

Abstract
Background and aims
Helicobacter pylori invades the mucosal barrier and infects the mucins of gastric epithelial cells. However, whether gastric carcinogenesis caused by H. pylori infection involves the membrane-bound mucins is unclear. This study explored the role of mucin 17 (MUC17) in gastric cancer (GC) associated with H. pylori infection.
Methods
The expression of MUC17 and carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) was examined in human GC cells and tissues with H. pylori infection. Gain- and loss-of-function assays were performed to assess the role of MUC17 in regulating CEACAM1 in H. pylori-infected GC cells.
Results
MUC17 was downregulated in H. pylori-infected GC cells and tissues in association with poor survival of GC patients. Downregulation of MUC17 was attributable to MUC17 promoter methylation mediated by DNA methyltransferase 1 (DNMT1) H. pylori-enhanced GC cell proliferation and colony formation associated with MUC17 downregulation. Gain- and loss-of-function assays showed that MUC17 inhibited the H. pylori-enhanced GC cell growth by preventing the translocation of H. pylori CagA into GC cells. Moreover, MUC17 downregulated the expression of CEACAM1 variant 3S (CEACAM1-3S) in GC cells and tissues with H. pylori infection. Additionally, MUC17 downregulated CEACAM1 promoter activity via attenuation of NF-κB activation in GC cells.
Conclusions
MUC17 was epigenetically downregulated in GC with H. pylori infection. MUC17 inhibited H. pylori CagA translocation via attenuation of NF-κB-mediated expression of CEACAM1-3S in GC cells. Thus, MUC17 may serve as a valuable prognostic biomarker for H. pylori-associated GC.
Keywords: H. pylori, Mucin 17, CEACAM1, CagA, Gastric cancer
Introduction
Gastric cancer (GC) is highly heterogeneous and seriously threatens human health worldwide. The 5-year survival rate of GC patients remains poor [1–3]. However, early diagnosis and treatment of GC may prolong the survival time and improve the life quality of the patients [4, 5]. Therefore, it is desirable to delineate the mechanisms of GC progression and develop efficacious therapeutic intervention.
The gastric pathogen Helicobacter pylori (H. pylori) colonizes the mucus niche of the gastric mucosa of more than a half human population [6, 7], invading gastric mucosal barrier. H. pylori is the most frequent etiological cause of inflammation-associated gastric carcinogenesis, responsible for approximately 90% of noncardia GC. GC patients with H. pylori infection showed poor GC-related survival and low relapse-free survival [8, 9].
Mucins (MUCs) are main components of gastric mucosal barrier, consisting of a family of high molecular-weight glycoproteins expressed by specialized epithelial cells as secreted or membrane-bound mucins [10, 11]. H. pylori colonizes gastric mucosa by utilizing adhesins or non-adhesins to bind mucins on epithelial cells. This process causes alterations in mucin subtypes, which on the one hand facilitate the major virulence proteins expressed by H. pylori, such as cytotoxin-associated gene A (CagA), to invade gastric mucosa, on the other hand limit the adherence of bacteria to epithelial cell surface, hampering the spread of H. pylori along the mucosal surface [12, 13]. The dual roles of mucins in the development of GC may depend on differentially expressed mucin subtypes in response to microenvironment changes caused by H. pylori infection, suggesting a complicated relationship between mucins and H. pylori during gastric carcinogenesis.
In this study, we investigated the expression and function of mucins in GC during H. pylori infection. We found that H. pylori epigenetically downregulated the expression of membrane-bound MUC17 in GC in favor of its infection characterized by H. pylori CagA translocation and GC cell proliferation. Gain-and loss-of-function assays showed that MUC17 prevented CagA translocation and GC cell proliferation in association with suppression of the expression of CEACAM1 variant 3S (CEACAM1-3S) via attenuation of NF-κB pathway. The results suggest that the capacity of H. pylori on GC cell proliferation is attributed to its impact on the expression of membrane-bound MUC17, resulting in NF-κB-mediated expression of CEACAM1-3S.
Materials and methods
Bacterial strain and cell culture
H. pylori 26695 was cultured at 37 °C in microaerobic atmosphere (5% O2, 10% CO2 and 85% N2) in Columbia agar plates with Helicobacter pylori selective supplement (0.025 mg/ml) and 10% horse blood (Oxoid, Cambridge, UK). Plates were incubated for 48 h under microaerobic conditions.
Human GC cell lines (BGC823, SGC7901) were grown in 90% DMEM (Gibco, Life technologies, Grand Island, NY, USA) with 10% fetal bovine serum (PAN Biotech, Adenbach, GER). GC cell lines BGC823 and SGC7901 were established in China and obtained from the tissue bank of Shanghai (Shanghai, China). GC cells were spilt to low density (30% confluence) with antibiotic-free cell culture medium. H pylori bacteria were co-cultured with GC cells at multiplicity of infection (MOI) 5, 25, 50 or 100. Cells were collected at 6, 12 or 18 h after H. pylori infection. Before collection, cells were exposed to 100 μg/μl of gentamicin (Sigma, St Louis, MO, USA) for 6 h. Cells were also treated with 5-Aza (5-aza-2′-dexocytidine) for 96 h with the growth medium being changed every 24 h.
DNA constructs and transfection
DNA constructs and transfection were performed as previously described [14, 15]. Details were provided in the Supplementary Materials and Methods.
Patients and specimens
67 cases of GC tissues (Supplementary Table 1) were obtained from Peking University Cancer Hospital from 2002 to 2007, patient’s name was removed from these case descriptions. This study was approved by the institutional Ethical Review Board for human investigation at Peking University. Endoscopical biopsy specimens of primary GC were obtained from patients and stored at the Tissue-Bank of Peking University Cancer Hospital according to the standard procedures of the Ethics Committee of Peking University Cancer Hospital.
RNA isolation, reverse transcription (RT), and polymerase chain reaction (PCR)
RNA Isolation, reverse transcription (RT), and polymerase chain reaction (PCR) were performed as previously described [14, 15]. Cells were harvested for RNA isolation using TRIzol Reagent (Invitrogen, Grand Island, NY, USA) and first-strand cDNA was synthesized with the Superscript First-Strand Synthesis System (Invitrogen). PCR was performed using primers listed in Supplementary Table 2. qPCR was performed using 2× SYBR Green-based qPCR reagent on ABI 7500 fast qPCR machine (Applied Biosystems, California, USA). The relative expression level of each gene was normalized to the amount of the same cDNA using the 2−ΔΔCt method and was further compared to β-actin.
DNA extraction, bisulfite modification, methylation-specific PCR (MSP-PCR) and bisulfite sequencing (BS)
DNA extraction, bisulfite modification, and methylation-specific PCR (MSP-PCR) were performed as previously described [14, 15]. Total DNA was extracted from tissues using the genomic DNA rapid extraction kit (Transgen Biotech, Beijing, China). Bisulfite modification of DNA was performed using Zymo DNA Methylation Kit (Zymo Research, Irvine, CA, USA). The in vitro methylation DNA (IVD) serving as the positive control was the A&D Human Methylated DNA Standard (A&D Technology, Beijing, China), and the negative control was the genomic DNA from normal human peripheral lymphocytes as described [14]. MSP-PCR was performed using primer pairs that specifically amplify either methylated or unmethylated sequences of the MUC17 gene as Kitamoto et al. described [16] (the primers were listed in Supplementary Table 2). MSP products were analyzed using a 2% agarose gel electrophoresis. Bisulfite-treated DNA was amplified using BS primers as described in Supplementary Table 2. PCR products were gel purified and cloned into pGEM-T vectors (Promega, Madison, WI, USA). Colonies were randomly selected for plasmid isolation using EasyPure Plasmid MiniPrep Kit (TransGen Biotech, Beijing, China) and subjected to sequencing with the SP6 reverse primer via automated sequencing (Beijing AUGCT Biotechnology Co. Ltd., Beijing, China) as previously described [14, 15].
Western blot analysis
SDS-PAGE and western blots were performed as previous described [14, 15]. Details were provided in the Supplementary Materials and Methods.
Luciferase assay
Luciferase reporter assay was performed using a dual-luciferase reporter assay system (Promega, Madison, WI, USA) with the pGL3 basic luciferase reporter system. Promoter-specific luciferase constructs and controls were transfected with into cells using Lipofectamine 2000 (Invitrogen). The luciferase signal was first normalized to the control luciferase signal and then normalized to the signal from control group.
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed by following EpiTech ChIPOne-Day Kit protocol (QIAGEN) as described [14, 15]. Details were provided in the Supplementary Materials and Methods.
Immunofluorescence staining (IF)
Immunofluorescence staining was performed as previously described [14, 15]. Cells with different treatments were harvested and fixed with 4% paraformaldehyde at room temperature for 10 min. After washing and pre-blocking, the cells were incubated at 4 °C overnight with antibodies against MUC17 (1:25), CEACAM1 (1:100), NF-κB (1:50) and Helicobacter pylori CagA (1:500), respectively. After washing, the cells were incubated with FITC-conjugated or TRITC-conjugated secondary antibody (1:100; Santa Cruz, CA, USA) for 1 h, and then were analyzed by Vectra3 automated quantitative pathology system from PerkinElmer Inc. (Boston, MA, USA). DAPI was used for nuclei staining (10 μg/ml in PBS, Invitrogen, Life Technologies, and Grand Island, NY, USA).
Immunohistochemistry (IHC)
IHC was performed on 5 mm thick serial sections of formaldehyde fixed as previously described [14, 15]. The sections were incubated with antibodies against MUC17 (1:25), CEACAM1 (1:100), H pylori CagA (1:500), and H pylori (1:100) overnight at 4 °C. The sections were incubated with Opal 4-Color Manual IHC Kit according to the manufacturer’s instructions or DAB, and then were analyzed by Vectra3 automated quantitative pathology system from PerkinElmer Inc. (Boston, MA, USA). GC patient tumor specimens were analyzed and scores were described previously [14, 15].
Cell viability and colony formation
GC cells were seeded into 96-well plates at 1 × 103 cells/well and cell viability was determined everyday by IncuCyte Zoom (Essen Bioscience, MI, USA) or using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay kit (KeyGEN Biotech, Jiangsu, China). The absorbance for MTT assay at 490 nm/570 nm wavelength was detected using a microplate reader (Thermo Multiskan MK3,Thermo Fisher Scientific Inc., USA). GC cells were seeded into 35 mm cell culture dishes (200 cells/well) with H. pylori infection (MOI 50, 12 h) every 4 days. All dishes were incubated at 37 °C in a humidified atmosphere of 5% CO2. Colonies were scored in 2 weeks. The cloning cells were fixed with 4% paraformaldehyde for 10 min and stained with 0.2% crystal violet (Beyotime Ltd., Jiangsu Province, China) for 20 min.
Statistical analysis
The data are expressed as means ± standard deviation (SD) of at least three independent experiments. The DNA methylation in human GC was analyzed by the Student’s t test. Overall survival was analyzed using Kaplan–Meier method and compared using log-rank tests. The relationship between MUC17 and H. pylori infection with clinic pathologic characteristics in GC patients was measured by χ2 or Fisher’s exact tests. Pearson correlation test was used to analyze two continuous value correlations. All statistical analyses were performed using SPSS version 23.0 (SPSS Inc., Chicago, IL, USA).
Result
MUC17 is reduced in H. pylori-infected GC in association with poor survival of GC patients
Experiments were performed using the GC cells treated with different MOI (multiplicity of infection) of H. pylori, resulting in enhanced cell proliferation, with optional effects at MOI 50 of the bacteria (Supplementary Fig. 1a). The proliferation on promoted effect of H. pylori on GC cells was confirmed by colony formation assay, in which GC cells treated with H. pylori formed significantly increased colonies than the cells treated with saline (Supplementary Fig. 1b). Western blotting showed the expression of H. pylori CagA in GC cells at a pathogen MOI of 50 (Supplementary Fig. 1c), indicating that GC cells were infected with H. pylori, since H. pylori promoted cell proliferation by translocating H. pylori CagA into the cytoplasm of target cells. By transfection of ectopic H. pylori CagA into BGC823 and SGC7901 cells (Supplementary Fig. 1d), we showed that overexpression of CagA in GC cells significantly promoted the viability of GC cells as compared with control vector transfected cells (Supplementary Fig. 1e). These results showed that H. pylori promoted GC cell proliferation mainly through CagA, suggesting a successful infection by H. pylori.
We next determined the expression of membrane-bound mucins in GC cells upon H. pylori infection. As shown in Fig. 1a, H. pylori infection caused significant reduction of mucin family members, MUC13, MUC17 and MUC20, in particular MUC17, although mRNA of MUC17 was barely detectable in BGC823 and weakly expressed in SGC7901 GC cells (Supplementary Fig. 2). Moreover, the mRNA expression of MUC17 was reduced upon H. pylori infection in a dose- and time-dependent manner (Fig. 1b, c).
Fig. 1.
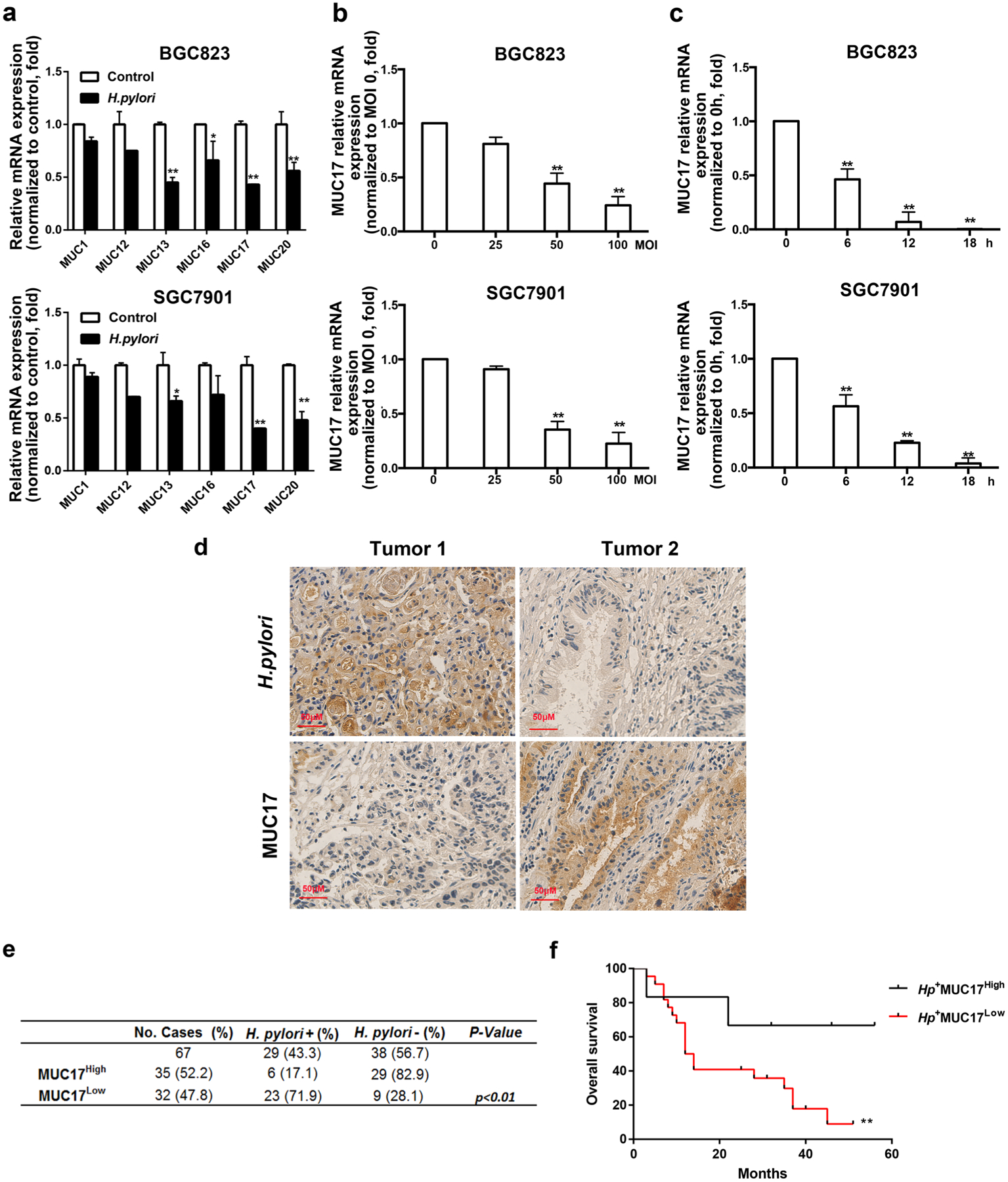
Decreased expression of MUC17 in H. pylori-infected GC. a RT-qPCR of the mRNA expression of membrane-bound mucins (MUC1, MUC12, MUC13, MUC16, MUC17, and MUC20) in GC cell BGC823 and SGC7901 after infected H. pylori at multiplicity of infection of 50 for 12 h (MOI 50, 12 h). Each gene was compared with its own untreated control after normalization to β-actin. b RT-qRCR of MUC17 expression in BGC823 and SGC7901 cells with H. pylori infection at different multiplicity of infection (MOI 5, 25, 50, 100) for 12 h. c RT-qRCR of MUC17 expression in BGC823 and SGC7901 cells with H. pylori infection (MOI 50) at different time points (0, 6, 12, 18 h). d Representative immunohistochemical staining of MUC17 expression in human GC tissues with or without H. pylori infection. Original microphotographs at magnification of × 400. Scale bar, 50 μM. e Analysis of MUC17 expression in association with H. pylori infection in the GC tissues by IHC (n = 67). *p < 0.05, **p < 0.01by χ2 test. f Kaplan–Meier analysis of overall survival until 120 months for the GC patients in accordance with H. pylori infection and the MUC17 expression level. *p < 0.05, **p < 0.01. Hp: H. pylori
We then validated MUC17 expression in a cohort of 67 patients with GC (Table 1), among which, 29 (43.3%) of patients were H. pylori positive. As shown in Fig. 1d, IHC staining demonstrated decreased expression of MUC17 (MUC17Low) in Hp+ gastric tumors in contrast to higher MUC17 expression (MUC17High) in Hp− tumors. Although there appeared no difference in the expression of MUC17 in the cohort, MUC17Low GC cases with Hp+ in tumor tissues (Hp+ MUC17Low) were about fourfold of Hp+ tumor cases (Hp+ MUC17High) (p < 0.01, Fig. 1e; Table 1). Follow-up of 60 months showed that the GC patients with Hp+MUC17High had significantly better survival time than that of Hp+MUC17Low GC patients (p < 0.01, Fig. 1f). These results suggest that MUC17 expression is decreased in GC with H. pylori infection, which is associated with the poor outcome of the GC patients.
Table 1.
MUC17 expression associated with clinicopathologic parameters in GC
| Characteristics | No. of cases | MUC17High (%) | MUC17Low (%) | p value |
|---|---|---|---|---|
| Total | 67 | 35 (52.2) | 32 (47.8) | |
| Gender | ||||
| Male | 52 | 29 (55.8) | 23 (44.2) | |
| Female | 15 | 6 (40.0) | 9 (60.0) | 0.281 |
| Age | ||||
| ≥ 60 | 20 | 10 (50.0) | 10 (50.0) | |
| < 60 | 47 | 25 (53.2) | 22 (46.8) | 0.811 |
| GC type | ||||
| Diffuse | 40 | 21 (52.5) | 19 (47.5) | |
| Intestinal | 27 | 14 (51.9) | 13 (48.1) | 0.958 |
| Differentiation Poor-moderate | 58 | 29 (50.0) | 29 (50.0) | |
| Well | 9 | 6 (66.7) | 3 (33.3) | 0.352 |
| TNM stage | ||||
| I/II | 21 | 13 (61.9) | 8 (38.1) | |
| III/IV | 46 | 22 (47.8) | 24 (52.2) | 0.285 |
| H. pylori infection | ||||
| Yes | 29 | 6 (20.7) | 23 (79.3) | |
| No | 38 | 29 (76.3) | 9 (23.7) | < 0.01 |
Downregulation of MUC17 expression by DNA hypermethylation in GC upon H. pylori infection
In 30 cases of human GC tissues available with RNA and DNA samples, the expression of MUC17 in tumor tissues was significantly decreased upon H. pylori infection (Fig. 2a). MSP analysis revealed that 93.75% (15 of 16) of Hp+ GC tissues were partially or heavily methylated in the promoter region of MUC17, significantly different from Hp− samples, in which methylation occurred in 42.86% (6 of 14) (p < 0.01, Fig. 2b and Supplementary Fig. 3). The representative GC tissue samples (Hp+: 5T and 13T; Hp−: 21T and 27T) were subjected to BS analysis of methylation status in the promoter region of MUC17 gene, showing heavily methylated in 5T, partial methylated in 13T and 21T, or unmethylated in 27T, respectively (Fig. 2c). To explore whether the decreased expression of MUC17 with H. pylori infection was attributable to epigenetic alteration, H. pylori-infected GC cells were treated with 5-Aza. As shown in Fig. 2d, MUC17 expression was increased in BGC823 and SGC7901 cells after 5-Aza treatment. Moreover, treatment of BGC823, and SGC7901 cells with 5-Aza significantly reduced the proliferation of H. pylori-infected GC cells (Fig. 2e). To determine the involvement of DNA methylation in the regulation of MUC17 expression, analysis of the TCGA (the cancer genome atlas) database showed that among the 372 cases of stomach adenocarcinoma (STAD) recorded with both the methylation and transcripts data, methylation of MUC17 was correlated with the downregulation of MUC17 expression (Fig. 2f, p < 0.05, r = − 0.5867). Bioinformatic analysis of the database also showed that in these 372 cases of STAD, patients with MUC17Low had a poor prognosis (Supplementary Fig. 4). Western Blotting analysis of DNA methyltransferase 1 (DNMT1), a preferred methylator involved in tumor suppressor genes hypermethylation [17, 18], showed that the expression of DNMT1 was increased in BGC823 and SGC7901 cells upon H. pylori infection (Fig. 2g). ChIP-qPCR analysis of genomic DNA immunoprecipitated with anti-DNMT1 antibody in GC cells showed that DNMT1 enriched more genomic DNA in MUC17 promoter region (Fig. 2h), a methylated region upon H. pylori treatment (Fig. 2c). These findings suggest downregulation of MUC17 expression in GC upon H. pylori infection in association with DNA hypermethylation regulated by DNMT1.
Fig. 2.
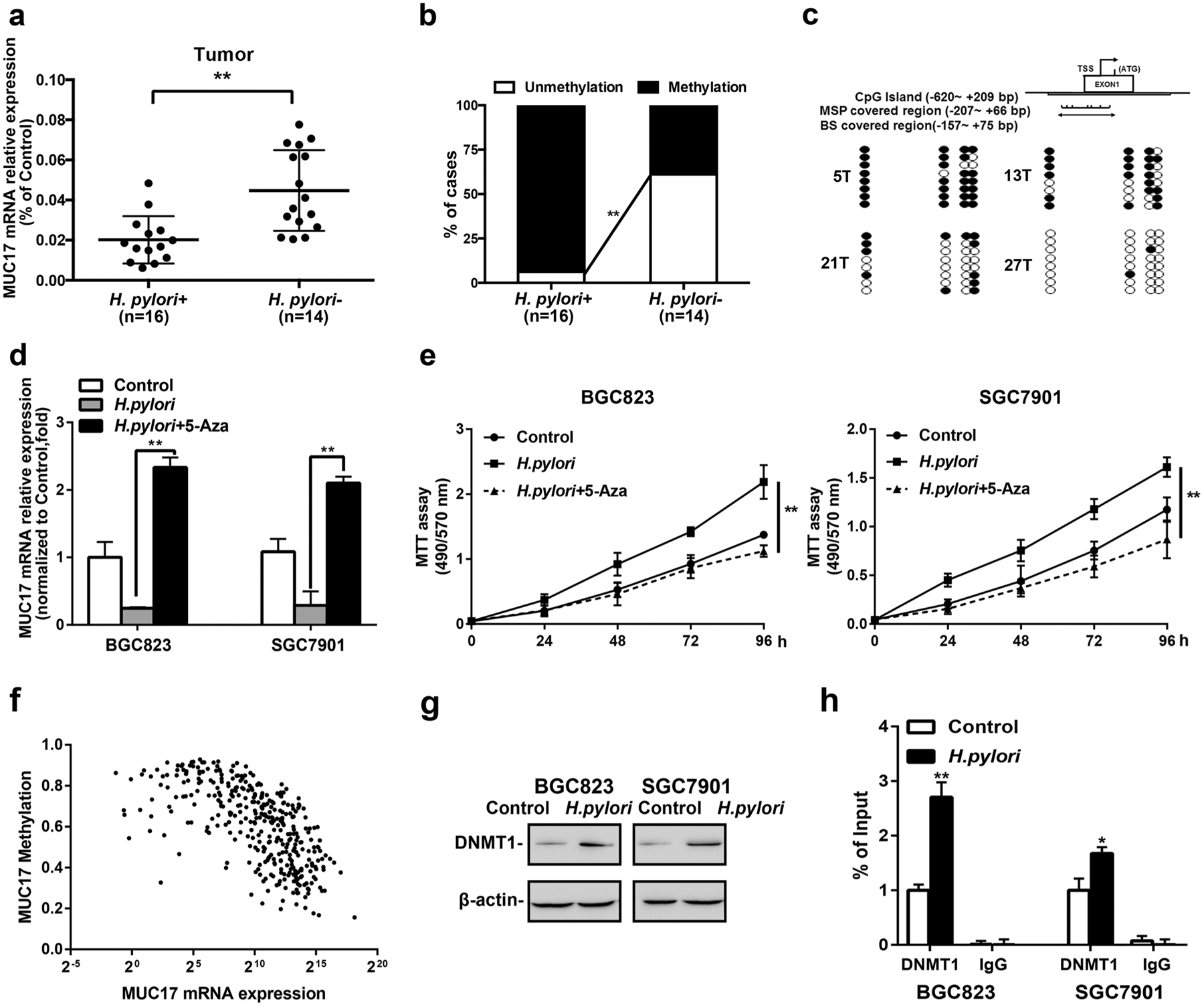
Epigenetic silencing of MUC17 by H. pylori in GC. a RT-qRCR of MUC17 expression in human GC tissues with respect to H. pylori infection (n = 30). b Methylated PCR analysis of the promoter methylation of MUC17 in the human GC tissues upon H. pylori infection (n = 30). c BS confirmation of MUC17 gene methylation. Representative GC tissue samples from Hp+ (5T and 13T) or Hp− (21T and 27T). Filled circles represent methylated CpG dinucleotides, and open circles indicate unmethylated sites. d GC cell lines with H. pylori infection were treated with 5-Aza (5 μM, 96 h). MUC17 mRNA expression, relative to β-actin as internal control, was measured by quantitative RT-PCR using the 2−ΔΔCt method. Results represent the mean ± SD (standard deviation). *p < 0.05, **p < 0.01 (by t test) compared with untreated control. e The effects of 5-Aza on GC cell proliferation. MTT assays monitoring the cell variability of H. pylori-infected GC cells treated with 5 μM 5-Aza daily for 3 days. Left panel: BGC823. Right panel: SGC7901. f Association between methylation and expression of MUC17 in GC by analyzing TCGA database. g Western blotting of DNMT1 expression in BGC823 and SGC7901 cells after H. pylori infection (MOI 50, 12 h), β-actin served as protein loading control. (h) ChIP-qPCR to analyze the enrichment of the MUC17 promoter region by DNMT1 in BGC823 and SGC7901 cells after H. pylori infection (MOI 50, 12 h). The specific enrichment was normalized to nonspecific control and was represented as the percentage of input. Data are representative of at least three independent experiments. Error bars indicate standard deviation (SD). *p < 0.05, **p < 0.01, significant differences from the control
MUC17 suppresses H. pylori-mediated CagA translocation in GC
We then knocked down MUC17 by shRNA in BGC823 and SGC7901 cell lines (Fig. 3a and Supplementary Fig. 5a). As shown in Fig. 3b, GC cell proliferation was significantly increased by transfection with MUC17 shRNA (p < 0.01), with a further enhancing effect when infected with H. pylori. Colony formation assays also showed that GC cells with MUC17 knockdown formed more colonies than control cells, and the effect was further increased upon H. pylori infection (Fig. 3c). These data suggest disruption of MUC17 in favor of H. pylori-mediated GC cell proliferation.
Fig. 3.
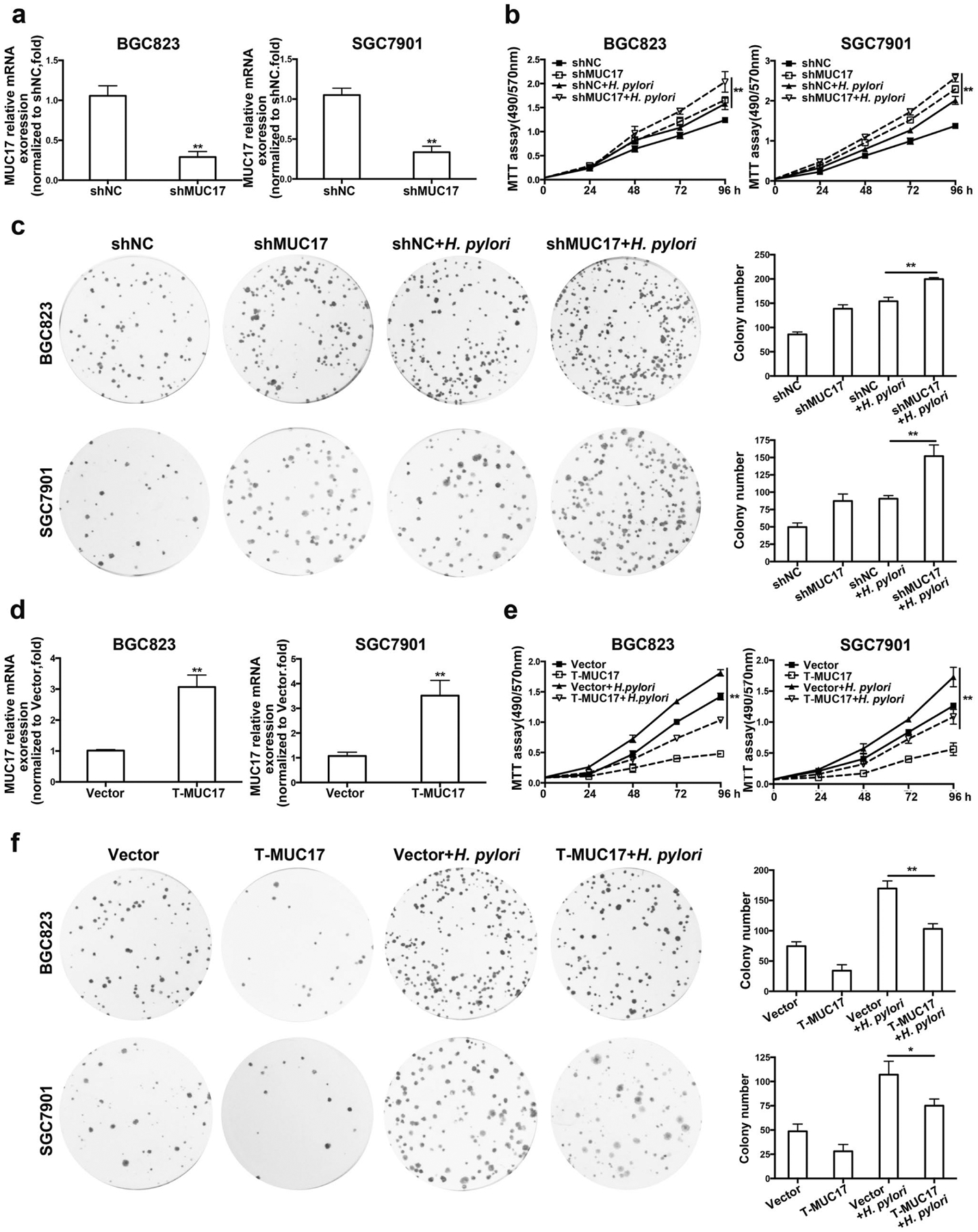
Effects of MUC17 on proliferation of H. pylori-infected GC cells. a–c BGC823 and SGC7901 cells were stably transfected with shMUC17 or nonspecific control (shNC). After validation of the mRNA expression level of MUC17 by RT-qPCR (a), the GC cells were infected with H. pylori (MOI 50,12 h). MTT assay to monitor the viability of the cells over 4 days (b). Colony formation was performed in every 4 days until 2 weeks (c). d–f After stably expression of ectopic truncated MUC17 (T-MUC17) in BGC823 and SGC7901 cells were validated by RT-qPCR (d), the GC cells were infected with H. pylori (MOI 50,12 h), and were subjected to MTT assays (e) and colony formation assays (f). Data are presented as the mean ± standard deviation (n = 3), *p < 0.05, **p < 0.01, significant differences from the control
Data from TCGA database indicates that mutations were highly frequent in MUC17 gene, including in-frame mutation, missense mutation, and truncating mutation. However, there was no mutation observed in EGF like domain of MUC17 and its subsequent region, a conserved domain that is critical for MUC17 to exert its functions. To explore whether the effect of MUC17 on H. pylori infection is mediated by this conserved domain, we overexpressed truncated MUC17 (T-MUC17) in BGC823 and SGC7901 cell lines (Fig. 3d, Supplementary Fig. 5b and 5c). As shown in Fig. 3e, with H. pylori infection, the proliferation of BGC823 and SGC7901 cells with overexpression of truncated MUC17 significantly decreased compared to control cells. These results were confirmed by colony formation assays (Fig. 3f). Thus, MUC17 inhibits H. pylori-mediated GC cell proliferation possibly through its EGF such as domain and the subsequent region. These data suggest an inhibitory effect of MUC17 on GC cell proliferation promoted by H. pylori infection.
By gain- and loss-of-function assays, we showed that H. pylori CagA protein in GC cells upon H. pylori treatment was reduced by MUC17 (Supplementary Fig. 6a). Immunofluorescence staining showed that H. pylori CagA in the cytoplasm of H. pylori-treated GC cells was reduced by MUC17 (Supplementary Fig. 6b and 6c). Furthermore, H. pylori CagA was present in MUC17Low, but not MUC17High tumor tissues (Supplementary Fig. 6d). These data suggest the inhibitory effect of MUC17 on GC with H. pylori infection by blocking translocation of H. pylori CagA.
MUC17 inhibits CEACAM1-3S expression in GC
Helicobacter pylori CagA translocation into gastric epithelial cells was possibly conducted through H. pylori HopQ interacting specifically with CEACAM1 expressed in target cells [19]. We, therefore, examined the effect of MUC17 on CEACAM1 expression in GC cells. As shown in Fig. 4a, in BGC823 and SGC7901 cells, knockdown of MUC17 increased the expression of CEACAM1 at both mRNA and protein levels. Overexpression of truncated MUC17 reduced CEACAM1 expression in GC cells (Fig. 4b). Immunofluorescence staining showed that the expression of CEACAM1 was inversely correlated with MUC17 levels in both Hp+ and Hp− GC tissues (Fig. 4c). For validation, we analyzed the RNA-Seq data of STAD recorded in TCGA database. A total of 186 cases was recorded with the clinical data, including 18 cases of H. pylori-positive patients and 168 of H. pylori-negative patients. The analysis showed that the expression of MUC17 was inversely correlated with CEACAM1 in both Hp+ and Hp− GC tissues (Supplementary Fig. 7a and 7b, p < 0.05). CEACAM1 has many variants through alternative splicing. We examined the expression of four main splice variants of CEACAM1, CEACAM1-4L, -4S, -3L, and -3S in GC cells and tissues. RT-qPCR showed that CEACAM1-3S was the most expressed compared with other variants in GC cells (Fig. 4d). Consistently, the expression of CEACAM1-3S was higher than any other variant in GC tissues (Fig. 4e). We then examined the expression of CEACAM1-3S along with MUC17 in GC cells. The mRNA of CEACAM1-3S was significantly increased or decreased upon MUC17 knockdown or overexpression, respectively (Fig. 4f, g). In GC tissues, increased expression of CEACAM1-3S is associated with decreased MUC17 (Fig. 4h, p < 0.01, r = − 0.6282). We further determined the expression of CEACAM1-3S and MUC17 in response to H. pylori infection. In BGC823 and SGC7901 cells, H. pylori infection significantly reduced MUC17, but increased CEACAM1-3S expression (Fig. 4i). These results suggest an inhibitory role of MUC17 in CEACAM1-3S expression possibly associated with its prevention of H. pylori infection in GC.
Fig. 4.
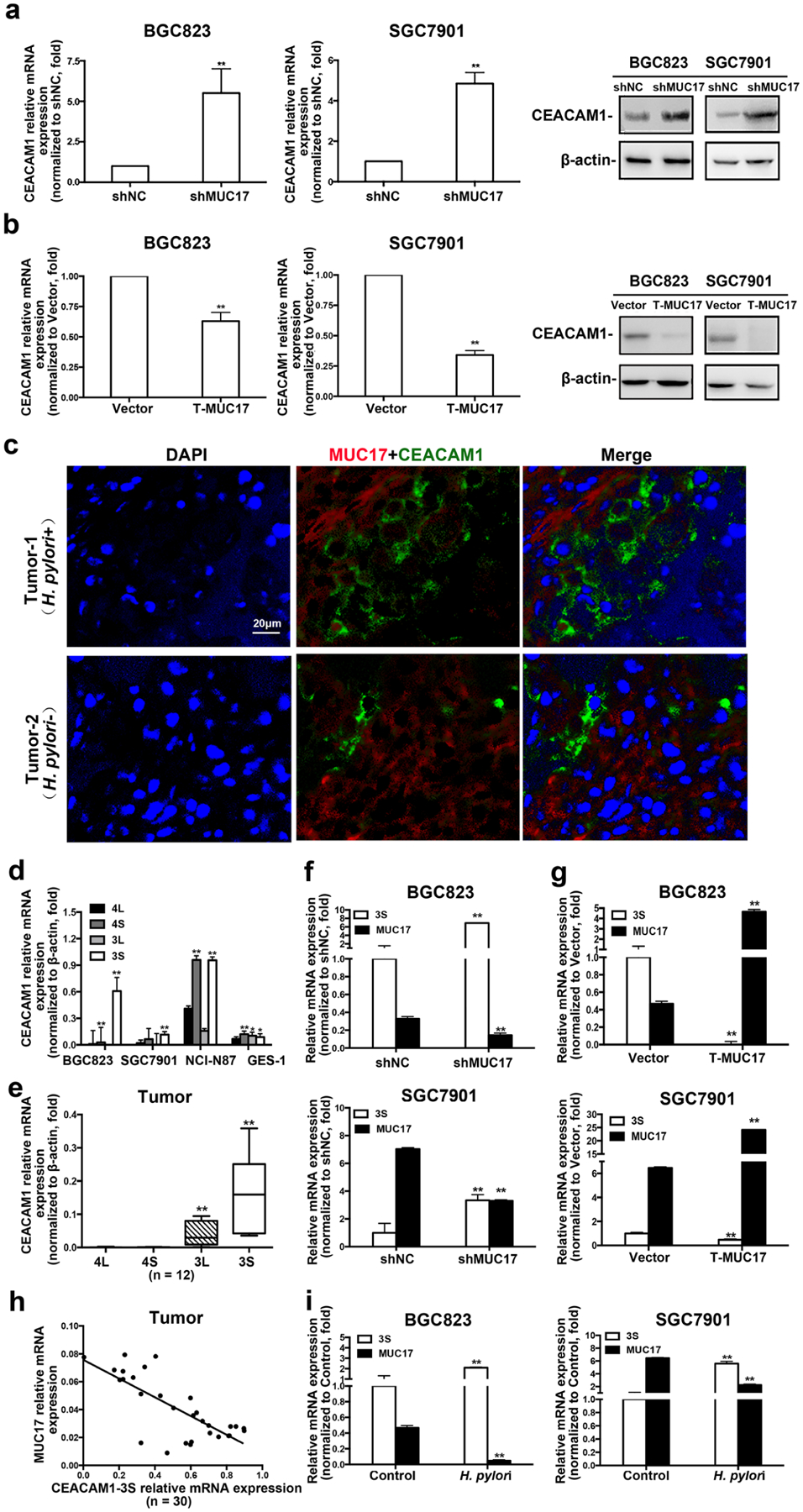
MUC17 downregulated the expression of CEACAM1-3S in human GC cells and tissues. a, b RT-qRCR and Western blot analysis of CEACAM1 expression in BGC823 and SGC7901 cells with knockdown of MUC17 (a) or ectopic expression of T-MUC17 (b). c Immunofluorescence staining of MUC17 (red) and CEACAM1 (green) in GC tissues. Nuclei shown by DAPI staining (blue). Shown are results from one of two comparable experiments. Scale bars, 20 μM. d RT-qRCR of the expression levels of CEACAM1 variants (CEACAM1-4L, -4S, -3L, and -3S) in BGC823, SGC7901, NCI-N87 and GES-1 cells. e RT-qRCR of the expression of CEACAM1 variants in GC tissues (n = 12). Analysis of CEACAM1-3S expression in BGC823 and SGC7901 cells by RT-qRCR after knockdown of MUC17 (f) or ectopic expression of T-MUC17 (g). h Correlation between mRNA of MUC17 and CEACAM1-3S in human GC tissues determined by RT-qPCR o (n = 30, p < 0.001, r = − 0.6282). i RT-qPCR of the expression of MUC17 and CEACAM1-3S in GC cells with H. pylori infection (MOI 50,12 h)
MUC17 inhibits H. pylori CagA translocation by downregulating CEACAM1-3S expression in GC cells
MUC17 or CEACAM1-3S was either knocked down or overexpressed transiently in BGC823 and SGC7901 cells (Fig. 5a, b). Upon H. pylori treatment, Western blotting showed that knockdown of both MUC17 and CEACAM1-3S attenuated H. pylori CagA translocation into GC cells compared with MUC17 single knocked down (Fig. 5c). Conversely, H. pylori CagA detected by Western blotting was increased in H. pylori-infected GC cells with overexpression of both MUC17 and CEACAM1-3S, compared with MUC17 single-overexpression in GC cells (Fig. 5d), suggesting the capacity of CEACAM1-3S to diminish the inhibitory effect of MUC17 on H. pylori CagA translocation. Furthermore, knockdown of both MUC17 and CEACAM1-3S resulted in cell proliferation similar to H. pylori-infected control cells, but significantly lower than infected GC cells with MUC17 single knockdown (Fig. 5e), whereas overexpression of both MUC17 and CEACAM1-3S significantly enhanced H. pylori-mediated GC cell proliferation at the level comparable to H. pylori-infected control cells (Fig. 5f). Thus CEACAM1-3S is capable of reversing the inhibitory effect of MUC17 on H. pylori CagA translocation.
Fig. 5.
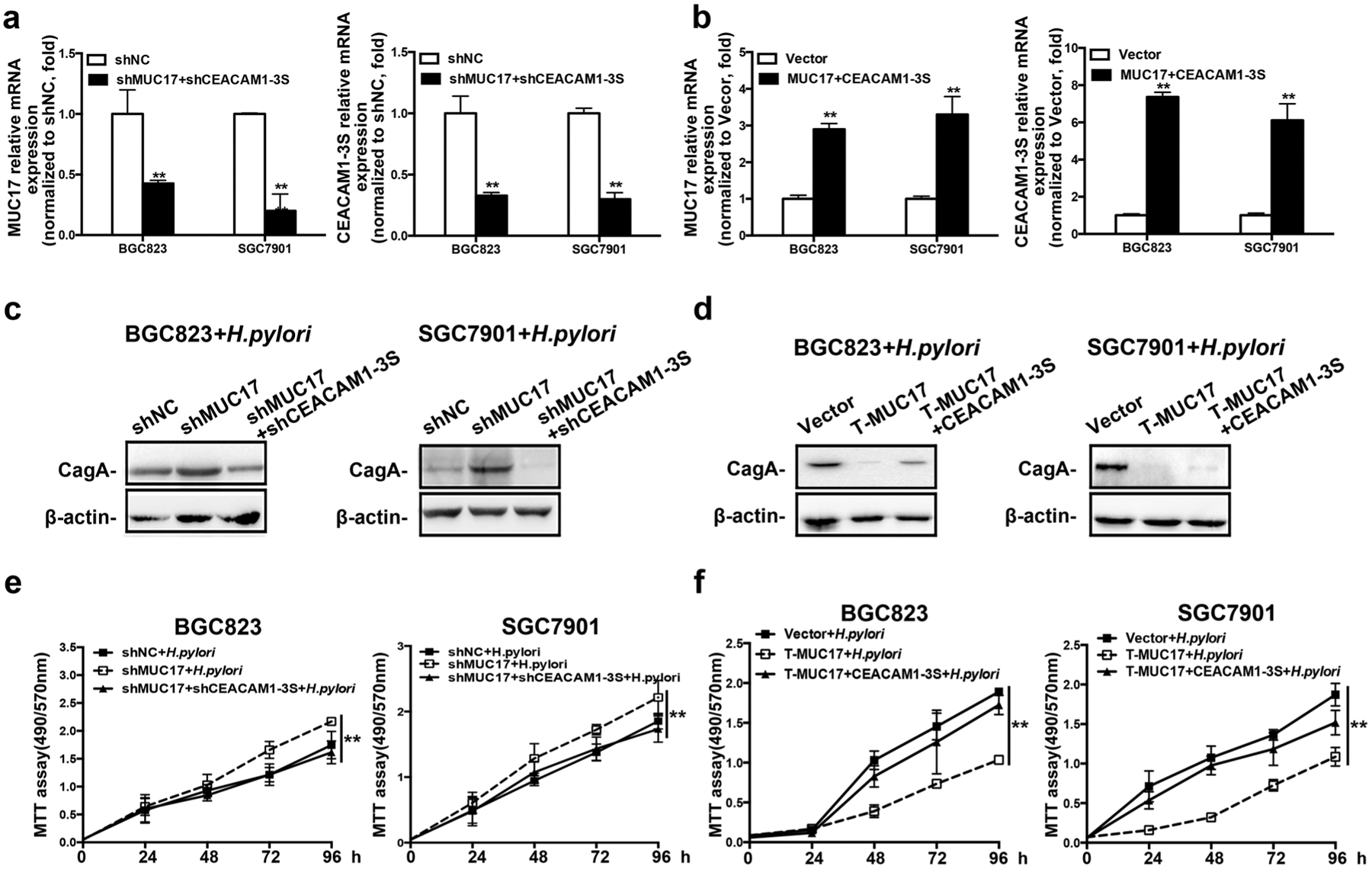
MUC17 inhibited CagA translocation by downregulating CEACAM1-3S expression. BGC823 and SGC7901 cells were transiently transfected or co-transfected with shMUC17 and/or shCEACAM1-3S (shNC transfected as control), or T-MUC17 and/or CEACAM1-3S (Vector as control), respectively. The expression MUC17 and CEACAM1-3S in the GC cells analyzed by RT-qRCR to validate the efficiency of knockdown (a), or overexpression (b). The transfected GC cells were further infected with H. pylori. Western blot analysis of H. pylori CagA expression in BGC823 and SGC7901 cells with knockdown of MUC17 and/or CEACAM1-3S (c) or the overexpression (d), β-actin as protein loading control. MTT assay showed the viability of the GC cells upon H. pylori infection after the knockdown (e) or the overexpression (f). Data are representative of at least three independent experiments. Error bars indicate standard deviation (SD). *p < 0.05, **p < 0.01, significant differences from the control
MUC17 downregulates CEACAM1 promoter activity via attenuation of NF-κB signaling
Aberrant activation of NF-κB was shown upon infection with CagA-positive H. pylori, attributable to gastric carcinogenesis. We hypothesized that MUC17 may downregulate CEACAM1 via NF-kB pathway. Although total p65 subunit of NF-κB showed no obvious change at protein level in whole-cell lysates (Fig. 6a), MUC17 knockdown in BGC823 and SGC7901 cells resulted in augmentation of p65 in the nuclei with concomitant diminution in the cytoplasm. Truncated MUC17 augmented p65 in the cell cytoplasm with a concomitant decrease in the nuclei in the GC cells (Fig. 6b). These data suggest that MUC17 inhibits NF-κB activation in GC cells.
Fig. 6.
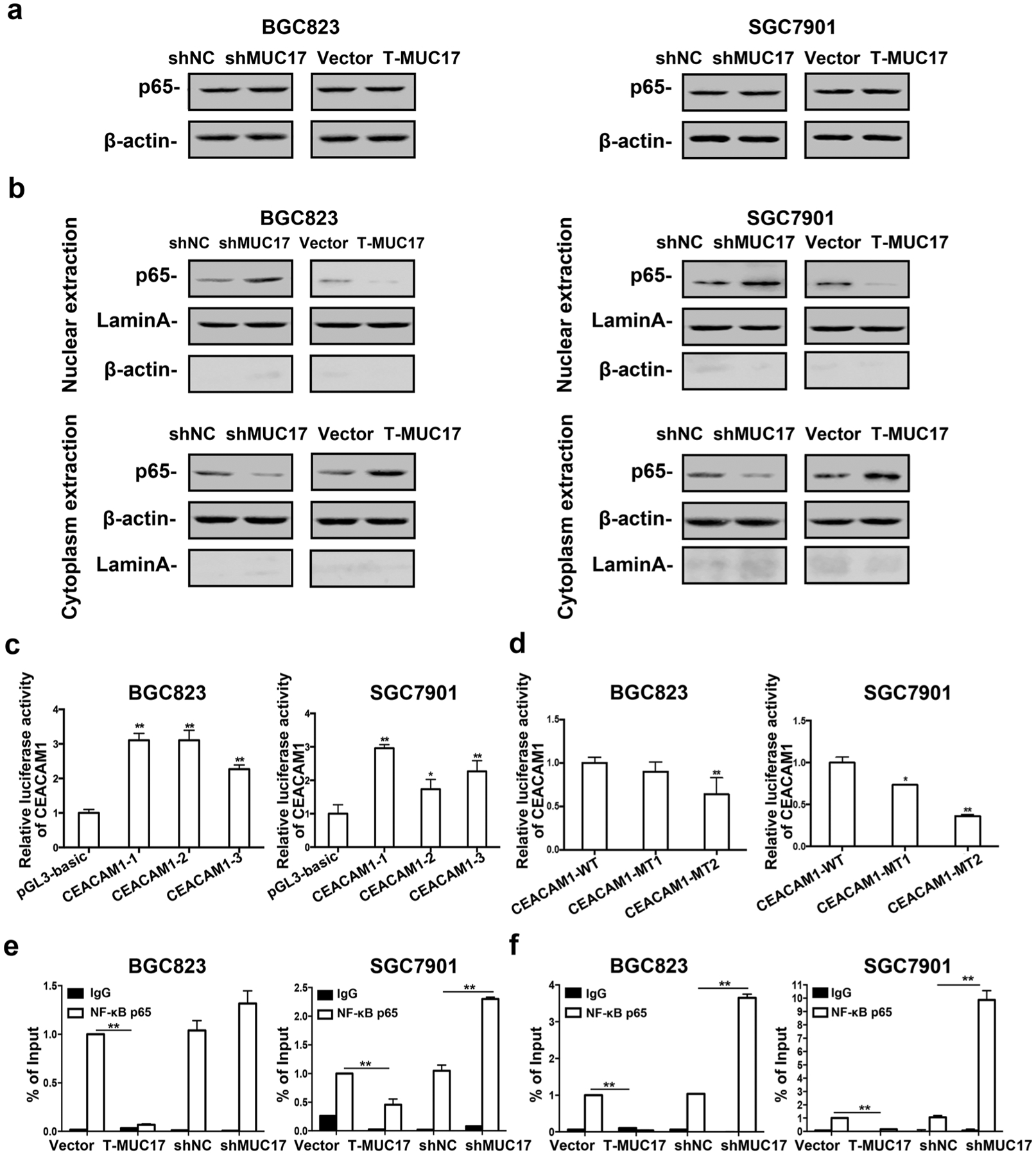
Downregulation of CEACAM1 by MUC17 through attenuation of NF-κB activation. a, b Western blotting analysis of the protein level of NF-κB p65 subunit in BGC823 and SGC7901 cells with knockdown of MUC17, or with T-MUC17 overexpression; a whole cell lysates, b nuclear and cytoplasmic extracts. β-actin served as cytoplasmic and Lamin A as nuclear protein loading control, respectively. c Luciferase reporter assay showing CEACAM1 activities in BGC823 and SGC7901 cells transiently transfected with CEACAM1 promoter construct, CEACAM1-1, CEACAM1-2, and CEACAM1-3 for 36 h. pGL-TK served as internal controls and pGL3-basic vector served as negative controls. d GC cells were transiently transfected with mutant CEACAM1 promoter constructed, CEACAM1-MT1 and CEACAM1-MT2 for 36 h. Luciferase reporter assay showed the CEACAM1 activities. pGL-TK serve as internal controls and CEACAM1 wild type serve as negative controls. e, f ChIP assay was used to identify association of NF-κB p65 with CEACAM1 promoter using antibody specific for NF-κB p65 or normal IgG, followed by qPCR amplification with primers specific for different CEACAM1 promoter regions P1 (e) and P2 (f). Data are representative of at least two independent experiments. Error bars indicate standard deviation (SD). *p < 0.05, **p < 0.01, significant differences from the control
Analysis of JASPAR database for putative transcription factors binding to CEACAM1 revealed consensus sequences of 5′-GGAGGGTCCCCCT-3′ located in the − 741/− 729 region and 5′ GGGGGATCCTCCT 3′ located in the − 311/− 299 region of CEACAM1 promoter represents a typical binding motif for NF-κB p65 (Supplementary Fig. 8). We cloned three DNA fragments in the CEACAM1 promoter region containing two or either one of p65 binding site into pGL3-basic plasmid, termed pCEACAM1-1, -2 and -3, respectively (Supplementary Fig. 8). As shown in Fig. 6c, upon transfection, the luciferase activity of pCEACAM1-1, -2 or -3 was significantly increased compared with pGL3-basic control vector in BGC823 and SGC7901 cells. We then constructed pCEACAM1 mutants by mutation of p65 binding site 1 in pCEACAM1-2 (pCEACAM1-MT1), or p65 binding site 2 in CEACAM1-3 (pCEACAM1-MT2), respectively (Supplementary Fig. 8). After transfection, the luciferase activity of CEACAM1-MT1, or -MT2 was decreased as compared with the wild type construct in BGC823 and SGC7901 cells (Fig. 6d). ChIP assay showed that anti-p65 antibody did not enrich DNA fragments upon MUC17 overexpression of truncated MUC17, but with higher enrichment when knockdown of MUC17 (Supplementary Fig. 8, Fig. 6e, f), suggesting physical association of p65 protein with the proximal promoter region in CEACAM1 gene from − 359 to − 240 of the transcription start site. Thus, MUC17 downregulates CEACAM1 expression via attenuation of NF-κB activation in GC cells.
Discussion
Gastric mucosal barrier disrupted by chronic inflammation, is thought to be the major cause of carcinogenesis [6]. However, the primary role of membrane-bound mucins in gastric carcinogenesis induced by H. pylori is unclear. In the present study, we found epigenetic downregulation of MUC17 by H. pylori in GC. Gain- and loss-of-function assays showed that MUC17 plays an inhibitory role in H. pylori-induced gastric cell proliferation associated with its CagA translocation in GC cells by suppressing the expression of CEACAM1-3S via inactivation of NF-κB.
According to the expression patterns by epithelial cells, mucins are divided into membrane-bound and secretory types [11, 20]. The specificity of the distribution of mucins constitutes a barrier on mucosal defense system and protects the mucosa from invasion by foreign substances including microorganism [13]. Studies have shown that H. pylori is an important factor leading to the damage of gastric mucosal barrier [21–23]. However, studies so far on association of mucins with H. pylori mostly focused on secretory mucins. For instance, H. pylori infection of gastric mucosal leads to downregulation of MUC5AC and abnormal expression of MUC6 on surface epithelium [24–26]. As a membrane-bound mucins, MUC17 plays an important role in epithelial protection [27] by limiting adherence of bacteria to the epithelial cell surface, and hampering the ability of bacteria to colonize and invade epithelial cells as well as blocking spread of bacteria along mucosal surfaces [10, 12]. One study showed that both native and exogenous MUC17 reduced attachment and invasion of E. coli to maintain epithelial barrier function [13]. Our results found that H. pylori infection resulted in decreased expression of membrane-bound mucins, MUC13, MUC16, MUC17, and MUC20, among which changes in MUC17 was most significantly associated with clinical outcome, in line with the studies showing that patients with H. pylori-induced GC showed worst survival time [8, 9]. Therefore, MUC17 has a prognostic value for GC infected by H. pylori.
Host gastric epithelial cells infected by H. pylori is attributed to a variety of bacterial virulence factors including, but not limited to, cag pathogenicity island, cytotoxin-associated gene A (CagA), peptidoglycan outer membrane proteins (e.g., BabA, SabA, OipA), among which, CagA is one of the major virulence proteins expressed by H. pylori and a major determinant of gastric cancer development [28]. CagA directly injected by the bacterium into epithelial cells, leads to cellular changes such as proliferation and cell motility, and stimulates gastric carcinogenesis [29]. A number of studies have shown that adhesin is the major determinant of H. pylori adhesion to cell surface [30, 31]. CagA requires to adhere to epithelial cells to exert its toxicity. Removal of BabA and SabA, two adhesins of H. pylori, had a much greater effect on the apoptosis in human cells induced by H. pylori infection than removal of CagA [32, 33]. Therefore, the adhesin of H. pylori determines its effect on the vitality of the epithelial cells [34, 35]. Recent studies indicate that H. pylori HopQ, as a major adhesin, specifically interacts with CEACAMs [19]. HopQ–CEACAM1 interaction enables translocation of CagA into host cells and enhances the release of pro-inflammatory mediators. Subsequent activation of NF-κB pathway promotes tumor development [36]. In this study, we demonstrated that MUC17 prevented H. pylori CagA translocation by inhibiting the expression of CEACAM1. MUC17 downregulated CEACAM1 promoter activity by inactivating NF-κB. CEACAM1 consists of many variants and transcriptional products through alternative splicing [19, 37]. We examined the expression of four main splice variants containing N-domain, CEACAM1-4L, -4S, -3L and -3S in GC cells and tissues. We found that CEACAM1-3S was the most expressed compared with other variants in tumors. The expression of CEACAM1-3S was negatively correlated with MUC17. Rescue experiment showed that MUC17 inhibits CagA translocation by downregulating CEACAM1-3S. However, overexpressing truncated MUC17 and CEACAM1-3S, does not well restore CagA, possibly because MUC17 also inhibits CagA translocation by other mechanisms. For example, H. pylori adhesin BabA and SabA could bind MUC1 in gastric fluid and MUC1 acts as a decoy covering H. pylori BabA and SabA, or releasing the associated proteolytic enzymes, thereby preventing further adhesion of H. pylori to epithelial cell [32, 38].
Helicobacter pylori infection is a high risk factor for development of precancerous lesions such as chronic atrophic gastritis (CAG), gastric intestinal metaplasia (IM), and cancer [39, 40]. GC development triggered primarily by H. pylori infection is mediated through multistep mechanisms [41]. Data from TCGA database indicates that mutations were highly frequent in MUC17 gene, including inframe mutation, missense mutation and truncating mutation (data not shown). Besides genetic and environmental factors, epigenetic alterations due to H. pylori infection appear to be crucial during chronic infection and gastric carcinogenesis [42]. In this study, we found that downregulation of MUC17 expression upon H. pylori infection was attributable to hypermethylation of MUC17 promoter regulated by DNMT1. The result of epigenetic silencing of MUC17 was further confirmed by analysis of TCGA database, which showed a correlation of DNA methylation with MUC17 downregulation. Since downregulation of MUC 17 was associated with poor overall survival of GC patients, these findings indicate that epigenetic downregulation of MUC17 expression upon H. pylori infection may participate in gastric carcinogenesis. Future translational research may need to clarify epigenetic silencing of MUC gene in context of gastric precancerous lesions upon H. pylori infection, which may provide new tools for the early prediction of GC [16, 43].
In summary, our study showed that membrane-bound MUC17 was epigenetically silenced in GC by H. pylori infection to facilitate its induction of GC cell proliferation and oncoprotein CagA translocation. Our results reveal an anti-H. pylori role of MUC17 by downregulation of CEACAM1 expression in GC cells, via attenuation of NF-κB. We, therefore, propose a pathway by which H. pylori to augments GC growth by downregulation of membrane-bound mucins (Supplementary Fig. 9). Therefor MUC17 appears to be a promising prognostic biomarker for GC with translational potential approach for the treatment of H. pylori-induced GC.
Supplementary Material
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (Grant nos. 81872021, 81572346, 81772502), Natural Science Foundation of Beijing Municipality (Grant no. 7182027), National Key Research and Development Program of China (2017YFC1308900), Beijing Municipal Commission of Health and Family Planning Project (PXM2018_026279_000005), National Bio-Tech 863 Program (no. 2012AA02A203), Beijing Nova Program (Z151100000315069), Beijing Talent Fund, and Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (ZYLX201701). And we also thank the tissue bank of Peking University Cancer Hospital/Institute for providing gastric specimens.
Footnotes
Conflict of interest None of the authors have conflicts of interest that could potentially influence.
Ethics approval This study was conducted with the approval of the Institutional Ethical Standards Committee.
Human rights statement and informed consent All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964 and later versions. Informed consent or substitute for it was obtained from all patients for being included in the study.
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s10120-019-00932-0) contains supplementary material, which is available to authorized users.
References
- 1.Bray F, Jemal A, Grey N, et al. Global cancer transitions according to the human development index (2008–2030): a population-based study. Lancet Oncol. 2012;13(8):790–801. [DOI] [PubMed] [Google Scholar]
- 2.Mahipal A, Choi M, Kim R. Second-line treatment of advanced gastric cancer: where do we stand? J Natl Compr Cancer Netw JNCCN. 2015;13(10):1281. [DOI] [PubMed] [Google Scholar]
- 3.Huang B, Sun Z, Wang Z, et al. Factors associated with peritoneal metastasis in non-serosa-invasive gastric cancer: a retrospective study of a prospectively-collected database. BMC Cancer. 2013;13(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sitarz R, Skierucha M, Mielko J, et al. Gastric cancer: epidemiology, prevention, classification, and treatment. Cancer Manag Res. 2018;10:239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee KJ, Inoue M, Otani T, et al. Gastric cancer screening and subsequent risk of gastric cancer: a large-scale population-based cohort study, with a 13-year follow-up in Japan. Int J Cancer. 2006;118(9):2315–21. [DOI] [PubMed] [Google Scholar]
- 6.Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136(6):1863–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sigal M, Rothenberg ME, Logan CY, et al. Helicobacter pylori activate and expand Lgr5(+) stem cells through direct colonization of the gastric glands. Gastroenterology. 2015;148(7):1392–404. [DOI] [PubMed] [Google Scholar]
- 8.Li G, Wang Z, Wang Z, et al. Gastric cancer patients with Helicobacter pylori infection have a poor prognosis. J Surg Oncol. 2013;108(7):421–6. [DOI] [PubMed] [Google Scholar]
- 9.Qu M, Li L, Zheng WC. Reduced miR-490–3p expression is associated with poor prognosis of Helicobacter pylori induced gastric cancer. Eur Rev Med Pharmacol Sci. 2017;21(15):3384–8. [PubMed] [Google Scholar]
- 10.Moniaux N, Junker WM, Singh AP, et al. Characterization of human mucin MUC17. J Biol Chem. 2006;281(12):23676–85. [DOI] [PubMed] [Google Scholar]
- 11.Dhanisha SS, Guruvayoorappan C, Drishya S, et al. Mucins: Structural diversity, biosynthesis, its role in pathogenesis and as possible therapeutic targets. Crit Rev Oncol Hematol. 2018;122:98–122. [DOI] [PubMed] [Google Scholar]
- 12.Senapati S, Sharma P, Bafna S, et al. The MUC gene family: their role in the diagnosis and prognosis of gastric cancer. Histol Histopathol. 2008;23(12):1541–52. [DOI] [PubMed] [Google Scholar]
- 13.Resta-Lenert S, Das S, Batra SK, et al. Muc17 protects intestinal epithelial cells from enteroinvasive E. coli infection by promoting epithelial barrier integrity. Am J Physiol Gastrointest Liver Physiol. 2011;300(6):G1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan Y, Lin S, Xing R, et al. Epigenetic upregulation of metal-lothionein 2A by diallyl trisulfide enhances chemosensitivity of human gastric cancer cells to docetaxel through attenuating NF-κB activation. Antioxid Redox Signal. 2016;24(15):839–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin S, Lin B, Wang X, et al. Silencing of ATP4B of ATPase H+/K+ transporting beta subunit by intragenic epigenetic alteration in human gastric cancer cells. Oncol Res. 2017;25(3):317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sho K, Norishige Y, Seiya Y, et al. DNA methylation and histone H3-K9 modifications contribute to MUC17 expression. Glycobiology. 2011;21(2):247–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang BG, Hu L, Zang MD, et al. Helicobacter pylori CagA induces tumor suppressor gene hypermethylation by upregulating DNMT1 via AKT-NFκB pathway in gastric cancer development. Oncotarget. 2016;7(9):9788–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan J, Zhang M, Zhang J, et al. Helicobacter pylori infection promotes methylation of WWOX gene in human gastric cancer. Biochem Biophys Res Commun. 2011;408(1):99–102. [DOI] [PubMed] [Google Scholar]
- 19.Javaheri A, Kruse T, Moonens K, et al. Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs. Nat Microbiol. 2016;2:16243. [DOI] [PubMed] [Google Scholar]
- 20.Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004;4(1):45–60. [DOI] [PubMed] [Google Scholar]
- 21.Wang F, Meng W, Wang B, et al. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014;345(2):196–202. [DOI] [PubMed] [Google Scholar]
- 22.Hagymási K, Tulassay Z. Helicobacter pylori infection: new pathogenetic and clinical aspects. World J Gastroenterol. 2014;20(21):6386–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noto JM, Peek RM. Helicobacter pylori: an overview. Methods Mol Biol. 2012;921(2):7–10. [DOI] [PubMed] [Google Scholar]
- 24.Shi D, Qiu XM, Bao YF. Effects of Helicobacter pylori infection on MUC5AC protein expression in gastric cancer. Future Oncology. 2013;9(1):115–20. [DOI] [PubMed] [Google Scholar]
- 25.Park JS, Yeom JS, Seo JH, et al. Immunohistochemical expressions of MUC2, MUC5AC, and MUC6 in normal, Helicobacter pylori infected and metaplastic gastric mucosa of children and adolescents. Helicobacter. 2015;20(4):260–8. [DOI] [PubMed] [Google Scholar]
- 26.Shi D, Qiu XM, Yan XJ. The changes in MUC5AC expression in gastric cancer before and after Helicobacter pylori eradication. Clin Res Hepatol Gastroenterol. 2014;38(2):235–40. [DOI] [PubMed] [Google Scholar]
- 27.Crawley GJ Jr, Hicks SC. JW, et al. MUC17, a novel membrane-tethered mucin. Biochem Biophys Res Commun. 2002;291(3):466–75. [DOI] [PubMed] [Google Scholar]
- 28.Backert S, Blaser MJ. The role of CagA in the gastric biology of Helicobacter pylori. Can Res. 2016;76(14):4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoon JH, Seo HS, Choi SS, et al. Gastrokine 1 inhibits the carcinogenic potentials of Helicobacter pylori CagA. Carcinogenesis. 2014;35(11):2619–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aspholmhurtig M, Dailide G, Lahmann M, et al. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science. 2004;305(5683):519–22. [DOI] [PubMed] [Google Scholar]
- 31.Evans DJ Jr, Evans DG. Helicobacter pylori adhesins: review and perspectives. Helicobacter. 2000;5(4):183–95. [DOI] [PubMed] [Google Scholar]
- 32.Lindén SK, Sheng YH, Every AL, et al. MUC1 limits Helicobacter pylori infection both by steric hindrance and by acting as a releasable decoy. PLoS Pathog. 2009;5(10):e1000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bugaytsova JA, Björnham O, Chernov YA, et al. Helicobacter pylori adapts to chronic infection and gastric disease via pH-responsive BabA-mediated adherence. Cell Host Microbe. 2017;21(3):376–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu J, Leung WK, Go MY, et al. Relationship between Helicobacter pylori babA2 status with gastric epithelial cell turnover and premalignant gastric lesions. Gut. 2002;51(4):480–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yanai A, Maeda S, Hikiba Y, et al. Clinical relevance of Helicobacter pylori sabA genotype in Japanese clinical isolates. J Gastroenterol Hepatol. 2007;22(12):2228–32. [DOI] [PubMed] [Google Scholar]
- 36.Feige MH, Sokolova O, Pickenhahn A, et al. HopQ impacts the integrin α5β1-independent NF-κB activation by Helicobacter pylori in CEACAM expressing cells. Int J Med Microbiol IJMM. 2018;308(5):527–33. [DOI] [PubMed] [Google Scholar]
- 37.Grayowen SD, Blumberg RS. CEACAM1: contact-dependent control of immunity. Nat Rev Immunol. 2006;6(6):433–46. [DOI] [PubMed] [Google Scholar]
- 38.Thathiah A, Blobel CP, Carson DD. Tumor necrosis factor-α converting enzyme/ADAM 17 mediates MUC1 shedding. J Biol Chem. 2003;278(5):3386. [DOI] [PubMed] [Google Scholar]
- 39.Watari J, Chen N, Amenta PS, et al. Helicobacter pylori associated chronic gastritis, clinical syndromes, precancerous lesions, and pathogenesis of gastric cancer development. World J Gastroenterol. 2014;20(18):5461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Correa P. Helicobacter pylori and gastric carcinogenesis. J Gastroenterol. 2009;44(4):239–48. [DOI] [PubMed] [Google Scholar]
- 41.Díaz P, Valenzuela MV, Bravo J, et al. Helicobacter pylori and gastric cancer: adaptive cellular mechanisms involved in disease progression. Front Microbiol. 2018;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maeda M, Moro H, Ushijima T. Mechanisms for the induction of gastric cancer by Helicobacter pylori infection: aberrant DNA methylation pathway. Gastric Cancer. 2016;20(Suppl 1):1–8. [DOI] [PubMed] [Google Scholar]
- 43.Yonezawa S, Higashi M, Yamada N, et al. Significance of mucin expression in pancreatobiliary neoplasms. J Hepato Biliary Pancreat Sci. 2010;17(2):108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.