Abstract
Bacteria regulate their metabolism to adapt and survive adverse conditions, in particular to stressful downshifts in nutrient availability. These shifts trigger the so-called stringent response, coordinated by the signaling molecules guanosine tetra and pentaphosphate collectively referred to as (p)ppGpp. In Escherichia coli, accumulation of theses alarmones depends on the (p)ppGpp synthetase RelA and the bifunctional (p)ppGpp synthetase/hydrolase SpoT. A tight regulation of these intracellular activities is therefore crucial to rapidly adjust the (p)ppGpp levels in response to environmental stresses but also to avoid toxic consequences of (p)ppGpp over-accumulation. In this study, we show that the small protein NirD restrains RelA-dependent accumulation of (p)ppGpp and can inhibit the stringent response in E. coli. Mechanistically, our in vivo and in vitro studies reveal that NirD directly binds the catalytic domains of RelA to balance (p)ppGpp accumulation. Finally, we show that NirD can control RelA activity by directly inhibiting the rate of (p)ppGpp synthesis.
Research organism: E. coli
Introduction
Bacteria have evolved numerous molecular mechanisms to detect and cope with environmental stress, including the use of nucleotide-based signaling pathways to efficiently coordinate cellular processes and provide a fast response. Among these signaling pathways, the stringent response is a general stress response that is mediated by the accumulation of the nucleotides guanosine 5′-diphosphate 3′-diphosphate (ppGpp) and guanosine 5′-triphosphate 3′-diphosphate (pppGpp), collectively known as (p)ppGpp (Potrykus and Cashel, 2008). These alarmones allow bacteria to rapidly respond and adapt to various conditions of nutritional and environmental stress by affecting gene expression and metabolism. In Gram-negative bacteria, (p)ppGpp binds RNA polymerase, thereby altering its promoter selectivity, which results in genome-wide transcriptional reprograming (Durfee et al., 2008; Ross et al., 2013; Ross et al., 2016; Sanchez-Vazquez et al., 2019; Traxler et al., 2008). Additionally, (p)ppGpp also binds directly and alters the activity of several enzymes including DNA primase, translation factors, lysine decarboxylase, and polyphosphate kinase (Corrigan et al., 2016; Kanjee et al., 2012; Wang et al., 2019a; Zhang et al., 2018). Importantly, this rewiring of cell physiology also appears to play a critical role in the regulation of bacterial virulence, survival during host invasion, and antibiotic resistance and tolerance (Hauryliuk et al., 2015; Hengge, 2020; Irving et al., 2021; Potrykus and Cashel, 2008).
In Escherichia coli, (p)ppGpp synthesis and hydrolysis are regulated by RelA and SpoT, which are members of RelA/SpoT homologue (RSH) (Atkinson et al., 2011) enzymes and share similar domain architecture. The enzymatic N-terminal half (NTD) has the synthetase (SYN) and the hydrolase (HYD) domains, and the C-terminal half (CTD) of the protein contains conserved domains, which play a critical role in sensing and transducing stress signals to the catalytic domains (Gratani et al., 2018; Hauryliuk et al., 2015; Hogg et al., 2004; Mechold et al., 2002; Pausch et al., 2020; Takada et al., 2020; Tamman et al., 2020). SpoT is a bifunctional enzyme with both hydrolytic and synthetic activities while RelA is a monofunctional (p)ppGpp synthetase. Indeed, despite close sequence similarity between RelA and SpoT, RelA maintains a ‘pseudo’-hydrolase domain that is structurally conserved but enzymatically inactive. The regulation of RelA and SpoT activities represents an obvious checkpoint for (p)ppGpp levels which reveals many complexities (reviewed in Irving and Corrigan, 2018; Ronneau and Hallez, 2019).
Activation of the (p)ppGpp synthetase RelA occurs via a ribosomal mechanism in response to amino acid starvation or other stressful conditions, which ultimately result in amino acid starvation, for example, fatty acid starvation, that leads to lysine starvation (Haseltine and Block, 1973; Sinha et al., 2019). Under this condition, deacylated tRNAs accumulate and RelA interacts with uncharged tRNA at the vacant ribosomal A-site causing the activation of its (p)ppGpp synthetic activity (Arenz et al., 2016; Brown et al., 2016; Loveland et al., 2016; Winther et al., 2018). Inversely, RelA is suggested to be inhibited by intra- and/or inter-molecular interactions under replete conditions (Gropp et al., 2001; Turnbull et al., 2019; Yang and Ishiguro, 2001), even though it retains residual activity (Murray and Bremer, 1996). Importantly, the hydrolysis function of SpoT provides an opposing activity that is crucial for balancing the basal activity of RelA. Indeed, disruption of the spoT gene results in a toxic accumulation of (p)ppGpp and is therefore lethal (Xiao et al., 1991).
The regulation of SpoT activities involves sources of nutritional stress other than amino acid starvation. These include the deficiency of fatty acid (Seyfzadeh et al., 1993), phosphate (Spira et al., 1995), carbon (Xiao et al., 1991), and iron (Vinella et al., 2005). Interaction of SpoT with other factors seems to regulate the catalytic balance between the synthetic and hydrolytic activities. During fatty acid starvation, the accumulation of (p)ppGpp required a specific interaction between the acyl carrier protein and SpoT (Battesti and Bouveret, 2006). Similarly, the binding of the protein YtfK with the catalytic domains of SpoT triggers (p)ppGpp accumulation during phosphate or fatty acid starvation (Germain et al., 2019). In addition, stimulation of the SpoT hydrolase activity is driven through direct interaction with the anti-σ70 factor Rsd upon carbon downshift (Lee et al., 2018).
Given the importance of (p)ppGpp in stress survival, virulence, and antibiotic tolerance, we have designed a genetic assay for the identification of new protein candidates that can modulate (p)ppGpp homeostasis in E. coli. We thereby discovered that overproduction of NirD, the small subunit of the nitrite reductase, decreases intracellular levels of (p)ppGpp. Importantly, our results show that overexpression of nirD limits RelA-dependent accumulation of (p)ppGpp in vivo and can prevent activation of the stringent response during amino acid starvation. Moreover, our results show that NirD is not involved in (p)ppGpp breakdown. Rather we convincingly show that, mechanistically, NirD inhibits the (p)ppGpp synthetase activity of RelA through a specific and direct interaction with the catalytic domains of RelA in vivo and in vitro.
Results
Identification of genes counteracting the toxicity of (p)ppGpp over-accumulation mediated by RelA
At high concentrations, (p)ppGpp becomes a potent inhibitor of bacterial cell growth. Indeed, rising (p)ppGpp levels by overexpression of the relA gene has been shown to inhibit E. coli growth in amino acid-rich media (Schreiber et al., 1991). To gain further insights into (p)ppGpp homeostasis, we developed a screening assay to identify genes that in multiple copies could reverse this toxicity (Figure 1A). In this study, the toxic accumulation of (p)ppGpp is achieved by induction of the relA gene cloned in the low-copy-number plasmid pBbS2k under the control of the anhydrotetracycline (aTc)-inducible Ptet promoter (Figure 1A). E. coli MG1655 cells harboring the pBbS2k-relA were transformed with a pooled collection of plasmids obtained from the ASKA library containing almost all E. coli K-12 genes (Kitagawa et al., 2005), each cloned into the high-copy-number vector pCA24N downstream of the isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible PT5-lac promoter (Figure 1A). This genetic screen led us to identify 88 clones that repetitively grow on non-permissive conditions. Sequence analysis of the pCA24N derivatives from these 88 clones enabled the identification of four different genes. The spoT, gppA, and nirD genes were predominant with each one accounting for almost a third of the genes carried by the sequenced plasmids (33.0, 36.4, and 29.5%, respectively) while uhpA was only identified once (1.1%). Our observations that spoT and gppA revert toxicity associated to (p)ppGpp accumulation is consistent with previous observations (Germain et al., 2019; Murray and Bremer, 1996; Sanyal and Harinarayanan, 2020). Considering the gppA and spoT genes as internal controls of the screen, we further focused our work on nirD.
Figure 1. NirD can limit the RelA-dependent accumulation of (p)ppGpp.
(A) Schematic representation of the genetic assay using ASKA library to identify genes that in multiple copies would suppress the growth defect associated with toxic over-accumulation of (p)ppGpp. Question marks denote a putative candidate that limits RelA-dependent accumulation of (p)ppGpp. (B) NirD suppresses the growth defect associated with RelA overproduction. Wild-type E. coli MG1655 cells were co-transformed with the plasmid derivatives pBbS2k-relA and pEG25-nirD inducible by anhydrotetracycline (aTc) and isopropyl β-D-1-thiogalactopyranoside (IPTG), respectively. Serial dilutions of stationary-phase cultures were spotted on NA plates containing the indicated concentrations of inducers. Additional controls and inducer concentrations are shown in Figure 1—figure supplement 1A. The results are representative of three independent experiments with similar results. (C, D) In vivo (p)ppGpp dynamic in E. coli MG1655 cells carrying pBbS2k-relA and pEG25-nirD after consecutive expression of the relA and nirD genes using 100 ng/mL aTc and 1 mM IPTG, respectively. Nucleotides extracted from samples collected at the indicated times were separated by thin layer chromatography. The autoradiogram (C) is representative of three independent experiments, and the curves of the relative levels of (p)ppGpp (D) are represented as the means of the three independent experiments, the error bars depicting the SDs.

Figure 1—figure supplement 1. NirD can inhibit RelA-dependent (p)ppGpp synthesis.

NirD balances (p)ppGpp level produced by RelA
nirD encodes the small subunit of the cytoplasmic nitrite reductase (Harborne et al., 1992; Peakman et al., 1990a) and has never been linked to the (p)ppGpp metabolism. We first confirmed the result obtained with the high-copy-number vector pCA24N-nirD from the ASKA library by re-cloning the coding region of nirD (untagged) in a more suitable physiological plasmid harboring a tight IPTG-inducible PT5-lac promoter (pEG25). Indeed, as shown in Figure 1B, NirD can robustly overcome the toxicity associated to (p)ppGpp over-accumulation mediated by RelA. To further assess the role of NirD in the stringent response, we monitored (p)ppGpp levels in vivo after consecutive inductions of RelA and NirD in a double expression system assay. As shown in Figure 1C, D, induction of RelA is associated with a massive accumulation of (p)ppGpp, which is sharply stopped and then decreased upon NirD induction.
The rapid (p)ppGpp decay observed following NirD induction raises the possibility that NirD may trigger SpoT-dependent (p)ppGpp hydrolysis. However, NirD still suppresses toxicity associated to RelA overproduction when the double expression assay experiment is performed in a ΔrelA spoT background (Figure 1—figure supplement 1A). These results show that the hydrolase activity of SpoT is not involved in the reduced (p)ppGpp accumulation observed upon NirD induction. Importantly, overexpression of nirD in a ΔrelA strain failed to phenocopy the well-known amino acid auxotrophic characteristic of a ppGpp0 strain (ΔrelA spoT mutant) (Xiao et al., 1991), strongly arguing that NirD does not act as a small alarmone hydrolase (Figure 1—figure supplement 1B). Taken together, our results show that NirD impairs RelA-dependent (p)ppGpp synthesis in E. coli.
NirD can prevent the stringent response in E. coli
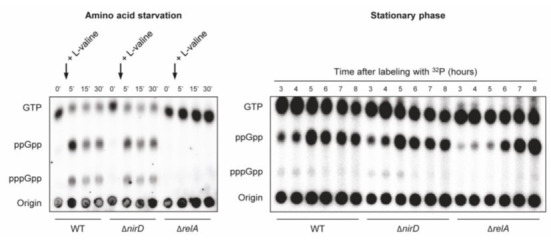
Since NirD can efficiently overcome toxicity associated to artificial level of (p)ppGpp when RelA is overproduced (Figure 1B), we next address whether similar effect is observed during physiological condition known to activate RelA. In natural environment, RelA synthesizes the alarmones in response to amino acid deprivation. Indeed during amino acid starvation, deacylated tRNAs accumulate and RelA interacts with uncharged tRNA at the vacant ribosomal A-site causing the activation of its (p)ppGpp synthetic activity (Arenz et al., 2016; Brown et al., 2016; Loveland et al., 2016; Winther et al., 2018). RelA activity is therefore required to survive under amino acid starvation, and as a result a ΔrelA mutant is not able to grow in the presence of 1 mM serine, methionine, and glycine (SMG), which induces isoleucine starvation (Uzan and Danchin, 1978). Interestingly, we observed that overexpression of nirD in wild-type (WT) strain phenocopies the growth defect of a ΔrelA mutant on SMG plate (Figure 2A), suggesting that nirD also prevents (p)ppGpp accumulation from endogenous RelA under these conditions. To further address this possibility, we monitored the dynamics of (p)ppGpp levels under amino acid starvation upon the addition of L-valine (which also induces isoleucine starvation). As shown in Figure 2B, C, and as previously observed (p)ppGpp rapidly increases in WT strain and reaches maximum levels within 5 min (Cashel and Gallant, 1969). Remarkably, when NirD is induced prior to amino acid starvation, no significant accumulation of (p)ppGpp is detected (Figure 2B, C). Taken together, our results show that once induced NirD abolishes the stringent response in E. coli.
Figure 2. NirD inhibits accumulation of (p)ppGpp upon amino acid starvation.

(A) NirD inhibits cell growth on M9-glucose minimal medium supplemented with serine, methionine, and glycine (SMG). Stationary-phase cultures of wild-type E. coli MG1655 strain harboring pEG25-nirD or pEG25-spoT were spotted on NA and SMG plates containing the indicated concentrations of isopropyl β-D-1-thiogalactopyranoside (IPTG). The results are representative of three independent experiments. (B, C) Exponentially growing cells of wild-type E. coli MG1655 harboring pEG25 or pEG25-nirD were challenged for amino acid starvation by addition of 500 µg/mL of L-valine. nirD is induced by the addition of 1 mM of IPTG. Nucleotides were extracted and separated by thin layer chromatography. The autoradiogram (B) is representative of three independent experiments, and the curves of the relative levels of (p)ppGpp (C) are represented as the means of the three independent experiments, the error bars depicting the SDs.
NirD functionally interacts with RelA in vivo
Regulation of RSH activities through protein–protein interactions has been previously reported (Battesti and Bouveret, 2006; Germain et al., 2019; Hahn et al., 2015; Karstens et al., 2014; Krüger et al., 2020; Lee et al., 2018; Peterson et al., 2020; Raskin et al., 2007; Ronneau et al., 2016; Ronneau et al., 2019; Wout et al., 2004). We therefore tested whether NirD is able to interact with RelA in vivo using a bacterial two-hybrid (BTH) assay (Karimova et al., 1998). For that purpose, the complementary T18 and T25 domains of Bordetella pertussis adenylate cyclase (CyA) were fused to the N-terminus of NirD and RelA proteins using the two compatible vectors pUT18C and pKT25, respectively. When the E. coli BTH101 strain (cya deficient) is co-transformed with plasmids harboring T18 and T25 fusions, NirD exhibited a strong interaction in vivo with RelA (Figure 3A). However, despite the strong sequence homologies between the paralogous RelA and SpoT proteins (Metzger et al., 1989), no physical interaction is observed between SpoT and NirD in vivo by BTH assay (Figure 3A). This result therefore supports a strong specificity of the interaction between NirD and RelA.
Figure 3. NirD can inhibit RelA-dependent accumulation through physical interaction in vivo.
(A) Bacterial two-hybrid assay using E. coli BTH101 cells co-transformed with plasmid derivatives pKT25-relA or pKT25-spoT and pUT18C-nirD or pUT18C-nirDE50K. Stationary-phase cultures were spotted on NA plates containing X-Gal as a blue color reporter for positive interaction. The bars showing β-galactosidase activity are represented as the means of three independent experiments, and the error bars depict the SDs. (B) Growth assay of E. coli MG1655 cells co-transformed with the plasmid derivatives pBbS2k-relA and pEG25-nirD or pEG25-nirDE50K inducible by anhydrotetracycline (aTc) and isopropyl β-D-1-thiogalactopyranoside (IPTG), respectively. Serial dilutions of stationary-phase cultures were spotted on NA plates containing the indicated concentrations of inducers. The results are representative of three independent experiments. (C, D) In vivo (p)ppGpp level dynamic in E. coli MG1655 cells carrying pBbS2k-relA and pEG25-nirD or pEG25-nirDE50K after successive inductions of expression of the relA and nirD or nirDE50K genes using 100 ng/mL aTc and 1 mM IPTG, respectively. Nucleotides extracted from samples collected at the indicated times were separated by thin layer chromatography. The autoradiogram (C) is representative of three independent experiments, and the curves of the relative levels of (p)ppGpp (D) are represented as the means of the three independent experiments, the error bars depicting the SDs. (E) Growth curves of wild-type (WT) cells, ΔnirD, and nirDE50K mutants under anaerobiosis in M9-glucose minimal medium without amino acids (left panel) or supplemented with 1 mM serine, methionine, and glycine (SMG) (right panel) (see Materials and methods). The growth curves are representative of at least three independent experiments, and the Δ(lag phase) are represented as the means (± SD) of the independent experiments.

Figure 3—figure supplement 1. NirDE50K functionally interacts with NirB.

To further assess the significance of the NirD-RelA interaction in vivo and its role in (p)ppGpp homeostasis, we randomly mutagenized nirD and searched for defective mutants unable to interact with RelA. One of such mutants that had charge reversal amino acid substitution, glutamate 50 to lysine (E50K), displayed compromised interaction with RelA (Figure 3A). Importantly, we confirm that both T18-NirD and T18-NirDE50K recombinant proteins were correctly expressed and folded as shown by their ability to strongly interact with the catalytic subunit of the nitrite reductase NirB (Figure 3—figure supplement 1) (Harborne et al., 1992). Importantly and on the contrary to the results obtained with the WT copy of nirD, we observed that overexpression of nirDE50K is not able to suppress toxicity associated to RelA overproduction on plate (Figure 3B). This result predicts that breaking the interaction with RelA abolishes the inhibitory effect of NirD on (p)ppGpp accumulation. Indeed, as shown in Figure 3C, D, nirDE50K failed to stop the massive accumulation of (p)ppGpp observed in vivo after RelA overproduction as compared to the fast decay of (p)ppGpp observed when the WT copy of nirD is expressed (Figure 3C and D).
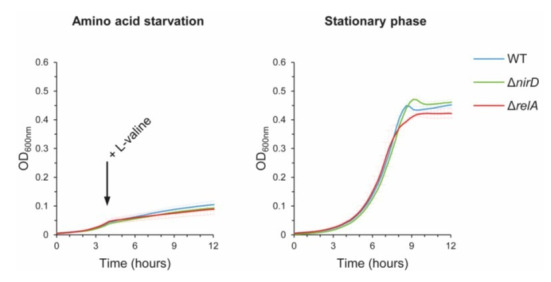
nirD is a second gene of a four-gene operon (nirBDC-cysG) strongly induced under anaerobic growth (Harborne et al., 1992; Peakman et al., 1990b). Interestingly, we observed that ΔnirD mutant presents an extended growth recovery in anaerobic SMG liquid medium consistent with a toxic over-accumulation of (p)ppGpp in this strain (Figure 3E). Indeed, a chromosomal encoded nirDE50K mutant, which is functional for the nitrite reductase activity (Figure 3—figure supplement 1C) but failed to interact with RelA, displays the same growth defect, suggesting that the NirD-RelA interaction is important to adjust proper (p)ppGpp level under this specific growth condition. Taken together, our results show that a specific functional interaction between RelA and NirD can lower RelA-dependent accumulation of (p)ppGpp in vivo.
NirD targets the catalytic N-terminal region of RelA
RelA consists of several protein domains that have been encompassed in two functional regions, the catalytic NTD and the regulatory CTD. The N-terminal region includes the enzymatically inactive (p)ppGpp hydrolase domain (HYD) as well as the functional (p)ppGpp synthetase domain (SYN), while the CTD comprises the Thr-tRNA synthetase, GTPase and SpoT domain (TGS), the alpha helical domain (AH), the zinc finger domain (ZFD), and finally the RNA recognition motif domain (RRM) (Figure 4A) (Brown et al., 2016; Loveland et al., 2016). To determine which of these domains are required for the interaction with NirD, we assayed the interaction between NirD and several truncated RelA proteins using the BTH assay.
Figure 4. NirD can interact with the catalytic N-terminal region of RelA.
(A) Schematic representation of RelA and its protein domains. (B) Bacterial two-hybrid assay using E. coli BTH101 cells co-transformed with plasmid derivatives pUT18C-nirD and pKT25 with the full-length or truncated relA gene as indicated. Stationary-phase cultures were spotted on NA plates containing X-Gal as a blue color reporter for positive interaction. The bars showing β-galactosidase activity are represented as the means of three independent experiments, and the error bars depict the SDs.

Figure 4—figure supplement 1. The RelAN-terminal-NirD interaction appears specific.

Truncated RelA proteins were fused to the T25 domain of the B. pertussis adenylate cyclase and transformed together with NirD fused to the T18 domain in a cya-deficient E. coli strain (BTH101). BTH analysis revealed that NirD interacts with the RelAN-terminal fusion comprising the catalytic domains (Figure 4B). Moreover, careful quantification of β-galactosidase activity revealed that none of the four domains encompassing the CTD of RelA is required for this interaction in vivo. However, truncated RelA fusion lacking either the synthetase domain (RelA1-181) or the ‘pseudo’ hydrolase domain (RelA181-744) failed to interact in vivo with NirD. Therefore, the catalytic domains of RelA seem necessary and sufficient to interact with NirD in vivo. The specificity of the interaction between NirD and RelA was further reinforced by the absence of interaction observed between NirD and SpoTN-terminal as well as NirDE50K and RelAN-terminal (Figure 4—figure supplement 1A). Interestingly, the C-terminal region of RelA is proposed to play a key role in the autoregulation of the enzymatic activity, and as a result the RelAN-terminal truncated protein is constitutively active (Schreiber et al., 1991). Given that NirD interacts with RelAN-terminal, we therefore tested the intriguing possibility that NirD may also be able to inhibit this dysregulated variant. Remarkably, as shown in Figure 4—figure supplement 1B, we observed that NirD but not NirDE50K inhibits the toxicity associated with (p)ppGpp over-accumulation mediated by the constitutively active RelAN-terminal. Taken together, our results show that NirD can inhibit RelA activity by interacting with its catalytic N-terminal region in vivo.
NirD directly binds RelA to inhibit the rate of (p)ppGpp synthesis in vitro
To confirm the in vivo interaction of NirD at the catalytic N-terminal region of RelA, we used different complementary in vitro methods. For that purpose, the RelA catalytic domains (RelAN-terminal), NirD, and NirDE50K were individually produced and purified by two consecutive chromatographies, affinity and size-exclusion (Figure 5—figure supplement 1A–C). We first performed size-exclusion chromatography (SEC) experiment separating RelAN-terminal and NirD alone or pre-mixed. As shown in Figure 5—figure supplement 1D, co-elution resulted in an earlier eluting peak, suggesting the formation of a stable complex. Indeed, SDS-PAGE analysis confirmed that the two proteins were co-eluted from the column (Figure 5—figure supplement 1E).
To further characterize the NirD-RelAN-terminal interaction, we then used biolayer interferometry (BLI), an in vitro approach for measuring biomolecular interactions. Biotinylated NirD was immobilized on streptavidin biosensors as ligand, and RelAN-terminal was used as the analyte. By adding RelA at concentrations ranging from 0.1 to 30 µM, a dose-response association is recorded and decreased during the dissociation step corresponding to the washing of the sensor (Figure 5A), showing that NirD directly interact with the catalytic N-terminal region of RelA. Moreover, by plotting the maximum binding response against the corresponding concentrations of RelA, we calculated a dissociation constant (KD) of 0.55 µM (± 0.053 µM) (Figure 5A). In addition, and in agreement with the BTH assays (Figure 4—figure supplement 1A), RelAN-terminal did not interact with the NirDE50K variant in vitro (Figure 5A). Finally, to further determine the thermodynamic parameters of the NirD-RelAN-terminal interaction, we performed isothermal titration calorimetry (ITC) experiment. The assay first confirmed the direct interaction between the two proteins with an estimated KD of 0.34 µM (Figure 5B), very similar to that estimated by BLI (Figure 5A). This reaction is a favorable spontaneous exothermic reaction with a ΔH kcal/mol of −3.63 ± 0.208 and is enthalpy driven at 25°C. On the contrary and consistent with BLI experiments, no heat exchange was recorded upon titration of RelAN-terminal by NirDE50K, further supporting the strong specificity of the interaction between NirD and RelA (Figure 5B). Collectively, these results show that NirD interacts directly and specifically with the catalytic domains of RelA in vitro.
Figure 5. NirD directly interact with the catalytic N-terminal region of RelA.
(A) Biolayer interferometric assay of RelAN-terminal on NirD or NirDE50K. Biotinylated NirD or NirDE50K were immobilized on streptavidin biosensors and probed with RelAN-terminal at concentrations ranging from 0.1 to 30 µM. The curves are represented as the means of the subtracted reference binding responses during association and dissociation from three experiments, the error bars depicting the SDs. The inset curve shows the specific biolayer interferometry (BLI) response (nm) 10 s before the end of association as a function of RelAN-terminal concentration. Data are represented as the means of the three experiments, the error bars depicting the SDs. (B, C) Isothermal titration calorimetry profiles corresponding to the binding of RelAN-terminal 30–300 µM NirD (B) or 300 µM NirDE50K (C) at 25°C. The upper panels show raw data for titration of NirD or NirDE50K with RelAN-terminal, and the lower panels show the integrated heats of binding obtained from the raw data. The data were fitted using a ‘One Set of Sites’ model in the PEAQ-ITC Analysis Software.

Figure 5—figure supplement 1. NirD can directly interact with the catalytic N-terminal region of RelA.

Finally, these results are in line with our in vivo results showing that NirD inhibits accumulation of (p)ppGpp from the constitutively active RelAN-terminal protein (Figure 4—figure supplement 1B) and therefore support a rather simple model where NirD would directly inhibit RelA synthetic activity through direct binding. To challenge this hypothesis, we performed in vitro pppGpp synthesis assay with purified proteins. We observed that RelAN-terminal protein could efficiently synthetize [32P]-pppGpp, but the addition of NirD strongly affected the synthetase activity in vitro. Indeed, 80% of GTP are converted to pppGpp within 60 min by RelA, whereas only 30% are converted when NirD is added (Figure 6A, B). Importantly, addition of the NirDE50K does not affect the RelA synthetic activity (Figure 6A, B). Finally, careful examination of the initial rates (calculated over the first 150 s) shows a concentration-dependent inhibition of RelA’s (p)ppGpp synthetic activity by NirD with a half-inhibition occurring at ∼1:1 molar ratio and a near complete inhibition for the highest tested concentration of NirD (1:16 molar ratio) (Figure 6C). Taken together, these results show that NirD can block pppGpp synthetase activity of RelA through a direct interaction with the catalytic domains.
Figure 6. NirD inhibits RelA activity in vitro.

(A, B) In vitro pppGpp formation by 1 µM RelAN-terminal in the presence or absence of 4 µM NirD or NirDE50K. Samples collected at the indicated times were separated by thin layer chromatography (TLC). The autoradiogram (A) is representative of three experiments, and the curves of the relative levels of pppGpp (B) are represented as the means of the three experiments, the error bars depicting the SDs. (C) Relative initial velocity of RelA as a function of NirD:RelA molar ratio. The in vitro formation of pppGpp by 1 µM RelAN-terminal was assayed in the presence or absence of NirD at concentrations ranging from 0.5 to 16 µM. Samples were collected every 30 s over a period of 150 s and separated by TLC to determine the relative initial velocity (estimated by linear regression using five data points). Results are represented as the means of three experiments, and the error bars depict the SDs.
Discussion
E. coli contains two long RSH enzymes, RelA and SpoT, that synthesize the alarmones (p)ppGpp. While SpoT is a bifunctional enzyme, RelA is monofunctional with a degenerated hydrolase domain, making SpoT the primary source of hydrolysis (Xiao et al., 1991). Therefore, a fine-tune regulation of the intracellular (p)ppGpp synthetic and hydrolytic activities in bacteria is crucial to rapidly adjust (p)ppGpp level in response to environmental stress but also to prevent toxic consequences due to (p)ppGpp over-accumulation (Xiao et al., 1991). Over the past decade, several molecular mechanisms regulating the activity of (p)ppGpp synthetase and/or hydrolase have been characterized (Battesti and Bouveret, 2006; Germain et al., 2019; Hahn et al., 2015; Karstens et al., 2014; Krüger et al., 2020; Lee et al., 2018; Peterson et al., 2020; Raskin et al., 2007; Ronneau et al., 2016; Ronneau et al., 2019; Wout et al., 2004). In this study, we uncovered an unprecedented mode of regulation that can prevent accumulation of (p)ppGpp in E. coli. This regulation implicates physical interaction in vivo and in vitro between the catalytic domains of RelA and the protein NirD, the small subunit of the cytoplasmic NADH-dependent nitrite reductase complex NirBD (Harborne et al., 1992). We show that NirD can functionally and directly inhibit the RelA (p)ppGpp synthetic activity. RelA and SpoT are paralogous proteins; however, despite strong sequence homologies and similar domain architecture we were not able to notice physical interaction between NirD and SpoT (Figure 3A and Figure 4—figure supplement 1A), supporting a specific physiological role of NirD in inhibiting RelA synthetic activity. The absence of a functional link between NirD and SpoT is further reinforced by the observation that NirD does not seem to lower basal (p)ppGpp level produced by the weak (p)ppGpp synthetase activity encoded by SpoT in the absence of RelA (Figure 1—figure supplement 1B).
Stimulation of RelA synthetase activity in E. coli is driven by stalled ribosomes upon amino acid starvation (Haseltine and Block, 1973; Wendrich et al., 2002; Winther et al., 2018). In addition, NtrC-dependent transcription of relA takes an important role in (p)ppGpp accumulation (Brown et al., 2014). Therefore, the identification of a potent intracellular inhibitor of RelA (p)ppGpp synthetase represents an additional level of complexity in the regulation to the current stringent model mediated by RelA. Our results show that the NirD protein level can play an important role for adjusting intracellular (p)ppGpp levels. Indeed, ectopic induction of NirD is sufficient to prevent accumulation of (p)ppGpp mediated by RelA in the absence of nutritional stress (Figure 1C, D). Moreover, we show that once expressed, nirD can also totally abolish activation of the stringent response during amino acid starvation (Figure 2C, D). The estimated concentration of RelA is in the range of 100–400 nM in E. coli (Li et al., 2014; Pedersen and Kjeldgaard, 1977). Importantly, we found in vitro that the NirD-RelA affinity is characterized by a KD in this concentration range (300–550 nM) (Figure 5A, B). Therefore, at concentrations above the KD, NirD would interact with RelA to catalytically inhibit its activity. nirD is a second gene of a four-gene operon (nirBDC-cysG) expressed under anaerobic growth (Harborne et al., 1992; Peakman et al., 1990b) and maximally induced in the presence of glucose and nitrate (Tyson et al., 1997; Wang and Gunsalus, 2000). Under these conditions, NirD is predicted to reach µM range (Khlebodarova et al., 2016) and regulation of RelA by NirD would therefore become physiologically relevant. Importantly, while NirB and NirD can form a stable complex needed for the NADH-dependent nitrite reductase activity, we observed that coexpression of nirB and nirD can still counteract RelA-dependent (p)ppGpp accumulation (Figure 3—figure supplement 1B). The cytoplasmic nitrite reductase NirBD has emerged as a key regulator of the nitric oxide (NO) level during nitrate respiration (Bulot et al., 2019). Indeed, by reducing nitrite generated during nitrate reduction, NirBD would limit the level of toxic nitrite and thus its further reduction to NO (Bulot et al., 2019). In addition, our results present evidence that NirD can also promote bacterial fitness by adjusting (p)ppGpp levels in response to amino acid starvation under anaerobic glucose fermentation, a condition where the nitrite reductase is expressed but where the substrate is missing (Figure 3E). This regulation is dependent on the RelA-NirD interaction since NirDE50K, which is not affected for the nitrite reductase activity (Figure 3—figure supplement 1C) but failed to interact with RelA (Figure 3A), does not enhance bacterial fitness in response to amino acid starvation under anaerobic glucose fermentation (Figure 3E). Therefore, our results suggest that NirD can link (p)ppGpp homeostasis to anaerobic metabolism in E. coli. Additional analyses are currently underway to further investigate the physiological roles of the RelA-NirD interaction in E. coli.
The CTD of RSH enzymes is assumed to play key roles in sensing nutrient starvation and adjusting the enzymatic state of the NTD. In that sense, the RelA structures on stalled ribosomes emphasized the role of CTD in sensing nutrient availability. Once bound to the ribosome, RelA adopts an open extended conformation, which is thought to release the autoinhibitory effect of the CTD on the NTD, therefore promoting (p)ppGpp synthesis (Arenz et al., 2016; Brown et al., 2016; Loveland et al., 2016). Interestingly, we observed that the NTD region of RelA is necessary and sufficient for NirD binding (Figure 4B and Figure 5). Moreover, our in vivo and in vitro results convincingly show that NirD can inhibit the (p)ppGpp synthesis from the constitutively active truncated RelAN-terminal (Figure 6 and Figure 4—figure supplement 1B), supporting the idea of a ribosome-independent inhibition of the stringent response. Therefore, rather than preventing activation of RelA our results support a simple model of regulation in which NirD directly interacts with the catalytic domains of RelA to inhibit the synthetic activity. Such inhibition might be mediated by directly introducing structural changes in the catalytic domains. Alternatively, NirD may interact with RelA to avoid nucleotide binding by masking the nucleotide-binding site. Structural and functional analyses are currently under investigation to test whether direct binding of NirD inhibits RelA activity.
Finally, (p)ppGpp has emerged as an important regulator of not only the bacterial stress response but also of many aspects of bacterial physiology including virulence, immune evasion, and antibiotic tolerance. Therefore, strategies to inhibit RelA activity and the subsequent production of (p)ppGpp represent an attractive approach for the success of antimicrobial therapy. Currently, there are only a limited number of synthetic inhibitors known to target RelA, mainly based on the design of (p)ppGpp analogues (Wexselblatt et al., 2012; Wexselblatt et al., 2013). Hence, our identification that NirD constitutes a natural potent inhibitor of RelA in vivo and in vitro could represent an important resource for the future design of new drug compounds. Importantly, it is also likely to be a fascinating molecular tool to further tackle the functional and structural determinant of the stringent response.
Materials and methods
Experimental model and subject details
The E. coli strains and plasmids used in this study are listed in Appendix 1—key resources table. Cells were grown in lysogeny broth (LB) broth (Miller, 1972) with agitation or on NA (Thermo Fisher Scientific, MA). For cells containing plasmid(s), the media were appropriately supplemented with the antibiotic(s) kanamycin (25 µg/mL), chloramphenicol (50 µg/mL), and/or ampicillin (50 µg/mL).
Plasmids construction
The plasmid derivatives were constructed by cloning into a plasmid a gene amplified by PCR from template DNA (chromosomal or plasmid) using the primers listed in Appendix 1—key resources table. The restriction enzymes as well as Shine-Dalgarno sequence for the pEG25 and pBbS2k plasmids are also indicated (Appendix 1—key resources table).
Multicopy suppressor screen
The genetic assay for the identification of new regulator of (p)ppGpp homeostasis in E. coli was carried out as follows. A pool of plasmids from the ASKA library (Kitagawa et al., 2005) was introduced by electroporation into E. coli MG1655 cells containing the pBbS2k-relA. The transformed cells were then spread on NA plates containing the appropriate antibiotics and the inducers aTc (0 or 100 ng/mL) and IPTG (0, 100, or 200 µM) for induction of pBbS2k-relA and pCA24N derivatives from the ASKA library, respectively. After 36 hr of incubation at 37°C, clones that were able to form colonies under these non-permissive conditions were chosen for the plasmid preparation and sequencing. The pool of plasmids from the ASKA library was obtained by Germain et al., 2019.
Growth assay
The ability of E. coli cells to grow under specific conditions was tested as follows. Single colonies were inoculated into 2 mL of LB broth, supplemented with the appropriate antibiotic(s), and cultured at 37°C until stationary phase (~8 hr). The cultures were then serially diluted and spotted on NA, M9-glucose minimal medium, or SMG plates (Supplementary file 1) containing the appropriate antibiotic(s) and plasmid inducer(s). For growth assays on M9-glucose minimal medium or SMG, the cultures were washed twice and diluted in PBS before being serially diluted and spotted on agar plates. The cells were finally incubated at 37°C overnight or ~36 hr depending on whether they were spotted on NA or minimal medium, respectively.
In vivo (p)ppGpp measurement
The levels of (p)ppGpp in the cells were determined as described by Germain et al., 2019. Briefly, 100 µL of 32P-labeled cell samples were taken at set times and added to 40 µL of ice-cold 21 M formic acid to stop the reactions. The mixtures were then placed on ice for 20 min before being centrifuged at 4°C for 20 min at 14,000 g to pellet cell debris and avoid them on chromatograms. 5 µL of each were loaded onto PEI-Cellulose thin layer chromatography (TLC) plates (Merck-Millipore) prior to ascending development with 1.5 M KH2PO4 solution (pH 3.4). Once fully developed, the TLC plates were dry at room temperature, separated from their upper part containing free 32P, and exposed overnight with a phosphor screen. The signals from the phosphor screen were then captured and quantified using an Amersham Typhoon Biomolecular Imager and ImageQuant TL 8.1 (GE Healthcare). We normalized the amount of (p)ppGpp to the total amount of G nucleotides observed in each sample, the total G being the sum of GTP, ppGpp, and pppGpp detected.
Regarding the preparation of the media, the labeling with 32P, and the induction of stress or starvation, they were carried out as follows. For overnight cultures, cells were grown in MOPS minimal medium (Neidhardt et al., 1974) supplemented with 0.2% glucose, 2 mM phosphate, and amino acids at 40 µg/mL (except for amino acid starvation experiment). Cultures were diluted 100 times in the same medium with 0.4 mM phosphate and incubated at 37°C with shaking. At an OD600nm of ~0.5, they were diluted to an OD600nm of 0.05, labeled with 150 µCi of 32P, and grew to an OD600nm of ~0.20. At this point and 30 min later, inducers (100 ng/mL aTc or 1 mM IPTG) or 500 µg/mL L-valine were added for gene overexpression or induction of isoleucine starvation, respectively.
Bacterial two-hybrid assay
In vivo protein-protein interactions were tested using the bacterial two-hybrid system (Karimova et al., 1998). Briefly, protein pairs were fused to the T18 and T25 domains of B. pertussis adenylate cyclase using the two compatible vectors pUT18C and pKT25, respectively. Co-transformed with the pairs of plasmids, single colonies of the cya-deficient strain BTH101 were inoculated into 2 mL of LB broth supplemented with ampicillin and kanamycin, and grown at 30°C to stationary phase (~12 hr). 5 µL of the cultures were then spotted on NA plates containing the two antibiotics and 40 µg/mL X-gal as a color reporter for β-galactosidase and adenylate cyclase activities. The cells were finally incubated at 30°C ~36 hr. To quantify the strength of the interaction, β-galactosidase activity was determined as described by Miller, 1992 using stationary phase culture (~12 hr).
Screening for loss of interaction by nirD random mutagenesis
nirD DNA coding sequence was mutagenized randomly through low-fidelity GoTaq polymerase amplification from pUT18C-nirD plasmid using 441 and 443 oligonucleotides (Appendix 1—key resources table). After 30 PCR cycles, amplified DNA was purified, diluted to 1:100,000 and further amplified for 30 cycles. Purified DNA were cloned into pUT18C plasmid. BTH101 cells harboring pKT25-relA were then transformed with the resulting pUT18C-nirD mutated library and screened on X-Gal plates. White colonies were selected and sequenced.
MG1655 nirDE50K construction
The chromosomal mutant strain nirDE50K was constructed by scarless mutagenesis using a two-step lambda Red system adapted from Blank et al., 2011. DNA fragments containing a chloramphenicol resistance marker and the meganuclease I-SceI recognition site were amplified by PCR using primers 629 and 630 with pWRG100 as template. The resulting PCR product was electroporated into MG1655 cells, which had induced lambda recombinase expression for 1 hr from plasmid pKD46 (Datsenko and Wanner, 2000). After 1 hr of phenotypic expression, the cells were plated on NA plates containing 25 µg/mL chloramphenicol. Selected colonies contained the nirD::cat I-SceI allele, which was subsequently transduced in MG1655 cells harboring the pWRG99 plasmid (Blank et al., 2011). The mutant allele nirDE50K was amplified from pEG25-nirDE50K using primers 655 and 656. The PCR product was then electroporated into MG1655 nirD::cat I-SceI expressing lambda recombinase from pWRG99 and after 1 hr of phenotypic expression, cells were serially diluted and plated on NA plates containing 100 µg/mL ampicillin and 1 µg/mL aTc. pWRG99 also encoded for the meganuclease I-SceI under the control of an aTc-inducible promoter. The proper integration of the nirDE50K was confirmed by diagnostic PCR and sequencing.
Growth curves
Growth of WT cells, ΔnirD, and nirDE50K mutants was measured under anaerobiosis as follows. Stationary-phase cultures were washed in PBS, diluted to an OD600nm of ~0.03 in M9-glucose minimal medium without amino acids or supplement with 1 mM SMG in Hungate tubes, and air was evacuated and replaced with N2. The cultures were then incubated at 37°C and growth was followed by measurement of OD600nm. Although at least three independent experiments were performed, representative growth curves are shown. Indeed, day-to-day variations in medium composition shifted the curves slightly, but the sample-to-sample comparisons were invariable.
For the functional test of the NADH-dependent nitrite reductase activity, the cultures were prepared as described above except that they were diluted in a defined medium supplemented with 140 mM glycerol and 100 mM nitrate. The defined medium is composed of Na2HPO4 (60 mM), KH2PO4 (22 mM), NaCl (8 mM), MgSO4 (1 mM), CaCl2 (100 µM), thiamine (1 µg/mL), sodium molybdate (5 µM), sodium selenite (1 µM), and FeSO4 (10 µM).
Protein expression and production
BL21 (DE3)-competent cells were transformed with the pEG25-relAN-terminal, pET28a(+)-nirD or pET28a(+)-nirDE50K and plated on selective NA. For RelAN-terminal production, several colonies were picked up and inoculated into 100 mL LB containing 100 µg/mL ampicillin. The cultures were grown with shaking at 30°C overnight. Overnight cultures were dispersed as inoculum of 20 mL aliquots into 1 L Terrific Broth media with 100 µg/mL ampicillin and shaken at 30°C until OD600nm ~0.5, then cells were induced with 0.5 mM IPTG and incubated at 30°C for 4 hr. For NirD and NirDE50K production, several colonies were picked up and inoculated into 100 mL LB containing 50 µg/mL kanamycin. The cultures were grown with shaking at 30°C overnight. Overnight cultures were dispersed as inoculum of 20 mL aliquots into 1 L LB media with 50 µg/mL kanamycin and shaken at 30°C until OD600nm ~0.5, then cells were induced with 0.5 mM IPTG and incubated at 30°C for 4 hr. Finally, cells were centrifuged at 9000 g for 20 min at 4°C. Dry cell pellet was stored at −80°C.
Protein purification
For NirD and NirDE50K purification, cells were suspended in lysis buffer (50 mM Tris-HCl [pH 8.0], 300 mM NaCl, 1 mM EDTA, 0.5 mg/mL lysozyme, 1 mM phenylmethylsulfonyl fluoride [PMSF], 20 µg/mL DNase, and 20 mM MgCl2), incubated for 1 hr at 4°C with gentle shaking, and then subjected to three cycles of French-press lysis steps. The soluble fraction was obtained by centrifugation for 20 min at 200,000 g. Recombinant proteins were purified by ion metal affinity chromatography using a 5 mL nickel (HiTrapHP) column on an ÄKTA pure 25 (GE Healthcare) pre-equilibrated in 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, and 10 mM imidazole (buffer A). After several washes in buffer A, His-tagged proteins were eluted in buffer A supplemented with 250 mM imidazole and directly desalted using HiPrep 26/10 Desalting column pre-equilibrated with buffer A. The desalted proteins were mixed with 0.2 mg/mL of TEV protease, incubated for 2 hr at 25°C, and then loaded onto a HisTrap column pre-equilibrated in buffer A to retain TEV, TRX, uncleaved proteins, and contaminants. Untagged NirD or NirDE50K were collected in the flow-through, concentrated on a Centricon (Millipore; cutoff of 3 kDa), and passed through a HiLoad 16/600 Superdex 200 column pre-equilibrated with 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, 2 mM β-mercaptoethanol, and 2% glycerol. The purity of NirD preparations was assessed by SDS-PAGE (Figure 5—figure supplement 1B, C).
For RelAN-terminal purification, cells were suspended in lysis buffer (50 mM Tris-HCl [pH 8.0], 500 mM NaCl, 10 mM imidazole, 2 mM β-mercaptoethanol, 0.5% CHAPS, 2% glycerol, 1 mM EDTA, 0.5 mg/mL lysozyme, 1 mM PMSF, 20 µg/mL DNase, and 20 mM MgCl2), incubated for 1 hr at 4°C with gentle shaking, and then subjected to three cycles of French-press lysis steps. The soluble fraction was obtained by centrifugation for 20 min at 200,000 g. Recombinant proteins were purified by ion metal affinity chromatography using a 5 mL nickel (HiTrapHP) column on an ÄKTA pure 25 (GE Healthcare) pre-equilibrated with equilibrium buffer (50 mM Tris-HCl [pH 8.0], 500 mM NaCl, 10 mM imidazole, 2 mM β-mercaptoethanol, and 2% glycerol), eluted in elution buffer (50 mM Tris-HCl [pH 8.0], 250 mM NaCl, 500 mM imidazole, 2 mM β-mercaptoethanol, and 2% glycerol), and directly desalted using HiPrep 26/10 Desalting column pre-equilibrated with buffer containing 50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 500 mM KCl, 2 mM β-mercaptoethanol, and 2% glycerol. Desalted proteins were concentrated on a Centricon (Millipore; cutoff of 10 kDa) and then subjected to SEC purification using a HiLoad 16/600 Superdex 200 column pre-equilibrated with 50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 500 mM KCl, 2 mM β-mercaptoethanol, and 2% glycerol. The purity of RelA preparations was assessed by SDS-PAGE (Figure 5—figure supplement 1A).
Protein-protein interaction analysis by SEC
Purified RelAN-terminal and NirD were either alone or pre-mixed in a 1:1 ratio (10 µM each) in buffer containing 50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 500 mM KCl, 2 mM β-mercaptoethanol, and 2% glycerol prior separation on a Superdex 200 10/300 GL using an ÄKTA pure 25 apparatus (GE Healthcare) pre-equilibrated with the same buffer. Elution fractions were then analyzed with SDS-PAGE.
Biolayer interferometry
NirD or NirDE50K were biotinylated using the EZ-Link NHS-PEG4-Biotin (Thermo Scientific) at 4°C for 2 hr. The reaction was stopped by removing the excess of the biotin using a Zeba Spin Desalting column (Thermo Scientific). BLI studies were performed at 25°C using the Blitz apparatus (FortéBio) with shaking at 2200 rpm with the following steps: 30 s baseline, 120 s association, and 120 s dissociation. Streptavidin biosensor tips (FortéBio) were hydrated with 0.350 mL of 50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 500 mM KCl, 2 mM β-mercaptoethanol, and 2% glycerol for 10 min and then loaded with 3 µM of biotinylated NirD or NirDE50K in the same buffer. The biosensors were then incubated with 10 µg/mL biocytin in interaction buffer (50 mM Tris-HCl [pH 8.0], 500 mM NaCl, 500 mM KCl, 2 mM β-mercaptoethanol, 2% glycerol, and 1 mg/mL) for 90 s to avoid the non-specific binding RelAN-terminal to the streptavidin biosensors. To study the binding of NirD or NirDE50K to RelAN-terminal, increasing concentrations RelAN-terminal (1–30 µM) in interaction buffer were used and the association and dissociation phases were monitored, respectively. In all experiments, the BLItz Pro Software performed a reference subtraction of the RelAN-terminal protein response on the uncoated biosensors for each tested concentration. The dissociation constant (KD), that is affinity of NirD to RelAN-terminal, was estimated using the GraphPad Prism 5.0 software on the basis of the steady-state-level responses in nm at equilibrium. The KD was estimated by plotting on x axis the different concentrations and the different responses of RelAN-terminal at the saturation (10 s before the end of the association) on the y axis. For KD calculation, a nonlinear regression fit for xy analysis was used and one site (specific binding) as a model [corresponding to the equation y=Bmax*x/(KD+x)].
Isothermal titration calorimetry
ITC was performed to determine the affinity between NirD and RelAN-terminal. In a typical setup, RelAN-terminal (30 µM) in buffer containing 50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 500 mM KCl, 2 mM β-mercaptoethanol, and 2% glycerol was placed in the cell, and NirD (or NirDE50K) ligand (300 μM in the same buffer) was placed in the titration syringe. All experiments were carried out at 25°C with a stirring speed of 750 rpm and a reference power of 5 µcal/s using the MicroCal PEAQ-ITC (Malvern Panalytical) with 19 injections (0.4 µL for the initial injection and 2 µL for the next 18). Each injection lasted for 4 s with a space of 150 s between each injection. ITC measures the energy released or absorbed by a chemical reaction, and the energy of the interaction is proportional to the amount of ligand that binds to the protein. These heat pulses are recorded for each injection and then integrated with respect to time and are normalized to the concentration of the reaction to generate kcal/mol versus the molar ratio (ligand/sample). The enthalpy of the reaction is obtained, and with a ‘One Set of Sites’ model in the PEAQ-ITC Analysis Software, the KD and stoichiometry is determined. The fitted offset option in the PEAQ-ITC Analysis Software was used to correct for the heat of the dilution.
pppGpp synthesis assay
The pppGpp synthetic activity of RelAN-terminal was assayed at 37°C in a buffer containing 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, 2 mM β-mercaptoethanol, 2% glycerol, and 15 mM Mg2+. Typically, reaction mixtures contained 8 mM ATP, 6 mM GTP and [α-32P]GTP, 1 µM RelAN-terminal with or without NirD (or NirDE50K) at concentrations ranging from 0.5 to 16 µM, and reactions were started by adding pre-warmed nucleotides to the protein(s). During the time course of the reaction, 20 µL samples were taken and added to 8 µL of ice-cold 21 M formic acid to stop the reactions. The time points were resolved by TLC, and radio-labeled nucleotides were quantified as described in the ‘In vivo (p)ppGpp measurement’ section.
Acknowledgements
This work was supported by the European Research Council starting grant (ERC StG) under the European Union’s Horizon 2020 and innovation program grant agreement no. 714934 ‘Stringency’ to EM. We thank Farida Seduk, Aurore Jacq-Bailly, Gaël Brasseur, Axel Magalon, and Benjamin Ezraty for advices and stimulating discussions for anaerobic experiments.
Appendix 1
Appendix 1—key resources table.
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| strain, strain background (Escherichia coli) | BTH101 | Karimova et al., 1998 doi: 10.1073/pnas.95.10.5752 | F– cya-99 araD139 galE15 galK16 rpsL(StrR) hsdR2 mcrA1 mcrB1 relA1 | |
| strain, strain background (E. coli) | BL21 (DE3) | New England Biolabs | B F– ompT gal | |
| strain, strain background (E. coli) | MG1655; WT | Lab collection | Wild-type | |
| strain, strain background (E. coli) | nirDE50K | This paper | MG1655 nirDE50K | |
| strain, strain background (E. coli) | ΔnirD | This paper | MG1655 nirD::FRT; P1 from KEIO collection (resistance cassette has been flipped out) (Baba et al., 2006) | |
| strain, strain background (E. coli) | ΔrelA | Germain et al., 2019 doi: 10.1038/s41467-019-13764-4 | MG1655 relA::FRT | |
| strain, strain background (E. coli) | ΔrelA spoT | This paper | MG1655 relA::FRT spoT207::cat; P1 transduction from CF1693 (Baba et al., 2006) in MG1655 relA::FRT | |
| recombinant DNA reagent | pBbS2k-rfp | Lee et al., 2011 doi: 10.1186/1754-1611-5-12 | KanR, Ptet, rfp | |
| recombinant DNA reagent | pBbS2k-relA | This paper | KanR, Ptet, relA; primers 16/17; Maisonneuve lab | |
| recombinant DNA reagent | pBbS2k-relA (sd4) | This paper | KanR, Ptet, sd4, relA; primers 163/17; Maisonneuve lab | |
| recombinant DNA reagent | pBbS2k-relAN-terminal | This paper | KanR, Ptet, relAN-terminal; primers 16/557; Maisonneuve lab | |
| recombinant DNA reagent | pEG25 | Germain et al., 2019 doi: 10.1038/s41467-019-13764-4 | AmpR, PT5-lac | |
| recombinant DNA reagent | pEG25-nirD | This paper | AmpR, PT5-lac, nirD; primers 386/387; Maisonneuve lab | |
| recombinant DNA reagent | pEG25-spoT (sd8U) | Germain et al., 2019 doi: 10.1038/s41467-019-13764-4 | AmpR, PT5-lac, sd8U, spoT | |
| recombinant DNA reagent | pEG25-nirDE50K | This paper | AmpR, PT5-lac, nirDE50K; primers 386/387; Maisonneuve lab | |
| recombinant DNA reagent | pEG25-nirD (WTsd) | This paper | AmpR, PT5-lac, WTsd, nirD; primers 555/387; Maisonneuve lab | |
| recombinant DNA reagent | pEG25-nirBD (WTsd) | This paper | AmpR, PT5-lac, WTsd, nirBD; primers 553/544; Maisonneuve lab | |
| recombinant DNA reagent | pEG25-relAN-terminal | Germain et al., 2019 doi: 10.1038/s41467-019-13764-4 | AmpR, PT5-lac, relAN-terminal | |
| recombinant DNA reagent | pKD46 | Datsenko and Wanner, 2000 doi: 10.1073/pnas.120163297 | AmpR, Para, λ Red | |
| recombinant DNA reagent | pKT25 | Karimova et al., 1998 doi: 10.1073/pnas.95.10.5752 | KanR | |
| recombinant DNA reagent | pKT25-zip | Karimova et al., 1998 doi: 10.1073/pnas.95.10.5752 | KanR, zip | |
| recombinant DNA reagent | pKT25-relA | Germain et al., 2019 doi: 10.1038/s41467-019-13764-4 | KanR, relA | |
| recombinant DNA reagent | pKT25-spoT | Germain et al., 2019 doi: 10.1038/s41467-019-13764-4 | KanR, spoT | |
| recombinant DNA reagent | pKT25-nirB | This paper | KanR, nirB; primers 617/618; Maisonneuve lab | |
| recombinant DNA reagent | pKT25-relA1-663 | This paper | KanR, relA1-663; primers 497/502; Maisonneuve lab | |
| recombinant DNA reagent | pKT25-relA1-580 | This paper | KanR, relA1-580; primers 497/501; Maisonneuve lab | |
| recombinant DNA reagent | pKT25-relA1-470 | This paper | KanR, relA1-470; primers 497/500; Maisonneuve lab | |
| recombinant DNA reagent | pKT25-relAN-terminal | This paper | KanR, relAN-terminal; primers 497/499; Maisonneuve lab | |
| recombinant DNA reagent | pKT25-relA1-181 | This paper | KanR, relA1-181; primers 497/498; Maisonneuve lab | |
| recombinant DNA reagent | pKT25-relA181-744 | This paper | KanR, relA181-744; primers 531/533; Maisonneuve lab | |
| recombinant DNA reagent | pKT25-spoTN-terminal | Germain et al., 2019 doi: 10.1038/s41467-019-13764-4 | KanR, relAN-terminal | |
| recombinant DNA reagent | pUT18C | Karimova et al., 1998 doi: 10.1073/pnas.95.10.5752 | AmpR | |
| recombinant DNA reagent | pUT18C-zip | Karimova et al., 1998 doi: 10.1073/pnas.95.10.5752 | AmpR, zip | |
| recombinant DNA reagent | pUT18C-nirD | This paper | AmpR, nirD; primers 441/443; Maisonneuve lab | |
| recombinant DNA reagent | pUT18C-nirDE50K | This paper | AmpR, nirDE50K; primers 441/443; Maisonneuve lab | |
| recombinant DNA reagent | pET28a(+) | Germain et al., 2019 doi: 10.1038/s41467-019-13764-4 | KanR, Trx, His | |
| recombinant DNA reagent | pET28a(+)-nirD | This paper | KanR, Trx, His, TEV, nirD; primers 558/544; Maisonneuve lab | |
| recombinant DNA reagent | pET28a(+)-nirDE50K | This paper | KanR, Trx, His, TEV, nirDE50K; primers 558/544; Maisonneuve lab | |
| recombinant DNA reagent | pWRG99 | Blank et al., 2011 doi: 10.1371/journal.pone.0015763 | pKD46 with Ptet, I-SceI | |
| recombinant DNA reagent | pWRG100 |
Karimova et al., 1998 doi: 10.1073/pnas.95.10.5752 doi: 10.1371/journal.pone.0015763 |
pKD3 with I-SceI recognition site | |
| sequence-based reagent | Primer 16 | This paper | CCCCGAATTCGTCGACTCAAGGAGGTTTTATAAATGGTTGCGGTAAGAAGTGCA; restriction enzyme EcoRI; Maisonneuve lab | |
| sequence-based reagent | Primer 17 | This paper | CCCCGGATCCCTAACTCCCGTGCAACCGAC; restriction enzyme BamHI; Maisonneuve lab | |
| sequence-based reagent | Primer 163 | This paper | CCCCGAATTCGTCGACTCAAGGATTAAATGGTTGCGGTAAGAAGTGCA; restriction enzyme EcoRI; Maisonneuve lab | |
| sequence-based reagent | Primer 386 | This paper | CCCCGAATTCGTCGACTCAAGGAGGTTTTATAAATGAGCCAGTGGAAAGACAT; restriction enzyme EcoRI; Maisonneuve lab | |
| sequence-based reagent | Primer 387 | This paper | CCCCGGATCCTTAACCGCGCAGCTGCACCA; restriction enzyme BamHI; Maisonneuve lab | |
| sequence-based reagent | Primer 441 | This paper | CCCCTCTAGAAATGAGCCAGTGGAAAGACAT; restriction enzyme XbaI; Maisonneuve lab | |
| sequence-based reagent | Primer 443 | This paper | CCCCGGTACCTTAACCGCGCAGCTGCACCA; restriction enzyme KpnI; Maisonneuve lab | |
| sequence-based reagent | Primer 497 | This paper | CCCCTCTAGAAATGGTTGCGGTAAGAAGTGC; restriction enzyme XbaI; Maisonneuve lab | |
| sequence-based reagent | Primer 498 | This paper | CCCCGGTACCGATGTTGGTACACTCTTTTG; restriction enzyme KpnI; Maisonneuve lab | |
| sequence-based reagent | Primer 499 | This paper | CCCCGGTACCTTCGTCGAGCATTTCGCCGG; restriction enzyme KpnI; Maisonneuve lab | |
| sequence-based reagent | Primer 500 | This paper | CCCCGGTACCGTTCGGCTGTTTCTGGGTGA; restriction enzyme KpnI; Maisonneuve lab | |
| sequence-based reagent | Primer 501 | This paper | CCCCGGTACCAAGTTGCTTCAGCGCGGCGG; restriction enzyme KpnI; Maisonneuve lab | |
| sequence-based reagent | Primer 502 | This paper | CCCCGGTACCGGCGGAGTAGCTCTCACCCC; restriction enzyme KpnI; Maisonneuve lab | |
| sequence-based reagent | Primer 531 | This paper | CCCCTCTAGAATACGCACCGCTGGCTAACCG; restriction enzyme XbaI; Maisonneuve lab | |
| sequence-based reagent | Primer 533 | This paper | CCCCGGTACCCTAACTCCCGTGCAACCGAC; restriction enzyme KpnI; Maisonneuve lab | |
| sequence-based reagent | Primer 544 | This paper | CCCCAAGCTTTTAACCGCGCAGCTGCACCA; restriction enzyme HindIII; Maisonneuve lab | |
| sequence-based reagent | Primer 553 | This paper | CCCCGAATTCAATAGAAAAGAAATCGAGGCAAAAATGAGCAAAGTCAGACTCGC; restriction enzyme EcoRI; Maisonneuve lab | |
| sequence-based reagent | Primer 555 | This paper | CCCCGAATTCAGTAACTCTGGTGGAGGACAACGCATGAGCCAGTGGAAAGACAT; restriction enzyme EcoRI; Maisonneuve lab | |
| sequence-based reagent | Primer 557 | This paper | CCCCGGATCCTTATTCGTCGAGCATTTCGCCGG; restriction enzyme BamHI; Maisonneuve lab | |
| sequence-based reagent | Primer 558 | This paper | CCCCGGATCCGAGAACCTGTACTTCCAATCAATGAGCCAGTGGAAAGACAT; restriction enzyme BamHI; Maisonneuve lab | |
| sequence-based reagent | Primer 617 | This paper | CCCCTCTAGAAATGAGCAAAGTCAGACTCGC; restriction enzyme XbaI; Maisonneuve lab | |
| sequence-based reagent | Primer 618 | This paper | CCCCGGTACCTCATGCGTTGTCCTCCACCA; restriction enzyme KpnI; Maisonneuve lab | |
| sequence-based reagent | Primer 629 | This paper | ATGAGCCAGTGGAAAGACATCTGCAAAATCGATGACATCCTGCCTGAAACCGCCTTACGCCCCGCCCTGC; Maisonneuve lab | |
| sequence-based reagent | Primer 630 | This paper | TTAACCGCGCAGCTGCACCACGCCGTCTTTCACTCGCGCTTCGTAATGTTCTAGACTATATTACCCTGTT; Maisonneuve lab | |
| sequence-based reagent | Primer 655 | This paper | ATGAGCCAGTGGAAAGACAT; Maisonneuve lab | |
| sequence-based reagent | Primer 656 | This paper | TTAACCGCGCAGCTGCACCA; Maisonneuve lab | |
| chemical compound, drug | [α32P]GTP | PerkinElmer | Catalog number: BLU006H250UC | |
| chemical compound, drug | 32P | PerkinElmer | Catalog number: NEX053H001MC | |
| chemical compound, drug | EZ-Link NHS-PEG4-Biotin | Thermo Scientific | Catalog number: 21330 | |
| software, algorithm | BLItz Pro Software | https://www.sartorius.com/en/products/protein-analysis | ||
| software, algorithm | MicroCal PEAQ-ITC Analysis Software | Malvern Panalytical | https://www.malvernpanalytical.com/fr/products/product-range/microcal-range/microcal-itc-range/microcal-peaq-itc | |
| software, algorithm | Prism 5.0 software | GraphPad | https://www.graphpad.com/scientific-software/prism/ | |
| other | HiLoad 16/600 Superdex 200 column | GE Healthcare | Catalog number: GE28-9893-35 | |
| other | HiPrep 26/10 Desalting column | GE Healthcare | Catalog number: GE17-5087-01 | |
| other | HisTrap column | GE Healthcare | Catalog number: GE17-5248-02 | |
| other | Streptavidin Biosensors | FortéBio | Catalog number: 18-5117 | |
| other | Superdex 200 10/300 GL | GE Healthcare | Catalog number: GE28-9909-44 | |
| other | Zeba Spin Desalting column | Thermo Scientific | Catalog number: 89882 |
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Etienne Maisonneuve, Email: emaisonneuve@imm.cnrs.fr.
Sophie Helaine, Harvard Medical School, United States.
Gisela Storz, National Institute of Child Health and Human Development, United States.
Funding Information
This paper was supported by the following grant:
H2020 European Research Council 714934 to Etienne Maisonneuve.
Additional information
Competing interests
No competing interests declared.
Author contributions
Conceptualization, Data curation, Formal analysis, Validation, Investigation, Visualization, Methodology, Writing - original draft, Writing - review and editing.
Data curation, Formal analysis, Methodology, Writing - review and editing.
Data curation, Formal analysis, Methodology, Writing - review and editing.
Conceptualization, Supervision, Validation, Methodology, Project administration, Writing - review and editing.
Conceptualization, Data curation, Supervision, Funding acquisition, Validation, Investigation, Writing - original draft, Project administration, Writing - review and editing.
Additional files
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files. Source data files have been provided for Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 3-figure supplement 1, Figure 4-figure supplement 1 and Figure 5-figure supplement 1.
References
- Arenz S, Abdelshahid M, Sohmen D, Payoe R, Starosta AL, Berninghausen O, Hauryliuk V, Beckmann R, Wilson DN. The stringent factor RelA adopts an open conformation on the ribosome to stimulate ppGpp synthesis. Nucleic Acids Research. 2016;44:6471–6481. doi: 10.1093/nar/gkw470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson GC, Tenson T, Hauryliuk V. The RelA/SpoT homolog (RSH) superfamily: distribution and functional evolution of ppGpp synthetases and hydrolases across the tree of life. PLOS ONE. 2011;6:e23479. doi: 10.1371/journal.pone.0023479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the keio collection. Molecular Systems Biology. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battesti A, Bouveret E. Acyl carrier protein/SpoT interaction, the switch linking SpoT-dependent stress response to fatty acid metabolism. Molecular Microbiology. 2006;62:1048–1063. doi: 10.1111/j.1365-2958.2006.05442.x. [DOI] [PubMed] [Google Scholar]
- Blank K, Hensel M, Gerlach RG. Rapid and highly efficient method for scarless mutagenesis within the Salmonella enterica chromosome. PLOS ONE. 2011;6:e15763. doi: 10.1371/journal.pone.0015763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DR, Barton G, Pan Z, Buck M, Wigneshweraraj S. Nitrogen stress response and stringent response are coupled in Escherichia coli. Nature Communications. 2014;5:4115. doi: 10.1038/ncomms5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Fernández IS, Gordiyenko Y, Ramakrishnan V. Ribosome-dependent activation of stringent control. Nature. 2016;534:277–280. doi: 10.1038/nature17675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulot S, Audebert S, Pieulle L, Seduk F, Baudelet E, Espinosa L, Pizay MC, Camoin L, Magalon A. Clustering as a means to control nitrate respiration efficiency and toxicity in Escherichia coli. mBio. 2019;10:e01832-19. doi: 10.1128/mBio.01832-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashel M, Gallant J. Two compounds implicated in the function of the RC gene of Escherichia coli. Nature. 1969;221:838–841. doi: 10.1038/221838a0. [DOI] [PubMed] [Google Scholar]
- Corrigan RM, Bellows LE, Wood A, Gründling A. ppGpp negatively impacts ribosome assembly affecting growth and antimicrobial tolerance in Gram-positive bacteria. PNAS. 2016;113:E1710–E1719. doi: 10.1073/pnas.1522179113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. PNAS. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durfee T, Hansen AM, Zhi H, Blattner FR, Jin DJ. Transcription profiling of the stringent response in Escherichia coli. Journal of Bacteriology. 2008;190:1084–1096. doi: 10.1128/JB.01092-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain E, Guiraud P, Byrne D, Douzi B, Djendli M, Maisonneuve E. YtfK activates the stringent response by triggering the alarmone synthetase SpoT in Escherichia coli. Nature Communications. 2019;10:1–12. doi: 10.1038/s41467-019-13764-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratani FL, Horvatek P, Geiger T, Borisova M, Mayer C, Grin I, Wagner S, Steinchen W, Bange G, Velic A, Maček B, Wolz C. Regulation of the opposing (p)ppGpp synthetase and hydrolase activities in a bifunctional RelA/SpoT homologue from Staphylococcus aureus. PLOS Genetics. 2018;14:e1007514. doi: 10.1371/journal.pgen.1007514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gropp M, Strausz Y, Gross M, Glaser G. Regulation of Escherichia coli RelA requires oligomerization of the C-terminal domain. Journal of Bacteriology. 2001;183:570–579. doi: 10.1128/JB.183.2.570-579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn J, Tanner AW, Carabetta VJ, Cristea IM, Dubnau D. ComGA-RelA interaction and persistence in the Bacillus subtilis K-state. Molecular Microbiology. 2015;97:454–471. doi: 10.1111/mmi.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harborne NR, Griffiths L, Busby SJ, Cole JA. Transcriptional control, translation and function of the products of the five open reading frames of the Escherichia coli nir operon. Molecular Microbiology. 1992;6:2805–2813. doi: 10.1111/j.1365-2958.1992.tb01460.x. [DOI] [PubMed] [Google Scholar]
- Haseltine WA, Block R. Synthesis of guanosine tetra- and pentaphosphate requires the presence of a codon-specific, uncharged transfer ribonucleic acid in the acceptor site of ribosomes. PNAS. 1973;70:1564–1568. doi: 10.1073/pnas.70.5.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauryliuk V, Atkinson GC, Murakami KS, Tenson T, Gerdes K. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nature Reviews Microbiology. 2015;13:298–309. doi: 10.1038/nrmicro3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge R. Linking bacterial growth, survival, and multicellularity - small signaling molecules as triggers and drivers. Current Opinion in Microbiology. 2020;55:57–66. doi: 10.1016/j.mib.2020.02.007. [DOI] [PubMed] [Google Scholar]
- Hogg T, Mechold U, Malke H, Cashel M, Hilgenfeld R. Conformational antagonism between opposing active sites in a bifunctional RelA/SpoT homolog modulates (p)ppGpp metabolism during the stringent response [corrected] Cell. 2004;117:57–68. doi: 10.1016/S0092-8674(04)00260-0. [DOI] [PubMed] [Google Scholar]
- Irving SE, Choudhury NR, Corrigan RM. The stringent response and physiological roles of (pp)pGpp in bacteria. Nature Reviews Microbiology. 2021;19:256–271. doi: 10.1038/s41579-020-00470-y. [DOI] [PubMed] [Google Scholar]
- Irving SE, Corrigan RM. Triggering the stringent response: signals responsible for activating (p)ppGpp synthesis in bacteria. Microbiology. 2018;164:268–276. doi: 10.1099/mic.0.000621. [DOI] [PubMed] [Google Scholar]
- Kanjee U, Ogata K, Houry WA. Direct binding targets of the stringent response alarmone (p)ppGpp. Molecular Microbiology. 2012;85:1029–1043. doi: 10.1111/j.1365-2958.2012.08177.x. [DOI] [PubMed] [Google Scholar]
- Karimova G, Pidoux J, Ullmann A, Ladant D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. PNAS. 1998;95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karstens K, Zschiedrich CP, Bowien B, Stülke J, Görke B. Phosphotransferase protein EIIANtr interacts with SpoT, a key enzyme of the stringent response, in Ralstonia eutropha H16. Microbiology. 2014;160:711–722. doi: 10.1099/mic.0.075226-0. [DOI] [PubMed] [Google Scholar]
- Khlebodarova TM, Ree NA, Likhoshvai VA. On the control mechanisms of the nitrite level in Escherichia coli cells: the mathematical model. BMC Microbiology. 2016;16 Suppl 1:7. doi: 10.1186/s12866-015-0619-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Research. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- Krüger L, Herzberg C, Wicke D, Bähre H, Heidemann JL, Dickmanns A, Schmitt K, Ficner R, Stülke J. A rendezvous of two second messengers: the c-di-AMP receptor protein DarB controls (p)ppGpp synthesis in Bacillus subtilis. bioRxiv. 2020 doi: 10.1101/2020.08.27.268672. [DOI] [PMC free article] [PubMed]
- Lee TS, Krupa RA, Zhang F, Hajimorad M, Holtz WJ, Prasad N, Lee SK, Keasling JD. BglBrick vectors and datasheets: A synthetic biology platform for gene expression. Journal of Biological Engineering. 2011;5:12. doi: 10.1186/1754-1611-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Park YH, Seok YJ. Rsd balances (p)ppGpp level by stimulating the hydrolase activity of SpoT during carbon source downshift in Escherichia coli. PNAS. 2018;115:E6845–E6854. doi: 10.1073/pnas.1722514115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GW, Burkhardt D, Gross C, Weissman JS. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014;157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveland AB, Bah E, Madireddy R, Zhang Y, Brilot AF, Grigorieff N, Korostelev AA. Ribosome•RelA structures reveal the mechanism of stringent response activation. eLife. 2016;5:e17029. doi: 10.7554/eLife.17029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechold U, Murphy H, Brown L, Cashel M. Intramolecular regulation of the opposing (p)ppGpp catalytic activities of Rel(Seq), the Rel/Spo enzyme from Streptococcus equisimilis. Journal of Bacteriology. 2002;184:2878–2888. doi: 10.1128/JB.184.11.2878-2888.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger S, Sarubbi E, Glaser G, Cashel M. Protein sequences encoded by the relA and the spoT genes of Escherichia coli are interrelated. Journal of Biological Chemistry. 1989;264:9122–9125. doi: 10.1016/S0021-9258(18)60500-9. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1972. [Google Scholar]
- Miller JH. A Short Course in Bacterial Genetics: A Laboratory Manual and Handbookfor Escherichia Coli and Related Bacteria. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- Murray KD, Bremer H. Control of spoT-dependent ppGpp synthesis and degradation in Escherichia coli. Journal of Molecular Biology. 1996;259:41–57. doi: 10.1006/jmbi.1996.0300. [DOI] [PubMed] [Google Scholar]
- Neidhardt FC, Bloch PL, Smith DF. Culture medium for enterobacteria. Journal of Bacteriology. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pausch P, Abdelshahid M, Steinchen W, Schäfer H, Gratani FL, Freibert SA, Wolz C, Turgay K, Wilson DN, Bange G. Structural Basis for Regulation of the Opposing (p)ppGpp Synthetase and Hydrolase within the Stringent Response Orchestrator Rel. Cell Reports. 2020;32:108157. doi: 10.1016/j.celrep.2020.108157. [DOI] [PubMed] [Google Scholar]
- Peakman T, Crouzet J, Mayaux JF, Busby S, Mohan S, Harborne N, Wootton J, Nicolson R, Cole J. Nucleotide sequence, organisation and structural analysis of the products of genes in the nirB-cysG region of the Escherichia coli K-12 chromosome. European Journal of Biochemistry. 1990a;191:315–323. doi: 10.1111/j.1432-1033.1990.tb19125.x. [DOI] [PubMed] [Google Scholar]
- Peakman T, Busby S, Cole J. Transcriptional control of the cysG gene of Escherichia coli K-12 during aerobic and anaerobic growth. European Journal of Biochemistry. 1990b;191:325–331. doi: 10.1111/j.1432-1033.1990.tb19126.x. [DOI] [PubMed] [Google Scholar]
- Pedersen FS, Kjeldgaard NO. Analysis of the relA gene product of Escherichia coli. European Journal of Biochemistry. 1977;76:91–97. doi: 10.1111/j.1432-1033.1977.tb11573.x. [DOI] [PubMed] [Google Scholar]
- Peterson BN, Young MKM, Luo S, Wang J, Whiteley AT, Woodward JJ, Tong L, Wang JD, Portnoy DA. (p)ppGpp and c-di-AMP Homeostasis Is Controlled by CbpB in Listeria monocytogenes. mBio. 2020;11:11. doi: 10.1128/mBio.01625-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potrykus K, Cashel M. (p)ppGpp: still magical? Annual Review of Microbiology. 2008;62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- Raskin DM, Judson N, Mekalanos JJ. Regulation of the stringent response is the essential function of the conserved bacterial G protein CgtA in Vibrio cholerae. PNAS. 2007;104:4636–4641. doi: 10.1073/pnas.0611650104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronneau S, Petit K, De Bolle X, Hallez R. Phosphotransferase-dependent accumulation of (p)ppGpp in response to glutamine deprivation in Caulobacter crescentus. Nature Communications. 2016;7:11423. doi: 10.1038/ncomms11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronneau S, Caballero-Montes J, Coppine J, Mayard A, Garcia-Pino A, Hallez R. Regulation of (p)ppGpp hydrolysis by a conserved archetypal regulatory domain. Nucleic Acids Research. 2019;47:843–854. doi: 10.1093/nar/gky1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronneau S, Hallez R. Make and break the alarmone: regulation of (p)ppGpp synthetase/hydrolase enzymes in bacteria. FEMS Microbiology Reviews. 2019;43:389–400. doi: 10.1093/femsre/fuz009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W, Vrentas CE, Sanchez-Vazquez P, Gaal T, Gourse RL. The magic spot: a ppGpp binding site on E. coli RNA polymerase responsible for regulation of transcription initiation. Molecular Cell. 2013;50:420–429. doi: 10.1016/j.molcel.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross W, Sanchez-Vazquez P, Chen AY, Lee JH, Burgos HL, Gourse RL. ppGpp Binding to a Site at the RNAP-DksA Interface Accounts for Its Dramatic Effects on Transcription Initiation during the Stringent Response. Molecular Cell. 2016;62:811–823. doi: 10.1016/j.molcel.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vazquez P, Dewey CN, Kitten N, Ross W, Gourse RL. Genome-wide effects on Escherichia coli transcription from ppGpp binding to its two sites on RNA polymerase. PNAS. 2019;116:8310–8319. doi: 10.1073/pnas.1819682116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal R, Harinarayanan R. Activation of RelA by pppGpp as the basis for its differential toxicity over ppGpp in Escherichia coli. Journal of Biosciences. 2020;45:2. doi: 10.1007/s12038-020-9991-2. [DOI] [PubMed] [Google Scholar]
- Schreiber G, Metzger S, Aizenman E, Roza S, Cashel M, Glaser G. Overexpression of the relA gene in Escherichia coli. Journal of Biological Chemistry. 1991;266:3760–3767. doi: 10.1016/S0021-9258(19)67860-9. [DOI] [PubMed] [Google Scholar]
- Seyfzadeh M, Keener J, Nomura M. spoT-dependent accumulation of guanosine tetraphosphate in response to fatty acid starvation in Escherichia coli. PNAS. 1993;90:11004–11008. doi: 10.1073/pnas.90.23.11004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha AK, Winther KS, Roghanian M, Gerdes K. Fatty acid starvation activates RelA by depleting lysine precursor pyruvate. Molecular Microbiology. 2019;112:1339–1349. doi: 10.1111/mmi.14366. [DOI] [PubMed] [Google Scholar]
- Spira B, Silberstein N, Yagil E. Guanosine 3',5'-bispyrophosphate (ppGpp) synthesis in cells of Escherichia coli starved for Pi. Journal of Bacteriology. 1995;177:4053–4058. doi: 10.1128/jb.177.14.4053-4058.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada H, Roghanian M, Murina V, Dzhygyr I, Murayama R, Akanuma G, Atkinson GC, Garcia-Pino A, Hauryliuk V. The C-Terminal RRM/ACT Domain Is Crucial for Fine-Tuning the Activation of 'Long' RelA-SpoT Homolog Enzymes by Ribosomal Complexes. Frontiers in Microbiology. 2020;11:277. doi: 10.3389/fmicb.2020.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamman H, Van Nerom K, Takada H, Vandenberk N, Scholl D, Polikanov Y, Hofkens J, Talavera A, Hauryliuk V, Hendrix J, Garcia-Pino A. A nucleotide-switch mechanism mediates opposing catalytic activities of Rel enzymes. Nature Chemical Biology. 2020;16:834–840. doi: 10.1038/s41589-020-0520-2. [DOI] [PubMed] [Google Scholar]
- Traxler MF, Summers SM, Nguyen HT, Zacharia VM, Hightower GA, Smith JT, Conway T. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Molecular Microbiology. 2008;68:1128–1148. doi: 10.1111/j.1365-2958.2008.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull KJ, Dzhygyr I, Lindemose S, Hauryliuk V, Roghanian M. Intramolecular Interactions Dominate the Autoregulation of Escherichia coli Stringent Factor RelA. Frontiers in Microbiology. 2019;10:1966. doi: 10.3389/fmicb.2019.01966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson K, Busby S, Cole J. Catabolite regulation of two Escherichia coli operons encoding nitrite reductases: role of the Cra protein. Archives of Microbiology. 1997;168:240–244. doi: 10.1007/s002030050494. [DOI] [PubMed] [Google Scholar]
- Uzan M, Danchin A. Correlation between the serine sensitivity and the derepressibility of the ilv genes in Escherichia coli relA- mutants. Molecular and General Genetics MGG. 1978;165:21–30. doi: 10.1007/BF00270372. [DOI] [PubMed] [Google Scholar]
- Vinella D, Albrecht C, Cashel M, D'Ari R, D’Ari R. Iron limitation induces SpoT-dependent accumulation of ppGpp in Escherichia coli. Molecular Microbiology. 2005;56:958–970. doi: 10.1111/j.1365-2958.2005.04601.x. [DOI] [PubMed] [Google Scholar]
- Wang B, Dai P, Ding D, Del Rosario A, Grant RA, Pentelute BL, Laub MT. Affinity-based capture and identification of protein effectors of the growth regulator ppGpp. Nature Chemical Biology. 2019a;15:141–150. doi: 10.1038/s41589-018-0183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Tamiev D, Alagurajan J, DiSpirito AA, Phillips GJ, Hargrove MS. The role of the NADH-dependent nitrite reductase, Nir, from Escherichia coli in fermentative ammonification. Archives of Microbiology. 2019b;201:519–530. doi: 10.1007/s00203-018-1590-3. [DOI] [PubMed] [Google Scholar]
- Wang H, Gunsalus RP. The nrfA and nirB nitrite reductase operons in Escherichia coli are expressed differently in response to nitrate than to nitrite. Journal of Bacteriology. 2000;182:5813–5822. doi: 10.1128/JB.182.20.5813-5822.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendrich TM, Blaha G, Wilson DN, Marahiel MA, Nierhaus KH. Dissection of the mechanism for the stringent factor RelA. Molecular Cell. 2002;10:779–788. doi: 10.1016/S1097-2765(02)00656-1. [DOI] [PubMed] [Google Scholar]
- Wexselblatt E, Oppenheimer-Shaanan Y, Kaspy I, London N, Schueler-Furman O, Yavin E, Glaser G, Katzhendler J, Ben-Yehuda S. Relacin, a novel antibacterial agent targeting the Stringent Response. PLOS Pathogens. 2012;8:e1002925. doi: 10.1371/journal.ppat.1002925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexselblatt E, Kaspy I, Glaser G, Katzhendler J, Yavin E. Design, synthesis and structure-activity relationship of novel Relacin analogs as inhibitors of Rel proteins. European Journal of Medicinal Chemistry. 2013;70:497–504. doi: 10.1016/j.ejmech.2013.10.036. [DOI] [PubMed] [Google Scholar]
- Winther KS, Roghanian M, Gerdes K. Activation of the Stringent Response by Loading of RelA-tRNA Complexes at the Ribosomal A-Site. Molecular Cell. 2018;70:95–105. doi: 10.1016/j.molcel.2018.02.033. [DOI] [PubMed] [Google Scholar]
- Wout P, Pu K, Sullivan SM, Reese V, Zhou S, Lin B, Maddock JR. The Escherichia coli GTPase CgtAE cofractionates with the 50S ribosomal subunit and interacts with SpoT, a ppGpp synthetase/hydrolase. Journal of Bacteriology. 2004;186:5249–5257. doi: 10.1128/JB.186.16.5249-5257.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Kalman M, Ikehara K, Zemel S, Glaser G, Cashel M. Residual guanosine 3',5'-bispyrophosphate synthetic activity of relA null mutants can be eliminated by spoT null mutations. Journal of Biological Chemistry. 1991;266:5980–5990. doi: 10.1016/S0021-9258(19)67694-5. [DOI] [PubMed] [Google Scholar]
- Yang X, Ishiguro EE. Dimerization of the RelA protein of Escherichia coli. Biochemistry and Cell Biology. 2001;79:729–736. doi: 10.1139/o01-144. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zborníková E, Rejman D, Gerdes K. Novel (p)ppGpp binding and metabolizing proteins of Escherichia coli. mBio. 2018;9:e02188-17. doi: 10.1128/mBio.02188-17. [DOI] [PMC free article] [PubMed] [Google Scholar]