

Abstract
Background:
With improvements in next-generation DNA sequencing technology, lower cost is needed to collect genetic data. More machine learning techniques can be used to help with cancer analysis and diagnosis.
Methods:
We developed an ensemble machine learning system named performance-weighted-voting model for cancer type classification in 6,249 samples across 14 cancer types. Our ensemble system consists of five weak classifiers (logistic regression, SVM, random forest, XGBoost and neural networks). We first used cross-validation to get the predicted results for the five classifiers. The weights of the five weak classifiers can be obtained based on their predictive performance by solving linear regression functions. The final predicted probability of the performance-weighted-voting model for a cancer type can be determined by the summation of each classifier’s weight multiplied by its predicted probability.
Results:
Using the somatic mutation count of each gene as the input feature, the overall accuracy of the performance-weighted-voting model reached 71.46%, which was significantly higher than the five weak classifiers and two other ensemble models: the hard-voting model and the soft-voting model. In addition, by analyzing the predictive pattern of the performance-weighted-voting model, we found that in most cancer types, higher tumor mutational burden can improve overall accuracy.
Conclusion:
This study has important clinical significance for identifying the origin of cancer, especially for those where the primary cannot be determined. In addition, our model presents a good strategy for using ensemble systems for cancer type classification.
Keywords: cancer type classification, ensemble method, performance-weighted-voting model, linear regression, single-nucleotide polymorphism
INTRODUCTION
It is generally accepted that tumorigenesis is a process of cell renewal, replacement and accumulation of a series of oncogenes, tumor suppressor genes and genetic instability [1], resulting in the collapse of controlling cell division and apoptosis. Studies of cancer genetics have shown that a few driver mutations are enough to cause cancer [2]. In addition to the driver mutations, neutral mutations (or “passenger” mutations) are believed to be common as well [3,4]. The accumulation of driver and passenger mutations is a marker that documents the evolutionary history of cancer [5].
The identification of tumorigenesis and the type of cancer is important. Once a cancer type is classified, the diagnosis can be determined from the prior experience. Studies have shown that cancer cell metastasis can occur at the early stages of cancer progression [6–8]. In addition, about 3% to 9% of all cancer diagnoses are cancer of unknown primary (CUP) [9]. Misclassification of a cancer type or misidentification of cancer of unknown primary usually results in a poor prognosis. Though full of challenges, the definition of the primary of cancer is important. In particular, it will provide significant information on therapeutic strategies that could improve the survival of patients.
Two decades ago, only clinical information was available regarding cancer type classification. Accompanied by the improvements in next-generation DNA sequencing technology, genomic data is growing rapidly. The recent large-scale whole-exome sequencing (WES) and whole-genome sequencing (WGS) projects have displayed different patterns of mutations across cancer types [10–12]. A recent study analyzed an extensive catalog of somatic mutations from 30 most common cancer types and uncovered 20 distinct mutational signatures, as a consequence of the intrinsic slight infidelity of the DNA replication machinery, exogenous or endogenous mutagen exposures, enzymatic modification of DNA, or defective DNA repair [13]. The prevalence of different mutational patterns makes cancer type classification and therapeutic strategies more accurate.
Due to the complexity and high intra-tumor heterogeneity (ITH) within cancer cells [14], it is not easy to determine a cancer type directly. Fortunately, a variety of machine learning techniques and deep-learning algorithms have been widely applied in the last three decades for cancer analysis [15–18]. Most of these studies apply methods for the definition of tumorigenesis, modeling the progression of cancer and determining informative factors that are utilized in the early detection of cancer [19,20]. Since the nineties of the 20th century, machine learning models have become widely used for molecular classification through microarray and oligonucleotide chip gene expression data [21–24]. In the meantime, more advanced methods use microarray data to select effective genes for cancer type classification [25–27]. Accompanied by the development of The Cancer Genome Atlas (TCGA) project, more related studies directly targeted WES [28] and RNA sequencing data [29], as well as the studies that utilized epigenetic profiling to classify cancer of unknown primary [30]. Zeng et al. used non-smooth non-negative matrix factorization (nsNMF) and support vector machine (SVM) to study the associations between somatic mutations and cancers [28]. Liang et al. used sparse logistic regression with an L-1/2 penalty for gene selection in cancer classification problems, and proposed a coordinate descent algorithm with a new univariate half thresholding operator to solve the L-1/2 penalized logistic regression [27]. Marquard et al. used a random forest method and multiple cancer genetic features to identify the primary site of the cancers of unknown primary origin [31]. More recently, Jiao et al. use neural networks model that integrate different features including single nucleotide variation (SNV), copy number alteration (CNA), structural variation (SV) from WGS data to classify the primary and metastasis of cancer cells [32].
Ensemble systems, also called multiple classifier systems, are becoming more and more popular as machine learning methods. They have demonstrated themselves to be very effective and extremely versatile in a broad spectrum of problem domains and real-world applications [33]. Ensemble systems are integrations of multiple machine learning classifiers whose decisions are combined [34]. In this study, we developed an ensemble machine learning model named performance-weighted-voting model based on the voting model. Our ensemble system consisted of five classifiers: logistic regression (LR), SVM, random forest (RF), extreme gradient boosting (XGBoost) and multilayer perceptron (MLP) neural network (NN). Unlike the basic voting model, the weights of the performance-weighted-voting model differ across the weak classifiers (Fig. 1). What’s more, each classifier’s weights across cancer types are different. The weights of the five weak classifiers can be obtained based on their predictive performance by solving linear regression functions. We applied our model to learn and predict 6,249 samples across 14 cancer types from the TCGA somatic mutation data and finally achieved an average accuracy of 71.46%, which was the among the eight models mentioned in our study. In addition, our model can theoretically promote any combination of weak classifiers with a high degree of accuracy.
Figure 1. The workflow of the performance-weighted-voting model.
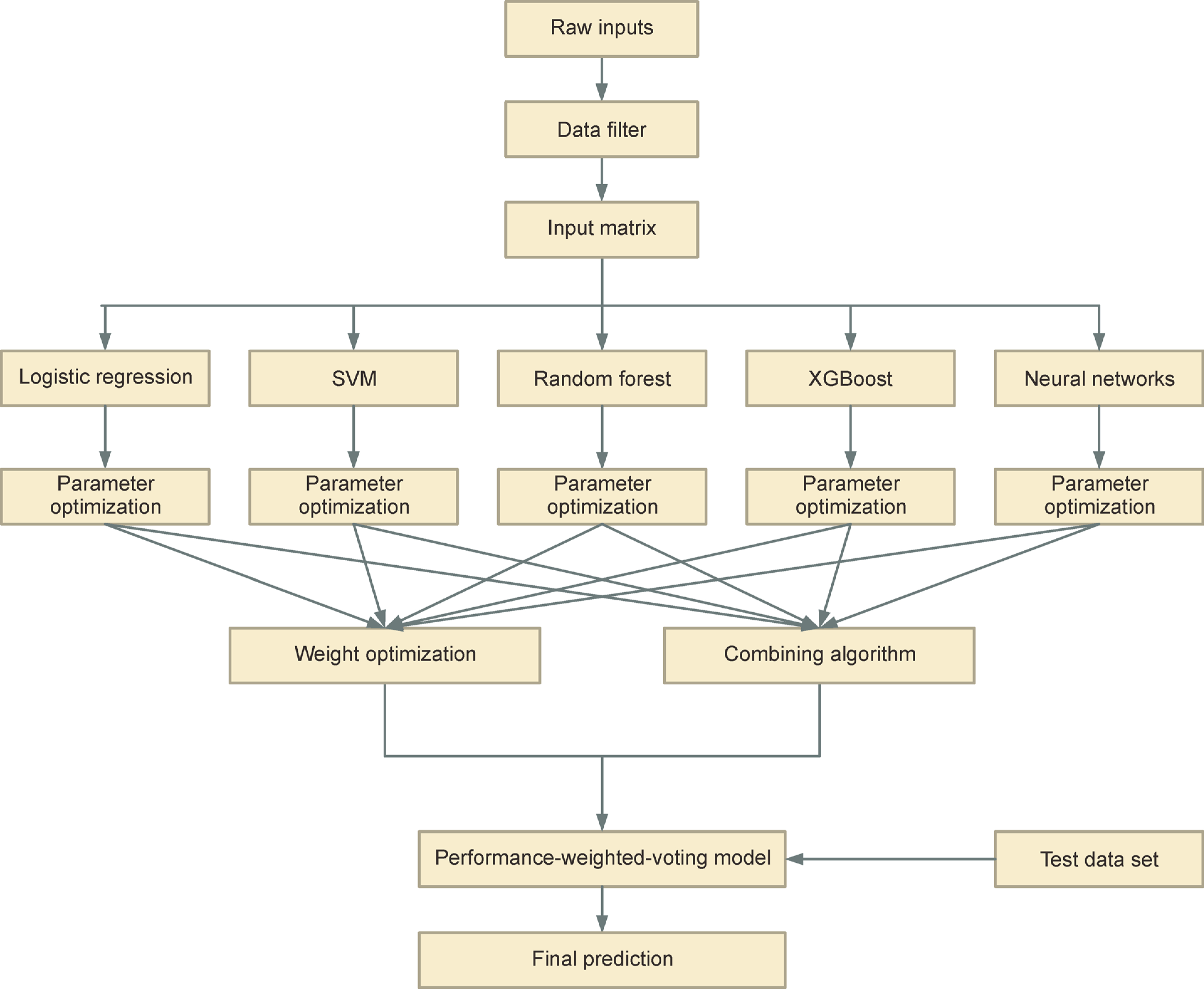
The performance-weighted-voting model integrates five classifiers including logistic regression, SVM, random forest, XGBoost and neural networks. We first used cross-validation to get the predicted results for the five classifiers. The weights of the five weak classifiers can be obtained based on their predictive performance by solving linear regression functions. The final predicted probability of the performance-weighted-voting model for a cancer type can be determined by the summation of each classifier’s weight multiplied by its predicted probability.
RESULTS
Data learning using five machine learning classifiers
We used mutation count per gene as the input feature to train the classifiers. The classifiers calculated the probability that belongs to each of the 14 cancer types through discriminative functions and output the cancer type that achieved the highest probability (see “Materials and Methods”). Figure 2 displays the overall predictive performance of the test set by the five classifiers with optimal parameters. Among the five classifiers, the logistic regression classifier (mean = 68.67%, SD = 1.21%) and the neural networks classifier (mean = 68.07%, SD = 0.94%) performed best, and the SVM (mean = 63.74%, SD = 0.72%) and XGBoost classifiers (mean = 62.89%, SD = 1.43%) followed closely. In contrast, the overall accuracy of the random forest classifier was only 54.79% (SD = 1.64%) that performed worse than the other classifiers.
Figure 2. The predictive performance for the five classifiers with optimal parameters.
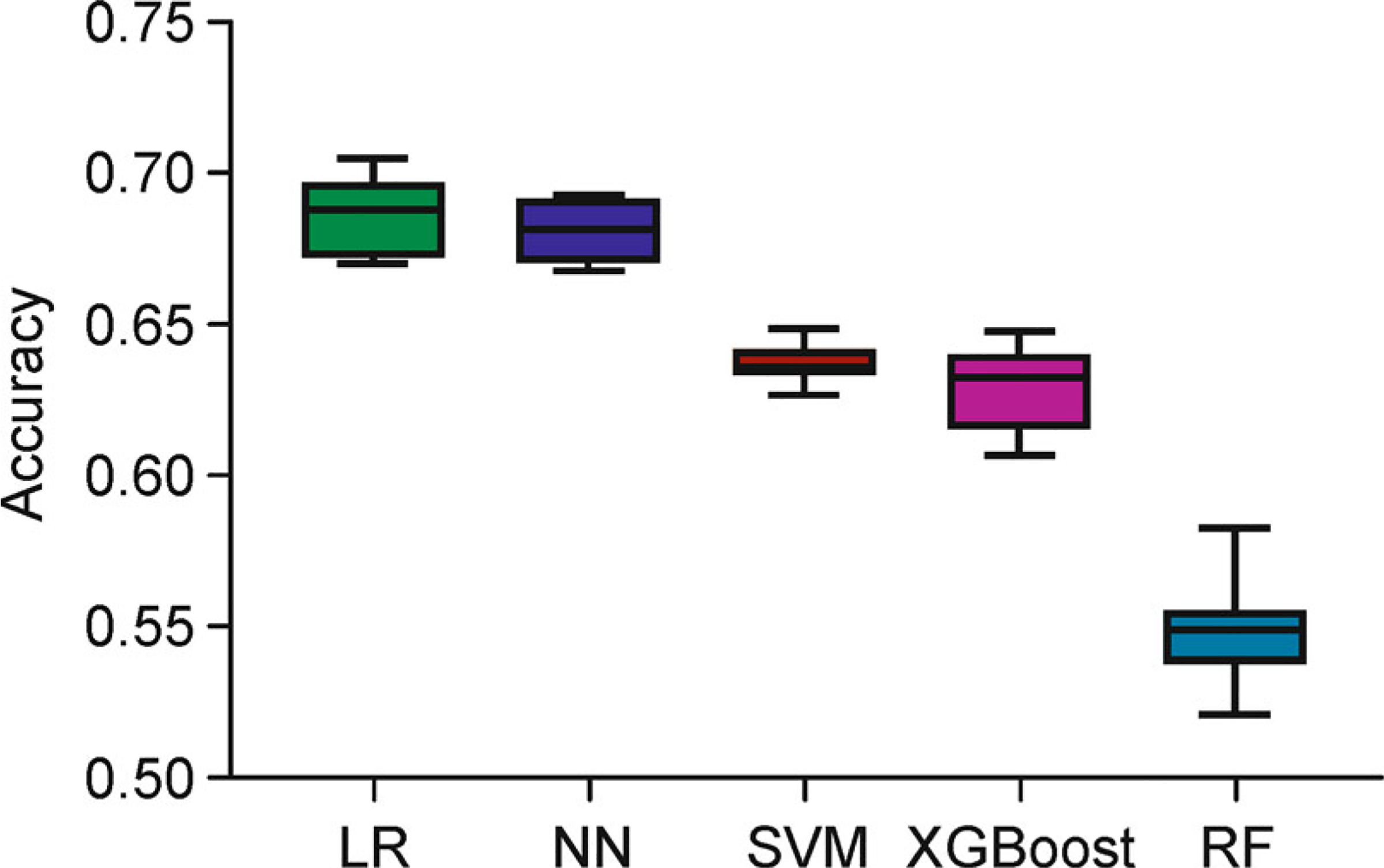
Five classifiers, logistic regression (LR, green box), neural networks (NN, blue box), support vector machine (SVM, brown box), extreme gradient boosting (XGBoost, purple box) and random forest (RF, steel blue box) were selected. Three-fold cross-validation was used to optimize the top parameters of each classifier. Each model was trained and predicted 10 times independently.
The precision, recall and F1-score among the five classifiers are similar to their overall accuracies (Supplementary Table S1). The logistic regression classifier (precision = 71.13%, recall = 68.08%, F1-score = 68.84%) and neural networks (precision = 69.80%, recall = 67.65%, F1-score = 68.14%) achieved higher scores than SVM (precision = 70.73%, recall = 62.28%, F1-score = 64.39%), XGBoost (precision = 64.83%, recall = 61.50%, F1-score = 62.40%) and random forest (precision = 60.47%, recall = 54.36%, F1-score = 53.00%). In particular, in most cancer types, the average F1-scores of logistic regression classifier and neural network classifier are the top two highest. Additionally, the F1-scores vary largely among different cancer types. Three cancer types, LGG, SKCM and THCA, achieved F1-scores greater than 80% in at least one classifier. In contrast, another three cancer types, HNSC, PRAD and STAD, performed poorly in all five classifiers, with no classifier achieving an F1-score greater than 60%.
Data learning using upgraded machine learning models
To improve the predictive accuracy, we considered to use ensemble methods by integrating the five classifiers for further prediction. We first applied two well-known models: the hard-voting model and the soft-voting model. Comparing the predictive performance of the two voting models with the five classifiers, the overall accuracy of both hard-voting model (69.06%; SD = 1.33%) and the soft-voting model (69.66%; SD = 1.37%) were significantly higher (P-value<0.05, Wilcoxon rank-sum test) than any of the five classifiers. In both the two voting models, their weight to the weak classifiers are equal. To address this issue, we developed a weighted voting model: the performance-weighted-voting model. The performance-weighted-voting model can strengthen the power of the weak classifier that has better predictive performance by allocating a higher weight (see “Materials and Methods”). The average overall accuracy of the performance-weighted-voting model reaches 71.46% (SD = 1.02%), which is significantly higher (P-value = 2.5×10−3, Wilcoxon rank-sum test) than the soft-voting model, the second highest model (Table 1 and Supplementary Fig. S1). The average precision, recall and F1-score of the performance-weighted-voting model are 72.67%, 70.97% and 72.02%, which is also significantly higher than the hard-voting model (72.25%, 68.35%, 69.24%), soft-voting model (72.08%, 68.49%, 69.36%) and five weak classifiers (Supplementary Table S1). In particular, the F1-score of the performance-weighted-voting model reached 60% across all cancer types except STAD.
Table 1.
The predictive results for the eight models
| Classifier | Accuracy |
|---|---|
| Logistic regression | 68.67%a±1.21%b |
| SVM | 63.74%±0.72% |
| Random forest | 54.79%±1.64% |
| XGBoost | 62.89%±1.43% |
| Neural network | 68.07%±0.94% |
| Hard-voting | 69.06%±1.33% |
| Soft-voting | 69.66%±1.37% |
| Performance-weighted-voting | 71.46%±1.02% |
The average number of 10 repeats.
The standard deviation of 10 repeats.
Theoretically, the hard-voting and the soft-voting models perform well only when all weak classifiers can achieve high overall accuracies. In contrast, the performance-weighted-voting model only relies on the highest accuracy of the weak classifiers in each specific cancer type. The model can filter the classifiers automatically by allocating different weights using a linear regression model. The performance-weighted-voting model would perform better if more weak classifiers were integrated.
Predictive pattern analysis of performance-weighted-voting model across cancer types
To evaluate the causes of misclassification predicted by the performance-weighted-voting model, we compared the different mutation count per sample between the correctly classified group and the misclassified group. The average mutation count per sample of the correctly classified group (215.18) is significantly higher (P-value = 5.4×10−3, Wilcoxon rank-sum test) than the misclassified group (142.27). More specificity, in 10 of 14 cancer types the average mutation count per sample of correctly classified groups was significantly higher (P-value < 0.05, Wilcoxon rank-sum test) than misclassified groups (Fig. 3). By contrast, in only 3 cancer types the average mutation count per sample of correctly classified groups was significantly lower (P-value < 0.05, Wilcoxon rank-sum test) than misclassified groups. The different mutation count between two groups implies that tumor mutation burden (TMB) as a cancer type-specific feature reveals a positive correlation to the predictive accuracy [35]. A deeper understanding and utilization of the inner relationships will help to improve the predictive accuracy of our model.
Figure 3. Mutation count comparison between correctly classified samples and misclassified samples.
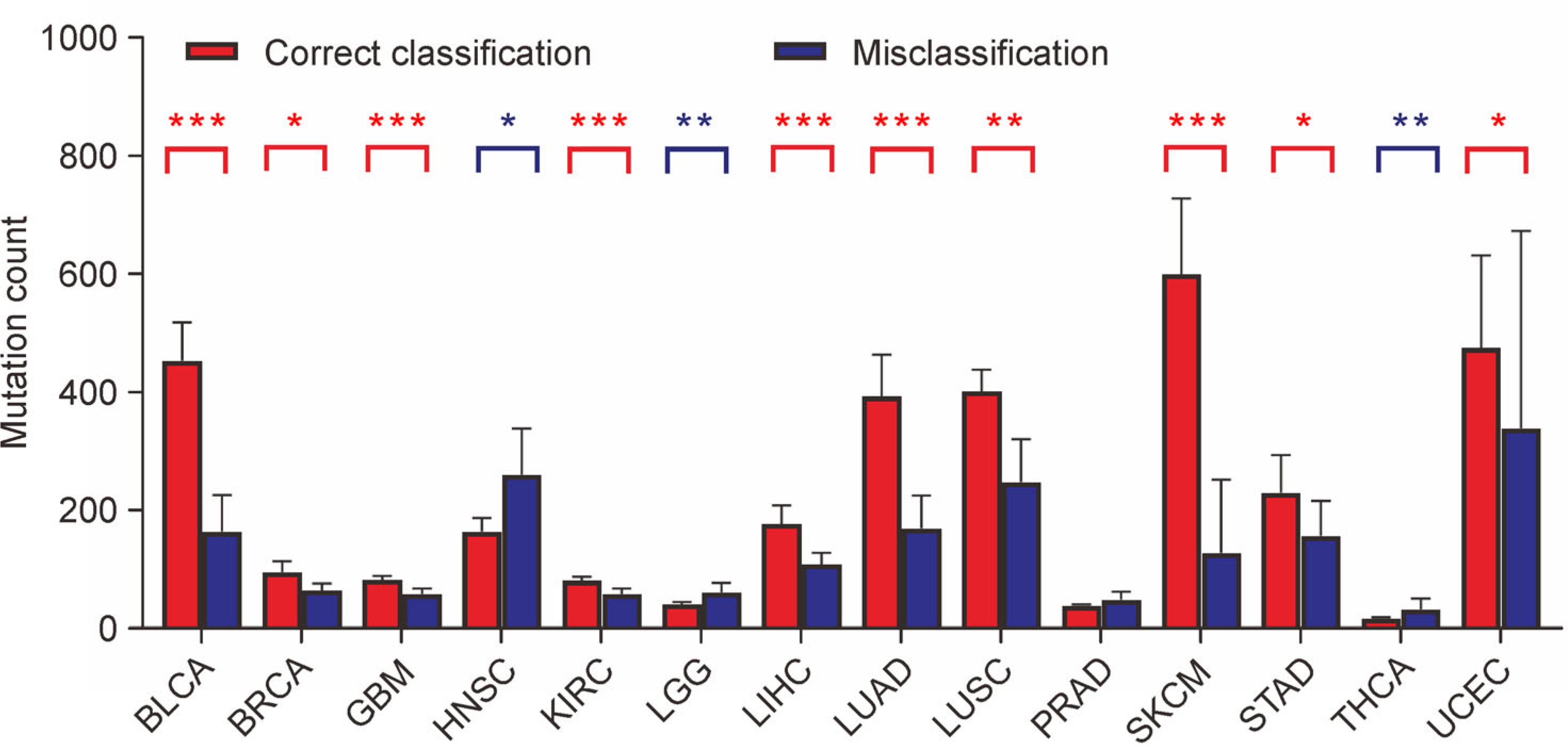
In 10 of the 14 cancer types, the average mutation count of correctly classified samples is significantly higher than misclassified samples (red asterisks above the bars). In contrast, in 3 of the 14 cancer types, the average mutation count of correctly classified samples is significantly lower than misclassified samples (blue asterisks above the bars). The height of each bar represents the average mutation count, and the error bar is the 95% confidence interval. *: P < 0.05, **: P < 0.01, ***: P < 0.001.
We explored the confusion matrix for the performance-weighted-voting model to analyze the patterns of misclassification (Table 2). Based on the confusion matrix, the model is most confused in distinguishing between BRCA ~ PRAD, LUSC ~ LUAD and HNSC ~ LUSC, which at least eight samples were misclassified to the other cancer type in both of the two cancer types. The confusions in distinguishing between GBM ~ LGG and TRAD ~ PHCA are intermediate. Some of these confusions are informative. The LUAD and LUSC are both lung cancers and the LGG and GBM are both brain cancers, these cancer cells share common developmental origins [36]. Though BRCA and PRAD cancer cells arise in organs that are different, they are more similar than different and driven by steroid hormone signaling [37,38]. In addition, we found that the proportion of the patients who are current smoker or current reformed smoker for no more than 15 years of the misclassified samples in misclassified samples is significantly higher (P-value = 0.0249, Chi-squared test) than correctly classified samples between HNSC and LUSC cancer pair, and similarly for LUSC ~ LUAD cancer pair (P-value = 0.0076, Chi-squared test). Tobacco smoking as an externality can change the characteristics and mutation signature [39] of these cancer types which may interfere with the prediction of classifiers.
Table 2.
The confusion matrix of the test set using performance-weighted-voting modela
| BLCA | BRCA | GBM | HNSC | KIRC | LGG | LIHC | LUAD | LUSC | PRAD | SKCM | STAD | THCA | UCEC | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BLCA | 47 | 3 | 0 | 10 | 0 | 0 | 4 | 1 | 2 | 5 | 0 | 6 | 1 | 1 |
| BRCA | 1 | 106 | 2 | 8 | 1 | 1 | 9 | 2 | 0 | 19 | 0 | 2 | 6 | 5 |
| GBM | 0 | 2 | 36 | 2 | 0 | 6 | 0 | 0 | 0 | 2 | 2 | 0 | 0 | 0 |
| HNSC | 3 | 4 | 1 | 68 | 0 | 1 | 1 | 1 | 10 | 5 | 1 | 6 | 0 | 1 |
| KIRC | 2 | 3 | 1 | 0 | 50 | 0 | 1 | 0 | 0 | 12 | 1 | 0 | 0 | 1 |
| LGG | 0 | 1 | 9 | 1 | 0 | 78 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| LIHC | 1 | 5 | 0 | 6 | 4 | 3 | 47 | 0 | 0 | 5 | 0 | 2 | 0 | 2 |
| LUAD | 1 | 4 | 1 | 9 | 1 | 3 | 1 | 55 | 11 | 12 | 0 | 3 | 2 | 0 |
| LUSC | 2 | 1 | 1 | 9 | 0 | 0 | 2 | 8 | 65 | 0 | 0 | 6 | 3 | 0 |
| PRAD | 0 | 10 | 2 | 1 | 0 | 2 | 0 | 0 | 0 | 80 | 0 | 3 | 6 | 0 |
| SKCM | 0 | 0 | 3 | 0 | 0 | 0 | 1 | 1 | 0 | 3 | 60 | 2 | 2 | 0 |
| STAD | 2 | 6 | 0 | 2 | 1 | 0 | 6 | 2 | 1 | 3 | 0 | 43 | 0 | 0 |
| THCA | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | 1 | 0 | 81 | 0 |
| UCEC | 3 | 8 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 2 | 0 | 2 | 0 | 70 |
Each row corresponds to the true cancer type; each column corresponds to the class predictions from performance-weighted-voting model.
We analyzed the final predictive probability across 14 cancer types for each of the samples, with 10 misclassified samples achieving a prediction probability greater than 70%. Among the 10 misclassified samples, four samples were GBM but predicted as LGG, three samples were LUAD but predicted as LUSC and three samples were HNSC but predicted as LUSC. We also scanned the predicted outcomes of five weak classifiers. To our surprise, in nearly 14% (52) of the misclassified cases, all five classifiers offered the same prediction (Supplementary Table S1). In particular, of the 52 samples, six HNSC samples were derived into LUSC, four GBM samples were derived into LGG, four LGG samples were derived into GBM and three LUAD samples were derived into LUSC. These errors are consistent with the results of confusion matrix.
The predictive performance across different mutation type subsets
The MAF file contains 16 types of somatic mutations flagged by various calling software packages. Typically, most driver mutations are nonsynonymous mutations. These mutations are believed to have greater effects than synonymous mutations for tumorigenesis and cell evolution. To assess whether using different mutation types can improve the accuracy in cancer type classification, we used four different subsets of mutations (“missense mutation group”, “synonymous nonsynonymous mutation group”, “high impact mutation group” and “total mutation group”) as input features based on their different impacts on cancer evolution (see “Materials and Methods”).
Figure 4 presents the predictive results for the four cancer type groups. The overall accuracy of the “missense mutation group” as well as the “synonymous nonsynonymous mutation group” are significantly lower than the other two groups. The overall accuracies of the eight classifiers range from 44.31% to 57.86% in “missense mutation group” and from 49.39% to 63.84% in “synonymous nonsynonymous mutation group”. The distributions of the “high impact mutation group” (55.64%–68.86%) are very close to the “total mutation group” (54.79%–71.46%), but still lower. In general, the test results have demonstrated that using more mutation types helps to improve the predictive accuracy. One interpretation is that though some passenger mutations (e.g., synonymous mutations) do not affect cancer growth directly, they still provide useful information for cancer type classification through hitchhiking effect. Usually, a driver mutation provides a fitness advantage to cancer cells. The frequency of the adaptive mutation can be high because of the positive selection. In the meantime, the frequencies of the genetically linked passenger mutations are also increased accompanied by the driver mutation due to the linkage disequilibrium [40], which will strengthen the genetic characteristics of a cancer type. Furthermore, the overall accuracies of the eight classifiers reveal that the performance-weighted-voting model performed better than the other classifiers across all mutation type groups (Fig. 4).
Figure 4. The overall accuracy of the input features using different groups of mutation types.
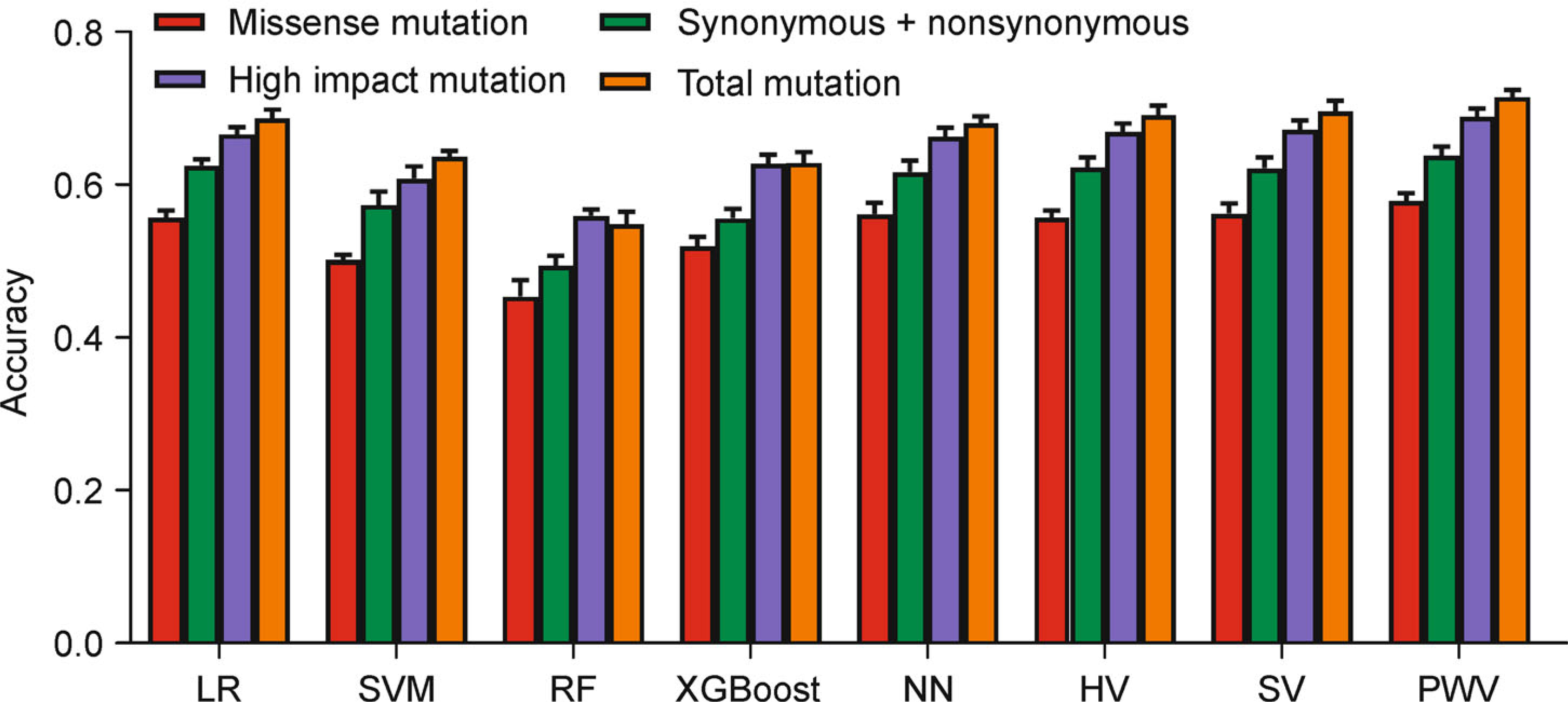
Four groups of mutation types, “missense mutation group” (red bar), “synonymous nonsynonymous mutation group” (green bar), “high impact mutation group” (blue bar) and “total mutation group” (orange bar), were selected as input features for cancer type classification. Each of the four groups of mutation types were used as input features predicted by the eight classifiers. The height of each bar represents the average number, and the error bar is the standard deviation.
The predictive performance using driver gene set
Studies are trying to use gene panels instead of total genes for cancer research. This trial will potentially enable cost-effective assessment of much larger numbers of samples for deeper biological and predictive insights. To this end, we aimed to test whether we can use less genes to improve the predictive performance of cancer type classification. Cancer driver genes are the genes whose mutations drive tumor growth. Herein, we used the mutation count of each of the 201 driver genes rather than the whole genes (see “Materials and Methods”) as input features. Unfortunately, all five weak classifiers, as well as the three voting models, failed to improve the overall accuracy. As the highest number of the eight classifiers, the overall accuracy models of performance-weighted-voting model (mean = 61.35%, SD = 0.50%) is more than ten percent below the accuracy using the mutation count of each of the total genes as input features (Fig. 5).
Figure 5. The overall accuracy of the input features using driver genes and total genes.
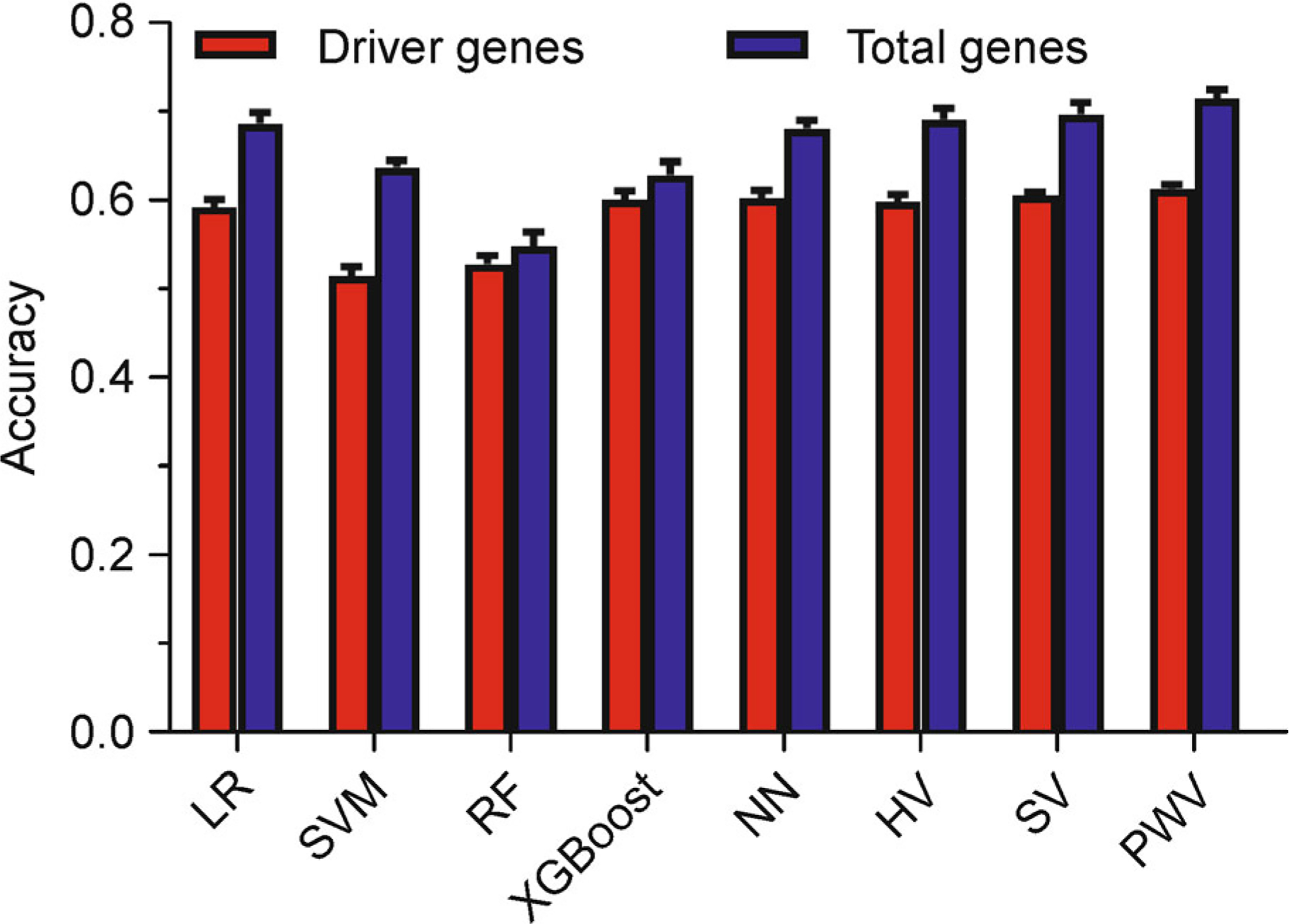
201 driver genes (red bar) were extracted in comparison with total genes (blue bar). Both of the two gene sets were used as input features to predict the cancer types by the eight models. The height of each bar represents the average number, and the error bar is the standard deviation.
DISCUSSION
This study used machine learning methods for cancer type classification for 6,249 samples across 14 cancer types. We attempt to assess, compare and analyze the performance of several classifiers that have been applied to, including logistic regression, support vector machine, random forest, neural networks and XGBoost. We used three-fold cross-validated grid-search over a parameter grid to optimize the parameters of the classifiers. To improve accuracy, we also employed three ensemble models, the hard-voting model, the soft-voting model and the performance-weighted-voting model, integrating the five weak classifiers. Relying on the performance-based methods to train the different weights of each weak classifier in the ensemble system, the overall accuracy of the performance-weighted-voting model reached 71.46%, which was significantly higher than the other classifiers. We used different mutation types based on their effect on cancer evolution for cancer type classification, and concluded that only using all mutation types yielded the highest accuracy (Fig. 4). We also attempted to use a set of driver genes [41] as the input feature, but found no improvement to the overall accuracy (Fig. 5). Our work on cancer type classification is similar to previous studies [32,42], but the two types of studies are different in a couple aspects. First, the machine learning classifiers they used (random forest, neural networks and soft-voting) are existing models, while we developed a new classifier and first proposed the performance-based idea to weight weak classifiers in the ensemble system. Second, the performance-weighted-voting model, as an improved voting model, has demonstrated to be superior to the two standard voting models (the hard-voting model and the soft-voting model) within the same dataset.
To analyze the patterns of misclassification in the performance-weighted-voting model, we divided the predicted data into correctly classified and misclassified subsets and compared the mutation count per sample between the two subsets. The average mutation count of the correctly classified subset is significantly higher than the misclassified subset. More specifically, the average mutation count of correctly classified samples is significantly higher than misclassified samples in 10 of 14 cancer types (Fig. 3). We also discovered that some misclassifications are possibly due to the common developmental origin (LUAD ~ LUSC and GBM ~ LGG) [36], steroid hormone signaling (BRCA ~ PRAD) [37,38], and tobacco smoking (LUAD ~ LUSC and HNSC ~ LUSC) [39]. Others may be due to the algorithms of the classifiers. To address this problem, we need to build more detailed training subsets or integrate more genetic or phenotypic data for cancer type classification.
Cancer of unknown primary site is a heterogeneous group of cancers for which the anatomical site of origin remains occult after detailed investigation [43]. The identification of the cancer of unknown primary, as well as the origin of metastasis, is important but challenging [44]. This study has important clinical significance for identifying the origin of cancer, especially for those where the primary cannot be determined [45]. Considering the occurrence of cancer cell dissemination at the early stages of cancer progression [6,46–49], our model can help to identify the primary of metastatic cancer cell types that are present in the cancer cell genome. In other fields, including circulating tumor cells (CTC) research for cancer metastatic detection, our model also presents the potential for cell detection and predicts the risk of cancer remission [50,51]. In addition, our finding of a positive correlation between TMB and prediction accuracy provides cancer type-specific features [35]. These features may be used to interpret the immunotherapy variances in different cancer types [52,53], which may provide new strategies for cancer therapy.
MATERIALS AND METHODS
TCGA mutation data
The MAF file containing WES somatic mutations from 10,295 samples across 33 cancer types was downloaded from TCGA. Mutations were called by seven software packages (MuTect, MuSE, VarScan2, Radia, Pindel, Somatic Sniper and Indelocator) from Multi-Center Mutation Calling in Multiple Cancers (MC3) working group [54]. All PASS somatic variants referred by two or more variant calling software packages were extracted. 344 hypermutator samples were excluded as artifactual sensitivity to high background mutation rates might perturb the prediction of classifiers. 705 samples marked as “mutation call filter”, 167 samples marked as “pathology review” and 75 samples marked as “RNA degradation” referred by Bailey et al. [41] were also excluded. To ensure a high quality of the learning dataset, we preferred a minimum cutoff of 300 samples per cancer type. Ultimately, our cancer type classification dataset consisted of 1,174,111 SNPs from 6,249 samples across 14 cancer types.
Machine learning classifiers
Five well-known machine learning classifiers were employed for cancer type classification, including logistic regression, support vector machine, random forest, extreme gradient boosting, and multilayer perceptron neural networks.
The logistic regression classifier is a classification method used to assign observations to a discrete set of classes. It builds a regression model to predict the probability that a given data entry belongs to the category using the sigmoid function. The dimension of the input vector is known as features or predictors. The model was implemented using the Python package sklearn with the LogisticRegression function.
Unlike the logistic classifier, a support vector machine [55] classifier can use a kernel function to map the input vectors into high-dimensional feature spaces implicitly and compute a maximum-margin hyperplane decision surface that separates the classes. This hyperplane has numerous statistical characteristics. Capabilities of SVM classifiers can be further expanded by kernel tricks by creating nonlinear decision boundaries [56]. The model was implemented using the Python package sklearn with the SVC function.
Random forest classifier [57] is a strong classifier named forest consisting of many weak decision trees that can obtain better performance than a single tree. Each decision tree is trained using a new training data set which is produced by random sampling with replacement from the original data set, i.e., a case may be sampled many times in a new training data set. The final decision is made via a majority vote from the decision trees in the forest. The model was implemented using the Python package sklearn with the RandomForestClassifier function.
Extreme gradient boosting [58] classifier is also a strong learner that combines a set of weak decision trees, but differs from random forest. In the random forest classifier, the training data set is randomly sampled as a replacement from the original data set. In contrast, in the extreme gradient boosting classifier, the training data set of the new decision tree is the residual between the predictive result of the previous decision trees and the correct result. The extreme gradient boosting is a computationally efficient variant of the gradient boosting algorithm. The model was implemented using the Python package xgboost with the XGBClassifier function.
Multilayer perceptron neural network classifier is a nonlinear model consisting of multiple neurons that can learn and generate a class of functions from the training data set. Each neuron weights the input nodes and generates the output by employing nonlinear activation mathematical functions. The linear combination is formed by perceptron through the computation of an output neuron from multiple real-valued inputs [59]. The model was implemented using the Python package sklearn with the MLPClassifier function.
Model training and parameter optimization
The 6,249 cases were split into a training set and a test set with a ratio of 80% to 20%. The training set was used for training the classifiers and optimizing the parameters while the test set was only used for final prediction. A three-fold cross-validated grid-search over a parameter grid was applied to optimize the parameters of a classifier. The training set was split into three subsets, two were used as training subsets and one was used as a validating subset by turns. The final prediction of the test set was based on the optimal parameters. The optimization was calculated using the Python package sklearn with the GridSearchCV function.
Evaluation metrics of classification performance
To evaluate the performance of the models, overall accuracy, precision, recall (sensitivity), and F1-score were applied to quantitatively assess the predictive performance. Accuracy measures the proportion of cases in correct assignments. Recall (also called true positive rate) measures the proportion of actual positives that are correctly identified to that type. Precision measures the proportion of samples assigned to a type that is correctly identified as that type. The F1-score is the harmonic means of recall and precision that combines precision and recall in a statistically more meaningful way. Let TP, TN, FP and FN denote the number of true positives, true negatives, false positives and false negatives, respectively. The evaluation metrics can be expressed as:
Upgraded ensemble machine learning models
An ensemble model can integrate multiple weak learning classifiers with the aim of obtaining better predictive performance than any of the constituent weak learning classifiers alone. Five classifiers, logistic regression, SVM, random forest, XGBoost and neural networks, were chosen as the weak classifiers in our ensemble model. The two voting models (hard-voting and soft-voting) were first considered as an ensemble classifier for cancer type classification. In both of the two voting models, the weights of the five classifiers are equal, indicating that we cannot make full use of the different predictive performances of the weak classifiers across the 14 cancer types. To address this issue, we developed a new ensemble model called the performance-weighted-voting model. The weights of the performance-weighted-voting model differ among the weak classifiers based on their predictive performance. Specifically, each weak classifier is allocated to different weights across the 14 cancer types, and each weight is dependent on the specific predictive performance of the corresponding cancer types.
The performance-weighted-voting model consists of three steps: parameter optimization, weights optimization and final prediction (Fig. 1). The first two steps can be learned by the training set and the final prediction is tested by the test set. In the parameter optimization step, we used the grid-search method by setting hyperparameters for each classifier and selecting the combination with the highest accuracy. To achieve the optimal weights of the five classifiers, we built a linear regression model. Let xi,j,n and yi,j,n denote the predicted probability and true state of classifier j, cancer type i and sample n, respectively. Here, 0⩽xi,j,n⩽1 and yi,n∈{0,1} is a one-hot matrix where yi,n=1 represents sample n belongs to cancer type i and for any n, . Let wi,j as the weight of classifier j and cancer type i. To obtain the five weights to cancer type i, we expect the weights satisfy linear regression functions,
for all sample n in training set and all i in cancer types. Using the least square method to solve the functions, the weight vector, Wi=(wi, LR, wi, SVM, wi, RF, wi, XGBoost, wi, NN), of cancer type i can be expressed as
The vector Yi=(yi, 1, yi, 2, ···, yi, N), where N is the sample size. The vector , where . And Zi is the normalization factor. The third step is the final prediction the test set. Denote pi, j as predicted probability of classifier j and cancer type i in test set. The score of the predicted cancer sample that belongs to cancer type i is
The predictive probability of the performance-weighted-voting model yields pi=Si/S, where is the summation of the scores that belong to the 14 cancer types.
Different mutation type subsets as input features
The MAF file contains 16 types of somatic mutations (Missense_Mutation, Silent, Nonsense_Mutation, Intron, 3′UTR, 5′UTR, Splice_Site, RNA, Frame_Shift_Ins, Frame_Shift_Del, In_Frame_Ins, Nonstop_Mutation, In_Frame_Del, 3′Flank, 5′Flank, Translation_Start_Site) flagged by variant calling software packages. Consider that most driver mutations are nonsynonymous mutations, and in most cases nonsynonymous mutations play more important roles than synonymous mutations in tumorigenesis. To evaluate whether these mutation types have positive or negative effects on cancer type classification, we selected four different subsets of mutations as input features according to their mutation types for cancer type classification. The four groups were “missense mutation group” (Missense_Mutation), “synonymous nonsynonymous mutation group” (Missense_Mutation, Silent, Nonsense_Mutation), “high impact mutation group” (Missense_Mutation, Nonsense_Mutation, Translation_Start_Site, Frame_Shift_Del, Nonstop_Mutation, Frame_Shift_Ins, In_Frame_Del, Splice_Site, In_Frame_Ins) and “total mutation group”.
Driver gene extraction as an input feature
The txt file (Mutation.CTAT.3D.Scores.txt) that characterizes the cancer driver genes of the mutations in the MAF file was downloaded from Bailey et al. [41]. Genes that were flagged as having at least two of the three columns “New_Linear (functional) flag”, “New_Linear (cancer-focused) flag” and “New_3D mutational hotspot flag” were selected as driver genes. A total of 201 genes met the requirement.
DATA AVAILABILITY
The TCGA MC3 Public MAF file and the txt file are available at https://gdc.cancer.gov/about-data/publications/pancan-driver. The codes of the models used in this study are available online at GitHub (https://github.com/duckliyawei/performance-weighted-voting).
Supplementary Material
Author summary:
The identification of the cancer of unknown primary is of clinical significance, and can provide important cancer behavioral therapeutic strategies. To achieve this, we developed an ensemble machine learning system called the performance-weighted-voting model for cancer type classification. The ensemble system can integrate weak classifiers and train the weights of the weak classifiers based on their predictive performance. The model has achieved the highest overall accuracy among the models mentioned in this study. Furthermore, the model can theoretically promote any combination of weak classifiers with a high degree of accuracy.
ACKNOWLEDGEMENTS
We thank Chengsheng Mao for the comments and suggestions during the preparation of the manuscript. We thank Xin Wu for their helps in the artwork of figures. This study is supported in part by NIH grant R21LM012618.
Footnotes
COMPLIANCE WITH ETHICS GUIDELINES
The authors Yawei Li and Yuan Luo declare that they have no conflict of interests.
This article does not contain any study materials with human or animal subjects performed by any of the authors.
SUPPLEMENTARY MATERIALS
The supplementary materials can be found online with this article at https://10.1007/s40484–020-0226–1.
REFERENCES
- 1.Vogelstein B and Kinzler KW (2004) Cancer genes and the pathways they control. Nat. Med, 10, 789–799 [DOI] [PubMed] [Google Scholar]
- 2.Knudson AG (2002) Cancer genetics. Am. J. Med. Genet, 111, 96–102 [DOI] [PubMed] [Google Scholar]
- 3.Ling S, Hu Z, Yang Z, Yang F, Li Y, Lin P, Chen K, Dong L, Cao L, Tao Y, et al. (2015) Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc. Natl. Acad. Sci. USA, 112, E6496–E6505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Li Y, Li T, Shen X, Zhu T, Tao Y, Li X, Wang D, Ma Q, Hu Z, et al. (2019) Genetic load and potential mutational meltdown in cancer cell populations. Mol. Biol. Evol, 36, 541–552 [DOI] [PubMed] [Google Scholar]
- 5.Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, Karchin R, Kinzler KW, Vogelstein B and Nowak MA (2010) Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA, 107, 18545–18550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu Z, Ding J, Ma Z, Sun R, Seoane JA, Scott Shaffer J, Suarez CJ, Berghoff AS, Cremolini C, Falcone A, et al. (2019) Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet, 51, 1113–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. (2010) Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature, 467, 1114–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yates LR, Knappskog S, Wedge D, Farmery JHR, Gonzalez S, Martincorena I, Alexandrov LB, Van Loo P, Haugland HK, Lilleng PK, et al. (2017) Genomic evolution of breast cancer metastasis and relapse. Cancer Cell, 32,169–184 eT [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varadhachary GR and Raber MN (2014) Cancer of unknown primary site. N. Engl. J. Med, 371, 757–765 [DOI] [PubMed] [Google Scholar]
- 10.Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabé RR, Bhan MK, Calvo F, Eerola I, Gerhard DS, et al. (2010) International network of cancer genome projects. Nature, 464, 993–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C and Stuart JM (2013) The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet, 45, 1113–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. (2020) Pan-cancer analysis of whole genomes. Nature, 578, 82–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. (2013) Signatures of mutational processes in human cancer. Nature, 500, 415–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burrell RA, McGranahan N, Bartek J and Swanton C (2013) The causes and consequences of genetic heterogeneity in cancer evolution. Nature, 501, 338–345 [DOI] [PubMed] [Google Scholar]
- 15.Cicchetti DV (1992) Neural networks and diagnosis in the clinical laboratory: state of the art. Clin. Chem, 38, 9–10 [PubMed] [Google Scholar]
- 16.Cochran AJ (1997) Prediction of outcome for patients with cutaneous melanoma. Pigment Cell Res, 10, 162–167 [DOI] [PubMed] [Google Scholar]
- 17.Cruz JA and Wishart DS (2007) Applications of machine learning in cancer prediction and prognosis. Cancer Inform, 2, 59–77 [PMC free article] [PubMed] [Google Scholar]
- 18.Kourou K, Exarchos TP, Exarchos KP, Karamouzis MV and Fotiadis DI (2015) Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J, 13, 8–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eraslan G, Avsec Ž, Gagneur J and Theis FJ (2019) Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet, 20, 389–403 [DOI] [PubMed] [Google Scholar]
- 20.Fakoor R, Ladhak F, Nazi A, Huber M (2013) Using deep learning to enhance cancer diagnosis and classification. In: 2018 IEEE International Conference on System, Computation, Automation and Networking (icscan), Pondicherry, pp.1–6 [Google Scholar]
- 21.Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RC, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, et al. (2002) Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat. Med, 8, 68–74 [DOI] [PubMed] [Google Scholar]
- 22.Brown MPS, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, Ares M Jr and Haussler D (2000) Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl. Acad. Sci. USA, 97, 262–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science, 286, 531–537 [DOI] [PubMed] [Google Scholar]
- 24.Ben-Dor A, Bruhn L, Friedman N, Nachman I, Schummer M and Yakhini Z (2000) Tissue classification with gene expression profiles. J. Comput. Biol, 7, 559–583 [DOI] [PubMed] [Google Scholar]
- 25.Danaee P, Ghaeini R and Hendrix DA (2017) A deep learning approach for cancer detection and relevant gene identification. Pac. Symp. Biocomput, 22, 219–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Tetko IV, Hall MA, Frank E, Facius A, Mayer KF and Mewes HW (2005) Gene selection from microarray data for cancer classification‒a machine learning approach. Comput. Biol. Chem, 29, 37–46 [DOI] [PubMed] [Google Scholar]
- 27.Liang Y, Liu C, Luan XZ, Leung KS, Chan TM, Xu ZB and Zhang H (2013) Sparse logistic regression with a L1/2 penalty for gene selection in cancer classification. BMC Bioinformatics, 14, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng Z, Vo AH, Mao C, Clare SE, Khan SA and Luo Y (2019) Cancer classification and pathway discovery using non-negative matrix factorization. J. Biomed. Inform, 96, 103247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milanez-Almeida P, Martins AJ, Germain RN and Tsang JS (2020) Cancer prognosis with shallow tumor RNA sequencing. Nat. Med, 26, 188–192 [DOI] [PubMed] [Google Scholar]
- 30.Moran S, Martínez-Cardús A, Sayols S, Musulén E, Balañá C, Estival-Gonzalez A, Moutinho C, Heyn H, Diaz-Lagares A, de Moura MC, et al. (2016) Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol, 17, 1386–1395 [DOI] [PubMed] [Google Scholar]
- 31.Marquard AM, Birkbak NJ, Thomas CE, Favero F, Krzystanek M, Lefebvre C, Ferté C, Jamal-Hanjani M, Wilson GA, Shafi S, et al. (2015) TumorTracer: a method to identify the tissue of origin from the somatic mutations of a tumor specimen. BMC Med. Genomics, 8, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiao W, Atwal G, Polak P, Karlic R, Cuppen E, Danyi A, de Ridder J, van Herpen C, Lolkema MP, Steeghs N, et al. (2020) A deep learning system accurately classifies primary and metastatic cancers using passenger mutation patterns. Nat. Commun, 11, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang C, Ma Y (2012) Ensemble Machine Learning: Methods and Applications. New York: Springer-Verlag [Google Scholar]
- 34.Tan AC and Gilbert D (2003) Ensemble machine learning on gene expression data for cancer classification. Appl. Bioinformatics, 2, S75–S83 [PubMed] [Google Scholar]
- 35.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, et al. (2017) Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med, 9, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al. (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell, 164, 550–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Risbridger GP, Davis ID, Birrell SN and Tilley WD (2010) Breast and prostate cancer: more similar than different. Nat. Rev. Cancer, 10, 205–212 [DOI] [PubMed] [Google Scholar]
- 38.Long MD and Campbell MJ (2015) Pan-cancer analyses of the nuclear receptor superfamily. Nucl. Receptor Res, 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, et al. (2016) Mutational signatures associated with tobacco smoking in human cancer. Science, 354, 618–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hartl DL and Clark AG (2007) Principles of Population Genetics. Sunderland: Sinauer Associates [Google Scholar]
- 41.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. (2018) Comprehensive characterization of cancer driver genes and mutations. Cell, 174, 1034–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee K, Jeong HO, Lee S and Jeong WK (2019) CPEM: Accurate cancer type classification based on somatic alterations using an ensemble of a random forest and a deep neural network. Sci. Rep, 9, 16927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.ESMO Guidelines Task Force. (2005) ESMO Minimum Clinical Recommendations for diagnosis, treatment and follow-up of cancers of unknown primary site (CUP). Ann. Oncol, 16, i75–i76 [DOI] [PubMed] [Google Scholar]
- 44.Mnatsakanyan E, Tung WC, Caine B and Smith-Gagen J (2014) Cancer of unknown primary: time trends in incidence, United States. Cancer Causes Control, 25, 747–757 [DOI] [PubMed] [Google Scholar]
- 45.Pavlidis N, Khaled H and Gaafar R (2015) A mini review on cancer of unknown primary site: A clinical puzzle for the oncologists. J. Adv. Res, 6, 375–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sänger N, Effenberger KE, Riethdorf S, Van Haasteren V, Gauwerky J, Wiegratz I, Strebhardt K, Kaufmann M and Pantel K (2011) Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. Int. J. Cancer, 129, 2522–2526 [DOI] [PubMed] [Google Scholar]
- 47.Hosseini H, Obradović MMS, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, Nanduri LK, Werno C, Ehrl C, Maneck M, et al. (2016) Early dissemination seeds metastasis in breast cancer. Nature, 540, 552–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, et al. (2012) EMT and dissemination precede pancreatic tumor formation. Cell, 148, 349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hüsemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmüller G, et al. (2008) Systemic spread is an early step in breast cancer. Cancer Cell, 13, 58–68 [DOI] [PubMed] [Google Scholar]
- 50.Svensson CM, Hübler R and Figge MT (2015) Automated classification of circulating tumor cells and the impact of interobsever variability on classifier training and performance. J. Immunol. Res, 2015, 573165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lannin TB, Thege FI and Kirby BJ (2016) Comparison and optimization of machine learning methods for automated classification of circulating tumor cells. Cytometry A, 89, 922–931 [DOI] [PubMed] [Google Scholar]
- 52.Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA and Kurzrock R (2017) Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther, 16, 2598–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A, et al. (2019) Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet, 51, 202–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. (2018) Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst, 6, 271–281.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cortes C and Vapnik V (1995) Support-vector networks. Mach. Learn, 20, 273–297 [Google Scholar]
- 56.Li A, Zhang J and Zhou Z (2014) PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics, 15, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Breiman L (2001) Random forests. Mach. Learn, 45, 5–32 [Google Scholar]
- 58.Chen T and Guestrin C (2016) XGBoost: A scalable tree boosting system. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pp.785–794 [Google Scholar]
- 59.Ting FF and Sim KS (2017) Self-regulated multilayer perceptron neural network for breast cancer classification. In: 2017 International Conference on Robotics, Automation and Sciences (Icoras), Melaka, pp.1–5 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The TCGA MC3 Public MAF file and the txt file are available at https://gdc.cancer.gov/about-data/publications/pancan-driver. The codes of the models used in this study are available online at GitHub (https://github.com/duckliyawei/performance-weighted-voting).