

Graphical abstract
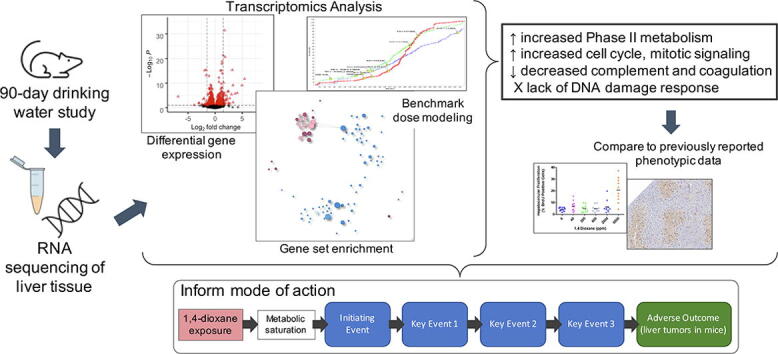
Keywords: 1,4-Dioxane; Transcriptomics; Mechanisms; Mode of action; Hepatotoxicity
Highlights
-
•
Minimal global gene expression changes at concentrations below 600 ppm of 1,4-dioxane.
-
•
Increased phase II metabolism genes and cellular cycle signaling at >600 ppm.
-
•
Increased cell cycle signaling occurred at 90 days, corroborating histopathology.
-
•
Data support the proposed cytotoxicity/regenerative hyperplasia mode of action (MOA)
-
•
Lack of DNA damage response at the mRNA level supports a non-mutagenic MOA.
Abstract
1,4-Dioxane is a volatile organic compound with industrial and commercial applications as a solvent and in the manufacture of other chemicals. 1,4-Dioxane has been demonstrated to induce liver tumors in chronic rodent bioassays conducted at very high doses. The available evidence for 1,4-dioxane-induced liver tumors in rodents aligns with a threshold-dependent mode of action (MOA), with the underlying mechanism being less clear in the mouse than in rats. To gain a better understanding of the underlying molecular mechanisms related to liver tumor development in mice orally exposed to 1,4-dioxane, transcriptomics analysis was conducted on liver tissue collected from a 90-day drinking water study in female B6D2F1/Crl mice (Lafranconi et al., 2020). Using tissue samples from female mice exposed to 1,4-dioxane in the drinking water at concentrations of 0, 40, 200, 600, 2,000 or 6,000 ppm for 7, 28, and 90 days, transcriptomic analyses demonstrate minimal treatment effects on global gene expression at concentrations below 600 ppm. At higher concentrations, genes involved in phase II metabolism and mitotic cell cycle checkpoints were significantly upregulated. There was an overall lack of enrichment of genes related to DNA damage response. The increase in mitotic signaling is most prevalent in the livers of mice exposed to 1,4-dioxane at the highest concentrations for 90 days. This finding aligns with phenotypic changes reported by Lafranconi et al. (2020) after 90-days of exposure to 6,000 ppm 1,4-dioxane in the same tissues. The transcriptomics analysis further supports overarching study findings demonstrating a non-mutagenic, threshold-based, mitogenic MOA for 1,4-dioxane-induced liver tumors.
1. Introduction
1,4-Dioxane is a volatile organic compound currently used in industrial processes as a solvent, in the manufacture of other chemicals, and as a laboratory reagent (ATSDR, 2012). Chronic exposure to high levels of 1,4-dioxane via the inhalation or oral routes has been observed to cause liver tumors in laboratory rodents (Argus et al., 1965, Argus et al., 1973, International Center for Medical et al., 1988; Kano et al., 2009; Kasai et al., 2009, Kociba et al., 1974; NCI, 1978). In recent years, investigators have put forth a hypothesized MOA for 1,4-dioxane-induced mouse liver tumors, with hepatic cytotoxicity and subsequent regenerative hyperplasia proposed as key events (KE) for tumor development, subsequent to metabolic saturation and consequential accumulation of the parent compound in the blood (Dourson et al., 2014, Dourson et al., 2017). However, questions remain as to whether there is sufficient information to understand early events in the development of hepatic tumors in mice exposed to 1,4-dioxane, and to support an initiating event of hepatotoxicity.
To further investigate the MOA related to 1,4-dioxane hepatocarcinogenicity in rodents, specifically mice, female B6D2F1/Crl mice were exposed to 0, 40, 200, 600, 2,000 or 6,000 ppm (approximately 0, 7.2, 37.3, 116, 364, and 979 mg/kg bw/day) 1,4-dioxane in drinking water for 7, 28, or 90 days (Lafranconi et al., 2020). The B6D2F1 mouse, which has been shown to be particularly susceptible to the development of liver tumors (Yamate et al., 1990, Katagiri et al., 1998), was selected to match as closely as possible the strain used in a previous study that demonstrated increased liver tumors at 66 mg/kg bw/day 1,4-dioxane (Kano et al., 2009). In the in-life portion of the present study reported by Lafranconi et al. (2020), the threshold for metabolic clearance was determined to be between 2000 and 6000 ppm, with pathological changes observed in the liver only after 90 days of exposure to the highest concentration (6000 ppm). The liver pathology was characterized as glycogen-like vacuolation, centrilobular hypertrophy, increased centrilobular GST-P staining, apoptosis, and a pan-lobular increase in cell proliferation (Lafranconi et al., 2020). These findings were concluded to demonstrate an early mitogenic response to 1,4-dioxane following sub-chronic (90 days) exposure to concentrations that exceeded the metabolic clearance threshold (Lafranconi et al., 2020). Mitogenesis is well-recognized as a nongenotoxic MOA for cancer (Cohen and Ellwein, 1990; U.S. EPA, 2005) with species differences (Elcombe et al., 2014), and is especially relevant to liver tumors in sensitive strains of mice (Maronpot, 2009).
As transcriptomic data can provide additional and/or supporting information regarding underlying mechanisms of effects associated with specific exposure scenarios (Gao et al., 2015, Dean et al., 2017, Joseph, 2017, Mulas et al., 2017), and can potentially be integrated into mode of action (MOA) analysis and human health risk or hazard assessments (Chepelev et al., 2015, Moffat et al., 2015, Johnson et al., 2020, LaRocca et al., 2020), whole transcriptome analyses were conducted on liver tissues from the 90-day drinking water study (7, 28, or 90 days of exposure). Transcriptomic signatures can also demonstrate adaptive, transient, and/or beneficial reactive responses to exposure. Considering the existing 1,4-dioxane evidence base, we hypothesized that genes related to xenobiotic metabolism, cell death, and cell proliferation would be altered by 1,4-dioxane exposure. Further, we sought to identify any additional molecular signaling alterations related to the liver effects seen in 1,4-dioxane-exposed mice. To address the question of genotoxicity, the presence of mRNA-level responses that may indicate enrichment of DNA damage and/or response pathways was specifically investigated. Gene set enrichment analysis and dose–response modeling were conducted to understand alterations in biological and disease processes across treatment groups. The transcriptomic signatures in the livers of exposed mice were also considered in relation to phenotypic data (i.e., apical endpoints) as determined by histopathological and immunohistochemical analyses of sections from the same liver tissue blocks, which demonstrated a significant increase in single-cell apoptosis and proliferation after 90 days of exposure, and an overall lack of significant treatment effect in the liver at concentrations of 1,4-dioxane below 6000 ppm (Lafranconi et al., 2020). The transcriptomic alterations were considered together with the phenotypic data reported by Lafranconi et al, (2020) to inform the MOA underlying the liver effects observed in female B6C2F1/Crl mice. This information is important for understanding the relevance of the findings and dose–response observed in sensitive strains of mice for assessing human health risks where potential exposure occurs with much lower dosages, such as via ingestion of contaminated drinking water.
2. Materials and methods
2.1. Animal husbandry and exposure conditions
The in-life study method details are described in Lafranconi et al. (2020). Briefly, the subchronic toxicity of 1,4-dioxane was evaluated in a 90-day study in female B6D2F1(BDF1)/Crl mice (Charles River Laboratories, Inc. [Raleigh, NC] aged between 5 and 8 weeks) exposed continuously to 0, 40, 200, 600, 2000, or 6000 ppm 1,4-dioxane in drinking water for 7, 28, or 90 days. The targeted mg/kg/day dose levels were 0, 10, 50, 150, 500, and 1500 mg/kg/day. The mouse strain and route of exposure (drinking water) were chosen to enable comparison to the results of the cancer bioassay findings reported by Kano et al. (Kano et al., 2009). Daily dosages at various time points were estimated using drinking water concentrations, body weights and average water consumption per group. Female mice were fed ad libitum LabDiet Certified Rodent Diet #5002 (PMI Nutrition International; St. Louis, MO). Animal care followed applicable animal welfare standards including the U.S. Department of Agriculture’s Animal Welfare Act (9) Code of Federal Regulations (CFR) Parts 1, 2 and 3, National Research Council Guide for the Care and Use of Laboratory Animals. Washington, DC (NRC, 2011), and the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals (AVMA, 2013). Non-fasted mice were euthanized by CO2 anesthesia and decapitation at 7, 28, and 90 days of exposure. Liver tissues were fixed in neutral, phosphate-buffered 10% formalin and embedded in paraffin.
2.2. RNA sequencing
Formalin-fixed paraffin embedded (FFPE) liver samples from each mouse (n = 5 per treatment group; i.e., each duration and concentration) were microtomed to obtain a single 4–6 µm liver section mounted on a glass slide (uncovered), yielding a total of 90 samples for RNA sequencing. Slides were shipped to BioSpyder Technologies (Carlsbad, CA) where the unstained liver sections were evenly scraped from the slides and processed according to the TempO-Seq protocol, as previously described (Yeakley et al., 2017). DNA libraries created from each liver sample were sequenced using a HiSeq 2500 Ultra-High-Throughput Sequencing System (Illumina, San Diego, CA). RNA sequencing data are publicly available at NCBI’s Gene Expression Omnibus1 (GEO series accession number GSE154899).
2.2.1. Data processing and analysis
Sequencing data were analyzed using multiple packages in the R software environment, version 4.0.2 (cran.r-project.org/). The number of sequenced reads per probe were extracted from the sequencing output files; a traditional alignment step was not required because TempO-Seq uses gene-specific probe sequences. The DESeq2 R package (version 1.28.1) (Love et al., 2014) was used to normalize data to account for sample-to-sample variation in sequencing depth. Samples with below-optimal sequencing depth or low representation of expressed genes were not included in the comparative analysis. This was characterized by a total number of sequence reads >2 standard deviations below the mean sequenced reads per sample (5,635,830 and 8,839,173 across two sequencing runs), or a total number of genes sequenced >2 standard deviations below the mean number of genes sequenced per sample (16,132 and 17,350 across two sequencing runs). Application of these criteria resulted in the removal of five samples from the total 90 samples that were sequenced. Removal of low-count probes was not conducted because it is not necessary when using the DESeq2 package, owing to the application of shrunken fold-changes and independent filtering to stabilize low-count probes (Love et al., 2014).
2.2.2. Identification of differentially expressed genes
Significant differentially expressed genes (DEGs) were identified for each concentration of 1,4-dioxane within DESeq2 based upon estimated variance-mean dependence in the TempO-Seq count and a model using the negative binomial distribution. DEGs for each concentration compared to controls within the same timepoint were determined using a Wald statistical test and betaPrior set to “false” within DESeq2. Genes were considered to be significant DEGs if one of their corresponding probes had a false discovery rate (FDR) <10% following adjustment for multiple testing using the Benjamini and Hochberg (BH) procedure (Love et al., 2014).
2.2.3. Biological pathway enrichment analysis across concentrations of 1,4-dioxane
Biological pathways associated with gene expression profiles were identified by pathway enrichment analysis. Mouse gene identifiers were converted to human identifiers using the biomaRt R package (v2.44.1) based on the Ensembl genome database (http://uswest.ensemble.org/index.html). The gene expression data were then queried for enrichment among gene sets in collections available in the Molecular Signatures Database (MSigDB) (http://software.broadinstitute.org/gsea/msigdb/index.jsp). The Canonical Pathways sub-collections were used (c2.cp.v6.2), which include gene sets from the following pathway databases: BioCarta online maps of metabolic and signaling pathways (BIOCARTA) (Nishimura, 2001), the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Ogata et al., 1999), the Pathway Interaction Database (PID) (Schaefer et al., 2009), and the Reactome database of reactions, pathways, and biological processes (REACTOME) (Croft et al., 2011).
Enrichment of gene sets and pathways was determined using two methods: the gene set enrichment analysis (GSEA) statistical method and a hypergeometric test. The GSEA method follows the analysis platform made available by the Broad Institute (http://software.broadinstitute.org/gsea/index.jsp); the second employed a simpler hypergeometric test (Falcon and Gentleman, 2008). The GSEA method (Subramanian et al., 2005) determines whether sets of genes (e.g., the members of a molecular signaling pathway) are significantly concordant between various defined groups (in the case presented herein, different doses and timepoints) based on a ranking metric (in this case, the Wald statistic for expression differences between the 1,4-dioxane concentrations and control mice). The GSEA method was applied within Platform for Integrative Analysis of Omics data (PIANO) R package (v2.4.0) (Väremo et al., 2013), with geneSetStat = “gsea” and significance calculated using permutation-based nominal P values based on weighted Kolmogorov-Smirnov test enrichment scores, adjusted for multiple hypothesis testing by calculating FDRs using the BH method (Subramanian et al., 2005). The second method, a hypergeometric test, considers only significant DEGs (i.e., FDR <10% by DESeq2 analysis) for overrepresentation among genes sets listed in the Canonical pathways sub-collections using the Fisher combined probability test function in the PIANO R package (using “runGSAhyper”). No fold-change criteria were set. For both analyses, a minimum of 5 and a maximum of 500 genes was set for the gene set size (number of member genes represented in the dataset tested, i.e., the results of the sequencing experiment presented herein) criteria for inclusion in the analysis. Gene sets with an FDR <10% were considered to be significantly enriched.
2.2.4. Investigation of DNA damage response
To further investigate enrichment of gene sets relevant torel DNA damage response and/or repair, a collection of gene sets was curated by searching through all gene sets in the MSigDB collections (v6.2) using key words related to DNA damage response. A total of 89 gene sets that are related to DNA damage and/or response were identified and then tested for enrichment among significant DEGs (i.e., FDR <10% by DESeq2 analysis) using a hypergeometric test for overrepresentation, using all genes among these 89 gene sets as the background (i.e., the gene “universe”). No fold-change criteria were set for the DEGs tested for enrichment. This targeted approach was conducted separately from the gene set enrichment analysis using the broader Canonical Pathways gene sets as a means to specifically evaluate enrichment of DNA damage-related gene sets. Some overlap exists in the gene sets from the Canonical Pathways collection and the curated list of DNA damage-specific list of 89 gene sets. A minimum of 5 and a maximum of 500 genes was set for the gene set size (number of member genes represented in the dataset tested, i.e., the results of the sequencing experiment presented herein) criteria for inclusion in the analysis. Gene sets with an FDR < 10% were considered to be significantly enriched.
Additionally, high-throughput screening (HTS) data available via the US EPA’s ToxCast downloadable data (invitroDBv3.2 database summary files2) were reviewed for 1,4-dioxane in a battery of nine assays (plus relevant viability or baseline assays) that are related DNA damage/repair (Hsieh et al., 2019).
2.3. Benchmark dose analysis
Dose-response modeling was conducted in using BMDExpress software (v2.2) (Phillips et al., 2019) using normalized expression data from DESeq2 without transformation. A Williams trend test (P value cutoff = 0.05) was employed to identify genes perturbed by 1,4-dioxane exposure. Fold-change filters and correction for multiple tests were not applied. Benchmark dose (BMD) analysis was conducted with linear, power, hill, 2° and 3° polynomial, and exponential models 2 to 5. The models were run assuming constant variance and a benchmark response (BMR) of 1 standard deviation. Functional classification of dose-responsive genes (genes with BMD P < 0.1) was conducted using the Gene Ontology (GO) and REACTOME gene sets available within BMDExpress. Genes were filtered from the analysis according to the default parameters within BMDExpress, as follows: genes with BMD/BMDL > 20, BMDU/BMDL > 40, BMDs above the highest dose (6000 ppm)), and/or genes with a BMD > 10-fold below the lowest positive dose were removed from functional classification analysis. No filters for minimum or maximum number of genes per gene set were used. Benchmark doses for the gene sets were also estimated. Additional parameters for the BMD modeling and pathway analyses can be found in Supplemental Materials.
3. Results
3.1. In-life summary
The results of the in-life portion of the study are described in Lafranconi et al. (2020). Briefly, there was no treatment-related effect on clinical signs, clinical chemistry parameters, body weights, or survival at any dose or timepoint in 1,4-dioxane-treated groups compared to controls. Liver weights were slightly increased in the 6000 ppm group at all timepoints; no changes were observed at lower concentrations. Histopathological analysis revealed minimal to mild vacuolation consistent with glycogen deposition in the centrilobular regions of the liver in animals exposed to 1,4-dioxane at concentrations ≥600 ppm after 7 days of exposure, which was nearly completely resolved by day 28. Minimal to mild centrilobular hypertrophy and centrilobular apoptosis was evident in the 2000 ppm and 6000 ppm groups at 28 and 90 days of exposure. There were no consistent treatment-related changes in hepatocellular proliferation in any dose group at 7 or 28 days according to immunohistochemical staining for bromodeoxyuridine (BrdU) incorporation, while there was a treatment-related increase at 90-days in the 6000 ppm group. The increase in BrdU incorporation corresponded with increased relative liver weights and blood levels of 1,4-dioxane (Lafranconi et al., 2020).
3.2. Transcriptomic changes associated with exposure to 1,4-dioxane
RNA sequencing was performed on liver samples to examine exposure effects of 1,4-dioxane on the hepatic gene expression of female mice compared to time-matched control mice. All sample libraries passed quality control measures necessary to be sequenced. As already noted in the Materials and Methods, following sequencings, five samples were removed from the analysis due to low sequencing depth or low gene diversity, from the 200 and 600 ppm groups across all three timepoints. There was an overall lack of transcriptomic response in the 40 and 200 ppm concentration groups, with the number of DEGs for these two concentrations across the three timepoints ranging from 0 to 22 (Table 1, Supplemental Table S1). At 600 ppm, an increase in the transcriptomic response was observed at both the 7 and 90 day timepoints, but not at 28 days. At 6000 ppm, the number of DEGs exhibited a similar pattern as the 600 ppm concentration, with a higher response at 7 and 90 days relative to 28 days (Table 1, Fig. 1). Approximately half of the DEGs for the 600 ppm group were also differentially expressed in the 6000 ppm group at 90 days, while at 7 days there was less overlap in the same genes being differentially expressed in the 600 vs. the 6000 ppm groups. At 2000 ppm, the transcriptomic response was similar across timepoints. The lower number of DEGs at 2000 ppm compared to 600 ppm at days 7 and 90 was an unexpected finding, and is without evidence of spurious origin. The overall relatively low number of DEGs across all experimental groups and timepoints likely contributed to this variability. The virtual lack of transcriptomic response following exposure to 1,4-dioxane concentrations below 600 ppm at all timepoints supports a conclusion that there is a threshold concentration for hepatic transcriptomic response to 1,4-dioxane in female mice somewhere in the range of 200 to 600 ppm. It is noted that the lowest dose tested in the Kano et al. (2009) bioassay was 500 ppm and the lowest dose in the NCI (1978) drinking water bioassay in mice was 5000 ppm (NCI, 1978, Kano et al., 2009).
Table 1.
Number of differentially expressed genes for each dose and length of exposure compared to time-matched control groups (shown as total DEG (Up-regulated [↑], Down-regulated [↓])). Full DESeq2 results can be found in Supplemental Table S1.
| Exposure Duration (days) | 1,4-Dioxane Concentration (ppm) |
||||
|---|---|---|---|---|---|
| 40 | 200 | 600 | 2000 | 6000 | |
| 7 | 0 (0) | 2 (↑0, ↓2) | 411 (↑165, ↓246) | 20 (↑6, ↓14) | 415 (↑180, ↓235) |
| 28 | 1 (↑0, ↓1) | 1 (↑0, ↓1) | 1 (↑0, ↓1) | 49 (↑21, ↓28) | 232 (↑87, ↓145) |
| 90 | 5 (↑1, ↓4) | 22 (↑11, ↓11) | 323 (↑165, ↓158) | 33 (↑25, ↓8) | 727 (↑352, ↓375) |
Fig. 1.

Volcano plots showing differentially expressed genes across concentrations at the 90-day timepoint (A), and across all three timepoints at the 6000 ppm concentration (B). Red points represent probes with an adjusted p-value ≤ 0.1; circles represent probes within a log2 (fold change) < 1.5, and red triangles represent probes with a log2 (fold change) ≥ 1.5. A: y-axis is scaled for all plots from 0 to 10 (resulting in some points cut off the plot for the 6000 ppm concentration). B: y-axis for all plots is scaled from 0 to 35. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3. Gene set enrichment analysis
3.3.1. Analysis by dose group relative to time-matched controls
The results of both gene set enrichment analysis methods were evaluated to further understand 1,4-dioxane treatment-related effects. Although the top-most significantly enriched pathways were similar across the two methods used for pathway enrichment analysis (Supplemental Tables S2 and S3), the hypergeometric method was determined to be less informative for the objective of the present study due to the minimal changes in gene expression at the lower concentrations (40–200 ppm), resulting in a complete lack of gene set enrichment at those concentrations. Thus, the results discussed below focus on the pre-ranked GSEA method for enrichment analysis. Due to the minimal treatment effect of 1,4-dioxane at any dose or timepoint on gene expression changes, liberal criteria were applied to identify DEGs and enriched signaling pathways. The full set of results for the hypergeometric test can be found in Supplemental Table S3. Pathway enrichment analysis of significantly differentially expressed genes at 40 ppm and 200 ppm 1,4-dioxane yielded very few significantly enriched gene sets/pathways using the pre-ranked GSEA test (Table 2, Supplemental Table S2).
Table 2.
Top most significantly enriched pathways for each treatment group according to the GSEA method (Subramanian et al., 2005). The top five most significantly enriched pathways for each direction of change are shown in the table; in cases where five gene sets were not significantly enriched, only those with an adjusted p-value < 0.1 are shown. Full results are presented in Supplemental Table S2.
| 1,4-Dioxane (ppm) | Duration (days) | Overall Direction | Gene set | Adjusted p-value |
|---|---|---|---|---|
| 40 | 7 | Up | None | NA |
| Down | None | NA | ||
| 28 | Up | REACTOME DEGRADATION OF THE EXTRACELLULAR MATRIX | 0.023524 | |
| REACTOME HS GAG DEGRADATION | 0.094038 | |||
| Down | None | NA | ||
| 90 | Up | None | 0.000913 | |
| Down | REACTOME SRP DEPENDENT COTRANSLATIONAL PROTEIN TARGETING TO MEMBRANE | 0.0015971 | ||
| REACTOME UNFOLDED PROTEIN RESPONSE | 0.0019783 | |||
| KEGG TERPENOID BACKBONE BIOSYNTHESIS | 0.0024729 | |||
| REACTOME TRANSLATION | 0.0031942 | |||
| REACTOME CHOLESTEROL BIOSYNTHESIS | 0.0032972 | |||
| 200 | 7 | Up | REACTOME TRANSLATION | < 0.0001 |
| REACTOME PEPTIDE CHAIN ELONGATION | < 0.0001 | |||
| REACTOME 3 UTR MEDIATED TRANSLATIONAL REGULATION | < 0.0001 | |||
| REACTOME SRP DEPENDENT COTRANSLATIONAL PROTEIN TARGETING TO MEMBRANE | < 0.0001 | |||
| KEGG RIBOSOME | < 0.0001 | |||
| Down | KEGG BIOSYNTHESIS OF UNSATURATED FATTY ACIDS | 0.019492 | ||
| KEGG PEROXISOME | 0.021777 | |||
| REACTOME SULFUR AMINO ACID METABOLISM | 0.022062 | |||
| KEGG ARGININE AND PROLINE METABOLISM | 0.028247 | |||
| REACTOME PYRUVATE METABOLISM AND CITRIC ACID TCA CYCLE | 0.029404 | |||
| 28 | Up | None | NA | |
| Down | None | NA | ||
| 90 | Up | None | NA | |
| Down | None | NA | ||
| 600 | 7 | Up | REACTOME 3 UTR MEDIATED TRANSLATIONAL REGULATION | < 0.0001 |
| REACTOME PEPTIDE CHAIN ELONGATION | < 0.0001 | |||
| REACTOME NONSENSE MEDIATED DECAY ENHANCED BY THE EXON JUNCTION COMPLEX | < 0.0001 | |||
| KEGG RIBOSOME | < 0.0001 | |||
| REACTOME INFLUENZA VIRAL RNA TRANSCRIPTION AND REPLICATION | < 0.0001 | |||
| Down | KEGG PROPANOATE METABOLISM | < 0.0001 | ||
| REACTOME FORMATION OF FIBRIN CLOT CLOTTING CASCADE | < 0.0001 | |||
| REACTOME COMMON PATHWAY | < 0.0001 | |||
| KEGG FATTY ACID METABOLISM | < 0.0001 | |||
| KEGG BUTANOATE METABOLISM | < 0.0001 | |||
| 28 | Up | None | NA | |
| Down | REACTOME TRNA AMINOACYLATION | 0.066645 | ||
| PID PLK1 PATHWAY | 0.098824 | |||
| 90 | Up | None | ||
| Down | REACTOME HEPARAN SULFATE HEPARIN HS GAG METABOLISM | 0.026099 | ||
| REACTOME A TETRASACCHARIDE LINKER SEQUENCE IS REQUIRED FOR GAG SYNTHESIS | 0.036539 | |||
| REACTOME HS GAG BIOSYNTHESIS | 0.095041 | |||
| 2000 | 7 | Up | REACTOME INFLUENZA VIRAL RNA TRANSCRIPTION AND REPLICATION | < 0.0001 |
| REACTOME INFLUENZA LIFE CYCLE | < 0.0001 | |||
| REACTOME PEPTIDE CHAIN ELONGATION | < 0.0001 | |||
| REACTOME TRANSLATION | < 0.0001 | |||
| REACTOME SRP DEPENDENT COTRANSLATIONAL PROTEIN TARGETING TO MEMBRANE | < 0.0001 | |||
| Down | KEGG VALINE LEUCINE AND ISOLEUCINE DEGRADATION | < 0.0001 | ||
| KEGG TRYPTOPHAN METABOLISM | < 0.0001 | |||
| KEGG PPAR SIGNALING PATHWAY | < 0.0001 | |||
| KEGG FATTY ACID METABOLISM | < 0.0001 | |||
| KEGG PROPANOATE METABOLISM | < 0.0001 | |||
| 28 | Up | REACTOME PEPTIDE CHAIN ELONGATION | < 0.0001 | |
| KEGG RIBOSOME | 0.0021614 | |||
| REACTOME INFLUENZA VIRAL RNA TRANSCRIPTION AND REPLICATION | 0.0023879 | |||
| REACTOME 3 UTR MEDIATED TRANSLATIONAL REGULATION | 0.012228 | |||
| REACTOME CELL DEATH SIGNALLING VIA NRAGE NRIF AND NADE | 0.014006 | |||
| Down | KEGG BIOSYNTHESIS OF UNSATURATED FATTY ACIDS | < 0.0001 | ||
| REACTOME FATTY ACYL COA BIOSYNTHESIS | 0.020023 | |||
| REACTOME POST TRANSLATIONAL PROTEIN MODIFICATION | 0.021946 | |||
| KEGG STEROID HORMONE BIOSYNTHESIS | 0.022519 | |||
| REACTOME METABOLISM OF AMINO ACIDS AND DERIVATIVES | 0.023419 | |||
| 90 | Up | REACTOME FORMATION OF TUBULIN FOLDING INTERMEDIATES BY CCT TRIC | 0.045617 | |
| BIOCARTA P53 PATHWAY | 0.059268 | |||
| SIG REGULATION OF THE ACTIN CYTOSKELETON BY RHO GTPASES | 0.068946 | |||
| REACTOME GLUTATHIONE CONJUGATION | 0.079748 | |||
| REACTOME POST CHAPERONIN TUBULIN FOLDING PATHWAY | 0.085827 | |||
| Down | REACTOME DEGRADATION OF THE EXTRACELLULAR MATRIX | < 0.0001 | ||
| BIOCARTA INTRINSIC PATHWAY | < 0.0001 | |||
| KEGG COMPLEMENT AND COAGULATION CASCADES | 0.0053303 | |||
| REACTOME LIPID DIGESTION MOBILIZATION AND TRANSPORT | 0.015464 | |||
| REACTOME FORMATION OF FIBRIN CLOT CLOTTING CASCADE | 0.016814 | |||
| 6000 | 7 | Up | BIOCARTA EIF PATHWAY | < 0.0001 |
| KEGG RIBOSOME | < 0.0001 | |||
| REACTOME PEPTIDE CHAIN ELONGATION | < 0.0001 | |||
| REACTOME INFLUENZA VIRAL RNA TRANSCRIPTION AND REPLICATION | < 0.0001 | |||
| REACTOME INFLUENZA LIFE CYCLE | < 0.0001 | |||
| Down | KEGG COMPLEMENT AND COAGULATION CASCADES | < 0.0001 | ||
| BIOCARTA COMP PATHWAY | < 0.0001 | |||
| NABA ECM REGULATORS | 0.0010723 | |||
| REACTOME FORMATION OF FIBRIN CLOT CLOTTING CASCADE | 0.0012178 | |||
| BIOCARTA CLASSIC PATHWAY | 0.0013393 | |||
| 28 | Up | REACTOME GLUTATHIONE CONJUGATION | 0.05646 | |
| Down | KEGG COMPLEMENT AND COAGULATION CASCADES | < 0.0001 | ||
| KEGG ARGININE AND PROLINE METABOLISM | 0.0027586 | |||
| NABA ECM REGULATORS | 0.0036022 | |||
| BIOCARTA INTRINSIC PATHWAY | 0.0036782 | |||
| REACTOME FORMATION OF FIBRIN CLOT CLOTTING CASCADE | 0.004578 | |||
| 90 | Up | PID AURORA B PATHWAY | 0.00041562 | |
| REACTOME FORMATION OF TUBULIN FOLDING INTERMEDIATES BY CCT TRIC | 0.00083123 | |||
| REACTOME GLUTATHIONE CONJUGATION | 0.017352 | |||
| REACTOME POST CHAPERONIN TUBULIN FOLDING PATHWAY | 0.026444 | |||
| REACTOME PREFOLDIN MEDIATED TRANSFER OF SUBSTRATE TO CCT TRIC | 0.043203 | |||
| Down | PID HNF3A PATHWAY | < 0.0001 | ||
| KEGG PANTOTHENATE AND COA BIOSYNTHESIS | < 0.0001 | |||
| REACTOME LIPID DIGESTION MOBILIZATION AND TRANSPORT | 0.0014751 | |||
| NABA ECM REGULATORS | 0.0055796 | |||
| REACTOME LIPOPROTEIN METABOLISM | 0.014064 |
The decreased regulation of complement and coagulation cascades, mitochondrial β-oxidation and several other fatty acid metabolism pathways observed in the 600 ppm group is consistent with other transcriptomic analyses of liver tissues from primate and mouse studies in which animals were treated with nuclear receptor agonists that induce mitosis and DNA synthesis (e.g., fibrates (Cariello et al., 2005, Lu et al., 2011, de la Rosa Rodriguez et al., 2018) (Table 2).
At the 2000 and 6000 ppm concentrations, significant enrichment of xenobiotic metabolism pathways was evident, which increased in significance with increasing time and dose. Examples of enriched up-regulated gene sets associated with phase II metabolism, specific to glutathione conjugation, include KEGG “glutathione metabolism” and REACTOME “glutathione conjugation” (Table 2). The enrichment of these gene sets was driven by altered genes that encode glutathione transferase isoforms. Additionally, similar to the pathway alterations observed at 600 ppm, complement and coagulation cascade pathways were enriched in the negative direction (down-regulated) at 2000 and 6000 ppm 1,4-dioxane due to decreased expression of genes encoding proteolytic subunits in the complement system and gene members of the serpin family (serine protease inhibitors) relative to controls (Table 2). The complement cascade is a part of the innate immune system and deficiency of certain serpins (e.g., Serpina1) has been associated with liver damage (Law et al., 2006), which is consistent with the reported increase in cell death in the highest 1,4-dioxane dose groups (Lafranconi et al., 2020). Down-regulation of extracellular matrix regulators is also related to the loss of genes related to clotting factors. The significant decrease in expression of lipid metabolism-related gene sets in the 2000 and 6000 ppm may be related to liver injury in these high 1,4-dioxane dose-groups following saturation of metabolism and accumulation of the parent compound (as described in Lafranconi et al., 2020).
At the 90-day timepoint, mitotic cell cycle and DNA synthesis pathways were significantly enriched in the 6000 ppm treatment group: aurora B kinase signaling, mitotic phase transition and checkpoint signaling, and general cell cycle (e.g., Reactome “Cell cycle, mitotic”) (Fig. 2, Table 2), indicating a mitogenic proliferative response that was not observed at earlier time points. Enriched tubulin folding pathways share many of the same gene members as the cell cycle gene sets (Fig. 2). This is consistent with biomarkers of proliferative liver response in the same tissues, as reported in Lafranconi et al. (2020). Specifically, a treatment-related pan-lobular increase in hepatocellular proliferation was observed at 90-days in animals exposed to 6,000 ppm, as measured by BrdU incorporation. The increase in BrdU incorporation corresponded with an increase in relative liver weight as well as blood levels of 1,4-dioxane. There were no consistent, treatment-related changes in hepatocellular proliferation at 7 or 28 days in any dose group (Lafranconi et al., 2020).
Fig. 2.

Network plots showing enriched gene sets at 6000 ppm relative to controls. (A/C/E: adjusted p-value ≤ 0.1, B/D/F: adjust p-value ≤ 0.05). Node size is scaled on number of member genes within the gene set, and node color is scaled according to significance (lighter blue/pink node color indicates more highly significant relevant to darker blue/pink node color). Nodes are spatially organized according to likeness, according to common individual genes within the gene sets. Lines connecting nodes represents common members, with thickness of the line scaled according to number of common gene members. Color of the nodes represents statistical significance as noted in the color bar key. For visualization, general descriptive categories are denoted for gene sets with common genes and, thus, similar functionality, as opposed to listing all actual gene set names. Select individual gene set of highest statistically significant enrichment are shown. Full results for all dose groups and timepoints are in Supplemental Table S2. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3.2. Targeted analysis of DNA damage response
According to the targeted analysis (hypergeometric test for overrepresentation) of changes in expression in genes included in a curated list of 89 gene sets related to DNA damage response and repair (Supplemental Table S4), there was no enrichment. A hypergeometric test was necessary for this assessment, due to the nature of the evaluation using a focused list of gene sets (Supplemental Table S5). Additionally, 1,4-dioxane was inactive in the battery of HTS assays used to identify compounds with genotoxic potential (Supplemental Table S6). It should be noted that challenges exist in testing volatile chemicals (such as 1,4-dioxane) in HTS assays, as these in vitro assays involve the use of open vessels with incubations carried out at temperatures ranging from 4 °C to 37 °C. In such conditions, a substance with a high vapor pressure can potentially volatilize during the course of the assay, thereby influencing the concentration of the test substance in the system. While 1,4-dioxane has a molecular weight less than 140 g/mol (88.11 g/mol), which indicates volatility, its vapor pressure and log octanol/water partition coefficients (38.1 mmHg and −0.27, respectively) are within suitable boundaries for ToxCast/Tox21 assays (Tice et al., 2013, Richard et al., 2016). Overall, these results are consistent with other findings indicating that 1,4-dioxane does not cause direct DNA damage in the liver in vivo in mice, nor does it cause changes in in vitro assays designed to detect DNA damaging agents (as reviewed in (EPA, 2010, ATSDR, 2012)). These findings support a non-mutagenic MOA.
3.3.3. Benchmark dose modeling
The dose–response for individual genes were analyzed using BMD modeling, and functional characterization of the dose-responsive genes was analyzed and visualized. The BMD results confirmed pathway enrichment results obtained for single dose groups. For example, similar to what was found at 90 days in the ≥600 ppm 1,4-dioxane groups, the REACTOME gene sets “glutathione conjugation” and “Phase II – Conjugation of compounds” were significantly enriched at 90 days with median BMD1SD values of 1548 and 1652 ppm, respectively, and median BMDLs of 1236 and 1251 ppm, respectively (Table 3, Fig. 3). In addition, “innate immune system”, a part of the complement and coagulation cascade pathway, was also significantly enriched in the negative direction with a median BMD1SD > 3200 ppm. Cell cycle and mitosis gene sets (median BMD1SD > 3400 ppm or higher, BMDLs > 2200 ppm or higher) along with a single “DNA repair” gene set (median BMD1SD > 4000 ppm) were enriched among dose-responsive genes at 90 days. Individual genes within the “DNA repair” gene set that were identified as dose-responsive are mainly histone encoding genes and DNA polymerase genes involved in DNA synthesis (Supplemental Table S7). An exception is the DNA repair gene Rad51, which was found to have a significant dose-responsive trend via BMDExpress (p = 0.0217 by Williams Trent Test) at 90 days. However, this gene had generally low expression in all treatment groups and was not significantly differentially expressed at any individual dose at any timepoint relative to time-respective controls according to DESeq2 analysis (adjusted p-value ≥ 0.1 for all probes for all concentrations and timepoints; Supplemental Table S1).
Table 3.
BMD modeling results for select up-regulated enriched gene sets related to xenobiotic metabolism and cell cycle. Full results are presented in Supplemental Table S7.
| Gene set | Exposure Duration (days) | Median BMD1SD (ppm) | Median BMDL (ppm) | Fisher’s exact two-tail test p-value |
|---|---|---|---|---|
| Glutathione Conjugation | 7 | 2305 | 1819 | 7.46x10-4 |
| 28 | 1682 | 1399 | 6.75x10-5 | |
| 90 | 1548 | 1236 | 6.16x10-4 | |
| Phase II - Conjugation of compounds | 7 | 2301 | 1696 | 4.11x10-5 |
| 28 | 1903 | 1401 | 9.28x10-9 | |
| 90 | 1652 | 1251 | 3.84x10-2 | |
| Cell Cycle | 7 | 3521 | 2333 | NS |
| 28 | 5455 | 2523 | NS | |
| 90 | 3874 | 2243 | 4.54x10-3 | |
| Cell Cycle Checkpoints | 7 | 3628 | 2428 | NS |
| 28 | 5455 | 2849 | NS | |
| 90 | 3474 | 2265 | 6.56x10-2 | |
| Cell Cycle, Mitotic | 7 | 3521 | 2333 | NS |
| 28 | 3639 | 2523 | 2.56x10-2 | |
| 90 | 3414 | 2242 | 9.44x10-3 |
NS, not significant for enrichment among dose-responsive genes.
Fig. 3.

BMDExpress analysis visualizations. A: Range plots for selected gene sets related to cell cycle. Data are shown for gene sets/timepoints with significant enrichment. B: Accumulation plot for all three timepoints, with select gene sets discussed herein annotated by text.
Similar to the gene sets that were determined as significantly enriched at 90 days according to the GSEA analysis at individual doses/timepoints, phase II metabolism, cell cycle and mitosis gene sets were up-regulated with comparable BMD values at 28 days (Fig. 3). The “DNA Repair” gene set was not significantly enriched according to BMD modeling at 28 days. Moreover, “fatty acid metabolism” and “immune system” were down-regulated at 28 days (median BMD1SD 3385 and 3367 ppm, respectively) but with less statistical significance compared to the results at 90 days (i.e., lower Fisher’s test p-values). Among the dose-responsive genes at 7 days, phase II metabolism gene sets were enriched, with much higher median BMD1SD values than those determined at 28 and 90 days. Although the gene ontology used in the BMDExpress software (REACTOME) was not an exact match to that of the GSEA analysis, the REACTOME gene sets were included in both analyses. Further, many similar gene sets exist across different ontologies, enabling a reasonable comparison of biological signals within the two analyses.
At 7 days, the top-most enriched gene sets among dose-responsive genes were related to signal transduction and were down-regulated, with median BMDs > 2000 ppm. This may represent an early stress response and/or cytotoxicity. “Glucuronidation” and “Phase II – Conjugation of compounds” were enriched, and up-regulated (median BMDs 1092 and 2301 ppm, respectively). The “DNA repair” gene set that was enriched at 90 days was also enriched at 7 days (BMD median of 3506 ppm). The histone and ubiquination genes underlying the enrichment of this gene set are involved in DNA synthesis and potentially cell proliferation.
Overall, BMD analysis confirmed the increase in phase II xenobiotic metabolism and a decrease in complement cascade and lipid metabolism pathways that was observed via analysis at each individual dose, as well as a significant increase mitotic cell cycle and cellular proliferation at concentrations above 2000 ppm at 90 days (Fig. 3). The BMD1SD and BMDLs were well above 600 ppm for some pathways that were significant at 600 ppm according to gene set enrichment analysis comparing each dose relative to the controls. This may be explained by the fact that BMD modeling analysis accounts for variability across the whole experiment, as well as the general dose–response curve information, as opposed to specifically comparing one dose group to the time-matched controls.
4. Discussion
Mechanistic data provide important information for human health risk assessment, in particular with respect to providing an understanding of the underlying mode/mechanisms of an adverse outcome. Such mechanistic data can inform the MOA of a chemical via the identification of specific key molecular or cellular events. Specifically, transcriptomic analysis can contribute to understanding drug- or chemical-induced liver toxicity by identifying biomarkers of effect or exposure, expression signatures, and/or changes in signaling (Merrick and Bruno, 2004, Cui and Paules, 2010). The identification of a MOA for a carcinogen is important for the selection of the risk assessment approach under current regulatory paradigms. Specifically, a mutagenic vs. a non-mutagenic MOA have historically been subject to linear low-dose extrapolation vs. a threshold approach, respectively, for risk assessment (U.S. EPA, 2005). In the case of 1,4-dioxane, several groups, including regulatory agencies, have applied a threshold approach (NICNAS, 1998, TNO/RIVM, 1999, Stickney et al., 2003; Health Canada, 2005), while others have applied a non-threshold approach (OEHHA, 2002; U.S. EPA, 2013). Previously, a MOA for rodent liver tumors was hypothesized that included metabolic saturation followed by cytotoxicity-induced regenerative repair (Dourson et al., 2014, Dourson et al., 2017). Biomarker analyses and histological examinations were conducted on the same liver tissues discussed herein, and reported by Lafranconi et al (2020). Collectively, these analyses demonstrated saturated metabolism of 1,4-dioxane in mice, as well as increased proliferation following 90 days of oral exposure via drinking water, at concentrations ≥2000 ppm. These results provide additional mechanistic information for 1,4-dioxane, informing potential key events in a MOA for liver cancer in a sensitive strain of mouse. The transcriptomic information also adds insights as to molecular events that explain these biomarker and histopathology findings. Moreover, the transcriptomic analyses serve to identify potential key events for further examination that were not visible with the more conventional histopathological observations.
As described in the Materials and Methods, gene expression data from the livers of 1,4-dioxane-exposed mice were analyzed for individual gene changes, gene set enrichment using two different statistical methods, and BMD modeling for individual genes, as well as functional classification of dose-responsive genes. The results demonstrate a generally low response to 1,4-dioxane in the livers of mice and the mRNA level. Overall, gene set enrichment demonstrated up-regulation of phase II metabolism in a dose–response manner. After 90 days of exposure, an increase in cell cycle signaling was evident in the highest concentration treatment group. Changes in individual genes that did not converge into gene set enrichment, and a general loss of signal transduction at the pathway level at the 7-day timepoint likely represents a non-specific adaptive and/or general stress response. Such changes were mitigated after 28 days of exposure, potentially related to the up-regulation of Phase II metabolism and, thus, detoxification. Importantly, transcriptomic profiling conducted to specifically query the enrichment of DNA damage response gene sets demonstrated a lack of DNA damage response at the mRNA level. The few enriched gene sets related to up-regulation of DNA damage response at 6000 ppm (i.e., p53 signaling pathways) according to the more lenient GSEA enrichment analysis and BMD functional classification analysis may be related to apical endpoints reported in Lafranconi et al. (2020): up-regulation of signaling pathways for cell cycle are potentially related to the reported increased BrdU labeling, and enrichment of cell death signaling potentially related to the increase in apoptosis as evidenced by Caspase 3 staining. While p53 signaling is known to be activated by DNA damage, it also can be activated by non-genotoxicants (Catizone et al., 2019). The individual genes driving the enrichment of p53-relevant pathways in the GSEA and BMDExpress analyses were regulators of apoptosis (e.g., Bax) and cytokines, without alteration to DNA repair enzymes nor the p53 gene itself. No individual genes for DNA damage repair enzymes were differentially expressed compared to controls at any dose or timepoint according to the DESeq2 analysis. This indicated that changes in cell cycle occurred in the high concentration group independent of DNA damage. This finding is corroborated by an overall negative profile for 1,4-dioxane in a set of HTS assays within the ToxCast/Tox21 database that are indicators of DNA damage and/or repair (Hsieh et al., 2019). This finding aligns with the proposed MOA for 1,4-dioxane rodent hepatotoxicity involving cytotoxicity and subsequent regenerative hyperplasia (Dourson et al., 2017), as well as with the mitogenic response reported for the same liver tissues evaluated herein (Lafranconi et al., 2020)
Transcriptomics data can provide important information for proposing potential key events for an alternative MOA or supporting existing key events in established MOAs. Signaling on the molecular level can demonstrate or inform underlying mechanisms of toxicity. Transcriptomic responses following relatively short exposures that are transient may represent a non-specific adaptive and/or stress response (Dean et al., 2017). For example, after 7 days of exposure to 1,4-dioxane in the present study, there were many more DEGs than following 28 days of exposure at the 600 and 6000 ppm concentrations. However, there were very few enriched gene sets at the 7-day timepoint, for any exposure concentration. This indicates that the altered genes are not members of a cohesive signaling pathway and may represent a transient response to the exposure scenario. After 28 days of exposure, the majority of the DEGs at the 7-day timepoint had returned to levels similar to the time-matched controls for the 600 and 6000 ppm groups. While the 2000 ppm group had overall more DEGs at 28 days compared to either 7 and 90 days, most of the DEGs at 7 days were not differentially expressed at 28 days. Following a sub-chronic exposure duration of 90 days, transcriptomic response was increased at the 600 and 6000 ppm concentrations. A 28-day “sub-acute” timepoint has been used to identify liver chemical carcinogenicity signatures in experimental animals (Waters et al., 2003), while 90-day exposures have been suggested to accentuate gene expression changes related to the carcinogenic activity of chemicals (Auerbach et al., 2010). Notably, transcriptomic analysis in target tissue following exposure durations of 14 days or less in in vivo models has been shown to be predictive of non-DNA-reactive mechanisms in hepatic tumors (Fielden et al., 2007). In the present study, the transcriptomic profiles at three different exposure durations were absent of a gene expression signal for DNA damage response or repair.
In addition to the pathway level enrichment of phase II metabolism and an increase in mitotic cell cycle at high concentrations and later timepoints, reduced expression of genes involved in coagulation and complement cascade, as well as extra-cellular matrix regulation, was a significant and transient signal in the present study; this signal was normalized at 90 days at all concentrations except for 6000 ppm. Although the significance of this finding is not fully known, downregulation of coagulation cascade proteins in the livers of mice with hyperplasia-mediated liver regeneration has been previously demonstrated (Tatsumi et al., 2009).
Although alterations to nuclear receptors involved in xenobiotic metabolism represents a known molecular initiating event for some cases of chemically-induced hepatotoxicity and/or hepatocarcinogenicity, in particular those with increased proliferation, the only general nuclear receptor gene set included in the analysis presented herein (“BIOCARTA_NUCLEARRS_PATHWAY”) was not significantly enriched. Thus, individual CYP-encoding genes that are considered indicators of several common nuclear receptors known to play a role in rodent liver pathogenesis (aryl hydrocarbon receptor [AhR], constitutive androstane receptor [CAR], peroxisome proliferator-activated receptor [PPAR], and pregnane X receptor [PXR]) were reviewed for treatment effect. The CYP-encoding genes were not differentially expressed in any dose group or timepoint, with the exception of the PXR-related Cyp3a11 (human homolog CYP3A4, Li et al., 2009), which was significantly up-regulated at 90 days in the 600 and 6000 ppm dose groups (Supplemental Table S1). The biological plausibility that PXR may be affected by 1,4-dioxane in mouse livers is supported by the fact that PXR regulates phase II conjugating enzymes. However; Cyp3a11 was only significantly up-regulated at the highest dose at 90 days, while phase II metabolism pathways were up-regulated at early timepoints as well as at the 600 ppm concentration, indicating that the two expression changes may not be dependent upon one another. PXR, among other xenobiotic-metabolizing nuclear receptors, is known to be differentially expressed across species, leading to species-specific liver effects in rodents (Luisier et al., 2014, Yamada et al., 2015). While this result suggests the possibility that 1,4-dioxane exposure affects the PXR, further investigation beyond Cyp3a11 mRNA level is necessary to confirm such a molecular event.
It should be noted that in the present study, due to the minimal treatment effect of 1,4-dioxane on gene expression at any dose or timepoint, liberal criteria were applied to identify DEGs and enriched signaling pathways. For example, no fold-change criterion was set for the identification of DEGs, and the use of the full complement of genes ranked by the Wald statistic for gene set enrichment rather than filtered by a significance cut-off was the approach emphasized herein (GSEA method as opposed to the hypergeometric test, with the exception of the DNA damage response analysis). These liberal criteria enabled identification of minimally altered genes and signaling networks and demonstrated that changes to signaling pathways were limited. Trends in changes to signaling pathways related to mechanisms of hepatoxicity and/or carcinogenesis were subtle and specific to high dose groups. This highlights the overall low effect of 1,4-dioxane on gene expression in the livers of mice, particularly at concentrations below 600 ppm. The results indicate that the threshold concentration for hepatic transcriptomic response to 1,4-dioxane in female mice, whether it be transient and/or adaptive or related to pathology, exists somewhere in the range of 600–2000 ppm.
In summary, the transcriptomic response in livers of mice exposed to 1,4-dioxane in a drinking water study demonstrates minimal treatment effects on global gene expression at concentrations below 600 ppm, with an increase in phase II metabolism and cellular cycle signaling in the absence of a significant increase in DNA damage response signaling at the mRNA level at 600 ppm and above. These findings align with the phenotypic findings of histopathological and biochemical analysis of the same liver tissues, and support the non-mutagenic, threshold-based mitogenic MOA for mouse liver tumors proposed by Lafranconi et al. (2020) based on all the study findings.
CRediT authorship contribution statement
G.A. Chappell: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Visualization, Writing - original draft, Writing - review & editing. M.M. Heintz: Formal analysis, Writing - original draft. L.C. Haws: Funding acquisition, Supervision, Project administration, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the American Chemistry Council (ACC) 1,4-Dioxane Panel (the Panel). The Panel was given the opportunity to review the manuscript and provide written or verbal comments prior to submission. The purpose of this review was for the authors to receive input on the clarity of the science presented but not on the interpretation of research results. The authors’ scientific conclusions and professional judgments were not subject to the funder's control; the contents of this manuscript reflect solely the view of the authors. The authors thank Dr. Chad Thompson for technical consultation regarding the benchmark dose modeling, and Drs. Robert Budinsky, Matthew LeBaron, Joanna Klapacz, and Susan Felter for helpful discussions and insight.
Footnotes
https://www.epa.gov/chemical-research/exploring-toxcast-data-downloadable-data (downloaded August 16th, 2019)
Supplementary data to this article can be found online at https://doi.org/10.1016/j.crtox.2021.01.003.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Argus MF, Arcos JC. Studies on the carcinogenic activity of protein-denaturing agents: Hepatocarcinogenicity of dioxane. J Natl Cancer Inst. 1965;35:949–958. [PubMed] [Google Scholar]
- Argus M.F., Sohal R.S., Bryant G.M., Hoch-Ligeti C., Arcos J.C. Dose-response and ultrastructural alterations in dioxane carcinogenesis. Eur. J. Cancer. 1973;9:237–243. doi: 10.1016/0014-2964(73)90088-1. [DOI] [PubMed] [Google Scholar]
- ATSDR . Division of Toxicology and Environmental Medicine/Applied Toxicology Branch, U.S. Department of Health and Human Services; 2012. Toxicological profile for 1,4-dioxane. Agency for Toxic Substances and Disease Registry (ATSDR) [PubMed] [Google Scholar]
- Auerbach S.S., Shah R.R., Mav D., Smith C.S., Walker N.J., Vallant M.K., Boorman G.A., Irwin R.D. Predicting the hepatocarcinogenic potential of alkenylbenzene flavoring agents using toxicogenomics and machine learning. Toxicol. Appl. Pharmacol. 2010;243(3):300–314. doi: 10.1016/j.taap.2009.11.021. Available from https://www.ncbi.nlm.nih.gov/pubmed/20004213. DOI 10.1016/j.taap.2009.11.021. [DOI] [PubMed] [Google Scholar]
- Cariello N.F., Romach E.H., Colton H.M., Ni H., Yoon L., Falls J.G., Casey W., Creech D., Anderson S.P., Benavides G.R., Hoivik D.J., Brown R., Miller R.T. Gene expression profiling of the ppar-alpha agonist ciprofibrate in the cynomolgus monkey liver. Toxicol. Sci. 2005;88(1):250–264. doi: 10.1093/toxsci/kfi273. Available from https://www.ncbi.nlm.nih.gov/pubmed/16081524. DOI 10.1093/toxsci/kfi273. [DOI] [PubMed] [Google Scholar]
- Catizone A.N., Good C.R., Alexander K.A., Berger S.L., Sammons M.A. Comparison of genotoxic versus nongenotoxic stabilization of p53 provides insight into parallel stress-responsive transcriptional networks. Cell Cycle. 2019;18(8):809–823. doi: 10.1080/15384101.2019.1593643. Available from https://www.ncbi.nlm.nih.gov/pubmed/30966857. DOI 10.1080/15384101.2019.1593643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chepelev N.L., Moffat I.D., Labib S., Bourdon-Lacombe J., Kuo B., Buick J.K., Lemieux F., Malik A.I., Halappanavar S., Williams A., Yauk C.L. Integrating toxicogenomics into human health risk assessment: Lessons learned from the benzo[a]pyrene case study. Crit. Rev. Toxicol. 2015;45(1):44–52. doi: 10.3109/10408444.2014.973935. Available from https://www.ncbi.nlm.nih.gov/pubmed/25605027. DOI 10.3109/10408444.2014.973935. [DOI] [PubMed] [Google Scholar]
- Cohen S.M., Ellwein L.B. Cell proliferation in carcinogenesis. Science. 1990;249(4972):1007–1011. doi: 10.1126/science.2204108. Available from https://www.ncbi.nlm.nih.gov/pubmed/2204108. DOI 10.1126/science.2204108. [DOI] [PubMed] [Google Scholar]
- Croft D., O'Kelly G., Wu G., Haw R., Gillespie M., Matthews L., Caudy M., Garapati P., Gopinath G., Jassal B., Jupe S., Kalatskaya I., Mahajan S., May B., Ndegwa N., Schmidt E., Shamovsky V., Yung C., Birney E., Hermjakob H., D'Eustachio P., Stein L. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39(Database issue):D691–D697. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y., Paules R.S. Use of transcriptomics in understanding mechanisms of drug-induced toxicity. Pharmacogenomics. 2010;11(4):573–585. doi: 10.2217/pgs.10.37. Available from https://www.ncbi.nlm.nih.gov/pubmed/20350139. DOI 10.2217/pgs.10.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa Rodriguez M.A., Sugahara G., Hooiveld G., Ishida Y., Tateno C., Kersten S. The whole transcriptome effects of the pparalpha agonist fenofibrate on livers of hepatocyte humanized mice. BMC Genomics. 2018;19(1):443. doi: 10.1186/s12864-018-4834-3. Available from https://www.ncbi.nlm.nih.gov/pubmed/29879903. DOI 10.1186/s12864-018-4834-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean J.L., Zhao Q.J., Lambert J.C., Hawkins B.S., Thomas R.S., Wesselkamper S.C. Editor's highlight: Application of gene set enrichment analysis for identification of chemically induced, biologically relevant transcriptomic networks and potential utilization in human health risk assessment. Toxicol. Sci. 2017;157(1):85–99. doi: 10.1093/toxsci/kfx021. Available from https://www.ncbi.nlm.nih.gov/pubmed/28123101. DOI 10.1093/toxsci/kfx021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dourson M., Reichard J., Nance P., Burleigh-Flayer H., Parker A., Vincent M., McConnell E.E. Mode of action analysis for liver tumors from oral 1,4-dioxane exposures and evidence-based dose response assessment. Regul. Toxicol. Pharmacol. 2014;68(3):387–401. doi: 10.1016/j.yrtph.2014.01.011. Available from https://www.ncbi.nlm.nih.gov/pubmed/24491968. DOI 10.1016/j.yrtph.2014.01.011. [DOI] [PubMed] [Google Scholar]
- Dourson M.L., Higginbotham J., Crum J., Burleigh-Flayer H., Nance P., Forsberg N.D., Lafranconi M., Reichard J. Update: Mode of action (moa) for liver tumors induced by oral exposure to 1,4-dioxane. Regul Toxicol Pharmacol. 2017;88:45–55. doi: 10.1016/j.yrtph.2017.02.025. Available from https://www.ncbi.nlm.nih.gov/pubmed/28366800. DOI 10.1016/j.yrtph.2017.02.025. [DOI] [PubMed] [Google Scholar]
- Elcombe C.R., Peffer R.C., Wolf D.C., Bailey J., Bars R., Bell D., Cattley R.C., Ferguson S.S., Geter D., Goetz A., Goodman J.I., Hester S., Jacobs A., Omiecinski C.J., Schoeny R., Xie W., Lake B.G. Mode of action and human relevance analysis for nuclear receptor-mediated liver toxicity: A case study with phenobarbital as a model constitutive androstane receptor (car) activator. Crit. Rev. Toxicol. 2014;44(1):64–82. doi: 10.3109/10408444.2013.835786. Available from https://www.ncbi.nlm.nih.gov/pubmed/24180433. DOI 10.3109/10408444.2013.835786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA, U., 2010. Toxicological review of 1,4-dioxane (cas no. 123-91-1) in support of summary information on the integrated risk information system (iris) (epa-635/r-09-005-f). . Washington, DC.
- Falcon S., Gentleman R. Bioconductor case studies. Springer; New York, NY: 2008. Hypergeometric testing used for gene set enrichment analysis; pp. 207–220. [Google Scholar]
- Fielden M.R., Brennan R., Gollub J. A gene expression biomarker provides early prediction and mechanistic assessment of hepatic tumor induction by nongenotoxic chemicals. Toxicol. Sci. 2007;99(1):90–100. doi: 10.1093/toxsci/kfm156. Available from https://www.ncbi.nlm.nih.gov/pubmed/17557906. DOI 10.1093/toxsci/kfm156. [DOI] [PubMed] [Google Scholar]
- Gao C., Weisman D., Lan J., Gou N., Gu A.Z. Toxicity mechanisms identification via gene set enrichment analysis of time-series toxicogenomics data: Impact of time and concentration. Environ Sci Technol. 2015;49(7):4618–4626. doi: 10.1021/es505199f. Available from https://www.ncbi.nlm.nih.gov/pubmed/25785649. DOI 10.1021/es505199f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Health Canada . Health Canada; Ottowa, CA: 2005. Drinking water guidance value for 1,4-dioxane. [Google Scholar]
- Hsieh J.H., Smith-Roe S.L., Huang R., Sedykh A., Shockley K.R., Auerbach S.S., Merrick B.A., Xia M., Tice R.R., Witt K.L. Identifying compounds with genotoxicity potential using tox21 high-throughput screening assays. Chem. Res. Toxicol. 2019;32(7):1384–1401. doi: 10.1021/acs.chemrestox.9b00053. Available from https://www.ncbi.nlm.nih.gov/pubmed/31243984. DOI 10.1021/acs.chemrestox.9b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Center for Medical Research., Y., National University of Singapore., O., ICMR Seminar., A., A, N., H, O., H, F., M, O., T, K., H, S., 1988. Asia-Pacific Symposium on Environmental and Occupational Toxicology, 4-7 October, 1987, Singapore : proceedings, Proceedings of the ICMR Seminar (International Center for Medical Research). International Center for Medical Research, Kobe University School of Medicine.
- Johnson K.J., Auerbach S.S., Costa E. A rat liver transcriptomic point of departure predicts a prospective liver or non-liver apical point of departure. Toxicol. Sci. 2020;176(1):86–102. doi: 10.1093/toxsci/kfaa062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph P. Transcriptomics in toxicology. Food Chem. Toxicol. 2017;109(Pt 1):650–662. doi: 10.1016/j.fct.2017.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano H., Umeda Y., Kasai T., Sasaki T., Matsumoto M., Yamazaki K., Nagano K., Arito H., Fukushima S. Carcinogenicity studies of 1,4-dioxane administered in drinking-water to rats and mice for 2 years. Food Chem Toxicol. 2009;47(11):2776–2784. doi: 10.1016/j.fct.2009.08.012. [DOI] [PubMed] [Google Scholar]
- Kasai T., Kano H., Umeda Y., Sasaki T., Ikawa N., Nishizawa T., Nagano K., Arito H., Nagashima H., Fukushima S. Two-year inhalation study of carcinogenicity and chronic toxicity of 1,4-dioxane in male rats 2-yr inhalation study of 1,4-dioxane in rats Tatsuya Kasai. et al. Inhal. Toxicol. 2009;21:889–897. doi: 10.1080/08958370802629610. [DOI] [PubMed] [Google Scholar]
- Katagiri T., Negano K., Aiso S., Senoh H., Sakura Y., Takeuchi T., Okudaira M. A pathological study on spontaneous hepatic neoplasms in bdf1 mice. Journal of Toxicologic Pathology. 1998;11(1):21–25. [Google Scholar]
- Kociba R.J., McCollister S.B., Park C., Torkelson T.R., Gehring P.J. 1,4-Dioxane. I. Results of a 2-year ingestion study in rats. Toxicol. Appl. Pharmacol. 1974;30:275–286. [Google Scholar]
- Lafranconi M., Budinsky R., Corey L., Klapacz J., Crissman J., LeBaron M., Golden R., Pleus R. A 90-day drinking water study in mice to characterize early events in the cancer mode of action of 1,4-dioxane (submitted) Toxicology and Applied Pharmacology. 2020 doi: 10.1016/j.yrtph.2020.104819. [DOI] [PubMed] [Google Scholar]
- LaRocca J., Costa E., Sriram S., Hannas B.R., Johnson K.J. Short-term toxicogenomics as an alternative approach to chronic in vivo studies for derivation of points of departure: A case study in the rat with a triazole fungicide. Regul. Toxicol. Pharmacol. 2020;113 doi: 10.1016/j.yrtph.2020.104655. [DOI] [PubMed] [Google Scholar]
- Law R.H.P., Zhang Q., McGowan S., Buckle A.M., Silverman G.A., Wong W., Rosado C.J., Langendorf C.G., Pike R.N., Bird P.I., Whisstock J.C. An overview of the serpin superfamily. Genome Biol. 2006;7(5):216. doi: 10.1186/gb-2006-7-5-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Ross-Viola J.S., Shay N.F., Moore D.D., Ricketts M.L. Human cyp3a4 and murine cyp3a11 are regulated by equol and genistein via the pregnane x receptor in a species-specific manner. J. Nutr. 2009;139(5):898–904. doi: 10.3945/jn.108.103572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Boekschoten M.V., Wopereis S., Muller M., Kersten S. Comparative transcriptomic and metabolomic analysis of fenofibrate and fish oil treatments in mice. Physiol. Genomics. 2011;43(23):1307–1318. doi: 10.1152/physiolgenomics.00100.2011. [DOI] [PubMed] [Google Scholar]
- Luisier R., Lempiainen H., Scherbichler N., Braeuning A., Geissler M., Dubost V., Muller A., Scheer N., Chibout S.D., Hara H., Picard F., Theil D., Couttet P., Vitobello A., Grenet O., Grasl-Kraupp B., Ellinger-Ziegelbauer H., Thomson J.P., Meehan R.R., Elcombe C.R., Henderson C.J., Wolf C.R., Schwarz M., Moulin P., Terranova R., Moggs J.G. Phenobarbital induces cell cycle transcriptional responses in mouse liver humanized for constitutive androstane and pregnane x receptors. Toxicol. Sci. 2014;139(2):501–511. doi: 10.1093/toxsci/kfu038. [DOI] [PubMed] [Google Scholar]
- Maronpot, R.R., 2009. Biological basis of differential susceptibility to hepatocarcinogenesis among mouse strains. J Toxicol Pathol, 22(1): 11-33. Available from https://www.ncbi.nlm.nih.gov/pubmed/22271974. DOI 10.1293/tox.22.11. [DOI] [PMC free article] [PubMed]
- Merrick B.A., Bruno M.E. Genomic and proteomic profiling for biomarkers and signature profiles of toxicity. Curr Opin Mol Ther. 2004;6(6):600–607. https://www.ncbi.nlm.nih.gov/pubmed/15663324 Available from. [PubMed] [Google Scholar]
- Moffat, I., N. Chepelev, S. Labib, J. Bourdon-Lacombe, B. Kuo, J.K. Buick, F. Lemieux, A. Williams, S. Halappanavar, A. Malik, M. Luijten, J. Aubrecht, D.R. Hyduke, A.J. Fornace, Jr., C.D. Swartz, L. Recio and C.L. Yauk, 2015. Comparison of toxicogenomics and traditional approaches to inform mode of action and points of departure in human health risk assessment of benzo[a]pyrene in drinking water. Crit Rev Toxicol, 45(1): 1-43. Available from https://www.ncbi.nlm.nih.gov/pubmed/25605026. DOI 10.3109/10408444.2014.973934. [DOI] [PMC free article] [PubMed]
- Mulas, F., A. Li, D.H. Sherr and S. Monti, 2017. Network-based analysis of transcriptional profiles from chemical perturbations experiments. BMC Bioinformatics, 18(Suppl 5): 130. Available from https://www.ncbi.nlm.nih.gov/pubmed/28361664. DOI 10.1186/s12859-017-1536-9. [DOI] [PMC free article] [PubMed]
- NCI, 1978. National cancer institute. Bioassay of 1,4-dioxane for possible carcinogenicity cas no. 123-91-1 technical report series number 80. [PubMed]
- NICNAS . Australia department of health and ageing. Australia Department of Health and Ageing; Sydney, Australia: 1998. National industrial chemicals notification and assessment scheme. Priority existing chemical assessment reports: 1,4-dioxane. [Google Scholar]
- Nishimura, D., 2001. Biocarta. Biotech Software & Internet Report, 2(3): 117-120. Available from https://www.liebertpub.com/doi/abs/10.1089/152791601750294344. DOI 10.1089/152791601750294344.
- OEHHA, 2002. Office of environmental health hazard assessment, california environmental protection agency. Air toxics hot spots program. Risk assessment guidelines. Part ii. Technical support document for describing available cancer potency factors. . OEHHA, Sacramento, CA.
- Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27(1):29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J.R., Svoboda D.L., Tandon A., Patel S., Sedykh A., Mav D., Kuo B., Yauk C.L., Yang L., Thomas R.S., Gift J.S., Davis J.A., Olszyk L., Merrick B.A., Paules R.S., Parham F., Saddler T., Shah R.R., Auerbach S.S. Bmdexpress 2: Enhanced transcriptomic dose-response analysis workflow. Bioinformatics. 2019;35(10):1780–1782. doi: 10.1093/bioinformatics/bty878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard A.M., Judson R.S., Houck K.A., Grulke C.M., Volarath P., Thillainadarajah I., Yang C., Rathman J., Martin M.T., Wambaugh J.F., Knudsen T.B., Kancherla J., Mansouri K., Patlewicz G., Williams A.J., Little S.B., Crofton K.M., Thomas R.S. Toxcast chemical landscape: Paving the road to 21st century toxicology. Chem. Res. Toxicol. 2016;29(8):1225–1251. doi: 10.1021/acs.chemrestox.6b00135. [DOI] [PubMed] [Google Scholar]
- Schaefer C.F., Anthony K., Krupa S., Buchoff J., Day M., Hannay T., Buetow K.H. Pid: The pathway interaction database. Nucleic Acids Res. 2009;37(Database issue):D674–D679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickney J.A., Sager S.L., Clarkson J.R., Smith L.A., Locey B.J., Bock M.J., Hartung R., Olp S.F. An updated evaluation of the carcinogenic potential of 1,4-dioxane. Regul. Toxicol. Pharmacol. 2003;38(2):183–195. doi: 10.1016/s0273-2300(03)00090-4. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumi K., Ohashi K., Taminishi S., Takagi S., Utoh R., Yoshioka A., Shima M., Okano T. Effects on coagulation factor production following primary hepatomitogen-induced direct hyperplasia. World J. Gastroenterol. 2009;15(42):5307–5315. doi: 10.3748/wjg.15.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tice R.R., Austin C.P., Kavlock R.J., Bucher J.R. Improving the human hazard characterization of chemicals: A tox21 update. Environ. Health Perspect. 2013;121(7):756–765. doi: 10.1289/ehp.1205784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TNO/RIVM, 1999. Netherlands organization for applied scientific research (tno) and the national institute of public health and the environment (rivm). Risk assessment: 1, 4-dioxane. Einecs-no.: 204-661-8. . M. o. H. Chemical Substances Bureau, Spatial Planning and the Environment (VROM) (Ed.). Netherlands.
- U.S. EPA, 2005. Guidelines for carcinogen risk assessment. Epa/630/p-03/001f (https://www.Epa.Gov/sites/production/files/2013-09/documents/cancer_guidelines_final_3-25-05.Pdf). US EPA, Washington, DC.
- U.S. EPA, 2013. U.S. Environmental protection agency; toxicological review of 1,4-dioxane (with inhalation update) (cas no. 123-91-1) in support of summary information on the integrated risk information system (iris) (epa-635/r-11/003-f). . US EPA, Washington, DC.
- Väremo L., Nielsen J., Nookaew I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013;41(8):4378–4391. doi: 10.1093/nar/gkt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters M.D., Olden K., Tennant R.W. Toxicogenomic approach for assessing toxicant-related disease. Mutat. Res. 2003;544(2–3):415–424. doi: 10.1016/j.mrrev.2003.06.014. [DOI] [PubMed] [Google Scholar]
- Yamada T., Cohen S.M., Lake B.G. The mode of action for phenobarbital-induced rodent liver tumor formation is not relevant for humans: Recent studies with humanized mice. Toxicol. Sci. 2015;147(2):298–299. doi: 10.1093/toxsci/kfv186. [DOI] [PubMed] [Google Scholar]
- Yamate J., Tajima M., Kudow S., Sannai S. Background pathology in bdf1 mice allowed to live out their life-span. Lab Anim. 1990;24(4):332–340. doi: 10.1258/002367790780865976. [DOI] [PubMed] [Google Scholar]
- Yeakley J.M., Shepard P.J., Goyena D.E., VanSteenhouse H.C., McComb J.D., Seligmann B.E. A trichostatin a expression signature identified by tempo-seq targeted whole transcriptome profiling. PLOS ONE. 2017;12(5) doi: 10.1371/journal.pone.0178302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.