

Abstract
Background
Pyridoxine-dependent epilepsy (PDE-ALDH7A1) is an inborn error of lysine catabolism that presents with refractory epilepsy in newborns. Biallelic ALDH7A1 variants lead to deficiency of α-aminoadipic semialdehyde dehydrogenase/antiquitin, resulting in accumulation of piperideine-6-carboxylate (P6C), and secondary deficiency of the important cofactor pyridoxal-5′-phosphate (PLP, active vitamin B6) through its complexation with P6C. Vitamin B6 supplementation resolves epilepsy in patients, but intellectual disability may still develop. Early diagnosis and treatment, preferably based on newborn screening, could optimize long-term clinical outcome. However, no suitable PDE-ALDH7A1 newborn screening biomarkers are currently available.
Methods
We combined the innovative analytical methods untargeted metabolomics and infrared ion spectroscopy to discover and identify biomarkers in plasma that would allow for PDE-ALDH7A1 diagnosis in newborn screening.
Results
We identified 2S,6S-/2S,6R-oxopropylpiperidine-2-carboxylic acid (2-OPP) as a PDE-ALDH7A1 biomarker, and confirmed 6-oxopiperidine-2-carboxylic acid (6-oxoPIP) as a biomarker. The suitability of 2-OPP as a potential PDE-ALDH7A1 newborn screening biomarker in dried bloodspots was shown. Additionally, we found that 2-OPP accumulates in brain tissue of patients and Aldh7a1-knockout mice, and induced epilepsy-like behavior in a zebrafish model system.
Conclusion
This study has opened the way to newborn screening for PDE-ALDH7A1. We speculate that 2-OPP may contribute to ongoing neurotoxicity, also in treated PDE-ALDH7A1 patients. As 2-OPP formation appears to increase upon ketosis, we emphasize the importance of avoiding catabolism in PDE-ALDH7A1 patients.
Funding
Society for Inborn Errors of Metabolism for Netherlands and Belgium (ESN), United for Metabolic Diseases (UMD), Stofwisselkracht, Radboud University, Canadian Institutes of Health Research, Dutch Research Council (NWO), and the European Research Council (ERC).
Keywords: Metabolism
Keywords: Diagnostics, Epilepsy, Genetic diseases
Introduction
Pyridoxine-dependent epilepsy due to biallelic ALDH7A1 variants (OMIM 266100, PDE-ALDH7A1) is an inborn error of metabolism (IEM) that presents with refractory seizures in early infancy. These seizures are typically unresponsive to anticonvulsant medications, but can be controlled with supplementation of pyridoxine (vitamin B6). ALDH7A1 encodes the enzyme α-aminoadipic semialdehyde (α-AASA) dehydrogenase, which is also known as antiquitin. Deficiency of this enzyme disrupts lysine catabolism at the level of the conversion of α-AASA to α-aminoadipic acid (Figure 1), leading to accumulation of α-AASA and pipecolic acid in body fluids (1, 2). α-AASA is found in equilibrium with its alternative molecular form Δ1-piperideine-6-carboxylic acid (P6C), which is formed by spontaneous cyclization. Subsequent condensation of accumulating P6C with pyridoxal 5′-phosphate (PLP) results in secondary deficiency of active vitamin B6, which is thought to be the main pathophysiological mechanism in PDE-ALDH7A1 (3). By supplementing patients with vitamin B6, this secondary deficiency can be corrected. Vitamin B6 supplementation is therefore a cornerstone of seizure treatment in PDE-ALDH7A1, although some patients may require add-on anticonvulsant medication for optimal seizure control (4). Recent studies have shown that a lysine-restricted diet can also be beneficial for ensuring optimal developmental outcome in these patients, but only if such a diet is started in the first year of life (5–8). Furthermore, the addition of arginine supplementation to this treatment regimen, referred to as triple therapy, has been shown to improve neurodevelopmental outcomes when started early in life (7, 8). Arginine competes with lysine for transport across the blood-brain barrier and therefore lowers the flux through lysine catabolism in the brain. At the biochemical level, triple therapy has been shown to lower levels of P6C, α-AASA, and pipecolic acid in plasma and cerebrospinal fluid (CSF) of treated PDE-ALDH7A1 patients (7, 8).
Figure 1. Overview of the L-lysine metabolic pathway involved in pyridoxine-dependent epilepsy.

Diminished function of the enzyme α-aminoadipic semialdehyde (α-AASA) dehydrogenase (antiquitin) encoded by the ALDH7A1 gene (indicated by red cross) leads to accumulation (indicated by red upward arrows) of α-AASA, its cyclic counterpart piperideine-6-carboxylic acid (P6C), and pipecolic acid.
In order to optimize clinical outcome, it is crucial that the diagnosis of PDE-ALDH7A1 is made as early as possible in life. The combination of readily available disease-modifying therapeutic options, the potential risk of additional brain damage upon delayed diagnosis, along with a reasonably high incidence of this rare disease (estimated 1:64,000 conceptions) (9) make PDE-ALDH7A1 an eligible condition for inclusion in newborn screening programs. However, this is presently not implemented, as optimal biomarkers for dried blood spot (DBS) analysis in newborn screening are not available. The current tools for rapid diagnostic screening for PDE-ALDH7A1 encompass analysis of α-AASA and P6C in urine (10). These compounds pose preanalytical challenges regarding stability, both in urine as well as in plasma and DBS, where the limited stability at room temperature is a bottleneck for including these markers in DBS-based newborn screening (11). Analytical methods have been published that attempt to circumvent this; however, these are incompatible with commonly used newborn screening workflows, which make use of direct-infusion mass spectrometry (MS) without separation by liquid chromatography (LC) in order to avoid time-consuming derivatization steps to enable high throughput (12–14). Therefore, there is a clear clinical need for other biomarkers to be able to include PDE-ALDH7A1 in newborn screening.
Additionally, clinical observations in PDE-ALDH7A1 patients suggest that our current understanding of disease pathophysiology is likely incomplete. Intellectual disability is present in many patients despite seizure control with pyridoxine, and in a subset of patients, progressive brain MRI abnormalities are observed with enlargement of ventricles and progressive white matter and corpus callosum abnormalities (15–19). In episodes of intercurrent febrile illness, breakthrough epileptic seizures can occur despite ongoing vitamin B6 treatment. In fact, according to data from the online PDE registry (5), breakthrough seizures manifest in almost 40% of the 116 patients enlisted in the registry. Also, a substantial percentage of patients still requires add-on classical anticonvulsant drugs (18%, based on PDE registry data; ref. 4). The molecular mechanisms underlying all these clinical observations are unknown, but they are suggestive of biochemical disease sequelae that go beyond our current understanding of disrupted lysine metabolism in PDE-ALDH7A1.
To address both the need for novel diagnostic biomarkers for PDE-ALDH7A1, as well as the need for deeper insights in the biochemical effects of α-AASA dehydrogenase deficiency, unbiased screening of the metabolic profile of patient body fluids is a crucial step. This can now be achieved through an innovative high-resolution MS–based technique referred to as untargeted metabolomics. In our laboratory, we have established an untargeted-metabolomics method for broad diagnostic screening of IEMs, coined next-generation metabolic screening (NGMS; ref. 20), which we have now applied in search of novel biomarkers for PDE-ALDH7A1. We have previously shown the potential of such an approach in the identification of novel IEM biomarkers and increasing pathophysiological insights in disease (21, 22). However, the structural elucidation of unknown features/novel metabolites from untargeted-metabolomics data remains a significant challenge. To overcome this challenge, we used a combination of NGMS and infrared ion spectroscopy (IRIS) for orthogonal structural identification (23–28). Using this innovative methodology, we were able to identify discriminative biomarkers for PDE-ALDH7A1, which can be measured in urine, plasma, and CSF, as well as in DBS. Our findings further pave the way for inclusion of PDE-ALDH7A1 in newborn screening, increase our insights into underlying disease mechanisms, and may pose clinical implications for patient management.
Results
Untargeted metabolomics/NGMS identifies biomarkers in body fluids of PDE-ALDH7A1 patients.
We performed untargeted metabolomics/NGMS in 11 plasma samples from 7 PDE-ALDH7A1 patients (3 female and 4 male patients, mean age at sampling 14 years, age range 3–28 years). Upon selection of features that were significantly increased in all patient plasma samples compared with non-IEM controls, we could readily identify features corresponding to the known biomarkers α-AASA, P6C, and pipecolic acid. Additionally, 2 isobaric features with a mass over charge ratio (m/z) of 186.1123 and retention times (RTs) of 2.33 minutes (biomarker A, mean 200-fold change compared with controls) and 2.55 minutes (biomarker B, mean 145-fold change compared with controls), as well as a third feature with an m/z of 144.0652 and RT of 3.05 minutes, (biomarker C, mean 45-fold change compared with controls) were identified as significantly increased in all patient samples. Subsequently, we confirmed increased levels of these unknown features in 2 CSF samples of PDE-ALDH7A1 patients. We could also detect these features in a PDE-ALDH7A1 urine sample. An overview of these findings is depicted in Table 1. Metabolite annotation of these features based on their accurate mass using the Human Metabolome Database (HMDB) only rendered ecgonine (HMDB0006548) and pseudoecgonine (HMDB0006348) as matches for biomarkers A and B. These are tropane alkaloids related to cocaine, so we considered this identification to be very unlikely. For biomarker C, HMDB indicated multiple possible matches to the exact mass; however, one of these matches, 6-oxopiperidine-2-carboxylic acid (6-oxoPIP, HMDB0061705), stood out as a likely candidate because of its chemical similarity to P6C.
Table 1. Untargeted metabolomics/NGMS results for biomarkers in PDE-ALDH7A1 patient body fluids.

For molecular identification of biomarkers A and B, we turned to a an approach using IRIS, which our group has recently successfully applied for metabolite identification (23, 24, 27). In short, MS fragmentation of biomarkers A and B gave a first hint to their chemical composition, as an MS fragment corresponding to P6C could be detected with an m/z of 128.0706. Through the comparison of in silico–generated IR spectra for putative chemical structures of biomarkers A and B to their experimental IR spectra as measured from PDE-ALDH7A1 patient plasma, we were able to further pinpoint their chemical structure without using reference standards. Based on this preliminary structural assignment using predicted IR spectra, we could focus on synthesis of only the predetermined compounds assigned to biomarkers A and B for use in final confirmation of their structures, and as analytical standards for the development of targeted MS–based diagnostics. Using this approach, we were able to identify biomarkers A and B as 2 diastereomers of 2-oxopropylpiperidine-2-carboxylic acid (2-OPP, 2S,6S and 2S,6R conformations, respectively; Figure 2, A and B). A detailed description of the IRIS strategy for molecular identification of the 2-OPP diastereomers goes beyond the scope of this current publication, but is elaborately described in a separate manuscript by van Outersterp et al. (29). For biomarker C, a reference standard of 6-oxoPIP was commercially available, and by comparison of its IR spectrum to the IR spectrum measured for the signal observed in patient body fluids, we were able to confirm the identity of biomarker C as 6-oxoPIP (Figure 2C). During the preparation of this manuscript, 6-oxoPIP was reported by 2 independent studies as a novel PDE-ALDH7A1 biomarker (30, 31). However, its suitability for detection in DBS using a direct-infusion MS method, which is commonly used for newborn screening, was not evaluated in these studies, as Wempe et al. (30) made use of an LC-MS/MS method for DBS measurement and Kuhara et al. (31) only evaluated urine.
Figure 2. Molecular identification of unknown biomarkers in PDE-ALDH7A1 from untargeted metabolomics/NGMS data using IRIS.

Matching overlay of the IR spectra of biomarker A (A, m/z 186.1123 and retention time [RT] 2.33 minutes), biomarker B (B, m/z 186.1123 and RT 2.55 minutes), and biomarker C (C, m/z 144.0652 and RT 3.05 minutes) measured in PDE-ALDH7A1 patient plasma (black traces) and the IR spectra of the protonated 2S,6S-2OPP (A), 2S,6R-2-OPP (B), and 6-oxoPIP (C) ions measured from synthetic reference standards (red traces). Molecular structures of the ions are inlaid in each panel.
Quantitative LC-MS measurements of 6-oxoPIP and 2-OPP in PDE-ALDH7A1 patient body fluids.
To further examine the suitability of 2-OPP and 6-oxoPIP as possible biomarkers for PDE-ALDH7A1, we developed quantitative targeted assays for these markers, using LC-MS/MS for analysis in plasma, urine, and CSF. These methods are further described in the Methods section and were validated according to ISO 15189 criteria. In brief, we analyzed 11 available plasma samples, 8 urine samples, and 9 CSF samples from a total of 15 PDE-ALDH7A1 patients (8 males, 7 females, mean age at sampling 13 years, age ranging from 1 month to 28 years) who all had a genetically confirmed diagnosis. These patients were on different treatment regimens (or untreated) and we compared 6-oxoPIP and 2-OPP levels (the sum of the 2-OPP 2S,6S and 2S,6R diastereomers was used) to anonymized, non-PDE control samples, which were age matched as much as possible (Figure 3). The quantitatively measured levels of 6-oxoPIP and 2-OPP in plasma showed satisfactory correlation to the corrected semiquantitative intensities measured using NGMS as described above (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI148272DS1). In urine of PDE-ALDH7A1 patients, mean 6-oxoPIP levels were substantially higher than 2-OPP levels (mean 6-oxoPIP 117.4 μmol/mmol creatinine versus 2-OPP 2.6 μmol/mmol creatinine in urine). The 6-oxoPIP levels in urine found in our study were comparable to those previously reported by Kuhara et al. (31). The difference in concentration between 6-oxoPIP and 2-OPP was also observed in plasma, though to a lesser extent as seen in urine (mean 6-oxoPIP 7.5 μM versus mean 2-OPP 3.1 μM in plasma), with 6-oxoPIP levels in plasma being comparable to those reported by Wempe et al. (30). However, in CSF, the difference in concentration was the other way around (mean 6-oxoPIP 0.5 μM versus mean 2-OPP 2.1 μM in CSF). Overall, we found no clear within-sample correlation between 6-oxoPIP and 2-OPP levels (Supplemental Figure 2); however, a positive correlation for 6-oxoPIP and 2-OPP in urine was found with the known PDE-ALDH7A1 urinary biomarker α-AASA (Supplemental figure 3). In all measured body fluids, PDE-ALDH7A1 patient samples could be distinguished from controls based on increased concentrations of the biomarkers 2-OPP and 6-oxoPIP.
Figure 3. 2-OPP and 6-oxoPIP concentrations in PDE-ALDH7A1 patient body fluids.

Absolute concentration of 6-oxoPIP and the sum of 2S,6S-2-OPP and 2S,6R-2-OPP in (A and B) plasma (n = 11), (C and D) urine (n = 8), and (E and F) CSF (n = 9) of PDE-ALDH7A1 patients (PDE) compared with non-IEM controls (n = 16 for plasma, n = 16 for urine, n = 13 for CSF), as measured by quantitative LC-MS/MS. Different treatment regimens in patients are coded as follows: open circles, untreated; filled squares, vitamin B6 supplementation; filled circles, vitamin B6 and arginine supplementation; filled triangles, vitamin B6 supplementation and lysine restriction; filled diamonds, vitamin B6 and arginine supplementation and lysine restriction; cross, therapy unknown. Data are shown as the mean and interquartile range. All patient 2-OPP and 6-oxoPIP concentration ranges shown were significantly increased compared with controls (Mann-Whitney U test; CSF 6-oxoPIP patients versus controls P ≤ 0.0008, all other conditions P ≤ 0.0001).
2-OPP and 6-oxoPIP are increased in PDE-ALDH7A1 patient brain tissue and in plasma and brain tissue Aldh7a1-knockout mice.
To further evaluate the biological relevance of the PDE biomarkers identified in this study, we analyzed an available brain tissue extract sample, obtained postmortem from a deceased PDE-ALDH7A1 patient, using our NGMS assay (Figure 4, A–E). The clinical details of this deceased patient have been described previously (18, 32). In this brain tissue extract, we were able to confirm the presence of features corresponding to the known PDE markers P6C, α-AASA, and pipecolic acid (Figure 4, A, B, and E). Additionally, the biomarker 2-OPP (2S,6S and 2S,6R diastereomers) and the recently described biomarker 6-oxoPIP could be observed in the PDE brain tissue extract (Figure 4, C and D). Upon quantitative measurement, we determined that 2-OPP levels were approximately 5 nmol/gram cortical tissue and 6-oxoPIP levels were approximately 29 nmol/gram. In a previous study, a P6C concentration of 8.14 nmol/gram was reported in this brain specimen, and α-AASA was determined to be 13.02 nmol/gram (32). In brain extracts of 4 non-PDE controls, 2-OPP and 6-oxoPIP levels were below the detection limit of our quantitative assay. In a recently described Aldh7a1-knockout (Aldh7a1-KO) mouse model (33), we were also able to detect increased concentrations of 2-OPP and 6-oxoPIP in plasma (Figure 5, A and B) and brain extracts (Figure 5, E and F), supporting our findings from the human situation. The known PDE-ALDH7A1 markers P6C, α-AASA, and pipecolic acid (Figure 5, C, D, and G) were also found to be increased in brain tissue of Aldh7a1-KO mice, with pipecolic acid accumulating at remarkably high levels.
Figure 4. NGMS analysis of PDE-ALDH7A1 patient brain tissue extract.

Extracted-ion chromatograms in brain tissue extract of a PDE-ALDH7A1 patient of (A) P6C, m/z 128.0706, retention time (RT) 0.92 minutes, (B) α-AASA, m/z 146.0812, RT 0.92 minutes, (C) 2S,6S-2-OPP and 2S,6R-2-OPP, m/z 186.1123, RT 2.42 and 2.67 minutes, respectively, (D) 6-oxoPIP, m/z 144.0652, RT 3.17 minutes, and (E) pipecolic acid, m/z 130.0863, RT 1.07 minutes. The y axis represents relative NGMS intensity, and the x axis represents RT (minutes). Note that an in-source P6C fragment of both 2-OPP enantiomers is also observed at RT 2.42 and 2.67 minutes (A).
Figure 5. Biomarker analysis of Aldh7a1-KO mouse plasma and brain.

Plasma concentrations in Aldh7a1-KO mice (n = 4) compared to WT mice (n = 4) of (A) 6-oxoPIP, and (B) the sum of 2S,6S-2-OPP and 2S,6R-2-OPP. Mean and interquartile ranges are shown. KO levels were significantly increased compared with WT (Mann-Whitney U test, P < 0.05). Extracted-ion chromatograms in brain tissue extract of Aldh7a1-KO mice of (C) P6C, m/z 128.0706, retention time (RT) 0.93 minutes, (D) α-AASA, m/z 146.0812, RT 0.93 minutes, (E) 2S,6S-2-OPP, m/z 186.1125, RT 2.46 minutes, (F) 6-oxoPIP, m/z 144.0652, RT 3.18 minutes, and (G) pipecolic acid, m/z 130.0863, RT 1.06 minutes. Representative data from 1 out of 2 Aldh7a1-KO brain samples is shown. The y axis represents relative NGMS intensity, and the x axis represents RT (minutes).
2-OPP is formed by reaction of P6C with acetoacetate under physiological conditions.
To gain further insight in the biochemical origin of 2-OPP as a biomarker for α-AASA dehydrogenase deficiency, we performed additional experiments to mimic physiological conditions for its formation. From the chemical structure of 2-OPP, as well as from the chemical synthesis of the 2-OPP model compound, we envisioned that 2-OPP could likely be formed in the body by the reaction of accumulating P6C with the ketone body acetoacetate (Figure 6H). To further substantiate this hypothesis, we incubated P6C with acetoacetate in different matrices: water, phosphate-buffered saline (PBS), human (non-IEM) control plasma, or human control urine (Figure 6, A–G). In all these incubation experiments we observed preferential formation of the 2S,6S-2-OPP enantiomer; however, in plasma there appeared to be comparable levels of both the 2S,6R- and 2S,6S-2-OPP enantiomers. Additionally, we incubated P6C with plasma and urine of a patient who was in a ketotic state (based on increased concentration of acetyl-carnitine in plasma and 3-hydroxy-butyric acid in urine; acetoacetate itself was not measured but was assumed to be elevated due to ketosis), and found that 2-OPP was readily formed in both samples, with the ratio of isomers similar to that found in the incubation experiments in control urine and plasma as described above. Finally, we incubated P6C in control plasma without the addition of acetoacetate and found detectable but significantly lower levels of 2-OPP formed. We therefore hypothesize that, even in non-ketotic circumstances, low amounts of acetoacetate present in body fluids of PDE-ALDH7A1 patients can react with accumulating P6C to form micromolar levels of 2-OPP. The identity of the observed features as 2-OPP was confirmed by obtaining IR spectra of the two 2-OPP features and matching those to reference IR spectra (Supplemental Figure 4). Additionally, we could confirm the specificity of 2-OPP as a PDE-ALDH7A1 biomarker, as we measured 2-OPP concentrations within the control range in highly ketotic plasma of non–PDE-ALDH7A1 controls, and showed no correlation between increased acetyl(C2)-carnitine and 2-OPP concentration in conditions of massive ketosis (Supplemental Figure 5).
Figure 6. 2-OPP is formed from P6C and acetoacetate (AcAc).

Upon incubation of 1 mM P6C with 1 mM AcAc in different matrices — (A) H2O, (B) PBS, (C) control plasma, and (D) control urine — 2-OPP formation was observed at elevated levels. Elevated 2-OPP formation was also observed upon incubation of 1 mM P6C with ketotic plasma (E) and urine (F). In the negative control incubation in plasma, including P6C but without the addition of AcAc, significantly lower 2-OPP signals were detected (G). The y axis represents relative NGMS intensity (104 or 106 scale), and the x axis represents RT (minutes). (H) The proposed chemical reaction for the formation of 2-OPP from AcAc and P6C (P6C is formed in equilibrium from α-AASA).
2-OPP shows epileptogenic potential in a zebrafish model system.
As we were able to confirm the presence of 2-OPP in CSF and brain extracts of PDE-ALDH7A1 patients and Aldh7a1-KO mice, we were interested to follow up on the hypothesis that this metabolite may contribute to the epilepsy phenotype in patients. Toward this end, we subjected zebrafish larvae to different concentrations of 2-OPP and compared induced behavior to that after exposure to pentylenetetrazole (PTZ), a positive-control compound known to induce epilepsy-like behavior in zebrafish (34).
Measurement of baseline behavioral activity of the zebrafish larvae 5 days post fertilization (dpf) revealed that 2-OPP significantly increased baseline activity at a concentration of 10 mM in the swimming water (Figure 7A). Upon testing of a 1 μM to 10 mM 2-OPP concentration range in the swimming water, this effect of increased locomotor activity was notable at a concentration of 1 mM, but statistical significance was only reached at 10 mM 2-OPP. This finding corresponded with detection of increasing 2-OPP intensities in zebrafish full-body lysates with higher concentrations of 2-OPP exposure (Supplemental Figure 6). All larvae survived for at least 24 hours at the tested doses, indicating that this effect does not result from immediate toxicity. To investigate whether 2-OPP induces seizure-like hyperactive behavior at 10 mM, zebrafish larvae were subjected to a previously published behavioral paradigm of light/dark cycles (35). After the addition of 2-OPP to the swimming water, larvae were allowed to acclimate in the behavioral chamber for 15 minutes, after which they were subjected to light/dark cycles. During the light flashes, 72.5% of 2-OPP–treated larvae displayed high-speed movements, as compared with 37.5% of control-treated larvae (Figure 7B). In addition, the cumulative duration of these high-speed movements was significantly increased upon treatment with 2-OPP. Further investigation of the observed hyperactive behavior during the light-on periods and the entire period of light/dark cycles revealed that larvae treated with 10 mM 2-OPP had higher movement frequency and travelled longer distances during these movements, as compared with control-treated larvae (Figure 7, C–F). Typical seizure behavior, with whole-body convulsions culminating in loss of posture, was only observed in PTZ-treated larvae. However, the hyperactivity upon 2-OPP treatment is in line with the seizure-like hyperactivity that is described in the genetic zebrafish PDE model (aldh7a1 mutant; ref. 35). This mutant displayed the first signs of seizure-like convulsions at 11 dpf, indicating that a certain buildup of metabolites might be required to induce this behavior.
Figure 7. Hyperactive behavior observed in zebrafish upon 2-OPP exposure.

(A) Distance moved by 5 dpf zebrafish larvae after 10-minute treatment with different concentrations of 2-OPP. PTZ was included as positive control of chemically induced seizure-like hyperactivity. (B) The percentage of zebrafish larvae that display high-speed movements after exposure to a series of light flashes increased upon treatment with 10 mM 2-OPP (left y axis). The duration of the high-speed movements was also significantly increased by 2-OPP (right y axis). (C–F) The frequency of all movements and the distance that the larvae moved were significantly increased during the light-on period (C and D) and during the entire 5-minute period of the light-flash paradigm (E and F), indicating that the observed increase in activity does not solely result from an increased response to the light flash, but is indicative of hyperactivity throughout the light-flash period. *P < 0.05; **P < 0.01; ***P < 0.001 by Kruskal-Wallis test (A) or Mann-Whitney U test (B–F).
Applicability of 2-OPP as a marker for PDE-ALDH7A1 in newborn screening.
To set the first steps toward newborn screening for PDE-ALDH7A1, we also evaluated whether 2-OPP and 6-oxoPIP could be detected in DBS through a direct-infusion MS method without derivatization. This MS method was developed to be compatible with reagents commonly used for neonatal screening (PerkinElmer NeoBase 2 Non-derivatized MS/MS kit, which is for example used in the Dutch screening program). During the development of this direct-infusion MS method for DBS, it was apparent that for 6-oxoPIP, isobaric compounds were present in control DBS that prevented adequate distinction of PDE-ALDH7A1 patients from controls, while for 2-OPP, this was not the case (Supplemental Figure 7). Using the ratio between 2-OPP and hexanoyl/C6-carnitine, a marker already included in the NeoBase kit, as a readout, we could clearly distinguish between 4 PDE-ALDH7A1 patients tested and non-PDE controls (Figure 8). Of note, 2 of these PDE DBS were actual neonatal screening samples from PDE-ALDH7A1 patients, and had been stored for 12 to 13 years at room temperature before the current analysis, indicating high stability of 2-OPP as a DBS biomarker for PDE-ALDH7A1.
Figure 8. 2-OPP–based diagnosis of PDE-ALDH7A1 in DBS using a direct-infusion MS method.
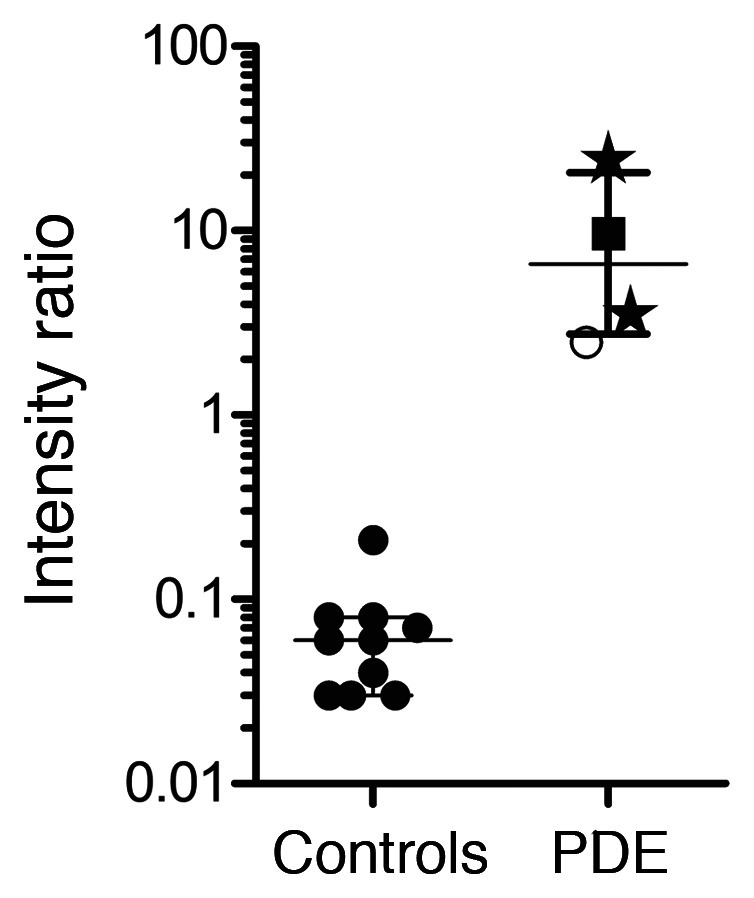
The y axis shows the relative intensity ratio between 2-OPP and C6-carnitine, while on the x axis the categories of non-PDE controls (n = 10) and PDE-ALDH7A1 patients (PDE, n = 4) are plotted. Patient results were significantly increased compared with controls (Mann-Whitney U test, P < 0.006). Patient results are marked as follows: stars, neonatal DBS, untreated patients; filled squares, DBS from 10-year-old patient on vitamin B6 supplementation; open circles, DBS from untreated 16-year-old patient.
Discussion
In this study, the power of combining untargeted metabolomics with IRIS for the identification of diagnostic biomarkers is illustrated. We applied this innovative technology to address the clinical need for novel biomarkers for the treatable metabolic epilepsy syndrome PDE-ALDH7A1 to enable its inclusion in newborn screening. This has allowed for the identification of 2-OPP and 6-oxoPIP as PDE-ALDH7A1 biomarkers accumulating in plasma, urine, CSF, and brain tissue. We have developed a quantitative LC-MS/MS assay for diagnostic application of 2-OPP and 6-oxoPIP measurement in body fluids, adhering to ISO 15189 criteria. Additionally, we have shown applicability of 2-OPP as a first-tier, PDE-ALDH7A1 newborn screening biomarker in DBS, through a direct-infusion MS method compatible with common newborn screening workflows.
Regarding the biochemical relevance of 2-OPP in PDE-ALDH7A1 pathology, we have obtained initial evidence for epileptogenic potential of this compound in a zebrafish model system. Even though the tested concentration of 2-OPP in the swimming water that induced epilepsy-like behavior in zebrafish larvae was significantly higher than the concentration measured in body fluids and brain extracts of PDE-ALDH7A1 patients, it is likely that the actual 2-OPP concentration in the zebrafish brain was much lower than the concentration added to the swimming water. In line with this, relative 2-OPP intensity measured with NGMS in full-body lysates of 10 mM 2-OPP–exposed zebrafish larvae was comparable to 2-OPP intensity measured in the human brain sample (intensity of ~7.5 × 105 in Supplemental Figure 6F versus intensity of ~3 × 106 in Figure 4C). As we have shown 2-OPP accumulation both in human patient and Aldh7a1-KO mouse brain tissue, it could be speculated that 2-OPP exerts neurotoxic effects on the brain and may in turn contribute to the ongoing-disease phenotype in treated PDE-ALDH7A1 patients. However, this hypothesis requires further experiments to determine the exact mechanism of action of 2-OPP in the brain, as well as how concurrent accumulation of α-AASA and P6C contributes to ongoing toxicity.
Of note, 2-OPP and 6-oxoPIP levels lacked a clear correlation with treatment status or patient genotype. In urine, overall a positive correlation between 2-OPP and 6-oxoPIP concentrations and levels of the known biomarker α-AASA was apparent. For most human plasma and urine samples obtained in an untreated state, the concentrations of 2-OPP and 6-oxoPIP were lower than those observed in patients treated solely with vitamin B6. This likely indicates that under treatment with vitamin B6 alone, substantial flux through the lysine pathway is ongoing and continues to lead to the formation of P6C conjugates such as 2-OPP. Unfortunately, CSF samples from untreated patients were not available for analysis and comparison to the other body fluids, but it was apparent that also in CSF, the highest 2-OPP and 6-oxoPIP levels were measured in patients on vitamin B6 monotherapy. We observed 2-OPP levels to be lowest in CSF for patients on B6 supplementation combined with arginine supplementation, with or without additional dietary lysine restriction. Supplementation with arginine competes with lysine for transport across the blood-brain barrier via cationic transporters CAT1 and CAT2, and shows an inverse relationship with CSF 2-OPP levels in the patients included this study. Whether this biochemical finding in CSF correlates with clinical improvement, and is the result of arginine supplementation or lysine-reduction therapy in general, requires further study of well-characterized PDE-ALDH7A1 patients. Indeed, we are planning such a study to evaluate the reliability of 2-OPP and 6-oxoPIP as biomarkers for treatment monitoring and risk stratification for PDE-ALDH7A1 patients.
Also, we identified additional unknown signals in the untargeted-metabolomics data that were significantly increased in PDE-ALDH7A1 patient body fluids. We will continue the process of identification of these unknown features to possibly yield additional insights into the biochemical consequences of α-AASA dehydrogenase deficiency. Of note, upon measurement of the 2-OPP model compound in the HPLC/electrochemical system in which previously an unknown compound (“Peak X”) was detected in CSF of PDE patients by Hyland et al. (36, 37), it was apparent that this “Peak X” could not be identified as 2-OPP (data not shown), indicating that the identity of “Peak X” may still be waiting to be uncovered from our untargeted-metabolomics data.
Another interesting finding from our study, with possible clinical implications, is the proposed mechanism of 2-OPP formation from the reaction of accumulating P6C with the ketone body acetoacetate. This would imply that, especially in a state of ketosis, the increased concentration of acetoacetate would further drive 2-OPP formation in PDE-ALDH7A1 patients. As was stated in the Introduction, in more than one-third of patients, breakthrough epilepsy occurs during intercurrent (febrile) illness, for example gastroenteritis, despite increasing vitamin B6 dosage (15, 16). Catabolism often occurs during such episodes and, as is also known for other IEMs, this can lead to increased flux through amino acid metabolism as well as to ketosis. Even though we have not measured patient samples taken during such an intercurrent illness or period of reduced food intake, we speculate that increased formation of 2-OPP due to ketosis may trigger breakthrough seizures, despite increased vitamin B6 dosage. Follow-up studies are required to further substantiate this hypothesis, and could support consideration of emergency plans for PDE-ALDH7A1 patients during intercurrent illness aimed at avoiding catabolism and ketosis. In the very recently updated consensus guidelines for the diagnosis and management of PDE-ALDH7A1 (38), it is already recommended to double the vitamin B6 dosage for a maximum of 3 days and maintain caloric intake to prevent catabolism during times of illness. Also, our current findings would again emphasize the importance of early diagnosis, as a ketogenic diet, which is a relatively common treatment in epilepsy, may be contraindicated in PDE-ALDH7A1 patients. This should be further studied, as there are currently no such cases reported in the literature.
Regarding the mechanism of formation of 2-OPP from P6C and acetoacetate, our incubation experiments point to a possible nonenzymatic process, as this reaction also takes place in water and PBS. The only apparent difference with the reaction in plasma was the observed ratio between 2S,6S-2-OPP and 2S,6R-2-OPP; in the plasma incubation both enantiomers appeared, while in other matrices the 2S,6S enantiomer was formed preferentially. While the precise mechanism underlying the specific stereochemistry involved in 2-OPP formation will require further study, variability in the 2-OPP enantiomer ratio will not interfere with diagnostic interpretation of results in plasma, urine, or CSF, as in these assays the sum of both 2-OPP enantiomers is used. Also for the DBS assay, the enantiomer ratio will not lead to diagnostic issues, as both enantiomers are detected simultaneously in the direct-infusion MS method, which does not involve LC separation.
In conclusion, our research has uncovered 2-OPP and confirmed 6-oxoPIP as diagnostic PDE-ALDH7A1 biomarkers and has also provided the first evidence to our knowledge for possible applicability of 2-OPP as a biomarker compatible with direct-infusion MS approaches for newborn screening. To our knowledge, our work represents the first example worldwide of the successful application of untargeted, high-resolution MS metabolomics in combination with IRIS for the identification of a candidate biomarker for newborn screening of a treatable neurometabolic disorder. Although whole-exome sequencing has been proposed as an alternative method for newborn screening, which may partly replace separate biochemical assays, a recent study emphasizes the crucial importance of biochemical testing to reach an acceptable level of sensitivity and specificity for newborn screening (39). To further confirm diagnostic specificity and sensitivity of 2-OPP as a first-tier PDE-ALDH7A1 screening biomarker, an extensive clinical validation study should be initiated as a next step, screening thousands of anonymized control neonatal DBS and additional samples from patients using the direct-infusion MS method we have developed. Based on our current results, we are now ready to take on this next step to bring PDE-ALDH7A1 into newborn screening. Additionally, our results pose potential clinical implications regarding the importance of avoiding ketosis in PDE-ALDH7A1 patients. We will further explore these insights in future studies together with the international PDE Consortium and the PDE registry, ultimately to reach optimal clinical outcomes for all PDE-ALDH7A1 patients through early diagnosis and personalized biochemical and clinical follow-up.
Methods
Sample collection.
Heparin-anticoagulated plasma, urine, CSF, and/or DBS of human PDE-ALDH7A1 patients had been previously collected for routine metabolic screening or treatment follow-up. For all PDE-ALDH7A1 patients included, the diagnosis had been genetically confirmed previously. Control samples were obtained from leftover material from non-IEM patients and which did not show abnormalities in targeted-metabolite analyses. All plasma, urine, and CSF samples were stored in a digital alarm–controlled freezer at –20°C before analysis, for a period ranging from 1 month to 9 years. The DBS were stored at room temperature before analysis; neonatal DBS were stored 12 to 13 years, the spot from an untreated adolescent patient was kept for 1.5 year at room temperature before analysis, while the spot of the vitamin B6–treated child was stored 1 month before analysis. Non-PDE control DBS had been stored at room temperature for a period of between 8 and 20 months.
With regard to the human brain tissue samples, full-thickness samples of the cerebral cortex from a 9-year-old female with PDE-ALDH7A1 were taken at autopsy 12 hours postmortem and placed at –80°C. Clinical details on this patient were previously described (18, 32). Control brain tissue was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, Maryland, USA. Cortical tissue was also obtained from epilepsy surgery procedures. After excision, the fresh tissue was frozen in liquid nitrogen and stored at –80° C. For preparation of cortical extracts, frozen cortical tissue (100–200 mg) was separated from underlying white matter and homogenized in 5 mM Tris/HCl (pH 7.4) with 0.32 M sucrose. The homogenates were centrifuged at 3000g and 4°C for 5 minutes, and the resulting supernatant was then centrifuged at 40,000g and 4°C for 1 hour to pellet cellular membranes. The supernatant from this centrifugation was used for subsequent sample preparation for NGMS analysis.
The Aldh7a1-KO mouse model used in this study was generated in accordance with guidelines of the Canadian Council on Animal Care and was previously published (33). Mice used in this study were housed in a pathogen-free barrier facility at the Centre for Molecular Medicine and Therapeutics, University of British Columbia, Canada and were given free access to food and water. Mice were fed a standard chow (Envigo 2018 Teklad Global 18% protein Rodent Diet) containing (w/w) 18% total protein, 0.9% lysine, and 18 ppm pyridoxine. Aldh7a1-KO mice were backcrossed for at least 5 generations onto the C57BL/6J background before generating cohorts for experiments, such that all mice used were incipient congenic on the C57BL/6J background. Plasma and brain tissue from 4 Aldh7a1-KO (1 female and 3 males) and 4 WT mice (2 females and 2 males), all age 14 months, were collected for metabolite analysis. Mice were anesthetized by Avertin (2,2,2-tribromoethanol) injection or isoflurane inhalation. The animals were then dissected, and blood was withdrawn directly from the heart by needle and syringe. Blood was collected in heparin tubes and plasma was separated by centrifuging the blood tubes at 3000 rpm for 10–15 minutes. Next, brain tissue samples were harvested. All samples were snap-frozen with isopentane in dry ice promptly after collection. Tissue and plasma samples were stored in a –80°C freezer until shipment.
Extractions were performed for 2 Aldh7a1-KO and 2 WT mouse brain tissues for NGMS analysis. For each tissue, a region containing mainly the cerebellum was dissected, weighing approximately 1030 mg (range 1003–1078 mg). In an Eppendorf tube, 400 μL of methanol was added and the samples were centrifuged for 30 minutes at 18,600g and room temperature. The tissues were then pulverized and centrifuged again for 20 minutes at room temperature. The resulting supernatant was used for subsequent sample preparation for NGMS analysis.
Sample preparation.
Frozen plasma, urine, CSF, or tissue extract samples were thawed at 4°C and mixed by vortexing. A sample aliquot of 100 μL was transferred into a 1.5 mL polypropylene microcentrifuge tube. Then, 400 μL ice cold methanol/ethanol (50:50 vol/vol) containing 5 internal standards (0.88 μmol/L caffeine-d3, 0.22 μmol/L hippuric-d5 acid, 0.88 μmol/L nicotinic-d4 acid, 0.22 μmol/L octanoyl-L-carnitine-d3, and 0.44 μmol/L L-phenyl-d5-alanine [all from C/D/N Isotopes]) was added to each aliquot. The samples were thoroughly mixed on a vortex mixer for 30 seconds. The samples were incubated at 4°C for 20 minutes, after which they were centrifuged at 18,600g and 4°C for 15 minutes. An aliquot of 350 μL of the supernatant was transferred into a 1.5 mL polypropylene microcentrifuge tube. Samples were dried in a centrifugal vacuum evaporator (Eppendorf) at room temperature. The samples were reconstituted in 100 μL of water containing 0.1% (vol/vol) formic acid, vortexed for 15 seconds, and centrifuged at 18,600g and room temperature for 15 minutes. An aliquot of 90 μL was transferred into 250 μL polypropylene autosampler vials. These samples were either placed in an autosampler operating at 4°C for direct analysis or stored at –80°C. Stored samples were thawed at room temperature and centrifuged at 18,600g and room temperature for 15 minutes before analysis.
NGMS.
NGMS was performed as previously described (20). In short, analyses were performed using an Agilent 1290 UHPLC system coupled to an Agilent 6545 QTOF mass spectrometer, equipped with a dual electrospray ionization (ESI) source. Each sample was run in duplicate in both positive and negative ionization modes. A 2.0 μL aliquot of extracted plasma sample was injected onto an Acquity HSS T3 (C18, 2.1 × 100 mm, 1.8 μm) column (Waters) operating at 40°C. Chromatographic separations were performed by applying a binary mobile phase system. Exact details of buffer composition and MS settings were as previously reported (20). Each analytical batch was composed of control samples, PDE-ALDH7A1 patient samples, analytical quality control (QC) samples, and a validation plasma pool to check for integrity of the automated data analysis pipeline. An analytical run consisted of a maximum of 150 samples. Control and patient samples were analyzed in duplicate. To correct for possible run-order influence on signal intensities, these duplicates were analyzed in antiparallel run order, meaning that duplicates of the first patient sample were analyzed in the first and last position in the analytical run, while duplicates of the last patient sample were analyzed in the 2 middle positions of the run. Eight random control plasma samples were injected at the start of each analytical batch in order to condition the analytical platform. For further details on the analytical QC procedure and requirements, please refer to Coene et al. (20).
NGMS data processing and statistics.
The output files of the NGMS runs were aligned using the open access software package XCMS (online version 3.7.1/2.7.2, XCMS version 1.47.3; https://xcmsonline.scripps.edu) in single-job modus (40). A complete description of XCMS online parameter settings applied is listed in Supplemental Table 1. Following alignment and feature extraction, features were annotated against the Human Metabolome Database (HMDB; ref. 41) for putative metabolite identification. For all features, 2-sided t tests were performed to identify significantly altered features between an individual patient and controls. These steps of alignment, annotation, and statistical testing were integrated in an in-house bioinformatic pipeline that was validated for diagnostic use against ISO 15189 standards. Because of the large number of features identified in an individual patient sample (~10,000), the Bonferroni-Holm procedure was used to correct for multiple testing to prevent false positives, i.e., features incorrectly marked as significantly different in a patient. Two types of t tests were applied to compare the intensity of each feature present in an individual patient sample to the intensities observed in the control samples, as previously described (20). Only features that were marked as significantly different by both t tests after Bonferroni-Holm correction (P value < 0.05) were retained for further analysis. Significantly different features were compared between PDE-ALDH7A1 patient samples to select common differential features.
IRIS MS.
Analyte separations for the IRIS experiments were performed with a Bruker Elute SP HPLC system consisting of a binary pump, cooled autosampler, and column oven using the exact same separation method as described above for NGMS. Fractions containing the metabolites of interest were obtained by collecting the eluent on 96-well plates using a Foxy R2 fraction collector. For these experiments, an injection volume of 20 μL was used.
IRIS experiments were performed in a quadrupole ion trap mass spectrometer (Bruker, AmaZon Speed ETD) modified for spectroscopy. Details of the hardware modifications and synchronization of the experiment with the infrared laser are described elsewhere (42). Collected LC fractions and solutions of reference compounds (~1 × 10−7 M in 50:50 methanol/water) were introduced at 80–180 μL/h flow rates to the electrospray source (+ESI). The ions of interest were mass isolated and irradiated by the Free Electron Laser for Infrared eXperiments (FELIX) for IR analysis. IR spectra were recorded with FELIX set to produce IR radiation in the form of approximately 10 μs macropulses of 50–150 mJ at a 10 Hz repetition rate (bandwidth ~0.4% of the center frequency).
When the laser is resonant with a vibrational transition of the ions, this leads to absorption of the IR photons, producing an increase in the ions’ internal energy and eventually leading to photodissociation. Thus, IR absorption can be observed by recording a fragmentation MS spectrum. IR spectra were constructed by plotting the fractional dissociation [IR yield = ΣI(fragment ions)/ΣI(parent + fragment ions)] as a function of IR laser frequency. The IR yield at each wavelength position of the laser was calculated from 4 to 8 averaged fragmentation mass spectra. The IR frequency was calibrated using a grating spectrometer, and the IR yield was linearly corrected for frequency-dependent variations in the laser pulse energy.
P6C incubation experiments.
P6C (from in-house synthesis, see Supplemental Methods) was incubated at a 1 mM concentration (based on Mills et al., ref. 3) with 1 mM acetoacetate (see Supplemental Figure 8 for synthesis of acetoacetate, not commercially available) for 24 hours at 37°C in different matrices: H2O, PBS (pH 7.5), a non-IEM control plasma pool, and a non-IEM control urine pool. The non-IEM control urine pool consisted of a mixture of 4 random, anonymized left-over samples from diagnostics, while the non-IEM control plasma pool consisted of a mixture of 7 random, anonymized left-over samples. Also, 1 mM P6C was incubated for 24 hours at 37°C with a ketotic urine sample (based on increased excretion of 3-hydroxybutyric acid) and a ketotic plasma sample (based on increased concentration of acetyl-carnitine). After the 24-hour incubation period, samples were put on ice, and a 100 μL aliquot was taken for further processing for NGMS analysis, equal to the sample preparation procedure described above. The resulting samples were also used to record IR spectra according to the procedure described above.
Zebrafish studies.
WT Tüpfel long fin (EZRC), F4 generation zebrafish were bred and raised in recirculation systems (temperature ~27°C, pH 7.5–8, conductivity ~320 μS/cm) under a 14-hour light/10-hour dark cycle. Fish were fed twice daily with Artemia sp. and Gemma Micro 300 (Skretting). Zebrafish eggs were obtained from natural spawning, and raised in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4, and 0.000013% methylene blue) at 28.5°C until experiments.
At 5 dpf, the zebrafish larvae were placed individually in a 96-well plate containing 100 μL E2 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, and 0.33 mM MgSO4). At this early developmental stage, zebrafish larvae are still asexual. Directly before larvae were subjected to behavioral analyses, PTZ (at a concentration of 15 mM) or 2S,6R-2-OPP (at a concentration of 1 μM, 10 μM, 100 μM, 1 mM, or 10 mM) was added to the wells via 100 μL of a 2× stock solution of compound in E2 medium. Video tracking of larval behavior was performed in a DanioVision system and tracked with EthoVision XT14 software from Noldus Information Technology at 28.5°C. The seizure-inducing protocol (35) consisted of 15 minutes of dark adaptation, followed by 5 cycles of 10 seconds of light (100%) and 50 seconds of darkness. After tracking, the 96-well plate was placed back in the incubator and subjected to the same behavioral paradigm after 24 hours of compound treatment. Behavioral responses were analyzed with EthoVision XT14 software, using start and stop velocities of 7.5 mm/s and 5 mm/s, respectively, to determine movement frequencies, and a lower limit of greater than 20 mm/s to determine high-speed movements.
Synthesis of 13C isotope–labeled 6-oxoPIP and (2S,6R)-2-OPP.
To enable quantitative MS, 13C isotope–labeled 6-oxoPIP and (2S,6R)-2-OPP derivatives were prepared, carrying 1 or 2 isotopic labels, respectively (Figure 9). Briefly, (S)-2-aminohexanedioic-6-acid carrying a single 13C label was dehydrated to produce an isotope-labeled 6-oxoPIP analog. The (2S,6R)-2-OPP isotope-labeled standard was prepared by reacting a protected P6C derivative with 13C-labeled acetone, followed by deprotection to produce the isotope-labeled derivative of (2S,6R)-2-OPP (see Supplemental Figures 8 and 9 and the Supplemental Methods for more details on the synthesis).
Figure 9. Isotope-labeled standards of 6-oxoPIP and 2-OPP synthesized for this study.

*Denotes the position of 13C.
Quantitative LC-MS measurement of 6-oxoPIP and 2-OPP in body fluids.
Levels of 6-oxoPIP and 2-OPP were determined in plasma, urine, and CSF using LC-MS/MS through a stable isotope dilution (SID) method. Ten microliters of body fluid (for urine an equivalent to a creatinine level of 0.1 mM) was diluted with 10 μL of stable isotope solution (5 μM 1,3-13C2-(2S,6R)-2-OPP and 0.5 μM 13C1-6-oxoPIP in H2O; see Figure 9 and Supplemental Information for more details), and 200 μL H2O. For plasma, SID was carried out in a 30 kDa centrifugal filter (Amicon Ultra 0.5 mL 30 kDa centrifugal filter regenerated cellulose 30,000 MWCO, Millipore) to remove protein (15 minutes, 14,000g, 15°C). One microliter of the isotope-diluted sample was injected into the HPLC-MS/MS system, which consisted of a Waters I-Class Acquity fitted with an Atlantis T3 (2.1 × 100 mm, particle size 3 μm) column connected to a Waters Xevo TQSμ mass spectrometer. The column was run at 400 μL/min in gradient mode using 0.5% acetic acid in H2O and acetonitrile (initial conditions: 100% 0.5% acetic acid in H2O; gradient: 7.5% acetonitrile per minute) The column flow was directed to the Xevo TQSμ fitted with an ESI probe operating in the positive mode at unit resolution. The capillary voltage was set at 0.35 kV. The temperature settings for the source and ion block were 550°C and 150°C, respectively. As drying gas, nitrogen was used at a flow rate of 800 L/h. The cone gas flow was set at 50 L/h. The collision cell was operated with argon as collision gas at a pressure of 0.35 Pa. For 6-oxoPIP and its stable isotope, a neutral loss of the carboxylic group as -HCOOH was monitored (m/z 144.1 → m/z 98.1). For 2-OPP and its stable isotope, the loss of the oxo-propyl group was monitored (m/z 186.1 → m/z 128.1). Quantification was done by comparison of the generated area response using the corresponding stable isotope as internal standard. This method was validated according to ISO 15189 standards for use in clinical diagnostic procedures.
Direct-infusion MS measurement of 6-oxoPIP and 2-OPP in DBS.
Analysis of 6-oxoPIP and 2-OPP in DBS was performed using a direct-infusion MS/MS method, similar to the NeoBase 2 non-derivatized MS/MS kit protocol. In short, DBS punches with a diameter of 6.35 mm were extracted with 300 μL methanol containing internal standards. Extraction was performed by shaking at 45°C and 100 rpm for 30 minutes. After a 30-minute cooling period, 50 μL of the extract was diluted with methanol and 0.2% formic acid in H2O at a ratio of 1:1:0.2. Using a Waters I-Class Acquity HPLC system, 10 μL of the diluted extract was infused into a Waters Xevo TQSμ at 40 μL/min using 80% acetonitrile and 20% 0.2% formic acid in H2O. The Xevo TQSμ, which was set to monitor not only multiple reaction monitoring (MRM) transitions that are already part of the NeoBase protocol but also the transitions 6-oxoPIP (m/z 144.1 → m/z 98.1) and 2-OPP (m/z 186.1 → m/z 128.1). For every compound, optimized cone and collision energies were used. The Xevo TQSμ was fitted with an electrospray probe and was operated at unit resolution. The capillary voltage was set at 3.0 kV. The temperature settings for the source and ion block were 250°C and 150°C, respectively. As a drying gas, nitrogen was used at a flow rate of 250 L/h. The cone gas flow was set at 50 L/h. The collision cell was operated with argon as the collision gas at a pressure of 0.35 Pa. MS/MS response for 2-OPP and 6-oxoPIP was normalized to the response for hexanoyl (C6)-carnitine, a metabolite that is already analyzed in the NeoBase 2 kit. Hexanoyl-carnitine levels in control DBS were comparable to control levels for 2-OPP in DBS and had a narrow control range, making hexanoyl-carnitine optimally suited for ratio-based normalization.
Statistics.
For comparison of concentration ranges of 2-OPP and 6-oxoPIP in body fluids between controls and PDE-ALDH7A1 patients, a Mann-Whitney U test was used with P less than 0.001 as an overall cut-off for statistical significance. For comparing Aldh7a1-KO mice to WT mice, a Mann-Whitney U test was used with P less than 0.05 as cutoff for statistical significance. For zebrafish experiments, distance moved upon increasing concentrations of 2-OPP exposure were compared with a Kruskal-Wallis test, with a cutoff of P less than 0.05 for statistical significance. A Mann-Whitney U test was used to compare frequency of movements and total moved distance of 2-OPP–exposed zebrafish, with P less than 0.05 considered statistically significant.
Study approval.
All patients and control subjects (or their guardians) registered their informed consent for the possible use of their leftover body fluid samples from clinical diagnostics for laboratory method validation purposes in their electronic patient record, in agreement with institutional and national legislation, as reviewed by the accredited Research Ethics Committee of Radboud University Medical Centre (file number 2021–7296).
Written informed consent for the use of the human brain tissue for research purposes was obtained from each subject or their legal guardian, and was approved by the Institutional Review Board of Seattle Children’s Hospital (application number 11756).
The Aldh7a1-KO mouse model used in this study was generated in accordance with guidelines of the Canadian Council on Animal Care under an approved protocol from the University of British Columbia Animal Care Committee (Animal Protocols numbers A15-0200, A15-0180, A14-0031, and A18-0117).
All zebrafish experiments were carried out in accordance with European guidelines on animal experiments (2010/63/EU).
Author contributions
KLMC, J Martens, JO, and RAW conceptualized the study. UFHE, KLMC, RAW, LAJK, MCDGH, and TMAP performed data interpretation of untargeted-metabolomics data. REVO, FAMGVG, GB, J Martens, and JO were responsible for IRIS-MS experiments and interpretation. J Merx, TJB, J Mecinović, and FPJTR designed and performed synthesis of model compounds and incubation experiments. The order of shared first authors UFHE, REVO, and J Merx was mutually agreed on, with UFHE as the first listed author to reflect the primarily clinical content of the paper. AVR designed and performed the targeted LC-MS/MS and direct-infusion MS experiments. HHAS and BRL provided Aldh7a1-KO mouse tissues and body fluids. Zebrafish studies were performed by SB, EDV, and EVW. LAJ and SMG prepared human PDE and control brain tissue extracts. EAS performed targeted analysis of α-AASA in urine. KH performed analysis of model compounds with HPLC/electrochemical analysis. LAT analyzed data from the PDE registry. LAT, SMA, MAAPW, LAB, and CDMVK were involved in clinical management of PDE-ALDH7A1 patients and translation of results to clinical implications. PK provided bioinformatic support for the untargeted-metabolomics data analysis pipelines. UFHE and KLMC wrote the initial draft of the manuscript. All authors critically revised the manuscript and gave final consent for its submission.
Supplementary Material
Acknowledgments
This research was partly funded by a Stimuleringsbeurs from the Society for Inborn Errors of Metabolism for Netherlands and Belgium (ESN), a catalyst grant from United for Metabolic Diseases (UMD-CG-2020-004), and a Stofwisselkracht grant under the project name “Innovative diagnostics in cerebrospinal fluid of patients with neurometabolic disorders” (all to KLMC). Also, parts of this work were financially supported by an Interfaculty Collaboration Grant from Radboud University Nijmegen (to KLMC, RAW, JO, and J Martens), and an Operating Grant from Canadian Institutes of Health Research (to HHAS and BRL). The authors also gratefully acknowledge the Dutch Research Council, division Natural Sciences, for the support of the FELIX Laboratory (grant numbers VICI 724.011.002, TTW 15769, TKI-LIFT 731.017.419, and Rekentijd 2019.062, all to JO). This work was also supported by an ERC-Stg grant (GlycoEdit, 758913) awarded to TJB. We are indebted to Siebolt de Boer, Joris Reintjes, and Ed van der Heeft for technical assistance, and to Dawn Cordeiro for assistance in CSF sample distribution. We would also like to thank our colleagues from the newborn screening laboratory at Elisabeth Tweesteden Ziekenhuis (ETZ) Tilburg, Netherlands, for helpful discussions on newborn screening methodology. This research made use of metabolomics infrastructure that is part of the NWO-funded Netherlands X-omics initiative, project 184.034.019.
Version 1. 06/17/2021
In-Press Preview
Version 2. 08/02/2021
Electronic publication
Funding Statement
Project ’Innovative diagnostics in cerebrospinal fluid of patients with neurometabolic disorders’
Stimuleringsbeurs project ‘Novel biomarkers for pyridoxine dependent epilepsy: functional characterization and clinical application’
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(15):e148272.https://doi.org/10.1172/JCI148272.
Contributor Information
Udo F.H. Engelke, Email: Udo.Engelke@radboudumc.nl.
Rianne E. van Outersterp, Email: rianne.vanoutersterp@ru.nl.
Jona Merx, Email: j.merx@science.ru.nl.
Fred A.M.G. van Geenen, Email: fredvangeenen@gmail.com.
Arno van Rooij, Email: arno.vanrooij@radboudumc.nl.
Giel Berden, Email: giel.berden@ru.nl.
Marleen C.D.G. Huigen, Email: marleen.huigen@radboudumc.nl.
Leo A.J. Kluijtmans, Email: Leo.Kluijtmans@radboudumc.nl.
Tessa M.A. Peters, Email: tessa.peters@radboudumc.nl.
Hilal H. Al-Shekaili, Email: halshekaili@bcchr.ca.
Blair R. Leavitt, Email: bleavitt@cmmt.ubc.ca.
Erik de Vrieze, Email: Erik.deVrieze@radboudumc.nl.
Sanne Broekman, Email: Sanne.Broekman@radboudumc.nl.
Erwin van Wijk, Email: erwin.vanwyk@radboudumc.nl.
Laura A. Tseng, Email: l.a.tseng@amsterdamumc.nl.
Purva Kulkarni, Email: purva.kulkarni@radboudumc.nl.
Floris P.J.T. Rutjes, Email: floris.rutjes@ru.nl.
Eduard A. Struys, Email: e.struys@amsterdamumc.nl.
Laura A. Jansen, Email: ljansen@wustl.edu.
Saadet Mercimek-Andrews, Email: saadet.andrews@sickkids.ca.
Keith Hyland, Email: khyland@mnglabs.com.
Levinus A. Bok, Email: l.bok@mmc.nl.
Clara D.M. van Karnebeek, Email: Clara.vanKarnebeek@radboudumc.nl.
Ron A. Wevers, Email: Ron.Wevers@radboudumc.nl.
Thomas J. Boltje, Email: t.boltje@ru.nl.
Jos Oomens, Email: j.oomens@science.ru.nl.
Jonathan Martens, Email: jonathan.martens@ru.nl.
Karlien L.M. Coene, Email: karlien.coene@radboudumc.nl.
References
- 1.Struys EA, Jakobs C. Metabolism of lysine in alpha-aminoadipic semialdehyde dehydrogenase-deficient fibroblasts: evidence for an alternative pathway of pipecolic acid formation. FEBS Lett. 2010;584(1):181–186. doi: 10.1016/j.febslet.2009.11.055. [DOI] [PubMed] [Google Scholar]
- 2.Crowther LM, et al. New insights into human lysine degradation pathways with relevance to pyridoxine-dependent epilepsy due to antiquitin deficiency. J Inherit Metab Dis. 2019;42(4):620–628. doi: 10.1002/jimd.12076. [DOI] [PubMed] [Google Scholar]
- 3.Mills PB, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006;12(3):307–309. doi: 10.1038/nm1366. [DOI] [PubMed] [Google Scholar]
- 4.Bok LA, et al. Long-term outcome in pyridoxine-dependent epilepsy. Dev Med Child Neurol. 2012;54(9):849–854. doi: 10.1111/j.1469-8749.2012.04347.x. [DOI] [PubMed] [Google Scholar]
- 5.van Karnebeek CDM, et al. Lysine restricted diet for pyridoxine-dependent epilepsy: first evidence and future trials. Mol Genet Metab. 2012;107(3):335–344. doi: 10.1016/j.ymgme.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Mercimek-Mahmutoglu S, et al. Novel therapy for pyridoxine dependent epilepsy due to ALDH7A1 genetic defect: L-arginine supplementation alternative to lysine-restricted diet. Eur J Paediatr Neurol. 2014;18(6):741–746. doi: 10.1016/j.ejpn.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Coughlin CR, et al. Triple therapy with pyridoxine, arginine supplementation and dietary lysine restriction in pyridoxine-dependent epilepsy: neurodevelopmental outcome. Mol Genet Metab. 2015;116(1–2):35–43. doi: 10.1016/j.ymgme.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 8.Al Teneiji A, et al. Phenotype, biochemical features, genotype and treatment outcome of pyridoxine-dependent epilepsy. Metab Brain Dis. 2017;32(2):443–451. doi: 10.1007/s11011-016-9933-8. [DOI] [PubMed] [Google Scholar]
- 9.Coughlin CR, et al. The genotypic spectrum of ALDH7A1 mutations resulting in pyridoxine dependent epilepsy: A common epileptic encephalopathy. J Inherit Metab Dis. 2019;42(2):353–361. doi: 10.1002/jimd.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Struys EA, et al. The measurement of urinary Δ1-piperideine-6-carboxylate, the alter ego of α-aminoadipic semialdehyde, in Antiquitin deficiency. J Inherit Metab Dis. 2012;35(5):909–916. doi: 10.1007/s10545-011-9443-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jung S, et al. Preliminary investigation of the use of newborn dried blood spots for screening pyridoxine-dependent epilepsy by LC-MS/MS. Mol Genet Metab. 2013;110(3):237–240. doi: 10.1016/j.ymgme.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 12.Yuzyuk T, et al. A novel method for simultaneous quantification of alpha-aminoadipic semialdehyde/piperideine-6-carboxylate and pipecolic acid in plasma and urine. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1017:145–152. doi: 10.1016/j.jchromb.2016.02.043. [DOI] [PubMed] [Google Scholar]
- 13.Xue J, et al. Simultaneous quantification of alpha-aminoadipic semialdehyde, piperideine-6-carboxylate, pipecolic acid and alpha-aminoadipic acid in pyridoxine-dependent epilepsy. Sci Rep. 2019;9(11371):1–10. doi: 10.1038/s41598-019-47882-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathew EM, et al. Biomarker profiling for pyridoxine dependent epilepsy in dried blood spots by HILIC-ESI-MS. Int J Anal Chem. 2018;2018(2583215):1–8. doi: 10.1155/2018/2583215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Karnebeek CDM, et al. Pyridoxine-dependent epilepsy: an expanding clinical spectrum. Pediatr Neurol. 2016;59:6–12. doi: 10.1016/j.pediatrneurol.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 16.Niermeijer JMF, et al. Pyridoxine dependent epilepsy: clinical features and progressive serial brain MRI abnormalities. Tijdschrift voor Kindergeneeskunde. 2013;81:88 [Google Scholar]
- 17.Friedman SD, et al. Callosal alterations in pyridoxine-dependent epilepsy. Dev Med Child Neurol. 2014;56(11):1106–1110. doi: 10.1111/dmcn.12511. [DOI] [PubMed] [Google Scholar]
- 18.Mills PB, et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency) Brain. 2010;133(pt 7):2148–2159. doi: 10.1093/brain/awq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oesch G, et al. Geometric morphometricsreveal altered corpus callosum shape in pyridoxine-dependent epilepsy. Neurology. 2018;91(1):E78–E86. doi: 10.1212/WNL.0000000000005748. [DOI] [PubMed] [Google Scholar]
- 20.Coene KLM, et al. Next-generation metabolic screening: targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J Inherit Metab Dis. 2018;41(3):337–353. doi: 10.1007/s10545-017-0131-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Karnebeek CDM, et al. NANS-mediated synthesis of sialic acid is required for brain and skeletal development. Nat Genet. 2016;48(7):777–784. doi: 10.1038/ng.3578. [DOI] [PubMed] [Google Scholar]
- 22.Vaclavik J, et al. Structural elucidation of novel biomarkers of known metabolic disorders based on multistage fragmentation mass spectra. J Inherit Metab Dis. 2018;41(3):407–414. doi: 10.1007/s10545-017-0109-4. [DOI] [PubMed] [Google Scholar]
- 23.Martens J, et al. Molecular identification in metabolomics using infrared ion spectroscopy. Sci Rep. 2017;7(3363):1–5. doi: 10.1038/s41598-017-03387-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Outersterp RE, et al. Reference-standard free metabolite identification using infrared ion spectroscopy. Int J Mass Spectrom. 2019;443:77–85. doi: 10.1016/j.ijms.2019.05.015. [DOI] [Google Scholar]
- 25.Martens J, et al. Unraveling the unknown areas of the human metabolome: the role of infrared ion spectroscopy. J Inherit Metab Dis. 2018;41(3):367–377. doi: 10.1007/s10545-018-0161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martens J, et al. Structural identification of electron transfer dissociation products in mass spectrometry using infrared ion spectroscopy. Nat Commun. 2016;7(11754):1–7. doi: 10.1038/ncomms11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martens J, et al. Infrared ion spectroscopy: new opportunities for small-molecule identification in mass spectrometry — a tutorial perspective. Anal Chim Acta. 2020;1093:1–15. doi: 10.1016/j.aca.2019.10.043. [DOI] [PubMed] [Google Scholar]
- 28.Martens J, et al. Combined liquid chromatography-infrared ion spectroscopy for identification of regioisomeric drug metabolites. Anal Chem. 2017;89(8):4359–4362. doi: 10.1021/acs.analchem.7b00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. doi: 10.26434/chemrxiv.14315579.v1. van Outersterp RE, et al. Metabolite identification using infrared ion spectroscopy – novel biomarkers for pyridoxine-dependent epilepsy [preprint]. Posted on ChemRxiv March 29, 2021. [DOI]
- 30.Wempe MF, et al. Identification of a novel biomarker for pyridoxine-dependent epilepsy: Implications for newborn screening. J Inherit Metab Dis. 2019;42(3):565–574. doi: 10.1002/jimd.12059. [DOI] [PubMed] [Google Scholar]
- 31.Kuhara T, et al. Identification of new biomarkers of pyridoxine-dependent epilepsy by GC/MS-based urine metabolomics. Anal Biochem. 2020;604(113739):1–10. doi: 10.1016/j.ab.2020.113739. [DOI] [PubMed] [Google Scholar]
- 32.Jansen LA, et al. Glial localization of antiquitin: implications for pyridoxine-dependent epilepsy. Ann Neurol. 2014;75(1):22–32. doi: 10.1002/ana.24027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al-Shekaili HH, et al. A novel mouse model for pyridoxine-dependent epilepsy due to antiquitin deficiency. Hum Mol Genet. 2020;29(19):3266–3284. doi: 10.1093/hmg/ddaa202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hortopan GA, et al. Zebrafish as a model for studying genetic aspects of epilepsy. Dis Model Mech. 2010;3(3–4):144–148. doi: 10.1242/dmm.002139. [DOI] [PubMed] [Google Scholar]
- 35.Pena IA, et al. Pyridoxine-dependent epilepsy in zebrafish caused by Aldh7a1 deficiency. Genetics. 2017;207(4):1501–1518. doi: 10.1534/genetics.117.300137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hyland K, et al. Folinic acid responsive seizures: a new syndrome? J Inherit Metab Dis. 1995;18(2):177–181. doi: 10.1007/BF00711760. [DOI] [PubMed] [Google Scholar]
- 37.Gallagher RC, et al. Folinic acid-responsive seizures are identical to pyridoxine-dependent epilepsy. Ann Neurol. 2009;65(5):550–556. doi: 10.1002/ana.21568. [DOI] [PubMed] [Google Scholar]
- 38.Coughlin CRI, et al. Consensus guidelines for the diagnosis and management of pyridoxine-dependent epilepsy due to α-aminoadipic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. 2021;44(1):178–192. doi: 10.1002/jimd.12332. [DOI] [PubMed] [Google Scholar]
- 39.Adhikari AN, et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat Med. 2020;26(9):1392–1397. doi: 10.1038/s41591-020-0966-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forsberg EM, et al. Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online. Nat Protoc. 2018;13(4):633–651. doi: 10.1038/nprot.2017.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wishart DS, et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018;46(D1):D608–D617. doi: 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martens J, et al. Infrared ion spectroscopy in a modified quadrupole ion trap mass spectrometer at the FELIX free electron laser laboratory. Rev Sci Instrum. 2016;87(10):1–8. doi: 10.1063/1.4964703. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.