

Abstract
Previously we identified the p.Thr61Ile mutation in coiled-coil-helix-coiled-coil-helix domain containing 2 (CHCHD2) in a Chinese family with autosomal dominant Parkinson’s disease. But the mechanism is still unclear. In this study, we explored the effects of CHCHD2 p.Thr61Ile mutation in cells and its association with coiled-coil-helix-coiled-coil-helix domain containing 10 (CHCHD10). We found that overexpression of Parkinson’s disease–associated T61I mutant CHCHD2 did not produce mitochondrial dysfunction. Rather, its protective effect from stress was abrogated. And, the level of the CHCHD2 protein and mRNA in patient fibroblasts was not significantly different from control. In addition, CHCHD2 T61I mutation caused increased interaction with CHCHD10 and reduced CHCHD10 level. The mitochondrial ultrastructural alterations in CHCHD2 T61I mutant patient fibroblasts are similar to that of CHCHD10 mutations. We therefore propose that CHCHD10 is involved in the development of Parkinson’s disease caused by CHCHD2 loss-of-function mutation p.T61I.
Keywords: Parkinson’s disease, CHCHD2, CHCHD10, Mitochondria
1. Introduction
Mutations and polymorphic risk variants of coiled-coil-helix-coiled-coil-helix domain containing 2 (CHCHD2) have been associated with Parkinson’s disease (PD) (Funayama et al., 2015). We have identified the p.Thr61Ile mutation in CHCHD2 in a Chinese family with autosomal dominant Parkinson’s disease (Mao et al., 2016; Shi et al., 2016). However, the exact role of CHCHD2 in the pathogenesis of PD is still unknown, and the pathogenicity of the reported mutations and risk variants has been debated (Tio et al., 2017).
CHCHD10 and CHCHD2 are homologous proteins with 58% sequence identity and belong to the twin CX9C family of proteins that mediate cellular stress responses. CHCHD10 genetic variants, such as p.Ser59Leu and p.Arg15Leu, were identified in patients with amyotrophic lateral sclerosis and/or frontotemporal dementia (Bannwarth et al., 2014). The missense mutations of CHCHD10 were also identified to be related to other human neurological disorders including late-onset spinal muscular atrophy, Jokela type (SMAJ) (p.Gly66Val); sensorimotor axonal Charcot-Marie-Tooth neuropathy (CMT2) (p.Gly66Val); and autosomal dominant mitochondrial myopathy (IMMD) (p.Arg15Ser and p.Gly58Arg) (Zhou et al., 2017). Recently, studies reported that loss of CHCHD10-CHCHD2 complexes underlies the pathogenicity of CHCHD10 mutation–related amyotrophic lateral sclerosis (Burstein et al., 2018; Straub et al., 2018). It is not yet known whether CHCHD10 is involved in the mechanism of CHCHD2 mutation in PD. In this study, we explored the effects of CHCHD2 p.Thr61Ile mutation in cells and its association with CHCHD10.
2. Materials and methods
2.1. Plasmids, cell lines, and antibodies
Full-length CHCHD2 cDNA was cloned into the pCMV6-Entry vector (Invitrogen). CHCHD2 T61I mutant was cloned in the vector using an overlap extension polymerase chain reaction (PCR). SHSY5Y cells stably overexpressing vector, wild-type (WT), or mutated (T61I) CHCHD2 were created. Stable knockdown of CHCHD2 or CHCHD10 was generated by transfecting SHSY5Y cells with CHCHD2 or CHCHD10 shRNA plasmid (Santa Cruz Biotechnology). Antibodies: anti-hCHCHD2 (Proteintech, 19424-1-AP), anti-hCHCHD10 (Proteintech, 25671-1-AP), anti-FLAG (Sigma-Aldrich, clone M2).
2.2. Flow cytometry, oxygen consumption, quantitative PCR, coimmunoprecipitation, and electron microscopy analysis
Mitochondrial membrane potential was measured using tetramethylrhodamine (Thermo Fisher) by flow cytometry, treated with MG-132(15 μM, Sigma). Cellular reactive oxidative species (ROS) measurements were performed using CM-H2DCFDA (Life Technologies), treated with rotenone (100 nmol/L, Sigma). Apoptosis was quantified by annexin V-FITC and PI double staining (Life Technologies), treated with staurosporine (1 μm, Sigma). Cellular oxygen consumption was measured with a Seahorse XFe96 Bioanalyzer, performed according to the manufacturer’s instructions. Primers for CHCHD2 in quantitative PCR: Fw primer: 5′-CACACATTGGGTCACGCCATTACT-3’; Rev primer: 5′-TTCTGGGCACACTCCAGAAACTGT-3’. Primers for CHCHD2 cDNA PCR: Fw primer: 5′-ACCTAGGATGCCGCGTGGAA-3’; Rev primer: 5′-GGTTCCCTGAGGCTCCTGGTAA-3’. Coimmunoprecipitation was performed using Dynabeads Protein G Kit (Invitrogen). Mitochondrial ultrastructure was analyzed by Hitachi H-8000 electron microscope.
3. Results
3.1. CHCHD2 p.Thr61Ile is a loss-of-function mutation
In stable transfected SH-SY5Y neuroblastoma cell lines, analysis of tetramethylrhodamine fluorescence revealed that T61I mutation had no significant effect on mitochondrial membrane potential under basal conditions (Fig. 1A). After stress with MG-132, there was a decrease in mitochondrial membrane potential from basal levels in all three groups. However, the decrease of WT CHCHD2 was the least, which was significantly different from the vector and T61I mutated groups (tetramethylrhodamine fluorescence intensity: Vector: 53.51% ± 9.39; WT: 72.58% ± 12.39; T61I: 52.25% ± 5.52). Furthermore, compared with the vector group, total cellular ROS in WT CHCHD2 cells showed a significant decreased fluorescent signal induced by rotenone, whereas the T61I mutant cells displayed a subtle reduction (Vector: 26.85% ± 8.01; WT: 14.24% ± 4.69; T61I: 21.34% ± 4.96) (Fig.1B). Consistent with the changes in ROS after stress, overexpression of WT CHCHD2 but not the T61I mutant significantly reduced the level of apoptotic cell death induced by staurosporine (Vector: 51.08% ± 7.98; WT: 37.84% ± 7.07; T61I: 54.74% ± 4.21) (Fig.1C). In addition, to determine if the p.T61I variant affected mitochondrial respiratory function, we measured oxygen consumption rates using an Extracellular Flux Analyzer (Seahorse) in naive, WT, T61I, and CHCHD2 knockdown (CHCHD2-KD) SHSY5Y cells. Basal mitochondrial respiration of CHCHD2-KD cell lines was reduced by 21.81% (p < 0.05) compared with controls and the maximal respiratory capacity in response to uncoupling agent FCCP was reduced by 35.78 % (p < 0.05) (Fig. 1D). However, compared with naive and WT groups, overexpression of CHCHD2 T61I in cells did not reduce oxygen consumption rates. These aforementioned findings suggest that WT CHCHD2 may protect neurons from stress-induced mitochondrial dysfunction, and that, this effect is abrogated by the T61I mutation.
Fig. 1.
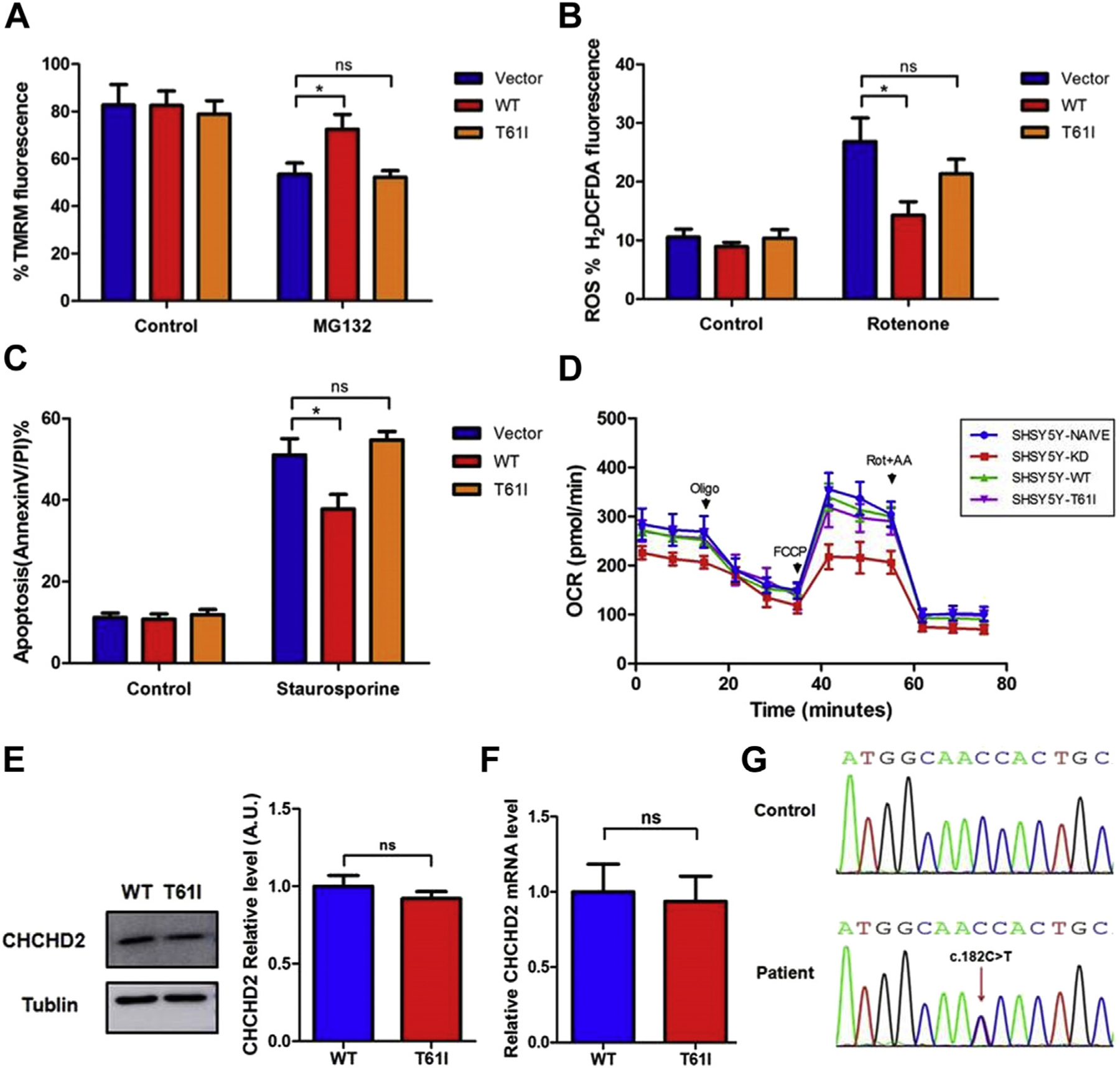
The effects of CHCHD2 p.Thr61Ile mutation in cells. (A) Effect of wild-type and T61I mutant CHCHD2 on mitochondrial membrane potential determined by FACS of SH-SY5Y cells treated with MG-132 (15 μM). (n = 5; *p < 0.05; ns, not significant) (B) Effect of wild-type and T61I mutant CHCHD2 on ROS determined by FACS of SH-SY5Y cells treated with rotenone (100 nmol/L). (n = 5; *p < 0.05; ns, not significant) (C) Effect of wild-type and T61I mutant CHCHD2 on apoptosis determined by FACS of SH-SY5Y cells treated with staurosporine (10020μm). (n = 5; *p < 0.05; ns, not significant). (D) Mitochondrial oxygen consumption was assessed as the oxygen consumption rate (OCR) by Seahorse XFe96 Bioanalyzer. (E) Immunoblot analysis and quantification of CHCHD2 levels in control and T61I mutant patient fibroblasts. (F) Quantitative PCR for CHCHD2 mRNA levels in control and patient fibroblasts. (G) cDNA analysis of patient fibroblasts. The DNA sequence chromatogram shows heterozygosity of the c.182C > T (p.T61I) mutation in patient fibroblasts. Abbreviations: CHCHD2, coiled-coil-helix-coiled-coil-helix domain containing 2; ROS, reactive oxidative species; PCR, polymerase chain reaction.
We further explored the expression of CHCHD2 in fibroblasts from a PD patient in the family (male, age of onset: 48 years, disease duration: 2 years, Hoehn and Yahr: 1.5), heterozygous for the p.T61I CHCHD2 variant. Immunoblot analysis showed that the difference of CHCHD2 levels between patient fibroblasts and control was not significant (Fig. 1E). In addition, the steady-state level of the CHCHD2 mRNA in patient fibroblasts was not significantly different from the control (Fig. 1F), and DNA sequencing of an reverse transcription-PCR product indicated similar contributions from the WT and mutant alleles, as would be expected for a heterozygous missense mutation (Fig. 1G). All evidence presented previously revealed that the CHCHD2 p.Thr61Ile mutation is a loss-of-function mutation.
3.2. CHCHD10 is involved in the development of PD caused by CHCHD2 p.T61I mutation
Immunoblot analysis indicated that when CHCHD2 was depleted, the levels of CHCHD10 decreased, whereas depletion of CHCHD10 did not affect the CHCHD2 level (Fig. 2A). Furthermore, because previous studies showed that CHCHD10 forms high molecular weight complexes with CHCHD2 (Burstein et al., 2018; Straub et al., 2018), we assessed the interaction of CHCHD10 with the CHCHD2 T61I mutant. We found that the mutant, although equally expressed (Flag-CHCHD2), showed increased interaction with CHCHD10 (Fig. 2B). In addition, interestingly, immunoblot analysis showed that CHCHD10 was reduced to 44% of control levels in patient fibroblasts (Fig. 2C).
Fig. 2.

CHCHD10 is involved in the mechanism of CHCHD2 p.T61I mutation in Parkinson’s disease. (A) Immunoblot analysis of CHCHD2 and CHCHD10 levels in control and CHCHD2/10-KD SHSY5Y cells. (B) Immunoblot analysis and quantification of CHCHD10 levels in control and T61I mutant patient fibroblasts. (C) The T61I mutant CHCHD2 shows increased interaction with CHCHD10. Flag-CHCHD2 plasmid was transfected in HEK293T cells. WT and Q112H CHCHD2 were immunoprecipitated with an anti-Flag antibody and probed for CHCHD10. (D) Ultrastructural analysis of control (a–c) and patient (d–e) fibroblasts. Scale bar = 500 nm. (a–c) Representative image of mitochondria with typical normal aspect found in control cells. (d) Moderate disorganization mainly found in patient cells. (e–f) Complete mitochondrial disorganization only found in patient cells. **p < 0.01. Abbreviations: CHCHD2, coiled-coil-helix-coiled-coil-helix domain containing 2; CHCHD10, coiled-coil-helix-coiled-coil-helix domain containing 10; WT, wild type.
As CHCHD10 mutations resulted into disorganized mitochondria with sparse or absent cristae, we therefore performed ultrastructural analysis of CHCHD2 T61I patient fibroblasts. Intact and healthy mitochondria of control cells had many well-defined and parallel cristae, running perpendicularly to the mitochondrial longitudinal axis (Fig. 2D a–c). They represented more than 90% of the mitochondrial profiles seen in control cells. Completely disorganized and swelling mitochondria with sparse or absent cristae without recognizable parallel orientation were only observed in patient fibroblasts (20%) (Fig. 2D e–f). Less disorganized mitochondria with sparse cristae represented about 50% of the mitochondrial pattern in patient cells and 10% in control cells (Fig. 2D d). The mitochondrial ultrastructural alterations in CHCHD2 T61I mutant patient fibroblasts are similar to that of CHCHD10 mutations (Bannwarth et al., 2014). Thus, these aforementioned results indicated that CHCHD10 is involved in the development of PD caused by CHCHD2 p.T61I mutation.
4. Discussion
In this study, we investigated the effects of CHCHD2 p.Thr61Ile mutation in cells and its association with CHCHD10 in PD. CHCHD2 contains a mitochondrial targeting sequence in the N-terminus and two cysteine-x9-cysteine (twin Cx9C) motifs at the C-terminus and has been localized to the intermembrane space of the mitochondria. Mutations in CHCHD2 have been identified in the autosomal dominant form of PD (Funayama et al., 2015), yet the pathogenesis remains unclear. Meng Hongrui et al. verified that these two missense mutations CHCHD2 T61I and R145Q are loss-of-function mutations (Meng et al., 2017). However, Tio Murni et al. demonstrated that Drosophila expressing mutant CHCHD2 proteins (T61I, R145Q, and P2L) displayed late-onset characteristics as seen in PD patients such as locomotor dysfunction (Tio et al., 2017). In this study, we found that overexpression of PD-associated CHCHD2 T61I mutant did not produce mitochondrial dysfunction. But, its protective effect from stress was abrogated. And, the level of the CHCHD2 protein and mRNA in patient fibroblasts was not significantly different from the control. These aforementioned results suggested that CHCHD2 T61I mutation leads to loss of function.
We found that CHCHD10 is involved in the development of PD caused by CHCHD2 p.T61I mutation. There are three lines of evidence. First, CHCHD2 is the major interactor of CHCHD10. We showed that CHCHD2 T61I mutant showed increased interaction with CHCHD10. Second, when CHCHD2 was depleted, the levels of CHCHD10 decreased, whereas depletion of CHCHD10 did not affect CHCHD2 level, which indicated that CHCHD2 regulated CHCHD10. Third, CHCHD10 was reduced to 44% of control levels in patient fibroblasts. And, the mitochondrial ultrastructural alterations in CHCHD2 T61I mutant patient fibroblasts are similar to that of CHCHD10 mutations. In addition, previous studies showed that CHCHD10 is a component of the mitochondrial contact site and cristae organizing system complex. The expression of CHCHD10 mutant alleles leads to mitochondrial contact site and cristae organizing system complex disassembly, loss of mitochondrial cristae, and nucleoid disorganization leading to a defect in mtDNA repair after oxidative stress (Genin et al., 2016). Thus, we further hypothesized that mutant CHCHD2 regulated CHCHD10 resulting into abnormal mitochondrial cristae and mitochondrial disfunction. Further studies are needed to support this hypothesis and clarify the specific mechanism.
5. Conclusions
Our results confirm that CHCHD10 is involved in the development of PD caused by CHCHD2 loss-of-function mutation p.T61I. Further functional and genetic studies are needed to elucidate the exact mechanism of CHCHD2 mutations.
Acknowledgements
This work is supported by National Key R&D Program of China [2017YFA0105000], the National Natural Science Foundation of China to YX [grant numbers 81530037,81471158] and the National Natural Science Foundation of China to CS [grant number 81771290]. The authors appreciate Dr. Jiansheng Kang, Dr. Zongping Xia, and Dr. Zhimin Wang from Zhengzhou University for their critical discussion.
Footnotes
Disclosure statement
The authors report no conflicts of interest.
References
- Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, Berg-Alonso L, Kageyama Y, Serre V, Moore DG, Verschueren A, Rouzier C, Le Ber I, Auge G, Cochaud C, Lespinasse F, N’Guyen K, de Septenville A, Brice A, Yu-Wai-Man P, Sesaki H, Pouget J, Paquis-Flucklinger V, 2014. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137, 2329–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein SR, Valsecchi F, Kawamata H, Bourens M, Zeng R, Zuberi A, Milner TA, Cloonan SM, Lutz C, Barrientos A, Manfredi G, 2018. In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum. Mol. Genet 27, 160–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama M, Ohe K, Amo T, Furuya N, Yamaguchi J, Saiki S, Li Y, Ogaki K, Ando M, Yoshino H, Tomiyama H, Nishioka K, Hasegawa K, Saiki H, Satake W, Mogushi K, Sasaki R, Kokubo Y, Kuzuhara S, Toda T, Mizuno Y, Uchiyama Y, Ohno K, Hattori N, 2015. CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: a genome-wide linkage and sequencing study. Lancet Neurol 14, 274–282. [DOI] [PubMed] [Google Scholar]
- Genin EC, Plutino M, Bannwarth S, Villa E, Cisneros-Barroso E, Roy M, Ortega-Vila B, Fragaki K, Lespinasse F, Pinero-Martos E, Auge G, Moore D, Burte F, Lacas-Gervais S, Kageyama Y, Itoh K, Yu-Wai-Man P, Sesaki H, Ricci JE, Vives-Bauza C, Paquis-Flucklinger V, 2016. CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med 8, 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao CY, Wu P, Zhang SY, Yang J, Liu YT, Zuo CT, Zhuang ZP, Shi CH, Xu YM, 2016. Brain glucose metabolism changes in Parkinson’s disease patients with CHCHD2 mutation based on (18)F-FDG PET imaging. J. Neurol. Sci 369, 303–305. [DOI] [PubMed] [Google Scholar]
- Meng H, Yamashita C, Shiba-Fukushima K, Inoshita T, Funayama M, Sato S, Hatta T, Natsume T, Umitsu M, Takagi J, Imai Y, Hattori N, 2017. Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun 8, 15500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CH, Mao CY, Zhang SY, Yang J, Song B, Wu P, Zuo CT, Liu YT, Ji Y, Yang ZH, Wu J, Zhuang ZP, Xu YM, 2016. CHCHD2 gene mutations in familial and sporadic Parkinson’s disease. Neurobiol. Aging 38, 217.e9–217.e13. [DOI] [PubMed] [Google Scholar]
- Straub IR, Janer A, Weraarpachai W, Zinman L, Robertson J, Rogaeva E, Shoubridge EA, 2018. Loss of CHCHD10-CHCHD2 complexes required for respiration underlies the pathogenicity of a CHCHD10 mutation in ALS. Hum. Mol. Genet 27, 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tio M, Wen R, Lim YL, Zukifli ZHB, Xie S, Ho P, Zhou Z, Koh TW, Zhao Y, Tan EK, 2017. Varied pathological and therapeutic response effects associated with CHCHD2 mutant and risk variants. Hum. Mutat 38, 978–987. [DOI] [PubMed] [Google Scholar]
- Zhou ZD, Saw WT, Tan EK, 2017. Mitochondrial CHCHD-containing proteins: physiologic functions and link with neurodegenerative diseases. Mol. Neurobiol 54, 5534–5546. [DOI] [PubMed] [Google Scholar]