

Abstract
Circadian rhythm evolved to allow organisms to coordinate intrinsic physiological functions in anticipation of recurring environmental changes. The importance of this coordination is exemplified by the tight temporal control of cardiac metabolism. Levels of metabolites, metabolic flux, and response to nutrients all oscillate in a time-of-day–dependent fashion. While these rhythms are affected by oscillatory behavior (feeding/fasting, wake/sleep) and neurohormonal changes, recent data have unequivocally demonstrated an intrinsic circadian regulation at the tissue and cellular level. The circadian clock — through a network of a core clock, slave clock, and effectors — exerts intricate temporal control of cardiac metabolism, which is also integrated with environmental cues. The combined anticipation and adaptability that the circadian clock enables provide maximum advantage to cardiac function. Disruption of the circadian rhythm, or dyssynchrony, leads to cardiometabolic disorders seen not only in shift workers but in most individuals in modern society. In this Review, we describe current findings on rhythmic cardiac metabolism and discuss the intricate regulation of circadian rhythm and the consequences of rhythm disruption. An in-depth understanding of the circadian biology in cardiac metabolism is critical in translating preclinical findings from nocturnal-animal models as well as in developing novel chronotherapeutic strategies.
Introduction
The heart beats relentlessly throughout life and consequently has a voracious demand for energy. This metabolic demand across a 24-hour cycle is affected by numerous influences, such as physical activity, feeding/fasting state, and neurohormonal tone. Consequently, optimal cardiac performance requires a high degree of metabolic flexibility. Importantly, the coordinated action of the circadian clock, both centrally and peripherally, allows the heart to anticipate energetic requirements instead of merely responding to metabolic needs. In this Review, we discuss evidence of circadian metabolic oscillation, mechanisms of its regulation, and the pathophysiological consequences when such rhythmicity is disrupted in the heart.
The core molecular clock is a set of transcription factors (TFs) that form an intricate feedback loop, in which the synthesis and degradation of these TFs takes about 24 hours (1). The molecular mechanism is discussed in greater detail below. The core clock factors are critical for rhymic output, and loss of function of core clock factors leads to an arrhythmic phenotype or a loss of period phenotype. While the same molecular clock is expressed and functional in virtually all cells except embryonic stem cells, the central clock refers to that in the suprachiasmatic nucleus of the hypothalamus (2). The central clock receives photic input from the melanopsin-expressing retinal cells and sends neural and hormonal output to other parts of the brain and other organs. The central clock is critical in coordinating behavior at the organismal level. The molecular clocks in other tissues and cell types are referred to as the peripheral clock. The peripheral clock receives neural and hormonal signals from the central clock and can be entrained to it; it may also receive local signals and act autonomously in dyssynchrony with the central clock (Figure 1A). In this Review, we mainly discuss the function of the peripheral clock independent of the neurohormonal input from the central clock; the main experimental paradigms include ex vivo working hearts and cardiac-specific clock mutants.
Figure 1. Basic concepts of circadian rhythm.

(A) The central clock exists in the suprachiasmatic nucleus (SCN) in the hypothalamus, receives optic input, and sends out neurohormonal signals to other peripheral tissues, such as the heart. The molecular clocks in the peripheral tissues (e.g., heart) are termed the peripheral clock; it receives entrainment signal both from the central clock and from additional external stimuli (e.g., diet and exercise). (B) The normal rhythm may be disrupted by a change in period, phase, or amplitude, or any combination of these. (C) The diurnal rhythm starts at zeitgeber time (ZT) 0, which is defined by the turning on of the light. In the light phase, diurnal animals (including humans) are active and nocturnal animals are resting. The opposite happens in the dark phase. When we refer to the active versus resting phases, we are describing the same biological phase and processes for any species studied.
The circadian rhythm traditionally refers specifically to the near-24-hour rhythm that is independent of external cues. In contrast, diurnal rhythm is more loosely used and refers to any 24-hour rhythm; as a result, it may not be directly regulated by the core clock and can be a reaction to cyclic stimuli. The circadian rhythm, like any other waveform, is defined by three key parameters: the period, the phase, and the amplitude (Figure 1B). The core clock defines the period, which evolved as a consequence of life on Earth. The phase is usually in relation to zeitgeber time (ZT). “Zeitgeber” is German for “time giver” and can refer to any environmental cue that synchronizes 24-hour rhythmic behavior. ZT0 now specifically refers to when the lights turn on in the facility in most experimental settings. As humans are diurnal and experimental rodent models (mice and rats) are nocturnal, referring to light versus dark phase is often confusing. Thus, we will use “active” versus “resting” phase in this Review, which may be in light or dark phase but always alludes to the same physiological activity and biological processes (Figure 1C).
Circadian metabolic oscillation in the heart
Metabolism refers to the chemical processes that convert food to energy or building blocks (e.g., amino acids, nucleotides) while eliminating waste through enzymatic reactions. Metabolism can be measured by flux or metabolites. The levels of rate-limiting enzymes also contribute significantly to flux and thus are often measured to provide additional information on metabolism. The rhythmicity of metabolism in the heart has been measured by all of these approaches, and we will summarize the key findings to date (below and in Figure 2).
Figure 2. Circadian rhythm of cardiac metabolism in rodents.

During the light phase, mice are resting, and the predominant processes in the heart are lipolysis, protein synthesis, and autophagy, representing a “remodeling and repair” phase. During the dark phase, mice are active, and the main processes in the heart are glucose uptake, glycolysis and oxidation, glycogenolysis, amino acid catabolism, ROS clearance, etc., representing a “catabolic” phase. The core clock factor BMAL1 reaches peak at ZT0 and nadir at ZT12.
Lipid metabolism.
While the mammalian heart is an omnivore, it uses fats as its main energy source. In fact, 70% of the energy in a healthy adult heart at rest comes from fatty acid oxidation (FAO), and esterified fatty acids (FAs) (e.g., triglyceride) are the major source of lipids for the human heart (3–5). Interestingly, while the plasma FA concentration varies with dietary availability, the catabolism of FA does not oscillate in a circadian fashion (6, 7). This is not completely surprising, as FAO is also not induced by changes in workload or fuel availability. In contrast, myocardial triglyceride (TG) levels show a diurnal variation with peak at the wake-to-sleep transition (ZT0) in WT mouse hearts. To exclude the effect of neurohumoral inputs, ex vivo–perfused hearts were studied, and, interestingly, both lipolysis and TG synthesis showed a diurnal variation, with a trough at the middle of the active phase (ZT18) and a peak at ZT6. However, net TG synthesis also showed peaks at ZT18, suggesting that oscillatory lipolysis is a driving force of the diurnal variation of myocardial net TG synthesis and TG levels (8). Multiple TG metabolic enzymes have shown circadian gene expression, e.g., Dgat2, Agpat2, and Hsl (encoding hormone-sensitive lipase). Interestingly, the lipolysis rate-liming enzyme adipose triglyceride lipase (ATGL) shows diurnal protein expression in the heart, although its RNA level does not oscillate, suggesting a posttranscriptional mechanism. Additionally, an AMPK-dependent phosphorylation site on HSL, which leads to enzymatic inhibition, is oscillatory with peak phosphorylation at ZT18 (8). Collectively, these studies emphasize that the circadian regulation of cardiac TG turnover occurs at multiple levels: transcriptional, translational, and posttranslational. However, given the importance and complexity of circadian control of cardiac TG, additional work is needed to understand regulation at the flux level under different feeding conditions along with greater insights into the molecular basis of these effects.
Carbohydrate metabolism.
Under basal conditions, about 20% of the heart’s energy is converted from carbohydrates. Increased workload and ischemia augment the use of glucose and other carbohydrates (including lactate and glycogen) to produce energy (9–11). Both glucose oxidation and glycolysis show diurnal variation in the heart, even when perfused ex vivo with medium containing radiolabeled isotopes, peaking during the dark phase at ZT18 (7, 12). This suggests that glucose uptake might be locally regulated in a circadian fashion that is independent of neurohormonal regulation. The glucose transporters GLUT1 and GLUT4 are abundant in the heart. While GLUT1 is responsible for low-level constant glucose uptake, GLUT4 is inducible and is insulin dependent in membrane translocation (13). Whether these transporters are expressed on the cellular surface in a circadian manner has not been assessed. Phospho-AMPK is known to promote glucose transport, and its level shows a modest diurnal variation with a peak also in the dark phase (ZT18), consistent with increased glucose uptake and increased utilization through both glycolysis and glucose oxidation at this time, when energy demand is high (Figure 2 and ref. 12).
Cardiac glycogen accounts for 2% of the cellular volume in adult cardiomyocytes, and its levels vary based on feeding status (14). Glycogen is 2-fold higher at ZT6 than at ZT18 after 40 minutes of glucose perfusion in ex vivo–perfused hearts, although they start with the same glycogen content. Given that the glucose incorporation to glycogen was identical as measured by 14C-glucose incorporation, this suggests that the glycogenolysis has a diurnal rhythm and is higher at ZT18 (7). Thus, ZT6, or the resting phase, is associated with reduced glucose uptake, reduced glycolysis, reduced glucose oxidation, and reduced glycogenolysis.
The hexosamine biosynthesis pathway (HBP) consumes 2%–5% of glucose uptake and is considered a glucose- or nutrient-sensing pathway (15, 16). Activation of HBP yields UDP-GlcNAc, which is the substrate for several glycosylation reactions, including O-GlcNAcylation. There is an approximately 20% diurnal variation in total cardiac O-GlcNAcylation levels, with a peak at ZT18 (12). While modest, O-GlcNAcylation mediates certain adverse effects of hyperglycemia, and as a posttranslational modification it may potentially affect many cellular processes, as detailed in the following section (15).
Protein and amino acid metabolism.
Early studies with radiolabeled isotopes showed that the incorporation of l-[3H]leucine into the rat heart, a measurement of protein synthesis, is greatest during the resting phase (Figure 1 and ref. 17). This observation is corroborated by more recent studies using transcriptomics, which showed that the heart prioritizes remodeling and repair at rest and catabolism during the active phase. In fact, most amino acid catabolic enzymes reach peak expression in the active phase (18). Future study of the metabolic flux of amino acids in a circadian fashion and its detailed molecular mechanism will be required to advance our understanding.
Nicotinamide adenine dinucleotide.
Nicotinamide adenine dinucleotide (NAD+) is a cofactor in reduction oxidation (redox) reactions that plays a central role in metabolism. The heart has an abundant NAD+ level to support its continuous high energy demand (19). NAD+ levels showed strong circadian oscillation with a peak at ZT14 in the heart, primarily driven by oscillatory levels of NAMPT, the rate-limiting enzyme of the NAD+ salvage pathway (8, 12, 20–23). The oscillatory NAD+ may affect the flux of numerous redox metabolic reactions in the heart. In addition, NAD+ serves as a cofactor for a number of non-redox reactions, such as those catalyzed by sirtuins (SIRTs) (24). Thus, oscillatory NAD+ levels in the myocardium may affect SIRT activities, which in turn exert a secondary effect on cardiac metabolism. This will be detailed in the following section.
Mitochondria.
Mitochondria are the key organelles that produce ATP from oxidative metabolism. The circadian regulation of mitochondrial biogenesis, respiration, and dynamics has been shown in multiple organ systems. Although circadian effects on mitochondria have not been directly functionally assessed, mitochondrial gene expression has been studied and showed a diurnal variation for both function (oxidative phosphorylation) (25) and dynamics (fission and fusion) (26). SIRT3 is the primary deacetylase in mitochondria and is involved in both metabolic homeostasis and stress response (27). While the protein level of SIRT3 is constant, its activity is regulated in a diurnal fashion due to oscillatory coenzyme NAD+ levels in the heart (20). This subsequently affects major mitochondrial functions, including metabolism and oxidation reduction. SIRT3-knockout cells show a more than 50% reduction of basal ATP levels (28). SIRT3-knockout mice show reduced FAO and glucose oxidation, but increased glycolysis in the heart; these changes are accompanied by hyperacetylation of many mitochondrial proteins, including enzymes involved in FA transport and oxidation, subunits of the electron transport chain, and enzymes involved in the tricarboxylic acid cycle (29). SIRT3 is known to deacetylate and activate forkhead transcription factor box O3a (FoxO3a), which further activates manganese superoxide dismutase (MnSOD) and catalase, thus leading to reduction of reactive oxygen species (ROS) in the heart (30–32). SIRT3 also directly deacetylates cyclophilin D, a component of the mitochondria permeability transition pore (mPTP), which leads to inhibition of mPTP opening (33). These diverse roles of SIRT3 directly contribute to the circadian cardiac metabolism and redox states in the heart.
Autophagy.
Autophagy is integral to cellular metabolic regulation. It provides energy and metabolites during periods of starvation, helps adapt to metabolic challenges, and maintains quality control of intracellular macromolecules and organelles (34). Autophagy is critical for the health of nonreplicating terminally differentiated cells such as cardiomyocytes and neurons. In the heart, autophagy is more active during the resting phase, as measured by both LC3-II cleavage and multiple gene expression (35, 36). More interestingly, autophagic vacuoles were found to fluctuate in volume in a diurnal fashion in the myocardium. The autophagosomes reached their largest volumes in the late resting phase and their smallest volumes in the early active phase (37). These observations are again supportive of the notion that the heart has a critical remodeling and repair function during the resting phase.
Molecular mechanisms of circadian regulation of cardiac metabolism
The molecular clock is a set of TFs that form a feedback loop and regulate their own expression. The CLOCK:BMAL1 complex activates Cry and Per gene expression. Accumulation of cytoplasmic CRY:PER complex leads to their nuclear translocation and inhibits the CLOCK:BMAL1-driven transcription of Cry and Per (38, 39). The CLOCK:BMAL1 complex also drives the transcription of Nr1d1/2 and Nr1f1, which encode the nuclear receptors REV-ERBα/β and RORα. REV-ERB competes with RORα at the ROR binding elements; REV-ERB and RORα then respectively suppress and activate the transcription of Bmal1 (40–42). These genes are components of the “core clock,” which resides and functions in almost every mammalian cell (38–42). Accumulating evidence has led to the appreciation that the clock also controls downstream cellular circuitry, which extends beyond the linear core clock–target gene transcriptional regulation. We will summarize these considerations in the section that follows.
Transcription regulation.
As in other organs, approximately 5%–10% of the genes expressed in the heart oscillate in a circadian fashion (23, 43–46). The core clock machinery directly regulates downstream targets, which are referred to as clock-controlled genes (CCGs), at the transcription level. This is achieved by direct DNA binding, recruitment of coregulators, and modification of the histone tails and chromatin state. The molecular details of the core clock’s regulation of transcription through epigenetic mechanisms were recently reviewed in great detail (47). For instance, BMAL1 and RORα have been reported as “pioneer” factors for their ability to access closed chromatin (48, 49). Both BMAL1 and RNAPII were found to be recruited to the chromatin in a circadian fashion, suggesting that in addition to direct regulation of individual target genes, the circadian core clock changes chromatin accessibility and transcription globally (50, 51). Despite the major role of the core clock in transcription, it is reported that only 22%–30% of the oscillating genes at the transcript level are driven by nascent transcription in Drosophila heads and mouse livers; other processes in RNA processing and stability likely contribute significantly (50, 52). Although this regulation has not been validated in the heart, presumably a similar percentage will hold true given the evolutionary divergence of flies and mice.
While all tissues have 5%–10% of genes oscillating (i.e., CCGs), the overlap between different tissues is very small (53). The concept of a so-called “slave clock” has long been proposed to mediate the tissue specificity of the oscillating genes (43, 54). The slave clocks may be tissue-specific TFs themselves that recruit clock factors; they may also be pioneering TFs that allow the accessibility to chromatin of certain clock factors; and they may themselves be CCGs directly driven by the clock. One key characteristic a slave clock must have, however, is that it must mediate the tissue-specific oscillation of other CCGs without affecting the rhythm from the clock. There are very few well-described examples. In the heart, the only confirmed slave clock to date is Krüppel-like factor 15 (KLF15). KLF15 expression (both RNA and protein) oscillates in a circadian fashion under the transcriptional control of BMAL1 (20, 55). Additionally, the polycomb protein enhancer of zeste homolog 2 (EZH2), which is bound to CLOCK:BMAL1 and modulates circadian rhythm, suppresses the KLF15 promoter by hypermethylation in patients with ischemic cardiomyopathy (56, 57). KLF15 in turn controls 75% of oscillating genes in the heart without affecting the core clock, particularly through direct binding to and regulation of catabolic gene expression in the active phase (18). Although the oscillatory expression of KLF15 is driven by the core clock, it is also regulated by various nutritional, physiological, and pathophysiological conditions. For example, fasting and exercise induce KLF15, while both ischemic and nonischemic cardiomyopathy has been associated with reduced KLF15 (18, 20, 58). Thus, instead of merely transmitting the oscillatory instruction from the core clock, the slave clock KLF15 allows integration of complex metabolic signals (Figure 3).
Figure 3. Integrated circadian cardiac metabolic regulation.
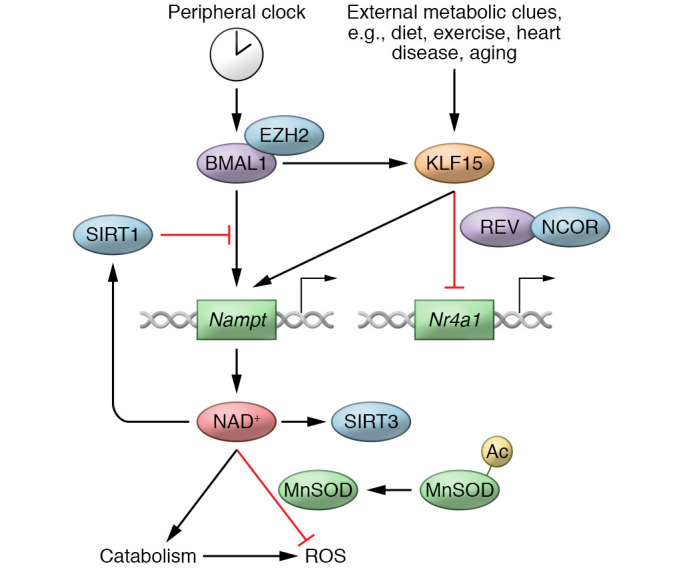
The slave clock KLF15 integrates signals from both the core clock (BMAL1) and external metabolic clues (diet, exercise, heart disease, and aging). In turn, KLF15 regulates downstream oscillatory target genes in the heart, both by activating them (e.g., Nampt) and by repressing their oscillation (e.g., Nr4a1). Critical pathways (e.g., NAD+) may be dually regulated by both the core clock and the slave clock to enable tight temporal control, as well as flexibility between anticipation and adaptation. Circadian regulation of NAD+ allows coordinated catabolism and ROS clearance, which is essential for myocardium homeostasis. REV, REV-ERB.
The core clock and slave clock may work in concert to regulate target gene expression. For example, both BMAL1 and KLF15 are required for the oscillatory expression of Nampt, encoding a rate-limiting enzyme for the NAD+ salvage pathway in the heart (20, 59). Somewhat surprisingly, KLF15 expression is antiphasic to BMAL1 expression in the heart. One plausible explanation might be that the expression of Nampt only occurs in the brief window when both BMAL1 and KLF15 are expressed. The dual regulation may be a mechanism to allow precise temporal control while also integrating additional physiological and nutritional status through KLF15 (Figure 3).
Most of the work in circadian biology has focused on the mechanisms that generate oscillation. The fact that each tissue has a unique set of oscillating genes suggests that mechanisms for tissue-specific suppression of oscillation may be equally important. KLF15 has been shown to recruit REV-ERB and nuclear receptor corepressor (NCoR) at hundreds of genes, preventing them from aberrant oscillation during the active phase. This is a critical mechanism allowing genes to remain nonresponsive to a fluctuating neurohormonal milieu that includes glucocorticoid and lipid species (Figure 3 and ref. 18). In fact, pharmacologically strengthening the repression of REV-ERB leads to cardiac protection while maintaining cardiac gene expression profile and metabolic homeostasis (60).
Thus, in contrast to what may be suggested by its name, a slave clock does not just passively transduce the oscillatory signal from the core clock to the tissue-specific CCGs. A slave clock allows integration of the perpetual rhythm with regulation of tissue-specific functions and adaptation to external stimuli, resulting in fine-tuning of the CCGs’ output to match the exact need of the moment. The presence of the slave clock is critical for both oscillation and quiescence, and the slave clocks are indispensable hubs of signal integration in the circadian regulatory network.
Translational regulation.
mTOR signaling senses energy and nutrient status, and it regulates cellular metabolism, growth, proliferation, and survival. The activity of mTOR signaling may directly affect clock function both in the suprachiasmatic nucleus and in the peripheral tissues (61, 62). mTOR signaling, as reported by its downstream targets phosphorylated ribosomal protein kinase S6K1 and S6, peaks at ZT2 in the heart in a BMAL1-dependent fashion (36, 63, 64). This correlates with reduced autophagy and increased protein synthesis during the resting phase (36). BMAL1 is thought to be a negative regulator of AKT signaling, which activates the mTOR signaling cascade. Loss of BMAL1 leads to persistent activation of mTOR signaling and increased protein synthesis together with reduced autophagy, which ultimately leads to cardiac hypertrophy (36).
Surprisingly, BMAL1 was found to be phosphorylated by S6K1, which then allows its association with translational machinery and promotes protein synthesis in the cytosol (65). Thus, in addition to its well-recognized role in circadian transcription, BMAL1 may also regulate protein synthesis both directly and indirectly via interaction with the mTOR signaling pathway.
Posttranslational regulation.
Posttranslational modification may greatly alter the localization, stability, and function of protein targets. While oscillatory phosphorylation of many specific targets has been found in the heart, a global trend has not been reported.
Cardiac NAD+ oscillates as a result of rhythmic expression of its rate-limiting enzyme, NAMPT. In addition to its roles in metabolic enzymatic reactions, NAD+ is also the coenzyme for SIRT. Activity of SIRT1, SIRT3, and SIRT5 is primarily determined by NAD+ levels, since their binding constant (Km) is close to physiological NAD+ levels. In particular, SIRT3 activity and mitochondrial global acetylation have been shown to oscillate in a circadian fashion, secondary to the rhythmic NAD+ levels in the heart (20). As a result, downstream protein function may be affected. For example, the hyperacetylation of MnSOD at lysine 122 inhibits its activity (Figure 3) and is associated with increased risk for ischemia/reperfusion injury of the heart (20, 66).
Global O-GlcNAcylation has a modest diurnal variation with a peak at ZT18 (12). Increased O-GlcNAcylation on AMPK has been associated with increased activity, while increased O-GlcNAcylation on AKT2 has been associated with attenuated activity (67, 68). Thus, increased O-GlcNAcylation at the active phase (ZT18) contributes to increased glucose uptake and utilization, as well as reduced glycogenolysis and protein synthesis. Acute O-GlcNAcylation has been shown to protect against calcium paradox and ischemia of the myocardium (69, 70).
Interestingly, phosphorylation, acetylation, and O-GlcNAcylation have all been found to modify the core clock machinery and thus directly affect clock function and timing (12, 20, 65). This highlights the interconnected relationship between circadian regulation and metabolism.
Disruption of circadian rhythm in mice and humans
The adverse effect of disrupted circadian rhythm in the cardiovascular system has long been documented in human epidemiological studies, which highlights the intimate relationship between circadian rhythm and cardiovascular health, particularly in cardiometabolic syndromes. In recent years, mice expressing tissue-specific mutations of the core clock machinery have allowed us to dissect the function of the cardiac peripheral clock independent of the systemic neurohormonal milieu and behavior. These findings will be summarized here.
Circadian disruption in mice.
The critical function of the peripheral core clock in cardiac metabolism has been exemplified by the study of two mutant mouse models: cardiomyocyte-specific Clock mutants (CCM mice) and cardiomyocyte-specific Bmal1 knockouts (CBK mice). CCM mice overexpress a dominant-negative CLOCK mutant in the cardiomyocytes that suppresses the function of wild-type CLOCK:BMAL1 complex and leads to increased Bmal1 expression with dampened amplitude in the heart. CCM mice have a reduced heart rate with a reduced diurnal heart rate variation and showed a reduced responsiveness to workload in the active phase (45). Metabolic studies showed that the diurnal variation of glucose oxidation, glycogen synthesis, and phosphorylation of AMPK is abolished in CCM mice because of inability to induce these parameters in the active phase (12). CCM mice also have reduced fasting myocardial TG, which is accompanied by increased Adpn (promoting lipolysis) and reduced Dgat2 (promoting lipogenesis) at the mRNA level (71). In contrast, CBK mice have floxed Bmal1 alleles, which are deleted in cardiomyocytes using α-MHC-Cre, and thus show reduced Bmal1 gene expression with dampened amplitude of oscillation. Similarly to the CCM mice, CBK mice show depressed glucose oxidation and reduced cardiac power when in a fed state, accompanied by reduced phosphorylation of AKT and GSK3β. This is thought to be due to reduced expression of Pik3r1 (encoding p85α, the regulatory subunit of PI3K) and its effects on insulin signaling (23). This effect may simulate a chronic fasting state in the CBK mice. The CBK mice develop systolic dysfunction at the age of 20 weeks and have a shortened lifespan overall due to dilated cardiomyopathy. They have also been reported to develop increased fibrosis, inflammation, and diastolic dysfunction at the age of 28 weeks and thus have been proposed to have an aging phenotype (72).
While both CCM and CBK mice have disrupted cardiac circadian rhythm, they are “paused” at different phases, one with a high BMAL1 level (beginning of the resting phase) and one with a low BMAL1 level (beginning of the active phase) (Figure 2). While their phenotypes are not identical, there are clearly similarities in cardiac metabolism, such as reduced glucose oxidation in the active/fed phase. This argues against a simple model of direct core clock regulation of CCG expression. While more detailed molecular studies, preferably using an unbiased approach, are required to unfold the exact mechanisms, it is possible that a complex system-level regulation is at play, and the lack of oscillation itself may be activating downstream signaling pathways. Alternatively, the similarities in these two core clock mutants may be driven by distinct molecular mechanisms.
The slave clock KLF15 directly regulates many catabolic enzymes’ expression during the active phase. While KLF15 deficiency does not change core clock gene expression or disrupt the clock per se, it leads to loss of oscillation for 75% of the oscillatory transcripts in the heart (18). Cardiac KLF15 deficiency leads to cardiac lipid utilization defects and cardiomyopathy (18, 58). NAMPT provides an interesting example of a rhythmically regulated catabolic enzyme target of KLF15; the loss of KLF15 in the myocardium leads to failed active-phase induction of NAMPT and cardiac NAD+ (Figure 3 and ref. 20). This likely evolved to allow numerous catabolic reactions requiring NAD+ as a coenzyme to take place in the active phase. Meanwhile, NAD+, which, as mentioned above, is a coenzyme for SIRT3, peaks during the catabolic phase, allowing deacetylation and activation of mitochondrial antioxidants such as MnSOD to effectively clear the increased ROS that accompany active oxidative metabolism. Consistent with this notion, cardiac KLF15 deficiency specifically led to NAD+ deficiency and increased susceptibility to ischemia/reperfusion injury during the active phase, which may be corrected by exogenous NAD+ precursor (20). Thus, KLF15 has been shown to be a key regulator of diurnal variation of cardiac susceptibility to ischemia and reperfusion. It does so secondarily to its evolutionary role of synchronizing catabolism and ROS clearance in the active phase via governing a key metabolite and coenzyme, NAD+.
REV-ERBα and REV-ERBβ are transcriptional repressors in the core clock. Pharmacological activation of REV-ERB using SR9009 showed cardiac protection against pressure overload. In particular, global gene expression toward suppressed FA activation, transportation, and oxidation due to cardiac stress was attenuated. This effect is thought to be significantly mediated by the master regulator of FA utilization, PDK4, which was shown to be a direct target of REV-ERB (60). Despite the controversy regarding the specificity of the widely used REV-ERB agonist SR9009, other agonists have shown a similar effect in cardiomyocytes, suggesting a REV-ERB–mediated effect (60, 73). Surprisingly, both REV-ERB agonism and antagonism have been shown to be cardiac protective after ischemia and reperfusion injury (74, 75). While reduced inflammasome activation has been associated with REV-ERB agonists, the molecular mechanism of REV-ERB antagonism during ischemia/reperfusion remains to be further elucidated. Given the metabolic regulation of NAD+ and its role in cardiac ischemia/reperfusion discussed above, it is tempting to speculate that REV-ERB antagonism leads to upregulation (disinhibition) of BMAL1, which allows increased expression of NAMPT and myocardial NAD+, thus resulting in cardiac protection from ischemia/reperfusion injury that is independent of REV-ERB–directed cardiac protection. If this is true, the REV-ERB antagonist–mediated cardiac protection after ischemia/reperfusion will be attenuated in cardiac-specific NAMPT-deficient mice. This remains to be tested.
Circadian disruption in humans.
The classic example of chronic circadian disruption is shift working. Early observations showed that shift workers have higher-than-average incidence of adverse cardiovascular events, including myocardial infarction, strokes, and sudden cardiac death (76–81), with a predilection for events occurring in the early morning hours. While behavior changes associated with shift working (and not the disruption of circadian clock per se) may also have contributed, it is of particular interest that the exposure time to shift working showed a linear correlation with the load of cardiac diseases (82).
There are also scenarios of acute rhythm disruption in modern life. “Social jet lag” refers to the timing difference in behavior between workdays and off days in a week. Seventy percent of the population reports social jet lag greater than 1 hour, and it has been associated with increased heart rate, increased cortisol levels, obesity, and diabetes — many of which are risk factors for cardiovascular diseases (83–85). Another interesting observation is that shifting to daylight saving time in the spring is associated with a modest transient increase in myocardial infarctions, which has been confirmed by a meta-analysis (86, 87). This increased risk is not observed in the winter shift back to standard time, suggesting that advancing behavior time (spring shift) may abolish the essential benefit of circadian anticipation.
While chronic circadian disruption relies on observational studies, acute disruption may be modeled in a controlled laboratory setting, although only a small number of healthy individuals have been studied to date. Laboratory human circadian misalignment protocol has been used to directly demonstrate the effect of disrupted circadian rhythm on human metabolism and physiological cardiovascular parameters (82, 88). While the physiological circadian variation of glucose tolerance is mainly a result of reduced insulin secretion in the evening, acute circadian disruption increases postprandial glucose mainly by decreasing insulin sensitivity (82). Also, a single rhythm shift led to elevated sleep systolic blood pressure and inflammatory markers, both of which have been associated with significant cardiovascular risks (88).
Future directions and opportunities for translation
Using traditional biochemical tools, we have captured many snapshots of circadian metabolism in the heart. It is time that we start to incorporate novel technologies such as artificial intelligence and machine learning to integrate these pieces of information and to try to understand circadian regulation at a system level. And a system-level blueprint of circadian metabolic regulation will allow us to better predict the flux outcome of each disruption and design more targeted and effective treatments for cardiovascular diseases.
The concept and the first example of chronotherapy, in which administration of medication at specific time points in the circadian cycle is used to achieve maximum efficacy and minimize side effects, were introduced when the alternate-morning oral corticosteroid regimen was proposed in the early 1960s (89). It is intuitive that the effectiveness and toxicity of many drugs vary depending on time of administration owing to the circadian rhythmicity of biochemical, physiological, and behavioral processes. One example is that a short-acting angiotensin-converting enzyme inhibitor (ACEI), captopril, shows cardiac remodeling benefits in mice only when given during the resting phase, not the active phase, despite having the same effect on blood pressure control (90). In fact, more than half of the 100 best-selling drugs in the United States target the product of an oscillatory gene, and half of these drugs have half-lives less than 6 hours (53). However, this information is rarely used in medical practice. Timing of medication may be just as important as dosing to optimize effectiveness while minimizing untoward effects. In general, medications protective against cardiac remodeling are likely best taken at bedtime (e.g., ACEIs and angiotensin receptor blockers), while medications that promote cardiac metabolism (e.g., NAD+ precursors) may be best taken early in the day. Pharmacokinetics studies have shown that twice-daily nicotinamide is able to increase the steady-state NAD+ level in the blood; however, whether the NAD+ level increases accordingly in the myocardium has yet to be examined (91, 92). Further, the window of therapy may also differ for the same medication depending on the different indications. Recent murine studies showed that NAD+ precursor supplementation improves the outcome of ischemia/reperfusion in mice only during the resting phase, when NAD+ is naturally low, and not during the active phase (20). Thus, for heart attack protection, NAD+ precursor may actually be best given at bedtime. While mice are nocturnal and humans are diurnal, most laboratory experiments are performed during the day for our convenience. Understanding the nuances of circadian variation will greatly enhance the design of human clinical studies to translate findings in the mouse models.
It takes at least 5 days for the heart to resynchronize after a reset of the cardiac peripheral clock (71). Strategies, whether pharmacological or behavioral/dietary, to facilitate the transition will shorten the window of dyssynchrony and potentially lower the risk of cardiometabolic disorder in shift workers, individuals who experience social jet lag, and frequent air travelers.
Aging attenuates circadian oscillation, although it is not clear whether this is due to an overall dampened oscillation or an inability to synchronize within the cell population (93). Single-cell sequencing may provide new insights into this phenomenon. Pharmacological and behavior modifications (lighting, exercise, and feeding schedules) reinforcing the oscillatory peripheral clock may deter the cardiovascular risk factors associated with aging (94–97).
Five to ten percent of genes oscillate in almost any tissue in the body, yet the overlaps between different tissue types are minimal (53). This observation attests to the fact that the peripheral circadian clock governs key functions of each tissue and allows upregulation of subsets of genes in a critical time window to optimize tissue function. Strategies to manipulate the core clock will allow synchronized regulation of this oscillatory gene set and may be a more efficient way of optimizing tissue function than altering each single gene. Indeed, treatment with a REV-ERB agonist was the first example of direct manipulation of the core clock to provide cardiac protection against pressure overload. Its mechanism is by synchronous regulation of a large number of target genes and the reprogramming of cardiac metabolism (60).
Conclusion
The cardiac peripheral clock has evolved to allow the heart to anticipate various energy demands during a 24-hour day. Much of the cardiac metabolism shows a diurnal rhythm under complex regulation at multiple molecular levels. Disruption of circadian clock function associated with modern lifestyle leads to increased risk of cardiovascular diseases. Understanding the molecular mechanisms and the intricate regulation of cardiac circadian gene regulation will allow us to modify our behavioral risk factors to reduce cardiovascular risks, and to develop effective treatments.
Acknowledgments
This work was supported by the National Heart, Lung, and Blood Institute (K08HL123551 to LZ, R01HL143067 to LZ, R35HL135789 to MKJ, and R01HL086548 to MKJ), an American Heart Association–Allen Frontiers Award (to MKJ), NIH grant R01DK111478 (to MKJ), and the Leducq Foundation Transatlantic Network of Excellence (to MKJ).
Version 1. 08/02/2021
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(15):e148276.https://doi.org/10.1172/JCI148276.
References
- 1.Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2017;18(3):164–179. doi: 10.1038/nrg.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hastings MH, et al. Generation of circadian rhythms in the suprachiasmatic nucleus. Nat Rev Neurosci. 2018;19(8):453–469. doi: 10.1038/s41583-018-0026-z. [DOI] [PubMed] [Google Scholar]
- 3.Neely JR, et al. Myocardial utilization of carbohydrate and lipids. Prog Cardiovasc Dis. 1972;15(3):289–329. doi: 10.1016/0033-0620(72)90029-1. [DOI] [PubMed] [Google Scholar]
- 4.Ballard FB, et al. Myocardial metabolism of fatty acids. J Clin Invest. 1960;39:717–723. doi: 10.1172/JCI104088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldberg IJ, et al. Lipid metabolism and toxicity in the heart. Cell Metab. 2012;15(6):805–812. doi: 10.1016/j.cmet.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stavinoha MA, et al. Diurnal variations in the responsiveness of cardiac and skeletal muscle to fatty acids. Am J Physiol Endocrinol Metab. 2004;287(5):E878–E887. doi: 10.1152/ajpendo.00189.2004. [DOI] [PubMed] [Google Scholar]
- 7.Durgan DJ, et al. Circadian rhythms in myocardial metabolism and contractile function: influence of workload and oleate. Am J Physiol Heart Circ Physiol. 2007;293(4):H2385–H2393. doi: 10.1152/ajpheart.01361.2006. [DOI] [PubMed] [Google Scholar]
- 8.Tsai JY, et al. Direct regulation of myocardial triglyceride metabolism by the cardiomyocyte circadian clock. J Biol Chem. 2010;285(5):2918–2929. doi: 10.1074/jbc.M109.077800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kemppainen J, et al. Myocardial and skeletal muscle glucose uptake during exercise in humans. J Physiol. 2002;542(pt 2):403–412. doi: 10.1113/jphysiol.2002.018135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodwin GW, et al. Regulation of energy metabolism of the heart during acute increase in heart work. J Biol Chem. 1998;273(45):29530–29539. doi: 10.1074/jbc.273.45.29530. [DOI] [PubMed] [Google Scholar]
- 11.Allard MF, et al. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994;267(2 pt 2):H742–H750. doi: 10.1152/ajpheart.1994.267.2.H742. [DOI] [PubMed] [Google Scholar]
- 12.Durgan DJ, et al. O-GlcNAcylation, novel post-translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J Biol Chem. 2011;286(52):44606–44619. doi: 10.1074/jbc.M111.278903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slot JW, et al. Translocation of the glucose transporter GLUT4 in cardiac myocytes of the rat. Proc Natl Acad Sci U S A. 1991;88(17):7815–7819. doi: 10.1073/pnas.88.17.7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider CA, et al. Feeding and fasting determine postischemic glucose utilization in isolated working rat hearts. Am J Physiol. 1991;260(2 pt 2):H542–H548. doi: 10.1152/ajpheart.1991.260.2.H542. [DOI] [PubMed] [Google Scholar]
- 15.Laczy B, et al. Protein O-GlcNAcylation: a new signaling paradigm for the cardiovascular system. Am J Physiol Heart Circ Physiol. 2009;296(1):H13–H28. doi: 10.1152/ajpheart.01056.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darley-Usmar VM, et al. Protein O-linked β-N-acetylglucosamine: a novel effector of cardiomyocyte metabolism and function. J Mol Cell Cardiol. 2012;52(3):538–549. doi: 10.1016/j.yjmcc.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rau E, Meyer DK. A diurnal rhythm of incorporation of L-[3H] leucine in myocardium of the rat. Recent Adv Stud Cardiac Struct Metab. 1975;7:105–110. [PubMed] [Google Scholar]
- 18.Zhang L, et al. KLF15 establishes the landscape of diurnal expression in the heart. Cell Rep. 2015;13(11):2368–2375. doi: 10.1016/j.celrep.2015.11.038. [DOI] [PubMed] [Google Scholar]
- 19.Mori V, et al. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS One. 2014;9(11):e113939. doi: 10.1371/journal.pone.0113939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L, et al. Kruppel-like factor 15 regulates the circadian susceptibility to ischemia reperfusion injury in the heart. Circulation. 2020;141(17):1427–1429. doi: 10.1161/CIRCULATIONAHA.119.041664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Um JH, et al. AMPK regulates circadian rhythms in a tissue- and isoform-specific manner. PLoS One. 2011;6(3):e18450. doi: 10.1371/journal.pone.0018450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peliciari-Garcia RA, et al. Altered myocardial metabolic adaptation to increased fatty acid availability in cardiomyocyte-specific CLOCK mutant mice. Biochim Biophys Acta. 2016;1861(10):1579–1595. doi: 10.1016/j.bbalip.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young ME, et al. Cardiomyocyte-specific BMAL1 plays critical roles in metabolism, signaling, and maintenance of contractile function of the heart. J Biol Rhythms. 2014;29(4):257–276. doi: 10.1177/0748730414543141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu W, et al. NAD+ metabolism as an emerging therapeutic target for cardiovascular diseases associated with sudden cardiac death. Front Physiol. 2020;11:901. doi: 10.3389/fphys.2020.00901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Goede P, et al. Circadian rhythms in mitochondrial respiration. J Mol Endocrinol. 2018;60(3):R115–R130. doi: 10.1530/JME-17-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohsaka A, et al. The circadian clock maintains cardiac function by regulating mitochondrial metabolism in mice. PLoS One. 2014;9(11):e112811. doi: 10.1371/journal.pone.0112811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dittenhafer-Reed KE, et al. SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab. 2015;21(4):637–646. doi: 10.1016/j.cmet.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahn BH, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105(38):14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koentges C, et al. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res Cardiol. 2015;110(4):36. doi: 10.1007/s00395-015-0493-6. [DOI] [PubMed] [Google Scholar]
- 30.Sundaresan NR, et al. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119(9):2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tseng AH, et al. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic Biol Med. 2013;63:222–234. doi: 10.1016/j.freeradbiomed.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 32.Tao R, et al. Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid Redox Signal. 2014;20(10):1646–1654. doi: 10.1089/ars.2013.5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hafner AV, et al. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2(12):914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lahiri V, et al. Watch what you (self-) eat: autophagic mechanisms that modulate metabolism. Cell Metab. 2019;29(4):803–826. doi: 10.1016/j.cmet.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma D, et al. Temporal orchestration of circadian autophagy rhythm by C/EBPβ. EMBO J. 2011;30(22):4642–4651. doi: 10.1038/emboj.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGinnis GR, et al. Genetic disruption of the cardiomyocyte circadian clock differentially influences insulin-mediated processes in the heart. J Mol Cell Cardiol. 2017;110:80–95. doi: 10.1016/j.yjmcc.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfeifer U, Strauss P. Autophagic vacuoles in heart muscle and liver. A comparative morphometric study including circadian variations in meal-fed rats. J Mol Cell Cardiol. 1981;13(1):37–49. doi: 10.1016/0022-2828(81)90227-3. [DOI] [PubMed] [Google Scholar]
- 38.Buhr ED, Takahashi JS Molecular components of the Mammalian circadian clock. Handb Exp Pharmacol. 2013(217):3–27. doi: 10.1007/978-3-642-25950-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee C, et al. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107(7):855–867. doi: 10.1016/S0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 40.Preitner N, et al. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110(2):251–260. doi: 10.1016/S0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- 41.Sato TK, et al. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron. 2004;43(4):527–537. doi: 10.1016/j.neuron.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, et al. Gene regulation. Discrete functions of nuclear receptor Rev-erbα couple metabolism to the clock. Science. 2015;348(6242):1488–1492. doi: 10.1126/science.aab3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Panda S, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109(3):307–320. doi: 10.1016/S0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- 44.Storch KF, et al. Extensive and divergent circadian gene expression in liver and heart. Nature. 2002;417(6884):78–83. doi: 10.1038/nature744. [DOI] [PubMed] [Google Scholar]
- 45.Bray MS, et al. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am J Physiol Heart Circ Physiol. 2008;294(2):H1036–H1047. doi: 10.1152/ajpheart.01291.2007. [DOI] [PubMed] [Google Scholar]
- 46.Podobed P, et al. The day/night proteome in the murine heart. Am J Physiol Regul Integr Comp Physiol. 2014;307(2):R121–R137. doi: 10.1152/ajpregu.00011.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim YH, Lazar MA. Transcriptional control of circadian rhythms and metabolism: a matter of time and space. Endocr Rev. 2020;41(5):707–732. doi: 10.1210/endrev/bnaa014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menet JS, et al. CLOCK:BMAL1 is a pioneer-like transcription factor. Genes Dev. 2014;28(1):8–13. doi: 10.1101/gad.228536.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu B, et al. Coactivator-dependent oscillation of chromatin accessibility dictates circadian gene amplitude via REV-ERB loading. Mol Cell. 2015;60(5):769–783. doi: 10.1016/j.molcel.2015.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koike N, et al. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338(6105):349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Partch CL, et al. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014;24(2):90–99. doi: 10.1016/j.tcb.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Menet JS, et al. Nascent-Seq reveals novel features of mouse circadian transcriptional regulation. Elife. 2012;1:e00011. doi: 10.7554/eLife.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang R, et al. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci U S A. 2014;111(45):16219–16224. doi: 10.1073/pnas.1408886111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.ATLAS Collaboration. et al. Muon reconstruction efficiency and momentum resolution of the ATLAS experiment in proton–proton collisions at √s = 7 TeV in 2010. Eur Phys J C Part Fields. 2014;74(9):3034. doi: 10.1140/epjc/s10052-014-3034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeyaraj D, et al. Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature. 2012;483(7387):96–99. doi: 10.1038/nature10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pepin ME, et al. Genome-wide DNA methylation encodes cardiac transcriptional reprogramming in human ischemic heart failure. Lab Invest. 2019;99(3):371–386. doi: 10.1038/s41374-018-0104-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Etchegaray JP, et al. The polycomb group protein EZH2 is required for mammalian circadian clock function. J Biol Chem. 2006;281(30):21209–21215. doi: 10.1074/jbc.M603722200. [DOI] [PubMed] [Google Scholar]
- 58.Prosdocimo DA, et al. Kruppel-like factor 15 is a critical regulator of cardiac lipid metabolism. J Biol Chem. 2014;289(9):5914–5924. doi: 10.1074/jbc.M113.531384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramsey KM, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324(5927):651–654. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang L, et al. REV-ERBalpha ameliorates heart failure through transcription repression. JCI Insight. 2017;2(17):e95177. doi: 10.1172/jci.insight.95177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang EE, et al. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell. 2009;139(1):199–210. doi: 10.1016/j.cell.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramanathan C, et al. mTOR signaling regulates central and peripheral circadian clock function. PLoS Genet. 2018;14(5):e1007369. doi: 10.1371/journal.pgen.1007369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khapre RV, et al. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging (Albany NY) 2014;6(1):48–57. doi: 10.18632/aging.100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang SW, et al. Circadian rhythm of intracellular protein synthesis signaling in rat cardiac and skeletal muscles. Biochem Biophys Rep. 2017;9:153–158. doi: 10.1016/j.bbrep.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lipton JO, et al. The circadian protein BMAL1 regulates translation in response to S6K1-mediated phosphorylation. Cell. 2015;161(5):1138–1151. doi: 10.1016/j.cell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tao R, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40(6):893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park SY, et al. O-GlcNAc modification on IRS-1 and Akt2 by PUGNAc inhibits their phosphorylation and induces insulin resistance in rat primary adipocytes. Exp Mol Med. 2005;37(3):220–229. doi: 10.1038/emm.2005.30. [DOI] [PubMed] [Google Scholar]
- 68.Luo B, et al. Chronic hexosamine flux stimulates fatty acid oxidation by activating AMP-activated protein kinase in adipocytes. J Biol Chem. 2007;282(10):7172–7180. doi: 10.1074/jbc.M607362200. [DOI] [PubMed] [Google Scholar]
- 69.Liu J, et al. Increased hexosamine biosynthesis and protein O-GlcNAc levels associated with myocardial protection against calcium paradox and ischemia. J Mol Cell Cardiol. 2006;40(2):303–312. doi: 10.1016/j.yjmcc.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 70.Laczy B, et al. Acute regulation of cardiac metabolism by the hexosamine biosynthesis pathway and protein O-GlcNAcylation. PLoS One. 2011;6(4):e18417. doi: 10.1371/journal.pone.0018417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Durgan DJ, et al. The circadian clock within the cardiomyocyte is essential for responsiveness of the heart to fatty acids. J Biol Chem. 2006;281(34):24254–24269. doi: 10.1074/jbc.M601704200. [DOI] [PubMed] [Google Scholar]
- 72.Ingle KA, et al. Cardiomyocyte-specific Bmal1 deletion in mice triggers diastolic dysfunction, extracellular matrix response, and impaired resolution of inflammation. Am J Physiol Heart Circ Physiol. 2015;309(11):H1827–H1836. doi: 10.1152/ajpheart.00608.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dierickx P, et al. SR9009 has REV-ERB-independent effects on cell proliferation and metabolism. Proc Natl Acad Sci U S A. 2019;116(25):12147–12152. doi: 10.1073/pnas.1904226116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reitz CJ, et al. SR9009 administered for one day after myocardial ischemia-reperfusion prevents heart failure in mice by targeting the cardiac inflammasome. Commun Biol. 2019;2:353. doi: 10.1038/s42003-019-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Montaigne D, et al. Daytime variation of perioperative myocardial injury in cardiac surgery and its prevention by Rev-Erbα antagonism: a single-centre propensity-matched cohort study and a randomised study. Lancet. 2018;391(10115):59–69. doi: 10.1016/S0140-6736(17)32132-3. [DOI] [PubMed] [Google Scholar]
- 76.Crnko S, et al. Circadian rhythms and the molecular clock in cardiovascular biology and disease. Nat Rev Cardiol. 2019;16(7):437–447. doi: 10.1038/s41569-019-0167-4. [DOI] [PubMed] [Google Scholar]
- 77.Abbott SM, et al. Circadian disruption and human health: a bidirectional relationship. Eur J Neurosci. 2020;51(1):567–583. doi: 10.1111/ejn.14298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jain M, et al. Circadian rhythm and cardiovascular disorders. ChronoPhysiol Ther. 2014;2014(12):27 [Google Scholar]
- 79.Zhang J, et al. Circadian regulation of cardiac physiology: rhythms that keep the heart beating. Annu Rev Physiol. 2020;82:79–101. doi: 10.1146/annurev-physiol-020518-114349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roenneberg T, Merrow M. The circadian clock and human health. Curr Biol. 2016;26(10):R432–R443. doi: 10.1016/j.cub.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 81.Walker WH, et al. Circadian rhythm disruption and mental health. Transl Psychiatry. 2020;10(1):28. doi: 10.1038/s41398-020-0694-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morris CJ, et al. Endogenous circadian system and circadian misalignment impact glucose tolerance via separate mechanisms in humans. Proc Natl Acad Sci U S A. 2015;112(17):E2225–E2234. doi: 10.1073/pnas.1418955112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roenneberg T, et al. Social jetlag and obesity. Curr Biol. 2012;22(10):939–943. doi: 10.1016/j.cub.2012.03.038. [DOI] [PubMed] [Google Scholar]
- 84.Rutters F, et al. Is social jetlag associated with an adverse endocrine, behavioral, and cardiovascular risk profile? J Biol Rhythms. 2014;29(5):377–383. doi: 10.1177/0748730414550199. [DOI] [PubMed] [Google Scholar]
- 85.Parsons MJ, et al. Social jetlag, obesity and metabolic disorder: investigation in a cohort study. Int J Obes (Lond) 2015;39(5):842–848. doi: 10.1038/ijo.2014.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jiddou MR, et al. Incidence of myocardial infarction with shifts to and from daylight savings time. Am J Cardiol. 2013;111(5):631–635. doi: 10.1016/j.amjcard.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 87.Manfredini R, et al. Daylight saving time and acute myocardial infarction: a meta-analysis. J Clin Med. 2019;8(3):404. doi: 10.3390/jcm8030404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morris CJ, et al. Circadian misalignment increases cardiovascular disease risk factors in humans. Proc Natl Acad Sci U S A. 2016;113(10):E1402–E1411. doi: 10.1073/pnas.1516953113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Harter JG, et al. Studies on an intermittent corticosteroid dosage regimen. N Engl J Med. 1963;269:591–596. doi: 10.1056/NEJM196309192691201. [DOI] [PubMed] [Google Scholar]
- 90.Martino TA, et al. The primary benefits of angiotensin-converting enzyme inhibition on cardiac remodeling occur during sleep time in murine pressure overload hypertrophy. J Am Coll Cardiol. 2011;57(20):2020–2028. doi: 10.1016/j.jacc.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 91.Airhart SE, et al. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS One. 2017;12(12):e0186459. doi: 10.1371/journal.pone.0186459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Trammell SA, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948. doi: 10.1038/ncomms12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kunieda T, et al. Cellular senescence impairs circadian expression of clock genes in vitro and in vivo. Circ Res. 2006;98(4):532–539. doi: 10.1161/01.RES.0000204504.25798.a8. [DOI] [PubMed] [Google Scholar]
- 94.Damiola F, et al. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000;14(23):2950–2961. doi: 10.1101/gad.183500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stokkan KA, et al. Entrainment of the circadian clock in the liver by feeding. Science. 2001;291(5503):490–493. doi: 10.1126/science.291.5503.490. [DOI] [PubMed] [Google Scholar]
- 96.Buxton OM, et al. Exercise elicits phase shifts and acute alterations of melatonin that vary with circadian phase. Am J Physiol Regul Integr Comp Physiol. 2003;284(3):R714–R724. doi: 10.1152/ajpregu.00355.2002. [DOI] [PubMed] [Google Scholar]
- 97.Scheer FA, et al. Daily nighttime melatonin reduces blood pressure in male patients with essential hypertension. Hypertension. 2004;43(2):192–197. doi: 10.1161/01.HYP.0000113293.15186.3b. [DOI] [PubMed] [Google Scholar]