

Abstract
Premature birth results in an increased risk of respiratory distress and often requires oxygen therapy. While the supplemental oxygen has been implicated as a cause of bronchopulmonary dysplasia (BPD), in clinical practice this supplementation usually only occurs after the patient’s oxygen saturation levels have dropped. The effect of hyperoxia on neonates has been extensively studied. However, there is an unanswered fundamental question: which has the most impact-hyperoxia, hypoxia or fluctuating oxygen levels? In this review, we will summarize the reported effect of hypoxia, hyperoxia or a fluctuation of oxygen levels (hypoxia/hyperoxia cycling) in preterm neonates, with special emphasis on the lungs.
Keywords: Bronchopulmonary dysplasia, oxidative stress, renin angiotensin system, lung injury
Bronchopulmonary dysplasia (BPD)
Bronchopulmonary dysplasia (BPD) is a form of chronic lung disease that affects premature newborns and infants and is the result of damage to the lungs caused by mechanical ventilation and long-term use of oxygen.1–4 Despite many advances in neonatal ventilation techniques and the widespread use of surfactant and antenatal corticosteroids, the incidence of BPD has been reported to be relatively stable at approximately 40% of surviving premature infants ≤28 weeks gestational age, with an estimated 10,000–15,000 new cases annually in the US alone.5–11 Mechanical ventilation and excessive oxygen supplementation are well-studied risk factors for BPD.12–14 Both the airway and parenchyma of the lung tissues are affected. These abnormalities have been attributed to ventilator induced injury as well as to oxygen therapy.15
BPD is recognized as a chronic lung disease of infancy that presents as a systemic syndrome and can be associated with neurodevelopmental deficits, cognitive impairments, failure to thrive, pulmonary hypertension and cor pulmonale.16 High rates of in utero and perinatal exposure to infection may be causally related to preterm delivery and subsequent lung injury.17 Over the past three decades, the histological presentation of BPD has changed from heterogeneous pulmonary inflammation and fibrosis (“Old BPD”) to uniform arrest of alveolar development and variable interstitial cellularity and/or fibroproliferation (“New BPD”).18,19
Oxidative injury and development of BPD
Reactive oxygen species (ROS) generation in the perinatal period
The fetus normally lives in a physiologically hypoxic environment, and relative hyperoxic exposure can cause an increase in the generation of “reactive oxygen species” (ROS). Preterm newborns are particularly vulnerable to oxygen toxicity due to inadequate levels of antioxidant enzymes, and hence to decreased protection from oxidative injury of rapidly growing tissues.20–24 The developing lung of the neonate is a perfect example of those vulnerable tissues; the endothelial cells and the alveolar type II cells are especially susceptible to oxidative injury. Oxidative stress in these cells activates transcription factors and pathways leading to cellular dysfunction, inactivation of surfactant, and impaired cell survival.23–27
Several pre- and post-natal adverse events generate ROS exposure and contribute to BPD pathogenesis; these include hypoxic, hyperoxic, and mechanical stimuli.28 Excess levels of oxygen (hyperoxia), as occur in supplemental oxygen administration, generate accumulation of ROS1. Hypoxia/hypoxemia, as in episodes of oxygen desaturations, also results in generation of ROS via a superoxide burst that occurs rapidly with hypoxic exposure.29–31 This ROS generation has been shown to be necessary for stabilization of Hypoxia Inducible Factor 1α (HIF-1α) and activation of the HIF system.27,29,30
The ROS generated by both hyperoxia and hypoxia can result in alterations in cellular proliferation and apoptosis. Mechanisms may involve dysregulation of key transcription factors involved in ROS signaling such as HIF, nuclear factor E2 related factor 2 (Nrf2), NF-κB, and activator protein-1 (AP-1).1,2,32 Altered regulation of these transcription factors and pathways can disrupt postnatal alveolar development, cause inflammation, and potentially lead to fibrosis in preterm infants.33,34 ROS generation activates the HIF system.27 Nrf2 deficiency was shown to augment lung injury and arrest of alveolarization caused by hyperoxia during the newborn period.32 Both NF-κB and AP-1 are activated in multiple cell types, as well as in lung, following hyperoxia.2 A summary of ROS generation and neonatal lung injury is summarized in Figure 1.
Figure 1.
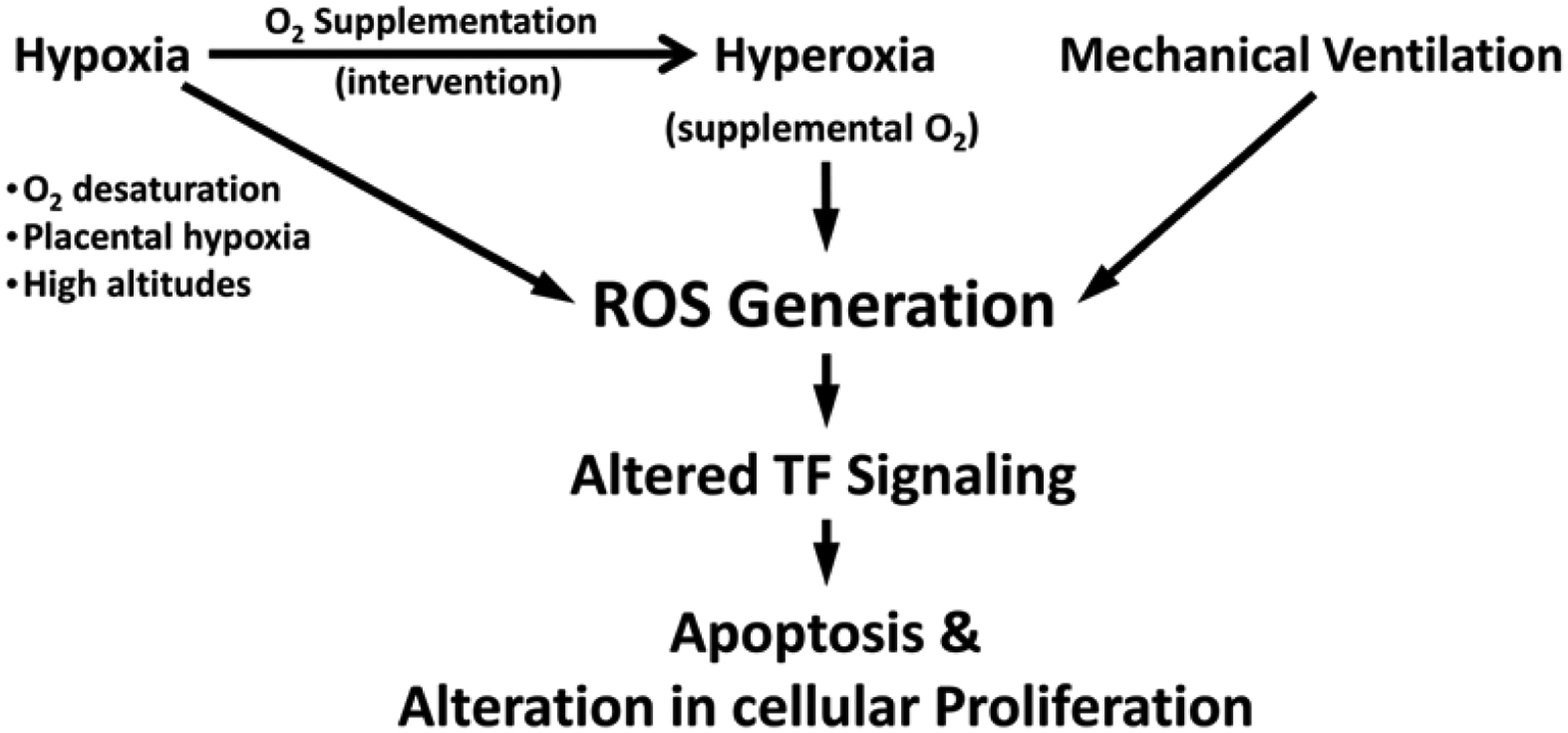
Schematic diagram illustrating reactive oxygen species (ROS) generation in neonatal pulmonary disease.
Prenatal hypoxia
Tissue injury due to oxidative stress is also noted in placental hypoxia, as is seen in preterm born neonates of pre-eclamptic mothers, where ROS are elevated and antioxidant levels are decreased in the maternal circulation.26,35,36 Placental hypoxia also induces an imbalance between pro-angiogenic and anti-angiogenic factors inhibiting VEGF signaling.37,38 This reduction in VEGF signaling in the developing lung was shown to result in impaired pulmonary vascular growth and alveolarization in neonatal rats.39 Maternal exposure to hypobaric hypoxia at high-altitudes can alter placental function, influence oxygen delivery to the fetus, result in lower birth weight, and increase the risk for pre-eclampsia.40,41
Postnatal hypoxia
Premature babies are exposed post-natally to chronic or intermittent hypoxia as a result of immature lungs, apnea of prematurity, inadequate ventilation, or persistence of intrapulmonary arteriovenous shunts causing hypoxemia.27,42 Hypoxia of the infant leads to generalized pulmonary vasoconstriction and increases the pulmonary vascular resistance, causing pulmonary hypertension. Sustained hypoxic pulmonary vasoconstriction causes vascular remodeling of the pulmonary vascular bed leading ultimately to right heart failure.43 Postnatal hypoxia causes impaired alveolarization and alveolar simplification with fewer and larger alveoli. The impaired alveolarization also impairs the vascular maturation in the alveolar wall via mechanisms involving. altered signaling of HIF-1α, VEGF, and TGF-β,27,44,45
Postnatal hyperoxic interventions
Preterm birth leads to premature transition of the pulmonary circulation from the physiologic hypoxic fetal environment to a relative hyperoxic postnatal environment (room air). In addition, oxygen supplementation is often required to fulfill the oxygen demands for adequate functioning of the body’s tissues and organs, in particular the brain, intestines, and kidneys.28 systemically circulating oxygen saturation levels (SPO2) is generally targeted to reach above 85%. However, the optimal systemic oxygen saturation in preterm infants is currently not clear. To compensate for the premature lung’s simplified alveolar structure and thick alveolar septae, higher levels of supplemental oxygen are often required to achieve the targeted intravascular oxygen levels, further augmenting the already existing (relative) postnatal hyperoxic state.25,27 Hyperoxia leads to impaired VEGF expression and disrupted angiogenesis and alveolarization due to rapid proteasomal degradation of HIF-1α.46–48 This induces vascular arrest, leading to pulmonary vascular diseases. Relative hyperoxia also increases generation of ROS and induces oxidative stress, an important contributor to the development of neonatal BPD. In addition, lower levels of antioxidants including vitamin E, transferrin, and superoxide dismutase, and higher levels of free iron further predispose preterm infants to oxidative injury.20,21,23,49,50
Mechanical ventilation in BPD
Although mechanical ventilation is often essential and life-saving, it can provoke ventilator-induced lung injury in severely premature infants mainly by over-stretching of the distal epithelium and capillary endothelium.25,51–55 The development of that injury is dependent on the developmental stage of the lung, and the type, duration, volume and pressure of the mechanical ventilation.52,54 Mechanical ventilation also results in down regulation of VEGF-1 and its receptor flt-1 and up-regulation of the TGF-β co-receptor endoglin. This imbalance in mechanically ventilated lungs likely contributes to altered alveolarization and angiogenesis.28,55,56
The renin angiotensin system (RAS) and BPD
RAS and local tissue injury
The RAS (Figure 2) is traditionally known to play a significant role in blood pressure regulation. Renin is produced by kidney and acts on circulating angiotensinogen (AGT) protein. Renin cleaves AGT to produce angiotensin I (Ang I). Ang I is converted by angiotensin-converting enzyme (ACE) to Ang II, which exerts its actions through binding to specific cell surface angiotensin receptors. Two main receptors to Ang II have been identified; AT1 and AT2; both belong to a super family of seven transmembrane G-protein coupled receptors. The AT1 receptor mediates all of the classical actions of Ang II (vasodilatation, sodium retention, cell growth and proliferation), while the AT2 receptor promotes vasodilatation, cell differentiation, inhibition of cell growth and apoptosis and may play a counterbalancing role to the effects of Ang II on the AT1 receptor.57 ACE-2 and its product angiotensin 1–7 (Ang 1–7) acting on its receptor Mas were shown to have counteracting effects against the adverse actions of the other RAS components. Findings from numerous experimental studies have suggested notable protective effects of ANG1–7/Mas activation in the cardiovascular system.58 Local tissue effects of RAS have been identified in a variety of tissues such as heart, kidney, liver, lung, brain, pancreas and adipose tissue, where RAS component expression has been detected.59,60 Local RAS is involved in injury and inflammatory and fibrogenic diseases of many organs including heart,61,62 lung,63–65 liver,66 pancreas,67 and kidneys,68,69 by mechanisms independent of the blood-derived RAS.
Figure 2.
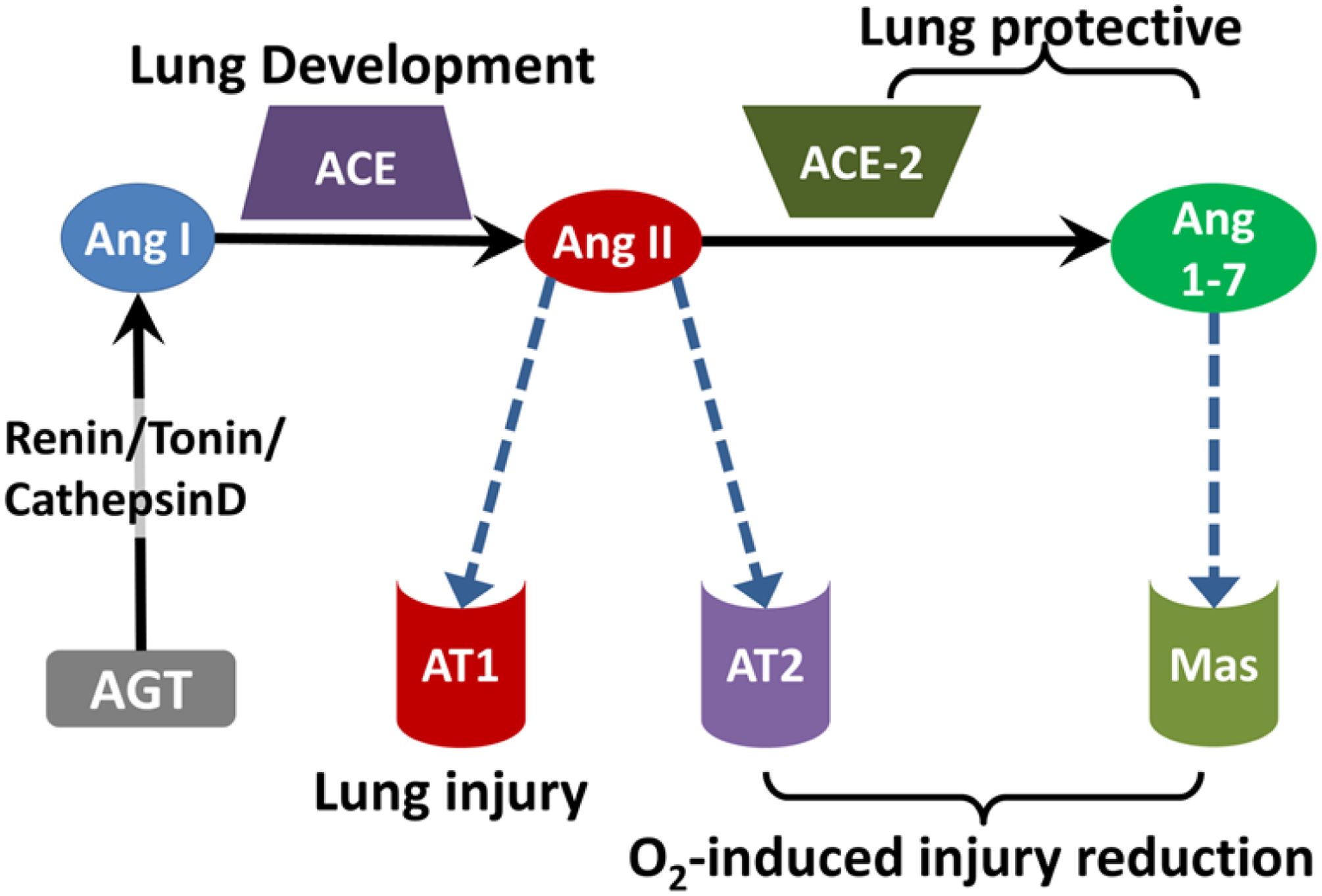
The renin angiotensin system components and its involvement in lung injury.
The role of RAS in BPD
RAS is believed to play a role in neonatal lung development and BPD pathogenesis (Figure 2). Perinatal exposure of animal models to ACE inhibitors was shown to disrupt normal alveolar and secondary septal formation during lung development in neonatal pups.70 In addition, neonatal rat ACE inhibition lowered the surface tension of bronchoalveolar lavage fluid and caused widening of respiratory airspaces and thinning of alveolar septa.71 Autopsy of human BPD patients showed decreased ACE expression in lungs as compared to controls without lung disease.72 Wagenaar et al.4 reported that Mas receptor and angiotensin receptor 2 (AT2) agonists reduced inflammation of oxygen-induced lung injury in rats. In this study, mRNA levels of the RAS component genes were measured during normal lung development. The mRNA levels of AT1, AT2, and ACE-2 decreased gradually, whereas expression of angiotensinogen and ACE-1 increased gradually as the neonatal rat lung develops. Exposure to 100% oxygen for 10 days resulted in an increase in expression of AT2 and a decrease in expression of AT1, angiotensinogen, and ACE.4 These studies suggest a critical role for angiotensin-angiotensin receptor signaling involving pulmonary alveolarization in the normal physiology of the neonatal lung and in the pathophysiology of BPD,. Angiotensinogen and ACE-1 play a role in alveolar development and septation, while the reported lower expression levels of ACE-2 in the neonatal lung could be facilitating the lung injury in the neonatal period.4,70–72
The protective potential of ACE-2 in BPD
ACE-2 has been shown to play a protective role in lung disease through effects mediated by the receptor Mas, the receptor for the ACE-2 peptide product ANG1–7.3,4,65,73 Previous studies from our lab and other groups suggest that ACE-2 is down-regulated in fibrotic conditions of the adult and neonatal human lung,3,65,73 via Mas receptor mechanisms.74 We have also demonstrated that ACE-2 regulates alveolar epithelial cell survival by balancing the proapoptotic Ang II and its antiapoptotic degradation product Ang 1–7, through Ang 1–7 action on its receptor Mas.74 Furthermore, we showed that ACE-2 is expressed in fetal human lung fibroblasts but is significantly decreased by hyperoxic lung injury in a cell culture model.3 Importantly, this effect was reversed by hypoxia preceding hyperoxia.65 Recombinant human ACE-2 has been tested in healthy individuals in clinical trials to determine medication pharmacokinetics and pharmacodynamics,75 and has been investigated as pipeline drug “GSK2586881” in a pilot clinical trial to treat adult acute lung injury.76 ACE-targeted therapies might be future beneficial treatments for BPD.77
Fluctuation of oxygen levels and BPD
Premature infants are known to experience intermittent episodes of hypoxemia lasting from a few seconds to several minutes78. Postnatal exposure to intermittent hypoxia followed by interventional hyperoxia induces oxidative stress and free radicals, which leads to direct cellular injury, oxidation of DNA, induction of cytokines, and recruitment of neutrophils and macrophages to the lung, manifested as pulmonary inflammation.20,21 Furthermore, ROS are released by immune cells resulting in epithelial and endothelial cell injury.26 Clinically unsuspected oxygen desaturation occurs frequently in preterm infants with and without bronchopulmonary dysplasia, and profound hypoxemia is claimed responsible for sudden unexplained deaths in these infants.79 Furthermore, infants who develop BPD experience more frequent episodes of oxygen desaturations than infants who recover from respiratory distress syndrome without developing BPD.19,80
BPD and Fluctuation of oxygen levels in animal models
Very few studies in literature directly compare the effects of hyperoxia, hypoxia, or their fluctuation on the neonatal lung development or injury. Table 1 summarizes the studies discussed below. Ratner et al.19 demonstrated that cycling hypoxia with hyperoxia episodes exacerbated lung injury in neonatal mice.19 In their study they tested the effects of hypoxic episodes on a normoxia background, continuous hyperoxia, and hypoxic episodes combined with a hyperoxia background on neonatal mice and compared the results with the normoxia group. Compared with the normoxia control, the hypoxic episodes on normoxia background had no significantchanges on radial alveolar count (RAC) as a marker of lung injury, on oxidized glutathione, or on protein carbonyls as markers of oxidative stress, while continuous hyperoxia significantly reduced RAC and increased oxidative stress markers compared with normoxia. Interestingly, the intermittent hypoxia on hyperoxia background group of mice showed significantly lower RAC and higher oxidative stress markers compared with continuous hyperoxia. Their study suggested that the combination of two oxidative stress mechanisms, hypoxia and hyperoxia, cause a more profound lung injury than either one alone. Furthermore, their study even suggests that hypoxic mice did not have any BPD-like changes.19
Table 1.
Summary of animal model studies for hypoxia-HYPEROXIA fluctuation (Cycling)
| Reference | Organism | Cycling conditions | Cycling effect | |
|---|---|---|---|---|
| hypoxia | Hyperoxia | |||
| Ratner et al.19 | Mouse, start at P3 | 8% O2 (N2 balanced) | 65% O2 | • Fewer alveoli compared to HYPEROXIA |
| 10 min/episode* | All other times when not in episode | • Decreased pulmonary total/oxidized glutathione ratio (anti- oxidative capacity) | ||
| 1 episode/day for 1 Wk.+ | • Elevation of protein carbonyl content (protein oxidation) | |||
| 1 episode/2days for 1 Wk. | ||||
| *monitor SO2 by pulse oximetry | ||||
| Total 4 Weeks | ||||
| Schmiedl et al.81 | Mouse, start with pregnant mothers E14 | 10% O2 | 75% O2 | • Higher volume of the parenchymal airspaces |
| Pregnant mice | Pups | • Higher wall thickness of septa (not significant) | ||
| E14->E18 | P1->P14 | • Lower volume of lamellar bodies in alveolar epithelial cells type II (by EM) | ||
| Valencia et al.82 | Rat, start at P0 | 12% O2 | 50% O2 | • At P23 and P45, compared with normoxia: |
| 1 min/episode | All other time | ∘ Intermittent hypoxia decreased pO2 | ||
| 3 episodes/cluster, 10 min apart | ∘ HYPEROXIA increased pO2 | |||
| 8 clusters/day for 2 Wks. | ∘ No significant difference in SaO2 in the HYPEROXIA group | |||
| Total 2 Weeks (P0->P14) | ||||
| Chang et al.84 | Rat, start at P0 (within 5hr of birth) | 12% O2 | 50% O2 | • No HYPEROXIA only group was done |
| 2 min/episode | All other time | • No histology | ||
| 3 episodes/cluster, 10 min apart | • Measured VEGF, MMP2,9, TIMP by immunoassays and reversal of the effects by SOD mimetic | |||
| 4 clusters/day (every 6hr) for 2 Wks. | ||||
| Total 2 Weeks (P0->P14) | ||||
| Sucre et al.83 | 3D organoid human fetal lung fibroblasts | 10% O2 | 70% O2 | • Markers of fibroblast activation were increased vs normoxia |
| 24 hr. then HYPEROXIA | 24 hr. then hypoxia | • No comparison to hyperoxia alone made | ||
| Total 4 days | ||||
A study by Schmiedl et al.82 on animal models of BPD compared the different effects of prenatal hypoxia and postnatal hyperoxia in neonatal mouse lung. Compared with normoxia controls, the lung volume, total air space volume and total septal surface were significantly reduced in the postnatal hyperoxia groups compared with either prenatal normoxia or prenatal hypoxia. The volume weighted mean volume of the parenchymal airspaces and the wall thickness of septa was significantly higher, and the volume density and the volume weighted mean volume of lamellar bodies in alveolar epithelial cells type II (AEII) were significantly lower in the prenatal hypoxia-postnatal hyperoxia group compared with normoxia controls, while prenatal normoxia-postnatal hyperoxia did not cause these changes. The study suggested that the prenatal hypoxia and postnatal hyperoxia model was found to best reflect morphological changes in lung development comparable with alterations found in BPD.81
A recent study by Valencia et al.82 examined the effects of intravitreal bevacizumab, a treatment for retinopathy of prematurity (ROP), on the lungs of a newborn rat pup model. The study showed long term effects of exposure to intermittent hypoxia and hyperoxia during the first 2 weeks of life. At postnatal days P23 and P45, both pO2 and SaO2 were lower in intermittent hypoxia exposure, while hyperoxia alone increased pO2 compared to the room air group. Interestingly, no significant difference was found in SaO2 in the hyperoxia group compared to room air.82 The results of this study suggest long-term effects of hypoxemia-hyperoxemia fluctuation on blood oxygenation and indicate the presence of a discrepancy between pO2 and SaO2 measures. In a 3-dimiensional cell culture organoid model of BPD, Sucre et al.83 showed that markers of fibroblast activation were increased by hypoxia-hyperoxia cycling as seen in BPD; α-SMA, Col1A1, TGFβ1, and PDE5a, and downstream targets of Wnt signaling (Cyclin D1, MMP2, and MMP9 RNA) were increased in hypoxia-hyperoxia relative to normoxia cultured organoids.83 A study by Chang et al.84 investigated the effects of hyperoxia with intermittent hypoxia on neonatal rats and found that repeated intermittent hypoxia during hyperoxia can alter biomarkers responsible for normal microvascular and alveolar development.84 However, both the Sucre et al.83 and Chang et al.84 studies have not compared the effects of hyperoxia alone to the effects of intermittent hypoxia during hyperoxia.
What is the safe oxygen target level for newborns?
Several clinical studies and randomized controlled trials (RCTs) were performed to determine the range of optimal saturation by pulse oximetry in preterm infants receiving supplemental oxygen.85–91 Many of these studies have been discussed in detail in other reviews,92,93 we summarize these in Table 2. The assessment outcomes for these studies involved mortality, morbidity, development of chronic lung disease, and ROP. The current 2016 update of the European Consensus Guidelines on the management of neonatal respiratory distress syndrome recommends an oxygen saturation target between 90 and 94% with suggested alarm limits of 89 and 95%.93 These guides are based on the NeOProM meta-analysis study of the three largest and most recent RCTs94; the Surfactant Positive Pressure and Pulse Oximetry Randomized Trial (SUPPORT),89 the Benefits of Oxygen Saturation Targeting II (BOOST II),90 and the Canadian Oxygen Trial (COT).91 In these trials oxygen saturation monitoring by pulse oximetry was the method used to monitor oxygen levels. Pulse oximetry is currently the prevalent monitoring technology to detect blood oxygenation.95 However, invasive blood gas analysis is needed in order to monitor the oxygenation status when SpO2 is close to saturation (≥97%). A new technological measure using multiwavelength pulse co-oximetry called oxygen reserve index (ORI) can be used to monitor oxygenation in the moderate hyperoxemic range (PaO2 100–200mmHg). The ORI is an index with a unit-less scale between 0.00 and 1.00, which is a relative indicator of changes in PaO2 in the moderate hyperoxemic range, and is used as a companion to pulse oximetry monitoring in patients receiving supplemental oxygen.96
Table 2.
Summary of clinical trials using oxygen saturation in neonatal care STOP-ROP, supplemental therapeutic oxygen for prethreshold retinopathy of prematurity; BOOST, benefits of oxygen saturation targeting; SUPPORT, surfactant positive pressure and pulse oximetry randomized trial; COT, canadian oxygen trial
| Study (ref) | High SO2 level | Low SO2 Level | Outcome(s) |
|---|---|---|---|
| Tin and Gupta,85 | 88–98% | 70–90% |
|
| Oxford Vermont Network,86 | >95% | <95% |
|
| STOP-ROP,87 | 96–99% | 89–94% |
|
| BOOST I,88 | 95–98% | 91–94% |
|
| SUPPORT,89 | 91–95% | 85–89% |
|
| BOOST II,90 | 91–95% | 85–89% |
|
| COT,91 | 91–95% | 85–89% |
|
Effects of altered oxygen levels on other organs
Retinopathy of prematurity
ROP is a disease affecting the development of the retinal vasculature characterized by abnormal growth of retinal blood vessels in the incompletely vascularized retina in preterm infants receiving supplemental oxygen therapy. ROP was first reported by Theodore L. Terry in 1942 and referred to as “retrolental fibroplasia”.97,98 An increase in arterial oxygen saturation, as when the preterm infant is resuscitated with high oxygen concentrations, is believed to be damaging to the newly developed retinal capillaries. After a preterm infant is no longer in supplemental oxygen, the avascular retina becomes hypoxic, leading to overexpression of angiogenic factors and vasoproliferation of intravitreal blood vessels.99–101 Penn et al.102 reported that exposure to variable hyperoxia (hypoxia/hyperoxia fluctuations) has been shown to be much more effective at producing proliferative retinopathy in neonatal rat than exposure to constant hyperoxia.102 Numerous animal studies established that intermittent hypox(em)ia cycling with hyperox(em)ia produces severe oxygen induced retinopathy.82,103–107 These studies emphasize the critical role of oxygen level fluctuations.
Heart
Several independent studies in humans and animal models have reported that chronic fetal hypoxia can trigger a fetal origin of cardiac dysfunction and increase the risk of cardiovascular disease in later life.108–115 A transcriptomic study of neonatal ventricular chamber growth and development during perinatal circulatory transition identified Wnt11 as a prominent regulator of chamber-specific proliferation. Perinatal hypoxia treatment in mice suppressed Wnt11 expression and was associated with cyanotic congenital heart defect (CHD) phenotypes and correlated with O2 saturation levels in hypoxemic infants with Tetralogy of Fallot (TOF).116 Several animal studies have shown cardiovascular adverse effects of neonatal hyperoxia with long-term consequences into adulthood. Mouse model studies on perinatal or maternal inflammation combined with neonatal hyperoxia exposure showed altered fetal development affecting cardiac structure and function, resulting in early cardiac dysfunction leading to cardiac failure in adulthood.117,118 In newborn pigs, hyperoxia was found to trigger oxygen free radical-mediated membrane injury together with an inability of the newborn heart to up-regulate its antioxidant enzyme defenses while impairing myocardial function and hemodynamics.119 In addition, neonatal hyperoxia exposure was shown to increase systemic blood pressure and impair vasoreactivity in adult rats,120 possibly due to developmental programming of endothelial nitric oxide synthase uncoupling and enhanced vascular oxidative stress.121 Neonatal hyperoxia exposure also increased adult airway reactivity and was associated with left ventricular (LV) dysfunction in adult mice.44 Cardiac effects of hyperoxia also affect adults. In adult patients with and without congestive heart failure (CHF), hyperoxia was associated with impairment of cardiac relaxation and increased LV filling pressures. These studies indicate that caution should be used in the administration of high inspired O2 fractions.122
Kidney
Oxygen supplementation and hyperoxic exposure of newborn animal models resulted in enlarged renal corpuscles and decreased number of nephrons in the kidneys in early adulthood.120,123 Neonatal hyperoxia exposure also resulted in impaired nephrogenesis causing reduction in both nephrogenic zone width and glomerular diameter and increased apoptotic cell count.124 Since the kidneys are highly vascular organs, the relative hyperoxia and oxygen supplementation in preterm infants could be responsible for the renal developmental abnormalities and for affecting glomerular vascularization, and should be examined in future studies.
Summary of clinical importance/correlation
Various groups are shedding light on the importance of events that are commonly seen in neonatal ICUs, particularly with extremely low birth weight preterm babies, and with how prematurity affects the lung as well as other organs. Hyperoxia might not be the only risk factor for worsening BPD. However, uncontrollable fluctuation/cycling between hypoxemia, normoxemia and hyperoxemia might be a higher risk than hyperoxemia alone. There are trials of simulations to those cycling effects in different animal models. In the neonatal ICU there is no clear cut-off to delineate the “sweet-spot” in the oxygen saturation zone; how much is too much has not yet been clearly defined.
In mice, the episodic “fluctuation” of hypoxia and hyperoxia during the induction of BPD potentiated the oxidative stress in lung tissue and exacerbated the alveolar developmental arrest.19These results suggest that the aggressive prevention of hypoxemic episodes in human neonates at risk for the development of BPD needs to be further investigated. Our group, along with other groups, has studied the role of RAS in local tissue injury,59,92,125–130 and showed that ACE-2 might be beneficial to attenuate lung injury.3,65,73 A future study by our group is planned to look at the effect of cycling of hypoxemia/normoxemia/hyperoxemia and the effect of ACE-2 in a small animal model of BPD. Measurement of partial pressure of oxygen and/or SPO2 is planned to better assess and quantify the effects of hypoxemia with that of hyperoxemia/hypoxemia.
Acknowledgments
This work was supported by the Department of Pediatrics and Human Development at Michigan State University through start-up funds awarded to Dr. Tarek Mohamed.
Abbreviations:
- BPD
bronchopulmonary dysplasia
- ROS
reactive oxygen species
- HIF
hypoxia inducible factor
- RAC
radial alveolar count
- AP-1
activator protein-1
- AGT
angiotensinogen
- RCTs
randomized controlled trials
- ACE
angiotensin-converting enzyme
Footnotes
Conflicts of interest
Authors declare that there is no conflict of interest.
References
- 1.Bhandari V Molecular mechanisms of hyperoxia-induced acute lung injury. Front Biosci. 2008;1:13:6653–6661. [DOI] [PubMed] [Google Scholar]
- 2.Lee PJ, Choi AM. Pathways of cell signaling in hyperoxia. Free Radic Biol Med. 2003;35(4):341–350. [DOI] [PubMed] [Google Scholar]
- 3.Oarhe CI, Dang V, Dang M, et al. Hyperoxia downregulates angiotensin-converting enzyme-2 in human fetal lung fibroblasts. Pediatr Res. 2015;77(5):656–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagenaar GTM, Laghmani EH, Fidder M, et al. Agonists of MAS oncogene and angiotensin II type 2 receptors attenuate cardiopulmonary disease in rats with neonatal hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2013;305(5):L341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith VC, Zupancic JAF, McCormick MC, et al. Trends in severe bronchopulmonary dysplasia rates between 1994 and 2002. J Pediatr. 2005;146(4):469–473. [DOI] [PubMed] [Google Scholar]
- 6.Davidson LM, Berkelhamer SK. Bronchopulmonary Dysplasia: Chronic Lung Disease of Infancy and Long-Term Pulmonary Outcomes. J Clin Med. 2017;6(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zysman-Colman Z, Tremblay GM, Bandeali S, et al. Bronchopulmonary dysplasia - trends over three decades. Paediatr Child Health. 2013;18(2):86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lapcharoensap W, Gage SC, Kan P, et al. Hospital Variation and Risk Factors for Bronchopulmonary Dysplasia in a Population-Based Cohort. JAMA Pediatr. 2015;169(2):e143676. [DOI] [PubMed] [Google Scholar]
- 9.Stoll BJ, Hansen NI, Bell EF, et al. Neonatal Outcomes of Extremely Preterm Infants From the NICHD Neonatal Research Network. Pediatrics. 2010;126(3):443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Day CL, Ryan RM. Bronchopulmonary dysplasia: new becomes old again!. Pediatr Res. 2017;81(1–2):210–213. [DOI] [PubMed] [Google Scholar]
- 11.Jensen EA, Schmidt B. Epidemiology of bronchopulmonary dysplasia. Birth Defects Res A Clin Mol Teratol. 2014;100(3):145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walsh MC, Yao Q, Gettner P, et al. Impact of a physiologic definition on bronchopulmonary dysplasia rates. Pediatrics. 2004;114(5):1305–1311 [DOI] [PubMed] [Google Scholar]
- 13.Deakins KM. Bronchopulmonary dysplasia. Respir Care. 2009;54(9):1252–1262. [PubMed] [Google Scholar]
- 14.Berkelhamer SK, Mestan KK, Steinhorn RH. Pulmonary hypertension in bronchopulmonary dysplasia. Semin Perinatol. 2013;37(2):124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kair LR, Leonard DT, Anderson JM, et all. Bronchopulmonary Dysplasia. Pediatrics in Review. 2012;33(6):255–264. [DOI] [PubMed] [Google Scholar]
- 16.Northway WH, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease. N Engl J Med. 1967;276(7):357–68. [DOI] [PubMed] [Google Scholar]
- 17.Payne MS, Goss KCW, Connett GJ, et al. Molecular microbiological characterization of preterm neonates at risk of bronchopulmonary dysplasia. Pediatr Res. 2010;67(4):412–8. [DOI] [PubMed] [Google Scholar]
- 18.Coalson JJ. Pathology of Bronchopulmonary Dysplasia. Semin Perinatol. 2006;30(4):179–184. [DOI] [PubMed] [Google Scholar]
- 19.Ratner V, Slinko S, Utkina-Sosunova I, et al. Hypoxic stress exacerbates hyperoxia-induced lung injury in a neonatal mouse model of bronchopulmonary dysplasia. Neonatology. 2009;95(4):299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Donovan DJ, Fernandes CJ. Free Radicals and Diseases in Premature Infants. Antioxid Redox Signal. 2004;6(1):169–176. [DOI] [PubMed] [Google Scholar]
- 21.Ozsurekci Y, Aykac K. Oxidative Stress Related Diseases in Newborns. Oxid Med Cell Longev. 2016:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saugstad OD. Mechanisms of tissue injury by oxygen radicals: implications for neonatal disease. Acta Paediatr. 1996;85(1):1–4. [DOI] [PubMed] [Google Scholar]
- 23.Saugstad OD. Bronchopulmonary dysplasia-oxidative stress and antioxidants. Semin Neonatol. 2003;8(1):39–49. [DOI] [PubMed] [Google Scholar]
- 24.Saugstad OD. Oxygen and oxidative stress in bronchopulmonary dysplasia. J Perinat Med. 2010;38(6):571–577. [DOI] [PubMed] [Google Scholar]
- 25.Stenmark KR, Abman SH. Lung vascular development: Implications for the Pathogenesis of Bronchopulmonary Dysplasia. Annu Rev Physiol. 2005;67(1):623–661. [DOI] [PubMed] [Google Scholar]
- 26.Dennery PA. Role of Redox in Fetal Development and Neonatal Diseases. Antioxid Redox Signal. 2004;6(1):147–153. [DOI] [PubMed] [Google Scholar]
- 27.Vogel ER, Britt RD, Trinidad MC, et al. Perinatal oxygen in the developing lung. Can J Physiol Pharmacol. 2015;93(2):119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.P D, Duncker DJ, Tibboel D, et al. Oxidative injury of the pulmonary circulation in the perinatal period: Short- and long-term consequences for the human cardiopulmonary system. Pulm Circ. 2017;7(1):55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006;91(5):807–819. [DOI] [PubMed] [Google Scholar]
- 30.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr Opin Cell Biol. 2009;21(6):894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hernansanz-Agustín P, Izquierdo-Álvarez A, Sánchez-Gómez FJ, et al. Acute hypoxia produces a superoxide burst in cells. Free Radic Biol Med. 2014;71:146–156. [DOI] [PubMed] [Google Scholar]
- 32.Cho H-Y, van Houten B, Wang X, et al. Targeted Deletion of Nrf2 Impairs Lung Development and Oxidant Injury in Neonatal Mice. Antioxid Redox Signal. 2012;17(8):1066–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hilgendorff A, Reiss I, Ehrhardt H, et al. Chronic Lung Disease in the Preterm Infant: Lessons Learned From Animal Models. Am J Respir Cell Mol Biol. 2013;50(2):233–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madurga A, Mizikova I, Ruiz-Camp J, et al. Recent advances in late lung development and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2013;305(12):L893–L905. [DOI] [PubMed] [Google Scholar]
- 35.Namdev S, Bhat V, Adhisivam B, et al. Oxidative stress and antioxidant status among neonates born to mothers with pre-eclampsia and their early outcome. J Matern Fetal Neonatal Med. 2014;27(14):1481–1484. [DOI] [PubMed] [Google Scholar]
- 36.Negi R, Pande D, Karki K, et al. Association of oxidative DNA damage, protein oxidation and antioxidant function with oxidative stress induced cellular injury in pre-eclamptic/eclamptic mothers during fetal circulation. Chem Biol Interact. 2014;208:77–83. [DOI] [PubMed] [Google Scholar]
- 37.Shah DA, Khalil RA. Bioactive factors in uteroplacental and systemic circulation link placental ischemia to generalized vascular dysfunction in hypertensive pregnancy and preeclampsia. Biochem Pharmacol. 2015;95(4):211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foidart JM, Schaaps JP, Chantraine F, et al. Dysregulation of anti-angiogenic agents (sFlt-1, PLGF, and sEndoglin) in preeclampsia—a step forward but not the definitive answer. J Reprod Immunol. 2009;82(2):106–111. [DOI] [PubMed] [Google Scholar]
- 39.Tang J-R, Karumanchi SA, Seedorf G, et al. Excess soluble vascular endothelial growth factor receptor-1 in amniotic fluid impairs lung growth in rats: linking preeclampsia with bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2012;302(1):L36–L46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilkins MR, Ghofrani H-A, Weissmann N, et al. Pathophysiology and Treatment of High-Altitude Pulmonary Vascular Disease. Circulation. 2015;131(6):582–590. [DOI] [PubMed] [Google Scholar]
- 41.Mortola JP, Frappell PB, Aguero L, et al. Birth weight and altitude: A study in Peruvian communities. J Pediatr. 2000;136(3):324–329. [DOI] [PubMed] [Google Scholar]
- 42.Lovering AT, Riemer RK, Thébaud B. Intrapulmonary Arteriovenous Anastomoses. Physiological, Pathophysiological, or Both?. Ann Am Thorac Soc. 2013;10(5):504–508. [DOI] [PubMed] [Google Scholar]
- 43.Sommer N, Dietrich A, Schermuly RT, et al. Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms. Eur Respir J. 2008;32(6):1639–1651. [DOI] [PubMed] [Google Scholar]
- 44.Ramani M, Bradley WE, Dell’Italia LJ, et al. Early Exposure to Hyperoxia or Hypoxia Adversely Impacts Cardiopulmonary Development. Am J Respir Cell Mol Biol. 2015;52(5):594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berger J, Bhandari V. Animal models of bronchopulmonary dysplasia. The term mouse models. Am J Physiol Lung Cell Mol Physiol. 2014;307(12):L936–L947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Groenman F, Rutter M, Caniggia I, et al. Hypoxia-inducible Factors in the First Trimester Human Lung. J Histochem Cytochem. 2007;55(4):355–363. [DOI] [PubMed] [Google Scholar]
- 47.Rajatapiti P, de Rooij JD, Beurskens LWJE, et al. Effect of Oxygen on the Expression of Hypoxia-Inducible Factors in Human Fetal Lung Explants. Neonatology. 2010;97(4):346–354. [DOI] [PubMed] [Google Scholar]
- 48.Maxwell PH. Hypoxia-inducible factor as a physiological regulator. Exp Physiol. 2005;90(6):791–797. [DOI] [PubMed] [Google Scholar]
- 49.Saugstad OD. Oxidative Stress in the Newborn – A 30-Year Perspective. Biol Neonate. 2005;88(3):228–236. [DOI] [PubMed] [Google Scholar]
- 50.Rogers S, Witz G, Anwar M, et al. Antioxidant capacity and oxygen radical diseases in the preterm newborn. Arch Pediatr Adolesc Med. 2000;154(6):544–548. [DOI] [PubMed] [Google Scholar]
- 51.Plötz FB, Slutsky AS, van Vught AJ, et al. Ventilator-induced lung injury and multiple system organ failure: a critical review of facts and hypotheses. Intensive Care Med. 2004;30(10):1865–1872. [DOI] [PubMed] [Google Scholar]
- 52.Fan E, Villar J, Slutsky AS. Novel approaches to minimize ventilator-induced lung injury. BMC Med. 2013;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vlahakis NE, Schroeder MA, Limper AH, et al. Stretch induces cytokine release by alveolar epithelial cells in vitro. Am J Physiol. 1999;277(1 Pt 1):L167–73. [DOI] [PubMed] [Google Scholar]
- 54.Jobe AH, Hillman N, Polglase G, et al. Injury and Inflammation from Resuscitation of the Preterm Infant. Neonatology. 200;94(3):190–196. [DOI] [PubMed] [Google Scholar]
- 55.De Paepe ME, Greco D, Mao Q. Angiogenesis-related gene expression profiling in ventilated preterm human lungs. Exp Lung Res. 2010;36(7):399–410. [DOI] [PubMed] [Google Scholar]
- 56.De Paepe ME, Patel C, Tsai A, et al. Endoglin (CD105) Up-regulation in Pulmonary Microvasculature of Ventilated Preterm Infants. Am J Respir Crit Care Med. 2008;178(2):180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Filippatos G, Tilak M, Pinillos H, et al. Regulation of apoptosis by angiotensin II in the heart and lungs (Review). Int J Mol Med. 2001;7(3):273–80. [DOI] [PubMed] [Google Scholar]
- 58.Jiang F, Yang J, Zhang Y, et al. Angiotensin-converting enzyme 2 and angiotensin 1–7: novel therapeutic targets. Nat Rev Cardiol. 2014;11(7):413–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li X, Zhang H, Soledad-Conrad V, et al. Bleomycin-induced apoptosis of alveolar epithelial cells requires angiotensin synthesis de novo. Am J Physiol Lung Cell Mol Physiol. 2003;284(3):L501–L5017. [DOI] [PubMed] [Google Scholar]
- 60.Filippatos G, Uhal BD. Blockade of apoptosis by ACE inhibitors and angiotensin receptor antagonists. Curr Pharm Des. 2003;9(9):707–714. [DOI] [PubMed] [Google Scholar]
- 61.Hale TM. Persistent phenotypic shift in cardiac fibroblasts: impact of transient renin angiotensin system inhibition. J Mol Cell Cardiol. 2016;93:125–32. [DOI] [PubMed] [Google Scholar]
- 62.Simões E Silva AC, Teixeira MM. ACE inhibition, ACE2 and angiotensin-(1–7) axis in kidney and cardiac inflammation and fibrosis. Pharmacol Res. 2016;107:154–62. [DOI] [PubMed] [Google Scholar]
- 63.Uhal BD, Li X, Piasecki CC, et al. Angiotensin signalling in pulmonary fibrosis. Int J Biochem Cell Biol. 2012;44(3):465–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uhal BD, Abdul-Hafez A. Angiotensin II in apoptotic lung injury: potential role in meconium aspiration syndrome. J Perinatol. 2008;28 Suppl 3:S108–12. [DOI] [PubMed] [Google Scholar]
- 65.Mohamed TL, Nguyen HT, Abdul-Hafez A, et al. Prior hypoxia prevents downregulation of ACE-2 by hyperoxia in fetal human lung fibroblasts. Exp Lung Res. 2016;42(3):121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grace JA, Herath CB, Mak KY, et al. Update on new aspects of the renin-angiotensin system in liver disease: clinical implications and new therapeutic options. Clin Sci (Lond). 2012;123(4):225–239. [DOI] [PubMed] [Google Scholar]
- 67.Goossens GH. The renin-angiotensin system in the pathophysiology of type 2 diabetes. Obes Facts. 2012;5(4):611–624. [DOI] [PubMed] [Google Scholar]
- 68.Macconi D, Remuzzi G, Benigni A. Key fibrogenic mediators: old players. Renin-angiotensin system. Kidney Int Suppl (2011). 2014;4(1):58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chappell MC, Abadir P, Foster D, et al. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute?. Am J Physiol Heart Circ Physiol. 2016;310(2):H137–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Capelari DN, Sánchez SI, Ortega HH, et al. Effects of maternal captopril treatment during late pregnancy on neonatal lung development in rats. Regul Pept. 2012;177(1–3):97–106. [DOI] [PubMed] [Google Scholar]
- 71.Lasaitiene D, Chen Y, Nannmark U,et al. Neonatal ACE inhibition in rats interferes with lung development. Clin Physiol Funct Imaging. 2004;24(1):65–68. [DOI] [PubMed] [Google Scholar]
- 72.Castro EC, Parks WT, Galambos C. The ontogeny of human pulmonary angiotensin-converting enzyme and its aberrant expression may contribute to the pathobiology of bronchopulmonary dysplasia (BPD). Pediatr Pulmonol. 2014;49(10):985–990. [DOI] [PubMed] [Google Scholar]
- 73.Li X, Molina-Molina M, Abdul-Hafez A, et al. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L178–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Uhal BD, Li X, Xue A, et al. Regulation of alveolar epithelial cell survival by the ACE-2 / angiotensin 1 – 7 / Mas axis. Am J Physiol Lung Cell Mol Physiol. 2011;779(3):269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haschke M, Schuster M, Poglitsch M,et al. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin Pharmacokinet. 2013;52(9):783–792. [DOI] [PubMed] [Google Scholar]
- 76.Khan A, Benthin C, Zeno B, et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit Care. 2017;21(1):234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gandhi C, Uhal BD. Roles of the Angiotensin System in Neonatal Lung Injury and Disease. JSM Atheroscler. 2016;1(3):1014. [PMC free article] [PubMed] [Google Scholar]
- 78.Martin RJ, Wang K, Köroğlu O, et al. Intermittent hypoxic episodes in preterm infants: do they matter?. Neonatology. 2011;100(3):303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Garg M, Kurzner SI, Bautista DB, et al. Clinically Unsuspected Hypoxia During Sleep and Feeding in Infants With Bronchopulmonary Dysplasia. Pediatrics. 1988;81(5):635–642. [PubMed] [Google Scholar]
- 80.Jia L, Xu M, Zhen W, et al. Novel anti-oxidative role of calreticulin in protecting A549 human type II alveolar epithelial cells against hypoxic injury. Am J Physiol Cell Physiol. 2008;294(1):C47–C55. [DOI] [PubMed] [Google Scholar]
- 81.Schmiedl A, Roolfs T, Tutdibi E, et al. Influence of prenatal hypoxia and postnatal hyperoxia on morphologic lung maturation in mice. PLoS ONE. 2017;12(4):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Valencia AM, Cai CL, Tan J, et al. Intravitreal bevacizumab alters type IV collagenases and exacerbates arrested alveologenesis in the neonatal rat lungs. Exp Lung Res. 2017;43(3):1–14. [DOI] [PubMed] [Google Scholar]
- 83.Sucre JMS, Vijayaraj P, Aros CJ, et al. Posttranslational modification of β-catenin is associated with pathogenic fibroblastic changes in bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2017;312(2):L186–L195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chang M, Bany-Mohammed F, Cristina Kenney M, et al. Effects of a superoxide dismutase mimetic on biomarkers of lung angiogenesis and alveolarization during hyperoxia with intermittent hypoxia. Am J Transl Res. 2013;5(6):594–607. [PMC free article] [PubMed] [Google Scholar]
- 85.Tin W, Gupta S. Optimum oxygen therapy in preterm babies. Arch Dis Child Fetal Neonatal Ed. 2007;92(2):F143–F147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sun S Relation of target SpO (2) levels and clinical outcome in ELBW infants on supplemental oxygen. Pediatric Research. 2002. [Google Scholar]
- 87.Supplemental Therapeutic Oxygen for Prethreshold Retinopathy of Prematurity (STOP-ROP), A Randomized, Controlled Trial. I: Primary Outcomes. Pediatrics. 2000;105(2):295–310. [DOI] [PubMed] [Google Scholar]
- 88.Askie LM, Henderson-Smart DJ, Irwig L, et al. Oxygen-Saturation Targets and Outcomes in Extremely Preterm Infants. N Engl J Med. 2003;349(10):959–967. [DOI] [PubMed] [Google Scholar]
- 89.Carlo WA, Finer NN, Walsh MC, et al. Target Ranges of Oxygen Saturation in Extremely Preterm Infants. N Engl J Med. 2010;362(21):1959–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stenson BJ, Tarnow-Mordi WO, Darlow BA, et al. Oxygen Saturation and Outcomes in Preterm Infants. N Engl J Med. 2013;368(22):2094–2104. [DOI] [PubMed] [Google Scholar]
- 91.Schmidt B, Whyte RK, Asztalos EV, et al. Effects of Targeting Higher vs Lower Arterial Oxygen Saturations on Death or Disability in Extremely Preterm Infants : a randomized clinical trial. JAMA. 2013;309(20):2111–2120 [DOI] [PubMed] [Google Scholar]
- 92.Perrone S, Bracciali C, Di Virgilio N, et al. Oxygen Use in Neonatal Care: A Two-edged Sword. Front Pediatr. 2017;4:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sweet DG, Carnielli V, Greisen G, et al. European Consensus Guidelines on the Management of Respiratory Distress Syndrome - 2016 Update. Neonatology. 2017;111(2):107–125. [DOI] [PubMed] [Google Scholar]
- 94.Askie LM, Brocklehurst P, Darlow BA, et al. NeOProM: Neonatal Oxygenation Prospective Meta-analysis Collaboration study protocol. BMC Pediatr. 2011;11(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jubran A Pulse oximetry. Crit Care. 2015;19(1):272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Scheeren TWL, Belda FJ, Perel A. The oxygen reserve index (ORI): a new tool to monitor oxygen therapy. J Clin Monit Comput. 2018;32(3):379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Terry TL. Fibroblastic Overgrowth of Persistent Tunica Vasculosa Lentis in Infants Born Prematurely: II. Report of Cases-Clinical Aspects. Trans Am Ophthalmol Soc.1942;40:262–284. [PMC free article] [PubMed] [Google Scholar]
- 98.Coats DK, Reddy AK. Retinopathy of Prematurity. In: Wilson ME, Trivedi RH, Saunders RA, editors. Pediatric Ophthalmology: Current Thought and A Practical Guide. Berlin, Heidelberg: Springer Berlin Heidelberg; 2009. p. 375–386. [Google Scholar]
- 99.Hartnett ME, Lane RH. Effects of oxygen on the development and severity of retinopathy of prematurity. J AAPOS. 2013;17(3):229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Flynn JT, Bancalari E, Snyder ES, et al. A cohort study of transcutaneous oxygen tension and the incidence and severity of retinopathy of prematurity. Trans Am Ophthalmol Soc. 1991;89:77–92. [PMC free article] [PubMed] [Google Scholar]
- 101.Hartnett ME. Studies on the pathogenesis of avascular retina and neovascularization into the vitreous in peripheral severe retinopathy of prematurity (an american ophthalmological society thesis). Trans Am Ophthalmol Soc. 2010;108:96–119. [PMC free article] [PubMed] [Google Scholar]
- 102.Penn JS, Henry MM, Tolman BL. Exposure to Alternating Hypoxia and Hyperoxia Causes Severe Proliferative Retinopathy in the Newborn Rat. Pediatr Res. 1994;36(6):724–731. [DOI] [PubMed] [Google Scholar]
- 103.Coleman RJ, Beharry KDA, Brock RS, et al. Effects of Brief, Clustered Versus Dispersed Hypoxic Episodes on Systemic and Ocular Growth Factors in a Rat Model of Oxygen-Induced Retinopathy. Pediatr Res. 2008;64(1):50–55. [DOI] [PubMed] [Google Scholar]
- 104.Brock RS, Gebrekristos BH, Kuniyoshi KM, et al. Biomolecular Effects of Jb1 (an IGF-I Peptide Analog) in a Rat Model of Oxygen-Induced Retinopathy. Pediatric Research. 2011;69(2):135–141. [DOI] [PubMed] [Google Scholar]
- 105.Beharry KD, Cai CL, Sharma P, et al. Hydrogen peroxide accumulation in the choroid during intermittent hypoxia increases risk of severe oxygen-induced retinopathy in neonatal rats. Invest Ophthalmol Vis Sci. 2013;54(12):7644–7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jivabhai Patel S, Bany-Mohammed F, McNally L, et al. Exogenous Superoxide Dismutase Mimetic Without Scavenging H2O2 Causes Photoreceptor Damage in a Rat Model for Oxygen-Induced Retinopathy. Invest Ophthalmol Vis Sci. 2015;56(3):1665–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aranda JV, Cai CL, Ahmad T, et al. Pharmacologic synergism of ocular ketorolac and systemic caffeine citrate in rat oxygen-induced retinopathy. Pediatr Res. 2016;80(4):554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Itani N, Skeffington KL, Beck C, Niu Y, Giussani DA. Melatonin rescues cardiovascular dysfunction during hypoxic development in the chick embryo. J Pineal Res. 2016;60(1):16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Veille J, Hanson R, Sivakoff M, et al. Fetal Cardiac Size in Normal, Intrauterine Growth Retarded, and Diabetic Pregnancies. Am J Perinatol. 1993;10(04):275–279. [DOI] [PubMed] [Google Scholar]
- 110.Zhang L Prenatal Hypoxia and Cardiac Programming. J Soc Gynecol Investig. 2005;12(1):2–13. [DOI] [PubMed] [Google Scholar]
- 111.Williams SJ, Hemmings DG, Mitchell JM, et al. Effects of maternal hypoxia or nutrient restriction during pregnancy on endothelial function in adult male rat offspring. J Physiol. 2005;565(Pt 1):125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Akira M, Yoshiyuki S. Placental Circulation, Fetal Growth, and Stiffness of the Abdominal Aorta in Newborn Infants. J Pediatr. 2006;148(1):49–53. [DOI] [PubMed] [Google Scholar]
- 113.Camm EJ, Hansell JA, Kane AD, et al. Partial contributions of developmental hypoxia and undernutrition to prenatal alterations in somatic growth and cardiovascular structure and function. Am J Obstet Gynecol. 2010;203(5):495.e24–495.e34. [DOI] [PubMed] [Google Scholar]
- 114.Giussani DA, Camm EJ, Niu Y, et al. Developmental Programming of Cardiovascular Dysfunction by Prenatal Hypoxia and Oxidative Stress Calbet JAL, editor. PLoS ONE. 2012;7(2):e31017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Thompson LP, Al-Hasan Y. Impact of Oxidative Stress in Fetal Programming. J Pregnancy. 2012;2012:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Touma M, Kang X, Gao F, et al. Wnt11 regulates cardiac chamber development and disease during perinatal maturation. JCI Insight. 2017;2(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Velten M, Gorr MW, Youtz DJ, et al. Adverse perinatal environment contributes to altered cardiac development and function. American journal of physiology. Am J Physiol Heart Circ Physiol. 2014;306(9):H1334–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Velten M, Hutchinson KR, Gorr MW, et al. Systemic maternal inflammation and neonatal hyperoxia induces remodeling and left ventricular dysfunction in mice. PloS one. 2011;6(9):e24544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bandali KS, Belanger MP, Wittnich C. Hyperoxia causes oxygen free radical-mediated membrane injury and alters myocardial function and hemodynamics in the newborn. Am J Physiol Heart Circ Physiol. 2004;287(2):H553–H559. [DOI] [PubMed] [Google Scholar]
- 120.Yzydorczyk C, Comte B, Cambonie G, et al. Neonatal Oxygen Exposure in Rats Leads to Cardiovascular and Renal Alterations in Adulthood. Hypertension. 2008;52(5):889–895. [DOI] [PubMed] [Google Scholar]
- 121.Yzydorczyk C, Comte B, Huyard F, et al. Developmental Programming of eNOS Uncoupling and Enhanced Vascular Oxidative Stress in Adult Rats After Transient Neonatal Oxygen Exposure. J Cardiovasc Pharmacol. 2013;61(1):8–16. [DOI] [PubMed] [Google Scholar]
- 122.Mak S, Azevedo ER, Liu PP, et al. Effect of hyperoxia on left ventricular function and filling pressures in patients with and without congestive heart failure. Chest. 2001;120(2):467–73. [DOI] [PubMed] [Google Scholar]
- 123.Sutherland MR, O’Reilly M, Kenna K, et al. Neonatal hyperoxia: effects on nephrogenesis and long-term glomerular structure. Am J Physiol Renal Physiol. 2013;304(10):F130–16. [DOI] [PubMed] [Google Scholar]
- 124.Popescu CR, Sutherland MR, Cloutier A, et al. Hyperoxia Exposure Impairs Nephrogenesis in the Neonatal Rat: Role of HIF-1α Ashton N, editor. PLoS ONE. 2013;8(12):e82421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Uhal BD, Joshi I, True AL, et al. Fibroblasts isolated after fibrotic lung injury induce apoptosis of alveolar epithelial cells in vitro. Am J Physiol. 1995;269(6 Pt 1):L819–28. [DOI] [PubMed] [Google Scholar]
- 126.Wang R, Zagariya A, Ibarra-Sunga O, et al. Angiotensin II induces apoptosis in human and rat alveolar epithelial cells. Am J Physiol. 1999;276(5 Pt 1):L885–9. [DOI] [PubMed] [Google Scholar]
- 127.Papp M, Li X, Zhuang J, et al. Angiotensin receptor subtype AT(1) mediates alveolar epithelial cell apoptosis in response to ANG II. Am J Physiol Lung Cell Mol Physiol. 2002;282(4):L713–8. [DOI] [PubMed] [Google Scholar]
- 128.Li X, Molina-Molina M, Abdul-Hafez A, et al. Extravascular sources of lung angiotensin peptide synthesis in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2006;291(5):L887–95. [DOI] [PubMed] [Google Scholar]
- 129.Hillman NH, Gisslen T, Polglase GR, et al. Ventilation-induced increases in EGFR ligand mRNA are not altered by intra-amniotic LPS or ureaplasma in preterm lambs. PloS one. 2014;9(4):e96087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Poisner AM, Adler F, Uhal B, et al. Persistent and progressive pulmonary fibrotic changes in a model of fat embolism. J Trauma Acute Care Surg. 2012;72(4):992–998. [DOI] [PubMed] [Google Scholar]