

ABSTRACT
Crohn’s disease (CD) is a major form of inflammatory bowel disease characterized by transmural inflammation along the alimentary tract. Changes in the microbial composition and reduction in species diversity are recognized as pivotal hallmarks in disease dynamics, challenging the gut barrier function and shaping a pathological immune response in genetically influenced subjects. The purpose of this review is to delve into the modification of the gut microbiota cluster network during CD progression and to discuss how this shift compromises the gut barrier integrity, granting the translocation of microbes and their products. We then complete the scope of the review by retracing gut microbiota dysbiosis interactions with the main pathophysiologic factors of CD, starting from the host’s genetic background to the immune inflammatory and fibrotic processes, providing a standpoint on the lifestyle/exogenous factors and the potential benefits of targeting a specific gut microbiota.
KEYWORDS: Crohn’s disease, microbiota, inflammation, fibrosis, dysbiosis, bacterial translocation
1. Dysbiosis and bacterial translocation in CD
The intraindividual intestinal microbiota profile is extremely diverse and relatively stable over years with some minor reversible disruptions. Bacteria from Bacteroidetes (~15-50%) and Firmicutes (~20-50%) phyla with fair deviation and to a lesser extent and from Actinobacteria (<5%) and Proteobacteria (<10%) phyla constitute the main taxa in the human gut; however, there is a great diversity at lower taxonomic levels1. Healthy conditions lacking proinflammatory responses against commensal bacteria underlines the mutualism and clearly defined lines of communication between the microbiome and the host. Many features of the modern lifestyle have a direct fingerprint on the gut microbial profile; those include medications, diets high in refined carbohydrates, illness, hospitalization, surgery, smoking, alcohol abuse, processed foods low in fermentable fibers, and chronic stress.2
Host–microbial interactions are detrimental in CD patients with dramatic changes at strain levels and reduced species richness as well as many defined microbial metabolites3–5 compared to non-IBD healthy subjects (Table 1). Some of these alterations are already present at the earliest stage in pediatric, treatment-naïve patients4,19 as well as in healthy first-degree relatives10 of CD patients. Of note, the gut microbiota of CD patients with active disease is comparatively more unstable over time as compared to CD patients with inactive disease or non-IBD healthy individuals.21 In these patients, the blooming of Proteobacteria, appearance of Fusobacterium, and reductions in Clostridia cluster IV of anaerobic bacteria are reported.1,22
Table 1.
Microbial alterations in CD patients. Relative changes in abundance of taxa in CD patients for Actinobacteria (A), Bacteroidetes (B), Firmicutes (F), Proteobacteria (P), Fusobacteria (Fu), and Euryarchaeota (E) phyla are categorized according to increases or reduction when compared to non-IBD subjects
| Studies Reporting Reduced Taxa in CD | Studies Reporting Increased Taxa in CD |
|---|---|
| Firmicutes (F)6–8 | Bacteroidetes (B)7–9 |
| F. prausnitzii (F) 1,3,4,6,9–15 |
Bacteroidaceae (B)7 |
| Lachnospira (F)1,6,9,15 | R. gnavus (F) 3,7,9,11,14–18 |
| Roseburia (F) 1,3,4,12,14,15 |
Oscillospira (F)1,9 |
| Lachnospiraceae (F)15 | [Eubacterium] (F)9 |
| Blautia (F)1,15,19 | Enterobacteriaceae1 |
| Bifidobacterium4,10,12,15,19 | Bifidobacterium (A)9 |
| Clostridium (F)1,3,8 | Clostridium (F)9 |
| Clostridia (F)8,19 | Enterobacter (P)7 |
| C. coccoides (F)13 and C. leptum (F)13 |
Enterococcaceae (F)4 |
| Prevotella3 | +Veillonellaceae (F)19,20 |
| Erysipelotrichales (F)19 | Proteobacteria (P)1,8 |
| Dorea (F)1,19 | Enterobacteriaceae (P) 3,7,12,15,18–20 |
| Anaerostipes3 | E. coli (P) 3,14,19 |
| Ruminococcaceae (F)3,4,19 | Pasteurellaceae (P)19 |
| Oscillospira (F)3,9,19 | Atopbobium (A)20 |
| Christensenellaceae (F)3 | +Fusobacteriales (Fu)3,12,19,20 |
| D. invisus* (F)10,11,19 | Escherichia (P)6 |
| Bacteroides (B)1,7,9,19 | |
| Alistipes (B)14 | |
| Actinomycetaceae (A)17 | |
| C. aeroffaciens (A)10 | |
| Methanobrevibacter (E)3 |
Critically, many studies relate to stool samples but mucosal and mucus-associated microbiota has been recently shown to harbor consortia that are different from the fecal microbiota in terms of abundance, metabolic functioning, behavior, and replication.23 Thus, this makes the mucus layer of particular interest in CD which has not been exhaustively characterized. In fact, several key functional pathways have been delineated being activated differently in IBD patients,24 but deep metagenomic and metatranscriptomic investigations focusing specifically on CD are relatively scarce.11 Predominant transcription of pathways by individual microbes within a host are known, and thus, loss of these organisms in CD can have more fare-reaching consequences than indicated by their genomic abundance.11 Even though many microbial organisms exhibited concordant DNA and RNA abundances, it is also reported that species-specific biases in the transcriptional activity reveals predominant transcription of pathways by individual microorganisms per host.11 For instance, Dialister invisus is metagenomically present with relatively reduced abundance; however, it does transcriptionally loss of the gene function.11 For example, all +-marked species in Table 1 are main producers of H2S through fermentation of sulfur-containing amino acids. Indeed, these genera have been suggested to predict the severity of CD (in conjunction with the concomitantly observed decrease in mitochondrial H2S detoxification capacity).20 Overall, it is important to include metagenomic and metatranscriptomic readouts that allow us to analyze both the activity and the presence of gut microorganisms, which ultimately provide better insight into the role of the microbiome in IBD.
Besides how each community member is abundant and functions in an intestinal ecological niche, it is important to understand their ecological roles which are fundamentally vital for the whole health biodiversity. The co-occurrence associations are beneficial to infer effects between taxa within the intestinal tract and are a key point to mechanistically examine the community structure and maintenance. Co-occurrence analysis by Yilmaz et al. revealed what could be considered as keystone species within the ecosystem of CD patients representing the most influential taxa such as Faecalibacterium and Ruminococcus.1 Together with Lachnospira, Blautia, Dorea, Coprococcus, Roseburia, Oscillospira, and Bilophila, Faecalibacterium and Ruminococcus build up the most prominent and influential taxa cluster in CD disease groups. Alterations of taxa within this network also characterize the risk of later disease recurrence of patients in remission after the active inflamed segment of CD has been surgically removed. The robustness of the microbial network as an interrelated cluster has been observed to associate with stable remission, whereas a loose structure of the microbial community and increases in abundance of Enterobacteriaceae is present in relapsing CD.25 In sum, the microbiota being present after surgery and/or lack of reestablishing a healthy robust cluster seems to be emblematic for the susceptibility of the patient to acquire CD manifestation again.
One important aspect that researchers still could not completely resolve is whether the disruption of the network leading to the dysbiosis is a cause or consequence of inflammation in CD. Inflammation is able to disrupt the stability of the microbial composition,26 and as a matter of fact, it has been proposed to drive dysbiosis and bacterial invasion in murine models of ileal CD.27 In addition, the extent of dysbiosis diminishes with decreasing inflammation and mucosal healing due to the beneficial effects of TNF inhibitors or exclusive enteral nutrition on patients underlining the potential effect of inflammation per se on the gut microbiota.28 However, it appears that the microbial compartment primarily and causatively contributes at least partly to inflammation when additional host factors are present since in animal experiments, i) dysbiotic gut microbiota can transmit Crohn’s like ileitis in mice independent of failure in antimicrobial defense29 ii) IL-2 and IL-10 deficient mice which are prone to develop colitis are protected when raised in germ-free conditions30,31 iii) susceptibility to CD is transmissible by stool microbiota of CD patients was shown in humanized gnotobiotic mice,31 and iv) gut contents elicit post-operative recurrence of CD in the terminal ileum proximal to the ileocolonic anastomosis after ileal resection.32 Thus, it is reasonable to propose that dysbiosis is also linked to treatment failure and postoperative recurrence in CD. Even though it is still not certain whether or to which extend this link is fundamental, one can argue that the failure to treatment lies within the underlying microbiota remaining as permanent sequelae in refractory CD which thus, can represent a promising target for treatment and/or preventive measures.33
Long-term effects of the bacterial network disruption with increased abundance of pathobionts within the gut might also influence the intestinal hyperpermeability even in the quiescent states of disease without any on-going inflammation.34 More recently and revealing, increases in intestinal permeability have been evidenced to determine later development of CD and thus, contributing to pathogenesis.35 The path of viable bacteria and/or their products such as outer membrane vesicles, lipopolysaccharides, peptidoglycans, muramyl-dipeptides, and bacterial DNA from the lumen through the gastrointestinal mucosa to normally sterile tissues, such as the mesenteric lymph nodes (MLN) and extra-nodal sites is referred to as bacterial translocation (BT).36,37 The link between dysbiosis and inflammation can be considered to be any pathological increase in BT due to alterations in intestinal hyperpermeability (Figure 1). Numerous studies have sampled MLN of patients suffering from IBD and conditions requiring surgery using culture-dependent techniques. Pathological bacteria translocation (PBT) to MLN has been realized for long in CD.36,38 Ambrose et al. showed that cultured bacteria from nodes were higher in numbers in involved CD segments than uninvolved ones where these bacteria were almost absent in control subjects.36 Takesue and colleagues obtained similar trends from isolated bacteria from MLN of subjects (CD patients (48%) and control (15%))39–41
Figure 1.
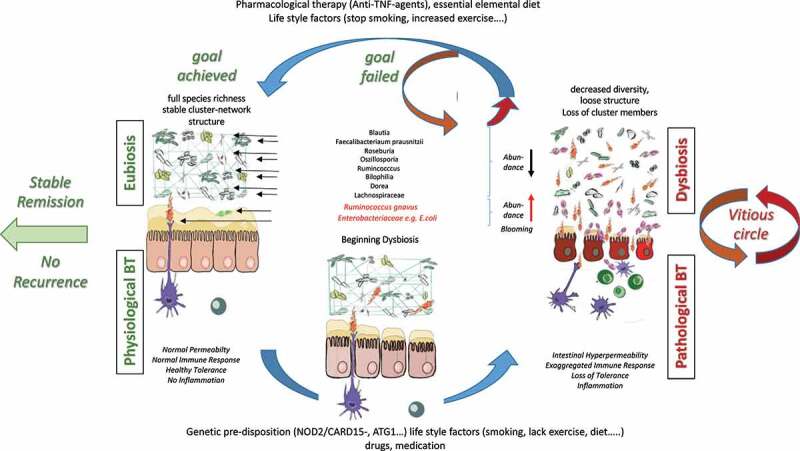
Interaction of microbiome, intestinal barriers, and translocating bacteria in healthy conditions and active CD. Eubiosis is characterized by stable cluster networks and full species richness which in concert with fully active healthy epithelial and secretory barriers (namely normal intestinal permeability) and normal immune responses to physiological bacterial translocation is the basis for stable remission. The proposed hypothetical scenario on pathophysiology of Crohn’s disease being at least partly primarily due to dysbiosis and loss of cluster network induced in individuals with genetic pre-disposition and/or by lifestyle/exogenous factors is visualized at the bottom. With increasing intestinal hyperpermeability and exaggerated pro-inflammatory immune response inflammation perpetuates fueling into a vitious circle of pathological bacterial translocation and further aggravation of dysbiosis. Thus, the goal of treatment put forward is restoration of eubiosis being the basis for stable remission or if not achieved for recurrence
Pathological increases in BT in contrast to physiological BT reflects sustained increases in quantity (rate and/or degree) and/or type (composition) of translocating agents. In that regard, as the first step across the epithelial layer increased adherence and endocytosis into enterocytes of commensal bacteria, particularly E. coli is observed in CD patients.39,42 Although any specific microorganism was absent in bacterial DNA positive nodes of CD patients or associated mucosa, Escherichia/Shigella (CD – 75% and healthy subjects – 60%) was detected at high levels in patients with DNA-positive lymph nodes. Of note, patients with terminal ileal CD had a greater proportion of E. coli.39 In fact, not only for E. coli but per se an increase in presence of adhesins and other virulence factors has been demonstrated in feces from CD patients.12
The follicle-associated epithelium lining Peyer’s patches is characterized as the site of increased translocation of bacteria to the underlying lymphoid tissue. Earliest observable lesions of recurrent CD are frequently microscopic erosions at the FAE, and an increased load of commensal bacteria at this vulnerable and inductive site of mucosal immunity has been reported in CD.42 Moreover, bona fide invasive pathogens, such as Salmonella and Shigella, are unable to invade via normal colon epithelial cells. However, they enter via the specialized M cells which lack fuzzy glycocalyx within the dome epithelium overlying Peyer’s patches in the distal ileum and lymphoid follicles in the colon.43 In addition, one of the most translocating bacterial strains is adherent-invasive E. coli (AIEC) pathovar which is increased in mucosa of CD patients.44 AIEC of CD patients lacking conventional pathogenicity genes might penetrate through M cells which fits well with evidence suggesting that the earliest lesions in CD occur at PP in the ileum and lymphoid follicles in the colon.45 Moreover, resident dendritic cells accumulate in an increased number in the subepithelial dome of Peyer’s patches in ileal CD and present with pronounced propensity to internalize translocated E. coli.46 These E. coli also cause IL-8 to be released from intestinal epithelial cells and induces granuloma formation after internalization by cultured macrophages.47
Another important target site for PBT from the gut is the mesenteric adipose tissue when the leaky gut occurs in CD.48,49 BT to mesenteric adipocytes i) occurs at a rate similar to the translocation to MLN;50 ii) could represent a bacterial reservoir51 iii) stimulating visceral fat for CRP-production;50 and iv) in terms of the microbiota profile dependent on the clinical status in CD patients.51 In addition, Ha et al. characterized the subset of mucosal-associated gut bacteria that consistently translocated and remained viable in “creeping fat” defined as an expansion of mesenteric adipose tissue around the inflamed and fibrotic intestine in CD ileal surgical resections.52 Translocation of C. innocuum was identified as a signature of this consortium i) with strain variation between mucosal and adipose isolates and ii) stimulating tissue remodeling via M2 macrophages leading to an adipose tissue barrier that serves to prevent systemic spread of translocated bacteria.52 Thus, in conjunction with the well-known cytokine production and immune cell infiltration in adjacent tissue in intestinal inflammation, the mesenteric fat might cooperate with the gut in a proinflammatory feedback loop, generating a specific microbiome signature in this tissue to influence the CD status and/or disease progression.51,52
PBT to the systemic compartment reflected by detection of bacterial DNA in serum has recently gained much attention53,54 revealing Enterobacteriaceae as a major source of bacterial DNA53 within actively smoking CD patients harboring NOD2-, CARD15- and ATG16L1-variants.55 Hence, these genetic risk factors for CD not only inhibit beneficial effects of microbial products during physiological BT to keep up mucosal tolerance but also drive pathology. PBT to the blood stream is evident almost twice as frequent in patients with clinically active disease as compared to patients with recurring disease.55 In fact, bacterial DNA translocation was significantly and independently related to disease activity. In a prospective, multicenter study on CD patients with CDAI<150, bacterial DNA translocation to serum was demonstrated to represent an independent risk factor for relapse and hospitalization within 6 months.53 Potential mechanisms explaining the impact of BT on the disease activity and aggravating course of disease are the reported consequences of translocating bacterial DNA on systemic and local inflammatory response. Bacterial DNA in serum has been found to associate with increased antimicrobial peptide (β-defensin 2, cathelecidin LL-37) and pro-inflammatory cytokine levels in CD patients in a concentration-dependent manner.54 In addition, the increased proinflammatory activity in the presence of bacterial DNA translocation is particularly pronounced in CD patients carrying mutated variants of NOD2/CARD15. Indeed, neutrophils from CD patients with variant NOD2-status release significantly more TNF when exposed to E. coli DNA in vitro as compared to CD patients with wild-type NOD2 status.55 Moreover, the presence of bacterial DNA in serum seems to decrease the availability of free anti-TNF in CD patients treated with biologicals.55 It is tempting to speculate that this is caused by pronounced TNF secretion in response to bacterial DNA in CD with NOD2-variant genotype due to faster consumption of free anti-TNF.55 Thus, PBT in subgroups of CD patients could (at least partly) cause the requirement of intensified anti-TNF therapy.
The observation that PBT per se disrupts the epithelial barrier and increases intestinal permeability56,57 strongly indicates the perpetuating potency of bacterial translocation as driving force of a vicious cycle in CD (Figure 1). This even more though considering that AIEC type 1 pili-mediated interaction with cell-adhesion molecules abnormally expressed in the quiescent phase of CD may disrupt intestinal barrier integrity before the onset of inflammation.56 Accumulating evidence shows that pathobionts by virtue of their possession of adhesion mechanisms and secretion of proteases, may have a causative role in CD. In CD, they most likely invade the tissue, perhaps as a result of defective clearance by neutrophils and macrophages. In addition, inflammation itself and disruption of the epithelial barrier increases intraluminal oxygen availability which leads to an outgrowth of pathobionts. This example highlights the complex interaction between intestinal barrier, gut microbiota and BT which when delineated in more detail will ultimately lead to improvements in preventive and therapeutic measures in CD.
2. Interaction between microbiota, genetics, and epigenetics in CD
IBD risk genes alter the gut microbiota composition
As stated above, genetic risk factors influence the interaction between gut microbiota and mucosal microenvironment in CD. Genetics is a well-known susceptibility component for IBD development, with more than 240 genomic IBD susceptibility loci identified in the GWAS.58 Data analyzed from several GWAS identified 30 CD-specific loci, 23 UC-specific loci, and 110 loci that play a role in both forms of IBD.59 Interestingly, genes involved in the epithelial barrier function were found to be more associated with UC and genes engaged in cellular innate immunity with CD. Additionally, more than 8% of variance in UC susceptibility and more than 13% of the variance in CD susceptibility might be explained by currently identified genetic variants.60
The host genetics, similar to environmental factors, have been shown to influence the gut microbiome composition and diversity61 (Table 2). NOD2, the first IBD susceptibility gene identified, is a NOD-like receptor that binds bacterial muramyl dipeptide.72 Studies involving NOD2-deficient mice established that the absence of NOD2 increases the relative abundance of Bacteroidetes and impairs the intestinal microbiota in several mouse models.73,74 Similarly, NOD2-risk allele-related increases in Enterobacteriaceae family was observed in three IBD cohorts.75
Table 2.
Influence of IBD-associated genes on the microbial composition
| Gene symbol | Influence on microbial composition | Reference |
|---|---|---|
| NOD2 | Enterobacteriaceae, Bacteroidetes | 62 |
| ATG16L1 | Fusobacteriaceae, Bacteroidaceae, Enterobacteriaceae | 63 |
| FUT2 | decreased microbial diversity and changes in several taxa | 64 |
| SLC39A8 | Anaerostipes, Coprococcus, Lachnospira | 65 |
| CARD9 |
Citrobacter rodentium infection More intestinal fungi |
66 |
| NLRP12 | Lachnospiraceae, Erysipelotrichaceae | 67 |
| TNFSF15 | Prevotella | 68 |
| BANK1, EFR3B, IL1R2, POMC | Β-diversity | 69 |
| IL6 | Helicobacter pylori | 70 |
| 11 functional genetic variants in genes: NOD2, CARD9, ATG16L1, IRGM and FUT2 | decrease in abundance of butyrate producing bacteria | 71 |
NOD2 (Nucleotide-binding Oligomerization Domain Containing protein 2), ATG16L1 (Autophagy-Related 16 Like 1), FUT2 (Fucosyltransferase 2), SLC39A8 (Solute Carrier Family 39 Member 9), CARD9 (Caspase Recruitment Domain Family Member 9), NLRP12 (NLR Family Pyrin Domain Containing 12), BANK1 (B Cell Scaffold Protein with Ankyrin Repeats 1), EFR3B (EFR3 Homolog B), IL1R2 (Interleukin 1 Receptor Type 2), POMC (proopiomelanocortin), IRGM (Immunity Related GTPase M), IL6 (Interleukin-6).
Next, ATG16L1, a gene identified by GWAS studies as IBD risk gene, regulates the development of T cells and cell autophagy.76 In mouse models, deletion of ATG16L1 triggers spontaneous intestinal inflammation with severely reduced CD4 + T cells.77 In ATG16L1 homozygous CD patient’s, enrichment of Fusobacteriaceae, Bacteroidaceae, Enterobacteriaceae in ileal tissue was observed as compared to control patients.63 Fucosyltransferase 2 (FUT2), an enzyme that regulates intestinal epithelial cells-microbe interaction is another factor that lost was found to increase susceptibility to CD development. Patients lacking functional FUT2 alleles have abnormal mucosal barrier with decreased microbial diversity and changes in several taxa.64,78 Additionally, the association between risk locus SLC39A8 and the abundance of Anaerostipes, Coprococcus, and Lachnospira was found in CD patients.65
Furthermore, combined genetic risk scores from 11 functional genetic variants associated with IBD susceptibility were directly involved in the bacterial composition in the gut and were associated with a decrease in the abundance of butyrate-producing bacteria.71
Interaction between the microbiota and epigenetics in IBD
Genetic susceptibility to develop disease might explain only part of disease risk and its heritability.79 The contribution of genetic risk alone to disease development was suggested to be around 20%.80 Epigenetic regulation of gene expression was proposed to play an additional role in the development and control of IBD.81 According to the current definition, epigenetic mechanisms are all heritable alternations of gene expression that are caused independently of changes in the primary DNA sequence.81 DNA methylation, histones modifications modulating chromatin structure, microRNA interference, and positioning of nucleosomes are the main epigenetic mechanisms that control gene expression.82
DNA methylation is a process characterized by the covalent addition of a methyl group to 5` carbon of cytosines in cytosine-guanine dinucleotides (CpG).83 This process is catalyzed by DNA methyltransferases (DNMTs). DNMT1, DNMT2, DNMT3a, DNMT3b, and DNMT3L are five members of DNMT protein family.84 As a result, hypermethylation of CpG island in promoters is transcriptionally repressive and genes are silenced.85
In addition, genes expression might be regulated by posttranslational histone modifications that include acetylation, methylation, ubiquitination, phosphorylation, sumoylation, and citrullination of histone tails.86 Acetylation is the best characterized process, and histone acetyl transferases (HATs) are the enzymes responsible for addition of acetyl groups to lysine residues in histones. Histone acetylation is associated with chromatin opening and transcriptional gene activity. On the other hand, histone deacetylation by histone deacetylases (HDACs) triggers chromatin compacting and gene silencing.87
The epigenetic machinery is influenced by environmental factors including intestinal microbiota and their metabolites. The influence of bacteria on the epigenetic control of gene expression is well characterized for SCFAs, a bacterial metabolite formed during anaerobic fermentation of dietary fibers. SCFAs, namely acetate, propionate, butyrate, produced by Firmicutes and Bacteroides phyla, inhibits HDAC activity.88 Interestingly, reduced numbers of SCFA produces have been observed in IBD patients.89 There are two possible mechanisms of SCFA action. The first mechanism involves regulation of naïve CD4 + T cells differentiation into T regulatory cells (T regs). It has been shown that acetylation of H3 histone within Foxp3 loci that is required for T regs differentiation is increased by butyrate.88 The second mechanism involves macrophages, the most abundant lamina propria cells. It has been shown that ther production of IL-6 and IL-12 from lipopolysaccharide stimulated macrophages was downregulated by addition of butyrate.90 Downregulation of proinflammatory cytokines produced by macrophages suggests that butyrate might induce hyporesponsiveness of macrophages and maintain tolerance in the gut.
As the epigenetic signature is profoundly cell specific, another cell type – intestinal epithelial cells (IECs), gained significant attention in studies focused on epigenetic regulation in relation to IBD. In their work on IECs from CD patients and mouse modes, Denizot et al. suggested that demethylation of CEACAM6 gene promoter in IECs correlated to high protein expression and favored gut inflammation mediated by colonization by AIEC. For the first time, due to epigenetic machinery, diet and the presence of methyl-donor ingredients were linked to the microbiota and inflammation.91
The influence of the microbiota on transcriptome and DNA methylation in IECs during postnatal development was investigated by Pan at el. Interestingly, differences in DNA methylation that are depended on the microbiota were detected early after birth, in contrast to transcriptome, where bacterial influence increased over time.92 Moreover, authors reveal 126 genomic loci where the presence of the intestinal microbiota was associated with combined differential RNA transcription and DNA methylation. The most important conclusion from this study is that during development, microbiota modulates epithelial cells transcriptome and functionally methylate genes and, on long term, change their expression signature in IEC.92
In summary, epigenetic modifications connect genetic susceptibility with environmental factors via the intestinal microbiota. As epigenetic changes are reversible in nature, from clinical perspective, it is necessary to understand how it would be possible to control or even reverse disease associated changes through epigenetic mechanism.
3. Dysbiotic microbiota effect on local and systemic inflammation in CD
As shown, the genetic background may influence the microbiota composition, which, in turn, tightly shapes the immune pathogenic mechanisms present in CD. During homeostasis, thus immune quiescence, microbial members of the commensal microbiota contribute to the maintenance of the gut barrier integrity and the optimal mucosal composition. They collaborate in the prevention of microorganisms crossing through the intestinal epithelium93 (Figure 2).
Figure 2.
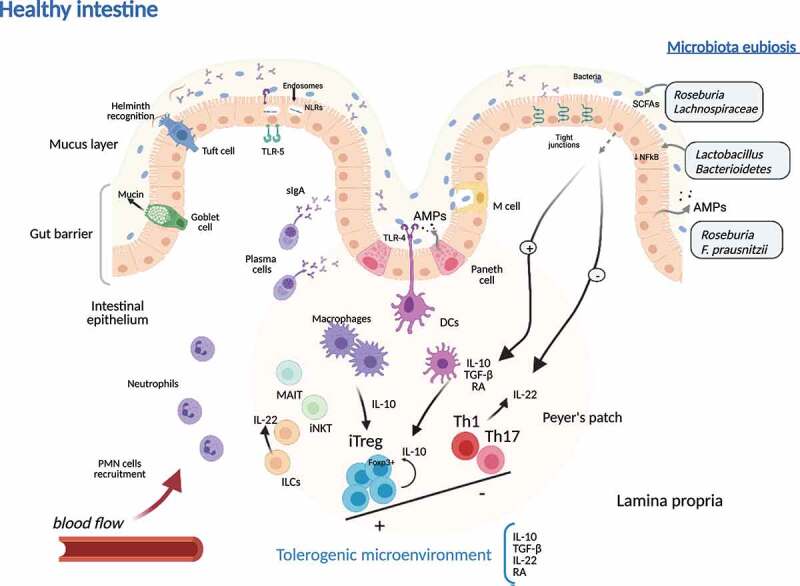
Tolerogenic microenvironment during microbiota eubiosis in healthy intestinal tissue in Crohn’s disease. In healthy colonic tissue, the gut barrier is made up of a thick mucus layer containing mucin produced by Goblet cells, sIgA produced by plasma cells, AMPs secreted by epithelial cell, and the cellular immune system (ILCs, MAIT cells, NKTs, macrophages, dendritic cells and T cells), mainly organized in Peyer’s patches. DCs and macrophages produce anti-inflammatory mediators that promote iTreg Foxp3+ expansion. Moreover, the eubiotic microbiota contributes to reduction of proinflammatory substrates induction (reduced activation of NFkB in epithelial cells) and produces SCFAs and subsequent inhibition of proinflammatory (Th1 and Th17) cell lineage activation. iTregs together with eubiotic microbiota are responsible of the predominant immunotolerance in intestinal tissue microenvironment, while ILC-derived IL-22 contributes to keep gut barrier integrity. sIgA, soluble IgA; AMPs, antimicrobial peptides; ILC, innate lymphoid cells; MAIT, mucosal associated invariant T cell; iNKT, invariant Natural Killer T cell; DC, dendritic cell; iTreg, induced regulatory T cells; NFkB, nuclear factor kappa-B; Th, T helper cell; SCFAs, short chain fatty acids, PMN, polymorphonuclear cells; TLRs, Toll-like receptors; NLRs, Nod-like receptors; IL, interleukin; RA, retinoic acid; TGF-β, transforming growth factor-β
Regulatory T (Treg) cells interact with the microbiota, and their expansion is controlled by products of commensal bacteria,94 and their balance with adaptive Th17 cells (main productors of IL-17 and IL-22) and the gut microbiota is directly implicated in illness developing or worsening.95 As an example, Roseburia and taxa from Lachnospiraceae family are in charge of producing short-chain fatty acids (butyrate, acetate, and propionate) from fermentable dietary fiber as a carbon supply for intestinal colonocytes and differentiation of Tregs.88,96 Also, polysaccharide-A (PSA) of Bacterioides fragilis induces IL-10-expressing Foxp3+ Treg cells,97 and Toll-like receptor (TLR)-2 internalization of PSA by dendritic cells induces the subsequent differentiation of IL-10-producing Treg cells in the colon.98,99 Moreover, Roseburia can improve the innate response in the gut by inducing antimicrobial peptide production and increasing the gut barrier function.100 Also, Bacteroidetes and Lactobacillus spp. can block NFκB signaling pathway, reducing proinflammatory microenvironment in the intestine. In vitro experiments using intestinal cell lines, Faecalibacterium prausnitzii can inhibit NFκB pathway through the microbial anti-inflammatory molecule MAM.101 Moreover, dendritic cells production of IFN-γ and IL-12 is also downregulated by the presence of F. prausnitzii that promotes production of IL-10 in homeostatic conditions,102 together with the expression of CD39, programmed death-ligand 1, and production of indoleamine 23-dioxygenase 1 (IDO-1) and IL-27. This dendritic cell phenotype can induce skewing of Th cells to Treg cells differentiation.101 IL-6 and TLR-4 expression levels on dendritic cells negatively correlated with that of F. prausnitzii, as well as IL-12p40 in same cells correlated with Bacteroides ratio.103
As previously stated, intestinal Paneth cells can sense bacteria by intracellular NOD-like receptors (NLRs), and they exert their control through production of antimicrobial peptides. Downregulation of antimicrobial peptides production may alter the composition of the microbiota and lead to an increase in susceptibility to inflammation93 (Figure 3). In fact, Paneth cells have a reduced production of α defensins in CD.104 Also, a reduction of mucosal-associated invariant T (MAIT) cells is observed in peripheral blood of CD, but an increase in their accumulation in intestinal inflamed areas. These MAIT cells can be activated by microbial-derived metabolites presented by dendritic cells in the context of MR-1 molecules and subsequently induce a proinflammatory tissue response.105
Figure 3.
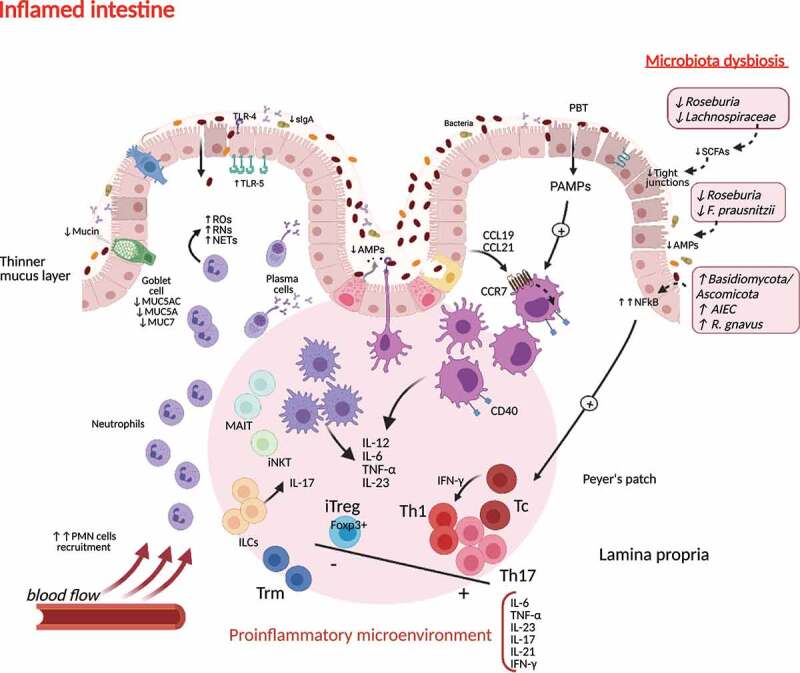
Immunogenic microenvironment during microbiota dysbiosis in inflamed intestinal tissue in Crohn’s disease. Crohn’s disease triggers induce a proinflammatory response orchestrated by tissue resident APCs activation, driving neutrophil recruitment, oxidative damage, the expansion of Th1 and Th17 populations and suppression of the regulatory T cell milieu. This environment favors the access of Tc and Trm cells to the damaged tissue. Innate lymphoid subpopulations cooperate in maintaining this immunogenic profile. Changes in gut microbiota composition and the reduction of SCFAs production contribute to the loss of tight junctions and the decrease of AMPs in the epithelial barrier, favoring an increased permeability (“leaky gut”). sIgA, soluble IgA; AMPs, antimicrobial peptides; ILC, innate lymphoid cells; MAIT, mucosal associated invariant T cell; iNKT, invariant Natural Killer T cell; DC, dendritic cell; iTreg, induced regulatory T cells; NFkB, nuclear factor kappa-B; Th, T helper cell; SCFAs, short-chain fatty acids, PMN, polymorphonuclear cells; TLRs, Toll-like receptors; Tc, T cytotoxic cell; NLRs, Nod-like receptors; IL, interleukin; ROs, reactive oxygen species; RNs, reactive nitrogen species; NETs, neutrophil extracellular traps; TNF-α, tumor necrosis factor-α; IFN-γ, interferon-gamma; CCL, chemokine ligand; CCR, chemokine receptor; PBT, pathological bacterial translocation; PAMPs; pathogen-associated molecular patterns; AIEC, adherent invasive Escherichia coli.
Moreover, the pathological variant of E. coli AIEC promotes Th17 differentiation in the intestine, and also remains functional inside macrophages, which elicit the release of high amount of TNF-α.106 This proinflammatory cytokine is also produced in a TLR-4 dependent way in response to Ruminococcus gnavus, a bacterial species related to CD among other inflammatory diseases.16 Also, the expression of PRRs TLR-5 in basolateral area of epithelium allows Clostridium spp. flagellin recognition, and this is one of the most representative antigens found in CD patients.107
As part of the innate immune response, innate lymphoid cell 3 (ILC3) subpopulation interacts with the intestinal microbiota and produces GM-CSF that helps maintain food tolerogenic responses and IL-22 that induces the production of antimicrobial peptides by epithelial cells. ILC3s express MHC-II that can control T CD4+ cell activation.108 In murine models, reduction of MHC-II in ILC3 induces inflammatory microbiota-dependent responses directed by T helper cells.108,109
Considering the adaptive intestinal T cell, Th CD4+ are highly abundant in the intestine and their response is mediated by crosstalk with local microbiota.93 Gut microbiota directs the balance of Th17 and Treg cells, and the altered response of T CD4+ to microbiota-derived antigens may give rise to a proinflammatory response in the intestinal microenvironment. Induction of Th1 and Th17 can be triggered by segmented filamentous bacteria in mice.110 These adherent bacteria can induce the production of IL-8/CCL20 chemokines and feedback the inflammatory response.111 Moreover, a biased microbiota-reactive Th17 response has been observed in IBD patients with a higher production of IL-17A.112 Blocking of IL-17A in CD patients has been showed to rise disease symptoms.113
In the case of fungal microbiota, the Basidiomycota/Ascomycota ratio has been shown increased in the intestinal mucosa of CD patients with active disease.114 Furthermore, Malasezia species have been probed to induce the production of inflammatory cytokines by dendritic cells in patients carrying the CARD9S12N gene allele SNP, worsening colitis in models for CD.115 Also, antibodies recognizing Saccharomyces cerevisiae mannan (ASCA) have been detected in approximately 50% of CD patients, while these antibodies are measured in around 8%–20% of the healthy subjects.116
4. Impact of the microbiota on fibrosis in CD patients
While at least 10% of CD patients have a clinically apparent fibrostenosing phenotype at diagnosis, the majority of patients initially exhibits a “pure” inflammatory phenotype without complications (strictures or fistulae).117,118 In population-based studies, the rate of patients progressing to fibrostenosis is approximately 20% at 20 years,119 a number reaching more than 30% within 10 years of diagnosis in tertiary referral centers.117,118 The location of strictures seems to be determined by the segmental location of inflammation, the most common site being the terminal ileum or the ileocecal region. Strictures, however, can occur in any segment of the intestine, including the upper gastrointestinal tract.120
Concepts on the pathogenesis of intestinal fibrosis in IBD and especially in CD patients have significantly changed over the last decade.121 For many years, intestinal strictures were seen as a consequence of long-standing inflammation.122 IBD-associated intestinal wall fibrosis was seen as an irreversible process frequently followed by stricture formations and subsequently intestinal obstruction requiring surgical intervention.121 However, despite the improved control of inflammation with new drugs such as biologics, the progression from inflammation to fibrosis in many CD patients has remained largely unaffected123 and the rate of surgery due to strictures remains high.124
The impact of the microbiota for tissue fibrosis was recognized in recent years and has been addressed by different experimental and clinical approaches. There seems to be both an impact of live bacteria as well as PAMPs. These microbial patterns, as well as “damage-associated molecular patterns” (DAMPs), bind or ligate to PRRs,122 thereby triggering tissue fibrosis. Interestingly, single-nucleotide polymorphisms affecting the function of PRRs and subsequently affecting bacterial sensing, recognition, or processing, such as the previously mentioned variants of NOD2, are associated with fibrostenotic CD.55
Mesenchymal cells such as collagen-forming fibroblasts express multiple PRRs such as TLRs and NLRs.125 Activation of those PRRs by bacterial wall products, bacterial DNA,, or proteins may drive mesenchymal cells into differentiation into a profibrotic phenotype. In addition, IECs may undergo epithelial-to-mesenchymal transition and contribute to intestinal fibrosis.126 The presence of EMT-associated molecules was demonstrated in fibrotic lesions of CD patients.126
Genetic variants encoding bacterial sensing PRRs as well as serum antibodies against microbial components have been associated with the risk of developing intestinal fibrosis.121,125 Animal models do not develop intestinal fibrosis in the absence of a microbiota.125 In vitro data suggest a specific action of flagellin in the profibrogenic response of intestinal mesenchymal cells.
In a situation of a “leaky barrier,” translocation of PAMPs may occur through the gut epithelium, leading to the activation of subepithelial myofibroblasts via PRRs (e.g. TLR1 to 9 and Nod1 and −2)125 followed by NFκB activation and expansion of fibroblasts (Figure 4). TLR5 ligands are known to induce the fibroblast cell cycle entry and proliferation and IECs undergo EMT after activation of TLR4.127 However, fibrosis may also occur independent of the signaling of PRRs. In a heterotopic transplant mouse model of intestinal fibrosis, the deletion of MyD88, the adaptor protein of TLRs, necessary for signal transduction did not alter development of fibrosis. Collagen deposition and transforming growth factor-beta 1 (TGF-β1) expression was equal in MyD88+/+ and MyD88-/-, indicating that MyD88 was not essential for fibrogenesis.128
Figure 4.
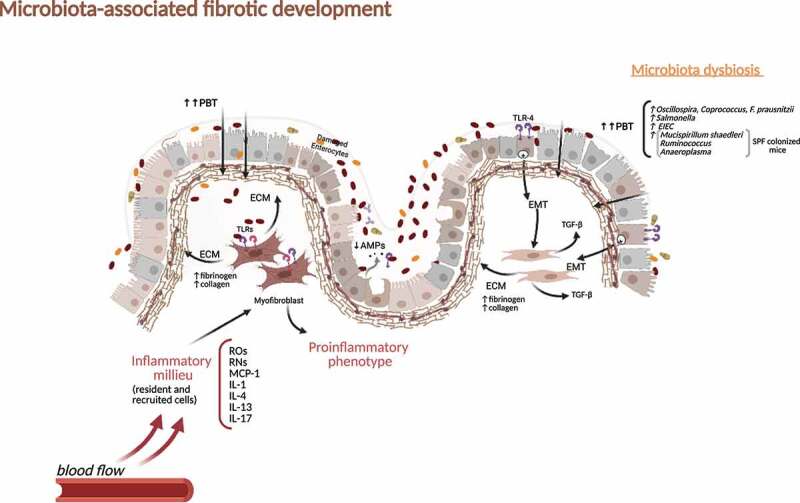
Intestinal fibrosis development during microbiota dysbiosis in Crohn’s disease. Mediators generated during sustained inflammation and continued PBT favored by gut barrier distortion induce myofibroblasts activation and extracellular matrix production with deposition of fibrinogen and collagen. Intestinal epithelial cells undergo EMT after activation of TLR-4 and contribute to the fibrotic context by inducing TGF-β. PBT, pathological bacterial translocation; TLRs, Toll-like receptors; ECM, extracellular matrix; EMT, epithelial to mesenchymal transition; ROs, reactive oxygen species; RNs, reactive nitrogen species; MCP-1, Monocyte Chemoattractant Protein-1; IL, interleukin; TGF-β, transforming growth factor-beta; SPF, Specific-pathogen-free; EIEC, Enteroinvasive Escherichia coli.
In addition to the discussion about a potential pathogenic role of bacterial proteins and bacterial wall compounds, there is evidence for a direct effect of certain microbes on fibrosis formation. When mice are infected and colonized with Salmonella (after pre-disposing streptomycin treatment), edema, mucosal ulcerations, and severe transmural inflammation is known to be a consequence of this colonization. However, besides inflammation, significantly increased expression of transforming growth factor-B, connective tissue growth factor, and insulin-like growth factor-I has been detected.129 This leads to collagen type I deposition in the mucosa, submucosa, and muscularis mucosa and subsequent persistent intestinal wall fibrosis.129 Besides Salmonella, the CD-associated pathobiont entero-invasive Escherichia coli (EIEC) was reported to cause fibrosis in mice via a flagellin-dependent mechanism via IL-33 induction and activation of the IL-33 receptor.130 Colitis-susceptible IL-10-deficient mice develop fibrosis when monoassociated with AIEC that harbors the yersiniabactin (Ybt) pathogenicity island.131 Inactivation of the Ybt siderophore production in AIEC nearly abrogated fibrosis.131 These findings may be specific for the genetic background of the host and the specific situation of IL-10 deficiency. On the other hand, it shows the impact of the microbiota on fibrosis development. This is further supported by the finding that the infection with chronic adherent-invasive E. coli in streptomycin-treated mouse strains (CD1, DBA/2, C3H, 129e and C57BL/6) induced profibrotic growth factors and fibrosis.132 Further evidence comes from another mouse model: tumor necrosis factor–like cytokine 1A (TL1A, TNFSF15) expression is increased in the inflamed gut mucosa and associated with fibrostenosing CD. Tl1a-overexpression in mice is followed by ileitis and fibrosis.133 When germ-free wild-type and Tl1a-transgenic mice were inoculated intragastrically with stool from specific pathogen–free (SPF) mice and a healthy human donor, the reconstitution with SPF, but not human microbiota, resulted in increased intestinal collagen deposition and fibroblast activation in the Tl1a-transgenic strain.133 Microbiota strains that were associated with increased fibrosis in this mouse model were groups of mucolytic bacteria such as Mucispirillum schaedleri and Ruminococcus. Anaeroplasma were also significantly associated with fibrosis in the cecum of SPF-colonized mice. Oscillospira and Coprococcus were negatively correlated with fibrosis in the cecum.133 Furthermore, similar to other models, F. prausnitzii and Bacteroides were negatively associated with fibrosis.133
To further elucidate the impact of the microbiota on the risk of intestinal stricture formation, we recently investigated the post-operative microbiota in CD patients that had already undergone resection of the ileo-cecal region or a segment or small intestine, with CD patients that had not needed an operation.1 Parabacteroides and Clostridiales were reduced in inactive postsurgical patients, and Enterobacteriaceae were significantly and reproducibly increased.1 All these findings point to a specific role of host–bacteria interaction for intestinal fibrosis. However, further research will be necessary to understand the mechanisms behind in more detail to be able to use this knowledge for therapeutic interventions.
5. Microbiota modulation targeting dysbiosis and potential beneficial effects in CD
Once reviewed the impact of the genetic background and the mucosal immunity dynamic influence in microbiota and disease progression, exogenous factors must be also considered when drawing the global picture of gut microbiota biological interactions and their relevance in CD pathogenesis.
Diet has substantial influence on the microbiome, intestinal permeability, and development of intestinal inflammation.134 For instance, western diet induces a shift in the microbiota composition, enhancing susceptibility to AIEC infection and intestinal inflammation.135 Also, smoking has been detected as the single-most important lifestyle factor impacting on the gut microbiome in CD patients and represents a well-accepted risk factor for a more complicated disease course in CD.136 In addition, exercise has been reported to deliver health benefit to CD patients,137 and in terms of impact on the microbiome associates with taxa also represented mainly in the CD cluster of taxa that if reduced in abundance associates with worse outcome. In other words, it is tempting to speculate that many lifestyle factors such as diet, smoking and exercise mediate their impact on the course of CD at least partly via changes in gut microbiota. Vice versa a change in diet, stop smoking and boosting exercise may well shift the microbiome to eubiosis and thus, at least contribute to achieve stable remission in CD.
The intestinal microbiota can be also modulated in order to improve the inflammatory response observed in CD, as well as considered a potential marker for disease state and progression. The knowledge of specific microbial drivers of pathogenic immune responses is crucial for achieving an effective microbial therapy.138 Rebalancing the intestinal microbiota through the enrichment of a specific anti-inflammatory bacterium, such as F. prausnitzii, is a potential strategy for CD treatment.139 An increase in Bifidobacterium, Collinsella, Lachnospira, Lachnospiraceae, Roseburia, and Eggerthella taxa has been linked to a strong response to anti-TNF-α treatment in CD.1 From a molecular point of view, the delivery of L. lactis containing a plasmid encoding for the anti-inflammatory molecule MAM has been shown effective in prevention of dinitrobenzene sulfonic acid-induced colitis in mice.101
Furthermore, the use of probiotics has been considered in CD. M2 macrophages, cells with immune anti-inflammatory properties, are induced by probiotic cocktail VSL#3 (four strains of Lactobacillus, three strains of Bifidobacterium and one of Streptococcus).140 This probiotic treatment helped to maintained remission in patients with CD.141 Dendritic cells with tolerogenic activities (CD103+) can induce inhibition of Th1/Th17 immune responses by promoting iTreg differentiation and by producing different intermediaries such as TGF-β or retinoic acid.142 Although IL-17 production is elevated in CD patients, treatment with antibodies anti-IL17 has been shown in the past to exacerbate the disease.143 However, microbiota products have been shown to ameliorate Th17 pathway, as polysaccharides from B. fragilis and protein fractions from F. prausnitzii can induce IL-10-producing Treg in mice.97
At present, fecal microbiota transplantation (FMT) in CD treatment is under study, with different clinical trials recently finished or in progress. Xiang et al. have reported clinical symptoms improvement in 43.7% of 174 CD patients treated and clinical remission in 20.1%.144 Also, Sokol et al. led a randomized controlled study of FMT in CD patients. They did not find differences in clinical remission for FMT-treated patients, but they observed that the incidence of flare in the FMT group was lower than in the control group of CD patients.145 Later, Li et al. noticed that adult population with the second dose of FMT at four months of receiving first, achieved long-term good clinical benefits.146 Moreover, a protocol for a double-blind, randomized, placebo-controlled pilot study in CD pediatric population is under development (Trial registration number NCT03378167).147
While conclusive clinical benefits yet require further study, expanding data on FMT anti-inflammatory effects are also emerging. The ongoing IMPACT-Crohn study145 shows a tendency toward decreased circulating white blood cells in fecal versus sham transplanted patients. In addition, Burrello et al. have reported the modulation of T cell phenotypes and the significant reduction of pro-inflammatory cytokines in colonic tissue after FMT transplant in a murine model of DSS-induced colitis.148
6. Future perspective
The modulation of intestinal microbiota and its communication with host’s regional and systemic immune behavior constitute a major research venue and a translational opportunity for improving CD outcomes. Considering that CD progresses mainly due to a sustained immune dysfunction and the central role of microbiota dysbiosis in negatively shaping immune homeostasis profile, the accurate accomplishment of microbiota eubiosis, bearing in mind the genetic, epigenetic, and environmental factors, is of outmost relevance for disease management.
In order to come to planned interventions, it becomes necessary to develop a much deeper mechanistical understanding of the ways microbiota and their derivatives contribute to mucosal immunity and how dysbiosis modifies their crosstalk, eventually comprising a CD pathogenesis hallmark. The identification of relevant specific bacterial metabolic pathways that may provide targeted interventions, as well as new insight on epithelial and immune cells biology, interaction and response to such highly dynamic microbiome, should be considered in all further research efforts.
The availability and use of new sophisticated technologies such as in vitro systems with complex organoids, analysis of gene expression, and epigenetic modifications regulating colonic tissue response or CRISPR-based new mutations screenings, among others, will help advance in our knowledge on immune mechanistic interactions with dysbiotic microbiota, their role in disease pathophysiology and how to intervene on them to personalize CD management.
Funding Statement
This study has been partially funded by PID2019-107036RB-I00, from Ministerio de Ciencia, Innovación y Universidades, Madrid, Spain, and 2020-0287, from IIS ISABIAL, Hospital General Universitario, Alicante, Spain;Ministerio de Ciencia, Innovacion y Universidades [PID2019-107036RB-I00];
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Yilmaz B, Juillerat P, Øyås O, Ramon C, Bravo FD, Franc Y, Fournier N, Michetti P, Mueller C, Geuking M, et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat Med. 2019;25(2):323–19. [DOI] [PubMed] [Google Scholar]
- 2.Conlon MA, Bird AR.. The impact of diet and lifestyle on gut microbiota and human health. Nutrients. 2014;7(1):17–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pascal V, Pozuelo M, Borruel N, Casellas F, Campos D, Santiago A, Martinez X, Varela E, Sarrabayrouse G, Machiels K, et al. A microbial signature for Crohn’s disease. Gut. 2017;66(5):813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kowalska-Duplaga K, Gosiewski T, Kapusta P, Sroka-Oleksiak A, Wędrychowicz A, Pieczarkowski S, Ludwig-Słomczyńska AH, Wołkow PP, Fyderek K et al. Differences in the intestinal microbiome of healthy children and patients with newly diagnosed Crohn’s disease. Sci Rep. 2019;9(1):18880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lavelle A, Sokol H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17(4):223–237. [DOI] [PubMed] [Google Scholar]
- 6.Suchodolski JS, Dowd SE, Wilke V, Steiner JM, Jergens AE. 16S rRNA gene pyrosequencing reveals bacterial dysbiosis in the duodenum of dogs with idiopathic inflammatory bowel disease. PloS One. 2012;7(6):e39333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rehman A, Lepage P, Nolte A, Hellmig S, Schreiber S, Ott SJ. Transcriptional activity of the dominant gut mucosal microbiota in chronic inflammatory bowel disease patients. J Med Microbiol. 2010;59(9):1114–1122. [DOI] [PubMed] [Google Scholar]
- 8.Gophna U, Sommerfeld K, Gophna S, Doolittle WF. Veldhuyzen van Zanten SJ differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol. 2006;44:4136–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobs JP, Goudarzi M, Singh N, Tong M, McHardy IH, Ruegger P, Asadourian M, Moon BH, Ayson A, Borneman J, et al. A disease-associated microbial and metabolomics state in relatives of pediatric inflammatory bowel disease patients. Cell Mol Gastroenterol Hepatol. 2016;2(6):750–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, Vandamme P, Vermeire S, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60(5):631–637. [DOI] [PubMed] [Google Scholar]
- 11.Schirmer M, Franzosa EA, Lloyd-Price J, McIver LJ, Schwager R, Poon TW, Ananthakrishnan AN, Andrews E, Barron G, Lake K, et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat Microbiol. 2018;3(3):337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vich Vila A, Imhann F, Collij V, Jankipersadsing SA, Gurry T, Mujagic Z, Kurilshikov A, Bonder MJ, Jiang X, Tigchelaar EF, et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci Transl Med. 2018;10:eaap8914. [DOI] [PubMed] [Google Scholar]
- 13.Rajca S, Grondin V, Louis E, Vernier-Massouille G, Grimaud JC, Bouhnik Y, Laharie D, Dupas JL, Pillant H, Picon L, Veyrac M, et al. Alterations in the intestinal microbiome (dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohn’s disease. Inflamm Bowel Dis. 2014;20:978–986. [DOI] [PubMed] [Google Scholar]
- 14.Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, Andrews E, Ajami NJ, Bonham KS, Brislawn CJ, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569(7758):655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Wang W, Zhou R, Ng SC, Li J, Huang M, Zhou F, Wang X, Shen B, A. Kamm M, et al. Characteristics of fecal and mucosa-associated microbiota in Chinese patients with inflammatory bowel disease. Medicine (Baltimore). 2014;93(8):e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henke MT, Kenny DJ, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc Natl Acad Sci U S A. 2019;116(26):12672–12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strombeck A, Lasson A, Strid H, Sundin J, Stotzer PO, Simrén M, Magnusson MK, Öhman L, et al. Fecal microbiota composition is linked to the postoperative disease course in patients with Crohn’s disease. BMC Gastroenterol. 2020;20(1):130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sokol H, Brot L, Stefanescu C, Auzolle C, Barnich N, Buisson A, Fumery M, Pariente B, Le Bourhis L, Treton X, et al. Prominence of ileal mucosa-associated microbiota to predict postoperative endoscopic recurrence in Crohn’s disease. Gut. 2020;69(3):462–472. [DOI] [PubMed] [Google Scholar]
- 19.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15(3):382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mottawea W, Chiang CK, Mühlbauer M, Starr AE, Butcher J, Abujamel T, Deeke SA, Brandel A, Zhou H, Shokralla S, et al. Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn’s disease. Nat Commun. 2016;7(1):13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sepehri S, Kotlowski R, Bernstein CN, Krause DO. Microbial diversity of inflamed and noninflamed gut biopsy tissues in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13(6):675–683. [DOI] [PubMed] [Google Scholar]
- 22.Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2(2):119–129. [DOI] [PubMed] [Google Scholar]
- 23.Zoetendal EG, von Wright A, Vilpponen-Salmela T, Ben-Amor K, Akkermans AD, de Vos WM. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ Microbiol. 2002;68(7):3401–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Collij V, Jaeger M, van den Munckhof ICL, Vich Vila A, Kurilshikov A, Gacesa R, Sinha T, Oosting M, Joosten LAB, et al. Gut microbial co-abundance networks show specificity in inflammatory bowel disease and obesity. Nat Commun. 2020;11(1):4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mondot S, Lepage P, Seksik P, Allez M, Tréton X, Bouhnik Y, Colombel JF, Leclerc M, Pochart P, Doré J, et al. Structural robustness of the gut mucosal microbiota is associated with Crohn’s disease remission after surgery. Gut. 2016;65(6):954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiely CJ, Pavli P, O’Brien CL. The role of inflammation in temporal shifts in the inflammatory bowel disease mucosal microbiome. Gut Microbes. 2018;9:477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Craven M, Egan CE, Dowd SE, McDonough SP, Dogan B, Denkers EY, et al. Inflammation drives dysbiosis and bacterial invasion in murine models of ileal Crohn’s disease. PloS One. 2012;7(7):e41594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, Bittinger K, Bailey A, Friedman ES, Hoffmann C, et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in Pediatric Crohn’s Disease. Cell Host Microbe. 2015;18(4):489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaubeck M, Clavel T, Calasan J, Lagkouvardos I, Haange SB, Jehmlich N, Basic M, Dupont A, Hornef M, von Bergen M, et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut. 2016;65(2):225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66(11):5224–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagao-Kitamoto H, Shreiner AB, Gillilland MG 3rd, Kitamoto S, Ishii C, Hirayama A, Kuffa P, El-Zaatari M, Grasberger H, Seekatz AM, et al. Functional characterization of inflammatory bowel disease-associated gut dysbiosis in gnotobiotic mice. Cell Mol Gastroenterol Hepatol. 2016;2(4):468–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D’Haens GR, Geboes K, Peeters M, Baert F, Penninckx F, Rutgeerts P. Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology. 1998;114(2):262–267. [DOI] [PubMed] [Google Scholar]
- 33.Monif GRG. Are the permanent sequelae of Crohn’s disease a failure to treat the gastrointestinal microbiota? Med Hypotheses. 2019;122:198–199. [DOI] [PubMed] [Google Scholar]
- 34.Vivinus-Nebot M, Frin-Mathy G, Bzioueche H, Dainese R, Bernard G, Anty R, Filippi J, Saint-Paul MC, Tulic MK, Verhasselt V, et al. Functional bowel symptoms in quiescent inflammatory bowel diseases: role of epithelial barrier disruption and low-grade inflammation. Gut. 2014;63(5):744–752. [DOI] [PubMed] [Google Scholar]
- 35.Turpin W, Lee SH, Raygoza Garay JA, Madsen KL, Meddings JB, Bedrani L, Power N, Espin-Garcia O, Xu W, Smith MI, et al. Increased Intestinal Permeability Is Associated With Later Development of Crohn’s Disease. Gastroenterology. 2020;159(6):2092–100 e5. [DOI] [PubMed] [Google Scholar]
- 36.Ambrose NS, Johnson M, Burdon DW, Keighley MR. Incidence of pathogenic bacteria from mesenteric lymph nodes and ileal serosa during Crohn’s disease surgery. Br J Surg. 1984;71(8):623–625. [DOI] [PubMed] [Google Scholar]
- 37.Lichtman SM. Bacterial [correction of baterial] translocation in humans. J Pediatr Gastroenterol Nutr. 2001;33(1):1–10. [DOI] [PubMed] [Google Scholar]
- 38.Takesue Y, Ohge H, Uemura K, Imamura Y, Murakami Y, Yokoyama T, Kakehashi M, Sueda T, et al. Bacterial translocation in patients with Crohn’s disease undergoing surgery. Dis Colon Rectum. 2002;45(12):1665–1671. [DOI] [PubMed] [Google Scholar]
- 39.O’Brien CL, Pavli P, Gordon DM, Allison GE. Detection of bacterial DNA in lymph nodes of Crohn’s disease patients using high throughput sequencing. Gut. 2014;63(10):1596–1606. [DOI] [PubMed] [Google Scholar]
- 40.Ward DM, Weller R, Bateson MM. 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature. 1990;345(6270):63–65. [DOI] [PubMed] [Google Scholar]
- 41.Browne HP, Forster SC, Anonye BO, Kumar N, Neville BA, Stares MD, Goulding D, Lawley TD, et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature. 2016;533(7604):543–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keita AV, Salim SY, Jiang T, Yang PC, Franzén L, Söderkvist P, Magnusson KE, Söderholm JD et al. Increased uptake of non-pathogenic E coli via the follicle-associated epithelium in longstanding ileal Crohn’s disease. J Pathol. 2008;215:135–144. [DOI] [PubMed] [Google Scholar]
- 43.Sansonetti PJ, Phalipon A. M cells as ports of entry for enteroinvasive pathogens: mechanisms of interaction, consequences for the disease process. Semin Immunol. 1999;11(3):193–203. [DOI] [PubMed] [Google Scholar]
- 44.Palmela C, Chevarin C, Xu Z, Torres J, Sevrin G, Hirten R, Barnich N, Ng SC, Colombel JF. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut. 2018;67(3):574–587. [DOI] [PubMed] [Google Scholar]
- 45.Fujimura Y, Kamoi R, Iida M. Pathogenesis of aphthoid ulcers in Crohn’s disease: correlative findings by magnifying colonoscopy, electron microscopy, and immunohistochemistry. Gut. 1996;38(5):724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salim SY, Silva MA, Keita AV, Larsson M, Andersson P, Magnusson KE, Perdue MH, Söderholm JD. CD83+CCR7- dendritic cells accumulate in the subepithelial dome and internalize translocated Escherichia coli HB101 in the Peyer’s patches of ileal Crohn’s disease. Am J Pathol. 2009;174(1):82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meconi S, Vercellone A, Levillain F, Payré B, Al Saati T, Capilla F, Desreumaux P, Darfeuille-Michaud A, Altare F. Adherent-invasive Escherichia coli isolated from Crohn’s disease patients induce granulomas in vitro. Cell Microbiol. 2007;9(5):1252–1261. [DOI] [PubMed] [Google Scholar]
- 48.Kong LC, Tap J, Aron-Wisnewsky J, Pelloux V, Basdevant A, Bouillot JL, Zucker JD, Doré J, Clément K. Gut microbiota after gastric bypass in human obesity: increased richness and associations of bacterial genera with adipose tissue genes. Am J Clin Nutr. 2013;98(1):16–24. [DOI] [PubMed] [Google Scholar]
- 49.Anhe FF, Jensen BAH, Varin TV, Servant F, Van Blerk S, Richard D, Marceau S, Surette M, Biertho L, Lelouvier B, et al. Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat Metab. 2020;2(3):233–242. [DOI] [PubMed] [Google Scholar]
- 50.Peyrin-Biroulet L, Gonzalez F, Dubuquoy L, Rousseaux C, Dubuquoy C, Decourcelle C, Saudemont A, Tachon M, Béclin E, Odou MF, et al. Mesenteric fat as a source of C reactive protein and as a target for bacterial translocation in Crohn’s disease. Gut. 2012;61(1):78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Serena C, Queipo-Ortuño M, Millan M, Sanchez-Alcoholado L, Caro A, Espina B, Menacho M, Bautista M, Monfort-Ferré D, Terrón-Puig M, et al. Microbial signature in adipose tissue of crohn’s disease patients. J Clin Med. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ha CWY, Martin A, Sepich-Poore GD, Shi B, Wang Y, Gouin K, Humphrey G, Sanders K, Ratnayake Y, Chan KSL, et al. Translocation of viable gut microbiota to mesenteric adipose drives formation of creeping fat in humans. Cell. 2020;183(3):666–83 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gutierrez A, Zapater P, Juanola O, Sempere L, García M, Laveda R, Martínez A, Scharl M, González-Navajas JM, Such J, et al. Gut Bacterial DNA translocation is an independent risk factor of flare at short term in patients with Crohn’s Disease. Am J Gastroenterol. 2016;111(4):529–540. [DOI] [PubMed] [Google Scholar]
- 54.Gutierrez A, Holler E, Zapater P, Sempere L, Jover R, Pérez-Mateo M, Schoelmerich J, Such J, Wiest R, Francés R. et al. Antimicrobial peptide response to blood translocation of bacterial DNA in Crohn’s disease is affected by NOD2/CARD15 genotype. Inflamm Bowel Dis. 2011;17(8):1641–1650. [DOI] [PubMed] [Google Scholar]
- 55.Gutierrez A, Scharl M, Sempere L, Holler E, Zapater P, Almenta I, González-Navajas JM, Such J, Wiest R, Rogler G, et al. Genetic susceptibility to increased bacterial translocation influences the response to biological therapy in patients with Crohn’s disease. Gut. 2014;63(2):272–280. [DOI] [PubMed] [Google Scholar]
- 56.Keita AV, Alkaissi LY, Holm EB, Heil SDS, Chassaing B, Darfeuille-Michaud A, McKay DM, Söderholm JD. Enhanced E. Enhanced E. coli LF82 translocation through the follicle-associated epithelium in Crohn’s disease is dependent on long polar fimbriae and ceacam6 expression, and increases paracellular permeability . J Crohn’s and Colitis. 2020;14(2):216–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Denizot J, Sivignon A, Barreau F, Darcha C, Chan HF, Stanners CP, Hofman P, Darfeuille-Michaud A, Barnich N. Adherent-invasive Escherichia coli induce claudin-2 expression and barrier defect in CEABAC10 mice and Crohn’s disease patients. Inflamm Bowel Dis. 2012;18(2):294–304. [DOI] [PubMed] [Google Scholar]
- 58.de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, Jostins L, Rice DL, Gutierrez-Achury J, Ji SG, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017;49(2):256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47(9):979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kurilshikov A, Wijmenga C, Fu J, Zhernakova A. Host Genetics and Gut Microbiome: challenges and Perspectives. Trends Immunol. 2017;38(9):633–647. [DOI] [PubMed] [Google Scholar]
- 62.Mondot S, Barreau F, Al Nabhani Z, Dussaillant M, Le Roux K, Doré J, Leclerc M, Hugot JP, Lepage P. Altered gut microbiota composition in immune-impaired Nod2−/−mice. Gut. 2012;61(4):634–635. [DOI] [PubMed] [Google Scholar]
- 63.Sadaghian Sadabad M, Regeling A, de Goffau MC, Blokzijl T, Weersma RK, Penders J, Faber KN, Harmsen HJ, Dijkstra G. The ATG16L1–T300A allele impairs clearance of pathosymbionts in the inflamed ileal mucosa of Crohn’s disease patients. Gut. 2015;64(10):1546–1552. [DOI] [PubMed] [Google Scholar]
- 64.Rausch P, Rehman A, Künzel S, Häsler R, Ott SJ, Schreiber S, Rosenstiel P, Franke A, Baines JF. Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (Secretor) genotype. Proc Natl Acad Sci U S A. 2011;108(47):19030–19035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li D, Achkar JP, Haritunians T, Jacobs JP, Hui KY, D'Amato M, Brand S, Radford-Smith G, Halfvarson J, Niess JH, et al. A pleiotropic missense variant in slc39a8 is associated with Crohn’s disease and human gut microbiome composition. Gastroenterology. 2016;151(4):724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lamas B, Michel ML, Waldschmitt N, Pham HP, Zacharioudaki V, Dupraz L, Delacre M, Natividad JM, Costa GD, Planchais J, et al. Card9 mediates susceptibility to intestinal pathogens through microbiota modulation and control of bacterial virulence. Gut. 2018;67(10):1836–1844. [DOI] [PubMed] [Google Scholar]
- 67.Chen L, Wilson JE, Koenigsknecht MJ, Chou WC, Montgomery SA, Truax AD, Brickey WJ, Packey CD, Maharshak N, Matsushima GK, et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat Immunol. 2017;18(5):541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakagome S, Chinen H, Iraha A, Hokama A, Takeyama Y, Sakisaka S, Matsui T, Kidd JR, Kidd KK, Said HS, et al. Confounding effects of microbiome on the susceptibility of TNFSF15 to Crohn’s disease in the Ryukyu Islands. Hum Genet. 2017;136(4):387–397. [DOI] [PubMed] [Google Scholar]
- 69.Wang J, Thingholm LB, Skiecevičienė J, Rausch P, Kummen M, Hov JR, Degenhardt F, Heinsen FA, Rühlemann MC, Szymczak S, et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat Genet. 2016;48:1396–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pohjanen VM, Koivurova OP, Niemela SE, Karttunen RA, Karttunen TJ. Role of Helicobacter pylori and interleukin 6 −174 gene polymorphism in dyslipidemia: a case–control study. BMJ Open. 2016;6(1):e009987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Imhann F, Vich Vila A, Bonder MJ, Fu J, Gevers D, Visschedijk MC, Spekhorst LM, Alberts R, Franke L, van Dullemen HM, et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut. 2018;67(1):108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Chamaillard M, Ogura Y, Henegariu O, Inohara N, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307(5710):731–734. [DOI] [PubMed] [Google Scholar]
- 73.Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123:700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rehman A, Sina C, Gavrilova O, Häsler R, Ott S, Baines JF, Schreiber S, Rosenstiel P. Nod2 is essential for temporal development of intestinal microbial communities. Gut. 2011;60(10):1354–1362. [DOI] [PubMed] [Google Scholar]
- 75.Knights D, Silverberg MS, Weersma RK, Gevers D, Dijkstra G, Huang H, Tyler AD, van Sommeren S, Imhann F, Stempak JM, et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med. 2014;6(12):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40(8):955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cummings JR, Cooney R, Pathan S, Anderson CA, Barrett JC, Beckly J, Geremia A, Hancock L, Guo C, Ahmad T, et al. Confirmation of the role of ATG16L1 as a Crohn’s disease susceptibility gene. Inflamm Bowel Dis. 2007;13(8):941–946. [DOI] [PubMed] [Google Scholar]
- 78.Tong M, McHardy I, Ruegger P, Goudarzi M, Kashyap PC, Haritunians T, Li X, Graeber TG, Schwager E, Huttenhower C, et al. Reprograming of gut microbiome energy metabolism by the FUT2 Crohn’s disease risk polymorphism. ISME J. 2014;8(11):2193–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brant SR. Promises, delivery, and challenges of inflammatory bowel disease risk gene discovery. Clin Gastroenterol Hepatol. 2013;11(1):22–26. [DOI] [PubMed] [Google Scholar]
- 80.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42(12):1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jenke AC, Zilbauer M. Epigenetics in inflammatory bowel disease. Curr Opin Gastroenterol. 2012;28(6):577–584. [DOI] [PubMed] [Google Scholar]
- 82.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–1068. [DOI] [PubMed] [Google Scholar]
- 83.Chen T, Li E. Establishment and maintenance of DNA methylation patterns in mammals. Curr Top Microbiol Immunol. 2006;301:179–201. [DOI] [PubMed] [Google Scholar]
- 84.Reik W. Epigenetic reprogramming in mammalian development. Science. 2001;293(5532):1089–1093. [DOI] [PubMed] [Google Scholar]
- 85.Hughes T, Webb R, Fei Y, Wren JD, Sawalha AH. DNA methylome in human CD4+ T cells identifies transcriptionally repressive and non-repressive methylation peaks. Genes Immun. 2010;11(7):554–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep. 2015;16(11):1467–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Henikoff S, Shilatifard A. Histone modification: cause or cog? Trends Genet. 2011;27(10):389–396. [DOI] [PubMed] [Google Scholar]
- 88.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504(7480):451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, Cosnes J, Corthier G, Marteau P, Doré J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15(8):1183–1189. [DOI] [PubMed] [Google Scholar]
- 90.Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111(6):2247–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Denizot J,Desrichard A, Agus A, Uhrhammer N, Dreux N, Vouret-Craviari V, Hofman P, Darfeuille-Michaud A, Barnich N. Diet-induced hypoxia responsive element demethylation increases CEACAM6 expression, favouring Crohn’s disease-associated Escherichia coli colonisation. Gut. 2015;64(3):428–437. [DOI] [PubMed] [Google Scholar]
- 92.Pan WH, Sommer F, Falk-Paulsen M, Ulas T, Best P, Fazio A, Kachroo P, Luzius A, Jentzsch M, Rehman A, et al. Exposure to the gut microbiota drives distinct methylome and transcriptome changes in intestinal epithelial cells during postnatal development. Genome Med. 2018;10(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. 2012;489(7415):231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, Umetsu DT, Rudensky AY. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature. 2012;482(7385):395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yue B, Luo X, Yu Z, Mani S, Wang Z, Dou W. Inflammatory bowel disease: a potential result from the collusion between gut microbiota and mucosal immune system. Microorganisms. 2019;7(10):440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev. 2001;81(3):1031–1064. [DOI] [PubMed] [Google Scholar]
- 97.Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332(6032):974–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122(1):107–118. [DOI] [PubMed] [Google Scholar]
- 99.Shen Y, Giardino Torchia ML, Lawson GW, Karp CL, Ashwell JD, Mazmanian SK. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe. 2012;12(4):509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Patterson AM, Mulder IE, Travis AJ, Lan A, Cerf-Bensussan N, Gaboriau-Routhiau V, Garden K, Logan E, Delday MI, Coutts AG, et al. Human gut symbiont roseburia hominis promotes and regulates innate immunity. Front Immunol. 2017;8:1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Quevrain E, Maubert MA, Michon C, Chain F, Marquant R, Tailhades J, Miquel S, Carlier L, Bermúdez-Humarán LG, Pigneur B, et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut. 2016;65(3):415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105(43):16731–16736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ng SC, Benjamin JL, McCarthy NE, Hedin CR, Koutsoumpas A, Plamondon S, Price CL, Hart AL, Kamm MA, Forbes A, et al. Relationship between human intestinal dendritic cells, gut microbiota, and disease activity in Crohn’s disease. Inflamm Bowel Dis. 2011;17(10):2027–2037. [DOI] [PubMed] [Google Scholar]
- 104.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A. 2005;102(50):18129–18134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee SH, Kwon JE, Cho ML. Immunological pathogenesis of inflammatory bowel disease. Intest Res. 2018;16(1):26–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Barnich N, Darfeuille-Michaud A. Adherent-invasive Escherichia coli and Crohn’s disease. Curr Opin Gastroenterol. 2007;23(1):16–20. [DOI] [PubMed] [Google Scholar]
- 107.Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, Fort M, Hershberg RM. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest. 2004;113:1296–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, Mantegazza AR, Ma HL, Crawford A, Angelosanto JM, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. 2013;498(7452):113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, Laufer T, Ignatowicz L, Ivanov I. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 2014;40:594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, Pisi A, De Paepe M, Brandi G, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31(4):677–689. [DOI] [PubMed] [Google Scholar]
- 111.Ryan FJ, Ahern AM, Fitzgerald RS, Laserna-Mendieta EJ, Power EM, Clooney AG, O'Donoghue KW, McMurdie PJ, Iwai S, Crits-Christoph A, et al. Colonic microbiota is associated with inflammation and host epigenomic alterations in inflammatory bowel disease. Nat Commun. 2020;11(1):1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hegazy AN, West NR, Stubbington MJT, Wendt E, Suijker KIM, Datsi A, This S, Danne C, Campion S, Duncan SH, et al. Circulating and Tissue-Resident CD4(+) T Cells With reactivity to intestinal microbiota are abundant in healthy individuals and function is altered during inflammation. Gastroenterology. 2017;153(5):1320–37 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang J, Sundrud MS, Skepner J, Yamagata T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol Sci. 2014;35(10):493–500. [DOI] [PubMed] [Google Scholar]
- 114.Sokol H, Leducq V, Aschard H, Pham HP, Jegou S, Landman C, Cohen D, Liguori G, Bourrier A, Nion-Larmurier I, et al. Fungal microbiota dysbiosis in IBD. Gut. 2017;66(6):1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Limon JJ, Tang J, Li D, Wolf AJ, Michelsen KS, Funari V, Gargus M, Nguyen C, Sharma P, Maymi VI, et al. Malassezia is associated with crohn’s disease and exacerbates colitis in mouse models. Cell Host Microbe. 2019;25(3):377–88 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vermeire S, Peeters M, Vlietinck R, Joossens S, Den Hond E, Bulteel V, Bossuyt X, Geypens B, Rutgeerts P, et al. Anti-Saccharomyces cerevisiae antibodies (ASCA), phenotypes of IBD, and intestinal permeability: a study in IBD families. Inflamm Bowel Dis. 2001;7(1):8–15. [DOI] [PubMed] [Google Scholar]
- 117.Cosnes J, Cattan S, Blain A, Beaugerie L, Carbonnel F, Parc R, Gendre JP. Long-term evolution of disease behavior of Crohn’s disease. Inflamm Bowel Dis. 2002;8(4):244–250. [DOI] [PubMed] [Google Scholar]
- 118.Louis E, Collard A, Oger AF, Degroote E. Aboul Nasr El Yafi FA, Belaiche J Behaviour of Crohn’s disease according to the vienna classification: changing pattern over the course of the disease . Gut. 2001;49:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Thia KT, Sandborn WJ, Harmsen WS, Zinsmeister AR, Loftus EV Jr.. Risk factors associated with progression to intestinal complications of Crohn’s disease in a population-based cohort. Gastroenterology. 2010;139(4):1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Freeman HJ. Natural history and clinical behavior of Crohn’s disease extending beyond two decades. J Clin Gastroenterol. 2003;37(3):216–219. [DOI] [PubMed] [Google Scholar]
- 121.Rieder F, Fiocchi C, Rogler G. Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases. Gastroenterology. 2017;152(2):340–50e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lawrance IC, Rogler G, Bamias G, Breynaert C, Florholmen J, Pellino G, Reif S, Speca S, Latella G, et al. Cellular and Molecular Mediators of Intestinal Fibrosis. Journal of Crohn’s & Colitis. pp.j.crohns.2014.09.008. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jeuring SF, van den Heuvel TR, Liu LY, Zeegers MP, Hameeteman WH, Romberg-Camps MJ, Oostenbrug LE, Masclee AA, Jonkers DM, Pierik MJ. Improvements in the long-term outcome of crohn’s disease over the past two decades and the relation to changes in medical management: results from the population-based IBDSL Cohort. Am J Gastroenterol. 2017;112(2):325–336. [DOI] [PubMed] [Google Scholar]
- 124.Pittet V, Rogler G, Michetti P, Fournier N, Vader JP, Schoepfer A, Mottet C, Burnand B, Froehlich Fal. Penetrating or stricturing diseases are the major determinants of time to first and repeat resection surgery in Crohn’s disease. Digestion. 2013;87(3):212–221. [DOI] [PubMed] [Google Scholar]
- 125.Rieder F. The gut microbiome in intestinal fibrosis: environmental protector or provocateur? Sci Transl Med. 2013;5(190):190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Scharl M, Huber N, Lang S, Furst A, Jehle E, Rogler G. Hallmarks of epithelial to mesenchymal transition are detectable in Crohn’s disease associated intestinal fibrosis. Clin Transl Med. 2015;4(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Masszi A, Fan L, Rosivall L, McCulloch CA, Rotstein OD, Mucsi I, Rotstein OD, Mucsi I, Kapus A. Integrity of cell-cell contacts is a critical regulator of TGF-beta 1-induced epithelial-to-myofibroblast transition: role for beta-catenin. Am J Pathol. 2004;165(6):1955–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lutz C, Weder B, Hunerwadel A, Fagagnini S, Lang B, Beerenwinkel N, Rossel JB, Rogler G, Misselwitz B, Hausmann M. Myeloid differentiation primary response gene (MyD) 88 signalling is not essential for intestinal fibrosis development. Sci Rep. 2017;7(1):17678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Grassl GA, Valdez Y, Bergstrom KS, Vallance BA, Finlay BB. Chronic enteric salmonella infection in mice leads to severe and persistent intestinal fibrosis. Gastroenterology. 2008;134(3):768–780. [DOI] [PubMed] [Google Scholar]
- 130.Imai J, Kitamoto S, Sugihara K, Nagao-Kitamoto H, Hayashi A, Morhardt TL, Kuffa P, Higgins PDR, Barnich N, Kamada N. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. 2019;12(3):632–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ellermann M, Gharaibeh RZ, Fulbright L, Dogan B, Moore LN, Broberg CA, Lopez LR, Rothemich AM, Herzog JW, Rogala A, et al. Yersiniabactin-producing adherent/invasive escherichia coli promotes inflammation-associated fibrosis in gnotobiotic Il10(-/-) mice. Infect Immun. 2019;87(11):e00587-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Small CL, Reid-Yu SA, McPhee JB, Coombes BK. Persistent infection with Crohn’s disease-associated adherent-invasive escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat Commun. 2013;4(1):1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jacob N, Jacobs JP, Kumagai K, Ha CWY, Kanazawa Y, Lagishetty V, Altmayer K, Hamill AM, Von Arx A, Sartor RB, et al. Inflammation-independent TL1A-mediated intestinal fibrosis is dependent on the gut microbiome. Mucosal Immunol. 2018;11(5):1466–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Llewellyn SR, Britton GJ, Contijoch EJ, Vennaro OH, Mortha A, Colombel JF, Grinspan A, Clemente JC, Merad M, Faith JJ. Interactions between diet and the intestinal microbiota alter intestinal permeability and colitis severity in mice. Gastroenterology. 2018;154(4):1037–46e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Agus A, Denizot J, Thevenot J, Martinez-Medina M, Massier S, Sauvanet P, Bernalier-Donadille A, Denis S, Hofman P, Bonnet R, et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E coli infection and intestinal inflammation. Sci Rep. 2016;6:19032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.To N, Ford AC, Gracie DJ. Systematic review with meta-analysis: the effect of tobacco smoking on the natural history of ulcerative colitis. Aliment Pharmacol Ther. 2016;44(2):117–126. [DOI] [PubMed] [Google Scholar]
- 137.Engels M, Cross RK, Long MD. Exercise in patients with inflammatory bowel diseases: current perspectives. Clin Exp Gastroenterol. 2018;11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mishima Y, Sartor RB. Manipulating resident microbiota to enhance regulatory immune function to treat inflammatory bowel diseases. J Gastroenterol. 2020;55(1):4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Glassner KL, Abraham BP, Quigley EMM. The microbiome and inflammatory bowel disease. J Allergy Clin Immunol. 2020;145(1):16–27. [DOI] [PubMed] [Google Scholar]
- 140.Biagioli M, Capobianco D, Carino A, Marchiano S, Fiorucci C, Ricci P, Distrutti E, Fiorucci S. divergent effectiveness of multispecies probiotic preparations on intestinal microbiota structure depends on metabolic properties. Nutrients. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fedorak RN, Feagan BG, Hotte N, Leddin D, Dieleman LA, Petrunia DM, Enns R, Bitton A, Chiba N, Paré P, et al. The probiotic VSL#3 has anti-inflammatory effects and could reduce endoscopic recurrence after surgery for Crohn’s disease. Clin Gastroenterol Hepatol. 2015;13(5):928–35e2. [DOI] [PubMed] [Google Scholar]
- 142.Bain CC, Montgomery J, Scott CL, Kel JM, Girard-Madoux MJH, Martens L, Zangerle-Murray TFP, Ober-Blöbaum J, Lindenbergh-Kortleve D, Samsom JN, et al. TGFbetaR signalling controls CD103(+)CD11b(+) dendritic cell development in the intestine. Nat Commun. 2017;8(1):620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, Wehkamp J, Feagan BG, Yao MD, Karczewski M, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61(12):1693–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Xiang L, Ding X, Li Q, Wu X, Dai M, Long C, He Z, Cui B, Zhang F. Efficacy of faecal microbiota transplantation in Crohn’s disease: a new target treatment? Microb Biotechnol. 2020;13(3):760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sokol H, Landman C, Seksik P, Berard L, Montil M, Nion-Larmurier I, Bourrier A, Le Gall G, Lalande V, De Rougemont A, et al. Fecal microbiota transplantation to maintain remission in Crohn’s disease: a pilot randomized controlled study. Microbiome. 2020;8(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li P, Zhang T, Xiao Y, Tian L, Cui B, Ji G, Liu YY, Zhang F. Timing for the second fecal microbiota transplantation to maintain the long-term benefit from the first treatment for Crohn’s disease. Appl Microbiol Biotechnol. 2019;103(1):349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pai N, Popov J, Hill L, Hartung E. Protocol for a double-blind, randomised, placebo-controlled pilot study for assessing the feasibility and efficacy of faecal microbiota transplant in a paediatric Crohn’s disease population: pediCRaFT Trial. BMJ Open. 2019;9:e030120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Burrello C, Giuffre MR, Macandog AD, Diaz-Basabe A, Cribiu FM, Lopez G, Borgo F, Nezi L, Caprioli F, Vecchi M, et al. Fecal microbiota transplantation controls murine chronic intestinal inflammation by modulating immune cell functions and gut microbiota composition. Cells. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]