

Graphical abstract

Keywords: PPARγ, Ciglitazone, GW 9662, HepG2 cells, Systems biology
Highlights
-
•
Human (HepG2) cells were exposed to PPARγ ligands to induce systems-level effects.
-
•
Ciglitazone decreases HepG2 cell viability while GW 9662 had no effect.
-
•
Ciglitazone and GW 9662 increase neutral lipids as a function of concentration.
-
•
Cholesterol biosynthesis transcripts are affected by ciglitazone and GW 9662.
-
•
Ciglitazone alters lipid profiles but GW 9662 was similar to vehicle-exposed cells.
Abstract
Peroxisome proliferator-activated receptor γ (PPARγ) is a nuclear receptor that, upon activation by ligands, heterodimerizes with retinoid X receptor (RXR), binds to PPAR response elements (PPREs), and activates transcription of downstream genes. As PPARγ plays a central role in adipogenesis, fatty acid storage, and glucose metabolism, PPARγ-specific pharmaceuticals (e.g., thiazolidinediones) have been developed to treat Type II diabetes and obesity within human populations. However, to our knowledge, no prior studies have concurrently assessed the effects of PPARγ ligand exposure on genome-wide PPARγ binding as well as effects on the transcriptome and lipidome within human cells at biologically active, non-cytotoxic concentrations. In addition to quantifying concentration-dependent effects of ciglitazone (a reference PPARγ agonist) and GW 9662 (a reference PPARγ antagonist) on human hepatocarcinoma (HepG2) cell viability, PPARγ abundance in situ, and neutral lipids, HepG2 cells were exposed to either vehicle (0.1% DMSO), ciglitazone, or GW 9662 for up to 24 h, and then harvested for 1) chromatin immunoprecipitation-sequencing (ChIP-seq) to identify PPARγ-bound regions across the entire genome, 2) mRNA-sequencing (mRNA-seq) to identify potential impacts on the transcriptome, and 3) lipidomics to identify potential alterations in lipid profiles. Following exposure to ciglitazone and GW 9662, we found that PPARγ levels were not significantly different after 2–8 h of exposure. While ciglitazone and GW 9662 resulted in a concentration-dependent increase in neutral lipids, the magnitude and localization of PPARγ-bound regions across the genome (as identified by ChIP-seq) did not vary by treatment. However, mRNA-seq and lipidomics revealed that exposure of HepG2 cells to ciglitazone and GW 9662 resulted in significant, treatment-specific effects on the transcriptome and lipidome. Overall, our findings suggest that exposure of human cells to PPARγ ligands at biologically active, non-cytotoxic concentrations results in toxicity that may be driven by a combination of both PPARγ-dependent and PPARγ-independent mechanisms.
1. Introduction
Peroxisome proliferator-activated receptor γ (PPARγ) is a nuclear receptor and transcription factor that is activated by both endogenous and exogenous ligands (Issemann and Green, 1990, Tontonoz et al., 1994, Martin et al., 1998). Upon activation by ligand binding, PPARγ heterodimerizes with retinoid X receptor (RXR) and then binds to PPAR response elements (PPREs) as PPARγ:RXR heterodimers across the genome, resulting in transcription of genes involved in lipid/glucose metabolism and adipogenesis (Tontonoz et al., 1994, Martin et al., 1998, Chawla et al., 1994). Polyunsaturated fatty acids are endogenous, low-affinity PPARγ ligands and include prostaglandin PGJ2, linolenic acid, eicosapentaeonic acid, docosahexaenoic acid, and arachidonic acids (Forman et al., 1995, Kliewer et al., 1995, Nagy et al., 1998). Based on structural studies, the PPARγ binding site accommodates lipophilic carboxylic acids and other acidic ligands that can bind to polar residues, consistent with its proposed physiological role as a fatty acid sensor (Velkov, 2013). Exogenous PPARγ ligands include pharmaceuticals (e.g., thiazolidinediones) developed to treat Type II diabetes and obesity within human populations as well as environmental chemicals that have the ability to bind and activate PPARγ (Nolan et al., 1994, Lehmann et al., 1995, Hurst and Waxman, 2003, Riu et al., 2011, Wang et al., 2016).
Activation of PPARγ results in transcription of downstream genes that vary by tissue and cell type. Within liver and adipose tissue, genes transcribed are involved in lipid metabolism and adipogenesis. For example, liver tissue from PPARγ knockout mice are deficient in lipid transport-related transcripts such as fatty acid translocase (CD36) and low-density lipoprotein receptor (LDLR) (Gavrilova et al., 2003). Within adipose tissue, PPARγ induces expression of cytosolic glycerol 3-phosphate dehydrogenase (cGPDH), which converts glucose into glycerol 3-phosphate that is incorporated into triglycerides (Patsouris et al., 2004). Human meibomian gland epithelial cells exposed to rosiglitazone (a thiazolidinedione-based PPARγ agonist) results in an increase in lipid transport and biosynthesis-related transcripts including angiopoietin-related protein 4 (ANGPTL4), perilipin-2 (PLIN2), CD36, CCAAT/enhancer-binding protein alpha (CEBPA), elongation of very long chain fatty acids protein 4 (ELOVL4), and ELOVL7 (Kim et al., 2019).
As many PPARγ endogenous ligands are derived from dietary sources and PPARγ plays a role in maintaining lipid homeostasis, prior studies have utilized lipidomics to identify changes in lipid profiles upon activation in different tissue types and disease states. While PPARγ is mainly expressed in adipose tissue and regulates adipogenesis, it is also expressed in liver tissue, with elevated levels found in steatotic liver (Pettinelli and Videla, 2011). Lipidome analysis in patients with nonalcoholic fatty liver or nonalcoholic steatohepatitis revealed a significant increase in diacylglycerol and triacylglycerol content relative to healthy patients without liver disease (Puri et al., 2007). Within liver-specific PPARγ knockout mice, hepatic PPARγ was shown to play a major role in fatty acid uptake and monoacylglycerol pathway-mediated fatty acid esterification (Greenstein et al., 2017). Based on these results and other studies in the literature, one of the primary physiological roles for PPARγ within adipocytes and liver tissue includes lipid storage in the form of fatty acids and triglycerides (Wang et al., 2013).
While animal models (e.g., mice and rats) are critical tools for understanding the effects of exogenous PPARγ ligands within physiologically intact systems, the complementary use of human cell-based models provide direct translational relevance and enhance our understanding of PPARγ signaling at the cellular-level. Previous studies have investigated the role of PPARγ as a transcription factor and its effects on transcription and cell physiology. However, to our knowledge no prior studies have systematically used a systems-level approach to simultaneously assess the effects of PPARγ ligand exposure on genome-wide PPARγ binding and downstream effects on the transcriptome and lipidome within human cell-based models – an approach that is needed for determining whether genome-wide PPARγ binding has the potential to predict systems-level effects at higher levels of biological organization. Therefore, using ciglitazone and GW 9662 as a reference PPARγ agonist and antagonist, respectively, the overall objective of this study was to determine whether exposure of human cells to biologically active, non-cytotoxic concentrations of PPARγ ligands results in systems-level effects on PPARγ binding (using ChIP-seq), transcription (using mRNA-seq), and lipid composition (using lipidomics) that are consistent with the known mechanism of action for both compounds. Our overall hypothesis was that ciglitazone- and GW 9662-induced effects on cell viability and lipid homeostasis were strongly associated with PPARγ-mediated alterations to the cellular transcriptome. Specifically, we hypothesized that ciglitazone (a PPARγ agonist) and GW 9662 (a PPARγ antagonist) would increase and decrease the magnitude and extent of genome-wide PPARγ binding, respectively, relative to vehicle control-treated cells, leading to opposing effects on cellular transcription and physiology.
For this study, we relied on hepatocellular carcinoma (HepG2) cells, as these cells express baseline levels of PPARγ and are widely used as models to understand DNA damage (Yang et al., 1999), regulation of drug metabolizing enzymes (Wilkening et al., 2003), and lipoprotein metabolism (Meex et al., 2011). By analyzing data generated at the genomic-, transcriptomic-, and lipidomic-level under the same conditions and within the same model system, we can begin to understand the relationship and potential association of alterations at each of these levels of biological organization following exposure to reference PPARγ ligands. Moreover, through careful identification of biologically active concentrations in the absence of cytotoxicity, this study enabled us to eliminate the potential for false negative findings resulting from limited to no chemical uptake while, at the same, providing the foundation for exploring whether systems-level effects induced by exposure to ciglitazone and GW 9662 may be driven by a combination of both PPARγ-dependent and PPARγ-independent mechanisms.
2. Materials and methods
2.1. Chemicals
Ciglitazone (>99.4% purity) was purchased from Tocris Biosciences (Bristol, UK) and GW 9662 (>98% purity) was purchased from Enzo Life Sciences (Farmingdale, NY USA). For both chemicals, stock solutions were prepared in high-performance liquid chromatography (HPLC)-grade dimethyl sulfoxide (DMSO) and stored in 2-mL amber glass vials with polytetrafluoroethylene-lined caps. Working solutions were prepared by spiking stock solutions into sterile cell culture media immediately prior to each experiment, resulting in 0.1% DMSO within all treatment groups.
2.2. PPARγ ligand exposures and cell viability assays
HepG2 cells were purchased from American Type Culture Collection (Manassas, VA, USA) and grown within T75 cell culture flasks (Millipore Sigma, St. Louis, MO, USA) containing 15 mL of Eagle’s Minimum Essential Medium supplemented with 10% fetal bovine serum (FBS) (ATCC, Manassas, VA, USA) at 37 °C and 5% CO2. Media was changed within each flask every other day and cells were split every four days using 0.25% Trypsin/0.53 mM EDTA (ATCC, Manassas, VA, USA) after reaching ~70–90% confluency.
HepG2 cells were plated at a concentration of 2 × 104 cells per well in a clear, polystyrene 96-well plate (ThermoFisher, Waltham, MA, USA) and allowed to adhere overnight. Media was removed and replaced with 200 µL media spiked with either vehicle (0.1% DMSO), ciglitazone (52, 65, 82, 102, 128, 160, or 200 µM), or GW 9662 (41, 51, 64, 80, or 100 µM) and incubated at 37 °C and 5% CO2 for 24 h (4 replicate wells per treatment). At the end of the exposure duration, treatment solution was removed and replaced with 100 µL of clean cell culture media and 20 µL of CellTiter-Blue (Promega, Madison, WI, USA), and then allowed to incubate for 2 h at 37 °C and 5% CO2. Fluorescence was then quantified using a GloMax Multi + Detection System (Promega, Madison, WI, USA).
2.3. PPARγ immunohistochemistry
To confirm the presence of PPARγ protein in situ across treatments, cells were exposed to either vehicle (0.1% DMSO), ciglitazone, or GW 9662 as described above for either 2, 4, 6, 8, or 24 h. At exposure termination, cells were fixed with 4% formaldehyde at room temperature for 10 min. Cells were then rinsed three times with 1X phosphate-buffered saline (PBS) and incubated in blocking buffer [1X PBS + 0.1% Tween-20 (PBST), 2 mg/mL bovine serum albumin, and 2% sheep serum] at room temperature for 1 h by shaking gently. Blocking buffer was then replaced with a 1:100 dilution of a human PPARγ-specific antibody (E-8, sc-7273; Santa Cruz Biotechnology, Dallas, TX USA) diluted in blocking buffer and allowed to incubate overnight at 4 °C. Cells were then incubated with a 1:500 dilution of Alexa Fluor 488-conjugated goat anti-mouse IgG1 antibody (ThermoFisher Scientific, Waltman, MA USA) overnight at 4 °C. Cells were then counterstained with a 1:3 solution of DAPI Fluoromount-G (Southern Biotechnology, GA) for 5 min, rinsed with 1X PBS three times, and then imaged (at 10X magnification) and analyzed using our ImageXpress Micro XLS Widefield High-Content Screening System (Molecular Devices, Sunnyvale, CA USA).
2.4. Oil Red O staining
To determine whether exposure to ciglitazone or GW 9662 affected neutral lipid abundance, HepG2 cells were stained for neutral lipids using Oil Red O (ORO) (Sigma-Aldrich, St. Louis, MO, USA) following exposure to vehicle (0.1% DMSO), ciglitazone, or GW 9662 as described above. Briefly, cells were fixed using 4% paraformaldehyde for 20 min at room temperature. Cells were then rinsed with 60% isopropanol and stained with ORO working solution (1.8 mg ORO per 1 mL 60% isopropanol) for 10 min at room temperature. Cells were then rinsed four times with molecular biology-grade water for 5 min at room temperature. After the final wash, cells were counterstained with a 1:3 solution of DAPI Fluoromount-G (Southern Biotech) for 5 min at room temperature. Cells were washed three more times with molecular biology-grade water and imaged (at 10X magnification) and analyzed using our ImageXpress Micro XLS Widefield High-Content Screening System (Molecular Devices, Sunnyvale, CA USA).
2.5. Chromatin immunoprecipitation-sequencing (ChIP-seq)
HepG2 cells were plated and exposed for 8 h to either vehicle (0.1% DMSO), 128 μM ciglitazone, or 100 μM GW 9662 (32 wells pooled per replicate; 3 replicates per treatment). Cells were fixed and lysed using the truChIP Chromatin Shearing Kit with Formaldehyde (Covaris, Woburn, MA USA), and chromatin was then sheared using a Covaris S220 Focused-Ultrasonicator (Peak Incident Power: 175 W, Duty Factor: 10%, Cycles per Burst: 200, Time: 500 s, Temperature: 3–6 °C). An aliquot of sheared chromatin was treated with 10 mg/mL RNase A and 10 mg/mL Proteinase K to reverse crosslinks, confirm shearing efficiency, and confirm DNA quantity and quality using a Qubit 4.0 Fluorometer and 2100 Bioanalyzer system, respectively. After confirming that chromatin was sheared to the optimal size range (150–700 bp), sheared chromatin was processed for immunoprecipitation using an Imprint Chromatin Immunoprecipitation Kit (Sigma-Aldrich) and ChIP-grade, human PPARγ-specific antibody (sc-7273X) (Santa Cruz Biotechnology, Dallas, TX USA).
An EpiNext ChIP-Seq High Sensitivity Kit (Epigentek, Farmingdale, NY USA) was then used to prepare sequencing libraries per the manufacturers’ instructions; treatment replicates were indexed using EpiNext NGS Barcodes (Epigentek, Farmingdale, NY USA). Library quantity and quality were confirmed using a Qubit 4.0 Fluorometer and Bioanalyzer 2100 system, respectively. Libraries (9 total) were then pooled, diluted to a concentration of 1.3 pM (with 1% PhiX control) and paired-end (2 × 150) sequenced on our Illumina MiniSeq Sequencing System (San Diego, California, USA) using a 300-cycle High-Output Reagent Kit. Raw Illumina (fastq.qz) sequencing files (9 total) are available via NCBI’s BioProject database under BioProject ID PRJNA681430, and a summary of sequencing run metrics are provided in Table S1.
After completion of the sequencing run, reads passing filter were aligned to the human genome (GRCh37/hg19) using a BWA Aligner application within Illumina’s BaseSpace to generate BAM files for each treatment replicate. BAM files were downloaded from BaseSpace and then uploaded into Galaxy (usegalaxy.com). Within Galaxy, MACS2 callpeak was run on pooled treatments to identify significant narrow peaks (i.e., transcription factor binding sites) and to generate BED files. All defaults were used for each MACS2 callpeak run, including a q-value = 0.05 as a cutoff for peak detection. BED files were then used to run ChIPseeker within Galaxy to annotate identified peaks using GRCh37/hg19 as a reference genome, and TFmotifView (http://bardet.u-strasbg.fr/tfmotifview/) (Leporcq et al., 2020) was used to identify PPARG-RXRA-specific motifs (i.e., PPREs) within ChIP-seq peaks.
2.6. mRNA-sequencing
HepG2 cells were plated and exposed as described above to either vehicle (0.1% DMSO), 128 μM ciglitazone, or 100 μM GW 9662 (2 wells pooled per replicate; 3 replicates per treatment). After 24 h, total RNA from each replicate was isolated using a Promega SV Total RNA Isolation System (Promega, Madison, WI, USA) following the manufacturer’s protocol. RNA quantity and quality were confirmed using a Qubit 4.0 Fluorometer and Bioanalyzer 2100 system, respectively. Based on sample-specific Bioanalyzer traces, the RNA Integrity Number (RIN) was >9 for all RNA samples used for library preparations.
Library preps were performed using a QuantSeq 3′ mRNA-Seq Library Prep Kit FWD for Illumina (Lexogen, Vienna, Austria) and indexed by treatment replicate per manufacturer’s instructions. Library quantity and quality were confirmed using a Qubit 4.0 Fluorometer and 2100 BioAnalyzer system, respectively. Raw Illumina (fastq.qz) sequencing files (9 total) are available via NCBI’s BioProject database under BioProject ID PRJNA681430, and a summary of sequencing run metrics are provided in Table S2. All nine raw and indexed Illumina (fastq.gz) sequencing files were downloaded from Illumina’s BaseSpace and uploaded to Bluebee’s genomics analysis platform (www.bluebee.com) to align reads against the human genome (GRCh38/hg38). After combining treatment replicate files, a DESeq2 application within Bluebee (Lexogen Quantseq DE1.2) was used to identify significant treatment-related effects on transcript abundance (relative to vehicle) based on a false discovery rate (FDR) p-adjusted value ≤ 0.05. Significantly affected transcripts were imported into the Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.8 for Gene Ontology (GO) enrichment analysis. Individual transcripts from significant GO terms (Benjamini score ≤ 0.05) were consolidated into a list of unique transcripts.
2.7. Lipidomics
Cells were plated and exposed as described above. Cells were exposed to either vehicle (0.1% DMSO), 128 μM ciglitazone, or 100 μM GW 9662 (6 wells pooled per replicate; 4 replicates per treatment). After 24 h, the exposure solution was removed, cells were rinsed with Hank’s Balance Salt Solution warmed to 37 °C, replaced with 50 μL of 100% methanol, and incubated at −80 °C for 60 min. LC-MS-based lipidomics analysis was performed as described previously with minor modifications (Reddam et al., 2019). Briefly, analysis was performed on a G2-XS quadrupole time-of-flight mass spectrometer (Waters Corp., Milford, MA USA) coupled to an H-class UPLC system (Waters Corp., Milford, MA USA). Separations were carried out on a CSH C18 column (2.1 × 100 mm, 1.7 µM) (Waters Corp., Milford, MA USA). The mobile phases were (A) 60:40 acetonitrile:water with 10 mM ammonium formate and 0.1% formic acid and (B) 90:10 isopropanol:acetonitrile with 10 mM ammonium formate and 0.1% formic acid. The flow rate was 400 µL/min and the column was held at 65 °C. The injection volume was 4 µL. The gradient was as follows: 0 min, 15% B; 2 min, 30% B; 3 min, 50% B; 10 min, 55% B; 14 min, 80% B; 16 min, 100% B; 20 min 100% B; 20.5 min, 15% B.
The MS was operated in positive ion mode (50 to 1600 m/z) with a 100-ms scan time. Source and desolvation temperatures were 150 °C and 600 °C, respectively. MS/MS was acquired in a data-dependent fashion. Desolvation gas was set to 1100 L/h and cone gas to 150 L/h. All gases were nitrogen except the collision gas, which was argon. Capillary voltage was 1 kV. A quality control sample, generated by pooling equal aliquots of each sample, was analyzed every 4–5 injections to monitor system stability and performance. Samples were analyzed in random order. Leucine enkephalin was infused and used for mass correction.
Untargeted data processing (peak picking, alignment, deconvolution, integration, normalization, and spectral matching) was performed in Progenesis Qi software (Nonlinear Dynamics). Data were normalized to total ion abundance. Features with a CV>30% were removed. To aid in the identification of features that belong to the same metabolite, features were assigned a cluster ID using RAMClust (Broeckling et al., 2014). An extension of the metabolomics standard initiative guidelines was used to assign annotation level confidence (Sumner et al., 2007, Schymanski et al., 2014). Annotation level 1 indicates an MS and MS/MS match or MS and retention time match to an in-house database generated with authentic standards. Level 2a indicates an MS and MS/MS match to an external database. Level 2b indicates an MS and MS/MS match to the Lipiblast in-silico database (Kind et al., 2013) or an MS match and diagnostic evidence, such as the dominant presence of an m/z 85 fragment ion for acylcarnitines. Level 3 indicates an MS match, though some additional evidence is required, such as adducts were detected to sufficiently deduce the neutral mass or the retention time is in the expected region. Several mass spectral metabolite databases were searched against including Metlin, Mass Bank of North America, and an in-house database.
2.8. Statistical analyses
For cell viability, ORO staining, and immunohistochemistry data, a general linear model (GLM) analysis of variance (ANOVA) (α = 0.05) was performed using SPSS Statistics 27 (IBM, Armonk, NY USA), as data did not meet the equal variance assumption for non-GLM ANOVAs. Treatment groups were compared with vehicle controls using pair-wise Tukey based multiple comparisons of least square means to identify significant treatment-specific differences.
3. Results
3.1. Ciglitazone decreases HepG2 cell viability at concentrations > 128 µM while GW 9662 had no effect on cell viability up to the limit of solubility
Relative to HepG2 cells exposed to vehicle (0.1% DMSO) for 24 h, HepG2 cells exposed to 128 µM ciglitazone for 24 h resulted in a slight (albeit non-significant) decrease in fluorescence as measured by CellTiter Blue Assay (~70% cell viability), whereas exposure to ciglitazone above 128 µM resulted in a significant increase in cell death (Fig. 1). HepG2 cells exposed to GW 9662 for 24 h did not result in decreased fluorescence up to its limit of solubility (100 µM) (Fig. 1). Based on cell viability data, the maximum tolerated concentrations (MTCs) for ciglitazone and GW 9662 were 128 µM and 100 µM, respectively. Therefore, samples for ChIP-seq, mRNA-seq, and lipidomics were generated following exposure to these MTCs.
Fig. 1.

Mean (±standard deviation) of fluorescence of HepG2 cells exposed to vehicle (0.1% DMSO) (black circle), 50–200 µM ciglitazone (red triangles), or 41–100 µM GW 9662 (blue squares) for 24 h as measured by a CellTiter Blue assay. Asterisk (*) indicates a significant difference (p < 0.05) in cell viability relative to vehicle-exposed cells. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. PPARγ levels in situ are not affected after 8 h of exposure to ciglitazone and GW 9662
A human PPARγ-specific antibody was used to quantify PPARγ protein levels within exposed cells. Exposure to ciglitazone (52–128 µM) or GW 9662 (41–80 µM) for 24 h did not affect PPARγ levels detected in situ relative to vehicle-exposed cells (Fig. 2A). However, exposure to 100 µM GW 9662 for 24 h resulted in a statistically significant decrease in PPARγ levels (Fig. 2A). PPARγ levels were also measured at 2, 4, 6, and 8 h after exposure to either 128 µM ciglitazone or 100 µM GW 9662. For both treatment groups, PPARγ levels were not significantly different from 2 to 8 h following initiation of exposure (Fig. 2B).
Fig. 2.

Mean (±standard deviation) of PPARγ immunofluorescence area divided by DAPI stained area of HepG2 cells exposed to vehicle (0.1% DMSO), 52–128 µM ciglitazone, or 41–100 µM GW 9662(A) for 24 h. Mean (±standard deviation) of PPARγ immunofluorescence area divided by DAPI stained area of HepG2 cells exposed to vehicle, 128 µM ciglitazone, or 100 µM GW 9662 for 2, 4, 6, 8, or 24 h (B).
3.3. Ciglitazone and GW 9662 increases neutral lipids in a concentration-dependent manner
ORO staining revealed a statistically significant increase in neutral lipids in HepG2 cells exposed to 128 µM ciglitazone relative to vehicle-exposed cells (Fig. 3A). In order to account for differences in neutral lipid staining as a function of differences in cell number, ORO staining was normalized to DAPI staining within each well. While there was a slight concentration-dependent increase in DAPI-normalized ORO staining in cells exposed to GW 9662, these results were not statistically significant up to the highest GW 9662 concentration tested. While concentrations of ciglitazone and GW 9662 were not cytotoxic, fixation of cells for ORO staining also revealed that exposed cells were more spherical in shape as opposed to a normal, epithelial-like shape observed following exposure to vehicle (Fig. 3B–G).
Fig. 3.
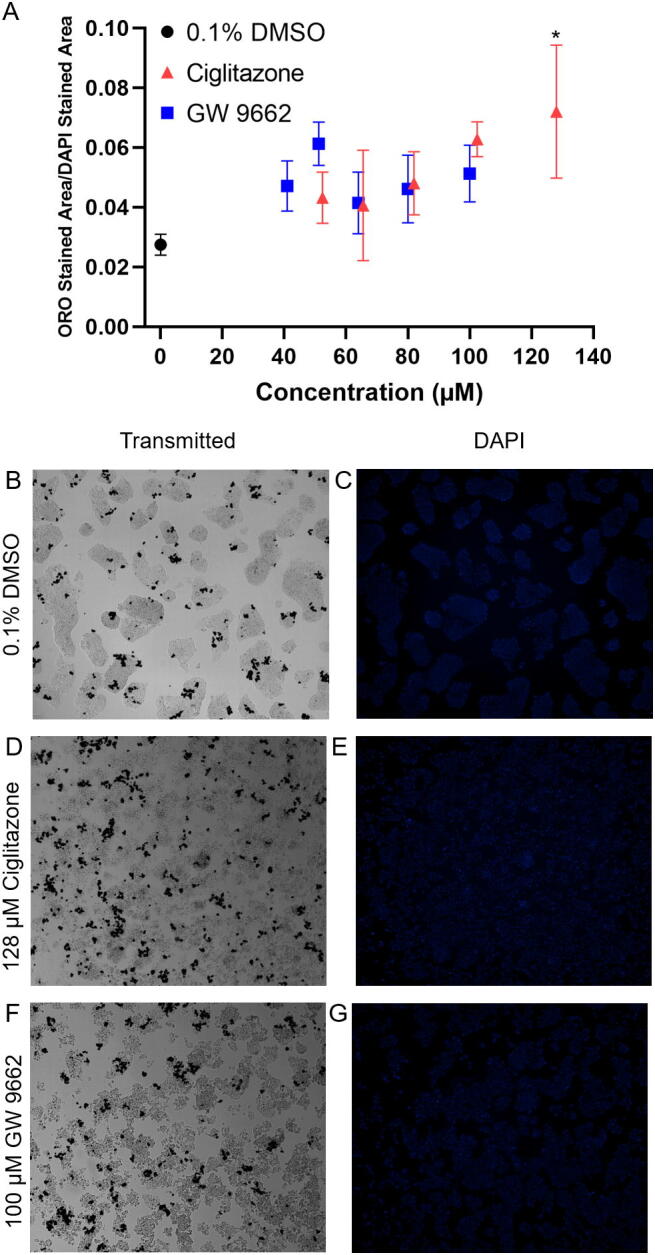
Mean (±standard deviation) of HepG2 cells stained with Oil Red O neutral lipid stain and normalized to DAPI staining after exposure to vehicle (0.1% DMSO), 52–128 µM ciglitazone, or 41–100 µM GW 9662 (A). Representative images taken under transmitted light for vehicle (0.1% DMSO) (B), 128 µM ciglitazone (D), or 100 µM GW 9662 (F), and under DAPI filter for vehicle (C), 128 µM ciglitazone (E), or 100 µM GW 9662 (G). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.4. PPARγ binding occurs genome-wide and the majority of PPARγ-bound PPARG:RXRA motifs are found within distal intergenic or intron regions
Cells were only exposed for 8 h since we hypothesized that binding of PPARγ to PPREs would precede effects on the transcriptome and lipidome at 24 h. Using MACS2 analysis, there were a total of 125, 145, and 89 ChIP peaks identified across the genome within HepG2 cells exposed for 8 h to vehicle (0.1% DMSO), ciglitazone, or GW 9662, respectively (Fig. 4A; Tables S3-S5). Each ChIP peak represented a region where reads were significantly abundant (or stacked) relative to a reference genome, indicating that these reads were derived from genomic DNA fragments pulled down following immunoprecipitation. Within MACS2-identified ChIP peaks, TFmotifView then revealed that 29, 39, and 25 PPARG-RXRA (PPRE) motifs were identified across the genome within HepG2 cells exposed for 24 h to vehicle (0.1% DMSO), ciglitazone, or GW 9662, respectively (Fig. 4B and C; Tables S6-S8). While PPARG-RXRA (PPRE) motifs were distributed among distal intergenic regions, introns, and promoter regions (Fig. 4D; Tables S6-S8), the majority of PPARγ-bound PPARG:RXRA motifs were found within distal intergenic or intron regions.
Fig. 4.

Number of ChIP peaks by chromosome number identified from ChIP-Seq MACS analysis (A). Number of ChIP peaks by chromosome number containing PPARG-RXRA (PPRE) motifs after analysis by TFmotifView (B). Venn diagram showing overlap of ChIP peaks between vehicle (0.1% DMSO), 128 µM ciglitazone, and 100 µM GW 9662 treatment (C). Bolded text indicates the number of peaks identified through MACS analysis. Text within parentheses indicate the number of PPARG-RXRA (PPRE) motifs within each treatment group. Distribution of annotated motif locations by treatment group (D).
3.5. Transcripts involved in cholesterol biosynthesis are oppositely affected by ciglitazone and GW 9662 exposure
Exposure of HepG2 cells to 128 µM ciglitazone resulted in significant effects on the abundance of 4146 transcripts (Fig. 5A; Table S9), while exposure to 100 µM GW 9662 resulted in significant effects on the abundance of 3704 transcripts (Fig. 5B; Table S10). Interestingly, a heat map based on significantly affected transcripts revealed that the transcriptome within ciglitazone- and GW 9662-exposed cells were similar, with only a subset of transcripts that were oppositely affected by ciglitazone vs. GW 9662 (Fig. 5C). To determine which biological processes were significantly affected due to ciglitazone or GW 9662 exposure, differentially affected transcripts relative to vehicle-exposed cells were analyzed using DAVID to identify top biological processes. To identify biological processes that were oppositely affected by ciglitazone and GW 9662 exposure, significantly affected transcripts were sorted by fold-change for each treatment and then compared to identify transcripts that were either 1) decreased by ciglitazone exposure and increased by GW 9662 exposure (Fig. 5D) or 2) increased by ciglitazone exposure and decreased by GW 9662 exposure (Fig. 5E). DAVID gene ontology analysis identified nine transcripts involved in cholesterol biosynthesis that were decreased by ciglitazone exposure and increased by GW9662 exposure (Fig. 5F and G; Table S11-S12).
Fig. 5.

Volcano plots indicating number of significantly affected transcripts for ciglitazone (A) or GW 9662 (B) relative to vehicle-exposed cells. Heat map of significantly affected transcripts organized by hierarchical clustering using Euclidean distance and complete linkage method (C). Venn diagrams showing overlap in transcripts between ciglitazone and GW 9662 (D, E). Gene ontology analysis of biological processes identified by DAVID based on decreased transcripts following ciglitazone exposure and increased transcripts following GW 9662 exposure or increased transcripts following ciglitazone exposure and decreased transcripts following GW 9662 exposure (E). Transcripts within cholesterol biosynthetic process plotted by log2(fold change) in GW 9662-exposed cells along the Y-axis, and log2(fold change) in ciglitazone-exposed cells along the x-axis (F). MVD: Mevalonate Diphosphate Decarboxylase, NSDHL: Sterol-4-alpha-caroxylate 3-dehydrogenase (NAD(P) dependent steroid dehydrogenase-like), LBR: Lamin B receptor, DHCR7: 7-Dehydrocholesterol Reductase, MSMO1: Methylsterol Monooxygenase 1, FDFT1: Farnesyl-Diphosphate Farnesyltransferase 1, SQLE: Squalene Epoxidase, LSS: Lanosterol synthase, FDPS: Farnesyl pyrophosphate synthase.
3.6. Ciglitazone alters lipid composition across several lipid classes while the lipid profile of GW 9662-exposed cells was more similar to vehicle-exposed cells
Lipid profile analysis revealed that, relative to vehicle-exposed cells, 1075 lipids were significantly altered after 24 h of exposure to ciglitazone whereas 498 lipids were significantly altered after exposure to GW 9662 (Fig. 6A; Table S13). The total abundance of lipids in ciglitazone- or GW 9662-exposed cells were not significantly altered compared to vehicle-exposed cells (Fig. 6C; Table S13). However, the relative composition of lipids within exposed cells were different relative to vehicle-exposed cells (Fig. 6B; Table S13). Within ciglitazone-exposed cells, there was a significant increase in lipids from the acylcarnitine, ceramide, lyso phosphatidylcholine (PC), spermidine, sterol, sterol ester, and triglyceride classes, and a significant decrease in lipids from the choline, lyso phosphatidylethanolamine (PE), PC, PE, and sphingomyelin (SM) classes. Although the lipid profile of GW 9662-exposed cells was more similar to vehicle-exposed cells (Fig. 6A), there was a significant increase in lyso PE lipids and significant decrease in SM and spermidine lipid classes. Cholesterol and two different sterol esters (16.0 and 16.3) were significantly increased following exposure to ciglitazone, whereas sterol ester (16.0) was significantly increased following exposure to GW 9662 (Fig. 6D).
Fig. 6.

Heat map of significantly altered lipids (data shown as z-scores) following exposure to 128 µM ciglitazone or 100 µM GW 9662 relative to vehicle-exposed cells (A). Relative lipid abundance divided into lipid class and treatment (B). Asterisk (*) indicates a significant difference (p < 0.05) in lipid abundance relative to vehicle. Sum of relative lipid abundance by treatment (C). Relative abundance of cholesterol and sterol esters following exposure to vehicle (0.1% DMSO), 128 µM ciglitazone, or 100 µM GW 9662 (D). PC: Phosphatidylcholine, PE: Phosphatidylethanolamine, PS: Phosphatidylserine, SM: Sphingomyelin, TG: Triacylglyceride.
4. Discussion
A prior study that exposed HepG2 cells to ciglitazone estimated an IC50 value of 46 µM based on a 16-h exposure and cell viability as an endpoint using a 3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide (MTT) assay (Guo et al., 2006), whereas our study identified 128 µM ciglitazone as the MTC following a 24-h exposure – differences that may be attributable to the type of assay (CellTiter-Blue vs. MTT) used to quantify cell viability. Within the same study, the IC50 value of ciglitazone relative to other thiazolidinedione compounds was the second lowest (Guo et al., 2006), indicating that higher nominal concentrations required within our study were not driven by a lack of potency of ciglitazone. To our knowledge, no studies have been conducted to determine the IC50 or MTC for GW 9662 within HepG2 cells. Within other human cell-based models, the IC50 for ciglitazone and GW 9662 based on cell viability as an endpoint ranged from 12 to 230 µM and 20–30 µM, respectively (Eibl et al., 2001, Strakova et al., 2004, Strakova et al., 2005, Vignati et al., 2006, Seargent et al., 2004).
While HepG2 cells express PPARγ, expression within liver tissue is lower relative to adipocytes (Elbrecht et al., 1996), suggesting that lower expression may account for the lower sensitivity of HepG2 cells to PPARγ ligands. As there are studies showing that PPARγ regulates adipogenesis in 3 T3-L1 cells through a positive feedback loop (Wakabayashi et al., 2009), we wanted to determine if ciglitazone or GW 9662 also regulated the accumulation of neutral lipids in this manner within HepG2 cells. Despite the higher nominal concentrations of ciglitazone and GW 9662 used within our study, there was a concentration-dependent increase in neutral lipids following exposure to either compound – a finding that may be PPARγ-independent since 1) we observed a similar response even though both compounds have opposing mechanisms of action (PPARγ agonist vs. antagonist) and 2) we did not observe a concentration-dependent effect on PPARγ protein levels in situ.
While previous PPARγ-specific ChIP-seq studies have been utilized to map PPARγ binding during adipogenesis (Nielsen et al., 2008) or understand differences in cell-specific PPARγ binding (Lefterova et al., 2010), to our knowledge our study was the first to utilize ChIP-seq to correlate phenotypic changes after PPARγ agonist exposure to genome binding within human cell-based models. As expected, PPARγ-bound DNA fragments sequenced after ChIP revealed binding of PPARγ across the genome. While we expected a strong increase in ChIP peaks following exposure to ciglitazone (a PPARγ agonist), the number of ChIP peaks within ciglitazone-exposed cells were similar relative to vehicle-exposed cells. As GW 9662 is a PPARγ antagonist that irreversibly binds to the ligand binding pocket of PPARγ (Leesnitzer et al., 2002), we expected GW 9662 to result in a decrease in ChIP peaks relative to vehicle-exposed cells. However, contrary to our hypothesis, we detected numerous ChIP peaks across the genome within GW 9662-exposed cells. Indeed, chromosome-specific effects were not detected following exposure to vehicle, ciglitazone, nor GW 9662, as the distribution of ChIP peaks were similar across all three treatment groups. Likewise, there was no clear treatment-dependent pattern of PPARG:RXRA (PPRE) motifs, and the majority of PPARG:RXRA (PPRE) motifs identified were located within intron and distal intergenic regions rather than within promoter regions that regulate transcription, a finding that is consistent with prior PPARγ-specific ChIP-seq studies (Lefterova et al., 2008) and other studies proposing that transcription factor binding to distal regions directs DNA looping as well as recruits coactivators and chromatin remodelers to the transcription start site of target genes (West and Fraser, 2005). Overall, these data suggest that the phenotypic effects of ciglitazone and GW 9662 on HepG2 cells were not associated with PPARγ binding to PPRE motifs within promoter regions across the genome.
Consistent with our finding that genome-wide PPARγ binding was similar following exposure to ciglitazone or GW 9662, both ciglitazone and GW 9662 also induced a similar magnitude of effect on transcripts that were either increased or decreased. While we expected ciglitazone and GW 9662 to act as a PPARγ agonist and antagonist, respectively, we found that the majority of transcripts were similarly affected by both compounds, suggesting that these overlapping transcriptional responses may be PPARγ-independent. To identify transcriptional responses that may be driven by PPARγ activation or inactivation, we identified transcripts that were oppositely affected by ciglitazone and GW 9662 exposure. Based on this analysis, cholesterol biosynthesis was the most significant pathway identified within the group of transcripts that were decreased and increased by ciglitazone and GW 9662, respectively. To our knowledge, our study was the first to identify cholesterol biosynthesis as a significantly affected process in human cell-based models after ciglitazone or GW 9662 exposure.
Interestingly, lipidomics revealed a unique lipid profile within ciglitazone-exposed cells compared to GW 9662- or vehicle-exposed cells, suggesting that ciglitazone exposure resulted in significant effects on the lipidome. Moreover, while total lipid abundance among all three treatment groups was not different, the abundance of certain lipid-specific classes was altered within ciglitazone- and GW 9662-exposed cells. Based on our mRNA-seq data, we expected to detect a decrease in cholesterol abundance, as cholesterol biosynthesis was predicted to be significantly decreased due to ciglitazone exposure. However, in both ciglitazone- and GW 9662-exposed cells, there was a significant increase in cholesterol and other sterol lipids relative to vehicle-exposed cells – differences that may have been attributable to the timing of chemically-induced effects on the transcriptome vs. lipidome. As samples for mRNA-seq and lipidomics were both derived from cells that were exposed for 24 h, it is possible that effects on the transcriptome observed at 24 h may have been associated with effects on the lipidome before or after 24 h of exposure. To our knowledge, our study was the first to associate transcriptional responses to lipidomic responses after exposure to a PPARγ agonist or antagonist within human cell-based models.
While ciglitazone and GW 9662 are marketed and used as a PPARγ agonist and antagonist, respectively, it is unclear whether the effects observed within our study were due to direct PPARγ modulation and/or off-target effects. While the mechanisms of PPARγ activation have been well studied and compounds within the thiazolidinedione class have been widely used as reference chemicals for PPARγ activation, previous studies have identified PPARγ-independent mechanisms of ciglitazone such as activation of MAP kinase cascades in human preadipocytes (Lennon et al., 2002) as well as regulation of cell cycle proteins in human prostate cancer cells (Lyles et al., 2009). Although thiazolidinediones have previously been used to treat Type 2 diabetes mellitus, these compounds were subsequently removed from the market due to adverse effects suggesting that other targets may be present (Nesto et al., 2003, Nissen and Wolski, 2007, Lewis et al., 2011). Our study relied on the MTC of ciglitazone based on cell viability, and the maximum concentration of GW 9662 based on its solubility in DMSO. Based on ORO staining, the concentrations of ciglitazone and GW 9662 used resulted in phenotypic effects on neutral lipids and cell morphology – effects which were not detected based on cell viability alone. Gene ontology analysis of mRNA-seq data for ciglitazone identified apoptotic processes, suggesting that ciglitazone may have resulted in systemic toxicity and, as such, PPARγ-driven effects may have been masked by off-target effects of ciglitazone within HepG2 cells.
In conclusion, our study systematically deployed multiple large-scale, high-resolution approaches to enhance our understanding of the effects of PPARγ ligand exposure within human cells at the systems-level. Moreover, our study was the first to 1) utilize ChIP-seq to correlate phenotypic changes after PPARγ agonist exposure to genome binding within human cell-based models; 2) identify cholesterol biosynthesis as a significantly affected process in human cell-based models after ciglitazone or GW 9662 exposure; and 3) associate transcriptional responses to lipidomic responses after exposure to a PPARγ agonist or antagonist within human cell-based models. Specifically, we found that 1) ciglitazone decreased HepG2 cell viability at concentrations >128 µM while GW 9662 had no effect on cell viability up to the limit of solubility; 2) ciglitazone and GW 9662 increased neutral lipids in a concentration-dependent manner; 3) PPARγ binding occurred genome-wide and the majority of PPARγ-bound PPARG:RXRA motifs were found within distal intergenic or intron regions; 4) transcripts involved in cholesterol biosynthesis were oppositely affected by ciglitazone and GW 9662 exposure; and 5) ciglitazone altered lipid composition across several lipid classes while the lipid profile of GW 9662-exposed cells was more similar to vehicle-exposed cells. Overall, our data suggest that exposure of human cells to PPARγ ligands at biologically active, non-cytotoxic concentrations results in effects on the transcriptome and lipidome that may be driven by a combination of both PPARγ-dependent and PPARγ-independent mechanisms. As such, our findings demonstrate that systems-level responses to PPARγ ligand exposure within human cells are complex and concentration-dependent, providing the foundation for continuing to investigate the specificity of PPARγ ligands within intact cells as well as discover novel mechanisms of action for reference PPARγ ligands such as ciglitazone and GW 9662.
Funding
This work was supported by a NRSA T32 Training Program [T32ES018827] to V.C, and a National Institutes of Health grant [R01ES027576] and USDA National Institute of Food and Agriculture Hatch Project [1009609] to D.C.V.
CRediT authorship contribution statement
Vanessa Cheng: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Writing - original draft, Writing - review & editing, Visualization. Aalekhya Reddam: Methodology, Investigation. Anil Bhatia: Methodology, Validation, Formal analysis, Investigation. Manhoi Hur: Methodology, Validation, Formal analysis, Investigation. Jay S. Kirkwood: Conceptualization, Resources, Writing - original draft, Supervision, Project administration. David C. Volz: Conceptualization, Resources, Writing - review & editing, Supervision, Project administration, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.crtox.2021.03.003.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Broeckling C.D., Afsar F.A., Neumann S., Ben-Hur A., Prenni J.E. RAMClust: a novel feature clustering method enables spectral-matching-based annotation for metabolomics data. Anal. Chem. 2014;86(14):6812–6817. doi: 10.1021/ac501530d. [DOI] [PubMed] [Google Scholar]
- Chawla A., Schwarz E.J., Dimaculangan D.D., Lazar M.A. Peroxisome proliferator-activated receptor (PPAR) gamma: adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology. 1994;135:798–800. doi: 10.1210/en.135.2.798. [DOI] [PubMed] [Google Scholar]
- Eibl G., Wente M.N., Reber H.A., Hines O.J. Peroxisome proliferator-activated receptor γ induces pancreatic cancer cell apoptosis. Biochem. Biophys. Res. Commun. 2001;287(2):522–529. doi: 10.1006/bbrc.2001.5619. [DOI] [PubMed] [Google Scholar]
- Elbrecht A., Chen Y., Cullinan C.A., Hayes N., Leibowitz M.D., Moller D.E., Berger J. Molecular cloning, expression and characterization of human peroxisome proliferator activated receptors γ1 and γ2. Biochem. Biophys. Res. Commun. 1996;224(2):431–437. doi: 10.1006/bbrc.1996.1044. [DOI] [PubMed] [Google Scholar]
- Forman B.M., Tontonoz P., Chen J., Brun R.P., Spiegelman B.M., Evans R.M. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- Gavrilova O., Haluzik M., Matsusue K., Cutson J.J., Johnson L., Dietz K.R., Nicol C.J., Vinson C., Gonzalez F.J., Reitman M.L. Liver peroxisome proliferator-activated receptor γ contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003;278(36):34268–34276. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- Greenstein A.W., Majumdar N., Yang P., Subbaiah P.V., Kineman R.D., Cordoba-Chacon J. Hepatocyte-specific, PPARγ-regulated mechanisms to promote steatosis in adult mice. J. Endocrinol. 2017;232:107–121. doi: 10.1530/JOE-16-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L., Zhang L.u., Sun Y., Muskhelishvili L., Blann E., Dial S., Shi L., Schroth G., Dragan Y.P. Differences in hepatotoxicity and gene expression profiles by anti-diabetic PPAR γ agonists on rat primary hepatocytes and human HepG2 cells. Mol Divers. 2006;10(3):349–360. doi: 10.1007/s11030-006-9038-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst C.H., Waxman D.J. Activation of PPARα and PPARγ by environmental phthalate monoesters. Toxicol. Sci. 2003;74:297–308. doi: 10.1093/toxsci/kfg145. [DOI] [PubMed] [Google Scholar]
- Issemann I., Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347(6294):645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- Kim S.W., Brown D.J., Jester J.V. Transcriptome analysis after PPARγ activation in human meibomian gland epithelial cells (hMGEC) Ocul Surf. 2019;17(4):809–816. doi: 10.1016/j.jtos.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind T., Liu K.-H., Lee D.Y., DeFelice B., Meissen J.K., Fiehn O. LipidBlast - in-silico tandem mass spectrometry database for lipid identification. Nat. Methods. 2013;10(8):755–758. doi: 10.1038/nmeth.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer S.A., Lenhard J.M., Willson T.M., Patel I., Morris D.C., Lehmann J.M. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- Leesnitzer L.M., Parks D.J., Bledsoe R.K., Cobb J.E., Collins J.L., Consler T.G., Davis R.G., Hull-Ryde E.A., Lenhard J.M., Patel L., Plunket K.D., Shenk J.L., Stimmel J.B., Therapontos C., Willson T.M., Blanchard S.G. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator Activated receptors by GW9662. Biochemistry. 2002;41(21):6640–6650. doi: 10.1021/bi0159581. [DOI] [PubMed] [Google Scholar]
- Lefterova M.I., Steger D.J., Zhuo D., Qatanani M., Mullican S.E., Tuteja G., Manduchi E., Grant G.R., Lazar M.A. Cell-specific determinants of peroxisome proliferator-activated receptor gamma function in adipocytes and macrophages. Mol. Cell. Biol. 2010;30:2078–2089. doi: 10.1128/MCB.01651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefterova M.I., Zhang Y., Steger D.J., Schupp M., Schug J., Cristancho A., Feng D., Zhuo D., Stoeckert C.J., Liu X.S., Lazar M.A. PPAR and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes & Development. 2008;22:2941–2952. doi: 10.1101/gad.1709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann J.M., Moore L.B., Smith-Oliver T.A., Wilkison W.O., Willson T.M., Kliewer S.A. An Antidiabetic Thiazolidinedione Is a High Affinity Ligand for Peroxisome Proliferator-activated Receptor γ (PPARγ) J. Biol. Chem. 1995;270(22):12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- Lennon A.M., Ramaugé M., Dessouroux A., Pierre M. MAP kinase cascades are activated in astrocytes and preadipocytes by 15-Deoxy-Δ12–14-prostaglandin J2 and the Thiazolidinedione Ciglitazone Through Peroxisome Proliferator Activator Receptor γ-independent mechanisms involving reactive oxygenated species*. J. Biol. Chem. 2002;277(33):29681–29685. doi: 10.1074/jbc.M201517200. [DOI] [PubMed] [Google Scholar]
- Leporcq C., Spill Y., Balaramane D., Toussaint C., Weber M., Bardet A.F. TFmotifView: a webserver for the visualization of transcription factor motifs in genomic regions. Nucleic Acids Res. 2020;48:W208–W217. doi: 10.1093/nar/gkaa252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J.D., Ferrara A., Peng T., Hedderson M., Bilker W.B., Quesenberry C.P., Vaughn D.J., Nessel L., Selby J., Strom B.L. Risk of bladder cancer among diabetic patients treated with pioglitazone: interim report of a longitudinal cohort study. Diabetes Care. 2011;34(4):916–922. doi: 10.2337/dc10-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyles B.E., Akinyeke T.O., Moss P.E., Stewart L.V. Thiazolidinediones regulate expression of cell cycle proteins in human prostate cancer cells via PPARγ-dependent and PPARγ-independent pathways. Cell Cycle. 2009;8(2):268–277. doi: 10.4161/cc.8.2.7584. [DOI] [PubMed] [Google Scholar]
- Martin G., Schoonjans K., Staels B., Auwerx J. PPARγ activators improve glucose homeostasis by stimulating fatty acid uptake in the adipocytes. Atherosclerosis. 1998;137:S75–S80. doi: 10.1016/S0021-9150(97)00315-8. [DOI] [PubMed] [Google Scholar]
- Meex S.J.R., Andreo U., Sparks J.D., Fisher E.A. Huh-7 or HepG2 cells: which is the better model for studying human apolipoprotein-B100 assembly and secretion? J. Lipid Res. 2011;52(1):152–158. doi: 10.1194/jlr.D008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy L., Tontonoz P., Alvarez J.G., Chen H., Evans R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- Nesto R.W., Bell D., Bonow R.O., Fonseca V., Grundy S.M., Horton E.S., Le Winter M., Porte D., Semenkovich C.F., Smith S., Young L.H., Kahn R. Thiazolidinedione Use, Fluid Retention, and Congestive Heart Failure. Circulation. 2003;108(23):2941–2948. doi: 10.1161/01.CIR.0000103683.99399.7E. [DOI] [PubMed] [Google Scholar]
- Nielsen R., Pedersen T.Å., Hagenbeek D., Moulos P., Siersbæk R., Megens E., Denissov S., Børgesen M., Francoijs K.-J., Mandrup S., Stunnenberg H.G. Genome-wide profiling of PPARγ:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008;22:2953–2967. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen S.E., Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007;356(24):2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- Nolan J.J., Ludvik B., Beerdsen P., Joyce M., Olefsky J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N. Engl. J. Med. 1994;331(18):1188–1193. doi: 10.1056/NEJM199411033311803. [DOI] [PubMed] [Google Scholar]
- Patsouris D., Mandard S., Voshol P.J., Escher P., Tan N.S., Havekes L.M., Koenig W., März W., Tafuri S., Wahli W., Müller M., Kersten S. PPARα governs glycerol metabolism. J. Clin. Invest. 2004;114(1):94–103. doi: 10.1172/JCI20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinelli P., Videla L.A. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J. Clin. Endocrinol. Metab. 2011;96:1424–1430. doi: 10.1210/jc.2010-2129. [DOI] [PubMed] [Google Scholar]
- Puri P., Baillie R.A., Wiest M.M., Mirshahi F., Choudhury J., Cheung O., Sargeant C., Contos M.J., Sanyal A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46(4):1081–1090. doi: 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- Reddam A., Mitchell C.A., Dasgupta S., Kirkwood J.S., Vollaro A., Hur M., Volz D.C. mRNA-sequencing identifies liver as a potential target organ for triphenyl phosphate in embryonic zebrafish. Toxicol. Sci. 2019;172(1):51–62. doi: 10.1093/toxsci/kfz169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riu A., Grimaldi M., le Maire A., Bey G., Phillips K., Boulahtouf A., Perdu E., Zalko D., Bourguet W., Balaguer P. Peroxisome proliferator-activated receptor γ is a target for halogenated analogs of bisphenol A. Environ. Health Perspect. 2011;119(9):1227–1232. doi: 10.1289/ehp.1003328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymanski E.L., Jeon J., Gulde R., Fenner K., Ruff M., Singer H.P., Hollender J. Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ. Sci. Technol. 2014;48(4):2097–2098. doi: 10.1021/es5002105. [DOI] [PubMed] [Google Scholar]
- Seargent J.M., Yates E.A., Gill J.H. GW9662, a potent antagonist of PPARγ, inhibits growth of breast tumour cells and promotes the anticancer effects of the PPARγ agonist rosiglitazone, independently of PPARγ activation. Br J. Pharmacol. 2004;143:933–937. doi: 10.1038/sj.bjp.0705973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strakova N., Ehrmann J., Bartoš J., Malikova J., Dolezel J., Kolar Z. Peroxisome proliferator-activated receptor (PPAR) agonists affect cell viability, apoptosis and expression of cell cycle related proteins in cell lines of glial brain tumors. Neoplasma. 2005;52:126–136. [PubMed] [Google Scholar]
- Strakova N., Ehrmann J., Dzubak P., Bouchal J., Kolar Z. The synthetic ligand of peroxisome proliferator-activated receptor-γ ciglitazone affects human glioblastoma cell lines. J. Pharmacol. Exp. Ther. 2004;309(3):1239–1247. doi: 10.1124/jpet.103.063438. [DOI] [PubMed] [Google Scholar]
- Sumner L.W., Amberg A., Barrett D., Beale M.H., Beger R., Daykin C.A., Fan T.-M., Fiehn O., Goodacre R., Griffin J.L., Hankemeier T., Hardy N., Harnly J., Higashi R., Kopka J., Lane A.N., Lindon J.C., Marriott P., Nicholls A.W., Reily M.D., Thaden J.J., Viant M.R. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI) Metabolomics. 2007;3(3):211–221. doi: 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P., Hu E., Spiegelman B.M. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–1156. doi: 10.1016/0092-8674(94)90006-X. [DOI] [PubMed] [Google Scholar]
- Velkov, T., 2013. Interactions between Human Liver Fatty Acid Binding Protein and Peroxisome Proliferator Activated Receptor Selective Drugs [WWW Document]. PPAR Research. 10.1155/2013/93840. [DOI] [PMC free article] [PubMed]
- Vignati S., Albertini V., Rinaldi A., Kwee I., Riva C., Oldrini R., Capella C., Bertoni F., Carbone G.M., Catapano C.V. Cellular, molecular consequences of peroxisome proliferator- activated receptor-δ activation in ovarian cancer cells. Neoplasia. 2006;8(10):851–IN12. doi: 10.1593/neo.06433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K.-I., Okamura M., Tsutsumi S., Nishikawa N.S., Tanaka T., Sakakibara I., Kitakami J.-I., Ihara S., Hashimoto Y., Hamakubo T., Kodama T., Aburatani H., Sakai J. The Peroxisome proliferator-activated receptor γ/Retinoid X receptor α heterodimer targets the histone modification enzyme PR-Set7/Setd8 gene and regulates adipogenesis through a positive feedback loop. Mol. Cell. Biol. 2009;29(13):3544–3555. doi: 10.1128/MCB.01856-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Mullican S.E., DiSpirito J.R., Peed L.C., Lazar M.A. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARγ. PNAS. 2013;110(46):18656–18661. doi: 10.1073/pnas.1314863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Kwon G., An L., Holmes C.N., Haeba M., LeBlanc G.A. Differential interactions of the flame retardant triphenyl phosphate within the PPAR signaling network. MOJ Toxicol. 2016;2 [Google Scholar]
- West A.G., Fraser P. Remote control of gene transcription. Hum. Mol. Genet. 2005;14:R101–R111. doi: 10.1093/hmg/ddi104. [DOI] [PubMed] [Google Scholar]
- Wilkening S., Stahl F., Bader A. Comparison of primary human hepatocytes and hepatoma cell line Hepg2 with regard to their biotransformation properties. Drug Metab. Dispos. 2003;31(8):1035–1042. doi: 10.1124/dmd.31.8.1035. [DOI] [PubMed] [Google Scholar]
- Yang C.-F., Shen H.-M., Ong C.-N. Protective effect of ebselen against hydrogen peroxide-induced cytotoxicity and DNA damage in HepG2 cells. Biochem. Pharmacol. 1999;57(3):273–279. doi: 10.1016/S0006-2952(98)00299-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.