

Abstract
MicroRNAs (miRNAs) are important gene expression regulators implicated in many biological processes, but we lack a global understanding of how miRNA genes evolve and contribute to developmental canalization and phenotypic diversification. Whole-genome duplication events likely provide a substrate for species divergence and phenotypic change by increasing gene numbers and relaxing evolutionary pressures. To understand the consequences of genome duplication on miRNA evolution, we studied miRNA genes following the teleost genome duplication (TGD). Analysis of miRNA genes in four teleosts and in spotted gar, whose lineage diverged before the TGD, revealed that miRNA genes were retained in ohnologous pairs more frequently than protein-coding genes, and that gene losses occurred rapidly after the TGD. Genomic context influenced retention rates, with clustered miRNA genes retained more often than nonclustered miRNA genes and intergenic miRNA genes retained more frequently than intragenic miRNA genes, which often shared the evolutionary fate of their protein-coding host. Expression analyses revealed both conserved and divergent expression patterns across species in line with miRNA functions in phenotypic canalization and diversification, respectively. Finally, major strands of miRNA genes experienced stronger purifying selection, especially in their seeds and 3′-complementary regions, compared with minor strands, which nonetheless also displayed evolutionary features compatible with constrained function. This study provides the first genome-wide, multispecies analysis of the mechanisms influencing metazoan miRNA evolution after whole-genome duplication.
Keywords: spotted gar Lepisosteus oculatus, zebrafish Danio rerio, three-spined stickleback Gasterosteus aculeatus, blackfin icefish Chaenocephalus aceratus, Japanese medaka Oryzias latipes, arm-switching
Introduction
MicroRNAs (miRNAs) are small noncoding RNAs that regulate protein-coding genes posttranscriptionally by binding to the 3′-UTR of messenger RNAs (mRNAs) when incorporated into the RNA-induced silencing complex (RISC) (Jonas and Izaurralde 2015; Bartel 2018). In metazoans, miRNAs are implicated in virtually every biological process, including cell proliferation and differentiation, development, physiology, and various pathologies (Sun and Lai 2013; Bartel 2018). In addition, miRNAs are hypothesized to provide robustness to embryonic development by buffering genetic noise, especially in stressful conditions (Mendell and Olson 2012; Cassidy et al. 2013; Schmiedel et al. 2015; Kasper et al. 2017; Liufu et al. 2017), which contributes to phenotypic canalization (Hornstein and Shomron 2006; Wu et al. 2009; Ebert and Sharp 2012; Posadas and Carthew 2014; Alberti and Cochella 2017). Conversely, miRNAs are hypothesized to promote the emergence of new phenotypes by differentially modulating developmental and physiological pathways (Niwa and Slack 2007; Berezikov 2011; Arif et al. 2013), which can influence adaptation, diversification, and speciation (Loh et al. 2011; Somel et al. 2011; Li and Zhang 2013; Quah et al. 2015; Franchini et al. 2016, 2019; Kittelmann and McGregor 2019; Xiong et al. 2019; Kelley et al. 2020). The finding that miRNA genes and their sequences are generally well conserved in evolution, especially in their seed (nucleotides 2–8), supports the crucial role of miRNAs in metazoan biology (Bartel 2009; Wheeler et al. 2009; Ameres and Zamore 2013; Fromm et al. 2015). Furthermore, the increase in miRNA gene numbers over time correlates with an increase in metazoan organismal complexity and bursts of miRNA gene repertoire expansion correlate with specific lineage divergence events (Sempere et al. 2006; Lee et al. 2007; Wheeler et al. 2009; Berezikov 2011; Guerra-Assunção and Enright 2012; Hertel and Stadler 2015), supporting a role of miRNAs in lineage diversification.
Whole-genome duplication events (WGD) had profound impacts on metazoan evolution and are thought to have provided a substrate for species and phenotypic diversification by increasing gene numbers and relaxing evolution (Ohno 1970; Force et al. 1999; Van de Peer et al. 2009; Ravi and Venkatesh 2018). The most studied metazoan WGDs include two rounds of vertebrate genome duplications (VGD1 and VGD2) that occurred at the origin of the vertebrate radiation (Dehal and Boore 2005; Smith et al. 2013; Simakov et al. 2020) and the teleost genome duplication (TGD) at the base of the teleost radiation (Amores et al. 1998; Taylor et al. 2003; Jaillon et al. 2004; Braasch et al. 2016). Teleosts form the most species-rich vertebrate group, accounting for more than half of living vertebrate species. Teleosts colonized virtually all aquatic habitats from pole to pole, surface to hadal zones, and from hot springs to subzero Antarctic seas (Nelson 2006; Fricke et al. 2021). The consequences of WGD on vertebrate genome organization and protein-coding genes have been relatively well characterized (Nakatani et al. 2007; Braasch et al. 2016; Pasquier et al. 2017), but with still many open questions. Notably, genes encoding regulatory and signal transduction system components accumulate proportionally more during genome evolution compared with genes involved in conserved functions such as translation systems and metabolic enzymes (Nimwegen 2003; Koonin 2011; Warnefors and Eyre-Walker 2011). In agreement, after the TGD, protein-coding genes involved in biological functions prone to spurring phenotypic diversification in fish (i.e., pigment and cognition-related genes) were retained in duplicates more often than protein-coding genes involved in well-conserved functions, such as liver genes (Braasch et al. 2009; Schartl et al. 2013). In striking contrast, an understanding of the consequences of WGDs on metazoan miRNA gene evolution and their genomic accumulation is lacking and only a few genome-wide examples have been reported (Berthelot et al. 2014; Braasch et al. 2016).
miRNA genes occupy several different types of genomic locations. Some miRNA genes are intergenic and others reside within protein-coding genes, either on the same or the opposite DNA strand as the host gene and generally in introns and rarely in exons (Campo‐Paysaa et al. 2011; Fromm et al. 2015; Bartel 2018); intragenic miRNA genes may or may not be functionally associated with the host gene (Rodriguez et al. 2004; Lutter et al. 2010; França et al. 2016). miRNA genes can also be organized in clusters characterized by the presence on the same DNA strand of successive miRNA precursors within a genomic region compatible in size with the expression of a polycistronic transcript (e.g., up to 50 kb apart) (Altuvia et al. 2005; Baskerville and Bartel 2005; Chan et al. 2012; Guerra-Assunção and Enright 2012; Bartel 2018). Initial estimates suggested that miRNA clusters were often retained during evolution (Marco et al. 2010; Sun et al. 2013) but to our knowledge, no genome-wide information exists on the co-conservation of intragenic miRNA genes and their protein-coding host gene after WGD except for a few anecdotal cases (Bhuiyan et al. 2013; Tani et al. 2013; Desvignes et al. 2014).
The evolution of miRNA gene expression across species also remains unresolved. miRNAs are generally considered to be specialized in function and to be developmental stage-specific, organ-specific, and even cell type-specific, therefore participating in cellular identity and tissue specificity (Lee et al. 2007; Christodoulou et al. 2010; Ludwig et al. 2016; de Rie et al. 2017; Juzenas et al. 2017; McCall et al. 2017; Avital et al. 2018; Halushka et al. 2018). Most of these data, however, originate from mammals and the extent to which conservation of expression is similar across all vertebrates is unknown.
Finally, miRNAs regulate gene expression largely by target recognition through the miRNA seed and 3′-complementary region (3′-CR) (Bartel 2018; McGeary et al. 2019; Sheu-Gruttadauria et al. 2019; Bofill-De Ros et al. 2020). Changes in the sequences of the seed or of the 3′-CR might lead to undetectable effects on regulatory efficiency or might cause dramatic alterations in the set of targeted transcripts (Neilsen et al. 2012; Ameres and Zamore 2013; Tan et al. 2014). Across species, the seed and the 3′-CR of orthologous miRNAs appear to be under selective pressure for sequence preservation (Wheeler et al. 2009; Fromm et al. 2015), arguing for conserved regulatory functions if miRNA binding sites in the 3′-UTRs of targeted transcripts are also preserved. Sequence evolution of duplicated miRNAs, however, has never been studied following WGD. Furthermore, it remains unknown whether miRNA genes that are retained in two copies produce mature miRNAs that tend to have identical sequences or whether, following an initial period of neutral drift or selection, duplicated copies diverged from one another in sequence or expression. And, if they diverged, whether mutations more often affected the seed and/or the 3′-CR, therefore potentially driving miRNA function towards novel genetic and developmental pathways.
To address these open questions, we studied the evolution of miRNAs after the TGD by analyzing miRNAs in the nonteleost actinopterygian spotted gar Lepisosteus oculatus, representing the sister lineage to duplicated teleost genomes, and in four teleosts: zebrafish Danio rerio, Japanese medaka Oryzias latipes, three-spined stickleback Gasterosteus aculeatus, and Antarctic blackfin icefish Chaenocephalus aceratus. Species were chosen based on their position in the tree of life and their importance in developmental, genetic, and phenotypic research. We hypothesized that 1) because miRNA genes are important regulators of gene expression similar to protein-coding genes with developmental and regulatory functions, they are more likely to be retained in duplicate after WGD than genes without known functions in gene expression regulation; 2) because miRNA genes are small genetic elements, their rate and pattern of retention after WGD might depend not only on their regulatory functions but also on their genomic context; 3) because miRNAs have the potential to contribute both to phenotypic canalization and conversely to diversification, expression patterns of miRNAs providing robustness to development should be well conserved in evolution whereas expression patterns of miRNAs involved in phenotypic diversification are likely to be rapidly evolving; and 4) similar to functional domains in proteins, the most highly functional parts within a miRNA gene (i.e., the seed and the 3′-CR) are more likely to be evolutionarily preserved than parts with little or no apparent function.
These hypotheses make the testable predictions that after the TGD: 1) miRNA genes should be more likely to be retained in duplicates than protein genes in general, most of which do not directly regulate the activity of other genes; 2) different genomic contexts, such as gene clusters or genomic overlaps with protein-coding genes, will display different rates and patterns of miRNA gene retention; 3) miRNAs involved in organs performing highly conserved tasks through evolutionary time (e.g., brain and heart ventricle) will show greater conservation in expression patterns among fish species compared with miRNAs involved in organs participating in species diversification (e.g., gonads); and 4) under the assumption that the strand that is more highly expressed in a miRNA duplex (i.e., the major strand) is the most functional strand, its sequence conservation will be greater than the sequence of the more lowly expressed strand, and a corollary, the sequences of the most functional parts of the mature miRNAs will have greater sequence conservation than parts of the miRNA that play minor functional roles.
Results and Discussion
The miRNAome of the Teleost-Holostei Last Common Ancestor
To study the retention patterns of miRNA gene ohnologs (i.e., gene duplicates originating from a WGD event; Ohno 1970; Wolfe 2000; fig. 1B), we inferred a hypothetical pre-TGD miRNAome to compare with the miRNAomes of extant teleost species. We used the gar as a representative of the Holostei, the unduplicated sister group to the teleosts. And, for extant teleosts, we used the developmental genetic models zebrafish and medaka, the evolutionary model stickleback, and the ecologically specialized Antarctic blackfin icefish.
Fig. 1.

Evolution of the actinopterygian miRNA repertoire. (A) miRNA repertoire composition in spotted gar, zebrafish, medaka, stickleback, and blackfin icefish. Each pie chart represents the proportion of miRNA genes present in singleton (orange), in TGD ohnolog pairs (blue), or lost (gray). The last common ancestors of each lineage were reconstructed by maximum parsimony and divergence times were based on TimeTree (Kumar et al. 2017). The teleost genome duplication (TGD) was assumed to have happened at the estimated divergence time of Holostei and Teleosts. (B) Terminology and schematic representation of miRNA gene relationships between gar and teleosts following the TGD. (C) Overlap of miRNA gene orthologs across the four studied teleosts. (D) Color-coded miRNA gene loss rates in genes per 10 My mapped on each branch of a time-calibrated tree (Kumar et al. 2017).
Lacking a comprehensive miRNA annotation for the Japanese Medaka (Kozomara and Griffiths-Jones 2014; Fromm et al. 2020), we annotated miRNA genes and mature miRNAs in the medaka using publicly available small RNA-sequencing data (Gay et al. 2018) that we aligned to the HdrR strain medaka genome using Prost! (Desvignes et al. 2019). Analysis recovered a total of 289 miRNA genes and 427 individual mature miRNAs in medaka (supplementary files 1–3, Supplementary Material online). The published miRNA annotations for gar, zebrafish, stickleback, and blackfin icefish used in this study contained a comparable number of genes and mature miRNAs with 233, 332, 286, and 294 miRNA genes, respectively, and 362, 495, 396, and 408 individual mature miRNAs, respectively (Braasch et al. 2016; Desvignes et al. 2019; Kim et al. 2019).
To focus the study on the effects of the TGD on miRNA gene retention and evolution, we excluded from analysis teleost-specific miRNA genes (e.g., mir10544, mir10551, mir2191 to mir2198, and mir7553) (Kozomara and Griffiths-Jones 2014; Fromm et al. 2020) because genes unknown in any nonteleost species are unlikely to have been present in the pre-TGD common ancestor of teleosts and gar, which we refer to as the Teleost-Holostei last common ancestor or TH-LCA. Orthology relationships among miRNAomes were carefully assigned for each species by both sequence similarity and synteny conservation. These comparisons identified in medaka six additional orthologous miRNA gene loci that were not detected in expression data sets. The absence of expression data for these miRNA genes could be related to insufficient sequencing depth, the limited number of tissues and developmental stages studied, or to expression loss. Unfortunately, we cannot distinguish among these possibilities. To identify putative gene losses, we carefully checked for potential gaps in genome assemblies at genomic regions where the genes would have been expected from conserved synteny data, for absence of small RNA reads originating from genes that would be missing in the genome assembly but potentially present in the biological genome, and the shared absence of genes in genome assemblies of related species, together suggesting true losses. The mir430 gene family was also excluded from our analysis. Although mir430 genes have been well studied and are crucial for early embryonic development in zebrafish (Giraldez et al. 2006), the evolution of this family remains uncertain. miR-430-3p mature products show great sequence conservation across species, especially in their seed (supplementary file 4A, Supplementary Material online), their synteny is in contrast not conserved. We identified two clustered intragenic mir430 genes in the spotted gar genome assembly (supplementary file 4B, Supplementary Material online) and many intergenic mir430 genes and clusters in each of the genome assemblies of the four studied teleost species—at least 55 genes in zebrafish, 58 in medaka, 139 in stickleback and 157 in icefish—but none of these genomic regions displayed any conserved synteny with any other species in our data set (supplementary file 4B, Supplementary Material online). We therefore excluded mir430 genes from our analysis because it was impossible to establish reliable orthology relationships between its members across species, to infer the organization of mir430 genes in the TH-LCA, and thus to decipher the evolutionary history of this outlier, highly duplicated and apparently mobile miRNA gene family. Together, our analysis led to sets of 233, 308, 283, 275, and 290 miRNA genes in gar, zebrafish, medaka, stickleback, and blackfin icefish, respectively, whose orthologs can also be found in nonteleost vertebrate species even if no expression could be measured in the studied expression data sets (fig. 1A and supplementary table 1, Supplementary Material online).
Finally, to search for miRNA genes that might have been present in the TH-LCA but lost in the gar lineage, we searched for genes that are present in nonteleost vertebrates (e.g., chicken, Xenopus, mouse, human) and in at least one teleost. We identified seven pre-TGD miRNA genes that were secondarily lost in gar but conserved in teleosts and nonteleost vertebrates. The miRNAome of the TH-LCA for this study was therefore defined as a set of 240 evolutionarily conserved miRNA genes (fig. 1A and supplementary table 1 and file 5, Supplementary Material online).
The Frequency of miRNA Gene TGD Ohnolog Retention in Duplicates Suggests a Role in Phenotypic Canalization and Diversification
Having established the miRNAome of several teleosts and the hypothetical pre-TGD TH-LCA miRNAome, we could address the question of gene retention rates and patterns following the TGD.
Teleost miRNAomes tended to be larger than that of the inferred TH-LCA. Teleost genomes appeared to have on an average 50 ± 14 more miRNA genes than the TH-LCA (supplementary table 1, Supplementary Material online), representing a 21% increase in gene number. Orthology relationships (fig. 1B and supplementary file 5, Supplementary Material online) revealed that most (76%) miRNA genes were present in all four studied teleost species (fig. 1C). In addition, 31.3 ± 3.0% of the TH-LCA miRNA genes were present in two ohnologous copies in the genome assemblies and/or small RNA-sequencing data of the studied teleost species, ranging from 35.8% in zebrafish to 27.9% in stickleback (fig. 1A and supplementary table 1, Supplementary Material online). This retention rate of miRNA genes in duplicated copies is significantly higher than the estimated 12–24% retention rate for protein-coding genes (Braasch and Postlethwait 2012; Pasquier et al. 2017) (P < 2.22e-16 and P = 4.5e-6 for protein-coding gene retention rates of 12% and 24%, respectively, repeated G–tests of goodness-of-fit [RGT]). These results are in agreement with the previous report of a 39% retention rate of miRNA gene TGD ohnolog pairs in zebrafish that was performed on a smaller set of 208 TH-LCA genes (Braasch et al. 2016) compared with the 240 genes studied here. The high duplicate retention rate of miRNA genes after the TGD is also consistent with observations made in rainbow trout, where miRNA genes located in double-conserved synteny regions after the salmonid genome duplication (a more recent event, about 80 Ma, than the TGD, about 315 Ma; Allendorf and Thorgaard 1984; Macqueen and Johnston 2014; Kumar et al. 2017) were retained in duplicate at a rate of 2.02-fold higher than that for protein-coding genes (Berthelot et al. 2014). These results are also consistent with observations of higher retention rates of miRNA genes than protein-coding genes following WGDs in plants (Sun et al. 2015; Zhao et al. 2015; Shi et al. 2017).
Besides the 31.3% of ancestral miRNA genes retained in duplicates in teleosts, 58.3 ± 1.1% were retained as singletons and 10.4 ± 2.1% were lost in the studied teleost species (fig. 1A and supplementary table 1, Supplementary Material online). The precise fractions of TH-LCA protein-coding genes that were retained as singletons or lost after the TGD are unknown, and therefore cannot be directly compared with the situation analyzed here for miRNA genes. Similarly, no other study has estimated the retention and loss rates of miRNA genes following other metazoan WGD events.
Together, these results quantitatively demonstrate that the TGD significantly contributed to the expansion of teleost miRNA gene repertoires. This observation confirms previous qualitative conclusions that WGDs likely played significant roles in the amplification of miRNA gene repertoires (Hertel et al. 2006; Campo‐Paysaa et al. 2011; Hertel and Stadler 2015).
These results also demonstrate that miRNA genes were retained significantly more frequently in duplicated copies compared with protein-coding genes after the TGD. This difference could be related to the small size of miRNA genes providing a reduced target for deleterious nucleotide substitution mutations that might lead to a pseudogene compared with protein-coding genes. Studies of the molecular evolution of protein-coding genes, however, demonstrated that longer genes evolved at slower rates than shorter genes (Wolf et al. 2009; Lopes et al. 2020), throwing into question whether mutation target size is a major factor in miRNA duplicate gene retention. Nevertheless, discrepancies in gene lengths between miRNA and protein-coding genes might still contribute to the higher retention rate of miRNA genes compared with protein-coding genes. Protein-coding genes that function in regulatory and signal transduction, however, accumulate proportionally more during genome evolution compared with protein-coding genes involved in conserved functions such as the translation system and enzymes of metabolic pathways (Nimwegen 2003; Koonin 2011; Warnefors and Eyre-Walker 2011). In agreement, specific groups of protein-coding genes suggested to be involved in biological functions prone to spurring phenotypic diversification in fish (i.e., pigment and cognition-related genes) were, however, retained in duplicate at a higher rate than protein-coding genes involved in well-conserved functions, such as those mediated by liver genes (Brunet et al. 2006; Braasch et al. 2009; Schartl et al. 2013). The higher retention rate of miRNA gene duplicates compared with the overall retention of protein-coding genes might therefore be linked to the continuous accumulation during evolution of regulatory genes, both protein-coding and miRNA genes. In addition, miRNA regulation of gene expression operates in a dosage-sensitive manner because a cell has more potential binding sites for each miRNA than it has molecules of that miRNA (Denzler et al. 2014, 2016). Thus, duplicated miRNA genes with identical or overlapping functions could provide twice as many miRNA molecules, therefore increasing robustness of their regulatory function, or maintaining proper network balance and relative dosage if their targets were also duplicated. This enhanced robustness in genetic regulation could in turn improve phenotypic canalization and be selected upon for genomic conservation under the gene balance hypothesis (Birchler and Veitia 2010, 2012; Abrouk et al. 2012). Alternatively, duplicated miRNA genes that diverged and evolved different functions would increase the extent of protein-coding gene expression regulation by developing new sets of targeted genes and regulatory pathways, potentially contributing to phenotypic diversification and, if these were evolutionarily advantageous, they could be selected upon and preserved. Together, an increase in miRNA gene numbers can strengthen pre-existing regulatory functions or promote the development of new regulatory links, contributing to phenotypic canalization or diversification, respectively. We do not know, however, to what extent the evolutionary conservation or evolution of new targets influenced the retention of pre-existing miRNA genes or the origin of new ones. Analyses aimed at revealing the post-TGD fate of targeted protein-coding genes, the conservation of target sites, and the coevolution of target site and miRNA sequences are needed to address this standing question.
miRNA Gene Losses Occurred Rapidly after the TGD
By reconstructing a series of ancestors across the phylogeny of investigated species (fig. 1A and supplementary file 5, Supplementary Material online), we could enumerate gene losses on each branch on a time-calibrated species tree (Kumar et al. 2017) and calculate gene loss rates (fig. 1D). Analysis revealed first that gar experienced few gene losses over about 315 My since divergence from the TH-LCA (seven genes; 0.22 gene losses per 10 My; fig. 1D). This result is in line with previous observations that genomes in the gar lineage evolved slowly (Braasch et al. 2016).
Second, evidence from the tree showed that during the approximately 85 My between the TGD and the stem Clupeocephalan teleost (including all four of our teleosts, i.e., between 315 and 230 Ma), miRNA genes were lost at a higher rate (15.88 genes per 10 My, fig. 1D) than in any other period in the 315 My encompassed by the phylogeny. This quantitative measure of gene loss agrees with previous qualitative statements (Hertel and Stadler 2015), suggesting that most WGD-related miRNA gene ohnologs became singletons or were both lost shortly after the duplication event. This finding for miRNA genes also mimics models and observations for protein-coding genes following the TGD (Braasch et al. 2009; Inoue et al. 2015).
Third, in contrast to the period immediately following the TGD, branches leading to extant species experienced limited gene losses, ranging from the low rate of 1.18 gene losses per 10 My in the cold-adapted Antarctic blackfin icefish over the 76 My of divergence from the Perciformes hypothetical ancestor to the moderate rate of 3.68 gene losses per 10 My over the same 76 My of divergence for stickleback (fig. 1D). Notably, the blackfin icefish lineage did not experience accelerated losses of miRNA genes that one might have hypothesized given that the frigid Antarctic environment could have impaired miRNA biogenesis and function due to increased nucleotide base-pairing strength that accompanies cold temperatures. In addition, although icefishes have pseudogenized hemoglobin genes and lack mature red blood cells (Near et al. 2006), miRNA genes known to be involved in erythropoiesis in at least some teleosts and other vertebrates are present in both the icefish genome assembly and small RNA-sequencing data (Desvignes et al. 2016; Kim et al. 2019), suggesting that these miRNA genes may have additional functions independent of erythropoiesis or have been genomically retained even in the absence of their original erythropoietic function.
Finally, these data revealed that the branch leading from the hypothesized Clupeocephala ancestor to the hypothesized Percomorphaceae ancestor experienced moderate gene loss with an average loss of 3.92 genes per 10 My over a period of 102 My (fig. 1C). This rate could be related to genome evolution concomitant with the diversification of the enormous Acanthomorpha group, which counts about 16,000 marine species and represents about 85% of marine ray-finned fish species and about one-third of all vertebrate species (Wainwright and Longo 2017).
The reconstruction of hypothetical ancestor miRNAomes does not account for possible convergent losses across lineages. Our data set, however, revealed four examples of asymmetrical ohnolog retentions (e.g., retention of the “a” copy in one lineage and of the “b” copy in another), representing 1.67% of the 240 TH-LCA genes. In one case, the TH-LCA intronic miRNA gene mir7b was retained as mir7ba within the ohnolog hnrnpkl in zebrafish but was lost at that locus in studied acanthomorphs (fig. 2A). Conversely, in acanthomorphs, the ancestral mir7b was conserved as mir7bb within hnrnpk but was lost at that locus in zebrafish (fig. 2A).
Fig. 2.

Examples of miRNA retention after the TGD. (A) Alternative retention of intronic miRNA mir7b between zebrafish and acanthomorphs. (B) Alternative retention of miRNA clusters mir30a/d-mir30b between zebrafish and acanthomorphs. (C) Alternative retention of intronic miRNA mir338-2 and host gene lmtk2 in teleosts. Synteny analyses were performed using Genomicus version 96.01 (Muffato et al. 2010). The blackfin icefish was manually added using the annotation of the published genome (Kim et al. 2019).
The second case is the asymmetric retention of the ancestral cluster mir30a/d-mir30b comparing zebrafish and acanthomorphs. At one ohnologous locus, zebrafish retained only mir30a whereas acanthomorphs retained the entire two-gene cluster mir30a-mir30bb, and at the other ohnologous locus, acanthomorphs lost the entire cluster whereas zebrafish retained the entire cluster (fig. 2B). The third and fourth identified cases of asymmetrical ohnolog resolution are the alternative retention of the mir124-5/mir124-6 and mir153aa/mir153bb ohnologous pairs present in medaka and in the hypothetical Percomorphaceae ancestor. In stickleback, mir124-6 and mir153aa were retained and mir124-5 and mir153ab were lost, whereas in the blackfin icefish, mir124-6 and mir153aa were lost and mir124-5 and mir153ab were retained (supplementary file 5, Supplementary Material online).
Together, these results demonstrate that the gar miRNAome remained highly similar to the TH-LCA miRNAome and that the highest rate of miRNA gene loss occurred during the 85 My following the TGD. The study of additional Holostei species, especially the bowfin Amia calva, would be necessary to better understand whether the high conservation of miRNA genes in gar is specific to this species, to Lepisosteiformes, or to the whole Holostei group. Similarly, the study of additional teleosts, especially the early diverging lineages of Elopomorpha and Osteoglossiforms, would help us understand the dynamics of gene resolution following the TGD.
Clustered miRNA Genes and miRNA Clusters Were Retained More Often Than Solo miRNAs
Metazoan miRNA genes are often organized in clusters that can be transcribed in a single transcript (Altuvia et al. 2005; Baskerville and Bartel 2005; Chan et al. 2012; Bartel 2018) and are often evolutionarily conserved (Chan et al. 2012; Marco et al. 2013; Sun et al. 2013; Wang et al. 2016). We know little, however, about the evolution of miRNA clusters and their clustered miRNA genes after any WGD event. To address this issue, we studied miRNA clusters themselves, which we refer to as “cluster loci” opposed to “solo loci” (fig. 3A), and the several miRNA hairpin genes within the clusters, which we refer to as “clustered miRNAs” opposed to “solo miRNAs” (fig. 3A).
Fig. 3.

miRNA gene retention rates and patterns following the TGD varied depending on genetic context. (A) Schematic representation of terminology used for clustered miRNAs and solo miRNAs, cluster loci and solo loci, intergenic and intragenic miRNAs. (B) Retention rates of post-TGD miRNAs depending on genomic context. (C) Retention patterns of post-TGD miRNAs depending on genomic context. (D) Patterns of retention and loss of miRNAs and host genes compared with a model of independent retention. (E) Influence of the retention or the loss of a member of the miRNA/host pair on the retention of the second member of the pair. Significant differences: *P < 0.05; **P < 0.01; ***P < 0.001.
Analysis revealed first that about half of the total number of miRNA genes were found in clusters both in the TH-LCA and in teleosts (47.1% and 49.6 ± 0.9%, respectively, supplementary table 2, Supplementary Material online), similar to other metazoans (Griffiths-Jones et al. 2007; Marco et al. 2010; Kabekkodu et al. 2018). In addition, we found a significantly higher overall retention rate (+11.5%) for clustered miRNA genes (63.7 ± 4.3%) compared with solo miRNA genes (52.2 ± 5.6%; P = 0.006079, Cochran–Mantel–Haenszel test [CMHT]) (fig. 3B and supplementary table 2, Supplementary Material online). Clustered miRNA genes were also retained in two copies significantly more frequently (+8.9%) than solo miRNA genes (36.1 ± 4.5% and 27.2 ± 3.3%, respectively; Padj = 0.0113, CMHT) (fig. 3C and supplementary table 3, Supplementary Material online). Clustered miRNA genes and solo miRNA genes, however, were retained as singletons at approximately similar rates (55.3 ± 2.1% and 60.6 ± 3.3%, respectively; Padj = 0.1098, CMHT) (fig. 3C and supplementary table 3, Supplementary Material online). Clustered miRNA genes and solo miRNA genes were fully lost (i.e., both copies were lost) at similar rates (8.6 ± 4.2% and 12.2 ± 1.4%, respectively; Padj = 0.1097, CMHT) (fig. 3C and supplementary table 3, Supplementary Material online). Mirroring the overall miRNA gene loss dynamic (fig. 1D), the highest rate of solo and clustered miRNA gene loss occurred within the 85 My following the TGD (supplementary file 6, Supplementary Material online).
We then asked whether the retention rate of miRNA cluster loci was greater than that of solo miRNA loci. The TH-LCA and gar both possess the same 46 miRNA clusters, whereas our four teleost species possess on an average 62 ± 4 clusters derived from the TH-LCA, ranging from 58 clusters in stickleback to 67 clusters in zebrafish (supplementary table 4, Supplementary Material online). In all four species, miRNA clusters are usually small and composed of two to six hairpins (supplementary table 4 and file 5, Supplementary Material online). Cluster loci represent 27.2% of miRNA loci in the TH-LCA and 29.9 ± 0.9% of miRNA loci in teleosts (supplementary table 4, Supplementary Material online). The overall retention rate of cluster loci was significantly higher (+15.5%) than that of solo miRNA loci (67.7 ± 4.1% and 52.2 ± 5.6%, respectively; P = 0.000794, CMHT) (fig. 3B and supplementary table 4, Supplementary Material online) and cluster loci were retained in two copies significantly more frequently (+13.6%) than solo miRNA loci (40.8 ± 4.5% and 27.2 ± 3.3%, respectively; Padj = 0.0026, CMHT) (fig. 3C and supplementary table 5, Supplementary Material online). In contrast, miRNA cluster loci and solo loci were retained in singletons at similar rates (52.7 ± 3.3% and 60.6 ± 3.3%; Padj = 0.0758, CMHT). miRNA cluster loci and solo loci were also lost at similar rates (6.5 ± 4.3% and 12.2 ± 1.4%; Padj = 0.0691, CMHT) (fig. 3C and supplementary table 5, Supplementary Material online). Similar to the loss dynamic of clustered miRNA genes and of miRNA genes overall, the highest rate of solo and cluster miRNA loci loss occurred within the 85 My following the TGD (fig. 1D and supplementary file 6, Supplementary Material online).
These results represent the first genome-wide quantitative demonstration that clustered miRNA genes are more likely to be retained after WGD, and confirm previous observations made on subsets of teleost clusters (Chan et al. 2012; Marco et al. 2013; Sun et al. 2013; Wang et al. 2016). Several possible hypotheses might help explain this finding. First, clustered miRNA genes are generally co-expressed in a polycistronic transcript (Altuvia et al. 2005; Baskerville and Bartel 2005; Chan et al. 2012; Bartel 2018); therefore mutations that alter the regulation of cluster expression might simultaneously nonfunctionalize the entire cluster. Second, clustering of miRNA genes improves the processing of nonoptimal hairpins within the cluster (Fang and Bartel 2020; Shang et al. 2020); therefore, mutations that alter a hairpin within a cluster might alter processing of other members of the cluster and modify the entire cluster function. These two types of alterations (i.e., locus control mutations and hairpin processing mutations) would likely be selected against to preserve existing regulation, favoring the maintenance of the entire cluster by tight genomic linkage (Marco et al. 2013). Third, if clustered miRNA genes encode several miRNAs that cooperatively regulate genetic pathways, which is still debated (Wang et al. 2016; Kabekkodu et al. 2018; Marco 2019), then a mutation that eliminates part of a cluster might lead to imbalanced regulation of genetic processes modulated by the functionally coadapted members of the cluster, which in turn could lead to the elimination of the entire cluster and prevent aberrant regulation. This type of alteration would also likely be selected against, favoring the retention of all the functional elements of the cluster and its established regulatory network. Together, these results demonstrate that miRNA genes assembled in clusters benefit from their association which increases their retention rates in evolution after genome duplication, suggesting that genetic linkage reduces the likelihood of cluster losses and that clustering is a way to maintain together a group of miRNAs that had already been individually selected for their regulatory functions and as a whole coordinately regulate a cohort of biologically related targeted genes as previously hypothesized (Mohammed et al. 2014).
Intergenic miRNA Genes Were Retained More Often Than Intragenic miRNA Genes
This analysis of clustered versus solo miRNA genes revealed that genomic context influenced the retention of miRNA genes and miRNA clusters. We then questioned how the intragenic or intergenic genomic context of a miRNA gene influences its retention after a WGD event. Many metazoan miRNA genes lie within protein-coding genes and are usually intronic and on the same strand as their host gene, but they can be on the opposite strand from their host gene and rarely be exonic (Campo‐Paysaa et al. 2011; Meunier et al. 2013; Hinske et al. 2014; Boivin et al. 2018). Despite a few individual case studies (Bhuiyan et al. 2013; Desvignes et al. 2014; Siddique et al. 2016), no genome-wide information has been available on the effect of WGD events on the retention of intragenic and intergenic metazoan miRNA genes.
More miRNA genes were intergenic than intragenic in the five studied fish genomes. Intragenic miRNA genes represented 34.6% of TH-LCA miRNA genes, 35.2% of gar miRNA genes, and 28.3 ± 0.9% on an average of miRNA genes in the four studied teleosts (supplementary table 6, Supplementary Material online). In the TH-LCA, gar, and teleosts, most intragenic miRNAs (88.0%, 87.8%, and 86.3 ± 1.0%, respectively) have the same orientation as their host gene (supplementary table 6, Supplementary Material online). In addition, independent of orientation, nearly all intragenic miRNA genes were within introns of the host gene in the TH-LCA, gar, and on an average in teleosts (94%, 96.3%, and 96.0 ± 1.2%, respectively) (supplementary table 6, Supplementary Material online). These proportions are in agreement with the abundance and orientation of intragenic miRNA genes in other metazoans (Griffiths-Jones et al. 2007; Chiang et al. 2010; Marco et al. 2010; Meunier et al. 2013; Hinske et al. 2014) and with the fact that intragenic miRNA genes are usually transcribed with their host gene (Baskerville and Bartel 2005; França et al. 2016).
We then studied the retention rates of post-TGD intragenic versus intergenic miRNA genes, independent of their strand and location in their host gene. The overall retention rate of intragenic miRNA genes was significantly lower (−7.4%) than of intergenic miRNA genes in teleosts (55.6 ± 3.2% and 63.0 ± 3.4%, respectively; P = 0.0019, CMHT) (fig. 3B and supplementary table 6, Supplementary Material online). Intragenic miRNA genes were also retained in two copies significantly less frequently (−11.4%) than intergenic miRNA genes (23.8 ± 3.3% and 35.2 ± 3.7%, respectively; Padj = 0.0012, CMHT) (fig. 3C and supplementary table 7, Supplementary Material online). Conversely, intragenic miRNA genes were retained as singletons significantly more frequently (+8%) than intergenic miRNA genes (63.6 ± 3.6% and 55.6 ± 1.5%, respectively; Padj = 0.0270, CMHT) (fig. 3C and supplementary table 7, Supplementary Material online). Intragenic and intergenic miRNA genes were, however, lost at similar rates (12.7 ± 4.0% and 9.2 ± 3.2%, respectively; Padj = 0.1243, CMHT) (fig. 3C and supplementary table 7, Supplementary Material online). Similar to the overall miRNA gene loss dynamic and that of clustered miRNA genes and cluster loci, most intragenic and intergenic miRNA gene losses occurred within the 85 My following the TGD (fig. 1D and supplementary file 6, Supplementary Material online). We conclude that the retention of miRNA genes after the TGD was significantly influenced by their genomic context and that inclusion within a protein-coding gene negatively influenced the retention in duplicate of a miRNA gene.
We then queried the effect of clustering on retention rates and patterns of intragenic versus intergenic miRNA genes. Results did not reveal any significant differences in retention rates between combinations of clustered versus solo and intragenic versus intergenic miRNA genes (supplementary file 7A, Supplementary Material online). Intergenic clustered miRNA genes were, however, retained in duplicate significantly more often than intragenic clustered miRNA genes (Padj = 0.0382, CMHT), and were retained as singletons significantly less frequently than intragenic clustered miRNA genes (Padj = 0.0382, CMHT) (supplementary file 7B, Supplementary Material online). At the miRNA locus level, intergenic cluster loci were retained more frequently than intergenic solo loci (P = 0.0282, CMHT), but this effect of clustering was not significant between intragenic cluster and intragenic solo loci (P = 0.0869, CMHT) (supplementary file 7C, Supplementary Material online). In contrast to clustered miRNA genes, intragenic and intergenic cluster loci were retained in statistically similar proportions (P = 0.0869, CMHT) (supplementary file 7D, Supplementary Material online), however, the low number of intragenic cluster loci (11) and intergenic cluster loci (35) could explain this lack of statistical difference. Intergenic cluster loci were retained in duplicate significantly more frequently than intergenic solo loci (Padj = 0.0204, CMHT) and were retained significantly less frequently as singletons than intergenic solo loci (Padj = 0.0440, CMHT). These results support the observation that clustering and association with a host gene both influence the retention of miRNA genes, with clustered intergenic miRNA genes and loci being the most frequently retained and intragenic solo miRNA genes and loci being the least frequently retained.
Evolutionary Association between Intragenic miRNA Genes and Host Genes
To address the mechanisms for the somewhat surprising finding that intragenic miRNA genes are less likely to be preserved than intergenic miRNA genes, we tested the hypothesis that the retention or loss of intragenic miRNA genes and their host gene are not independent. In a simplified model where intragenic miRNA genes and host gene fates were completely independent (i.e., no effect of genetic linkage and no transcriptional or functional associations), the probability that both are retained is 25%, the probability neither is retained is 25%, the probability only the miRNA gene is retained is 25%, and the probability only the host gene is retained is 25% (fig. 3D). Results showed, however, significant deviations from this independent retention model (P = 1.77e-67, RGT) (fig. 3D). Retention of both genes occurred significantly more often than expected by independent retention (50.2 ± 2.8% vs. 25%; Padj = 1.99e-40, RGT) (fig. 3D and supplementary table 8, Supplementary Material online). The converse situation, the loss of both genes, also significantly differed from expected (32.2 ± 1.4% vs. 25%; Padj = 0.0013, RGT) (fig. 3D and supplementary table 8, Supplementary Material online). Together, intragenic miRNA genes and their host genes shared the same fate in 82.4 ± 2.0% of the cases, significantly more often than by chance (vs. 50%; P = 1.88e-64, RGT) (supplementary table 8, Supplementary Material online), with the coretention situation occurring significantly more often than the co-loss situation (50.2 ± 2.8% vs. 32.2 ± 1.4%; P = 2.03e-5, RGT).
Conversely, the alternative retention of intragenic miRNA genes and host genes was significantly lower than expected in a scenario of independent evolution: the miRNA gene was retained and the host gene was lost in only 5.4 ± 0.9% of the cases rather than the expected 25% (Padj = 5.54e-38, RGT) (fig. 3D and supplementary table 8, Supplementary Material online), and the miRNA gene was lost whereas the host gene was retained in only 12.2 ± 2.1% instead of 25% of the situations (Padj = 4.02e-14, RGT) (fig. 3D and supplementary table 8, Supplementary Material online). Together, intragenic miRNA genes and their host genes have different fates significantly less often than by chance (17.6 ± 2.0% rather than 50%, P = 1.88e-64, RGT) (supplementary table 8, Supplementary Material online). Notably, the situation of a lost miRNA gene and a retained host gene occurred significantly more often than the opposite (12.2 ± 2.1% vs. 5.4 ± 0.9%; P = 0.0008, RGT) (supplementary table 8, Supplementary Material online). This lack of independence is expected under the hypothesis that the fates of intragenic miRNA genes and their host genes are related, likely through genetic linkage and potentially through biological function or biogenesis association.
Among the few cases of alternative retentions of intragenic miRNA genes and host genes, mir338-2 provides an example shared by all studied teleosts (fig. 2C). In gar, and presumably in the TH-LCA, mir338-2 was on the same DNA strand and in an intron of the protein-coding gene lmtk2. In our four teleosts, however, the miRNA gene and the host gene were alternatively retained between the two ohnologous regions. In one ohnologous region, mir338-2 was retained along with many surrounding genes whereas the host gene was lost; in the second ohnologous region, the miRNA gene was lost whereas the host gene was retained also with multiple surrounding genes (fig. 2C). In another case, both teleost copies of the gar host gene dnah3 appear to have been lost whereas the intragenic mir135a was retained in duplicate in all three Acanthomorphs and as a singleton in zebrafish (supplementary file 4, Supplementary Material online). Finally, mir103b, which is intronic in pank3 in gar and human, was found between different genes in teleosts (i.e., slc30a9 and grxcr1a in teleosts, and ctnna1 and sncb in gar) whereas pank3 was lost in teleosts (supplementary file 4, Supplementary Material online), suggesting that a translocation of a pank3-mir103b genomic region between slc30a9 and grxcr1a in early teleosts preserved the miRNA gene but not the host gene. All other cases of decoupled miRNA gene and host gene fate were species-specific, with for example, the alternative retention of the myomiR mir499 and its host gene myh14 that occurred independently in medaka and stickleback (Bhuiyan et al. 2013).
Having shown that the retention of miRNA genes and host genes was not independent, we questioned the fate of miRNA genes when their host gene was retained or lost, and the reciprocal. When the host gene was retained, the intragenic miRNA gene was also retained 80.4 ± 3.5% of the time, whereas when the host gene was lost, the intragenic miRNA gene was retained significantly less frequently, in only 14.4 ± 2.4% of the cases (P < 2.2e-16, CMHT) (fig. 3E and supplementary table 9, Supplementary Material online). Reciprocally, when the miRNA gene was retained, the host gene was also retained 90.3 ± 2.4% of the time, whereas when the miRNA gene was lost, the host gene was retained significantly less frequently, in only 27.3 ± 2.7% of the cases (P < 2.2e-16, CMHT) (fig. 3E and supplementary table 9, Supplementary Material online). These results confirm that the fates of intragenic miRNA genes and of the host genes are evolutionarily associated. Furthermore, we found that the retention of the miRNA gene when the host gene was kept was significantly less frequent than the retention of the host gene when the miRNA gene was kept (80.4% vs. 90.3%; P = 0.0002, CMHT) (fig. 3E and supplementary table 9, Supplementary Material online). Similarly, the retention of the miRNA gene when the host was lost was significantly less frequent than the retention of the host gene when the miRNA gene was lost (14.4% vs. 27.3%; P = 0.0003, CMHT) (fig. 3E and supplementary table 9, Supplementary Material online). This result could be explained by the genetic linkage mechanism that intragenic miRNA genes probably depend on the host protein-coding gene’s regulatory network more than the host gene depends on the miRNA gene regulatory network. In addition, due to the physical overlap of the two genes, the loss of a host gene by deletion might alter the regulatory elements of the miRNA gene and/or simultaneously delete the part of the protein-coding gene in which the miRNA gene resides. In contrast, the loss of an intronic miRNA gene by the remodeling of the intron might have only limited or no impact on the regulation or the preservation of the host gene, enabling the host gene to persist in the absence of the miRNA gene. In addition to genetic linkage between intragenic miRNA genes and hosts genes, an exciting but still speculative mechanism behind that observation could be that intragenic miRNA genes and host genes function cooperatively as suggested by other analyses (Lutter et al. 2010; Mandemakers et al. 2013; França et al. 2016; Boivin et al. 2018), including pathological situations (Hinske et al. 2010; França et al. 2017; Liu et al. 2019). This evolutionary association calls for additional large-scale co-expression studies of intragenic miRNA genes and their host genes to uncover whether cooperative modes of action of the miRNA gene and host gene pair in developmental, physiological, or pathological contexts, or if the related fates of intragenic miRNA genes and host genes only rely on genetic linkage devoid of functional association.
Evolution of miRNA Expression Patterns across Species and Organs
Conservation of Organ-Enriched miRNAs in Brain and Heart
The hypothesis that miRNAs perform evolutionarily conserved functions in development predicts not only that miRNA genes would be well preserved after the TGD as shown above, but also that miRNA expression patterns, which reflect their functions, would also be well conserved after the TGD. To test this prediction, we reanalyzed small RNA-sequencing data from gar, zebrafish, and stickleback from adult brains, heart ventricles, ovaries, and testes, all using the same number of biological replicates, library preparation protocol, and sequencing platform (Braasch et al. 2016; Desvignes et al. 2019) and added small RNA-sequencing data available for the same organs from medaka, although they provide a single sample per organ and were generated using a different library preparation protocol (Gay et al. 2018).
To analyze organ-specific enrichment of miRNAs in spotted gar, we followed the expression of mature miRNAs that displayed an overall expression of at least five reads-per-million (RPM) averaged over the entire data set; 284 of the 329 gar miRNAs that Prost! detected (86.3%) met this criterion. Organ pairwise differential expression analysis using DESeq2 (Love et al. 2014) showed that the brain displayed the greatest number of differentially expressed (DE) miRNAs among the four studied organs; the gonads (testis and ovary) displayed the fewest DE miRNAs and showed the largest intragroup variability (fig. 4A and B), in agreement with previous findings for zebrafish and stickleback (Desvignes et al. 2019). Across the six pairwise DE analyses of the four organs, 42 miRNAs were consistently overexpressed in the gar brain compared with each of the three other organs, and 30, 12, and ten miRNAs were consistently overexpressed in the heart, ovary, and testis, respectively (fig. 4C and supplementary files 8 and 9, Supplementary Material online). Because gar testis and ovary each showed few organ-enriched miRNAs, we looked at miRNAs that were overexpressed in both testis and ovary compared with heart and brain; these miRNAs might be implicated in shared gametogenesis processes like meiosis and cell proliferation. This approach revealed 20 additional miRNAs, bringing the total number of miRNAs that were enriched in one or both gonads to 42 (supplementary file 9, Supplementary Material online). Together, 114 miRNAs (i.e., 40% of the 284 minimally expressed miRNAs) displayed organ enrichment in either brain, heart, testis, ovary, or both gonads (fig. 4C). This result shows that miRNAs in gar display many organ-enriched miRNAs as previously shown for zebrafish and stickleback (Desvignes et al. 2019). More organ-enriched miRNAs would likely be identified with the study of more organs and with additional biological replicates to increase statistical power.
Fig. 4.
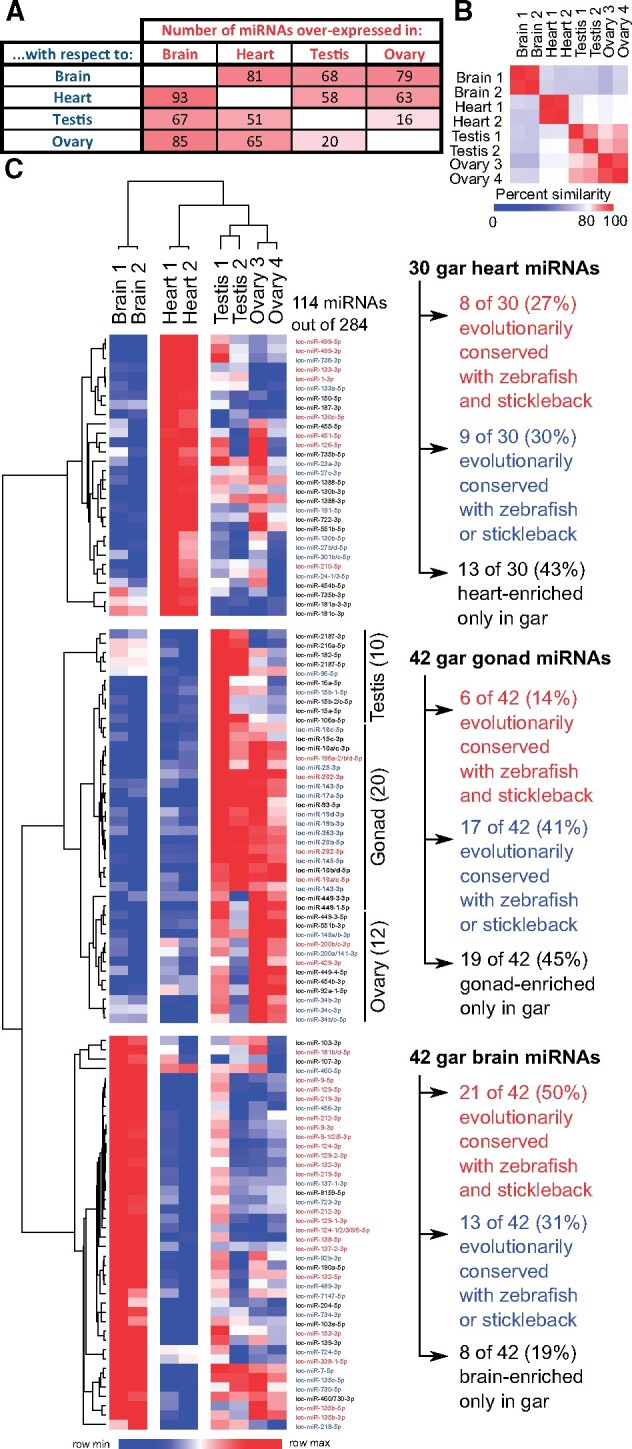
miRNA differential expression and organ-enrichment conservation in spotted gar brain, heart, testis, and ovary. (A) Heat map showing the number of gar mature miRNAs overexpressed in each organ compared with each other organ along with (B) a sample-similarity plot that compares each sample with the other seven samples tested. (C) Heat map of 114 gar mature miRNAs (in rows) that were consistently enriched in one organ (in columns) compared with the three other organs, or in gonads compared with brain and heart. Names of gar organ-enriched miRNAs that are enriched in the same organ in zebrafish and stickleback are labeled in red, gar organ-enriched miRNAs that are enriched in the same organ in zebrafish or stickleback are labeled in blue, and miRNAs that are organ-enriched only in gar are labeled in black.
The hypothesis that miRNA functions were conserved in evolution predicts that organ-enriched miRNAs in gar would tend to be enriched in the same organs in teleosts. To test this prediction, we compared the organ-enriched miRNAs in gar with the organ-enriched miRNAs in zebrafish and stickleback. Note that, because mature miRNAs from some ohnolog or other paralog miRNA genes have identical sequences, it was not always possible to assign locus origin unambiguously. We thus considered mature miRNAs to be orthologous between gar and teleost species if they could originate from orthologous genes even if their sequence could also originate from other loci that are not orthologous. The comparison of organ-enriched miRNAs between the three species revealed that 50% (21 of 42) of the brain-enriched gar miRNAs had at least one orthologous miRNA that was also brain-enriched in both zebrafish and stickleback (fig. 4C and supplementary file 9, Supplementary Material online), with many, such as miR-9-5p, miR-124-3p, and miR-138-5p (fig. 5A–C), already known to be brain-associated miRNAs in several fish species (Kitano et al. 2013; Vaz et al. 2015; Andreassen et al. 2016; Desvignes et al. 2019) and to be necessary for nervous system development in metazoans (Miska et al. 2004; Makeyev et al. 2007; Leucht et al. 2008; Cheng et al. 2009; Christodoulou et al. 2010; Yoo et al. 2011; Coolen et al. 2012; Jung et al. 2012; Ludwig et al. 2016). Eight of 42 gar brain-enriched miRNAs (19%) were brain-enriched only in gar (supplementary file 9, Supplementary Material online). However, 13 brain-enriched miRNAs in gar (31%) were brain enriched in either zebrafish or stickleback (supplementary file 9, Supplementary Material online). Together, 34 of the 42 (81%) gar brain-enriched miRNAs were brain-enriched in either or both zebrafish and stickleback.
Fig. 5.

Expression patterns of selected miRNAs in spotted gar, zebrafish, medaka, and stickleback. Average expression of evolutionarily conserved, organ-enriched miRNAs. Expression levels are given in RPM (reads per million) for the four organs studied in gar, zebrafish, medaka, and stickleback. Associated SDs across biological replicates are provided for gar, zebrafish, and stickleback.
Although gar miR-2188-5p was not enriched in any organ, we observed frequent A-to-I edition of its seed sequence at nucleotides 2 and 8, with organ-specific editing rates highest in the brain (23.6% in brain compared with 12.9%, 13.3%, and 13.7% edition in heart, ovary, and testis, respectively). The same pattern of brain-enriched seed sequence editing of miR-2188-5p was previously observed in zebrafish and stickleback and was shown to dramatically alter the sets of predicted targets in each species, revealing the potential importance of specific miRNA posttranscriptional editing in evolution (Desvignes et al. 2019).
The heart ventricle also displayed a substantial number of evolutionarily conserved, organ-enriched miRNAs, with 27% (8 of 30) of gar heart-enriched miRNAs being also heart-enriched in both zebrafish and stickleback (fig. 4C and supplementary file 9, Supplementary Material online). Heart-enriched miRNAs included well-described myomiRs, such as miR-1-3p, miR-133-3p, and miR-499-5p (fig. 5D–F), which are crucial regulators of muscle formation and function (Sokol and Ambros 2005; Chen et al. 2006; Zhao et al. 2007; Mishima et al. 2009; Christodoulou et al. 2010; Lin et al. 2014; Horak et al. 2016; Ludwig et al. 2016) and the erythromiR miR-451-5p necessary for maturation of red blood cells (Pase et al. 2009; Cifuentes et al. 2010; Rasmussen et al. 2010; Yang et al. 2010), many of which were likely present in the heart ventricle at the time of RNA extraction. Although 13 of 30 gar heart-enriched miRNAs (43%) were heart-enriched only in gar (supplementary file 9, Supplementary Material online), nine of 30 gar heart-enriched miRNAs (30%) were heart-enriched in either stickleback or zebrafish (supplementary file 9, Supplementary Material online). Together, 17 of the 30 (57%) gar heart-enriched miRNAs were heart-enriched in either or both zebrafish and stickleback.
Poor Evolutionary Conservation of Organ-Enriched miRNAs in Gonads
The hypothesis that miRNAs also perform functions in phenotypic evolution predicts that the expression patterns of miRNAs enriched in organs showing great interspecies variability would change after the TGD or in a lineage-specific manner. Gonads are an interesting model to study the role of miRNAs in diversification because fishes display a wide variety of reproductive features, including different sex determination mechanisms and modes of reproduction (Devlin and Nagahama 2002; Wootton and Smith 2014; Ortega-Recalde et al. 2020).
Supporting the hypothesis that gonad miRNAs evolve with reproductive features in lineage-specific ways—and in striking contrast to brain and heart-enriched miRNAs—conservation of organ-enrichment for miRNAs in gonads was poor. Our expression analysis identified ten testis-enriched miRNAs in gar, but none were testis-enriched in either zebrafish or stickleback (supplementary file 9, Supplementary Material online). We had previously identified a single miRNA, miR-31a-5p, that was testis-enriched in both zebrafish and stickleback (Desvignes et al. 2019). In gar, although miR-31-5p was not statistically testis-enriched, it was predominantly expressed in testis compared with the other tissues (fig. 5G), whereas in medaka, miR-31-5p expression in testis was low compared with other organs (fig. 5G). These observations suggest a conserved function of miR-31-5p in fish testis physiology that was lost in medaka.
For ovary-enriched miRNAs, 12 were significantly enriched compared with the other three organs in gar. Among these 12, eight were ovary-enriched only in gar, four were also ovary-enriched in zebrafish (miR-34b-3p, miR-34b/c-5p, miR-34c-3p, and miR-429-3p), and none were enriched in the stickleback ovary (supplementary file 9, Supplementary Material online). Interestingly, gar mir34b and mir34c compose a cluster that was retained in a single copy in zebrafish and lost in the acanthomorphs stickleback and medaka. Zebrafish miR-34b-5p and miR-34c-5p, which are co-orthologs of gar miR-34b/c-5p, both displayed an ovary-enriched expression pattern, but with expression levels 128 and 32 times less in zebrafish than in gar, respectively (fig. 5H). Apparently, zebrafish retained the miRNA genes but with greatly diminished relative expression, whereas acanthomorphs lost these genes entirely. Dysregulation of human MIR34 genes is associated with various cancers including ovarian cancer (Corney et al. 2010; Jia et al. 2019), for which a treatment using miR34 mimics was the first miRNA-based therapy to enter Phase 1 clinical trials (Agostini and Knight 2014; Zhang et al. 2019), suggesting a deep ancestral function of mir34 genes in ovarian function that was reduced or entirely lost in teleosts.
Twenty gar miRNAs were enriched in both testis and ovary compared with gar heart and brain. Among these 20 miRNAs, six were also enriched in either testis, ovary, or both gonads in both zebrafish and stickleback (fig. 4C and supplementary file 9, Supplementary Material online). Among the six miRNAs displaying gonad enrichment in all three species, two of the gar miRNAs, miR-10a/c-5p and miR-196a-2/b/d-5p, originate from miRNA genes located in Hox clusters. miR-10a/c-5p was equally enriched in both gar gonads whereas zebrafish co-orthologs miR-10a-5p and miR-10c-5p (Woltering and Durston 2006) showed the strongest expression in gonads with a testis-enrichment statistically significant for miR-10c-5p (fig. 5I). In medaka and stickleback, mir10a was lost, but miR-10c-5p showed enrichment in the ovary with moderate expression in testis in stickleback, whereas miR-10c-5p expression was almost null in all tested organs in medaka (fig. 5I). This observation suggests an evolutionarily conserved function of miR-10-5p miRNAs in gonads that was secondarily lost in medaka.
The Hox cluster miRNA gene family mir196 also showed gonad-enriched expression. In gar, the same mature miRNA sequence can be produced by two paralogous genes (miR-196a-2/b/d-5p from mir196a-2/d and mir196b) and orthologous mature miRNAs can originate from the three ohnologous genes in zebrafish (mir196a-2, mir196b, and mir196d) and two in stickleback (mir196a-2 and mir196d), as well as an additional paralogous gene (mir196a-1) that is not orthologous to either of the two gar genes. We can nonetheless cautiously observe that gar miR-196a-2/b/d-5p appeared to be equally enriched in testis and ovary, which was similar to the expression pattern of miR-196a-5p in stickleback and in medaka (fig. 5J). In zebrafish, all three miR-196-5p sequences (miR-196a-5p, miR-196b-5p, and miR-196d-5p) were enriched primarily in testis. In contrast, miR-196d-5p in stickleback was enriched in ovary with substantial expression in testis (fig. 5J). Together, these expression patterns point at an underlying role of the mir196 gene family in reproductive function that evolved in a lineage-specific manner towards one or the other gonad type.
The well-known gonad-enriched miR-202-5p was predominantly expressed in both gonads in all studied species (figs. 4C and 5K), confirming a broadly conserved role in gonadal biology (Wainwright et al. 2013; Zhang et al. 2017; Gay et al. 2018).
The last two cases of conserved gonad expression enrichment correspond to members of the mir8 gene family (figs. 5C and 6; supplementary file 9, Supplementary Material online). The gar mir8 gene family is composed of three genes, mir200b/c, mir200a/141, and mir429 (Kozomara and Griffiths-Jones 2014) organized in a single cluster. In gar, major products of all three genes displayed similar expression patterns, with the strongest expression in the ovary (fig. 6). In zebrafish, the cluster was retained in duplicate but in acanthomorphs, the cluster was retained as a singleton. Zebrafish and medaka preserved an expression pattern with enrichment in the ovary; in contrast, the clustered miRNAs in stickleback displayed significant enrichment in testis, with only weak expression in ovary (fig. 6). In zebrafish, miRNAs from the ohnologous cluster that was subsequently lost in acanthomorphs displayed comparable expression in both gonads, however, at lower expression levels than the ohnolog that was retained in all teleosts (fig. 6). Together these results suggest that all the miRNA genes in the cluster are transcribed together in a single transcript and that the mir8 family as a whole is important for fish reproduction. This conclusion is in agreement with the fact that members of the mir8 family were found to be essential for reproduction and reproductive success in both mouse and mosquito (Hasuwa et al. 2013; Lucas et al. 2015) and to be implicated in human ovarian cancer (Koutsaki et al. 2017), suggesting a prevertebrate origin of a function of the mir8 gene family in reproduction.
Fig. 6.

Expression patterns of major strands of miRNAs composing the mir200 clusters. (A) Orthology relationships (approximate genomic locations in parentheses) and (B) organization of mir200 clusters in each studied species aligned with (C) the average expression of each major strand given in RPM (reads per million) for the four organs studied in gar, zebrafish, medaka, and stickleback. Associated SDs across biological replicates are provided for gar, zebrafish, and stickleback.
These results confirmed previous observations that, although many miRNAs have crucial roles at all stages of gonadal physiology (Bizuayehu and Babiak 2014; Reza et al. 2019; Bhat et al., for reviews), miRNAs enriched in gonads seem to have relatively unstable expression patterns among fish species (Desvignes et al. 2019). Factors that contribute to this lack of conservation could include diversity of genetic sex determination systems, reproductive behaviors, or reproductive modes among teleost lineages (Devlin and Nagahama 2002; Wootton and Smith 2014; Ortega-Recalde et al. 2020). For example, the diversity in ovarian development types ranging from asynchronous in medaka and zebrafish to group-synchronous in gar and stickleback as well as the time of sampling during the day in zebrafish and medaka and season of the year in gar could dramatically impact the organ’s small RNA transcriptome by having oocytes at different stages and in different proportions in our samples.
Together, these expression data across four tissues revealed that miRNAs displaying a well-defined expression pattern in one species tend to conserve this expression pattern in other species, especially in organs that perform similar tasks through evolutionary time (i.e., the vertebrate brain and heart ventricle among our organs), suggesting functions of miRNAs in phenotypic and developmental canalization. In contrast, miRNAs strongly expressed in gonads, which have many rapidly evolving functions, tend to show less conservation of expression patterns across species, suggesting involvement of miRNAs in the diversification of these phenotypes. The study of more nonteleost and teleost species with contrasting organismal morphologies and reproductive physiologies as well as the study of the coevolution of targets and miRNAs should be performed to decipher the role of miRNAs in phenotypes stabilization and diversification throughout evolution.
Major Strands Displayed Greater Sequence Conservation than Minor Strands
The hypothesis that miRNAs have conserved functions after the TGD predicts that the most functionally important of the two mature miRNAs derived from each miRNA gene would experience the least sequence divergence comparing ohnologs within a species. To test this prediction, we focused in each species on miRNA genes retained in ohnologous pairs after the TGD. In each of the four teleosts, each miRNA gene was analyzed for the relative overall expression levels of 5p and 3p strands and each strand was assigned to one of four categories: 1) “Major strands” (sometime called “dominant” or “guide” strands), which are strands that are expressed at average levels more than twice as much as their complementary strands (a cut off favored by the field; Fromm et al. 2015) (fig. 7A1); 2) “Co-major” strands (sometime called “codominant” strands), which are expressed at average levels less than 2-fold differently (fig. 7A2); 3) “Minor” strands (sometime called “passenger” or “star” strands), which are strands that are expressed at average levels less than half the amount of their complementary strands (fig. 7A1); and 4) “Missing” strands, which are strands that were not expressed in our data sets or any public annotation databases that we examined (fig. 7A3). A few miRNA genes that had no expression for either strand were discarded because they might represent pseudogenized genes that would be under different selective pressure. Each pair of ohnologous strands within each species was then categorized as: 1) “major strand pair” when at least one of the corresponding strands from the two ohnologs is a major or co-major strand (fig. 7B1 and B2); 2) “minor strand pairs” when both strands are minor strands for both ohnologs (fig. 7B3); or 3) “missing strand pairs” when a corresponding strand is missing in one of the two ohnologs (fig. 7B4 and B5). Note that because the cases of corresponding strands being major or co-major in one hairpin and minor in its ohnologous hairpin (fig. 7B2) were rare and were not observed in all species, a specific category was not created for these few cases and these pairs were qualified as major strand pairs. This categorization led to sets of ohnologous miRNA pairs composed on an average across all four teleost species of 78 ± 7 major strand pairs, 54 ± 9 minor strand pairs, and 13 ± 4 missing strand pairs (supplementary table 10, Supplementary Material online).
We first asked how often sequences of corresponding strands in ohnologous miRNA genes remained identical to each other or evolved at least one single-nucleotide polymorphism (SNP) that differentiates them. Results showed that most (66.9%) of the mature strands of major strand pairs were identical to one another in sequence and fewer were different (33.1%, P = 2.00e-7, RGT) (fig. 7C and supplementary table 10, Supplementary Material online). In contrast, minor strand pairs were more likely to have evolved sequences different from each other than to have preserved identical sequences (only 32.8% identical sequences vs. 67.2% different, P = 3.46e-5, RGT) (fig. 7C and supplementary table 10, Supplementary Material online). For strand pairs that had a strand missing from expression data, we compared sequences of the expressed strand with the sequence of the corresponding strand of the ohnolog assuming that it would have been processed into a mature product with the same Dicer and Drosha cutting sites. Missing strand pairs displayed sequences different between the two ohnologs in 100% of the cases across all species (fig. 7C and supplementary table 10, Supplementary Material online). These results are predicted by the hypothesis that major strands are under more evolutionary constraints than minor strands or strands with expression levels undetectable in the organs sampled.
Fig. 7.
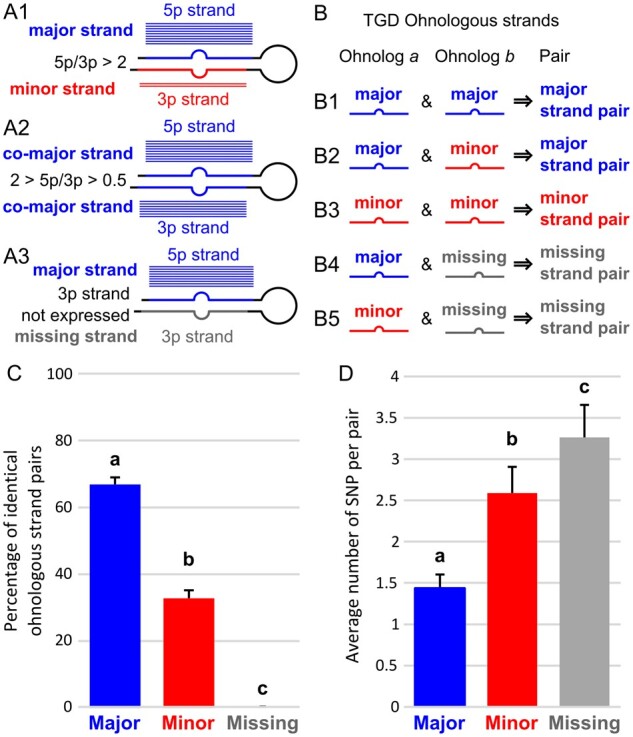
Global pattern of sequence evolution between ohnologous mature miRNAs. (A) Each strand in a hairpin was qualified as either major or minor depending on its relative expression compared with the complementary strand of the hairpin. Blue and red horizontal bars illustrate relative expression patterns (in analogy to aligned reads). Strands that were not expressed were qualified as missing. (B) Ohnologous mature miRNA pairs were further qualified as major strand pairs, minor strand pairs, and missing strand pairs depending on the qualification of each of the mature miRNA of the ohnologous pair. (C) Percentage of ohnologous miRNA strand pairs that were composed of identical miRNAs. Values represent teleost averages with SD for major, minor, and missing strand pairs. (D) Average number of single-nucleotide polymorphisms (SNPs) in ohnologous strand pairs that were not identical. Values represent teleost averages with SDs for major, minor, and missing strand pairs. Different letters signify significant differences at P < 0.05.
Among ohnologous pairs that have different sequences, we queried the number of SNPs differentiating each pair. Results showed that major strand pairs with different sequences displayed on an average 1.4 SNPs, whereas minor strands pairs with different sequences had on an average almost twice as many SNPs per pair (2.6 SNPs on an average, fig. 7D and supplementary table 10, Supplementary Material online), significantly more than major strand pairs (Padj = 2.50e-6, AOV). Missing strand pairs accumulated even more SNPs, 3.3 SNPs per pair (fig. 7D and supplementary table 10, Supplementary Material online) (Padj < 1.00e-7 and Padj = 0.0407 compared with major and minor strand pairs respectively, AOV). These results demonstrate that the most highly expressed strands of miRNA genes were under purifying selection for sequence conservation and that minor strands were under more relaxed purifying selection. In contrast, strands missing from our expression data experienced even more relaxed selection for sequence conservation, potentially only constrained to preserve folding of the precursor miRNA, which is crucial for biogenesis of the major strand (Fromm et al. 2015).
Sequence Conservation between TGD Ohnologs Identifies Functional Parts of Mature miRNAs
The hypothesis that miRNAs have conserved functions after the TGD predicts that fewer SNPs would affect the functional portions of the mature miRNA molecule, such as the seed (nucleotides 2–8 of the mature miRNA, fig. 8A), compared with less functional portions of the miRNA, so we checked whether the SNP distribution between ohnolog pairs was uniform across the mature miRNA.
Fig. 8.

Sequence evolution of ohnologous mature miRNAs. (A) Schematic representation of a generalized 22 nucleotide long miRNA with the seed and 3′-complementary sequence regions (3′-CR) marked by green and purple bars, respectively. (B–D) Single-nucleotide polymorphism (SNP) frequencies at each nucleotide position in major, minor, and missing ohnologous strand pairs. Values represent averages across all four studied teleost species with associated SDs. (E) Frequency of major, minor, and missing nonidentical strand pairs displaying seed-shifts, SNPs in their seed, and different seeds. Values represent averages across the four studied teleost species with associated SDs. (F) SNP frequency per nucleotide in the seed, the 3′-CR, and other nucleotides (i.e., nucleotides 1, 9–12, and 17–22) of major, minor, and missing nonidentical strand pairs. Values represent averages across the four studied teleost species with associated SDs. (G) SNP frequency per nucleotide in major, minor, and missing nonidentical strand pairs for the seed, the 3′-CR, and other nucleotides. Values represent averages across the four studied teleost species with associated SDs. Different letters signify significant differences at P < 0.05.
First, results showed significantly fewer cases of seed-shift (i.e., alternative cutting of the 5′ end of the miRNA) in major strand pairs compared with minor strand pairs (4.9% vs. 17.1%, P = 0.0077, CMHT) (fig. 8E). Excluding these seed-shifted pairs, major strand pairs displayed significantly less frequently SNPs in seed sequences compared with minor strand pairs (i.e., one or more SNPs in the seed) (7.5% vs. 44.8%, Padj = 1.48e-08, CMHT) (fig. 8E). Together, major strand pairs displayed significantly fewer changes (12.0%) affecting their seed (i.e., seed-shift or seed SNPs) compared with minor strand pairs (54.2%, Padj = 1.75e-10, CMHT) and to missing strand pairs (57.4%, Padj = 1.48e-06, CMHT). Missing strand pairs displayed levels of different seeds comparable to minor strand pairs (Padj = 0.9036, CMHT) (fig. 8E).
To examine other parts of the molecule, we analyzed miRNA ohnolog evolution at the nucleotide level across the entire length of the mature miRNA. For each ohnologous miRNA pair in each of the four species, we recorded SNP positions, then averaged frequencies across all four species (fig. 8B–D). Overall, major strands displayed a pattern of low SNP frequency in the seed but also in the 3′-CR compared with the central bulge and the 3′-end (fig. 8B). Notably, although the 3′-CR is proposed to be restricted to nucleotides 13 to 16 (Bartel 2018; Sheu-Gruttadauria et al. 2019; Bofill-De Ros et al. 2020), we observed that nucleotides 12 and 17 also displayed SNP frequencies as low as nucleotides of the 3′-CR, suggesting that the full stretch of nucleotides 12 through 17 may be important in the 3′-CR function. In contrast, in minor and missing strand pairs, SNPs were distributed throughout the molecule (fig. 8C and D). Major strand pairs showed lower SNP frequencies per nucleotide in their seeds compared with minor strand pairs, although not significantly lower (fig. 8F, Padj = 0.0562, AOV), similar to previous qualitative reports (Wheeler et al. 2009; Fromm et al. 2015). Major strand pairs, however, displayed significantly lower SNP frequencies per nucleotide in their seed compared with missing strand pairs (fig. 8F;Padj = 0.0102, AOV). Similarly, major strand pairs showed lower SNP frequencies per nucleotide in their 3′-CR compared with minor strand pairs, although not significantly lower (fig. 8F, Padj = 0.0549, AOV) but displayed significantly lower SNP frequencies per nucleotide in their 3′-CR compared with missing strand pairs (fig. 8F;Padj = 0.0190, AOV). Minor and missing strands displayed similar SNP frequency per nucleotide in their seed and their 3′-CR (Padj = 0.5338 and Padj = 0.7776, respectively, AOV) (fig. 8F). At other nucleotides of the mature miRNAs (i.e., nucleotides 1, 9–12, and 17–22), major strand pairs only trended to display lower SNP frequencies per nucleotide compared with minor and missing strands (fig. 8F, Padj = 0.3123 and Padj = 0.1315, respectively, AOV). This analysis also demonstrated that in major strand pairs, both the seed and the 3′-CR displayed on an average 4.7 times lower SNP frequency per nucleotide compared with the other nucleotides in the major strand miRNAs (fig. 8G) (Padj = 8.48e-5 and Padj = 7.20e-5, respectively, AOV). Similarly, in minor strands, nucleotides of the seed and 3′-CR had a significantly lower SNP frequency per nucleotide compared with the other nucleotides of the minor strand miRNAs (fig. 8G) (Padj = 0.0079 and Padj = 0.0160, respectively, AOV). The SNP frequency at nucleotides in the seed and 3′-CR in minor strand pairs was, however, only 1.6 times lower than for nucleotides outside the seed and the 3′-CR (fig. 8G). For missing strand pairs, the seed and 3′-CR did not show a statistically significant difference in SNP frequency compared with other nucleotides in the missing strands, although they had a trend in that direction (fig. 8G), suggesting fewer functional constraints throughout the entire molecule.
This analysis of SNP frequencies revealed several key features of miRNA function and sequence evolution. First, because the strand with the greatest relative expression within a miRNA hairpin is more likely to have greater sequence conservation, we conclude that major strands are under stronger purifying selection than minor or missing strands, maybe because major strands play more important roles in regulating specific mRNAs. Second, the selective pressure for sequence conservation of major strands was stronger in the seed and the 3′-CR, suggesting selective pressure towards function conservation in line with the role of the seed and 3′-CR in target recognition (Bartel 2018; Sheu-Gruttadauria et al. 2019; Bofill-De Ros et al. 2020). Third, strands that are missing expression in our data set displayed evenly distributed SNP frequencies throughout their sequence and may thus be evolutionarily constrained only to preserve folding of the miRNA hairpin and thus biogenesis of their major complementary strand (Fromm et al. 2015). Fourth, minor strands displayed an evolutionary pattern intermediate to that of major and missing strands with about a third of all minor strand pairs preserved as identical ohnologs, an average number of SNPs between ohnologous pairs intermediate between that of major and missing strands, and, similar to major strands, evidence of purifying selection in the seed and 3′-CR sequences compared with other nucleotides of the mature miRNA. Although the evolution of major strands would be under constraints to preserve mRNA regulation, and missing strands under constraints to preserve hairpins or duplex structures, the evolution of minor strand sequences reflects an interplay between purifying selection preserving miRNA biogenesis and existing regulatory functions and relaxed selection enabling the emergence of novel targets and thus novel roles (Guo and Lu 2010; Fromm et al. 2015). The balance between functional conservation and diversification would then be modulated by the level of incorporation of a minor strand into existing genetic regulatory pathways, and among the studied minor strands, some may have evolved similar to major strands, some similar to missing strands, and some across a spectrum between major and missing strand evolution, leading to the global intermediate evolutionary pattern we observed.
Arm-Switching Reveals Evolutionarily Unstable Strand Selection
The hypothesis that some minor strands are under relaxed selection predicts that minor strands may be freer than major strands to establish novel regulatory interactions. To test this prediction, we studied cases of “arm-switching,” in which one strand of a miRNA is the major strand in one set of organs, developmental stages, or species but the other strand is the major strand in a different set of organs, stages, or species (Okamura et al. 2008; Berezikov 2011; Griffiths-Jones et al. 2011). Although we did not detect any clear case of evolutionarily conserved arm-switching between the four organs that we studied, we did observe cases of changes in overall arm selection across species. For example, mir221 had co-major strands in gar and zebrafish but with miR-221-5p expression higher than miR-221-3p in gar (and mouse and human; Ludwig et al. 2016; de Rie et al. 2017) and miR-221-3p expression higher than miR-221-5p in zebrafish (fig. 9A). In contrast, in all stickleback and medaka organs, miR-221a-3p was the major strand, with a specifically marked reduction in miR-221-5p expression in medaka organs (fig. 9A). These results suggest a progressive switch in strand selection in teleosts compared with gar, with a further reduction of miR-221-5p function in medaka. In contrast to mir221, miR-129-5p was the dominant strand in all teleosts but miR-129-3p was the dominant strand in gar (fig. 9B) and human (Ludwig et al. 2016). The change in strand selectivity in teleosts appears to be mediated by an overall reduction in miR-129-3p expression and an increase in miR-129-5p expression in the brain.
Fig. 9.

Examples of arm-switching events between species. Average expression of each strand in each organ is given in RPM (reads per million) on logarithmic scales for the four organs studied in gar, zebrafish, medaka, and stickleback. The solid line represents equal expression of 5p and 3p strands. Dashed lines represent 2-fold expression difference between one strand and the other. On each graph, points in the top-left half represent organs in which the 3p strand is more expressed than the 5p strand, and points in the bottom-right half represent organs in which the 5p strand is more expressed than the 3p strand.
Following a different strand selection pattern, mir142 mature strand expression in gar was strongly biased towards miR-142-5p with only weak expression of miR-142-3p (fig. 9C). In zebrafish and stickleback, the relative expression of the mature strands remained biased towards miR-142-5p, however, the relative expression of miR-142-3p was much higher than in gar (fig. 9C). In contrast, in medaka, miR-142-5p and miR-142-3p appeared to be co-major strands with a higher expression of miR-142-3p than miR-142-5p seemingly due to a reduction in miR-142-5p expression compared with zebrafish and stickleback (fig. 9C). Finally, miR-21-3p was the major strand in gar, whereas in teleosts, it was miR-21-5p (fig. 9D), similar to human and mouse (Ludwig et al. 2016; de Rie et al. 2017). This pattern suggests that the arm-switch situation observed for mir21 resulted from a reduction of miR-21-5p function in gar compared with mammals and teleosts whereas the expression and potentially the function of miR-21-3p was conserved, rather than a gain of function of miR-21-5p in both mammals and teleosts. The study of additional organs and additional nonteleost actinopterygian species such as bowfin is necessary to understand the precise evolution of arm-switching events and their functional consequences.
These four examples confirm that strand selection is not an evolutionarily stable process and that minor strands can become major strands and major strands can become minor strands and even potentially lose their expression (Okamura et al. 2008; Berezikov 2011; Griffiths-Jones et al. 2011). This observation, along with the demonstration that minor strands were under purifying selection in their seed and 3′-CR regions, argues for continuing systematic consideration of minor strands as potential functional miRNAs. This analysis supports use of the terms 5p and 3p qualifiers for the two miRNA strands over the “dominant,” “passenger,” or “star” strands qualifiers, which can convey a misleading notion of function that may not be conserved across organs, developmental stages, or species.
Conclusions
These analyses of miRNA gene structure, evolution, and expression in gar and teleosts revealed mechanisms that underlie miRNA gene retention and function evolution after the TGD (fig. 10). First, we estimated that the pre-TGD miRNAome of the last common ancestor of Teleosts and Holostei was composed of 240 evolutionarily conserved miRNA genes. Quantitative results demonstrated that after the TGD, teleost miRNA gene repertoires expanded significantly more than did protein-coding gene repertoires. Similar to protein-coding genes, however, most miRNA gene losses occurred soon after the TGD rather than a constant lineage-specific resolution of duplicates over time. We conclude that an increase in miRNA gene numbers relative to protein numbers may strengthen pre-existing regulatory functions under the gene balance hypothesis or allow the development of new regulatory roles, contributing to phenotypic canalization and diversification, respectively.
Fig. 10.
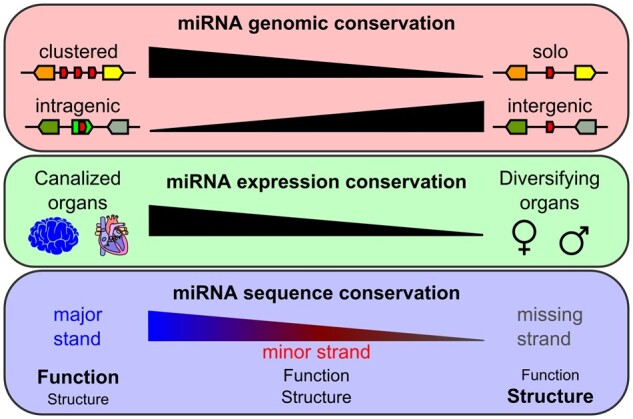
Factors influencing miRNA evolution after the TGD.
Second, results unambiguously demonstrated that the pre-TGD genomic context of a miRNA gene significantly influenced its retention pattern post-TGD. Specifically, the clustering of miRNA genes increased duplicate retention rates after the TGD, suggesting that clustering may maintain a cohort of miRNAs that were already regulating a group of biologically related targeted genes due to strong genetic linkage. Furthermore, we found that intergenic miRNA genes were more often retained in duplicate than intragenic miRNA genes, whose fates were evolutionarily associated with that of their host gene.
Third, our miRNA expression study revealed patterns of evolution compatible with phenotypic canalization or phenotypic diversification functions acting as modulators for the conservation of expression patterns. Specifically, results showed that miRNAs that display a well-defined expression pattern in one species tend to conserve this expression pattern in other species, especially in organs such as the brain and heart ventricle, in line with the important evolutionarily conserved role of miRNAs in the establishment of tissue identity and in the development of these organs. These results confirm the hypothesis that expression patterns of miRNAs providing robustness to organ development and physiology were preserved in evolution. In contrast, we observed that miRNAs enriched in gonads tend to show weak conservation of expression patterns across species, suggesting an involvement of these miRNAs in the diversification of reproductive phenotypes, such as sex determination pathways, egg-coat formation, sperm–egg interactions, reproductive timing, and other reproductive mechanisms that can rapidly evolve between species and potentially erect reproductive barriers.
Fourth, our analysis of SNP frequency in post-TGD miRNA pairs revealed the importance of functional conservation and diversification in modulating miRNA sequence evolution. Specifically, we demonstrated that major strands are under stronger purifying selection than minor or missing strands and that the selective pressure for sequence conservation of major strands was the strongest in the seed and the 3′-CR, as expected if major strands play a more important role in regulating specific messenger RNAs compared with minor strands and greater selective pressure for function conservation in line with the role of the seed and 3′-CR in target recognition. In contrast, strands that were missing expression in our data set may be evolutionarily constrained only to preserve the folding of the miRNA hairpin and thus the biogenesis of their major complementary strand. The finding that the evolutionary pattern of minor strand sequences was intermediate between major and missing strands suggests an interplay between purifying selection preserving existing regulatory functions and relaxed selection that enables the emergence of novel targets and thus novel roles. The balance between functional conservation and diversification could be modulated by the level of incorporation of a minor strand into existing genetic regulatory pathways.
Finally, these results suggest that all strands of a miRNA hairpin are functional, or have the potential to become functional, by changes in their sequences or modifications in strand selectivity, possibly resulting in the emergence of novel expression patterns in association with the establishment of novel genetic regulation that can be selected upon.
Together, our results revealed the determinants of miRNA evolution following the TGD, which likely reflect general mechanisms acting on miRNA evolution following WGDs in metazoans. These findings increase our understanding of the processes by which miRNA genes have increased in number and functional diversity during metazoan evolution and delineate their roles in phenotypic canalization and diversification.
Materials and Methods
Origin of Small RNA-Sequencing Data
Small RNA-sequencing data analyzed in this study are publicly available under accession numbers SRP063942, SRP039502, SRP151190, SRP157992, and SRP069031 for spotted gar, zebrafish (AB strain), medaka (Cab strain), stickleback (Boot Lake fresh water strain), and blackfin icefish (West Antarctic Peninsula), respectively (Desvignes et al. 2014, 2016, 2019; Braasch et al. 2016; Gay et al. 2018).
Small RNA Read Analysis Using Prost!
Raw Illumina-sequencing reads were processed as in Desvignes et al. (2019). For each species, libraries were simultaneously analyzed using Prost! (Desvignes et al. 2019) selecting for read length 17 to 25 nucleotides and aligning reads to the species’ reference genome using bbmapskimmer.sh version 37.85 of the BBMap suite (https://sourceforge.net/projects/bbmap/) with parameters identical to those published. Publicly available genome assemblies were used for gar (LepOcu1), zebrafish (GRCz10), Japanese medaka HdrR (ASM223467v1), three-spined stickleback (BROAD S1), and blackfin icefish (Chaenocephalus aceratus V1.0). We configured Prost! to retain only sequences with a minimum of five identical reads for the annotation of miRNA genes and mature miRNAs in medaka as previously described (Desvignes et al. 2019) and as detailed in the Prost! documentation page (https://prost.readthedocs.io). In the current study, we configured Prost! to use all mature and hairpin sequences from chordates in miRBase Release 22 (Kozomara and Griffiths-Jones 2014), as well as the published stickleback miRNA annotations (Desvignes et al. 2019), the published Blackfin Icefish annotation (Kim et al. 2019), the extended zebrafish miRNA annotation (Desvignes et al. 2014), and the published gar miRNA annotation (Braasch et al. 2016). All annotation data sets used in this study are available on the Prost! Github page (https://github.com/uoregon-postlethwait/prost). The miRNA and isomiR nomenclature used follows the rules established for zebrafish (Desvignes et al. 2015, 2020; Ruzicka et al. 2019). For differential expression analysis in gar, zebrafish, medaka, and stickleback, only sequences with a minimum of 30 reads were retained. From Prost! output, we used the nonnormalized read counts of annotated miRNA reads to perform differential expression analyses using DESeq2 (Love et al. 2014) with a cut-off adjusted P value of 0.01 to draw conclusions on differential expression as previously described (Desvignes et al. 2019). Heat maps and similarity matrices were generated using the Broad Institute Morpheus webserver (https://software.broadinstitute.org/morpheus/) and log2-transformed normalized counts from annotated miRNAs that displayed a minimum normalized average expression of five RPM over the entire data set. Hierarchical clustering on rows and columns was performed using the “one minus Pearson’s correlation” model and the “average” linkage method.
Genomic Context Analyses
Gene orthology and ohnology relationships across species were established first by sequence similarities of mature miRNAs and miRNA hairpins among species. Orthologies and ohnologies were then confirmed by conserved synteny analyses by examining the orthologies of surrounding protein-coding genes. Reciprocal best-BLAST hit analyses were performed on the miRNA hairpins when synteny analyses could not resolve orthology and ohnology relationships. Whether miRNA genes were in clusters or in introns was determined using genomic locations of miRNA genes and annotations of protein-coding genes available for each genome assembly (Aken et al. 2017; Kim et al. 2019).
To conclude that a miRNA gene has been lost in a species, we followed three lines of evidence. First, for each putatively missing miRNA gene, we searched by BlastN using hairpin sequences to see whether a hit could be found in the genome assembly. Each hit was manually checked for potentially being an already annotated paralogous gene or the missing miRNA gene, and small RNA-sequencing data were searched for expression of miRNAs from the identified locus. If no BlastN alignments were returned, an analysis of conserved syntenies was performed to determine the genomic region where the miRNA gene “should” have been compared with the outgroup (e.g., between two protein-coding genes or intragenic within a certain protein-coding gene). This genomic region was then searched for expressed reads in the small RNA-sequencing data set and if an expressed read displayed any features suggesting that it could originate from the searched miRNA gene. Second, it is possible that a miRNA gene is missing from a genome assembly due to assembly gaps and errors. To address this possibility, we searched for the potentially missing miRNA gene in the genome assemblies of closely related species available in Ensembl (Aken et al. 2017); shared absence suggested loss in a common ancestor. Third, because assembly errors might plague similar regions of the genome assembly of different species, we searched the small RNA-sequencing data for expressed reads that did not align to the genome assembly but were identical to known miRNAs in other species. If none of these three steps identified a missing miRNA gene, we annotated this miRNA gene as lost, recognizing that this conclusion rests on the criteria stated here and might be revised with more transcriptomic data or improved genome assemblies.
Statistical Analyses
For nominal variables, we used Cochran-Mantel-Haenszel Tests with continuity correction (CMHT). Homogeneity of odds ratios across strata were verified with a Woof test. In case of multiple testing (e.g., retention in duplicates, singleton, or loss), P values were adjusted using the Benjamini–Hochberg procedure for false discovery correction. When comparing nominal variables to expected results, we used Repeated G–tests of goodness-of-fit (RGT). For quantitative variables, we used a two-way analysis of variance followed by a Tukey’s HSD post hoc test (AOV). Conditions of applicability were verified by visually inspecting the residuals and using a Levene test for homogeneity of variances. All statistical analyses were performed using RStudio version 1.1.463.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (Grant Nos. NIH R24 OD011199, NIH 5R01 OD011116, and NIH R01 GM085318 to J.H.P.), the National Science Foundation Office of Polar Program (NSF OPP-1543383 to J.H.P. and T.D.), and Agence Nationale de la Recherche (ANR-18-CE20-0004 to J.B.). This work benefited from access to the University of Oregon high performance computers Talapas and ACISS (NSF Grant OCI-0960354). Authors also thank Clayton M. Small for advice on statistical analyses and the handling editor and three anonymous reviewers for their helpful comments.
Author Contributions
Study concept and design: T.D. and J.H.P.; Acquisition of data: T.D.; Analysis and interpretation of data: T.D., J.S., J.M., J.B., and J.H.P.; Wrote the manuscript: T.D.; Critical revision of the manuscript: T.D., J.B., and J.H.P.; Obtained funding: J.H.P., T.D., and J.B.; Study supervision: T.D. and J.H.P.
Data Availability
All data generated or analyzed during this study are included in the published article (and its supplementary files) and/or deposited in NCBI.
References
- Abrouk M, Zhang R, Murat F, Li A, Pont C, Mao L, Salse J.. 2012. Grass microRNA gene paleohistory unveils new insights into gene dosage balance in subgenome partitioning after whole-genome duplication. Plant Cell 24(5):1776–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostini M, Knight RA.. 2014. miR-34: from bench to bedside. Oncotarget 5(4):872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, Billis K, Carvalho-Silva D, Cummins C, Clapham P, et al. 2017. Ensembl 2017. Nucleic Acids Res. 45(D1):D635–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti C, Cochella L.. 2017. A framework for understanding the roles of miRNAs in animal development. Development 144(14):2548–2559. [DOI] [PubMed] [Google Scholar]
- Allendorf FW, Thorgaard GH.. 1984. Tetraploidy and the evolution of salmonid fishes. In: Turner B. J. (Ed.), Evolutionary genetics of fishes. Boston, MA: Springer US. p. 55–93. [Google Scholar]
- Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S, Aravin A, Brownstein MJ, Tuschl T, Margalit H.. 2005. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 33(8):2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, Zamore PD.. 2013. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 14(8):475–488. [DOI] [PubMed] [Google Scholar]
- Amores A, Force A, Yan Y-L, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang Y-L, et al. 1998. Zebrafish hox clusters and vertebrate genome evolution. Science 282(5394):1711–1714. [DOI] [PubMed] [Google Scholar]
- Andreassen R, Rangnes F, Sivertsen M, Chiang M, Tran M, Worren MM.. 2016. Discovery of miRNAs and their corresponding miRNA genes in Atlantic cod (Gadus morhua): use of stable miRNAs as reference genes reveals subgroups of miRNAs that are highly expressed in particular organs. PLoS One 11(4):e0153324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arif S, Murat S, Almudi I, Nunes MDS, Bortolamiol-Becet D, McGregor NS, Currie JMS, Hughes H, Ronshaugen M, Sucena É, et al. 2013. Evolution of miR-92a underlies natural morphological variation in Drosophila melanogaster. Curr Biol. 23(6):523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avital G, França GS, Yanai I.. 2018. Bimodal evolutionary developmental miRNA program in animal embryogenesis. Mol Biol Evol. 35(3):646–654. [DOI] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136(2):215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2018. Metazoan microRNAs. Cell 173(1):20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskerville S, Bartel DP.. 2005. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11(3):241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E. 2011. Evolution of microRNA diversity and regulation in animals. Nat Rev Genet. 12(12):846–860. [DOI] [PubMed] [Google Scholar]
- Berthelot C, Brunet F, Chalopin D, Juanchich A, Bernard M, Noël B, Bento P, Da Silva C, Labadie K, Alberti A, et al. 2014. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun. 5:3657. Available from: https://www.nature.com/articles/ncomms4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RA, Priyam M, Foysal MJ, Gupta SK, Sundaray JK. Role of sex-biased miRNAs in teleosts – a review. Rev Aquac.13:269–281. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/raq.12474 [Google Scholar]
- Bhuiyan SS, Kinoshita S, Wongwarangkana C, Asaduzzaman M, Asakawa S, Watabe S.. 2013. Evolution of the myosin heavy chain gene MYH14 and its intronic microRNA miR-499: muscle-specific miR-499 expression persists in the absence of the ancestral host gene. BMC Evol Biol. 13(1):142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA.. 2010. The gene balance hypothesis: implications for gene regulation, quantitative traits and evolution. New Phytol. 186(1):54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA.. 2012. Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc Natl Acad Sci U S A. 109(37):14746–14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizuayehu TT, Babiak I.. 2014. MicroRNA in teleost fish. Genome Biol Evol. 6(8):1911–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bofill-De Ros X, Yang A, Gu S.. 2020. IsomiRs: expanding the miRNA repression toolbox beyond the seed. Biochim Biophys Acta Gene Regul Mech. 1863(4):194373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin V, Deschamps-Francoeur G, Scott MS.. 2018. Protein coding genes as hosts for noncoding RNA expression. Semin Cell Dev Biol. 75:3–12.28811264 [Google Scholar]
- Braasch I, Brunet F, Volff J-N, Schartl M.. 2009. Pigmentation pathway evolution after whole-genome duplication in fish. Genome Biol Evol. 1:479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braasch I, Gehrke AR, Smith JJ, Kawasaki K, Manousaki T, Pasquier J, Amores A, Desvignes T, Batzel P, Catchen J, et al. 2016. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat Genet. 48(4):427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braasch I, Postlethwait JH.. 2012. Polyploidy in fish and the teleost genome duplication. In: Soltis PS, Soltis DE, editors. Polyploidy and genome evolution. Heidelberg (Berlin: ): Springer Berlin Heidelberg. p. 341–383. [Google Scholar]
- Brunet FG, Crollius HR, Paris M, Aury J-M, Gibert P, Jaillon O, Laudet V, Robinson-Rechavi M.. 2006. Gene loss and evolutionary rates following whole-genome duplication in teleost fishes. Mol Biol Evol. 23(9):1808–1816. [DOI] [PubMed] [Google Scholar]
- Campo-Paysaa F, Sémon M, Cameron RA, Peterson KJ, Schubert M.. 2011. microRNA complements in deuterostomes: origin and evolution of microRNAs. Evol Dev. 13(1):15–27. [DOI] [PubMed] [Google Scholar]
- Cassidy JJ, Jha AR, Posadas DM, Giri R, Venken KJT, Ji J, Jiang H, Bellen HJ, White KP, Carthew RW.. 2013. miR-9a minimizes the phenotypic impact of genomic diversity by buffering a transcription factor. Cell 155(7):1556–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan W-C, Ho M-R, Li S-C, Tsai K-W, Lai C-H, Hsu C-N, Lin W.. 2012. MetaMirClust: discovery of miRNA cluster patterns using a data-mining approach. Genomics 100(3):141–148. [DOI] [PubMed] [Google Scholar]
- Chen J-F, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang D-Z.. 2006. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 38(2):228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L-C, Pastrana E, Tavazoie M, Doetsch F.. 2009. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci. 12(4):399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, et al. 2010. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 24(10):992–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulou F, Raible F, Tomer R, Simakov O, Trachana K, Klaus S, Snyman H, Hannon GJ, Bork P, Arendt D.. 2010. Ancient animal microRNAs and the evolution of tissue identity. Nature 463(7284):1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, et al. 2010. A novel miRNA processing pathway independent of dicer requires argonaute2 catalytic activity. Science 328(5986):1694–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolen M, Thieffry D, Drivenes Ø, Becker TS, Bally-Cuif L.. 2012. miR-9 controls the timing of neurogenesis through the direct inhibition of antagonistic factors. Dev Cell. 22(5):1052–1064. [DOI] [PubMed] [Google Scholar]
- Corney DC, Hwang C-I, Matoso A, Vogt M, Flesken-Nikitin A, Godwin AK, Kamat AA, Sood AK, Ellenson LH, Hermeking H, et al. 2010. Frequent downregulation of miR-34 family in human ovarian cancers. Clin Cancer Res. 16(4):1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehal P, Boore JL.. 2005. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 3(10):e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M.. 2014. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell. 54(5):766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R, McGeary SE, Title AC, Agarwal V, Bartel DP, Stoffel M.. 2016. Impact of microRNA levels, target-site complementarity, and cooperativity on competing endogenous RNA-regulated gene expression. Mol Cell. 64(3):565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rie D, Abugessaisa I, Alam T, Arner E, Arner P, Ashoor H, Åström G, Babina M, Bertin N, Burroughs AM, Carlisle AJ.. 2017. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat Biotechnol. 35(9):872–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvignes T, Batzel P, Berezikov E, Eilbeck K, Eppig JT, McAndrews MS, Singer A, Postlethwait JH.. 2015. miRNA nomenclature: a view incorporating genetic origins, biosynthetic pathways, and sequence variants. Trends Genet. 31(11):613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvignes T, Batzel P, Sydes J, Eames BF, Postlethwait JH.. 2019. miRNA analysis with Prost! reveals evolutionary conservation of organ-enriched expression and post-transcriptional modifications in three-spined stickleback and zebrafish. Sci Rep. 9:3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvignes T, Beam MJ, Batzel P, Sydes J, Postlethwait JH.. 2014. Expanding the annotation of zebrafish microRNAs based on small RNA sequencing. Gene 546(2):386–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvignes T, Contreras A, Postlethwait JH.. 2014. Evolution of the miR199-214 cluster and vertebrate skeletal development. RNA Biol. 11(4):281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvignes T, Detrich III HW, Postlethwait JH.. 2016. Genomic conservation of erythropoietic microRNAs (erythromiRs) in white-blooded Antarctic icefish. Mar Genomics. 30:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvignes T, Loher P, Eilbeck K, Ma J, Urgese G, Fromm B, Sydes J, Aparicio-Puerta E, Barrera V, Espín R, et al. 2020. Unification of miRNA and isomiR research: the mirGFF3 format and the mirtop API. Bioinformatics 36(3):698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin RH, Nagahama Y.. 2002. Sex determination and sex differentiation in fish: an overview of genetic, physiological, and environmental influences. Aquaculture 208(3–4):191–364. [Google Scholar]
- Ebert MS, Sharp PA.. 2012. Roles for microRNAs in conferring robustness to biological processes. Cell 149(3):515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke R, Eschmeyer WN, & Van der Laan R. (Eds.), . 2015. Eschmeyer’s Catalog of fishes: genera, species, references. Available from: http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp. Electronic version accessed 22 Apr 2021.
- Fang W, Bartel DP.. 2020. MicroRNA clustering assists processing of suboptimal microRNA hairpins through the action of the ERH protein. Mol Cell. 78(2):289–302.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J.. 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151:1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- França GS, Hinske LC, Galante PAF, Vibranovski MD.. 2017. Unveiling the impact of the genomic architecture on the evolution of vertebrate microRNAs. Front Genet. 8. Available from: https://www.frontiersin.org/articles/10.3389/fgene.2017.00034/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- França GS, Vibranovski MD, Galante PAF.. 2016. Host gene constraints and genomic context impact the expression and evolution of human microRNAs. Nat Commun. 7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchini P, Xiong P, Fruciano C, Meyer A.. 2016. The role of microRNAs in the repeated parallel diversification of lineages of Midas cichlid fish from Nicaragua. Genome Biol Evol. 8(5):1543–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchini P, Xiong P, Fruciano C, Schneider RF, Woltering JM, Hulsey CD, Meyer A.. 2019. MicroRNA gene regulation in extremely young and parallel adaptive radiations of crater lake cichlid fish. Mol Biol Evol. 36(11):2498–2511. [DOI] [PubMed] [Google Scholar]
- Fromm B, Billipp T, Peck LE, Johansen M, Tarver JE, King BL, Newcomb JM, Sempere LF, Flatmark K, Hovig E, et al. 2015. A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu Rev Genet. 49(1):213–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm B, Domanska D, Høye E, Ovchinnikov V, Kang W, Aparicio-Puerta E, Johansen M, Flatmark K, Mathelier A, Hovig E, et al. 2020. MirGeneDB 2.0: the metazoan microRNA complement. Nucleic Acids Res. 48(D1):D132–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay S, Bugeon J, Bouchareb A, Henry L, Delahaye C, Legeai F, Montfort J, Cam AL, Siegel A, Bobe J, et al. 2018. MiR-202 controls female fecundity by regulating medaka oogenesis. PLoS Genet. 14(9):e1007593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Dongen SV, Inoue K, Enright AJ, Schier AF.. 2006. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science 312(5770):75–79. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Hui JHL, Marco A, Ronshaugen M.. 2011. MicroRNA evolution by arm switching. EMBO Rep. 12(2):172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ.. 2007. miRBase: tools for microRNA genomics. Nucleic Acids Res. 36(Database):D154–D158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra-Assunção JA, Enright AJ.. 2012. Large-scale analysis of microRNA evolution. BMC Genomics 13(1):218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Lu Z.. 2010. The fate of miRNA strand through evolutionary analysis: implication for degradation as merely carrier strand or potential regulatory molecule? PLoS One 5(6):e11387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halushka MK, Fromm B, Peterson KJ, McCall MN.. 2018. Big strides in cellular microRNA expression. Trends Genet. 34(3):165–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasuwa H, Ueda J, Ikawa M, Okabe M.. 2013. MiR-200b and miR-429 function in mouse ovulation and are essential for female fertility. Science 341(6141):71–73. [DOI] [PubMed] [Google Scholar]
- Hertel J, Lindemeyer M, Missal K, Fried C, Tanzer A, Flamm C, Hofacker IL, Stadler PF, Students of Bioinformatics Computer Labs 2004 and 2005. 2006. The expansion of the metazoan microRNA repertoire. BMC Genomics 7:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel J, Stadler PF.. 2015. The expansion of animal microRNA families revisited. Life Basel Switz. 5(1):905–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinske LC, França GS, Torres HAM, Ohara DT, Lopes-Ramos CM, Heyn J, Reis LFL, Ohno-Machado L, Kreth S, Galante PAF.. 2014. miRIAD—integrating microRNA inter- and intragenic data. Database 2014:bau099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinske LCG, Galante PA, Kuo WP, Ohno-Machado L.. 2010. A potential role for intragenic miRNAs on their hosts’ interactome. BMC Genomics 11(1):533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak M, Novak J, Bienertova-Vasku J.. 2016. Muscle-specific microRNAs in skeletal muscle development. Dev Biol. 410(1):1–13. [DOI] [PubMed] [Google Scholar]
- Hornstein E, Shomron N.. 2006. Canalization of development by microRNAs. Nat Genet. 38(S6):S20–S24. [DOI] [PubMed] [Google Scholar]
- Inoue J, Sato Y, Sinclair R, Tsukamoto K, Nishida M.. 2015. Rapid genome reshaping by multiple-gene loss after whole-genome duplication in teleost fish suggested by mathematical modeling. Proc Natl Acad Sci U S A. 112(48):14918–14923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, Aury J-M, Brunet F, Petit J-L, Stange-Thomann N, Mauceli E, Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A, et al. 2004. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431(7011):946–957. [DOI] [PubMed] [Google Scholar]
- Jia Y, Lin R, Jin H, Si L, Jian W, Yu Q, Yang S.. 2019. MicroRNA-34 suppresses proliferation of human ovarian cancer cells by triggering autophagy and apoptosis and inhibits cell invasion by targeting Notch 1. Biochimie 160:193–199. [DOI] [PubMed] [Google Scholar]
- Jonas S, Izaurralde E.. 2015. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 16(7):421–433. [DOI] [PubMed] [Google Scholar]
- Jung H-J, Coffinier C, Choe Y, Beigneux AP, Davies BSJ, Yang SH, Barnes RH, Hong J, Sun T, Pleasure SJ, et al. 2012. Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc Natl Acad Sci U S A. 109(7):E423–E431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juzenas S, Venkatesh G, Hübenthal M, Hoeppner MP, Du ZG, Paulsen M, Rosenstiel P, Senger P, Hofmann-Apitius M, Keller A, et al. 2017. A comprehensive, cell specific microRNA catalogue of human peripheral blood. Nucleic Acids Res. 45(16):9290–9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabekkodu SP, Shukla V, Varghese VK, Souza JD, Chakrabarty S, Satyamoorthy K.. 2018. Clustered miRNAs and their role in biological functions and diseases. Biol Rev Camb Philos Soc. 93(4):1955–1986. [DOI] [PubMed] [Google Scholar]
- Kasper DM, Moro A, Ristori E, Narayanan A, Hill-Teran G, Fleming E, Moreno-Mateos M, Vejnar CE, Zhang J, Lee D, et al. 2017. MicroRNAs establish uniform traits during the architecture of vertebrate embryos. Dev Cell. 40(6):552–565.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley JL, Desvignes T, McGowan KL, Perez M, Rodriguez LA, Brown AP, Culumber Z, Tobler M.. 2020. microRNA expression variation as a potential molecular mechanism contributing to adaptation to hydrogen sulphide. J Evol Biol. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/jeb.13727 [DOI] [PubMed] [Google Scholar]
- Kim B-M, Amores A, Kang S, Ahn D-H, Kim J-H, Kim I-C, Lee JH, Lee SG, Lee H, Lee J, et al. 2019. Antarctic blackfin icefish genome reveals adaptations to extreme environments. Nat Ecol Evol. 3(3):469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano J, Yoshida K, Suzuki Y.. 2013. RNA sequencing reveals small RNAs differentially expressed between incipient Japanese threespine sticklebacks. BMC Genomics 14:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittelmann S, McGregor AP.. 2019. Modulation and evolution of animal development through microRNA regulation of gene expression. Genes 10:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. 2011. Are there laws of genome evolution? PLoS Comput Biol. 7(8):e1002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsaki M, Libra M, Spandidos DA, Zaravinos A.. 2017. The miR-200 family in ovarian cancer. Oncotarget 8(39):66629–66640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S.. 2014. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42(Database Issue):D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Suleski M, Hedges SB.. 2017. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol. 34(7):1812–1819. [DOI] [PubMed] [Google Scholar]
- Lee C-T, Risom T, Strauss WM.. 2007. Evolutionary conservation of microRNA regulatory circuits: an examination of microRNA gene complexity and conserved microRNA-target interactions through metazoan phylogeny. DNA Cell Biol. 26(4):209–218. [DOI] [PubMed] [Google Scholar]
- Leucht C, Stigloher C, Wizenmann A, Klafke R, Folchert A, Bally-Cuif L.. 2008. MicroRNA-9 directs late organizer activity of the midbrain-hindbrain boundary. Nat Neurosci. 11(6):641–648. [DOI] [PubMed] [Google Scholar]
- Li J, Zhang Z.. 2013. miRNA regulatory variation in human evolution. Trends Genet. 29(2):116–124. [DOI] [PubMed] [Google Scholar]
- Lin C-C, Chang Y-M, Pan C-T, Chen C-C, Ling L, Tsao K-C, Yang R-B, Li W-H.. 2014. Functional evolution of cardiac microRNAs in heart development and functions. Mol Biol Evol. 31(10):2722–2734. [DOI] [PubMed] [Google Scholar]
- Liu B, Shyr Y, Cai J, Liu Q.. 2019. Interplay between miRNAs and host genes and their role in cancer. Brief Funct Genomics. 18(4):255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liufu Z, Zhao Y, Guo L, Miao G, Xiao J, Lyu Y, Chen Y, Shi S, Tang T, Wu C-I.. 2017. Redundant and incoherent regulations of multiple phenotypes suggest microRNAs’ role in stability control. Genome Res. 27(10):1665–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh Y-HE, Yi SV, Streelman JT.. 2011. Evolution of microRNAs and the diversification of species. Genome Biol Evol. 3:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes I, Altab G, Raina P, de Magalhães JP.. 2020. Gene size matters: What determines gene length in the human genome? bioRxiv:2020.01.10.901272. Available from: https://www.frontiersin.org/articles/10.3389/fgene.2021.559998/full
- Love MI, Huber W, Anders S.. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas KJ, Roy S, Ha J, Gervaise AL, Kokoza VA, Raikhel AS.. 2015. MicroRNA-8 targets the Wingless signaling pathway in the female mosquito fat body to regulate reproductive processes. Proc Natl Acad Sci U S A. 112(5):1440–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig N, Leidinger P, Becker K, Backes C, Fehlmann T, Pallasch C, Rheinheimer S, Meder B, Stähler C, Meese E, et al. 2016. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 44(8):3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutter D, Marr C, Krumsiek J, Lang EW, Theis FJ.. 2010. Intronic microRNAs support their host genes by mediating synergistic and antagonistic regulatory effects. BMC Genomics 11(1):224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macqueen DJ, Johnston IA.. 2014. A well-constrained estimate for the timing of the salmonid whole genome duplication reveals major decoupling from species diversification. Proc R Soc B. 281(1778):20132881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makeyev EV, Zhang J, Carrasco MA, Maniatis T.. 2007. The microRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell. 27(3):435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandemakers W, Abuhatzira L, Xu H, Caromile LA, Hébert SS, Snellinx A, Morais VA, Matta S, Cai T, Notkins AL, et al. 2013. Co-regulation of intragenic microRNA miR-153 and its host gene Ia-2β: identification of miR-153 target genes with functions related to IA-2β in pancreas and brain. Diabetologia 56(7):1547–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A. 2019. Comment on “microRNAs in the same clusters evolve to coordinately regulate functionally related genes.” Mol Biol Evol. 36(8):1843–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Hui JHL, Ronshaugen M, Griffiths-Jones S.. 2010. Functional shifts in insect microRNA evolution. Genome Biol Evol. 2:686–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Ninova M, Ronshaugen M, Griffiths-Jones S.. 2013. Clusters of microRNAs emerge by new hairpins in existing transcripts. Nucleic Acids Res. 41(16):7745–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MN, Kim M-S, Adil M, Patil AH, Lu Y, Mitchell CJ, Leal-Rojas P, Xu J, Kumar M, Dawson VL, et al. 2017. Toward the human cellular microRNAome. Genome Res. 27(10):1769–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeary SE, Lin KS, Shi CY, Pham TM, Bisaria N, Kelley GM, Bartel DP.. 2019. The biochemical basis of microRNA targeting efficacy. Science 366(6472):eaav1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT, Olson EN.. 2012. MicroRNAs in stress signaling and human disease. Cell 148(6):1172–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Lemoine F, Soumillon M, Liechti A, Weier M, Guschanski K, Hu H, Khaitovich P, Kaessmann H.. 2013. Birth and expression evolution of mammalian microRNA genes. Genome Res. 23(1):34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima Y, Abreu-Goodger C, Staton AA, Stahlhut C, Shou C, Cheng C, Gerstein M, Enright AJ, Giraldez AJ.. 2009. Zebrafish miR-1 and miR-133 shape muscle gene expression and regulate sarcomeric actin organization. Genes Dev. 23(5):619–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Townsend M, Yoshii A, Šestan N, Rakic P, Constantine-Paton M, Horvitz HR.. 2004. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 5(9):R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed J, Siepel A, Lai EC.. 2014. Diverse modes of evolutionary emergence and flux of conserved microRNA clusters. RNA 20(12):1850–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muffato M, Louis A, Poisnel C-E, Crollius HR.. 2010. Genomicus: a database and a browser to study gene synteny in modern and ancestral genomes. Bioinformatics 26(8):1119–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani Y, Takeda H, Kohara Y, Morishita S.. 2007. Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res. 17(9):1254–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Near TJ, Parker SK, Detrich HW.. 2006. A genomic fossil reveals key steps in hemoglobin loss by the Antarctic icefishes. Mol Biol Evol. 23(11):2008–2016. [DOI] [PubMed] [Google Scholar]
- Neilsen CT, Goodall GJ, Bracken CP.. 2012. IsomiRs – the overlooked repertoire in the dynamic microRNAome. Trends Genet. 28(11):544–549. [DOI] [PubMed] [Google Scholar]
- Nelson JS. 2006. Fishes of the world. John Wiley & Sons; [Database]. [Google Scholar]
- van Nimwegen E. 2003. Scaling laws in the functional content of genomes. Trends Genet. 19:479–484. [DOI] [PubMed] [Google Scholar]
- Niwa R, Slack FJ.. 2007. The evolution of animal microRNA function. Curr Opin Genet Dev. 17(2):145–150. [DOI] [PubMed] [Google Scholar]
- Ohno S. 1970. Evolution by gene duplication. Heidelberg (Berlin: ): Springer. [Google Scholar]
- Okamura K, Phillips MD, Tyler DM, Duan H, Chou Y, Lai EC.. 2008. The regulatory activity of microRNA species has substantial influence on microRNA and 3′ UTR evolution. Nat Struct Mol Biol. 15(4):354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Recalde O, Goikoetxea A, Hore TA, Todd EV, Gemmell NJ.. 2020. The genetics and epigenetics of sex change in fish. Annu Rev Anim Biosci. 8(1):47–69. [DOI] [PubMed] [Google Scholar]
- Pase L, Layton JE, Kloosterman WP, Carradice D, Waterhouse PM, Lieschke GJ.. 2009. miR-451 regulates zebrafish erythroid maturation in vivo via its target gata2. Blood 113(8):1794–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier J, Braasch I, Batzel P, Cabau C, Montfort J, Nguyen T, Jouanno E, Berthelot C, Klopp C, Journot L, et al. 2017. Evolution of gene expression after whole-genome duplication: new insights from the spotted gar genome. J Exp Zoolog B Mol Dev Evol. 328(7):709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posadas DM, Carthew RW.. 2014. MicroRNAs and their roles in developmental canalization. Curr Opin Genet Dev. 27:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quah S, Hui JHL, Holland PWH.. 2015. A burst of miRNA innovation in the early evolution of butterflies and moths. Mol Biol Evol. 32(5):1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen KD, Simmini S, Abreu-Goodger C, Bartonicek N, Di Giacomo M, Bilbao-Cortes D, Horos R, Von Lindern M, Enright AJ, O’Carroll D.. 2010. The miR-144/451 locus is required for erythroid homeostasis. J Exp Med. 207(7):1351–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi V, Venkatesh B.. 2018. The divergent genomes of teleosts. Annu Rev Anim Biosci. 6:47–68. [DOI] [PubMed] [Google Scholar]
- Reza AMMT, Choi Y-J, Han SG, Song H, Park C, Hong K, Kim J-H.. 2019. Roles of microRNAs in mammalian reproduction: from the commitment of germ cells to peri-implantation embryos. Biol Rev Camb Philos Soc. 94(2):415–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A.. 2004. Identification of mammalian microRNA host genes and transcription units. Genome Res. 14(10A):1902–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka L, Howe DG, Ramachandran S, Toro S, Van Slyke CE, Bradford YM, Eagle A, Fashena D, Frazer K, Kalita P, et al. 2019. The Zebrafish Information Network: new support for non-coding genes, richer Gene Ontology annotations and the Alliance of Genome Resources. Nucleic Acids Res. 47(D1):D867–D873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schartl M, Walter RB, Shen Y, Garcia T, Catchen J, Amores A, Braasch I, Chalopin D, Volff J-N, Lesch K-P, et al. 2013. The genome of the platyfish, Xiphophorus maculatus, provides insights into evolutionary adaptation and several complex traits. Nat Genet. 45(5):567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedel JM, Klemm SL, Zheng Y, Sahay A, Blüthgen N, Marks DS, Oudenaarden A van.. 2015. MicroRNA control of protein expression noise. Science 348(6230):128–132. [DOI] [PubMed] [Google Scholar]
- Sempere LF, Cole CN, Mcpeek MA, Peterson KJ.. 2006. The phylogenetic distribution of metazoan microRNAs: insights into evolutionary complexity and constraint. J Exp Zoolog B Mol Dev Evol. 306B(6):575–588. [DOI] [PubMed] [Google Scholar]
- Shang R, Baek SC, Kim K, Kim B, Kim VN, Lai EC.. 2020. Genomic clustering facilitates nuclear processing of suboptimal pri-miRNA loci. Mol Cell. 78(2):303–316.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu-Gruttadauria J, Xiao Y, Gebert LF, MacRae IJ.. 2019. Beyond the seed: structural basis for supplementary microRNA targeting by human Argonaute2. EMBO J. 38(13):e101153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Wang K, Yang P.. 2017. The evolution of plant microRNAs: insights from a basal eudicot sacred lotus. Plant J. 89(3):442–457. [DOI] [PubMed] [Google Scholar]
- Siddique BS, Kinoshita S, Wongkarangkana C, Asakawa S, Watabe S.. 2016. Evolution and distribution of teleost myomiRNAs: functionally diversified myomiRs in teleosts. Mar Biotechnol. 18(3):436–447. [DOI] [PubMed] [Google Scholar]
- Simakov O, Marlétaz F, Yue J-X, O’Connell B, Jenkins J, Brandt A, Calef R, Tung C-H, Huang T-K, Schmutz J, et al. 2020. Deeply conserved synteny resolves early events in vertebrate evolution. Nat Ecol Evol. 4:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Kuraku S, Holt C, Sauka-Spengler T, Jiang N, Campbell MS, Yandell MD, Manousaki T, Meyer A, Bloom OE, et al. 2013. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat Genet. 45(4):415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol NS, Ambros V.. 2005. Mesodermally expressed Drosophila microRNA-1 is regulated by Twist and is required in muscles during larval growth. Genes Dev. 19(19):2343–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somel M, Liu X, Tang L, Yan Z, Hu H, Guo S, Jiang X, Zhang X, Xu G, Xie G, et al. 2011. MicroRNA-driven developmental remodeling in the brain distinguishes humans from other primates. PLoS Biol. 9(12):e1001214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Wu J, Liang J, Schnable JC, Yang W, Cheng F, Wang X.. 2015. Impacts of whole-genome triplication on miRNA evolution in Brassica rapa. Genome Biol Evol. 7(11):3085–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Gao B, Zhou M, Wang Z, Zhang F, Deng J, Li X.. 2013. Comparative genomic analysis reveals evolutionary characteristics and patterns of microRNA clusters in vertebrates. Gene 512(2):383–391. [DOI] [PubMed] [Google Scholar]
- Sun K, Lai EC.. 2013. Adult-specific functions of animal microRNAs. Nat Rev Genet. 14(8):535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan GC, Chan E, Molnar A, Sarkar R, Alexieva D, Isa IM, Robinson S, Zhang S, Ellis P, Langford CF, et al. 2014. 5′ isomiR variation is of functional and evolutionary importance. Nucleic Acids Res. 42(14):9424–9435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani S, Kuraku S, Sakamoto H, Inoue K, Kusakabe R.. 2013. Developmental expression and evolution of muscle-specific microRNAs conserved in vertebrates. Evol Dev. 15(4):293–304. [DOI] [PubMed] [Google Scholar]
- Taylor JS, Braasch I, Frickey T, Meyer A, de Peer YV.. 2003. Genome duplication, a trait shared by 22,000 species of ray-finned fish. Genome Res. 13(3):382–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer Y, Maere S, Meyer A.. 2009. The evolutionary significance of ancient genome duplications. Nat Rev Genet. 10(10):725–732. [DOI] [PubMed] [Google Scholar]
- Vaz C, Wee CW, Lee GPS, Ingham PW, Tanavde V, Mathavan S.. 2015. Deep sequencing of small RNA facilitates tissue and sex associated microRNA discovery in zebrafish. BMC Genomics 16:950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainwright EN, Jorgensen JS, Kim Y, Truong V, Bagheri-Fam S, Davidson T, Svingen T, Fernandez-Valverde SL, McClelland KS, Taft RJ, et al. 2013. SOX9 regulates microRNA miR-202-5p/3p expression during mouse testis differentiation. Biol Reprod. 89(2):34. [DOI] [PubMed] [Google Scholar]
- Wainwright PC, Longo SJ.. 2017. Functional innovations and the conquest of the oceans by Acanthomorph fishes. Curr Biol. 27(11):R550–R557. [DOI] [PubMed] [Google Scholar]
- Wang Y, Luo J, Zhang H, Lu J.. 2016. microRNAs in the same clusters evolve to coordinately regulate functionally related genes. Mol Biol Evol. 33(9):2232–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnefors M, Eyre-Walker A.. 2011. The accumulation of gene regulation through time. Genome Biol Evol. 3:667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler BM, Heimberg AM, Moy VN, Sperling EA, Holstein TW, Heber S, Peterson KJ.. 2009. The deep evolution of metazoan microRNAs. Evol Dev. 11(1):50–68. [DOI] [PubMed] [Google Scholar]
- Wolf YI, Novichkov PS, Karev GP, Koonin EV, Lipman DJ.. 2009. The universal distribution of evolutionary rates of genes and distinct characteristics of eukaryotic genes of different apparent ages. Proc Natl Acad Sci U S A. 106(18):7273–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe K. 2000. Robustness – it’s not where you think it is. Nat Genet. 25(1):3–4. [DOI] [PubMed] [Google Scholar]
- Woltering JM, Durston AJ.. 2006. The zebrafish hoxDb cluster has been reduced to a single microRNA. Nat Genet. 38(6):601–602. [DOI] [PubMed] [Google Scholar]
- Wootton RJ, Smith C.. 2014. Reproductive biology of teleost fishes. John Wiley & Sons, Ltd. [Google Scholar]
- Wu C-I, Shen Y, Tang T.. 2009. Evolution under canalization and the dual roles of microRNAs—a hypothesis. Genome Res. 19(5):734–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong P, Schneider RF, Hulsey CD, Meyer A, Franchini P.. 2019. Conservation and novelty in the microRNA genomic landscape of hyperdiverse cichlid fishes. Sci Rep. 9(1):13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S Jr, Maurin T, Robine N, Rasmussen KD, Jeffrey KL, Chandwani R, Papapetrou EP, Sadelain M, O’Carroll D, Lai EC.. 2010. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc Natl Acad Sci U S A. 107(34):15163–15168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, Lee-Messer C, Dolmetsch RE, Tsien RW, Crabtree GR.. 2011. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476(7359):228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liu W, Jin Y, Jia P, Jia K, Yi M.. 2017. MiR-202-5p is a novel germ plasm-specific microRNA in zebrafish. Sci Rep. 7:7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Liao Y, Tang L.. 2019. MicroRNA-34 family: a potential tumor suppressor and therapeutic candidate in cancer. J Exp Clin Cancer Res. 38:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Meyers BC, Cai C, Xu W, Ma J.. 2015. Evolutionary patterns and coevolutionary consequences of miRNA genes and microRNA targets triggered by multiple mechanisms of genomic duplications in soybean. Plant Cell 27(3):546–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D.. 2007. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 129(2):303–317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in the published article (and its supplementary files) and/or deposited in NCBI.