

Abstract
Clear cell sarcoma (CCS) is a deadly malignancy affecting adolescents and young adults. It is characterized by reciprocal translocations resulting in expression of the chimeric EWSR1-ATF1 or EWSR1-CREB1 fusion proteins, driving sarcomagenesis. Besides these characteristics, CCS has remained genomically uncharacterized. Copy number analysis of human CCSs showed frequent amplifications of the MITF locus and chromosomes 7 and 8. Few alterations were shared with Ewing sarcoma or desmoplastic, small round cell tumors, which are other EWSR1-rearranged tumors. Exome sequencing in mouse tumors generated by expression of EWSR1-ATF1 from the Rosa26 locus demonstrated no other repeated pathogenic variants. Additionally, we generated a new CCS mouse by Cre-loxP–induced chromosomal translocation between Ewsr1 and Atf1, resulting in copy number loss of chromosome 6 and chromosome 15 instability, including amplification of a portion syntenic to human chromosome 8, surrounding Myc. Additional experiments in the Rosa26 conditional model demonstrated that Mitf or Myc can contribute to sarcomagenesis. Copy number observations in human tumors and genetic experiments in mice rendered, for the first time to our knowledge, a functional landscape of the CCS genome. These data advance efforts to understand the biology of CCS using innovative models that will eventually allow us to validate preclinical therapies necessary to achieve longer and better survival for young patients with this disease.
Keywords: Genetics, Oncology
Keywords: Cancer, Mouse models, Oncogenes

Introduction
Specific chromosomal translocations are associated with defined malignancies, especially among leukemias and sarcomas. In some of these translocation-associated cancers, few genetic perturbations other than the characteristic fusion oncogene have been identified. Although the scarcity of genetic perturbations provides strong circumstantial evidence that the fusion protein generated by each chromosomal translocation drives its associated cancer, experimental evidence is preferable.
Early attempts to create mouse models to test the oncogenicity of specific products of chromosomal translocations involved the expression of fusion gene products of chromosomal translocations as randomly inserted transgenes under the control of general promoters (1). Others placed fusion genes under Cre-loxP conditionally controlled expression from general promoters (2–5). Still others modified the 5′ parent locus by the targeted addition of a coding sequence from the 3′ parent locus, moved into position by Cre-mediated recombination (6). The Cre-loxP system has also been used to generate chromosomal translocations in mouse somatic cells (7–10), but only 1 resulted in the associated leukemia (9).
Clear cell sarcoma (CCS) is a rare but particularly deadly type of soft-tissue sarcoma that arises most commonly in adolescents and young adults. It was initially characterized by tissue features reminiscent of melanoma, but later found to associate consistently with either t(12;22) or t(2;22) balanced chromosomal translocations, generating EWSR1-ATF1 or EWSR1-CREB1, respectively. Each of these fusion oncogenes was later demonstrated to associate with a slightly broader range of neoplasias that did not consistently include the melanocytic differentiation of the original, clinically defined entity of CCS. A mouse model expressing the human fusion gene EWSR1-ATF1 was found to generate a faithful recapitulation of human CCS, as well as other variants of EWSR1-ATF1–related neoplasias, depending on the cell of origin selected for conditional expression of the fusion gene (5, 11). This observation made the strong argument that EWSR1-ATF1 is sufficient for induction of CCS and functions as a true oncogene. The presence of other secondary genetic changes in CCS has not been previously characterized.
As much as translocation-driven sarcomas are considered to be otherwise genetically quiet, those that have been better characterized than CCS are not completely lacking other alterations (12–21). For example, Ewing sarcoma (ES) is a pediatric bone tumor driven by a related chimeric fusion oncogene, EWSR1-FLI1. Beyond the pathognomonic fusion, a minority of tumors show a loss of STAG2 or TP53 (12, 16, 21). ESs also harbor recurrent copy number alterations (CNAs), such as amplification of 1q and trisomies of chromosomes 8 and 12 (22).
A number of questions arise when such secondary alterations are observed repeatedly in a single type of tumor. Do they represent factors that help drive tumor formation, or merely passenger mutations that accompany it? If these alterations are cofactors to transformation driven by a specific translocation fusion oncogene, do they overcome some vulnerability engendered by that fusion, or synergize with that fusion to drive oncogenesis? Are the specific secondary changes selected for by necessity or just by increasing the rate of cell proliferation? Can these accompanying genetic changes reveal details about the mechanism of the fusion oncoprotein’s function in a cell? Testing the necessity or sufficiency of these secondary changes requires a model for tumorigenesis to which the observed secondary change can be added or removed. Such a model is generally lacking for ES, but readily available for CCS.
We set out to profile the CNAs in a cohort of human CCS tumors and then compare these findings with both observations and experimental manipulations of the genome in mouse genetic models of CCS, including a new model driven by an induced chromosomal translocation.
Results
Human CCS tumors demonstrate recurrent CNAs.
We used formalin-fixed, paraffin-embedded (FFPE) human CCS tissue specimens and matched adjacent tissue controls to retrieve tumor and germline DNA. Thirteen of these pairs rendered sufficient quantities and quality of DNA to be hybridized onto the OncoScan CNV Plus array, a microarray-based assay of CNAs and loss of heterozygosity that is intended for FFPE-retrieved DNA. The assay enables the detection of relevant data across the whole genome and specifically checks for the presence of 64 hotspot mutations in 9 cancer-related genes (BRAF, EGFR, IDH1, IDH2, KRAS, NRAS, PIK3CA, PTEN, and TP53). Only a single tumor showed any of these point mutations, an activating mutation of EGFR (Figure 1A).
Figure 1. Human CCSs demonstrate a few repeated CNAs across the genome.

(A) The frequency of copy number gains (blue) or losses (red), LOH (yellow), and point mutations (cyan diamond), as called by BioDiscovery Nexus Express, is shown for CCSs (n = 13). (B) The frequency of copy number gains (blue) or losses (red) is shown for DSRCTs (n = 8), as analyzed in parallel with CCSs for an accurate comparison. chr., chromosome.
Across the CCS samples, we found that the most frequent large copy number increase (ranging in length from >10 Mb to an entire chromosome arm) occurred for chromosomes 8 and 9. Ten of the 13 samples (76.9%) demonstrated a copy number increase in 8q, and 7 of the 13 samples (53.8%) had either gains or deletions of 8p and 9p (Figure 1A). The long arm of chromosome 7 also had a high percentage of CNAs (61.5%, all gains), as did 7p, 9q, and 17q. Analysis for co-occurrence or exclusion revealed only 3 significant pairings or associations: co-occurrence of 9q and 11q (P = 0.035), 17q and age under 20 years (P = 0.035), and exclusion of 8q and 14q (P = 0.038). Copy number increases at MITF and FAT1 were observed in 6 and 5 of the 13 samples, respectively. We observed no focal loss of CDKN2A, although 5 of the 13 samples had deletions of 9p, which contains CDKN2A.
Some human CCS CNAs are shared with other tumors associated with EWSR1 fusions.
We performed the same OncoScan Plus array analysis on 8 desmoplastic small round cell tumors (DSRCTs) with matched control tissues. These tumors were chosen because they are characterized by the translocation-generated EWSR1-WT1 fusion oncogene and thus share with CCS the EWSR1 gene involvement. For DSRCT samples, 5 of the 8 (62.5%) had gains of chromosome 5 that were nearly absent in the CCS samples (Figure 1B). We also observed deletions of 8p or 16q (in 3 of the 8, 37.5%, incomplete 16q arm deletion in 1 of the 3) and copy number increases on 18p or chromosome 20 (also in 3 of the 8, 37.5%, incomplete 18p arm deletion in 1 of the 3). No co-occurrence or exclusion of gains or deletions were observed at an unadjusted P < 0.05 level. The gains at MITF and FAT1 observed in CCS were absent in the DSRCTs.
Overall, the frequencies of large (>10 Mb) increases in copy numbers on 8q and 8p, as well as focal gains involving MITF, were greater in CCS than DSRCTs, whereas gains of 5q, 5p, and 20p and loss of the X chromosome were substantially more frequent in the DSRCT samples.
We compared the frequency of common CNAs in ES with the frequencies seen in CCS and DSRCTs. Trisomy of chromosome 8 is reported in the majority of EWSR1-FLI1 tumors, along with amplifications of 1q and whole chromosome 12 (Figure 2). Using the data from Tirode et al. (21) on 112 tumors from patients with ES as a comparator, we found no significant difference in the prevalence of copy number increases in 1q and chromosomes 8 and 12 between ES and CCS tumors. The prevalence of deletions of CDKN2A and 16q in ES and CCS tumors were also not significantly different. The same findings of no difference in prevalence were observed when comparing DSRCTs with ESs for these specific alterations, except for a significantly higher prevalence of chromosome 8 copy number increases in ESs compared with DSRCTs (P = 0.02).
Figure 2. CCS shares few secondary genomic alterations with other EWSR1-rearranged cancers.
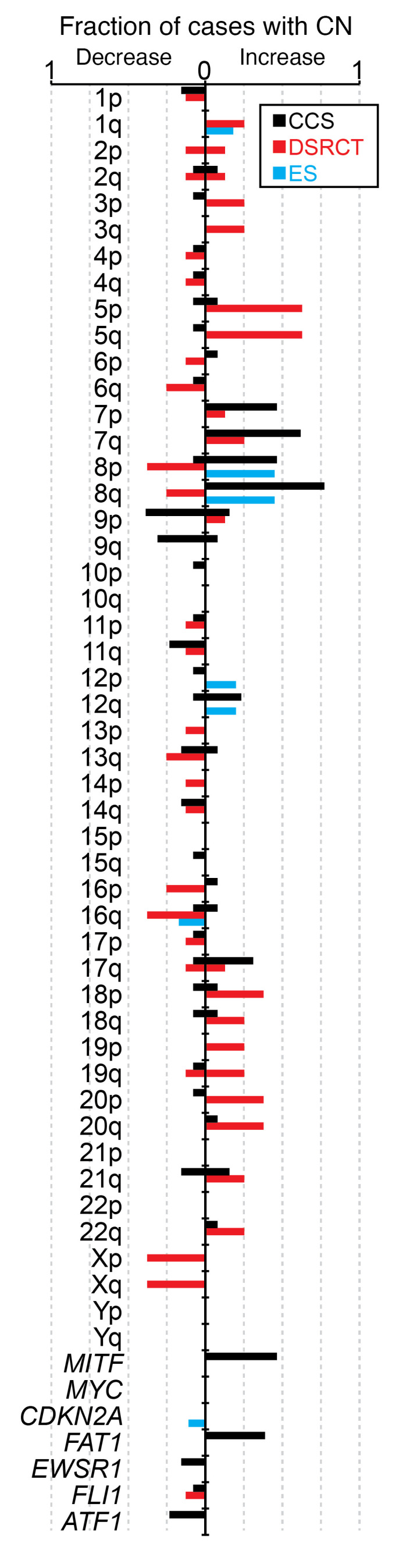
Summary of CNAs by chromosome arm and genes of interest, displayed as the fraction (0–1) for each sarcoma subtype: CCS (n = 13), DSRCT (n = 8), and ES (n = 112). The prevalence of gains and losses for CCSs and DSRCTs reflects the heterozygous or homozygous loss or copy number (CN) gains present with at least 10% estimated clonality.
EWSR1-ATF1 is sufficient for murine sarcomagenesis without additional protein-coding mutations.
Mice engineered to conditionally express human EWSR1-ATF1 from the Rosa26 locus were administered tamoxifen at various ages to induce Bmi1CreERT2 activity (Figure 3, A–C). This specific lineage is optimally suited to produce clonal sarcomas with clear cell morphology and the characteristic CCS melanoma–like features (5). When we sequenced exomes from 34 tumors harvested from 6 mice (Figure 3D, and each large enough to permit isolation of high-quality DNA and RNA samples) to a minimum depth of 20× coverage and compared them with matched host germline exomes sequenced to a minimum depth of 8× coverage, we identified 7846 variants. Of these, we defined high-quality variants as having no more than 1 variant read in the host germline and at least 2 variant reads in the tumor, yielding 265 potential somatic variant alleles (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI146301DS1). This list of potential somatic variants was subjected to targeted sequencing to a minimum depth of 100× coverage for both germline controls and tumors. A variant allele frequency (VAF) of no more than 0.01 in a matched control and at least 0.05 in a tumor yielded 107 variants in 26 of the tumors, with 8 tumors having no variants that reached these thresholds (Figure 3E and Supplemental Table 2).
Figure 3. Exome sequencing of EWSR1-ATF1 expression–initiated mouse tumors reveals that no secondary alterations are strictly required to complete sarcomagenesis.

(A) Schematic illustrating CreERT2 engineered at the Bmi1 locus in conjunction with Cre-inducible human EWSR1-ATF1 engineered at the Rosa26 locus. Cre recombinase removes the neoR-STOP cassette to induce expression of EWSR1-ATF1, and the GFP reporter confirms EWSR1-ATF1 expression. (B) Tamoxifen injection induces CreERT2 activity in Bmi1-expressing stem cells (blue), and then in turn, Cre recombinase induces Rosa26-mediated expression of EWSR1-ATF1 (EA1). (C) Time course showing age at injection and age at tumor harvesting (n = 6). (D) Summary of total number of tumors detected and harvested per mouse (n = 6). (E) Exome sequencing of 34 mouse tumors, with 1 tumor’s exome presented on each line, clustered by host mouse, and each variant allele denoted by a circle whose size corresponds to the VAF. All variants with a VAF of greater than 0.25 are also identified by corresponding colors in the list of gene symbols and protein amino acid substitutions below. (F) RNA-Seq rendered expression levels relative to Gapdh for the 8 genes with variants present at fractions higher than 0.4. The level of Mitf expression is included for reference. (G) Exome-wide CNV was inferred from exome sequencing data, as shown for 12 mouse tumors. Normal diploidy is represented by the x axis crossing at a value of y = 2.
KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis of the genes implicated by the 82 variants with a VAF of greater than 0.1 identified shared involvement in calcium signaling, chemical carcinogenesis, and aldosterone synthesis and secretion pathways, but none reached more than minimal significance. Because the tumor cell prevalence in mouse CCS tumor tissues was consistently greater than 80%, a VAF of 0.4 was considered the lowest that might represent a clonal variant across the tumor cells, which could potentially have contributed to the initial tumorigenesis. We performed RNA-Seq to measure expression levels of the 8 genes with a VAF of greater than 0.4 in a subset of 22 of these 34 tumors; only 3 genes showed substantial expression, a level set at 1% of Gapdh expression and comparable to that of Mitf (Figure 3F and Supplemental Tables 3 and 4). Thus, less than 10% of the tumors bore even a single mutation that was found to be clonal across the tumor cells and that affected a gene that was expressed generally by this tumor type.
We also analyzed exome data with regard to copy number variation (CNV). No repeated changes were identified across the group of tumors (Figure 3G). Further, no whole chromosome gains or losses were noted in the exome of any of the tumors, and no large fragment changes reached significance.
Induced chromosomal translocation in mice drives tumorigenesis and reveals other secondary changes.
We next sought to develop a new mouse model that could recapitulate CCS more closely at the genetic level, by an induced chromosomal translocation. In the mouse, both the Ewsr1 and Atf1 loci bear significant homology to each respective parent locus in the human genome (5). However, in contrast to their human counterparts, the mouse genes are in opposite orientation with respect to each host chromosome’s centromere, such that recombination generates an acentric chromosome bearing the Ewsr1-Atf1 fusion gene as well as most of chromosome 11 and a small fragment of chromosome 15, and a dicentric chromosome bearing most of chromosome 15 and a small fragment of chromosome 11 (Figure 4A). Although different from the human t(12;22) translocation, the mouse translocation would therefore permit the investigation of 2 important aspects of oncogene-driven sarcomagenesis. First, retention, or possibly amplification, of an acentric chromosome fragment with the fusion oncogene would indicate that either tumor formation and growth were dependent on the presence of the fusion oncogene, or that there was selection for tumor cells in which the fusion oncogene was present. Second, dicentric chromosome generation is thought to drive aneuploidy due to double-stranded breaks that develop during cytokinesis after the 2 centromeres are pulled to different daughter cells. This should encourage CNV and even chromothripsis, which might uncover secondary changes that accompany murine clear cell sarcomagenesis driven by a chromosomal translocation. Therefore, using homologous recombination, we targeted a single loxP site into the seventh intron of the Ewsr1 locus on mouse chromosome 11 and another into the fourth intron of the Atf1 locus on mouse chromosome 15 (Figure 4A).
Figure 4. Cre-mediated chromosomal translocation induces sarcomagenesis in the mouse.

(A) Schematic representation of the loxP sites targeted to Ewsr1 intron 7 and Atf1 intron 4, as well as the 2 products of Cre-mediated chromosomal translocation. (B) Schematics and Kaplan-Meier survival plots of tumorigenesis in mice heterozygous for Ewsr1-loxP and Atf1-loxP, induced by 3 different Cre recombinase delivery methods: a knockin allele (HprtCre), a transgenic allele (Prx1Cre), and injection of Cre recombinase protein (TATCre). (C) Gross photo of a hind-limb Prx1Cre-induced tumor (arrowhead) forming in a mouse. Scale bar: 10 mm. (D) Magnetic resonance images of HprtCre- or TATCre-induced tumors (indicated by arrowheads) forming in the thigh, dorsal pelvis, thigh, and both the pelvis and contralateral thigh of mice (upper panels, T2-weighted; lower panels, proton density–weighted). (E) Representative H&E-stained photomicrographs of histological sections of tumors from translocation (top panels), Rosa26-EA1–expressing (middle panels), and human CCS tumors (below), with (from left to right) short spindle cell, myxoid, and clear cell morphologies. Scale bars: 10 μm. (F) Schematic of the PCR amplification strategy with a schematic of primers that amplify across each translocation site, tested in genomic tumor DNA from a TATCre-induced tumor, with WT and lox alleles as well as each translocation product detected by specified primer combinations. ER, Ewsr1 reverse; EF, Ewsr1 forward; AF, Atf1 forward; AR, Atf1 reverse. (G) Graph showing the RNA-Seq–determined expression (in fragments per kilobase per million reads [FPKM]) of 2 melanocytic marker genes, with 8 HprtCre-induced translocation tumors and their mean indicated in black and the mean of 13 EA1-induced tumors in gray.
Double-heterozygous mice that also had HprtCre, Prx1Cre, or limb injection of the protein TATCre generated tumors at similar penetrance (Figure 4B). Tumors arose in mesenchymal tissues, as was seen in the Rosa26-EA1 models, but at longer latencies and a lower prevalence (Figure 4, C and D). Histologically, translocation-generated tumors matched the range of tumors produced in the Rosa26 models as well as the range of neoplasias associated with EWSR1-ATF1 in humans, showing the typical clear cytoplasm and enlarged, irregular hyperchromatic nuclei (Figure 4, E–G).
We isolated total RNA from 8 tumors, generated cDNA libraries using the RiboZero method, and then sequenced these on an Illumina HiSeq instrument to generate transcriptomes comparable to those from a prior sequencing effort of many mouse genetic cancer models, including the Rosa26-EA1 model initiated by a variety of drivers for Cre-recombinase, as well as a set of mixed-lineage mesenchymal controls from the chest wall (with skeletal muscle, bone, cartilage, tendon, and aponeurosis in each). CCS tumors from each induction method shared a somewhat similar expression profile overall and clustered near each other on principal component analysis (PCA) compared with the other tumor types (Figure 5, A–C).
Figure 5. Cre-mediated translocation model of clear cell sarcomagenesis mimics the transcriptome of the EWSR1-ATF1 expression model but has additional genome CNAs.

(A) Graph of the RPM that aligned onto the human EWSR1-ATF1 fusion oncogene cDNA coding sequence expressed conditionally from the Rosa26 locus and from the HprtCre-induced translocation tumor. For reference, also noted are the RT-qPCR–calculated RPM levels of EWSR1-ATF1 expression from the human SU-CCS1 cell line and from 3 FFPE human CCS tumor specimens, using B2M as a control in the human samples and its average expression across the mouse samples to calculate an RPM estimate. (B) RPM alignments across the genomic sequence for Ewsr1 and Atf1, averaged across 8 translocation-generated tumors (dark blue) and 12 Rosa26-EA1 tumors (cyan), demonstrating overall lower expression (compared with cDNA expression in the EA1 tumors) and a reduced 3′ bias in the exons 3′ to the translocation point (arrow) of Ewsr1 relative to the same in Atf1 in the translocation-generated tumors, which may represent reduced expression of the exons 3′ to the translocation in Ewsr1 or increased expression of the exons 3′ to the translocation in Atf1. (C) PCA demonstrating relative clustering of 8 translocation-driven sarcomas with Rosa26-EA1–driven comparators, separate from the clusterings of other mouse cancer subtypes sequenced in the same batch as the EA1 tumors. SS, synovial sarcoma; OS, osteosarcoma; mel, melanoma. (D) Heatmap of the Pearson’s correlation distance between single-batch–sequenced transcriptomes of the Rosa26-EA1–driven (cyan diamonds) or HprtCre-induced translocation–driven (blue circles) sarcomas, as well as 1 control tissue sample. (E) PCA of the same transcriptomes as in D. (F) CNA analysis on 4 HprtCre-induced translocation tumors, using a low-read-depth whole-genome sequencing approach. The upper row shows higher-resolution images of chromosome 15 from the sequencing of each tumor, the middle row shows the CNAs across the entire genome, and lower row shows chromosome 11 from each.
Next, for a tighter comparison, we sequenced, in a single batch, transcriptomes from 5 tumors induced by HprtCre-initiated translocations and 5 tumors induced by TATCre injection to activate the Rosa26-EA1 allele. Again, these clustered somewhat closely to each other, relative to a control, albeit with slightly wider variation among the translocation-initiated tumors by PCA and whole-transcriptome distance clustering (Figure 5, D and E).
We investigated CNAs in 4 of the translocation-generated tumor samples using a low-read-depth whole-genome sequencing approach (Figure 5F). The data demonstrated that the genome segments that comprised the dicentric chromosome (a very short segment immediately adjacent to the chromosome 11 centromere and most of the body of chromosome 15) had variable copy numbers, consistent with the expected breakage of this chromosome during unbalanced cytokinesis. However, rather than whole-genome chromothripsis, which might be the expected result of double-stranded breaks, the only other associated CNA was a copy number decrease of chromosome 6 in the translocation model. The bulk of chromosome 11, which accompanied the translocation-generated Ewsr1-Atf1 fusion on the acentric derivative chromosomal fragment maintained an approximately diploid copy number, showing neither significant amplification nor loss.
Mitf contributes modestly to EWSR1-ATF1–driven tumorigenesis.
Surprisingly, the chromosome 6 copy number decrease that was detected in the translocation-initiated mouse model mapped to syntenic portions of human chromosome 7 that showed copy number increases in human CCS. None of the chromosomal regions demonstrating copy number decreases in more than 1 human CCS tumor correlated with syntenic segments lost in chromosome 6 in the mouse.
The Mitf locus, homologous to the region of focal copy number increases in human CCS, is also on mouse chromosome 6 (Figure 6A). Prior research has identified melanocyte-inducing transcription factor (MITF) as being responsible for the pigment generation and melanocytic morphology of CCS (23). Work in cell lines suggested that MITF drives the progression of CCS, functioning as an additional oncogene, similar to a role defined for it in melanoma (24). We previously noted that EWSR1-ATF1–induced mouse sarcomas that exhibited melanocytic features expressed Mitf (5). In order to test the oncogenic contribution of Mitf, we procured a mouse strain with a natural loss-of-function point mutation in Mitf, termed Mitf vit, for the vitiligo (loss of pigment) that develops patchily in heterozygotes, but completely in homozygotes (25).
Figure 6. MITF contributes to oncogenesis driven by EWSR1-ATF1.

(A) Left: Human chromosome 3 is shown with the spike in amplification (in blue) that occurred in 6 of 13 human CCS tumors. Right: Copy number microarray data showing that the recurrent, focal amplification occurred at the MITF locus in all 6 tumors. (B) Mouse DNA sequences depicting the Mitf vitiligo (Mitf vit) mutation (top left panel, in blue) and the normal Mitf sequence. The BsiEI restriction enzyme was used to confirm Mitf vit after PCR, because it recognizes and cuts WT Mitf but not Mitf vit (right panel). (C) Mouse breeding schematic showing the strategy to generate EA1 heterozygotic mice with homozygous mutant or homozygous WT Mitf littermates, which we injected with TATCre to induce EWS-ATF1 expression. (D) Kaplan-Meier curve shows the achievement of morbidity of Mitf WT/WT mice (solid line) and Mitf vit/vit mice (dotted line) following TATCre injection at 28 days of age (n = 37 Mitf WT/WT mice and n = 15 Mitf vit/vit mice). The 50% median survival time to morbidity for Mitf vit/vit mice was 102 days compared with 87 days for WT mice. A log-rank test was performed, and the difference was deemed statistically significant (P < 0.0001, z score = 4.52). (E) Tumor mass measurements for Mitf WT/WT mice (black dots) and Mitf vit/vit mice (gray circles). The tumors were not significantly different in size (P = 0.6, by 2-tailed Student’s t test). (F) Graph of the blinded quantitation of histological features of tumors developing from EA1 expression in either Mitf WT/WT mice (black diamonds, n = 37) or Mitf vit/vit mice (gray diamonds, n = 15), with human CCSs on a tissue microarray (n = 20, blue diamonds). P = 0.05, by 2-tailed Student’s t test (none of these comparisons between the 2 mouse groups reached statistical significance). (G) Representative histomorphologies in H&E-stained tissue sections of EA1-expressing tumors. Each photomicrograph is a 100 μm square obtained with a 60× original magnification objective lens.
Fortuitously, in this naturally occurring point mutation, a BsiEI endonuclease digestion site was disrupted by the Mitf vit mutation, such that enzymatic digestion of a PCR-generated fragment incorporating this region could be used for genotyping (Figure 6B). The Mitf vit and Rosa26 loci are both on mouse chromosome 6 at a 15 Mb distance from one another. After 9 matings and nearly 100 progeny were screened, meiotic recombination generated a single chromosome 6 with both conditionally activatable EWSR1-ATF1 and Mitf vit segregating together. Mice bearing this doubly targeted chromosome 6 and a second chromosome 6 with only the Rosa26-EA1 allele were then bred with Mitf vit heterozygotes to generate littermate cohorts of mice heterozygous for Rosa26-EA1 and either homozygous for WT Mitf (Mitf WT/WT) or homozygous for the vitiligo allele (Mitf vit/vit). Mice in these cohorts were injected with TATCre protein at 4 weeks of age and monitored for tumorigenesis (Figure 6C). Sarcomagenesis to the point of morbidity occurred at a latency that was markedly longer in the homozygous vitiligo mice; similarly sized tumors were produced, but at longer latencies (Figure 6, D and E). A blinded histological assessment of the tumors revealed a subtly reduced frequency of the clear cell morphology that dominated the tumors in the WT Mitf group (Figure 6, F and G).
In order to test the potential contribution of higher baseline expression of Mitf in a potential cell of origin for EWSR1-ATF1–mediated tumorigenesis, we crossed mice bearing the conditionally expressed Rosa26-EA1 allele with mice bearing Cre recombinase under the control of the tyrosinase gene (TyrCreERT2) that is expressed in melanoblasts, which have very high native expression of Mitf. We found that topical administration of tamoxifen to induce Cre-mediated recombination in dermal melanoblasts generated no tumors. Systemic administration of tamoxifen to 4-week-old mice led to the development of a few tumors at a latency of more than 1 year. Although these tumors showed GFP signal, suggesting expression of the EA1 allele at Rosa26, they developed at a much longer latency than did any other model of Rosa26-induced CCS.
Myc overexpression contributes to EWSR1-ATF1–driven sarcomagenesis, but alters the tumor phenotype.
We also noted that the CNA distribution in the translocation-initiated tumors, across the length of mouse chromosome 15 included on the dicentric chromosome, was not random. Specific segments of mouse chromosome 15 were amplified in all 4 tumors, and others demonstrated consistent copy number losses. Approximately one-third of human chromosome 8, including the portion that was amplified in a majority of the human CCS tumors, was syntenic to the part of mouse chromosome 15 that underwent CNA. A subregion of this syntenic region was amplified in all 4 translocation tumors, but significantly so in 3 of them. This included several potential oncogenes, such as Fzd6, Angpt1, Borg, Baalc, Azin1, and Dcaf13 (Figure 7A). Myc was in a portion that had neither increased nor decreased copy numbers in mouse chromosome 15 (Figure 7B).
Figure 7. MYC stabilization enhances EWSR1-ATF1–induced sarcomagenesis but alters tumor phenotypes.

(A) BioDiscovery data on CNAs in regions of human chromosome 8 that are syntenic to amplified regions of mouse chromosome 15 and that flank MYC. (B) CNAs in regions of mouse chromosome 15 syntenic to human chromosome 8 and surrounding Myc. (C) Schematic showing the floxed stop cassette that allows for Cre-inducible expression of MycT58A at the Igs2 locus. (D) Breeding strategy to generate mice that express both EWSR1-ATF1 (EA1) and MycT58A upon TATCre injection, as well as Igs2WT/WT littermate controls. (E) Tumor growth curves for Rosa26EA1/WT mice with either Igs2LSL-Myc/WT (red) or Igs2WT/WT (black), following TATCre injection at 28 days of age. (F) Graph of the blinded quantitation of H&E-stained slides for histologic features distinguishing tumors expressing EA1 alone and tumors expressing EA1 plus MycT58A (EA1+Myc, red), with, for reference, the prevalence of the nested morphology observed in human CCSs on a tissue microarray (n = 20). (G) Photomicrographs of H&E-stained tissue sections demonstrating the 2 variants on myxoid features, 1 in each genotype, as well as the nested histomorphology observed focally in 7 human tumors (n = 20) and all Myc-activating mouse tumors (each photomicrograph is a 100 μm square obtained with a 60× original magnification objective lens).
We observed copy number amplification of the MYC locus in 9 of the 13 human CCS tumors (Figure 7A). In order to test the potential contribution of a locus commonly thought to be the reason for selection for chromosome 8 amplification in other tumors, we bred the Rosa26EA1/EA1 mice with mice heterozygous for a conditionally expressed activated mutant allele of Myc knocked into the Igs2 pseudogene locus (Figure 7, C and D). Littermate cohorts were injected with TATCre at 4 weeks of age, and sarcomagenesis was monitored. We noted a dramatic shortening of the latency to tumorigenesis in mice heterozygous for the conditionally activated Myc allele (Figure 7E). Histologically, however, tumors with added Myc overexpression had features that were strikingly dissimilar from those of the EA1-only tumors, including poor recapitulation of CCS characteristics, with predominance of a nested histomorphology that is very rarely observed in human CCS (Figure 7, F and G).
Discussion
The genome of human CCS is characterized most prominently by copy number increases at the MITF locus and whole chromosomes 7 and 8, with each of these CNAs identified in a majority of tumors. The functional character of these observed alterations in copy number was informed by comparisons with other groups of tumors: 2 human tumor types with alternative EWSR1 rearrangements and 2 mouse genetic models of neoplasia induced by expression of EWSR1-ATF1, either as a conditional transgene or the product of a conditional chromosomal translocation of the homologous mouse loci. The last of these, the successful generation of a sarcoma from a Cre-loxP–induced chromosomal translocation, was a singular achievement in itself, but also instructive with regard to the function of the CCS genome.
We believe these copy number data provide the first glimpse into the human CCS genome. Because of the small numbers of CCS tumors available at any single center, we used FFPE samples collected over 2 decades, and were thus limited to CNV assessments instead of full-genome or -exome sequencing. Given that sarcomas typically progress toward genomic complexity by copy number changes rather than point mutation burden (14), and that only a single event affecting any of the hot-spot mutations in known cancer genes was identified in a single CCS tumor, our copy number data may provide an acceptable genome landscape for this tumor type. Some altered copy number observations were shared with ESs and DSRCTs, both of which bear low mutational burdens (12, 13, 15, 16). Naturally, the significance of these comparisons was limited by small numbers in the CCS and DSCRT cohorts, calling for caution in overinterpreting the numbers.
Although we did not observe focal loss of CDKN2A in our cohort of 13 human samples, 5 of them showed deletions of 9p, which contains the CDKN2A locus. Three others (none overlapped with the 9p deletion) showed amplification of the portion of chromosome 12 that contains the MDM2 locus. Amplification of MDM2 or loss of CDKN2A can reduce the stability of p53. Disruption of p53 stability is considered an important step for many cells undergoing malignant transformation driven by the expression of an oncogene, as the cells must overcome oncogene-induced senescence. In particular, Komura and colleagues (26) demonstrated that escape from senescence is an important hurdle to clear during initial growth of EWSR1-ATF1–expressing cells. These findings from human CCS genomes are at least a nod in support of that line of thinking.
The most important conclusion from each of these assembled and compared data sets is that no single secondary genetic alteration was strictly required for EWSR1-ATF1–driven sarcomagenesis. In the human tumors, even the most prevalent alterations were still absent in a substantial minority of tumors. Whole-exome sequencing and copy number analyses of mouse sarcomas initiated from EWSR1-ATF1 expression revealed no single change that was repeated in more than 1 tumor. The observation that tumors developed at all in a mouse model in which an acentric chromosomal fragment was host to the Ewsr1-Atf1 fusion testifies to the profound transformational capacity of this particular oncogene.
While 2 solid tumor types have been generated previously in mice by CRISPR/Cas9-induced translocations (27, 28), we know of no prior solid tumors arising from Cre-loxP–induced translocations and no other previously reported opportunities to compare tumorigenesis from induced translocations with that from expression of the fusion gene–coding sequence alone.
A chromosomal translocation achieves multiple changes, any of which may contribute to oncogenesis. First, newly associated exons express a fusion oncoprotein that wields effects not shared with either parent locus. Second, translocation positions the new fusion gene under the genetic and epigenetic control of a specific promoter and enhancers, which regulate the level of expression and responsiveness to other novel conditions. Third, translocation disrupts 1 allele of each of the parent loci. Fourth, translocation may broadly affect other nearby genes by repositioning.
Obviously, the impact of changes other than the expression of the fusion oncogene will vary with each specific type. Not all parent alleles will have sufficient homology with their human counterparts to be oncogenic in the mouse. Further, the regional effects of the translocation may be more or less homologous depending on the extent of these effects. For example, Atf1 is on mouse chromosome 15, where only a small region immediately surrounding the locus is syntenic to the host chromosome of ATF1 on human chromosome 12 (29). The breadth of these syntenic regions will determine whether enhancer elements and other regionally important sequences are shared sufficiently to make the model of these larger shifts in mouse chromosomal segments a true mimic of the human translocation.
The translocation mouse model for CCS provided additional data. As noted, the importance of Ewsr1-Atf1 in sarcomagenesis was strengthened by its retention in cells even on an acentric chromosome that required the presence of selective pressure. Also of interest, however, was the fact that this acentric chromosome with the fusion was not amplified in the tumors. The Rosa26-driven model also showed no amplification of the fusion expression locus, which was noted as the only other secondary change in the exomes of a similar model of synovial sarcoma (4). This suggests that the continued presence of the fusion is critical in the tumor cells, but that there is no apparent selection for increased levels of expression.
The loss of 1 copy of chromosome 6 in the translocation tumors was especially interesting, because loss of chromosome 6 would have been artifactually affected in the Rosa26-driven model. Because the fusion expression locus maps to chromosome 6, negative selection pressure would likely prevent loss of 1 of the 2 copies of the chromosome in that model. The mouse translocation model’s copy number decrease of chromosome 6 and the tumors that formed even in mice homozygous for Mitfvit in the Rosa26 model suggest that melanocytic microphthalmia-associated transcription factor (MITF) is not strictly required for clear cell sarcomagenesis. However, evidence from another CCS mouse model, in which the fusion EWSR1-ATF1 is expressed from the Col1A1 locus on mouse chromosome 11 (26), corroborates our human CNA analysis showing MITF amplification and the subtle — but significant — increase in latency to morbid tumorigenesis that we observed in the Mitfvit experiments. MITF probably facilitates tumor formation from EWSR1-ATF1 expression, possibly also affecting the tumor phenotype. This last point is important, given that the human samples profiled in this group were tumors that fit the traditional pathological definition of CCS, including melanoma-related features. It is known that nonmelanocytic human EWSR1-ATF1–driven tumors are less dependent on MITF than EWSR1-ATF1–driven neoplasias that include melanocytic features (30). On mouse chromosome 6, there may be other tumor suppressors or factors that would prohibit maintenance of the acentric chromosomal fragment critical to this particular translocation model, but mouse chromosome 6 has no synteny with the few significant copy number decreases observed in human CCS. In spite of this subtle copy number loss of the chromosome, Mitf expression was strong in the translocation tumors by RNA-Seq.
The CNAs in mouse chromosome 15, syntenic to the regions of human chromosome 8 that were so frequently gained in human CCS tumors, also suggested additional drivers to consider. MYC is often considered a driving oncogene to lend selective pressure for human chromosome 8 copy number gains. However, this was not specifically amplified in the translocation-driven mouse tumors. Furthermore, while amplified Myc expression and stability were clearly capable of enhancing tumorigenesis when added to EWSR1-ATF1 expression, they did not faithfully maintain the tumor phenotype of CCS, suggesting that, although MYC is — not surprisingly — oncogenic, it may not specifically be a driver of clear cell sarcomagenesis, as it is a driving pathway in many types of cancer (31). The other genes in the cross-species–amplified regions deserve additional attention in future investigations.
Finally, the amplifications in chromosome 7 in human CCS also require additional study. Of note, EGFR, located in the consistently amplified regions of human chromosome 7, is also present on syntenic regions of the mouse acentric chromosomal fragment (including most of mouse chromosome 11) in the translocation-driven model. Again, this was specifically retained in tumors, but not amplified. The EGF receptor (EGFR) contained the only hot-spot mutation that was detected in any of the tested human tumors. This is another locus deserving specific future attention, especially given the potential for therapeutic targeting of the EGFR.
Overall, these tumors — like others in the family of translocation-associated sarcomas — have a relatively quiet genome that is quite distinct from that of melanomas, to which they are also related. While no other mutations are strictly required in the process of sarcomagenesis, some of the frequently observed secondary alterations may provide additional insights into relative genetic dependencies for this cancer or suggest means by which it can circumvent attempts to directly target the fusion biology. The EWSR1-ATF1–associated family of neoplasias includes very aggressive malignancies that have a predilection for adolescents and young adults. We have no targeted therapies available for these deadly sarcomas. More efforts to understand the biology of these tumors and innovative models with which to validate therapies preclinically are needed to move toward longer and better survival for the young patients with these diseases.
Methods
DNA microarray and CNA analyses of human FFPE tumors.
Extracted DNA was assessed using the OncoScan FFPE Assay Kit (Agilent Technologies). The raw microarray data for each sample were then used to generate OSCHP files for analysis by Nexus Express, version 10.0 (BioDiscovery). Initial calls of copy number gains and losses were made with the FASST2 method at default settings. Distributions of intensity values and B-allele frequency (BAF) for each sample were manually inspected to identify diploid chromosomes, and subsequent recentering of probes based on these diploid regions was performed. For each sample, the p and q arms of each chromosome were assessed by Nexus as well as by manual inspection for regions of gain or loss extending 10 Mb or more and present in an estimated 10% or more of the cells, allowing for analysis of the substantial presence of clones in addition to near-complete clonal expansion. Focal gains and deletions involving individual genes not part of the 10 Mb gains or deletions or frequent in other sarcomas were also determined, with additional manual inspection of the most prevalent genes. Concomitance of gain or loss across the p and q arms of each chromosome as well as associations with age, sex, primary tumor status, and metastatic tumor status were assessed with Fisher’s exact test and the Cochran-Mantel-Haenszel statistical test.
The quality of each processed sample was assessed by its osmapd score, which showed a clear separation of values. Samples having a median absolute pairwise difference (MAPD) score of greater than 0.5 were excluded, as were samples with a MAPD score near this threshold (0.41), which showed potential sample contamination. Paired germline samples were not available for all persons, but when present, the frequencies of copy number events were assessed and any aberrations present in both the germline and tumor were excluded. All samples passing these preliminary quality control checks were then confirmed to be from unique individuals by comparing 4948 common SNPs. The MITF region was found to have frequent copy number gain calls and also had frequent loss of heterozygosity. To determine whether loss of heterozygosity (LOH) in this region was specific to CCS or more likely to be a common germline event, 2 publicly available data sets processed on the identical microarray were also processed and compared (https://www.ncbi.nlm.nih.gov/gds; GSE147353 and GSE110026). LOH in the MITF region was found to be a common event in both of these sets, although gains were nearly absent in the Synchronous ColoRectal Cancer data, but found to be called somewhat frequently in the Prostate Needle Biopsy data. Assessment of significant concomitance or exclusion across arms and with covariates was performed with Fisher’s exact test. Only 3 tests were found to meet an unadjusted P < 0.05 threshold, with loss of significance when adjusted for multiple comparisons. As a result, we share these top 3 results with the caveat that they will need future replication before being considered bona fide.
Exome sequencing of Bmi1CreERT2-Rosa26 murine tumors.
Paired-end libraries were produced with enrichment using Agilent SureSelect, followed by alignment to the UCSC mm9 mouse reference (derived from the NCBI Mouse Build 37) with Burrows-Wheeler Aligner, version 0.5.9 (with parameters -t 4 -q 5). A total of 34 tumor samples were paired with 6 matched normal samples and processed through our somatic variation pipeline using Samtools, Varscan, GATK, and Pindel.
All putative somatic variants were annotated using transcripts identified in release 58 of Ensembl (58_37K). A single representative transcript was selected for annotation of each variant on the basis of the most extreme functional effect and longest transcript isoform. Filters were applied, removing variants that affected only the 3′-UTR, the 5′-UTR, the intronic sequence, and the intergenic sequence, in addition to any coding SNPs that affect transcripts without a complete open reading frame.
Using supporting read counts, 323 high-quality nonsynonymous somatic variants were identified. High-quality variants had a minimum of 8-read coverage in the normal sample and 20-read coverage in the tumor, a maximum of 1 supporting read in the normal sample or 1% VAF, and a minimum of 2 unique supporting reads in the tumor with a minimum tumor VAF of 10%.
KEGG pathway analysis was performed and corroborated by 2 programs — g:Profiler and Enrichr (32–34). Copy number analysis was derived from exome-sequencing data in the Bioconductor DNAcopy package (35).
Mouse studies and generation of loxP animals.
The Rosa26EA1 (5), HprtCre (36), and Prx1Cre (37) mice were all previously described. Mice for these experiments were obtained from the laboratory colony. The first line was generated in the laboratory, but the latter 2 lines were obtained originally from The Jackson Laboratory. All mice were maintained on a mixed 129/SvJ and BL/6 strain background, and littermate controls were used for any comparative experiments between groups. The sex of the mice was not intentionally varied between groups, and all groups had both sexes represented. The ages of the mice are noted in all tumorigenesis experiments.
Using gene targeting, we inserted a loxP element in intron 7 of the Ewsr1 gene and in intron 4 of the Atf1 gene. In this way, we generated 2 distinct animal lines bearing loxP-engineered chromosomes. Gene targeting vectors were designed using Gene Construction Kit software (GCK, TextCo Biosoftware). To generate the vectors, we used a combination of traditional cloning techniques and recombineering (38, 39). The procedures for the electroporation of mouse embryonic stem cells, the selection of positive clones, the blastocyst injection, the generation of chimeras, and the germline transmission were performed as previously described (39).
Heterozygous Ewsr1loxP/WT and Atf1loxP/WT animals were crossed to obtain double-heterozygous and, finally, double-homozygous Ewsr1loxP/loxP Atf1loxP/loxP animals. The genotype of the animals was confirmed by PCR using specific primers (Ewsr1 forward, 5′-TAGGGCTGGCATTCTTTAAG-3′ and Ewsr1 reverse, 5′-TTAAGAGCCTTAAGAGTCGG-3′; Atf1 forward, 5′-TGGATTGGTTATGGAGCAGG-3′ and Atf1 reverse, 5′-GTAGGCCTTGGATATTACCC-3′. For both Ewsr1 and Atf1, with these oligonucleotides the targeted allele gave an amplicon of 100 bp longer than did the WT allele.
TATCre injection.
TATCre is a soluble form of the Cre protein that can reach the nuclei of cells through the TAT epitope. TATCre injections into mice were performed as previously described (5).
MRI.
All imaging was performed on euthanized animals, using an HT Bruker BioSpec 7.1 T horizontal-bore MRI instrument.
RNA extraction.
One biopsy was immediately collected in RNAlater (Thermo Fisher Scientific) and stored at 4°C for 3 days to preserve RNA integrity until RNA-Seq was performed. RNA extraction was performed with the QIAGEN RNeasy kit (QIAGEN) following the manufacturer’s recommendations.
DNA extraction.
A small minced biopsy was treated with Proteinase K (GoldBio, Gold Biotechnology) at 37°C overnight, and DNA was extracted using a traditional phenol chloroform procedure. DNA was quantitated with a spectrophotometer, and 1 μL was run on an agarose gel for a quality check.
Histology.
Tumor biopsies were reduced to 1 cm3 pieces and fixed for 3 days in 4% formaldehyde in PBS solution. Samples for histological analysis were prepared using traditional techniques. Briefly, samples were washed of the fixative by rinsing them in PBS twice for 10 minutes with gentle rolling. The dehydration phase was performed with sequential incubation in 25%, 50%, 70%, 95%, and 100% ethanol solutions. A 2-hour incubation of xylene was performed to clarify the samples before the specimens were paraffin embedded. Paraffin blocks were then sectioned at 5–8 μm thickness with a microtome, mounted onto slides, and aged overnight at 40°C. The next day, the slides were washed in xylene to remove the paraffin and rehydrated through an ethanol series (100%, 95%, 70%, 50%, and 25% H2O). The samples were stained with H&E, and coverslip glass was mounted to preserve the integrity of the samples following standard protocols.
All assessments of H&E-stained sections were performed by a pathologist blinded to the genotype of the tumor. For human correlate pathology, we used a previously collected and H&E-stained human CCS tissue microarray, which included doubled 3 mm cores from 20 cases of molecularly confirmed CCS.
RNA-Seq.
RNA-Seq libraries were prepared using the RiboZero method, and then sequenced using either an Illumina HiSeq instrument as previously described (5) or an Illumina NovaSeq instrument.
EWSR1-ATF1 expression level calculation for human correlates.
Total RNA was isolated from 3 human tumor FFPE scrolls as well as from a frozen aliquot of the human SU-CCS-1 cell line (obtained from the American Type Culture Collection [ATCC]). The Recoverall Total Nucleic Acid Isolation kit (Thermo Fisher Scientific) was used for isolation of RNA from the scrolls. The SuperScript IV First-Strand Reverse Transcription kit (Thermo Fisher Scientific) was used to generate cDNAs, which were subjected to quantitative reverse transcription PCR (RT-qPCR) with primers specific for the fusion (forward, 5′-CATGAGCAGAGGTGGGCG-′3; reverse, 5′-CCCCGTGTATCTTCAGAAGATAAGTC-′3) and for the housekeeping gene B2m (forward, 5′-TGACTTTGTCACAGCCCAAGATA-′3; reverse, 5′-AATCCAAATGCGGCATCTTC-′3). The mean reads per million mapped reads (RPM) level was used for mouse B2m tumor RNA-Seq data, along with the fold expression of the fusion in human tumors and cell lines to present the reference levels in Figure 4A.
Low read-depth, whole-genome sequencing.
Genomic DNA was isolated and libraries generated using Nextera DNA Flex Library Prep with UDI (Illumina). Each library was sequenced on a NovaSeq instrument to a level of approximately 40 M reads in order to generate approximately 2–3 times the depth coverage of the whole genome. Data were aligned on the mm10 genome and run through the CNVkit with the circular binary segmentation (CBS) method and a bin size of 100 kb.
Raw data availability.
Sequencing data were deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE158726).
Statistics.
All data are presented as individual points, with the mean also indicated. Two-tailed Student’s t tests were performed to acquire P values. The evanmiller.org calculator was used for the log-rank tests. Sample sizes are either noted explicitly in the figure legend, clearly discernible by the dot-plot data presented, or both.
Study approval.
All of the animal studies were performed according to ethics standards and under the approval of the IACUC of the University of Utah. All the human samples used in this study were obtained from consenting patients with IRB approval of the MD Anderson Cancer Center.
Author contributions
This study was conceived of and supported by EP, KS, AB, SWAT, CCM, LD, AJL, MRC, and KBJ. Patients’ samples were collected by AJL. Mouse genetic experiments with the Rosa26 model were performed by KS, JJB, and YW. The translocation mouse model was designed and generated by EP, KS, and MRC. Experiments with the translocation mouse were performed by EP, AB, SWAT, MRC and KBJ. Mouse exome data were analyzed by KS, MX, LD, and KBJ. Human CNA data were analyzed by BBO, CCM, and KBJ. Transcriptomic data were analyzed by KS and LL. All authors reviewed, edited, and approved the final manuscript.
Supplementary Material
Acknowledgments
Brian Dalley from the High-Throughput Genomics core facility at the University of Utah assisted with transcriptome and genome sequencing, and Chris Stubben and Tim Parnell from the Bioinformatics Shared Resource at the University of Utah with analysis. BioDiscovery provided software and analysis support for the OncoScan array hybridizations. All animal imaging was performed at the Preclinical Imaging Core Facility at the University of Utah. This work was supported by the Cure Childhood Cancer Foundation, the Pablove Foundation, and Sara’s Cure (to KBJ), as well as by NIH grant R01 MH093595 (to MRC) and the Halt Cancer at X Foundation (to SWAT). CCM was supported by the Pediatric Cancer Program, which is funded by the Intermountain Healthcare and Primary Children’s Hospital Foundations and the Department of Pediatrics at the University of Utah.
Version 1. 06/22/2021
In-Press Preview
Version 2. 08/02/2021
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(15):e146301.https://doi.org/10.1172/JCI146301.
Contributor Information
Emanuele Panza, Email: emanuele.panza@unibo.it.
Benjamin B. Ozenberger, Email: ben.ozenberger@hci.utah.edu.
Krystal M. Straessler, Email: straessler3@gmail.com.
Jared J. Barrott, Email: barrjare@isu.edu.
Li Li, Email: li.li@hci.utah.edu.
Yanliang Wang, Email: yanliangwang75@gmail.com.
Mingchao Xie, Email: solunar.xie@gmail.com.
Anne Boulet, Email: aboulet@genetics.utah.edu.
Simon W.A. Titen, Email: stiten@genetics.utah.edu.
Clinton C. Mason, Email: Clint.Mason@hci.utah.edu.
Alexander J. Lazar, Email: alazar@mdanderson.org.
Li Ding, Email: lding@genome.wustl.edu.
Mario R. Capecchi, Email: capecchi@genetics.utah.edu.
Kevin B. Jones, Email: kevin.jones@hci.utah.edu.
References
- 1.Hanahan D, et al. The origins of oncomice: a history of the first transgenic mice genetically engineered to develop cancer. Genes Dev. 2007;21(18):2258–2270. doi: 10.1101/gad.1583307. [DOI] [PubMed] [Google Scholar]
- 2.Goodwin ML, et al. Modeling alveolar soft part sarcomagenesis in the mouse: a role for lactate in the tumor microenvironment. Cancer Cell. 2014;26(6):851–862. doi: 10.1016/j.ccell.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haldar M, et al. A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell. 2007;11(4):375–388. doi: 10.1016/j.ccr.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 4.Jones KB, et al. The impact of chromosomal translocation locus and fusion oncogene coding sequence in synovial sarcomagenesis. Oncogene. 2016;35(38):5021–5032. doi: 10.1038/onc.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Straessler KM, et al. Modeling clear cell sarcomagenesis in the mouse: cell of origin differentiation state impacts tumor characteristics. Cancer Cell. 2013;23(2):215–227. doi: 10.1016/j.ccr.2012.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keller C, et al. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004;18(21):2614–2626. doi: 10.1101/gad.1244004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchholz F, et al. Inducible chromosomal translocation of AML1 and ETO genes through Cre/loxP-mediated recombination in the mouse. EMBO Rep. 2000;1(2):133–139. doi: 10.1093/embo-reports/kvd027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins EC, et al. Inter-chromosomal recombination of Mll and Af9 genes mediated by cre-loxP in mouse development. EMBO Rep. 2000;1(2):127–132. doi: 10.1093/embo-reports/kvd021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forster A, et al. Engineering de novo reciprocal chromosomal translocations associated with Mll to replicate primary events of human cancer. Cancer Cell. 2003;3(5):449–458. doi: 10.1016/S1535-6108(03)00106-5. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka M, et al. Somatic chromosomal translocation between Ewsr1 and Fli1 loci leads to dilated cardiomyopathy in a mouse model. Sci Rep. 2015;5:7826. doi: 10.1038/srep07826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamada K, et al. EWS/ATF1 expression induces sarcomas from neural crest-derived cells in mice. J Clin Invest. 2013;123(2):600–610. doi: 10.1172/JCI63572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brohl AS, et al. The genomic landscape of the Ewing sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014;10(7):e1004475. doi: 10.1371/journal.pgen.1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulbul A, et al. Genomic and proteomic alterations in desmoplastic small round blue-cell tumors. JCO Precis Oncol. 2018;2:1–9. doi: 10.1200/PO.17.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research Network Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell. 2017;171(4):950–965. doi: 10.1016/j.cell.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chow WA, et al. Recurrent secondary genomic alterations in desmoplastic small round cell tumors. BMC Med Genet. 2020;21(1):101. doi: 10.1186/s12881-020-01034-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crompton BD, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326–1341. doi: 10.1158/2159-8290.CD-13-1037. [DOI] [PubMed] [Google Scholar]
- 17.Lagarde P, et al. Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol. 2013;31(5):608–615. doi: 10.1200/JCO.2012.46.0147. [DOI] [PubMed] [Google Scholar]
- 18.Orbach D, et al. Genomic complexity in pediatric synovial sarcomas (Synobio study): the European pediatric soft tissue sarcoma group (EpSSG) experience. Cancer Med. 2018;7(4):1384–1393. doi: 10.1002/cam4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vlenterie M, et al. Next generation sequencing in synovial sarcoma reveals novel gene mutations. Oncotarget. 2015;6(33):34680–34690. doi: 10.18632/oncotarget.5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor BS, et al. Advances in sarcoma genomics and new therapeutic targets. Nat Rev Cancer. 2011;11(8):541–557. doi: 10.1038/nrc3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tirode F, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014;4(11):1342–1353. doi: 10.1158/2159-8290.CD-14-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jahromi MS, et al. Molecular inversion probe analysis detects novel copy number alterations in Ewing sarcoma. Cancer Genet. 2012;205(7–8):391–404. doi: 10.1016/j.cancergen.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li KK, et al. The melanocyte inducing factor MITF is stably expressed in cell lines from human clear cell sarcoma. Br J Cancer. 2003;89(6):1072–1078. doi: 10.1038/sj.bjc.6601212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis IJ, et al. Oncogenic MITF dysregulation in clear cell sarcoma: defining the MiT family of human cancers. Cancer Cell. 2006;9(6):473–484. doi: 10.1016/j.ccr.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 25.Steingrimsson E, et al. Molecular basis of mouse microphthalmia (mi) mutations helps explain their developmental and phenotypic consequences. Nat Genet. 1994;8(3):256–263. doi: 10.1038/ng1194-256. [DOI] [PubMed] [Google Scholar]
- 26.Komura S, et al. Cell-type dependent enhancer binding of the EWS/ATF1 fusion gene in clear cell sarcomas. Nat Commun. 2019;10(1):3999. doi: 10.1038/s41467-019-11745-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lagutina IV, et al. Modeling of the human alveolar rhabdomyosarcoma Pax3-Foxo1 chromosome translocation in mouse myoblasts using CRISPR-Cas9 nuclease. PLoS Genet. 2015;11(2):e1004951. doi: 10.1371/journal.pgen.1004951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maddalo D, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516(7531):423–427. doi: 10.1038/nature13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolishovski G, et al. The JAX Synteny Browser for mouse-human comparative genomics. Mamm Genome. 2019;30(11–12):353–361. doi: 10.1007/s00335-019-09821-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hallor KH, et al. Fusion genes in angiomatoid fibrous histiocytoma. Cancer Lett. 2007;251(1):158–163. doi: 10.1016/j.canlet.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 31.Schaub FX, et al. Pan-cancer alterations of the MYC oncogene and its proximal network across the Cancer Genome Atlas. Cell Syst. 2018;6(3):282–300. doi: 10.1016/j.cels.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen EY, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raudvere U, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47(w1):W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seshan VE, Olshen A. DNAcopy: DNA copy number data analysis. R package, version 1.62.0, 2020. https://bioconductor.org/packages/release/bioc/html/DNAcopy.html Accessed July 6, 2021.
- 36.Tang SH, et al. A Cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis. 2002;32(3):199–202. doi: 10.1002/gene.10030. [DOI] [PubMed] [Google Scholar]
- 37.Logan M, et al. Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33(2):77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 38.Liu P, et al. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13(3):476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu S, et al. A protocol for constructing gene targeting vectors: generating knockout mice for the cadherin family and beyond. Nat Protoc. 2008;3(6):1056–1076. doi: 10.1038/nprot.2008.70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.