

Abstract
Gold redox chemistry holds the promise of unique reactivities and selectivities that are different to other transition metals. Recent studies have utilized strain release, ligand design, and photochemistry to promote the otherwise sluggish oxidative addition to Au(I) complexes. More details on the reductive elimination from Au(III) complexes have also been revealed. These discoveries have facilitated the development of gold redox catalysis and will continue to offer mechanistic insight and inspiration for other transition metals. This review highlights how research in organometallic chemistry has led to gold redox catalysis, as well as applications in materials science, bioconjugation, and radiochemical synthesis.
Why Is Homogeneous Gold Redox Chemistry Desirable?
The chemistry of gold has attracted tremendous research efforts over the past two decades. Despite rapid development, the majority of reactions discovered with homogeneous gold complexes involve the electrophilic activation of carbon–carbon π-bonds and/or the generation of electrophilic gold carbene intermediates (Figure 1A) [1–12]. Within these activation manifolds, the formal oxidation state of gold remains unchanged throughout the stoichiometric reaction or catalytic cycle. By comparison, relatively few transformations have been proposed to access gold intermediates with different oxidation states (e.g., I/II/III) [13,14].
Figure 1. Introduction to Gold Redox Chemistry.
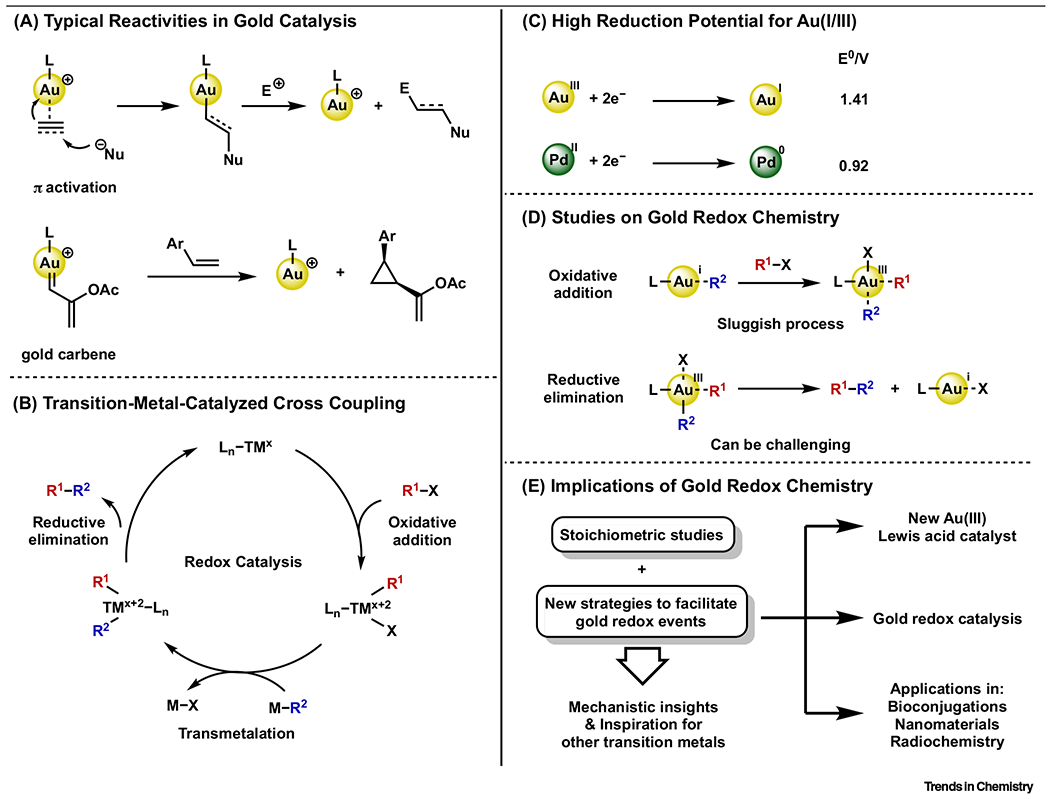
(A) Typical reactivities in gold catalysis; (B) transition-metal-catalyzed cross-coupling; (C) high reduction potential for Au(I/III); (D) studies on gold redox chemistry; and (E) implications of gold redox chemistry.
Transition-metal-catalyzed cross-coupling reactions (see Glossary) are among the most frequently used methods in synthetic chemistry [15]. These methodologies typically rely on the ability of transition metals (e.g., Pd, Ni, Cu) to effectively cycle between oxidation states (Figure 1B). The elementary steps bridging the oxidation states of metals are called oxidative addition and reductive elimination. Despite Au(I/III) and Pd(0/II) sharing the same d-electron counts, redox events are far less common for gold complexes, especially oxidative addition. Thermodynamically, this manifests as a redox potential of E0(PdII/0) = 0.92 V, whereas E0(AuIII/I) = 1.41 V (Figure 1C) [16]. Moreover, the symmetry and steric changes that result from oxidative addition to linear Au(I) complexes kinetically disfavor the process. The challenge of enabling redox processes with gold has raised significant interest in stoichiometric elementary oxidation-state conversions at gold and the mechanistic details of these transformations. Ultimately, these insights yield new catalytic transformations for chemical synthesis and/or applications in chemical biology, materials science, and radiochemistry (Figure 1D,E).
Although it is still in the early stage of discovery, notable advances in homogeneous gold redox chemistry have been made in the past 6 years. The reactivities revealed during this time already show features distinct from other transition metals and are the main focus of this review. A few examples from earlier years are also presented as the foundation for discussion. The studies are organized by oxidative addition or reductive elimination processes, with a focus on the fundamental organometallic studies and, where possible, the potentially useful chemical transformations arising from these findings.
Oxidative Addition of Strained Carbon–Carbon Bonds Led to Stable Au(III) Catalysts
Strong external oxidants (e.g., I(III) compounds and [X]+ reagents, X = halogen) are usually required to access Au(III) complexes or intermediates from Au(I) species [17–21]. Oxidative addition under milder conditions may lead to a broader scope and utility for the resulting Au(III) complexes. In 2013, Amgoune, Bourissou, and coworkers reported that cationic phosphine Au(I) complexes underwent oxidative additions of disilanes at −80°C [22]. The ensuing Au(III) complex 1 was characterized by nuclear magnetic resonance (NMR) spectroscopy; however, it was unstable even at low temperatures (−80 to −60°C) (Figure 2A).
Figure 2. Oxidative Addition Reactions Leading to Stable Au(III) Catalysts.
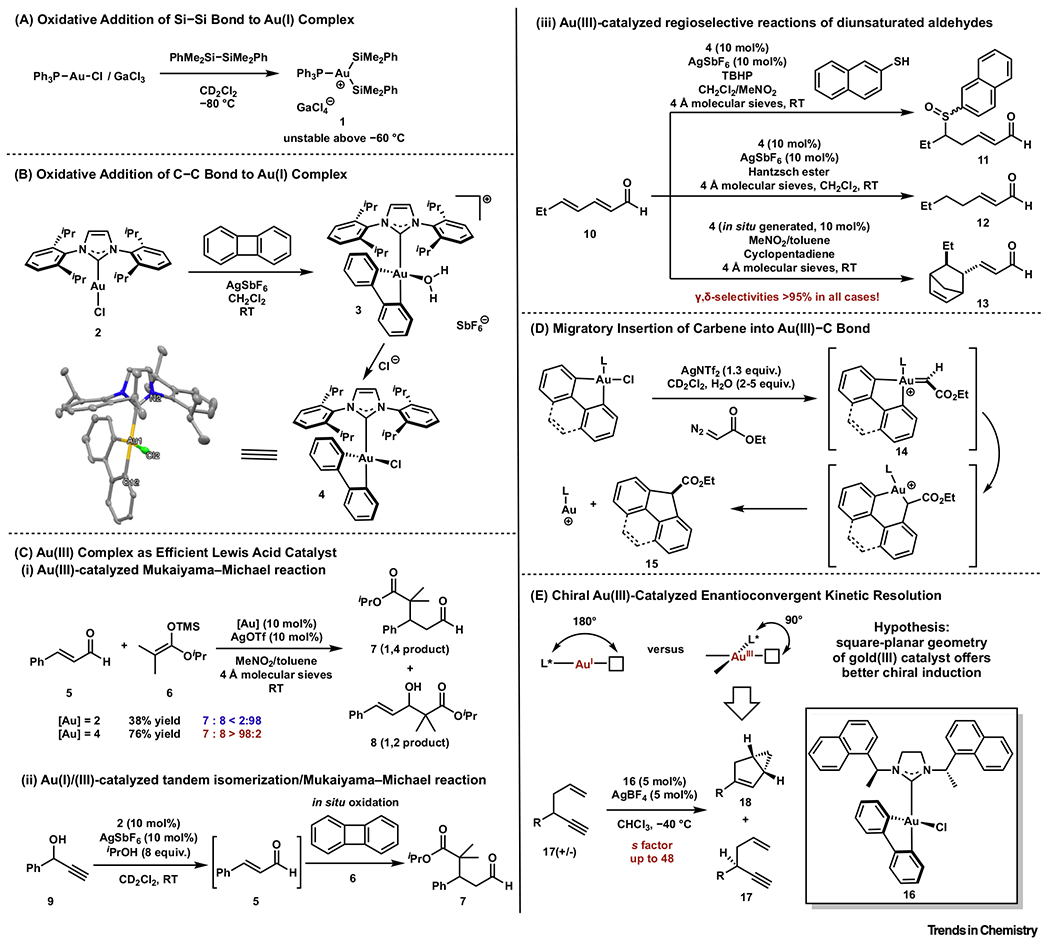
(A) Oxidative addition of an Si–Si bond to an Au(I) complex. (B) Oxidative addition of a C–C bond to an Au(I) complex. (C) Au(III) complexes as efficient Lewis acid catalysts: (i) an Au(III)-catalyzed Mukaiyama–Michael reaction; (ii) an Au(I)/(III)-catalyzed tandem isomerization/Mukaiyama–Michael reaction; and (iii) Au(III)-catalyzed regioselective reactions of unsaturated aldehydes. (D) Migratory insertion of a carbene into an Au(III)–C bond. (E) Chiral Au(III)-catalyzed enantioconvergent kinetic resolution.
In 2015, Toste and coworkers reported oxidative addition of the strained C–C bond in biphenylene to a cationic N-heterocyclic carbene (NHC) Au(I) complex 2 at room temperature (Figure 2B) [23]. The resulting cationic NHC biphenyl-Au(III) complex 3 was air and moisture stable. An X-ray structure of the corresponding Au(III) chloride complex 4 indicated a Cs-symmetric distorted square-planar geometry.
On chloride abstraction, 4 was also found to act as a Lewis acid catalyst. This feature is especially valuable since the number of stable and tunable Au(III) catalysts was limited compared with their Au(I) counterparts. Until this point, most Au(III)-catalyzed reactions employed Au(III) salts, or its ligated forms, as a catalysts or precatalysts [24–26]. Moreover, many putative Au(III) precatalysts undergo facile reduction to Au(I) or Au(0) species in the presence of other electron-rich reagents [27–29]. Stable, well-defined, and yet catalytically active Au(III) catalysts remain rare.
The cationic biphenylene-derived Au(III) complex catalyzed a Mukaiyama–Michael reaction of 5 and 6 with excellent 1,4-selectivity, in stark contrast to the high 1,2-selectivity rendered by the corresponding cationic Au(I) catalyst (Figure 2Ci). The mild conditions of the oxidative addition of biphenylene allowed the in situ oxidation of an Au(I) catalyst, leading to a tandem isomerization/Mukaiyama–Michael reaction (Figure 2Cii). Excellent remote selectivities were observed for reactions with α,β,γ,δ-unsaturated aldehyde 10: 1,6-addition/oxidation of naphthalene thiol, γ,δ-reduction by Hantzsch ester, and γ,δ-Diels–Alder addition with cyclopentadiene (Figure 2Ciii). The remote selectivities were attributed to the sterically hindered binding pocket of the complex. The potential reductive elimination of biphenyl ligand from this Au(III) catalyst was recently addressed by incorporation into metal–organic frameworks [30]. The activity and stability of this Au(III) catalyst across various reaction conditions (oxidative, reductive conditions) provides a promising platform to enable new catalytic reactions with new selectivities and thus invites further investigation.
In 2017, Toste and coworkers demonstrated that cationic ligand-supported biphenyl- or 4,5-phenanthryl-Au(III) carbene complexes 14 underwent sequential migratory insertions/reductive eliminations at temperatures ≥−40°C (Figure 2D) [31]. The effects of spectator ligands and counter anions on the migratory insertion yields were systematically studied. While catalytic processes involving bond-forming migratory insertion at gold remain rare, this work paves the way for Au(III)-catalyzed transformation involving the elementary step, such as carbene polymerization reactions. Moreover, it highlights the challenge of circumventing migratory insertion as a side reaction when developing Au(III)-catalyzed reaction involving a carbene intermediate in the future.
Another inherent advantage of this type of Au(III) complex in asymmetric catalysis is that, compared with its Au(I) counterpart, the square-planar geometry can more effectively transmit the chiral information from the ancillary ligand on the reaction site [32–37]. It has long been postulated that the linear geometry of Au(I) complexes renders the chiral induction less favorable (Figure 2E). This principle was validated by Toste and coworkers [36]. A series of chiral NHC-supported biphenyl-Au(III) complexes were synthesized and employed as catalysts for the cycloisomerization reaction of racemic 1,5-enyne 17 resulting in an enantioconvergent kinetic resolution with selectivity factors (s-factors) of up to 48 (Figure 2E). Using the corresponding chiral NHC Au(I) catalyst afforded the product with no enantiomeric excess (ee), which supported the aforementioned hypothesis that square-planar Au(III) catalysts can be superior in asymmetric catalysis. More recently, chiral NHC(biphenyl) Au(III)-catalyzed enantioselective γ,δ-Diels-Alder reaction of 2,4-dienals was also reported [38].
Ligand Designs to Facilitate Oxidative Addition to Au(I) Complexes
Ligand design is an attractive approach to tune the reactivities of transition-metal complexes [13, 15]. Several ligand designs have emerged over the years to facilitate the oxidative addition to Au(I) complexes; namely, the use of bimetallic Au(I) complexes, the use of Au(I) complexes chelated by small bite angle bidentate ligands, and the use of P,N-hemilabile bidentate ligand-supported Au(I) complexes. Some of the strategies have resulted in cross-coupling reactivities with stoichiometric or catalytic amounts of gold complexes.
The two-electron oxidation of bimetallic complexes has a lower barrier than a two-electron oxidation at a single metal center through electronic cooperation. Fackler and coworkers reported the oxidation of bimetallic Au(I) complexes by oxidants that are not reactive towards mononuclear Au(I) complexes [39]. More recently, Goddard, Toste, and coworkers conducted electrochemical and computational studies that indicated a reduced oxidation potential of bimetallic Au(I) species compared with that of mononuclear Au(I) complexes [40].
Inspired by these previous reports, Levin and Toste presented an Au(I)-catalyzed allylation of aryl boronic acids (Figure 3Ai) [41]. A bis(diphenylphosphino)amine ligand-supported Au(I) complex 19 was identified as the optimal catalyst, whereas mononuclear Au(I) catalysts led to much lower yields. The accelerated oxidative addition to bimetallic gold complexes was suggested to account for the higher yields. The method is orthogonal to traditional palladium-catalyzed cross-coupling reactions: substrate 20 containing boronic acid and aryl iodide was subjected to the allylation condition and 21 was obtained at a 94% yield with no competitive cyclization. This work demonstrated how stoichiometric transformations and mechanistic insights can inspire the design of a new catalytic system with unique selectivity.
Figure 3. Ligand Designs to Facilitate Oxidative Addition to Au(I) Complexes, Part 1.
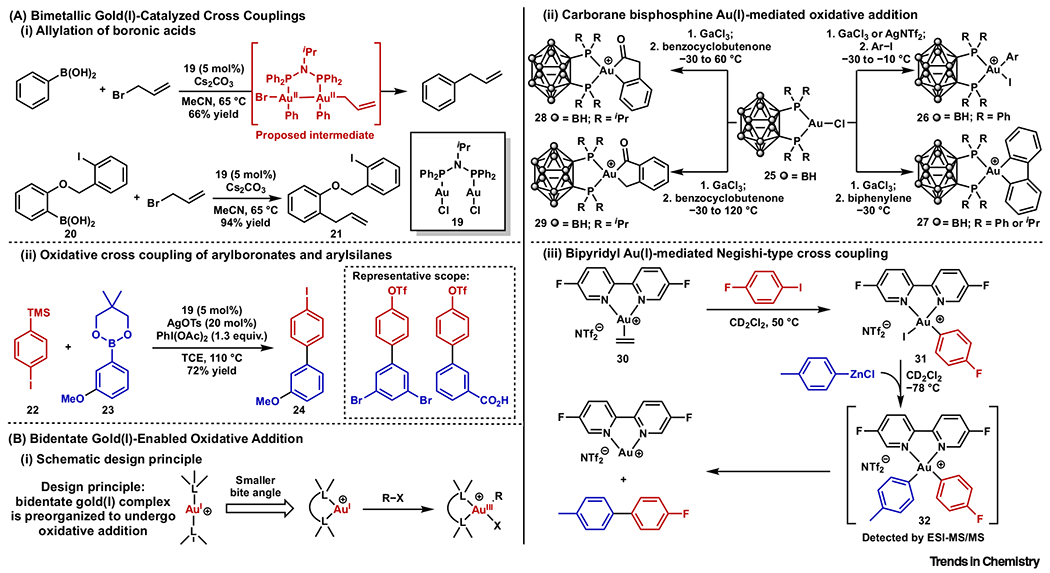
(A) Bimetallic Au(I)-catalyzed cross-couplings: (i) allylation of boronic acids; and (ii) oxidative cross-coupling of arylboronates and arylsilanes. (B) Bidentate Au(I)-enabled oxidative addition: (i) schematic design principle; (ii) carborane bisphosphine Au(I)-mediated oxidative addition; and (iii) bipyridyl Au(I)-mediated Negishi-type cross-coupling.
More recently, Xie and coworkers demonstrated that the same bimetallic Au(I) complex catalyzed a highly efficient oxidative cross-coupling reaction of arylboronate 22 and arylsilane 23 to furnish 24 (Figure 3Aii) [42]. By comparison, monomeric Au(I) catalysts showed extremely low activities despite their successes in other reported oxidative biaryl couplings [43–49]. Good-to-excellent yields were obtained for a wide range of substrates by employing this bimetallic Au(I) catalyst. Aryl halides and aryl triflates were untouched in the reaction, which again showed the unusual selectivity and functional group tolerance of gold catalysis compared with traditional cross-coupling catalysts.
A strategy using bidentate ligands with small bite angles was also postulated to facilitate oxidative addition to Au(I). It was previously demonstrated that distorting the L–M–L (M = Pd or Pt) bond angle raises the highest occupied molecular orbital (HOMO) energy of the transition-metal complex, preorganizing them towards the square-planar geometry of the oxidative addition products (Figure 3Bi) [50]. Amgoune, Bourissou, and coworkers demonstrated that this design also promoted oxidative addition to Au(I) complexes. Carborane diphosphino-chelated Au(I) triflimidate () complexes (PAuP bite angles = 90–100°) were treated with various aryl iodides and oxidative addition product 26 was observed under mild conditions (Figure 3Bii) [51]. The same groups later reported that oxidative addition of strained C–C bonds in benzocyclobutenone and biphenylene to a similar carborane diphosphino-chelated Au(I) complex [52].
Applying a similar strategy, McGrady, Bower, Russell, and coworkers recently demonstrated that a 2,2′-bipyridine (bipy) chelated Au(I) ethylene complex 30 participated in sequential oxidative addition, transmetalation, and reductive elimination to furnish biaryl coupling products (Figure 3Biii) [53]. The cationic (K2-bipy) biaryl Au(III) intermediate 32 was detected by mass spectrometry. Furthermore, this study may provide insights into the mechanism of previous examples of gold-catalyzed redox processes that were assisted by bipy-type ligands [54–57] and inspire future developments in this arena. More recently, Bower, Russell, and coworkers showed similar redox cycles of alkenyl and alkynyl iodides [58].
Despite the successes achieved by the chelation strategy, the strong preference of Au(I) to form bis(gold) complexes with most bidentate ligands limited its applicability [2]. To circumvent this limitation, in 2017 Amgoune, Bourissou, and coworkers proposed that hemilabile bidentate ligands could trigger oxidative addition by stabilizing the Au(III) product (Figure 4Ai–iii) [59]. By employing a hemilabile (P,N) bidentate ligand (Me-Dalphos), the Au(I) complex 33 underwent chloride abstraction followed by oxidative addition of aryl iodides or biphenylene under mild conditions. In the cases of aryl iodides, subsequent iodide abstraction, C–H auration, and reductive elimination afforded biaryl coupling products. A catalytic system was also developed for the C–H arylation of trimethoxybenzene. Several aryl iodides and bromides were successfully coupled under catalytic conditions. Mechanistic studies were detailed in a later publication, along with the C3-arylation of protected indoles (Figure 4Aiii) [60]. The high C3-selectivity and excellent functional group tolerance of this arylation reaction render it a useful synthetic method. This concept was further explored by the Bourissou group, as well as by Patil and coworkers, to realize C–N coupling reactions of aryl iodides (Figure 4Aiv) [61,62]. More recently, Patil and coworkers exploited the same Au(III) intermediate to achieve catalytic 1,2-diarylation of alkenes (Figure 4Av) [63].
Figure 4. Ligand Designs to Facilitate Oxidative Addition to Au(I) Complexes, Part 2.
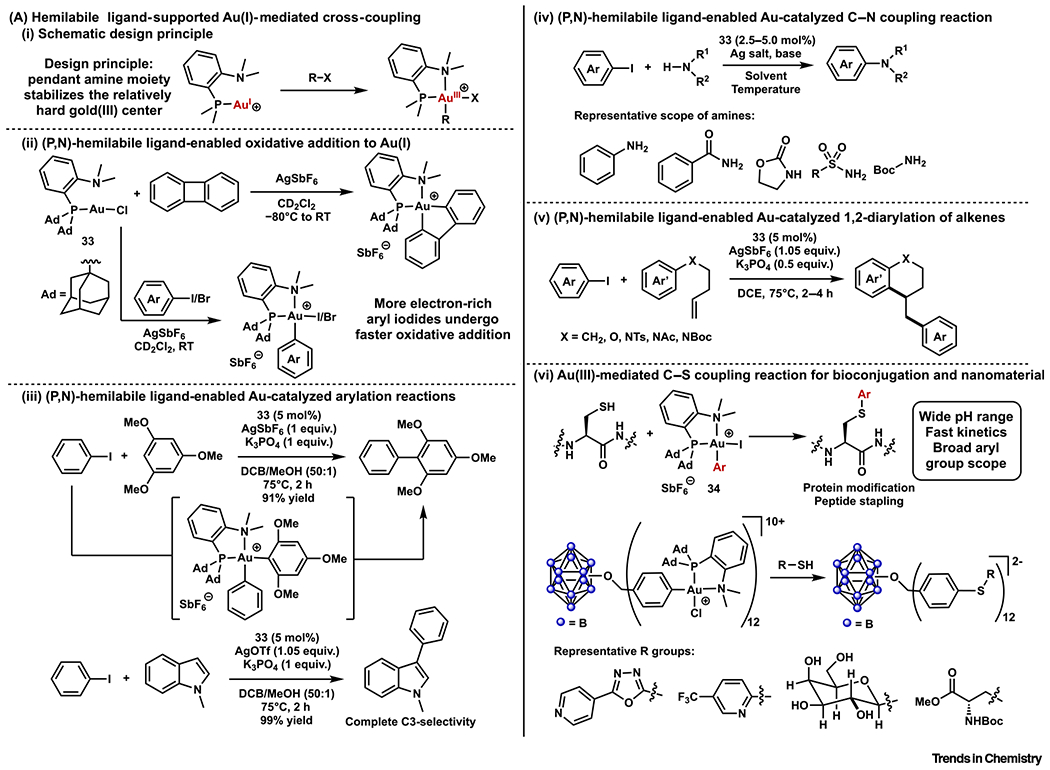
(A) Hemilabile ligand-supported Au(I)-mediated cross-coupling: (i) schematic design principle; (ii) (P,N)-hemilabile ligand-enabled oxidative addition to Au(I); (iii) (P,N)-hemilabile ligand-enabled Au-catalyzed arylation reactions; (iv) (P,N)-hemilabile ligand-enabled Au-catalyzed C–N coupling reaction; (v) (P,N)-hemilabile ligand-enabled Au-catalyzed 1,2-diarylation of alkenes; and (vi) Au(III)-mediated C–S coupling reaction for bioconjugation and nanomaterials.
The Au(III)-mediated arylation reactivity has found applications beyond synthetic organic chemistry. Given the previous examples of Pd(II)-mediated cysteine S-arylation [64], Maynard, Spokoyny, and coworkers explored similar transformations using Au(III) complexes 34 (Figure 4Avi) [65]. An efficient and chemoselective cysteine arylation procedure was developed employing [(Me-DalPhos)AuArCl][SbF6]. The protocol was further applied to protein labeling and peptide stapling. This methodology offered a much broader scope of aryl group than previous Au(III)-mediated S-arylation reactions [66–69]. Compared with Pd(II)-mediated cysteine S-arylations, the Au(III)-promoted version was effective at a wider pH range and displayed a faster reaction rate [70]. The mechanism of this methodology was studied later by Zhang and Dong [71]. More recently, this C–S coupling reactivity was used to construct atomically precise hybrid nanomaterials (Figure 4Avi) [72].
Photochemically Triggered Oxidative Addition to Au(I) Complexes
Photochemical conditions can allow access to high-energy intermediates (e.g., radicals) that are difficult to access under mild thermal conditions [73]. Naturally, these conditions been exploited to facilitate the otherwise sluggish oxidative addition to Au(I) complexes. This section covers examples of photoinitiated oxidative addition, photoredox-gold dual catalysis, and photosensitized oxidative addition.
In 2014, Toste and coworkers reported photoinitiated oxidative addition of CF3I to phosphine Au(I) aryl complex 35 (Figure 5A) [74]. The reaction proceeded through a radical chain process from light-promoted excitation of CF3I. Iodide abstraction from the resulting Au(III) complex 36 led to fast C(aryl)–CF3 reductive elimination from cationic tricoordinate Au(III) species. A closely related CF2H-containing complex was later reported by other groups to undergo fast C(aryl)–CF2H reductive elimination as well [75]. Derived from the iodide-containing complex, a series of Au(III) complexes 38 bearing other halides underwent thermal reductive elimination with product distributions [C(aryl)–CF3 versus C(aryl)–X formation, X = halide] that depended on the identity of the halide (Figure 5A) [76].
Figure 5. Photochemically Triggered Oxidative Addition to Au(I) Complexes.
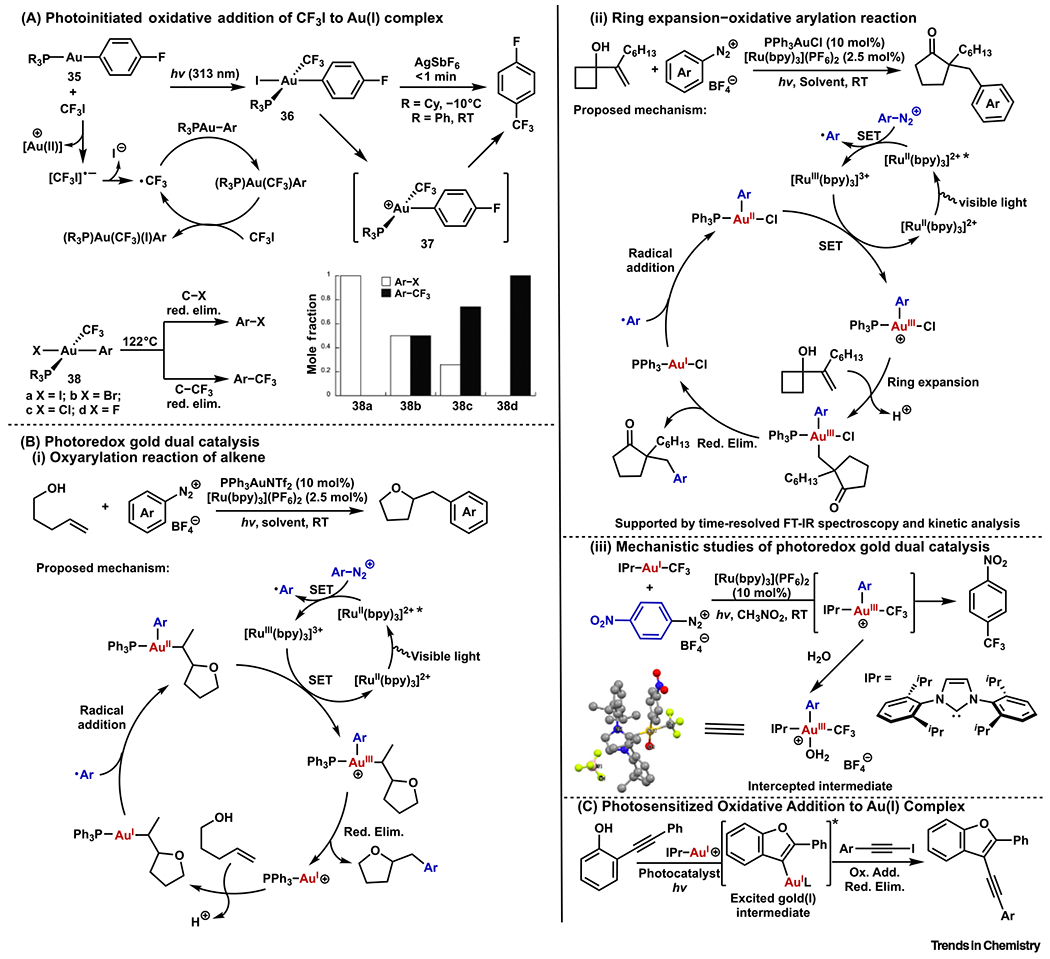
(A) Photoinitiated oxidative addition of CF3I to an Au(I) complex. (B) Photoredox gold dual catalysis: (i) an oxyarylation reaction of alkene; (ii) ring expansion–oxidative arylation reaction; and (iii) mechanistic studies of photoredox gold dual catalysis. (C) Photosensitized oxidative addition to an Au(I) complex.
Photoredox catalysis has also been combined with transition-metal catalysis to enable novel reactivities [77]. Inspired by the early examples of photoredox-enabled mild oxidative additions to Pd(II) and Cu(I) [78–80], Glorius and Toste independently reported a similar strategy for Au(I) catalysis. Glorius and coworkers demonstrated oxy- and aminoarylation reactions of alkenes (Figure 5Bi) [81]. Frei, Toste, and coworkers reported a ring expansion–oxidative arylation reaction (Figure 5Bii) [82]. In these publications, two different mechanisms were proposed, differing only with regard to the timing of the Au(I) intermediate undergoing oxidative addition. Glorius and coworkers proposed an Au(I)-mediated cyclization followed by photoredox-facilitated oxidative addition of the aryldiazonium salt (Figure 5Bi), whereas Frei, Toste, and coworkers suggested that photoredox-facilitated oxidative addition of the aryldiazonium salt occurred prior to the ring expansion (Figure 5Bii). The later mechanism was supported by time-resolved FT-IR spectroscopy and kinetic analysis.
Following these reports, many dual photoredox gold catalytic systems have been discovered. These reactions were reviewed elsewhere and are not elaborated here [83,84]; however, it is worth noting that examples of gold redox catalysis using aryl diazonium salts, without additional photocatalyst, have been reported under either photochemical or thermal conditions [85–94].
Given the proposed involvement of light-generated radical species in these dual gold-photoredox processes, another general mechanistic question was whether the bond-forming events were reductive eliminations from Au(III) or intermediates. To gain insights into this question, Kim and Toste recently reported the mechanistic studies of photoredox-initiated arylation of IPrAu(I)–CF3 and IPrAu(I)–succinimide (Figure 5Biii) [95]. Photochemical and electrochemical data, along with crystallographic characterization of key Au(III) intermediates, supported a mechanism involving reductive elimination from the Au(III) center as the bond-forming step. This study may also provide mechanistic insights into other metallaphotoredox catalysis.
Most recently, an Au(I)-catalyzed alkynylative cyclization reaction system using photocatalysts was disclosed (Figure 5C). Instead of the aforementioned electron-transfer mechanism for aryl diazonium salts, the authors proposed an energy-transfer mechanism based on experimental and theoretical evidence [96]. It is worth noting that another alkynylative cyclization was reported by Hashmi and coworkers at around same time [97]. Direct oxidative addition of bromoalkynes to Au(I) intermediates was proposed without invoking photosensitization. Further studies will help to elucidate the role and principle of photosensitization in these systems.
Other Reductive Elimination Reactions from Au(III) Centers
Many of the reductive elimination reactions presented above are subsequent steps to an oxidative addition. In this section, other examples of reductive elimination reactions of Au(III) complexes made from more conventional oxidation–transmetalation routes are discussed.
In 2014, Toste and coworkers reported that very rapid C(aryl)–C(aryl) reductive elimination from 39 occurred even below −20°C (Figure 6Ai) [98]. Kinetic studies indicated a unimolecular pathway with one of the fastest measured rates of transition-metal-mediated C–C reductive elimination. Surprisingly, a significant rate enhancement was observed in the presence of excess triphenylphosphine, while adding NBu4Cl had no impact on the rate. This implied a mechanism different from the ‘phosphine dissociation mechanism’ proposed by Kochi for C(alkyl)–C(alkyl) reductive elimination [99]. A mechanism was proposed to account for the rate acceleration by extra phosphine: a transient cationic (Ph3P)2bis(aryl)Au(III) species 40 was formed and rendered faster reductive elimination. More recently, Rocchigiani, Budzelaar, Bochmann, and coworkers also reported fast C(aryl)–C(aryl) reductive elimination of Au(III) complexes [100].
Figure 6. Other Reductive Elimination Reactions from Au(III) Centers.
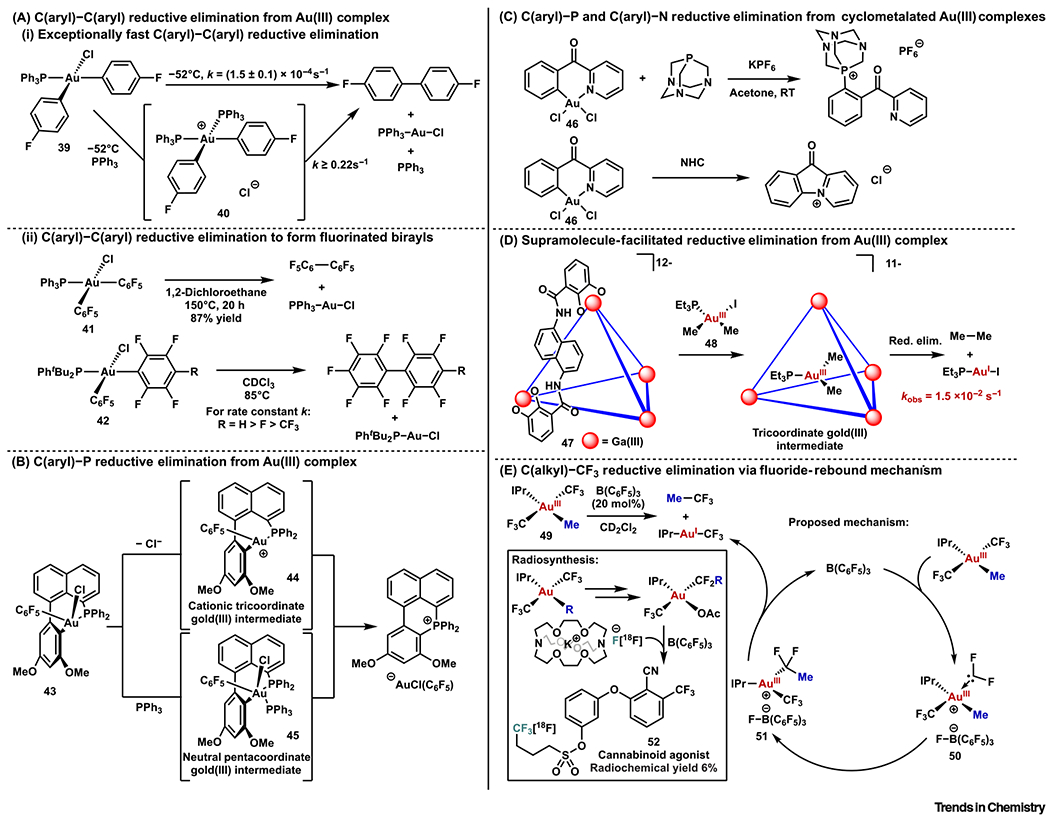
(A) C(aryl)–C(aryl) reductive elimination from an Au(III) complex: (i) exceptionally fast C(aryl)–C(aryl) reductive elimination; and (ii) C(aryl)–C(aryl) reductive elimination to form fluorinated birayls. (B) C(aryl)–P reductive elimination from an Au(III) complex. (C) C(aryl)–P and C(aryl)–N reductive elimination from cyclometalated Au(III) complexes. (D) Supramolecule-facilitated reductive elimination from an Au(III) complex. (E) C(alkyl)–CF3 reductive elimination via a fluoride-rebound mechanism.
Around the same time in 2014, the group of Nevado studied a related Au(III) complex 41 and reported an extremely slow C6F5–C6F5 reductive elimination even under elevated temperature (150°C, 20 h, 87% yield) (Figure 6Aii) [101]. More recently, Lan, Shen, and coworkers conducted systematic studies on a series of phosphine biarylAu(III) chloride complexes 42 [102]. While the electronic properties of ligands had a trivial effect on the reductive elimination rate, more electron-deficient arenes underwent slower reductive elimination (Figure 6Aii). These reports were consistent with the computational studies by Datta and coworkers [103].
In the course of studying cis-[(R3P)(Aryl)AuCl2], the precursor of 39 described above, Toste and coworkers reported that facile C(aryl)–P reductive elimination ensued from a related Au(III) complex 43 on heating/addition of AgSbF6 or addition of nucleophiles (Figure 6B) [104]. A cationic tricoordinate Au(III) intermediate 44 and a neutral pentacoordinate Au(III) specie 45 were proposed, respectively, as intermediates in these reductive elimination reactions. This study disclosed a decomposition pathway for phosphine-supported Au(III) catalysts, which was valuable for future Au(III) catalyst development. A recent study from Barone, Casini, and coworkers also showed C(aryl)–P coupling from a cyclometalated Au(III) complex 46 (Figure 6C) [105]. The same complex was reported to undergo C(aryl)–N reductive elimination on treatment with NHCs (Figure 6C) [106].
As noted above, reductive eliminations can be triggered by halide abstraction from neutral Au(III) complexes. Motivated by the previous work that halide dissociation was promoted by a highly anionic self-assembled tetrahedral host 47 [107], Bergman, Raymond, Toste, and coworkers envisioned the host catalyzing the reductive elimination from the Au(III) center [108, 109]. The strategy was applied to complex 48. On the addition of host 47, C(alkyl)–C(alkyl) reductive elimination was observed with a 1.9 × 107-times faster rate compared with the uncatalyzed reaction (Figure 6D). Notably, subsequent computational studies from several research groups unveiled the critical role of electrostatic interaction between the supramolecular assembly and the cationic Au(III) complex in facilitating the reductive elimination [110–112]. Since the development of alkyl–alkyl cross-coupling processes is hampered, in part, by the slow C(alkyl)–C(alkyl) reductive elimination [15], the successful implementation of the supramolecular-assembly-catalyzed reductive elimination provided a new strategy to resolve this challenging problem.
In 2017, O’Neil, Toste, and coworkers discovered a C(alkyl)–CF3 reductive elimination reaction that proceeded via a fluoride-rebound mechanism (Figure 6E). A trifluoromethylated product, trifluoroethane, was first obtained on treatment of complex 49 with B(C6F5)3. Mechanistic investigation pointed to a pathway in which fluoride abstraction from 49 formed a cationic difluorocarbene Au(III) complex 50, which underwent carbon–carbon bond-forming migratory insertion of a methyl group into difluorocarbene. Final recombination of fluoride to 51 furnished the final product. This understanding established the possibility of 18F incorporation into the product with external [18F]-fluoride addition. A protocol was developed to synthesize 18F-radiolabeled aliphatic CF3-containing compounds including a cannabinoid agonist 52 [113].
Concluding Remarks and Future Perspectives
As described above, many exciting details about the gold redox events were unfurled in the past 6 years. Retrospectively, detailed stoichiometric studies and mechanistic investigation were critical to the development of catalytic transformations, as well as applications of gold-promoted transformations aimed at biology or materials science. The importance of serendipity should also be noted, as many of the discoveries came unexpectedly.
It is becoming increasingly apparent that this field of research not just has the potential to replicate that of palladium or nickel cross-coupling catalysis, but instead can offer milder reaction conditions, complementary substrate scope, and unprecedented reactivities or selectivities. To reach this potential, many important questions and opportunities remain (see Outstanding Questions).
Outstanding Questions.
Can new platforms for the generation of stable and tunable Au(III) complexes that retain catalytic activity be discovered? If so, will the increased diversity of Au(III) catalysts result in new reactions that diverge from the reactivity of Au(I) and other transition metals?
How can more Au-catalyzed crosscoupling reactions, especially those that leverage the facile reductive elimination from gold(III), be developed? Will the development of these reactions complement existing technology or enable new applications?
Can mechanistic studies on elementary organometallic reaction classes common to other transition metals, such as migratory insertion, enable new reactivity in Au complexes and continue to enable new technologies? For example, can mechanistic studies improve the efficiency of Au-mediated 18F incorporation?
Can emerging technologies, such as electrochemistry, be deployed to facilitate gold redox events?
First, the utility of stable Au(III) complexes arising from the oxidative addition to Au(I) remains underexplored. Taking the biphenylene-supported Au(III) complex as an example, studies need to be conducted to understand the structural influence of both ancillary ligands (NHC ligand and substituted biphenylene) on the rate of oxidative addition. Moreover, the discovery of other thermodynamically favored oxidative additions to Au(I) that produce stable Au(III) complexes can lead to a library of Au(III) complexes, creating a larger chemical space for exploration. New catalysts in this area can reveal new opportunities in asymmetric catalysis, especially in cases where remote regioselectivities and high enantioselectivities are desired.
In addition to ligands that enable stable Au(III) catalysts, opportunities exist to discover new strategies that allow facile entry into Au(I)/Au(III) catalytic cycles. In this context, the P,N-hemilabile ligands inspire the search for additional hemilabile ligands that can be investigated to increase reactivity, and provide tunability, using this platform. Discoveries in this area are likely to lead to coupling reactivities that may be complimentary to existing transition-metal-catalyzed processes or further provide opportunities in chemical synthesis, biology, and materials science.
Much remains to be learned about migratory insertion processes at gold centers. For example, studies aimed at further understanding of carbene migratory insertion at gold can inform on how ligands can been chosen to either disfavor migratory insertion to avoid catalyst decomposition or support this step and lead to polymerization reactivity. As a specific example, ligand effects on Au(III)-mediated C(alkyl)–CF3 bond formation, via migratory insertion, can result in improved radiochemical yield and specific activity for 18F incorporation. Along these lines, a bonding model for Au(III) carbene complexes, compared with the Au(I) version [114–116], remains to be established.
Similarly, while photochemical gold catalysis also has enormous potential, much remains to be explored and developed. For example, photoredox gold-catalyzed aryl amination and trifluoromethylation have not been realized despite all of the elementary steps demonstrated. Finding reaction conditions in which transmetalation, reductive elimination, and other elementary processes at gold are compatible with photoredox catalysis is likely to reveal many new opportunities for reaction development. Additionally, outside combined photoredox/gold catalysis, additional examples of photosensitized oxidative addition must be discovered to establish this reaction manifold as a design principle for gold-only catalytic reactivity. Last, metallaelectrochemistry has experienced a renaissance over the past several years [117–119]. Its application to facilitate gold redox chemistry has also started to emerge in the literature [120]. With more detailed mechanistic studies, the development of new electro/gold catalysis is on the horizon.
Highlights.
Gold redox chemistry enables selective synthetic methods with a wide range of applications.
A number of strategies have been developed to facilitate redox reactions of gold complexes and mechanistic studies have guided their development.
The use of gold redox transformations in bioconjugation, materials sciences, and radiochemical synthesis has started to appear.
Acknowledgments
We acknowledge the National Institute of General Medical Science (grant R35 GM118190) for support of this work. We also thank Professor Mark Levin, Dr Suhong Kim, and Mr Edward Miller for helpful discussions.
Glossary
- C–H auration
a process in which a C–H bond is transformed into a C–Au bond.
- Cross-coupling reaction
the reaction that connects two different fragments together.
- Migratory insertion
a process in which one ligand of the metal center inserts into another metal–ligand bond.
- Oxidative addition
a process in which the metal inserts into a covalent bond and the oxidation state of the metal center increases by two.
- Photoredox catalysis
a form of catalysis that accelerates the reaction via light-promoted redox events.
- Redox chemistry
chemical transformations that involve changes in the oxidation states of atoms.
- Redox potential
an indicator of the tendency of chemical species to gain or lose electrons.
- Reductive elimination
a process in which the bonds between the metal center and two X-type ligands break and a covalent bond is formed between two ligands.
- Selectivity factor (s-factor)
a measure of relative reaction rates of enantiomers in a kinetic resolution.
- Transmetalation
a process in which an X-type ligand is transferred to the metal center.
References
- 1.Hashmi ASK (2007) Gold-catalyzed organic reactions. Chem. Rev. 107, 3180–3211 [DOI] [PubMed] [Google Scholar]
- 2.Gorin DJ and Toste FD (2007) Relativistic effects in homogeneous gold catalysis. Nature 446, 395–403 [DOI] [PubMed] [Google Scholar]
- 3.Fürstner A and Davies PW (2007) Catalytic carbophilic activation: catalysis by platinum and gold π acids. Angew. Chem. Int. Ed. 46, 3410–3449 [DOI] [PubMed] [Google Scholar]
- 4.Gorin DJ et al. (2008) Ligand effects in homogeneous Au catalysis. Chem. Rev. 108, 3351–3378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He C et al. (2008) Gold-catalyzed organic transformations. Chem. Rev. 108, 3239–3265 [DOI] [PubMed] [Google Scholar]
- 6.Jiménez-Núñez E and Echavarren AM (2008) Gold-catalyzed cycloisomerizations of enynes: a mechanistic perspective. Chem. Rev. 108, 3326–3350 [DOI] [PubMed] [Google Scholar]
- 7.Shapiro ND and Toste FD (2010) A reactivity-driven approach to the discovery and development of gold-catalyzed organic reactions. Synlett 2010, 675–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L (2014) A non-diazo approach to α-oxo gold carbenes via gold-catalyzed alkyne oxidation. Acc. Chem. Res. 47, 877–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng Z et al. (2016) Au-catalysed oxidative cyclisation. Chem. Soc. Rev. 45, 4448–4458 [DOI] [PubMed] [Google Scholar]
- 10.Dorel R and Echavarren AM (2015) Gold(I)-catalyzed activation of alkynes for the construction of molecular complexity. Chem. Rev. 115, 9028–9072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao X et al. (2019) Dual gold catalysis – an update. Chem. Commun. 55, 12127–12135 [DOI] [PubMed] [Google Scholar]
- 12.Nugent WA (2012) “Black swan events” in organic synthesis. Angew. Chem. Int. Ed. 51,8936–8949 [DOI] [PubMed] [Google Scholar]
- 13.Joost M et al. (2015) Reactivity of gold complexes towards elementary organometallic reactions. Angew. Chem. Int. Ed. 54, 15022–15045 [DOI] [PubMed] [Google Scholar]
- 14.Mertens RT and Awuah SG (2019) Gold catalysis: fundamentals and recent developments. ACS Symp. Ser. 1317,19–55 [Google Scholar]
- 15.Hartwig JF (2010) Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books [Google Scholar]
- 16.Bratsch SG (1989) Standard electrode potentials and temperature coefficients in water at 298.15 K. J. Phys. Chem. Ref. Data 18, 1–21 [Google Scholar]
- 17.Hopkinson MN et al. (2011) AuI/AuIII catalysis: an alternative approach for C–C oxidative coupling. Chem. Eur. J. 17, 8248–8262 [DOI] [PubMed] [Google Scholar]
- 18.Garcia P et al. (2010) Gold-catalyzed cross-couplings: new opportunities for C–C bond formation. ChemCatChem 2, 493–497 [Google Scholar]
- 19.Wegner HA and Auzias M (2011) Gold for C–C coupling reactions: a Swiss-army-knife catalyst? Angew. Chem. Int. Ed. 50, 8236–8247 [DOI] [PubMed] [Google Scholar]
- 20.Boorman TC and Larrosa I (2011) Gold-mediated C–H bond functionalisation. Chem. Soc. Rev. 40, 1910–1925 [DOI] [PubMed] [Google Scholar]
- 21.Hashmi ASK et al. (2009) Synthesis, structure and reactivity of organogold compounds of relevance to homogeneous gold catalysis. J. Organomet. Chem. 694, 592–597 [Google Scholar]
- 22.Joost M et al. (2014) Direct evidence for intermolecular oxidetive addition of σ(Si–Si) bonds to gold. Angew. Chem. Int. Ed. 53, 747–751 [DOI] [PubMed] [Google Scholar]
- 23.Wu CY et al. (2015) Stable gold(III) catalysts by oxidative addition of a carbon–carbon bond. Nature 517, 449–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidbaur H and Schier A (2012) Gold(III) compounds for homogeneous catalysis: preparation, reaction conditions, and scope of application. Arab. J. Sci. Eng. 37, 1187–1225 [Google Scholar]
- 25.Kumar R and Nevado C (2017) Cyclometalated gold(III) complexes: synthesis, reactivity, and physicochemical properties. Angew. Chem. Int. Ed. 56,1994–2015 [DOI] [PubMed] [Google Scholar]
- 26.Hashmi ASK et al. (2004) Gold catalysis: the benefits of N and N,O ligands. Angew. Chem. Int. Ed. 43, 6545–6547 [DOI] [PubMed] [Google Scholar]
- 27.Hashmi ASK et al. (2006) Gold catalysis: evidence for the in-situ reduction of gold(III) during the cyclization of allenyl carbinols. Eur. J. Org. Chem. 2006, 1387–1389 [Google Scholar]
- 28.Gaillard S et al. (2010) Synthetic and structural studies of [AuCl3(NHC)] complexes. Organometallics 29, 394–402 [Google Scholar]
- 29.De Frémont P et al. (2007) Synthesis, characterization and reactivity of N-heterocyclic carbene gold(III) complexes. Organometallics 26, 1376–1385 [Google Scholar]
- 30.Lee JS et al. (2020) Architectural stabilization of a gold(III) catalyst in metal–organic frameworks. Chem 6, 142–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhukhovitskiy AV et al. (2018) Migratory insertion of carbenes into Au(III)–C bonds. J. Am. Chem. Soc. 140, 466–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.López F and Mascareñas JL (2013) Gold(I)-catalyzed enantioselective cycloaddition reactions. Beilstein J. Org. Chem. 9, 2250–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang YM et al. (2014) Development of catalysts and ligands for enantioselective gold catalysis. Acc. Chem. Res. 47, 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zi W and Toste FD (2016) Recent advances in enantioselective gold catalysis. Chem. Soc. Rev. 45, 4567–4589 [DOI] [PubMed] [Google Scholar]
- 35.Li Y et al. (2017) Gold-catalyzed enantioselective annulations. Chem. Eur. J. 23, 467–512 [DOI] [PubMed] [Google Scholar]
- 36.Bohan PT and Dean Toste F (2017) Well-defined chiral gold(III) complex catalyzed direct enantioconvergent kinetic resolution of 1,5-enynes. J. Am. Chem. Soc. 139,11016–11019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez J and Bourissou D (2018) Well-defined chiral gold(III) complexes: new opportunities in asymmetric catalysis. Angew. Chem. Int. Ed. 57, 386–388 [DOI] [PubMed] [Google Scholar]
- 38.Reid JP et al. Strategies for remote enantiocontrol in chiral gold (III) complexes applied to catalytic enantioselective γ, δ-Diels–Alder reactions. Chem. Sci. 10.1039/D0SC00497A (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abdou HE et al. (2007) Oxidative addition of small molecules to a dinuclear Au(I) amidinate complex, Au2[2,6-Me2Ph2N2CH]2. Syntheses and characterization of Au(II) amidinate complexes including one which possesses Au(II)–oxygen bonds. Inorg. Chem. 46, 9692–9699 [DOI] [PubMed] [Google Scholar]
- 40.Tkatchouk E et al. (2011) Two metals are better than one in the gold catalyzed oxidative heteroarylation of alkenes. J. Am. Chem. Soc. 133, 14293–14300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levin MD and Toste FD (2014) Gold-catalyzed allylation of aryl boronic acids: accessing cross-coupling reactivity with gold. Angew. Chem. Int Ed. 53, 6211–6215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu K et al. (2019) Gold-catalyzed oxidative biaryl cross-coupling of organometallics. Chem 5, 2718–2730 [Google Scholar]
- 43.Ball LT et al. (2012) Gold-catalyzed direct arylation. Science 337, 1644–1648 [DOI] [PubMed] [Google Scholar]
- 44.Ball LT et al. (2014) Gold-catalyzed oxidative coupling of arylsilanes and arenes: origin of selectivity and improved precatalyst. J. Am. Chem. Soc. 136, 254–264 [DOI] [PubMed] [Google Scholar]
- 45.Cambeiro XC et al. (2015) Au-catalyzed cross-coupling of arenes via double C–H activation. J. Am. Chem. Soc. 137, 15636–15639 [DOI] [PubMed] [Google Scholar]
- 46.Robinson MP and Lloyd-Jones GC (2018) Au-catalyzed oxidative arylation: chelation-induced turnover of ortho-substituted arylsilanes. ACS Catal. 8, 7484–7488 [Google Scholar]
- 47.Fricke C et al. (2019) Gold-catalyzed C–H functionalization with aryl germanes. ACS Catal. 9, 9231–9236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W et al. (2019) Cooperative Au/Ag dual-catalyzed cross-dehydrogenative biaryl coupling: reaction development and mechanistic insight. J. Am. Chem. Soc. 141, 3187–3197 [DOI] [PubMed] [Google Scholar]
- 49.Dahiya A et al. (2020) Gold-catalyzed chemoselective couplings of polyfluoroarenes with aryl germanes and downstream diversification. J. Am. Chem. Soc. 142, 7754–7759 [DOI] [PubMed] [Google Scholar]
- 50.Van Leeuwen PWNM et al. (2000) Ligand bite angle effects in metal-catalyzed C–C bond formation. Chem. Rev. 100, 2741–2769 [DOI] [PubMed] [Google Scholar]
- 51.Joost M et al. (2014) Facile oxidative addition of aryl iodides to gold(I) by ligand design: bending turns on reactivity. J. Am. Chem. Soc. 136, 14654–14657 [DOI] [PubMed] [Google Scholar]
- 52.Joost M et al. (2015) Oxidative addition of carbon–carbon bonds to gold. Angew. Chem. Int. Ed. 54, 5236–5240 [DOI] [PubMed] [Google Scholar]
- 53.Harper MJ et al. (2018) Oxidative addition,transmetalation, and reductive elimination at a 2,2’-bipyridyl-ligated gold center. J. Am. Chem. Soc. 140, 4440–4445 [DOI] [PubMed] [Google Scholar]
- 54.Peng H et al. (2014) Gold-catalyzed oxidative cross-coupling of terminal alkynes: selective synthesis of unsymmetrical 1,3-diynes. J. Am. Chem. Soc. 136, 13174–13177 [DOI] [PubMed] [Google Scholar]
- 55.Cai R et al. (2015) Ligand-assisted gold-catalyzed cross-coupling with aryldiazonium salts: redox gold catalysis without an external oxidant. Angew. Chem. Int. Ed. 54, 8772–8776 [DOI] [PubMed] [Google Scholar]
- 56.Ye X et al. (2018) Gold-catalyzed oxidative coupling of alkynes toward the synthesis of cyclic conjugated diynes. Chem 4, 1983–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Y et al. (2019) Dual gold/silver catalysis involving alkynylgold(III) intermediates formed by oxidative addition and silver-catalyzed C–H activation for the direct alkynylation of cyclopropenes. Angew. Chem. Int. Ed. 58, 5129–5133 [DOI] [PubMed] [Google Scholar]
- 58.Cadge JA et al. (2020) Oxidative addition of alkenyl and alkynyl iodides to a Au’ complex. Angew. Chem. Int. Ed. 59, 6617–6621 [DOI] [PubMed] [Google Scholar]
- 59.Zeineddine A et al. (2017) Rational development of catalytic Au(I)/Au(III) arylation involving mild oxidative addition of aryl halides. Nat. Commun. 8, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodriguez J et al. (2019) Catalytic Au(I)/Au(III) arylation with the hemilabile MeDalphos ligand: unusual selectivity for electron-rich iodoarenes and efficient application to indoles. Chem. Sci. 10, 7183–7192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodriguez J et al. (2020) Au(I)/Au(III)-catalyzed C–N coupling. Chem. Commun. 56, 94–97 [Google Scholar]
- 62.Akram MO et al. (2019) Ligand-enabled gold-catalyzed C (sp2)–N cross-coupling reactions of aryl iodides with amines. Org. Lett. 21, 8101–8105 [DOI] [PubMed] [Google Scholar]
- 63.Patil NT et al. (2020) Gold-catalyzed 1,2-diarylation of alkenes. Angew. Chem. Int. Ed. Published online March 23, 2020. 10.1002/anie.202002141 [DOI] [PubMed] [Google Scholar]
- 64.Vinogradova EV et al. (2015) Organometallic palladium reagents for cysteine bioconjugation. Nature 526, 687–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Messina MS et al. (2018) Organometallic gold(III) reagents for cysteine arylation. J. Am. Chem. Soc. 140, 7065–7069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kung KKY et al. (2014) Cyclometalated gold(III) complexes for chemoselective cysteine modification via ligand controlled C–S bond-forming reductive elimination. Chem. Commun. 50, 11899–11902 [DOI] [PubMed] [Google Scholar]
- 67.de Paiva REF et al. (2018) Gold-catalyzed C–S aryl-group transfer in zinc finger proteins. Angew. Chem. Int. Ed. 57, 9305–9309 [DOI] [PubMed] [Google Scholar]
- 68.Wenzel MN et al. (2019) Cyclometalated AuIII complexes for cysteine arylation in zinc finger protein domains: towards controlled reductive elimination. Chem. Eur. J. 25, 7628–7634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Currie L et al. (2018) Carbon–sulfur bond formation by reductive elimination of gold(III) thiolates. Dalton Trans. 47, 6333–6343 [DOI] [PubMed] [Google Scholar]
- 70.Zhang C et al. (2019) Arylation chemistry for bioconjugation. Angew. Chem. Int. Ed. 58, 4810–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang SL and Dong JJ (2019) Mechanism and chemoselectivity origins of bioconjugation of cysteine with Au (III)-aryl reagents. Org. Biomol. Chem. 17, 1245–1253 [DOI] [PubMed] [Google Scholar]
- 72.Stauber JM et al. (2019) An organometallic strategy for assembling atomically precise hybrid nanomaterials. J. Am. Chem. Soc. 142, 327–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Turro NJ (1991) Modern Molecular Photochemistry, University Science Books [Google Scholar]
- 74.Winston MS et al. (2014) Photoinitiated oxidative addition of CF3I to gold(I) and facile aryl-CF3 reductive elimination. J. Am. Chem. Soc. 136, 7777–7782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu S et al. (2018) The difluoromethylated organogold(III) complex cis-[Au(PCy3)(4-F-C6H4)(CF2H)(Cl)]: preparation, characterization, and its C(sp2)-CF2H reductive elimination. Organometallics 37, 3901–3908 [Google Scholar]
- 76.Winston MS et al. (2015) Halide-dependent mechanisms of reductive elimination from gold(III). J. Am. Chem. Soc. 137, 7921–7928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Levin MD et al. (2016) Photoredox catalysis unlocks single-electron elementary steps in transition metal catalyzed cross-coupling. ACS Cent. Sci. 2, 293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kalyani D et al. (2011) Room-temperature C–H arylation: merger of Pd-catalyzed C–H functionalization and visible-light photocatalysis. J. Am. Chem. Soc. 133, 18566–18569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Neufeldt SR and Sanford MS (2012) Combining transition metal catalysis with radical chemistry: dramatic acceleration of palladium-catalyzed C–H arylation with diaryliodonium salts. Adv. Synth. Catal. 354, 3517–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ye Y and Sanford MS (2012) Merging visible-light photocatalysis and transition-metal catalysis in the copper-catalyzed trifluoromethylation of boronic acids with CF3I. J. Am. Chem. Soc. 134, 9034–9037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sahoo B et al. (2013) Combining gold and photoredox catalysis: visible light-mediated oxy- and aminoarylation of alkenes. J. Am. Chem. Soc. 135, 5505–5508 [DOI] [PubMed] [Google Scholar]
- 82.Shu XZ et al. (2014) Dual visible light photoredox and gold-catalyzed arylative ring expansion. J. Am. Chem. Soc. 136, 5844–5847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hopkinson MN et al. (2016) Merging visible light photoredox and gold catalysis. Acc. Chem. Res. 49, 2261–2272 [DOI] [PubMed] [Google Scholar]
- 84.Zidan M et al. (2018) Recent advances in mono and binuclear gold photoredox catalysis. Catal. Sci. Technol. 8, 6019–6028 [Google Scholar]
- 85.Huang L et al. (2016) Photosensitizer-free visible-light-mediated gold-catalyzed 1,2-difunctionalization of alkynes. Angew. Chem. Int. Ed. 55, 4808–4813 [DOI] [PubMed] [Google Scholar]
- 86.Deng JR et al. (2017) Photosensitizer-free visible light-mediated gold-catalysed: cis-difunctionalization of silyl-substituted alkynes. Chem. Sci. 8, 7537–7544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Witzel S et al. (2018) New transmetalation reagents for the gold-catalyzed visible light-enabled C(sp or sp2)–C(sp2) cross-coupling with aryldiazonium salts in the absence of a photosensitizer. Chem. Commun. 54, 13802–13804 [DOI] [PubMed] [Google Scholar]
- 88.Xie J et al. (2018) Light-induced gold-catalyzed Hiyama arylation: a coupling access to biarylboronates. Angew. Chem. Int Ed. 130, 16890–16895 [DOI] [PubMed] [Google Scholar]
- 89.Taschinski S et al. (2019) Light-induced mechanistic diver-gence in gold(I) catalysis: revisiting the reactivity of diazonium salts. Angew. Chem. Int. Ed. 58, 16988–16993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peng H et al. (2016) Nucleophile promoted gold redox catalysis with diazonium salts: C–Br, C–S and C–P bond formation through catalytic Sandmeyer coupling. Chem. Sci. 7, 6190–6196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dong B et al. (2017) Gold redox catalysis through base-initiated diazonium decomposition toward alkene, alkyne, and allene activation. Chem. Eur. J. 23, 11093–11099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Akram MO et al. (2018) Gold(I)-catalyzed cross-coupling reactions of aryldiazonium salts with organostannanes. Org. Biomol. Chem. 16, 2865–2869 [DOI] [PubMed] [Google Scholar]
- 93.Jimoh AA et al. (2019) Gold redox catalysis for cyclization/arylation of allylic oximes: synthesis of isoxazoline derivatives. Chem. Commun. 55, 8150–8153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Witzel S et al. (2017) Photosensitizer-free, gold-catalyzed C–C cross-coupling of boronic acids and diazonium salts enabled by visible light. Adv. Synth. Catal. 359, 1522–1528 [Google Scholar]
- 95.Kim S and Toste FD (2019) Mechanism of photoredox-initiated C–C and C–N bond formation by arylation of IPrAu(I)-CF3 and IPrAu(I)-succinimide. J. Am. Chem. Soc. 141,4308–4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xia Z et al. (2019) Photosensitized oxidative addition to gold (I) enables alkynylative cyclization of o-alkylnylphenols with iodoalkynes. Nat. Chem. 11, 797–805 [DOI] [PubMed] [Google Scholar]
- 97.Yang Y et al. (2019) Gold-catalyzed C(sp2)–C(sp) coupling by alkynylation through oxidative addition of bromoalkynes. Chem. Eur. J. 25, 9624–9628 [DOI] [PubMed] [Google Scholar]
- 98.Wolf WJ et al. (2014) Exceptionally fast carbon–carbon bond reductive elimination from gold(III). Nat. Chem. 6, 159–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Komiya S and Kochi JK (1976) Electrophilic cleavage of organogold complexes with acids. The mechanism of the reductive elimination of dialkyl(aniono)gold(III) species. J. Am. Chem. Soc. 98, 7599–7607 [Google Scholar]
- 100.Rocchigiani L et al. (2018) Reductive elimination leading to C–C bond formation in gold(III) complexes: a mechanistic and computational study. Chem. Eur. J. 24, 8893–8903 [DOI] [PubMed] [Google Scholar]
- 101.Hofer M et al. (2014) A neutral gold(III)–boron transmetalation. Organometallics 33, 1328–1332 [Google Scholar]
- 102.Kang K et al. (2017) C(sp2)–C(sp2) reductive elimination from well-defined diarylgold(III) complexes. Organometallics 36, 4727–4740 [Google Scholar]
- 103.Nijamudheen A et al. (2014) Understanding the mechanisms of unusually fast H–H, C–H, and C–C bond reductive eliminations from gold(III) complexes. Chem. Eur. J. 20,14650–14658 [DOI] [PubMed] [Google Scholar]
- 104.Kawai H et al. (2016) Phosphonium formation by facile carbon–phosphorus reductive elimination from gold(III). J. Am. Chem. Soc. 138, 587–593 [DOI] [PubMed] [Google Scholar]
- 105.Bonsignore R et al. (2020) Carbon–phosphorus coupling from C^N cyclometalated AuIII complexes. Chem. Eur. J. 26, 4226–4231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim JH et al. (2019) Direct intramolecular carbon(sp2)–nitrogen(sp2) reductive elimination from gold(III). Dalton Trans. 48, 6273–6282 [DOI] [PubMed] [Google Scholar]
- 107.Hong CM et al. (2018) Self-assembled tetrahedral hosts as supramolecular catalysts. Acc. Chem. Res. 51,2447–2455 [DOI] [PubMed] [Google Scholar]
- 108.Kaphan DM et al. (2015) A supramolecular microenvironment strategy for transition metal catalysis. Science 350, 1235–1238 [DOI] [PubMed] [Google Scholar]
- 109.Levin MD et al. (2016) Scope and mechanism of cooperativity at the intersection of organometallic and supramolecular catalysis. J. Am. Chem. Soc. 138, 9682–9693 [DOI] [PubMed] [Google Scholar]
- 110.Vaissier Welborn V and Head-Gordon T (2018) Electrostatics generated by a supramolecular capsule stabilizes the transition state for carbon–carbon reductive elimination from gold(III) complex. J. Phys. Chem. Lett. 9, 3814–3818 [DOI] [PubMed] [Google Scholar]
- 111.Norjmaa G et al. (2019) Microsolvation and encapsulation effects on supramolecular catalysis: C–C reductive elimination inside [Ga4L6]12-metallocage. J. Am. Chem. Soc. 141, 13114–13123 [DOI] [PubMed] [Google Scholar]
- 112.Welborn VV et al. (2020) Interplay of water and a supramolecular capsule for catalysis of reductive elimination reaction from gold. Nat. Commun. 11, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Levin MD et al. (2017) A catalytic fluoride-rebound mechanism for C(sp3)–CF3 bond formation. Science 356,1272–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Benitez D et al. (2009) A bonding model for gold(I) carbene complexes. Nat. Chem. 1, 482–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang Y et al. (2015) Gold carbene or carbenoid: is there a difference? Chem. Eur. J. 21,7332–7339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Harris RJ and Widenhoefer RA (2016) Gold carbenes, gold-stabilized carbocations, and cationic intermediates relevant to gold-catalysed enyne cycloaddition. Chem. Soc. Rev. 45, 4533–4551 [DOI] [PubMed] [Google Scholar]
- 117.Meyer TH et al. (2019) Resource economy by metallaelectrocatalysis: merging electrochemistry and C–H activation. Trends Chem. 1, 63–76 [Google Scholar]
- 118.Sauermann N et al. (2018) Electrocatalytic C–H activation. ACS Catal. 8, 7086–7103 [Google Scholar]
- 119.Ma C et al. (2018) Recent advances in C–H functionalization using electrochemical transition metal catalysis. ACS Catal. 8, 7179–7189 [Google Scholar]
- 120.Ye X et al. (2019) Facilitating gold redox catalysis with electrochemistry: an efficient chemical-oxidant-free approach. Angew. Chem. Int. Ed. 58, 17226–17230 [DOI] [PMC free article] [PubMed] [Google Scholar]