiple stimulatory epitopes could be identified in a single pulldown, the enrichment scores can be semi-quantitative, and this information taken together can be used to fine map epitopes.
Identification of epitopes targeted by orphan T cell receptors
Having established the ability to identify known epitopes, we set out to apply our method towards discovery of epitopes targeted by TCRs that are common in the population (i.e. “public” TCRs), but whose targets remain unknown (i.e. “orphan” TCRs). We started with a published database of 89,840,865 TCRβ sequences identified in the peripheral blood of 666 donors (10). Analysis of this dataset had shown that 164 TCRβ sequences were shared by multiple individuals and were statistically associated with CMV seropositive status (10). While prior data suggested that nine of the TCRβ sequences recognize known class I CMV epitopes, 155 of the epitopes remain unknown (10).
To identify targets of the orphan TCRs, we first needed to define (i) the TCRα genes whose protein products form heterodimers with the selected TCRβ proteins, (ii) the HLA presenting the epitope to the TCRs, and (iii) a putative epitope set (Fig. 6A). Because these are public TCRβ, we searched databases of single-cell TCR sequences (from peripheral blood, tumor, or normal tissue) (42–45) for the exact TCRβ genes, leading to identification of 11 of the 155 TCRβ sequences with paired TCRα sequences (fig. S6A and table S7). Candidate HLA were identified for 10 of 11 TCRs from available HLA typing (table S7), although full typing was not available, representing a potential source of negative screening results. For four TCRs (TCRs #1–4), candidate HLA were narrowed to a single type based on statistical association with the TCRβ (10); and for two TCRs (TCRs #5–6), candidate HLA were narrowed to two HLA types each (table S7).
Fig. 6. Identification of epitopes targeted by orphan T cell receptors.
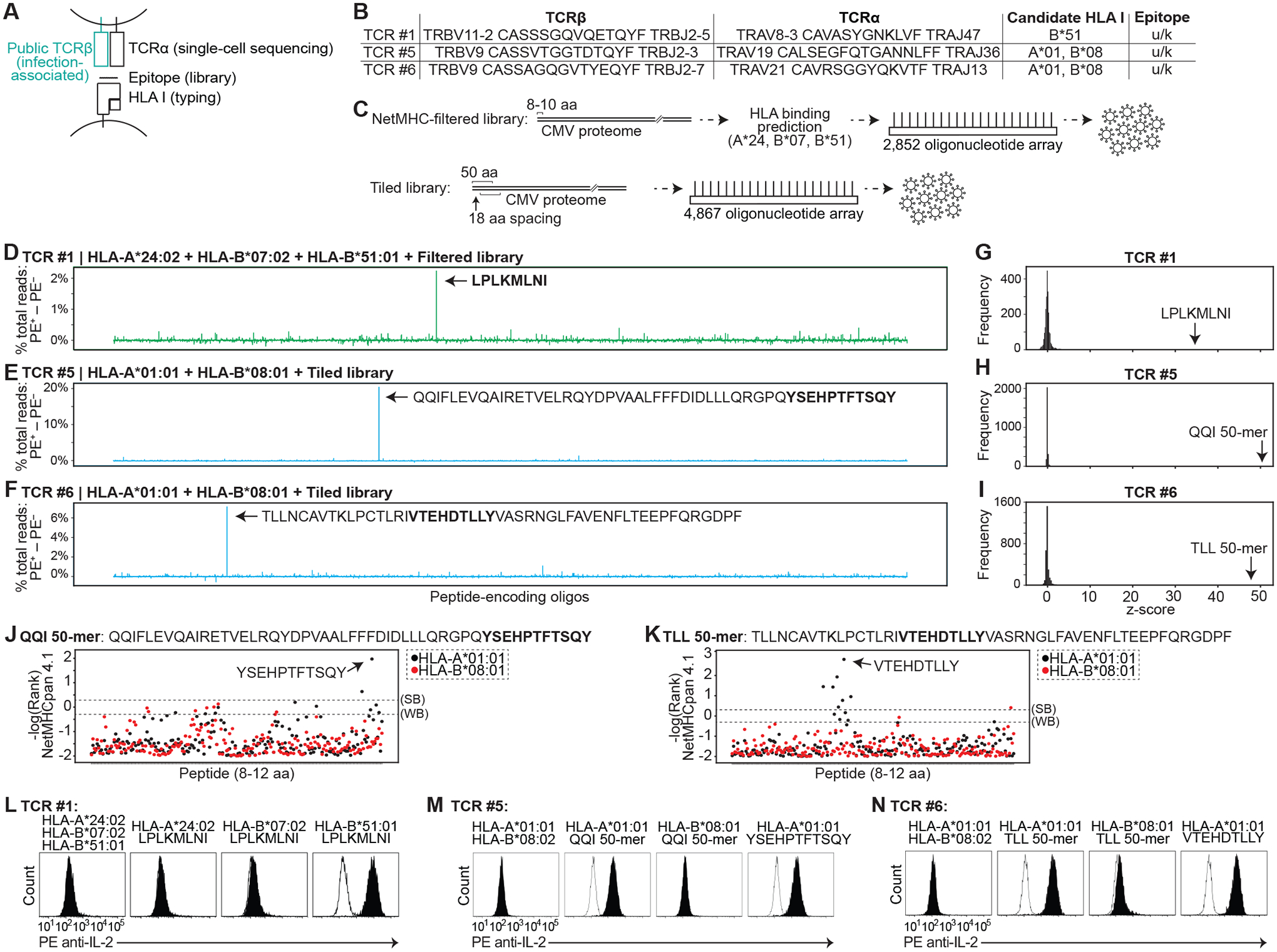
(A) Schematic showing sources of genes used to functionally test HLA-peptide-TCR interactions. (B) Table of TCRβ and TCRα genes screened below, with candidate HLA I. (C) Two CMV peptide-encoding libraries were synthesized. The “NetMHC-filtered” library consisted of 2,852 8–10 aa peptides selected for binding to HLA-A24:02, HLA-B07:02, or HLA-B51:01. The “tiled” library consisted of 4,867 50 aa peptides tiling the CMV proteome, each with 18 aa spacing from the adjacent peptide. (D to F) Difference between sorted and flow-through cells in percent total read count for each encoded peptide sequence screened with TCR #1 (D), TCR #5 (E), or TCR #6 (F). (G to I) Frequency of z-score measurements from screening data for TCR #1 (G), TCR #5 (H), and TCR #6 (I). (J and K) %Rank scores (-log) from NetMHCpan 4.1 for QQI (J) and TLL (K) 50-mers. Thresholds for strong binders (SB) and weak binders (WB) are marked. (L to N) Histogram of IL-2 cell surface capture after co-culture of TCR #1- (L), TCR #5- (M), and TCR #6 (N)-expressing T cells with APCs expressing indicated HLA with or without indicated encoded peptides. For each TCR, histogram of HLA alone is overlaid in white. Data are representative of two or three independent experiments.
As a source of putative epitopes, we focused on the 236 kbp CMV genome – the largest among viruses that cause disease in humans (46) (fig. S6B) – as the most likely antigen source given the TCRs’ association with CMV seropositivity (fig. S6A). We synthesized two libraries (Fig. 6C and table S6, F and G): (i) a library of 2,852 oligonucleotides, each encoding 8–10 aa peptides from CMV that were predicted to bind to HLA-A*24:02, HLA-B*07:02, or HLA-B*51:01 (our “NetMHC-filtered” library); and (ii) a library of 4,867 oligonucleotides, each encoding 50 aa peptides (with 32 aa overlaps) tiling the CMV proteome (our “tiled” library). We transduced the NetMHC-filtered library into HLA-A*24:02, HLA-B*07:02, and HLA-B*51:01 co-expressing APCs (to screen TCRs #1–4). We transduced the tiled library into APCs co-expressing HLA-A*01:01 and HLA-B*08:01 (to screen TCR #5–6); APCs co-expressing HLA-DRA*01:01, HLA-DRB1*04:01, HLA-DRB4*01:03, and HLA-DRB5*01:01 (to screen TCRs #7–9); or APCs co-expressing HLA-DRA*01:01, HLA-DRB1*01:03, HLA-DRB1*07:01, and HLA-DRB4*01:01 (to screen TCR #10). For class II presentation, encoded peptides were fused to CD74.
To screen all TCRs concurrently, we streamlined technical aspects of the workflow. We (i) transduced the oligonucleotide library at m.o.i. >1 (approximately 4–10) to increase the rate of positive clones; (ii) plated 300,000 APCs in a single 10-cm plate for each TCR tested (rather than partitioning the library among wells as we had done in earlier screens); and (iii) sorted the cells using an array of magnetic columns (fig. S6C). To further enrich for PE+ cells, we cultured the first round of sorted APCs, re-incubated them with T cells, performed a second sort, and then generated NGS libraries (fig. S6C). For the screens in which only few PE+ cells were pulled down (i.e. for TCRs #3, 4, and 9), NGS libraries were not prepared.
Four candidate epitopes were found in the class I screens (Fig. 6, D to N, and fig. S6, D to K). For TCR #1, using the NetMHC-filtered library, we identified LPLKMLNI from the CMV UL83 protein as the top candidate (Fig. 6D, fig. S6D, and table S6F), with a z-score of 34.1 (Fig. 6G). We confirmed that LPLKMLNI stimulates TCR #4-expressing T cells, and that recognition is restricted by HLA-B*51:01 (Fig. 6L).
For TCR #5, using the tiled library, we identified the 50 aa peptide CMV UL83 (324–373) (table S2) as the top candidate (Fig. 6E, fig. S6E, and table S6G), with a z-score of 50.3 (Fig. 6H). We confirmed that this peptide (QQI 50-mer) stimulates TCR #5-expressing T cells, and found that recognition is restricted by HLA-A*01:01 (Fig. 6M). Within the QQI 50-mer, the 11 aa peptide YSEHPTFTSQY was the top ranked peptide predicted to bind to HLA-A*01:01 (Fig. 6J and table S5). We confirmed that YSEHPTFTSQY stimulates TCR #29-expressing T cells (Fig. 6M and fig. S6G). In addition to TCR #5, we had identified another T cell (TCR #5b) in the single-cell data which expressed the same TCRβ but with an alternative TCRα (tables S4 and S7). TCR #5b did not show substantial activation by the QQI 50-mer (fig. S6H).
For TCR #6, we identified the 50 aa peptide CMV UL44 (229–278) (table S2) as the top candidate (Fig. 6F, fig. S5F, and table S6H) with a z-score of 48.2 (Fig. 6I). We confirmed that this peptide (TLL 50-mer) stimulates TCR #6-expressing T cells, and found that recognition is restricted by HLA-A*01:01 (Fig. 6N). Within the TLL 50-mer, the 9 aa peptide VTEHDTLLY was the top ranked peptide predicted to bind to HLA-A*01:01 (Fig. 6K and table S5). We confirmed that VTEHDTLLY stimulates TCR #1-expressing T cells when presented on HLA-A*01:01 (Fig. 6N).
For TCR #2, using the NetMHC-filtered library, we identified 18 candidates that appeared to be outliers relative to background (fig. S6, I and J, and table S6I). Replication of both rounds of screening showed that VYAIFIFQL from the CMV US20 protein was a candidate in both screens (fig. S6K and table S6I). We confirmed that VYAIFIFQL stimulates TCR #2-expressing T cells, and that recognition is restricted by HLA-A*24:02 (fig. S6L). Expression of a 50 aa peptide that contains the endogenous sequence context around VYAIFIFQL also stimulated TCR #2-expressing T cells (fig. S6M). We hypothesized that the candidates that were not reproducible in the original screen may be false positives caused by (i) transduction of the oligonucleotide library at m.o.i. >1 combined with (ii) selection of a small number of clones in the first round of the screen. This “bottleneck” effect would cause APCs expressing VYAIFIFQL to be selected, but “passenger” peptides within VYAIFIFQL-expressing APCs to also be enriched. With higher m.o.i., our screening workflow depended upon selection of many different clones, so that only the target epitope is enriched (Fig. 3B). Testing of several of the hypothesized passenger peptides confirmed that they did not stimulate TCR #2 (fig. S6N).
Altogether, we identified epitopes for four of six TCRs with unique TCRβ genes that were screened on class I libraries. Potential reasons for lack of identification of targets (e.g. for TCRs #3–4) are: (i) an absence of the epitope in the encoded peptide library secondary to filtering for predicted HLA binding, to strain-specific differences or unannotated ORFs, to inefficient processing of a long peptide, or to the absence of post-translational modifications; (ii) an incorrect TCRα due to promiscuity in TCRβ-TCRα pairing; or (iii) a lack of power secondary to variability of abundance of encoded peptides during input library preparation. Of the four epitopes we identified, three have been described. YSEHPTFTSQY and VTEHDTLLY have previously been shown to stimulate T cells from multiple individuals (47), consistent with the associated TCRβ genes being public. LPLKMLNI has previously been shown to stimulate T cells from at least one individual (48). In contrast, the VYAIFIFQL epitope has not been described previously.
Identification of epitopes targeted by orphan, class II-restricted T cell receptors
Three candidate epitopes were found in the class II screens (Fig. 7, C to J, and fig. S7, A to E). For TCR #7, using the tiled library, we identified the 50 aa peptide CMV UL86 (1217–1266) (table S2) as the top candidate (Fig. 7C, fig. S7A, and table S6J), with a z-score of 19.7 (Fig. 7E). We confirmed that this peptide (HRE 50-mer) stimulates TCR #7-expressing T cells, and found that recognition is restricted by HLA-DRB1*04:01 (Fig. 7I). Within the HRE 50-mer, AQTFAATHNPWASQA was the top ranked 15 aa peptide predicted to bind to HLA-DRB1*04:01 (Fig. 7G and table S5). We confirmed that AQTFAATHNPWASQA stimulates TCR #7-expressing T cells when presented on HLA-DRB1*04:01 (Fig. 7I and fig. S7, B and C).
Fig. 7. Identification of epitopes targeted by orphan, class II-restricted T cell receptors.
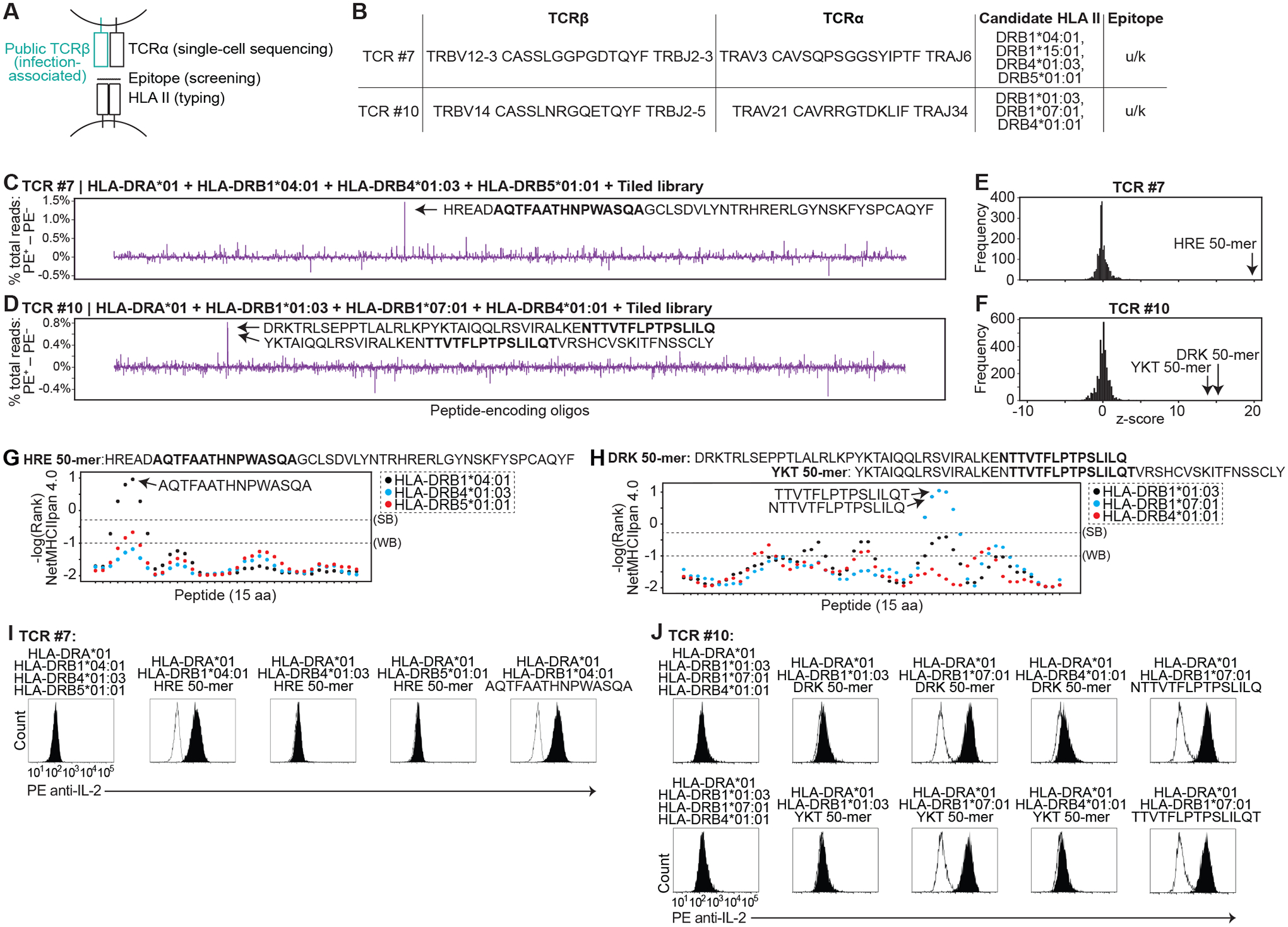
(A) Schematic showing sources of TCRβ and TCRα genes, encoded peptides, and HLA II genes used to functionally test HLA-peptide-TCR interactions. (B) Table of TCRβ and TCRα genes screened below, with candidate HLA II. (C and D) Difference between sorted and flow-through cells in percent total read count for each encoded peptide screened with TCR #7 (C) or TCR #10 (D). (E and F) Frequency of z-score measurements from screening data for TCR #7 (E) and TCR #10 (F). Z-scores were calculated from the screening data (left). (G and H) %Rank scores (-log) from NetMHCIIpan 4.0 for HRE (G) and DRK/YKT 50-mers (H). (I and J) Histogram of IL-2 cell surface capture after co-culture of TCR #7- (I) or TCR #10 (J)-expressing T cells with APCs expressing indicated HLA with or without indicated encoded peptides. For each TCR, histogram of HLA alone is overlaid in white. Data are representative of two or three independent experiments.
For TCR #8, we identified the same HRE 50-mer as the top candidate (fig. S7, D and E, and table S6K). This was not unexpected given sequence similarity between TCR #7 and TCR #8 (table S4). We confirmed that TCR #8 was similarly restricted by HLA-DRB1*04:01 (fig. S7F), and that AQTFAATHNPWASQA stimulates TCR #8-expressing T cells when presented on HLA-DRB1*04:01 (fig. S7G). Of note, TCR #7 and #8 use the TRBV12–3 gene segment. TCRβ sequencing (10, 42) had been unable to resolve TRBV12–3 from TRBV12–4 (table S7). In contrast to T cells expressing TCR #7 or #8, T cells expressing TCRs with the corresponding TRBV12–4-containing sequences (TCR #7–4 and TCR #8–4) (table S4) were not activated by the HRE 50-mer nor AQTFAATHNPWASQA (fig. S7G). This suggests that TRBV12–3 CASSLGGPGDTQYF TRBJ2–3 and TRBV12–3 CASSLGGAGDTQYF TRBJ2–3 are the TCRβ driving the statistical associations with CMV seropositivity (10).
For TCR #10, we identified the 50 aa peptide CMV UL44 (2–51) (table S2) as the top candidate (Fig. 7D, fig. S7H, and table S6L), with a z-score of 15.7 (Fig. 7F). The second highest-scoring candidate (z-score of 13.7) was the adjacent 50-mer, CMV UL44 (20–69) (Fig. 7, D and F, and tables S2 and S6L) (Fig. 7H). We confirmed that these peptides (DRK 50-mer and YKT 50-mer) stimulate TCR #10-expressing T cells, and found that recognition is restricted by HLA-DRB1*07:01 (Fig. 7J and fig. S7I). Within the DRK 50-mer, NTTVTFLPTPSLILQ was the top ranked 15 aa peptide predicted to bind to HLA-DRB1*07:01 (Fig. 7G); the overlapping peptide TTVTFLPTPSLILQT was the top ranked peptide within the YKT 50-mer (Fig. 7G). We confirmed that NTTVTFLPTPSLILQ and TTVTFLPTPSLILQT stimulate TCR #10-expressing T cells when presented on HLA-DRB1*07:01 (Fig. 7J and fig. S7, J and K).
Taken together, we identified epitopes for three of four TCRs with unique TCRβ genes that were screened on class II libraries. One previous report had described peptides containing the FAATHNPWA binding core (e.g. AQTFAATHNPWASQA) that stimulate CD4+ T cells in several individuals (47, 49), consistent with the associated TCRβ genes being public. Class II epitopes containing the FLPTPSLIL binding core (e.g. TTVTFLPTPSLILQT) have not been previously described. In total, out of the ten CD8+ or CD4+ TCRs with unique TCRβ genes, we identified CMV epitopes for seven, including both known and previously undescribed epitopes. These data validate the accuracy and robustness of our screening method.
DISCUSSION
Our key advance is the development of engineered APCs that link a T cell-secreted cytokine signal with the DNA that encodes the presented peptide, allowing oligonucleotide pools with complexities in at least the thousands to be filtered for T cell epitopes. We used this system to identify epitopes for CD8+ and CD4+ T cell receptors that are shared amongst many individuals, and whose peptide-HLA targets were previously unknown.
In contrast to other pooled DNA-based screening systems, signal generation occurs through an unmodified HLA molecule, and the signal (T cell activation-dependent cytokine) is common to both class I- and II-restricted T cells. These features have potential advantages over screening systems in which: optimization is needed to screen different HLA molecules (19) with potential intractability for certain alleles (24); tethered peptide-HLA complexes are used that may alter peptide binding affinities and necessitate encoded peptide libraries to be separately prepared with each HLA (19–21, 25); signal is generated solely through binding interactions (19, 20); or signal is restricted to certain T cell subsets (22). Previous technologies to screen HLA class II libraries have been limited to testing hundreds of peptides (50) or limited to screening single HLA alleles (19, 25). Our assay combines (i) inexpensive scaling to many thousands of encoded peptides; (ii) co-transduction of multiple class II genes into an immortal, class II-negative APC line; (iii) an encoded anti-cytokine antibody that can be interchanged (Fig. 1D), theoretically allowing selection of T cell subsets through their differential cytokine secretion (51); and (iv) a robust epitope selection strategy, which together could help greatly expand the number of known class II epitope/TCR pairings (52). For example, our identification of TTVTFLPTPSLILQT represents, to our knowledge, the first human CD4+ TCR for which an epitope has been identified using an oligonucleotide library-based functional screening approach. Such high-throughput screening for HLA class II epitopes is of particular need given the poorer accuracy of in silico HLA class II binding prediction algorithms (53) combined with the importance of CD4+ T cells in disease physiology.
The major advantage of our approach relative to traditional epitope identification methods (e.g. ELISPOT and ICS) is the greatly increased scale of peptides that could be tested, given orders of magnitude lower cost. This enables two opportunities. One is relatively cheap screening of otherwise very expensive peptide sets, e.g. peptides tiling entire proteomes of viruses. The second is construction of even larger peptide libraries that are difficult, if not impossible, to screen using chemically synthesized peptides due to cost and/or technical constraints; e.g. virome-wide libraries, proteome-wide bacterial libraries, or proteome-wide mammalian libraries. Here we screened libraries with complexities in the thousands, although increases of peptide library diversity beyond those described here should be possible as oligonucleotide libraries can be synthesized at orders of magnitude greater scale (18). The main drawback lies in the inherent counterbalance between tackling peptide complexity vs. T cell complexity, which is common to current epitope identification technologies (54). Screening efficiency may be improved by adding mixtures of T cells to the libraries, or by combining our method with high-throughput TCR cloning (55) and/or T cell selection strategies (12, 56).
We performed unbiased screening of the CMV proteome to identify the targets of TCRs that are common among humans. Our identification of CMV epitopes confirms the genetic associations between presence of these TCRβ genes and CMV seropositive status (10). As the epitope search space was unbiased for sequences within CMV, the identification of several known epitopes serves as evidence for the specificity of our method. We also identified class I- and II-presented CMV epitopes (VYAIFIFQL and TTVTFLPTPSLILQT, respectively) that have not been previously described. Because these epitopes are targeted by public TCRβ genes, these epitopes may be common, but previously unrecognized, antigens within CMV; population studies are needed to evaluate this possibility. Beyond public TCRs, our method could be used to identify antigens for orphan TCRs derived from, e.g.: T cells that are infiltrating into tissue (e.g. tumor-infiltrating T cells); T cells that are selected through in vitro activation studies (e.g. using whole virus) in which the minimal epitopes are unmapped; or individuals’ memory T cell repertoires for wide-ranging characterization of T cell specificities.
Knowledge of HLA-epitope-TCR complexes is increasingly being used to develop diagnostics (10) and therapeutics (6, 7) such as TCR-based cellular therapies, underscoring the growing need for improvements in HLA-epitope-TCR complex identification as described herein. The ability of our method to scale peptide testing at low cost may enable further discovery of class I and II epitopes in patients with infectious diseases, and suggests a path towards identification of novel tumor neoepitopes as well as elusive T cell targets, such as autoimmune epitopes (57).
MATERIALS AND METHODS
Study design
The goal of this study was to develop a method, based on capturing T cell-secreted cytokines on the APC cell surface, to identify T cell-targeted epitopes from peptide-encoding oligonucleotide pools. We genetically manipulated APCs to express defined HLA genes, libraries of peptide-encoding genes, and surface-bound anti-cytokine antibody-encoding genes. We used TCRs with known specificity to screen for their cognate epitope(s) as proof-of-principle, and then screened encoded peptide libraries for epitopes targeted by orphan TCRs. The number of independent experiments is described in the figure legends, where applicable.
Cells, viruses, and reagents
Jurkat cells were maintained in RPMI 1640 with 10% FBS. HeLa and 293T cells were maintained in DMEM with 10% FBS. Lentivirus was produced in 293T cells using pLX301 or pLX303 (Addgene #25895/25897), psPAX2, and pCMV-VSVG. PMA and ionomycin were obtained from Sigma-Aldrich. Recombinant IL-2 and IFN-γ were obtained from Peprotech. Antibodies were obtained from the following sources: PE- or Vio515-conjugated anti-IL-2 (N7.48 A) and PE-conjugated anti-IFN- γ (IFN-γ Secretion Assay-Detection Kit) from Miltenyi; APC-conjugated anti-CD45 (HI30), FITC-conjugated anti-HLA-A/B/C (W6/32), FITC-conjugated anti-HLA-DR (Tü36), FITC-conjugated anti-HLA-DP/DQ/DR (Tü39), PE-conjugated anti-HLA-DM (MaP.DM1), and PE-conjugated anti-TCR α/β (IP26) from BioLegend.
Antigen-presenting cell preparation
Genomic DNA from HeLa and HEK293T cells was extracted (DNeasy Blood & Tissue Kit; Qiagen), and HLA typed by NGS (CD Genomics). HeLa cells were typed as HLA-A*68:02 (homozygous), HLA-B*15:03 (homozygous), HLA-C*12:03 (heterozygous c.391G>A (p.G131R)), DPA1*02:01:08 (homozygous), DPB1*01:01:01 (homozygous), DQA1*01:01:01 (homozygous), DQB1*05:01:01 (heterozygous c.186C>T (p.H62Q)), DRB1*01:02:01 (homozygous), DRB345 not present. 293T cells were typed as homozygous HLA-A*02:01, HLA-B*07:02, HLA-C*07:02, DPA1*01:03:01, DPB1*04:02:01, DQA1*01:02:01, DQB1*06:02:01, DRB1*15:01:01, and DRB5*01:01:01. Two CRISPR/Cas9 cassettes directed to cleave sequences in all the above class I genes were cloned into pX330 (Addgene #42230) (table S1). The targeting vectors were transfected into HeLa or HEK293T cells using TransIT-LT1 (Mirus Bio), and single-cell clones were established. To create class I/ II knockout cells, CRISPR/Cas9 cassettes directed to cleave the HeLa class II genes were cloned into pXPR_001 (Addgene #49535) (table S1). The targeting vectors were transfected into a CIITA-expressing class I KO HeLa clone, and single-cell clones were established. Class I or I/II KO APCs were then transduced with selected HLA cDNAs.
To express surface-bound antibodies, anti-IL-2 (clone 16C3.1) (58) or anti-IFN-γ (clone NI-0501) (59) heavy and light chain cDNAs were cloned separately into pLX303; antibody heavy chains were fused to a GGSGGGSG linker followed by a transmembrane domain from PDGFRB (table S3). APCs were co-transduced with the heavy and light chain genes. Cells were used directly, or single-cell clones were established.
Encoded peptide library construction
Class I encoded peptides were cloned 3’ of the human IL2 signal sequence, into pLX301 using Gibson Assembly (New England Biolabs). Oligonucleotides were synthesized by IDT, CustomArray/GenScript, or Twist Bioscience. For class II-presented peptides, the invariant chain (CD74) gene was fused to a cathepsin S cleavage site (aa sequence: GRWHTVGL) followed by a peptide-encoding sequence. Alternatively, the CLIP-encoding sequence (aa sequence: MRMATPLLM) in CD74 was replaced with a peptide-encoding sequence. Long (50 aa) class I peptides were cloned 3’ of a methionine; the spacer sequence encoding HTVGLYM was added between the methionine and peptide to facilitate cloning. For long class II peptides, a YM spacer sequence was added between the cathepsin S cleavage site and the encoded peptide.
To construct the CMV libraries, we downloaded sequence data for all 169 annotated ORFs (open reading frames) in strain Merlin (NCBI AY446894 and UniProt UP000000938); 190 ORFs in strain AD169 (UniProt UP000008991); and 57 ORFs from mass spectrometry of CMV-infected cells (60). To construct the tiled library, for all ORFs, we tiled 50 aa peptides starting from the start codon, each with 32 aa overlaps. Full-length ORFs were used if ORF length was <50 aa. This resulted in 4,867 total unique peptides. To construct the NetMHC-filtered library, we ran all ORF sequences through the NetMHCpan 4.0 server (61), specifying peptide lengths of 8–10 aa, and alleles HLA-A*24:02, HLA-B*07:02, and HLA-B51:01. We obtained 1,407 peptides for HLA-A*24:02 (%Rank </= 0.5), 947 peptides for HLA-B*07:02 (%Rank </= 0.2), and 631 peptides for HLA-B*51:01 (%Rank </= 0.2); for 2,852 unique peptides.
T cell receptor expression
Jurkat T cells were transduced with lentivirus containing CRISPR/Cas9 cassettes (pXPR_001) directed to cleave the TCR constant regions (TRAC and TRBC1) (table S1), and single-cell clones were established.
TCR cDNAs were prepared as follows. The TCR constant regions were modified: silent mutations were introduced to avoid CRISPR/Cas9 targeting (table S1), and TRAC p.T48C and TRBC p.S57C mutations were introduced (62). Specific TCRα and TCRβ variable regions were fused in-frame of the respective TCR constant region, and cloned into pLX301. Lentivirus containing TCRα and TCRβ genes were co-transduced into TCR KO T cells.
APC cytokine capture assay
APCs expressing desired HLA, encoded peptides, and surface-bound anti-cytokine antibodies were seeded in 384-well, 96-well, or 10-cm plates, and cultured for 2–8 days. T cells were then added at a ratio between 2:1 and 16:1 along with 25–80 ng/mL PMA. After incubation at 37°C for 16–24 hours (or for time intervals indicated in text), cells were washed with phosphate buffered saline (PBS). Cells were dissociated with 0.25% trypsin-EDTA or enzyme-free cell dissociation buffer (Thermo Fisher Scientific), pooled, and stained with fluorescently-labeled anti-IL2 antibody; alternatively, cells were stained in the plate prior to dissociation. Stained cells were washed with PBS. A subset was imaged (Zeiss Axiovert 40 CFL, Olympus CK40, or Olympus IX73) to assess fluorescence, or analyzed by flow cytometry (BD LSRFortessa). For pulldowns, PE+ cells were labeled using Anti-PE MicroBeads (Miltenyi) or PE Positive Selection Kit (StemCell Technologies), and separated from unlabeled cells using a MACS Separator (Miltenyi) or EasySep Magnet (StemCell Technologies).
Amplification of encoded peptides and sequencing
Genomic DNA was extracted from PE+ (pulldown) and PE– cells (flow-through). The integrated peptide-encoding sequences were amplified by PCR (HiFi HotStart ReadyMix; Kapa Biosystems) using barcoded primers (table S1) complementary to sequences flanking the peptide-encoding sequences. NGS libraries were prepared from the PCR products and sequenced by MGH CCIB DNA Core (Boston, MA).
Data analysis
For each NGS read, the 5’ and 3’ barcodes were identified to demultiplex the reads into associated samples. For long encoded peptides, paired end reads were joined using fastq-join (63). For each read, common sequences flanking the peptide-encoding sequence were identified, and the intervening peptide-encoding sequences were enumerated for each sample. For each sample, the fractional abundance of each encoded peptide in the pulldown was calculated. The difference in fractional abundance of each encoded peptide between pulldown and flow-through samples (annotated as % enrichment) was calculated.
Peptide-HLA binding predictions were performed using NetMHCpan 4.0 (61), NetMHCpan 4.1 (64), or NetMHCIIpan 4.0 (65) servers using default parameters, including rank thresholds for strong binding (SB) peptides (0.5% for NetMHCpan, 2% for NetMHCIIpan) and weak binding (WB) peptides (2% for NetMHCpan, 10% for NetMHCIIpan).
Statistical analysis
Data are reported as mean ± standard deviation. P values were calculated using a paired, two-tailed Student’s t-test; *P<0.05, **P<0.005. Z-scores were calculated for encoded peptides identified in the pulldown or flow-through samples as the difference of each % enrichment value from the mean, divided by the standard deviation.
Supplementary Material
Table S7. Sources of putative CMV-targeting TCRs (Excel spreadsheet).
Table S6. Sequencing reads from screens (Excel spreadsheet).
Fig. S5. Fine mapping epitope sequences using tiled encoded peptides.
Fig. S1. Establishing a system for epitope identification by APC cytokine capture.
Fig. S2. Engineered APCs capture cytokine in an HLA class I or II epitope-specific manner.
Fig. S3. Identification of a target epitope from a pooled oligonucleotide library.
Fig. S4. Scale-up to identify a targeted neoepitope from an oligonucleotide array.
Fig. S6. Identification of epitopes targeted by orphan T cell receptors.
Fig. S7. Identification of epitopes targeted by orphan, class II-restricted T cell receptors.
Table S1. List of primers.
Table S2. List of encoded peptides.
Table S3. List of cDNA sequences.
Table S4. List of TCR sequences.
Table S5. HLA binding affinity predictions.
Table S8. Raw data file (Excel spreadsheet).
Acknowledgments:
We are grateful to members of the Meyerson laboratory and Dr. Peter Choi (Children’s Hospital of Philadelphia) for advice and valuable discussions.
Funding:
This work was supported by the US National Institutes of Health (NIH) grant R35 CA197568 (M.M.). M.N.L. was supported by the NIH T32 HL066987 and HL007627 training grants.
Competing interests:
M.N.L. and M.M. are co-inventors on a patent application concerning the described technology. M.M. is the scientific advisory board chair of OrigiMed; an inventor of a patent licensed to LabCorp for EGFR mutation diagnosis; and receives research funding from Bayer, Janssen, Novo, and Ono.
Data and materials availability:
All data and datasets generated in this study are present in the paper or the Supplementary Materials. Plasmids and cell lines generated in this work can be provided upon request to the senior author, subject to a Material Transfer Agreement and barring any restrictions that apply to material owned by third parties.
References and Notes:
- 1.International HIV Controllers Study et al. , The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 330, 1551–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyadera H, Tokunaga K, Associations of human leukocyte antigens with autoimmune diseases: Challenges in identifying the mechanism. J. Hum. Genet 60, 697–702 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Zaretsky JM et al. , Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med 375, 819–829 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chowell D et al. , Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 359, 582–587 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Latorre D et al. , T cells in patients with narcolepsy target self-antigens of hypocretin neurons. Nature. 562, 63–68 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Tran E et al. , T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med 375, 2255–2262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maus MV et al. , Adoptive immunotherapy for cancer or viruses. Annu. Rev. Immunol 32, 189–225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dendrou CA, Petersen J, Rossjohn J, Fugger L, HLA variation and disease. Nat. Rev. Immunol 18, 325–339 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Robins HS et al. , Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 114, 4099–107 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emerson RO et al. , Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat. Genet 49, 659–665 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Czerkinsky C et al. , Reverse ELISPOT assay for clonal analysis of cytokine production I. Enumeration of gamma-interferon-secreting cells. J. Immunol. Methods 110, 29–36 (1988). [DOI] [PubMed] [Google Scholar]
- 12.Jung T, Schauer U, Heusser C, Neumann C, Rieger C, Detection of intracellular cytokines by flow cytometry. J. Immunol. Methods 159, 197–207 (1993). [DOI] [PubMed] [Google Scholar]
- 13.Alspach E et al. , MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 574, 696–701 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Bert N et al. , SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 584, 457–462 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Hondowicz BD et al. , Discovery of T cell antigens by high-throughput screening of synthetic minigene libraries. PLoS One. 7 (2012), doi: 10.1371/journal.pone.0029949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siewert K et al. , Unbiased identification of target antigens of CD8 + T cells with combinatorial libraries coding for short peptides. Nat. Med 18, 824–828 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Tian J et al. , Accurate multiplex gene synthesis from programmable DNA microchips. Nature. 432, 1050–4 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Kosuri S, Church GM, Large-scale de novo DNA synthesis: Technologies and applications. Nat. Methods 11, 499–507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Birnbaum ME et al. , Deconstructing the peptide-MHC specificity of T cell recognition. Cell. 157, 1073–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li G et al. , T cell antigen discovery via trogocytosis. Nat. Methods 16, 183–190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joglekar AV et al. , T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat. Methods 16, 191–198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kula T et al. , T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell. 178, 1016–1028.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gee MH et al. , Antigen Identification for Orphan T Cell Receptors Expressed on Tumor-Infiltrating Lymphocytes. Cell. 172, 549–563.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saligrama N et al. , Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature. 572, 481–487 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kisielow J, Obermair FJ, Kopf M, Deciphering CD4 + T cell specificity using novel MHC–TCR chimeric receptors. Nat. Immunol 20, 652–662 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Martoglio B, Dobberstein B, Signal sequences: More than just greasy peptides. Trends Cell Biol. 8, 410–415 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Yang X et al. , Structural basis for clonal diversity of the public T cell response to a dominant human cytomegalovirus epitope. J. Biol. Chem 290, 29106–29119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stewart-Jones GBE, McMichael AJ, Bell JI, Stuart DI, Jones EY, A structural basis for immunodominant human T cell receptor recognition. Nat. Immunol 4, 657–663 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Chen J-L et al. , Structural and kinetic basis for heightened immunogenicity of T cell vaccines. J. Exp. Med 201, 1243–1255 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dössinger G et al. , MHC Multimer-Guided and Cell Culture-Independent Isolation of Functional T Cell Receptors from Single Cells Facilitates TCR Identification for Immunotherapy. PLoS One. 8 (2013), doi: 10.1371/journal.pone.0061384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schub A, Schuster IG, Hammerschmidt W, Moosmann A, CMV-Specific TCR-Transgenic T Cells for Immunotherapy. J. Immunol 183, 6819–6830 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Motozono C et al. , Molecular Basis of a Dominant T Cell Response to an HIV Reverse Transcriptase 8-mer Epitope Presented by the Protective Allele HLA-B*51:01. J. Immunol 192, 3428–3434 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B, Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science. 265, 106–9 (1994). [DOI] [PubMed] [Google Scholar]
- 34.van Bergen J et al. , Efficient loading of HLA-DR with a T helper epitope by genetic exchange of CLIP. Proc. Natl. Acad. Sci. U. S. A 94, 7499–502 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakano N, Rooke R, Benoist C, Mathis D, Positive selection of T cells induced by viral delivery of neopeptides to the thymus. Science. 275, 678–83 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Hennecke J, Carfi A, Wiley DC, Structure of a covalently stabilized complex of a human alphabeta T-cell receptor, influenza HA peptide and MHC class II molecule, HLA-DR1. EMBO J. 19, 5611–24 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wucherpfennig KW et al. , Structural requirements for binding of an immunodominant myelin basic protein peptide to DR2 isotypes and for its recognition by human T cell clones. J. Exp. Med 179, 279–90 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wucherpfennig KW et al. , Clonal expansion and persistence of human T cells specific for an immunodominant myelin basic protein peptide. J. Immunol 152, 5581–92 (1994). [PubMed] [Google Scholar]
- 39.Currier JR et al. , A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J. Immunol. Methods 260, 157–72 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Vita R et al. , The Immune Epitope Database 2.0. Nucleic Acids Res. 38 (2009), doi: 10.1093/nar/gkp1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Planas R et al. , GDP-l-fucose synthase is a CD4+ T cell-specific autoantigen in DRB3*02:02 patients with multiple sclerosis. Sci. Transl. Med 10, 1–16 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Tanno H et al. , Determinants governing T cell receptor α/β-chain pairing in repertoire formation of identical twins. Proc. Natl. Acad. Sci. U. S. A 117, 532–540 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo X et al. , Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med 24, 978–985 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Zheng C et al. , Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell. 169, 1342–1356.e16 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Sade-Feldman M et al. , Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell. 175, 998–1013.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sijmons S et al. , High-Throughput Analysis of Human Cytomegalovirus Genome Diversity Highlights the Widespread Occurrence of Gene-Disrupting Mutations and Pervasive Recombination. J. Virol 89, 7673–7695 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vita R et al. , The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nastke MD et al. , Major contribution of codominant CD8 and CD4 T cell epitopes to the human cytomegalovirus-specific T cell repertoire. Cell. Mol. Life Sci 62, 77–86 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Fuhrmann S, Streitz M, Reinke P, Volk HD, Kern F, T cell response to the cytomegalovirus major capsid protein (UL86) is dominated by helper cells with a large polyfunctional component and diverse epitope recognition. J. Infect. Dis 197, 1455–1458 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Linnemann C et al. , High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+T cells in human melanoma. Nat. Med 21, 81–85 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Haining WN, Travels in time: assessing the functional complexity of T cells. Proc. Natl. Acad. Sci. U. S. A 109, 1359–60 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shugay M et al. , VDJdb: A curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 46, D419–D427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.The problem with neoantigen prediction. Nat. Biotechnol 35, 97 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Joglekar AV, Li G, T cell antigen discovery. Nat. Methods (2020), doi: 10.1038/s41592-020-0867-z. [DOI] [PubMed] [Google Scholar]
- 55.Spindler MJ et al. , Massively parallel interrogation and mining of natively paired human TCRαβ repertoires. Nat. Biotechnol 38, 609–619 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maino VC, Suni MA, Ruitenberg JJ, Rapid flow cytometric method for measuring lymphocyte subset activation. Cytometry. 20, 127–133 (1995). [DOI] [PubMed] [Google Scholar]
- 57.Rosenblum MD, Gratz IK, Paw JS, Abbas AK, Treating human autoimmunity: Current practice and future prospects. Sci. Transl. Med 4, 1–10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rondon IJ, Crellin NK, Bessette P, Trotta E, Bluestone JA, Anti-IL-2 antibodies and compositions and uses thereof. Patent application WO2015109212A1 (2015).
- 59.Ferlin W, Fischer N, Elson G, Leger O, Anti-interferon gamma antibodies and methods of use thereof. U.S. Patent 9682142 (2017).
- 60.Stern-Ginossar N et al. , Decoding human cytomegalovirus. Science (80-.). 338, 1088–1093 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jurtz V et al. , NetMHCpan-4.0: Improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol 199, 3360–3368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuball J et al. , Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 109, 2331–8 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aronesty E, Comparison of Sequencing Utility Programs. Open Bioinforma. J 7, 1–8 (2013). [Google Scholar]
- 64.Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M, NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, W449–W454 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reynisson B et al. , Improved Prediction of MHC II Antigen Presentation through Integration and Motif Deconvolution of Mass Spectrometry MHC Eluted Ligand Data. J. Proteome Res 19, 2304–2315 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S7. Sources of putative CMV-targeting TCRs (Excel spreadsheet).
Table S6. Sequencing reads from screens (Excel spreadsheet).
Fig. S5. Fine mapping epitope sequences using tiled encoded peptides.
Fig. S1. Establishing a system for epitope identification by APC cytokine capture.
Fig. S2. Engineered APCs capture cytokine in an HLA class I or II epitope-specific manner.
Fig. S3. Identification of a target epitope from a pooled oligonucleotide library.
Fig. S4. Scale-up to identify a targeted neoepitope from an oligonucleotide array.
Fig. S6. Identification of epitopes targeted by orphan T cell receptors.
Fig. S7. Identification of epitopes targeted by orphan, class II-restricted T cell receptors.
Table S1. List of primers.
Table S2. List of encoded peptides.
Table S3. List of cDNA sequences.
Table S4. List of TCR sequences.
Table S5. HLA binding affinity predictions.
Table S8. Raw data file (Excel spreadsheet).
Data Availability Statement
All data and datasets generated in this study are present in the paper or the Supplementary Materials. Plasmids and cell lines generated in this work can be provided upon request to the senior author, subject to a Material Transfer Agreement and barring any restrictions that apply to material owned by third parties.