

Keywords: adhesion, inflammation, liver sinusoidal endothelial cells, nonalcoholic steatohepatitis
Abstract
Liver sinusoidal endothelial cells (LSECs) are distinct subtypes of endothelial cells lining a low flow vascular bed at the interface of the liver parenchyma and the circulating immune cells and soluble factors. Emerging literature implicates LSEC in the pathogenesis and progression of nonalcoholic fatty liver disease (NAFLD). During the evolution of NAFLD, LSEC dysfunction ensues. LSECs undergo morphological and functional transformation known as “capillarization,” as well as a pathogenic increase in surface adhesion molecules expression, referred to in this review as “endotheliopathy.” LSECs govern the composition of hepatic immune cell populations in nonalcoholic steatohepatis (NASH) by mediating leukocyte subset adhesion through specific combinations of activated adhesion molecules and secreted chemokines. Moreover, extracellular vesicles released by hepatocyte under lipotoxic stress in NASH act as a catalyst for the inflammatory response and promote immune cell chemotaxis and adhesion. In the current review, we highlight leukocyte adhesion to LSEC as an initiating event in the sterile inflammatory response in NASH. We discuss preclinical studies targeting immune cells adhesion in NASH mouse models and potential therapeutic anti-inflammatory strategies for human NASH.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD), characterized by hepatic steatosis in the absence of alcohol consumption, is the most common chronic liver disease worldwide (1). A subset of patients with NAFLD develop an inflammatory and fibrotic form known as nonalcoholic steatohepatitis (NASH) and eventually progress to end-stage liver disease. To date, available treatments are mainly geared toward controlling NASH-associated comorbidities (2). Thus, there is a critical need for mechanism-based therapeutic strategies to control the progression of NAFLD. A change in the name for NAFLD to metabolic (dysfunction)-associated fatty liver disease (MAFLD) was recently recommended by an international panel of experts (3). In the current review, we employed the term “NAFLD” in accordance with the referenced studies.
LSECs are unique endothelial cells lining the liver sinusoidal space (4). LSECs differ from other capillary endothelial cells by the presence of fenestrae and the absence of the basement membrane. Fenestrae are 50–200 nm dynamic pores occupying up to 20% of the LSEC surface. Fenestrae are clustered as sieve plates and extend from the basolateral to the apical membranes of the LSEC. Furthermore, fenestrae change their diameters in response to the local microenvironment signals. These features enhance the LSEC capacity to control the movement of macromolecules between the circulation and the liver parenchyma (5). Moreover, LSECs shape the hepatic immune cell populations by mediating diverse leukocyte subpopulation adhesion by distinct interaction of adhesion molecules and chemokines (4). Hammoutene et al. (6) have recently shown, using transmission electron microscopy, impaired LSEC autophagy in human NASH. In addition, authors employed mice deficient in autophagy (ATG5) in endothelial cells on high-fat diet and showed upregulation of adhesion molecules, proinflammatory and profibrogenic mediators, and striking presinusoidal fibrosis (6). These findings suggest that impaired LSEC autophagy in NASH may promote the endothelial-to-mesenchymal transition.
Toxic lipid-induced hepatocellular stress known as lipotoxicity has been recognized as a driver of the inflammatory response in the NASH liver (7). Hepatocytes under lipotoxic stress release extracellular vesicles (EVs) enriched with proinflammatory mediators. These EVs enhance the recruitment of proinflammatory monocytes into the liver and their adhesion to the LSECs (8–10). Furthermore, emerging studies suggest that the expression of adhesion and proinflammatory molecules is increased in LSEC under lipotoxic conditions (11, 12). In this review, we discuss the pathogenic roles of LSECs in NAFLD with special emphasis on LSEC adhesion molecule blockade, as a potential therapeutic strategy in NASH.
LIVER SINUSOIDAL ENDOTHELIAL CELLS CAPILLARIZATION AND DYSFUNCTION IN NAFLD
During the evolution of NASH, the LSECs undergo phenotypic changes referred to as capillarization consisting of reduced size and number of fenestrae and deposition of basement membrane on the abluminal side. An emerging body of literature supports the development of LSEC capillarization during the early stage of NAFLD (13). LSEC capillarization is partially attributed to excessive dietary fat and gut microbiota-related factors (14). LSEC capillarization in turn promotes hepatic steatosis, mainly secondary to reduced chylomicron remnant uptake by hepatocyte, and clearance from the circulation resulting in a compensatory increase in hepatocyte de novo lipogenesis (15).
Before the development of inflammation and fibrosis, hepatic steatosis induces a hemodynamically significant increase in intrahepatic vascular resistance. This process is partially attributed to the impaired microcirculation secondary to the compression of the sinusoidal space by steatotic and ballooned hepatocytes. The mechanical stress exerted by steatotic hepatocytes on LSECs may increase the intrahepatic vascular resistance and culminate in the development of a pathological condition known as endothelial dysfunction. Endothelial dysfunction is defined as impaired regulation of vasodilation by the endothelium in response to the environmental changes and is likely secondary to diminished production of the vasodilator nitric oxide (NO) by the LSECs and increased endothelin-1 (ET1) synthesis (16). More mechanistic insight into LSEC dysfunction was gained through work by Pasarín et al. (17). The authors employed a rat model of steatosis using a cafeteria diet (65% of fat, mostly saturated) for 1 mo and showed a link between increased hepatic vascular resistance and impaired insulin sensitivity and Akt signaling. These rats showed decreased Akt-dependent endothelial nitric oxide synthase (NOS) phosphorylation and NO synthesis and release (17). Hence, therapeutic agents geared toward enhancing insulin sensitivity may also improve endothelial dysfunction in NASH. In addition, Francque et al. (16) applied in situ liver perfusion in methionine choline-deficient (MCD)-fed rats for 4 wk and showed increased portal pressure, secondary to increased intrahepatic resistance in rats with steatosis. Rats with steatosis showed reduced vasodilation in response to acetylcholine. The increased vascular resistance was associated with increased liver thromboxane synthase and endothelin-1 (ET-1) and serum ET-1. However, no alterations in NO was identified in this study (16). Nonetheless, authors subsequently demonstrated hyporesponsiveness to exogenous NO in their liver steatosis rat model (18). They further explored the therapeutic benefit of blocking both ET-A and -B receptors by Bosentan. These data were reported in a poster presentation in 2020 and showed a preventive and therapeutic effect of Bosentan in their animal model of steatosis, as manifested by improving transhepatic pressure gradient and transaminases level. Taken together, these studies suggest that ET receptor blockade is a potential therapeutic target for LSEC dysfunction in NASH.
A recent study by Dr. Iwakiri group unraveled a new regulatory mechanism of NOS. The study showed increased heat shock protein 90 (Hsp90) acetylation in ethanol-fed mice. HSP90 enhanced endothelial NOS activity, and its acetylation impaired its interaction with endothelial NOS and decreased NO production. Histone deacetylase 6 (HDAC6) overexpression in LSECs with adeno-associated virus vector serotype 8 (AAV8)-mediated gene delivery system to mice led to Hsp90 deacetylation, restoration of Hsp90’s interaction with endothelial NOS, increased NO release, and improved alcohol-induced LSEC dysfunction (19). Collectively, these studies indicate that multiple druggable targets are involved in the regulation of NO and ET pathways and may have a role in maintaining LSEC homeostasis in chronic liver disease and NASH.
PROINFLAMMATORY ROLES OF LSECs IN NASH
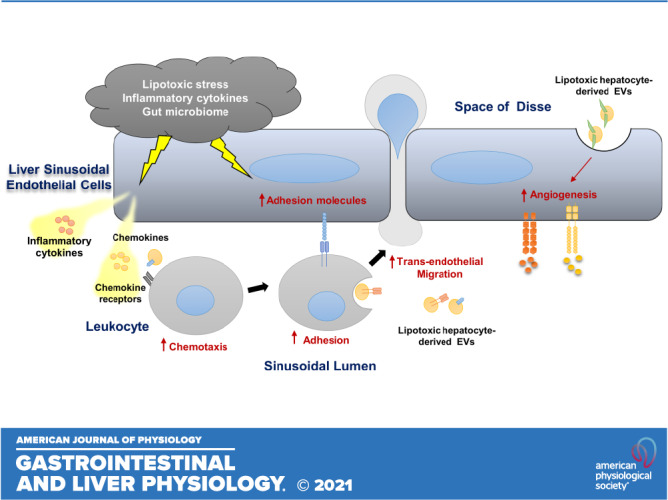
During NASH progression, LSECs play a key role in regulating the recruitment of leukocytes from the circulation and their homing into the liver (20). LSECs develop a proinflammatory phenotype that includes 1) secretion of proinflammatory chemokines and cytokines (LSECs can also present chemokines produced by neighboring cells to promote adhesion and migration of various immune cells); 2) enhanced adhesion molecules expression, a process referred to as “endotheliopathy” and also recognized in atherosclerosis progression; and 3) augmented angiogenesis (8, 21, 22) (Fig. 1).
Figure 1.

Liver sinusoidal endothelial cell (LSEC), a key player in hepatic inflammation in nonalcoholic steatohepatis (NASH). Lipotoxic stress, proinflammatory cytokines and chemokines, and the gut microbiome promote LSEC release of proinflammatory cytokines and chemokines and enhance the expression of adhesion molecules. Lipotoxic hepatocyte-derived extracellular vesicles (EVs) are enriched with CXCL10, ITGα9β1, and Vanin-1 and promote monocyte-derived macrophage chemotaxis and adhesion to LSECs. These EVs also elicit a proangiogenic signaling. Vascular cell adhesion molecule 1 (VCAM1), intercellular adhesion molecule 1 (ICAM-1), and the monoamine oxidase vascular adhesion protein-1 (VAP-1) on the LSEC surface mediate leukocyte adhesion and homing in the NASH liver. The vascular endothelial growth factor (VEGF) and angiopoietin/Tie2 pathways are activated in NASH and enhance angiogenesis.
Release and Transcytosis of Inflammatory Mediators by LSECs in NASH
LSECs are instrumental in the liver innate defense mechanism. For instance, LSECs express the pattern recognition receptor, toll-like receptor 9 (TLR9), in the lysosomal compartments, which is engaged by endocytosed bacterial DNA via scavenger receptors. TLR9 engagement results in the activation of the transcription factor NF-κB and the release of interleukin (IL)-6 and IL-1β among other cytokines (21). Furthermore, LSECs secrete in response to cytokine stimulation or lipotoxic stress, numerous chemokines including the C-X-C motif ligands, and the C-C motif chemokine ligands (11, 20). These chemokines enhance the trafficking of immune cells to the liver.
Using single-cell RNA-sequencing and spatial mapping, Ramachandran et al. (23) resolved the fibrotic niche consisting of nonparenchymal cells topographically located within areas of fibrosis in humans with cirrhosis. The authors identified expansion of endothelial cells positive for ACKR1, also known as Duffy Antigen Receptor for Chemokines (DARC) in the fibrotic niche in cirrhosis. In addition, they confirmed that ACKR1 enhances the transmigration of leukocytes (23). DARC is a silent chemokine receptor that internalizes chemokines and mediates their transcytosis, but does not effectively scavenge them or transmit intracellular signals (24). Hence, in the NASH liver, DARC may enhance the transport of chemokines produced by lipotoxic hepatocytes to the sinusoidal space and facilitate their engagement in circulating leukocyte chemotaxis and adhesion.
Moreover, Hilscher et al. (25) showed that LSECs release the neutrophil chemotactic ligand CXCL1 in response to mechanical stretch in a mouse model of hepatic congestion. In this study, C-X-C motif chemokine ligand 1 (CXCL1) enhanced the development of neutrophil extracellular traps (NETs). NETs consist of heterogeneous, filamentous structures of extracellular DNA, histones, and granular proteins extruded by neutrophils. Interestingly, NETs enhance the inflammation and the development of hepatocellular carcinoma in NASH (26). Future area of investigation includes the release of CXCL1 from LSEC under lipotoxic stress and the role of CXCL1 in the development of NETs in NASH.
The Role of LSEC Adhesion Molecules in Leukocyte Homing in the NASH Liver
Leukocyte trafficking to the inflamed tissue is tightly regulated and involves a sequence of rolling, adhesion, and transendothelial migration. These events are mediated by precise binding interactions between adhesion molecules on the endothelial cell surface and their corresponding partners on the leukocytes and are regulated by leukocyte stimulatory molecules, such as chemokines (20). Firm adhesion is mediated mainly by integrins (ITGs). For instance, neutrophils ITGαMβ2 (or Mac-1) and ITGαLβ2 [or lymphocyte function-associated (LFA)-1] bind to intercellular adhesion molecule-1 (ICAM-1) on LSECs. In addition, ITGα4β1 and ITGα4β7 for all other leukocytes bind mainly to vascular cell adhesion molecule (VCAM1) and mucosal address in cell adhesion molecule-1 (MAdCAM1) on LSECs. Tissue specificity of lymphocyte homing is mediated by distinct chemokine signaling via either C-C motif chemokine ligand (CCL25) or CXCL10. Chemokine signaling results in differential phosphorylation of the ITGβ7 tail, which regulates the binding affinity of the lymphocyte ITGα4β7 to its endothelial ligands MAdCAM1 and VCAM1. The integrins switch to high avidity and/or affinity status following leukocyte activation by chemokines allows firm adhesion to occur (27). Stable adhesion results in the retention of circulating leukocytes by the activated LSECs and their transendothelial migration to the inflammatory sentinels in the liver parenchyma (28).
Adhesion molecules expression in LSEC is distinguished from other endothelial cells, as discussed below. First, LSECs have low expression of the leukocyte rolling mediators E- and P-selectins. In line with this, leukocyte adhesion within sinusoids occurs independently of rolling (29, 30). Second, LSECs express atypical adhesion molecules such as vascular adhesion protein-1 (VAP-1) and scavenger receptor like Stabilin 1 and 2, in addition to the classical adhesion molecules VCAM-1 and ICAM-1(20). VAP-1 is a membrane bound amine oxidase, also recognized as an induced ectoenzyme at inflammatory sites. VAP-1 activates nuclear factor‐κB via a phosphatidylinositol‐3 kinase, and a mitogen‐activated protein kinase (MAPK) pathway and modulates the expression of chemokine (CXCL8) and adhesion molecules (Vcam1, Icam1, and E-selectin) in human LSECs (31). In addition to the typical paracellular course, circulating immune cells can directly traverse through the body of LSEC by “transcellular crawling.” This route of migration is facilitated by the translocation of clustered ICAM-1 upon leukocyte occupancy and the development of transcellular channels (32).
Clinical relevance in NASH of major adhesion molecules expressed on LSECs and their leukocyte counterpart ligands are shown in Table 1 and discussed below as follows. Weston et al. (33) showed that mice expressing a catalytically inactive form of VAP-1 or treated with anti-VAP-1 antibody were relatively protected against MCD diet-induced NASH, as manifested by reduced liver inflammation and fibrosis (33). Moreover, serum levels of soluble VAP-1 were elevated in NAFLD patients when compared with normal subjects (33). Likewise, we have recently reported that LSEC VCAM1 expression is augmented in mouse and human NASH. More importantly, VCAM1 pharmacological inhibition or conditional endothelial cell deletion in mice with diet-induced NASH attenuated liver injury, inflammation, and fibrosis (12). Neutralizing antibody against ITGβ1 (a VCAM1-binding partner) also ameliorated the hepatic inflammation, and fibrosis in mice with diet-induced NASH, likely secondary to reduced proinflammatory monocyte adhesion and hepatic infiltration (10). The human relevance of VCAM1 as a potential NASH biomarker was also explored by Lefere et al. (34). Authors reported that circulating soluble VCAM-1 level in patients with NAFLD was an independent predictor of liver fibrosis at stage 2 and above (34). Similarly, neutralizing antibody against ITGα4 (one of the VCAM-1 ligand) diminished the proinflammatory monocyte-associated liver inflammation in mice with NASH (11). These studies also noted that the expression of ICAM-1 and VCAM-1 was enhanced in LSEC treated with the saturated free fatty acid palmitate (11, 12). Similarly, Rai et al. (35) observed reduced liver inflammation, fibrosis, and metabolic dysfunction in mice with diet-induced NASH treated with the ITGα4β7 antibody. ITGα4β7 antibody abrogated CD4 T-cell homing to the liver (35). Recently, Drescher et al. (36) targeted L-selectin [also known as CD62 Ligand (CD62L)], a known lymphocyte ligand for ICAM1 and MadCAM1. In their study, CD62L knockout mice and anti-CD62L antibody-treated mice were protected against diet-induced NASH. Furthermore, serum levels of soluble CD62L was increased in NASH patients and hepatic expression of CD62L correlated with NASH activity (36). Collectively, these data suggest that lipotoxicity enhances the expression of LSEC adhesion molecules, mainly via a MAPK signaling pathway (12). Furthermore, CD62L, VCAM1, and/or VAP-1 among other adhesion molecules are candidate therapeutic targets and/or biomarkers in human NASH.
Table 1.
Mediators of immune cells adhesion and clinical relevance in NASH
| LSEC Adhesion Molecules |
Leukocyte Binding Partner |
Mice Studies |
Human Studies: | ||
|---|---|---|---|---|---|
| Inhibition | Mouse Model | Liver Histology | Adhesion Molecule Serum Level/Hepatic Expression | ||
| VCAM-1/MAdCAM-1 | ITGα4β1 ITGα4β7 ITGα9β1 |
Anti-ITGα4β1 Ab | HFD | Decreased myeloid cell hepatic infiltration (11) | VCAM-1 serum level was an independent predictor for ≥ F2 fibrosis in NAFLD (34). VCAM-1 hepatic expression was increased in NASH (12). MAdCAM-1 hepatic expression was elevated in NASH and correlated with ITGα4β7(35) |
| Anti-ITGα4β7 Ab | WD | Decreased CD4+ T-cell hepatic infiltration and fibrosis (35) |
|||
| Anti-ITGβ1 Ab | FFC | Decreased MoMF-associated liver inflammation and fibrosis (10) | |||
| Anti-VCAM1 Ab AGI-1067 |
FFC | Reduced MoMF-associated liver inflammation and fibrosis (12) | |||
| ICAM1/ MadCAM1 | L-Selectin (CD62L) | CD62L−/− Anti-CD62L Ab |
HFD or MCD | Decreased steatosis, increased Treg (36) | Soluble CD62L serum level was increased in NASH CD62L hepatic expression correlated with NASH activity (36) |
| VAP-1 | Unknown | VAP-1−/− Anti VAP-1 Ab |
WD, or MCD | Decreased hepaticinflammatory cell infiltrate and fibrosis(33) | VAP-1 serum level correlated with histological severity in NAFLD (33) |
FFC, fat, fructose- and cholesterol-rich diet; HFD, high-fat diet; ICAM1, intercellular adhesion molecule 1; ITG, integrin; LSEC, liver sinusoidal endothelial cell; MAdCAM-1, mucosal address in cell adhesion molecule-1; MoMF; monocyte-derived macrophage; Treg cell, regulatory T cell; VAP1, vascular adhesion protein; VCAM-1, vascular cell adhesion molecule 1; WD, Western diet.
Complement System Activation in NASH and the Role of LSEC C5a Receptor
The complement system is activated in human NAFLD, in part, secondary to the deposition of gut-derived lipopolysaccharides (LPS) in the NASH liver, and the engagement of the complement system in the clearance of apoptotic hepatocytes (37). However, the role of LSECs in regulating the complement-mediated immune response in NASH is unclear. Prior reports suggested that the human umbilical venous endothelial cells (HUVEC) are activated by the complement activation peptides, C3a and C5a. Activated HUVEC in this context upregulate the adhesion molecule VCAM1 and ICAM1 (38), and modulate the activation and polarization of trafficking lymphocytes (39). Likewise, C5a receptor (C5aR) expression in the dermal and lung microvascular endothelium is upregulated upon LPS or cytokines stimulation (40). These studies suggest that C5aR may, in part, mediate the proinflammatory effects of the endothelium. Whereas, blocking C5aR might dampen the inflammatory response in NASH; impaired regeneration with C5aR inhibition might offset the potential beneficial anti-inflammatory effect.
Enhanced Angiogenesis in NASH Culminates in Disease Progression
Angiogenesis in the liver is driven in part via engagement of the vascular endothelial growth factor (VEGF)/VEGF receptor (VEGFR) and the angiopoietin (Ang)/tyrosine kinase with immunoglobulin-like and EGF-like domains 2 (Tie2) (41, 42) (Fig. 1). Emerging literature links anomalous activation of the angiogenic signaling with the development of chronic inflammation in NASH. For instance, pharmacological inhibition of VEGFR2 attenuated liver inflammation in mice with MCD diet-induced NASH (43). However, the serum levels of soluble VEGFR1 but not VEGFR2 were increased in patients with NAFLD in comparison to normal subjects (44). Nonetheless, the role of serum VEGF or soluble VEGFR levels as NAFLD biomarkers requires further investigation. In addition, given the role of VEGF signaling in fibrosis resolution in the liver (45), the clinical application of VEGF blockade in NASH is controversial. Moreover, angiopoietin-2 (Ang-2)/Tie2 receptor inhibiting peptibody L1-10 ameliorated ballooning, fibrosis, and the microvascular architecture in mice with MCD diet and streptozotocin-western diet-induced NASH (46). Blocking the Ang-2 signaling also reduced VCAM-1 expression in endothelial cells treated with LPS (46). The human relevance of this observation was confirmed by increased circulating Ang-2 level in NASH patients and its correlation with the disease activity (46). Moreover, a recent study by Winkler et al. (47) showed that endothelial GATA binding protein 4 (GATA4) protected against perisinusoidal liver fibrosis by repressing MYC activation and profibrotic angiocrine signaling at the chromatin level in LSECs of mice with choline-deficient, l-amino acid-defined (CDAA) diet-induced NASH. Perisinusoidal fibrosis in this study was associated with LSEC capillarization and LSEC expression of the hepatic stellate cell-activating cytokine platelet-derived growth factor subunit B (PDGFB) (47). Taken together, these findings support a role for aberrant angiogenesis in endotheliopathy, perisinusoidal fibrosis, and NASH progression.
THE PATHOGENIC ROLE OF LIPOTOXIC EVs IN PROMOTING IMMUNE CELL ADHESION IN NASH
Extracellular vesicles (EVs) are membrane-bound, nanometer-sized microparticles released by different cells into the extracellular milieu under physiological conditions. The number and cargo of EVs are altered in response to various insults. More importantly, EVs are efficient intercellular messengers, with superior stability and bioavailability of their signature cargos implicated in the inflammatory response (48–50). Povero et al. (51) reported that palmitic acid-treated hepatocytes released EVs laden with the ectoenzyme Vanin-1 (VNN1). VNN1 facilitated EVs uptake by HUVEC and induced the formation of endothelial tubule (51). Moreover, MCD-fed mice had increased circulating EVs enriched with VNN1 (Fig. 1) (51). Hence, EVs released from hepatocytes under lipotoxic stress enhanced LSEC angiogenesis. Furthermore, we have reported that lipotoxic EVs released by hepatocyte treated with toxic lipid augmented monocyte-derived macrophage (MoMF) chemotaxis and adhesion. These EVs were enriched with the MoMF chemotactic ligand CXCL10 (8). Interestingly, CXCL10−/− mice were protected against high-fat, fructose, and cholesterol (FFC) diet-induced inflammation and fibrosis likely secondary to reduced proinflammatory MoMF trafficking into the NASH liver (52). Lipotoxic hepatocyte-derived EVs play a pleotropic role in inflammation and adhesion in NASH. These EVs are laden with ITGα9β1 and enhance monocyte adhesion to LSECs via an ITGα9β1/VCAM1 binding interaction (10). More importantly, anti-ITGβ1 antibody treatment attenuated FFC diet-induced NASH in mice (10). However, little is known about the biological role of LSEC-derived EVs especially in NASH, as well as the contribution of these EVs in long-range communications with the cardiovascular system and the adipose tissue in the metabolic syndrome.
IMMUNE CELL TRANSENDOTHELIAL MIGRATION
Leukocytes undergo alterations in their surface adhesion molecules expression during their migration through the endothelium (53). Hence, transmigration regulates the phenotype of emigrating cells. However, the sequelae of molecular interactions engaged in leukocyte transmigration through the LSECs are largely unknown. Similarly, the role of LSECs and perivascular basement membrane in leukocyte subpopulations activation and hepatic infiltration in NASH remains an unexplored area. Matrix metalloproteinases (MMPs) facilitate leukocyte migration through the vascular wall. Among other MMPs, MMP-9 is an inducible gelatinase expressed by leukocytes during hepatic ischemia reperfusion injury (IRI). MMP-9 has been recognized as a key mediator of focal matrix degradation and leukocyte trafficking to the inflamed liver (54). MMP-9 positive leukocytes infiltrate the liver after IR and cause hepatocyte anoikis or apoptosis secondary to detachment from the extracellular matrix. Whether MMP9 or another MMP has a similar role in NASH is an area ripe for future investigations.
CONCLUSION AND REFLECTION
LSEC capillarization and dysfunction occur early in NAFLD and might be the distinctive event that trigger the progression of simple steatosis to NASH. Promoting the LSEC immunomodulatory and homeostatic properties could serve as a preventive strategy in NASH. For example, the use of the endothelin receptor antagonists might be beneficial in halting the progression of human NASH. These agents are used for other indications including interstitial pulmonary fibrosis and can be repurposed for the use in NASH, especially the endothelin receptor antagonist ambrisentan that lacks the hepatic toxicity associated with others of its class (55). Therapies targeting the chemokine receptors might have merit in NASH. Hence, CXCR3 is an appealing, but unexplored candidate. Furthermore, targeting the LSEC endotheliopathy in NASH through the development of liver selective pharmacological inhibitors or neutralizing antibodies for candidate adhesion molecules is an area ripe for exploration (Table 1). This approach has been successful in the field of inflammatory bowel disease, where various adhesion molecules neutralizing antibody geared toward the leukocyte are employed. A common example is the Integrin α4β7 target vedolizumab. An alternative approach will be used to target the LSEC adhesion molecules, VCAM1, for example, is an appealing anti-inflammatory and antifibrotic target (56). This approach may allow the modulation of LSEC recruitment of harmful inflammatory leukocytes to the liver and attenuate the progression to end stage liver disease. Moreover, targeting the EVs as proinflammatory mediators and NASH catalysts may potentially dampen NASH progression. This can be achieved via either reducing EV release from lipotoxic hepatocytes using a Rho-associated protein kinase (ROCK) kinase inhibitor (57) or neutralizing a specific EV pathogenic cargo like CXCL10 or Vanin-1. In-depth insight into these various mechanisms is crucial for the development of targeted therapeutic approach that blocks the aberrant inflammatory response while preserving the host immune defense mechanism.
GRANTS
Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (NIH) under Award DK 122948 (to S. H. Ibrahim), the NIH Silvio O. Conte Digestive Diseases Research Core Centers P30 grant mechanism (DK084567), and the Mayo Clinic.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.H.I. conceived and designed research; prepared figures; drafted manuscript; edited and revised manuscript; and approved final version of manuscript.
REFERENCES
- 1.Younossi ZM. Non-alcoholic fatty liver disease—a global public health perspective. J Hepatol 70: 531–544, 2019. doi: 10.1016/j.jhep.2018.10.033. [DOI] [PubMed] [Google Scholar]
- 2.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 24: 908–922, 2018. doi: 10.1038/s41591-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eslam M, Sanyal AJ, George J; International Consensus Panel. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158: 1999–2014.e1, 2020. doi: 10.1053/j.gastro.2019.11.312. [DOI] [PubMed] [Google Scholar]
- 4.Knolle PA, Wohlleber D. Immunological functions of liver sinusoidal endothelial cells. Cell Mol Immunol 13: 347–353, 2016. doi: 10.1038/cmi.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorensen KK, Simon-Santamaria J, McCuskey RS, Smedsrod B. Liver sinusoidal endothelial cells. Compr Physiol 5: 1751–1774, 2015. doi: 10.1016/j.jhep.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Hammoutene A, Biquard L, Lasselin J, Kheloufi M, Tanguy M, Vion AC, Mérian J, Colnot N, Loyer X, Tedgui A, Codogno P, Lotersztajn S, Paradis V, Boulanger CM, Rautou P-E. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J Hepatol 72: 528–538, 2020. doi: 10.1016/j.jhep.2019.10.028. [DOI] [PubMed] [Google Scholar]
- 7.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 52: 774–788, 2010. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 8.Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, Goodfellow VS, Malhi H, Gores GJ. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology 63: 731–744, 2016. doi: 10.1002/hep.28252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomita K, Kabashima A, Freeman BL, Bronk SF, Hirsova P, Ibrahim SH. Mixed lineage kinase 3 mediates the induction of CXCL10 by a STAT1-dependent mechanism during hepatocyte lipotoxicity. J Cell Biochem 118: 3249–3259, 2017. doi: 10.1002/jcb.25973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo Q, Furuta K, Lucien F, Gutierrez Sanchez LH, Hirsova P, Krishnan A, Kabashima A, Pavelko KD, Madden B, Alhuwaish H, Gao Y, Revzin A, Ibrahim SH. Integrin beta1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J Hepatol 71: 1193–1205, 2019. doi: 10.1016/j.jhep.2019.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyachi Y, Tsuchiya K, Komiya C, Shiba K, Shimazu N, Yamaguchi S, Deushi M, Osaka M, Inoue K, Sato Y, Matsumoto S, Kikuta J, Wake K, Yoshida M, Ishii M, Ogawa1 Y. Roles for cell-cell adhesion and contact in obesity-induced hepatic myeloid cell accumulation and glucose intolerance. Cell Rep 18: 2766–2779, 2017. doi: 10.1016/j.celrep.2017.02.039. [DOI] [PubMed] [Google Scholar]
- 12.Furuta K, Guo Q, Pavelko KD, Lee JH, Robertson KD, Nakao Y, Melek J, Shah VH, Hirsova P, Ibrahim SH. Lipid-induced endothelial vascular cell adhesion molecule 1 promotes nonalcoholic steatohepatitis pathogenesis. J Clin Invest 131: e143690, 2021. doi: 10.1172/JCI143690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyao M, Kotani H, Ishida T, Kawai C, Manabe S, Abiru H, Tamaki K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab Invest 95: 1130–1144, 2015. doi: 10.1038/labinvest.2015.95. [DOI] [PubMed] [Google Scholar]
- 14.Furuta K, Guo Q, Hirsova P, Ibrahim SH. Emerging roles of liver sinusoidal endothelial cells in nonalcoholic steatohepatitis. Biology (Basel) 9: 395, 2020. doi: 10.3390/biology9110395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herrnberger L, Hennig R, Kremer W, Hellerbrand C, Goepferich A, Kalbitzer HR, Tamm ER. Formation of fenestrae in murine liver sinusoids depends on plasmalemma vesicle-associated protein and is required for lipoprotein passage. PloS One 9: e115005, 2014. doi: 10.1371/journal.pone.0115005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Francque S, Laleman W, Verbeke L, Van Steenkiste C, Casteleyn C, Kwanten W, Dyck CV, D'Hondt M, Ramon A, Vermeulen W, De Winter B, Van Marck E, Van Marck V, Pelckmans P, Michielsen P. Increased intrahepatic resistance in severe steatosis: endothelial dysfunction, vasoconstrictor overproduction and altered microvascular architecture. Lab Invest 92: 1428–1439, 2012. doi: 10.1038/labinvest.2012.103. [DOI] [PubMed] [Google Scholar]
- 17.Pasarín M, La Mura V, Gracia-Sancho J, Garcia-Caldero H, Rodriguez-Vilarrupla A, Garcia-Pagan JC, Bosch J, Abraldes JG. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS One 7: e32785, 2012. doi: 10.1371/journal.pone.0032785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van der Graaff D, Kwanten WJ, Couturier FJ, Govaerts JS, Verlinden W, Brosius I, D’Hondt M, Driessen A, De Winter BY, De Man JG, Michielsen P. Severe steatosis induces portal hypertension by systemic arterial hyporeactivity and hepatic vasoconstrictor hyperreactivity in rats. Lab Invest 98: 1263–1275, 2018. doi: 10.1038/s41374-017-0018-z. [DOI] [PubMed] [Google Scholar]
- 19.Yang Y, Sangwung P, Kondo R, Jung Y, McConnell MJ, Jeong J, Utsumi T, Sessa W, Iwakiri Y. Alcohol-induced Hsp90 acetylation is a novel driver of liver sinusoidal endothelial dysfunction and alcohol-related liver disease. J Hepatol, 2021. doi: 10.1016/j.jhep.2021.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol 15: 555–567, 2018. doi: 10.1038/s41575-018-0020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin-Armas M, Simon-Santamaria J, Pettersen I, Moens U, Smedsrød B, Sveinbjørnsson B. Toll-like receptor 9 (TLR9) is present in murine liver sinusoidal endothelial cells (LSECs) and mediates the effect of CpG-oligonucleotides. J Hepatol 44: 939–946, 2006. doi: 10.1016/j.jhep.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 22.Wu J, Meng Z, Jiang M, Zhang E, Trippler M, Broering R, Bucchi A, Krux F, Dittmer U, Yang D, Roggendorf M, Gerken G, Lu M, Schlaak JF. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology 129: 363–374, 2010. doi: 10.1111/j.1365-2567.2009.03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA, Taylor RS, Efremova M, Vento-Tormo R, Carragher NO, Kendall TJ, Fallowfield JA, Harrison EM, Mole DJ, Wigmore SJ, Newsome PN, Weston CJ, Iredale JP, Tacke F, Pollard JW, Ponting CP, Marioni JC, Teichmann SA, Henderson NC. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575: 512–518, 2019. doi: 10.1038/s41586-019-1631-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pruenster M, Mudde L, Bombosi P, Dimitrova S, Zsak M, Middleton J, Richmond A, Graham GJ, Segerer S, Nibbs RJB, Rot A. The Duffy antigen receptor for chemokines transports chemokines and supports their promigratory activity. Nat Immunol 10: 101–108, 2009. [Erratum in Nat Immunol 2009 Feb;10(2):223, 2009]. doi: 10.1038/ni.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hilscher MB, Sehrawat T, Arab JP, Zeng Z, Gao J, Liu M, Kostallari E, Gao Y, Simonetto DA, Yaqoob U, Cao S, Revzin A, Beyder A, Wang RA, Kamath PS, Kubes P, Shah VH. Mechanical stretch increases expression of CXCL1 in liver sinusoidal endothelial cells to recruit neutrophils, generate sinusoidal microthombi, and promote portal hypertension. Gastroenterology 157: 193–209 e9, 2019. doi: 10.1053/j.gastro.2019.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Windt DJ, Sud V, Zhang H, Varley PR, Goswami J, Yazdani HO, Tohme S, Loughran P, O'Doherty RM, Minervini MI, Huang H, Simmons RL, Tsung A. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 68: 1347–1360, 2018. doi: 10.1002/hep.29914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun H, Liu J, Zheng Y, Pan Y, Zhang K, Chen J. Distinct chemokine signaling regulates integrin ligand specificity to dictate tissue-specific lymphocyte homing. Dev Cell 30: 61–70, 2014. doi: 10.1016/j.devcel.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 28.Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity 41: 694–707, 2014. doi: 10.1016/j.immuni.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 29.Wong J, Johnston B, Lee SS, Bullard DC, Smith CW, Beaudet AL, Kubes P. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J Clin Invest 99: 2782–2790, 1997. doi: 10.1172/JCI119468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams DH, Hubscher SG, Fisher NC, Williams A, Robinson M. Expression of E-selectin and E-selectin ligands in human liver inflammation. Hepatology 24: 533–538, 1996. doi: 10.1002/hep.510240311. [DOI] [PubMed] [Google Scholar]
- 31.Lalor PF, Sun PJ, Weston CJ, Martin-Santos A, Wakelam MJ, Adams DH. Activation of vascular adhesion protein-1 on liver endothelium results in an NF-kappaB-dependent increase in lymphocyte adhesion. Hepatology 45: 465–474, 2007. doi: 10.1002/hep.21497. [DOI] [PubMed] [Google Scholar]
- 32.Patten DA, Wilson GK, Bailey D, Shaw RK, Jalkanen S, Salmi M, Rot A, Weston CJ, Adams DH, Shetty S. Human liver sinusoidal endothelial cells promote intracellular crawling of lymphocytes during recruitment: a new step in migration. Hepatology 65: 294–309, 2017. doi: 10.1002/hep.28879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weston CJ, Shepherd EL, Claridge LC, Rantakari P, Curbishley SM, Tomlinson JW, Hubscher SG, Reynolds GM, Aalto K, Anstee QM, Jalkanen S, Salmi M, Smith DJ, Day CP, Adams DH. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest 125: 501–520, 2015. doi: 10.1172/JCI73722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lefere S, Van de Velde F, Devisscher L, Bekaert M, Raevens S, Verhelst X, Van Nieuwenhove Y, Praet M, Hoorens A, Van Steenkiste C, Van Vlierberghe H, Lapauw B, Geerts A. Serum vascular cell adhesion molecule-1 predicts significant liver fibrosis in non-alcoholic fatty liver disease. Int J Obes 41: 1207–1213, 2017. doi: 10.1038/ijo.2017.102. [DOI] [PubMed] [Google Scholar]
- 35.Rai RP, Liu Y, Iyer SS, Liu S, Gupta B, Desai C, Kumar P, Smith TD, Singhi AD, Nusrat A, Parkos CA, Monga SS, Czaja M, Anania FA, Raeman R. Blocking integrin α4β7-mediated CD4 T cell recruitment to the intestine and liver protects mice from western diet-induced non-alcoholic steatohepatitis. J Hepatol 73: 1013–1022, 2020. doi: 10.1016/j.jhep.2020.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drescher HK, Schippers A, Rosenhain S, Gremse F, Bongiovanni L, de Bruin A, Eswaran S, Gallage SU, Pfister D, Szydlowska M, Heikenwalder M, Weiskirchen S, Wagner N, Trautwein C, Weiskirchen R, Kroy DC. L-Selectin/CD62L is a key driver of non-alcoholic steatohepatitis in mice and men. Cells-Basel 9: 1106, 2020. doi: 10.3390/cells9051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rensen SS, Slaats Y, Driessen A, Peutz-Kootstra CJ, Nijhuis J, Steffensen R, Greve JW, Buurman W. Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology 50: 1809–1817, 2009. doi: 10.1002/hep.23228. [DOI] [PubMed] [Google Scholar]
- 38.Albrecht EA, Chinnaiyan AM, Varambally S, Kumar-Sinha C, Barrette TR, Sarma JV, Ward PA. C5a-induced gene expression in human umbilical vein endothelial cells. Am J Pathol 164: 849–859, 2004. doi: 10.1016/S0002-9440(10)63173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shivshankar P, Li YD, Mueller-Ortiz SL, Wetsel RA. In response to complement anaphylatoxin peptides C3a and C5a, human vascular endothelial cells migrate and mediate the activation of B-cells and polarization of T-cells. FASEB J 34: 7540–7560, 2020. doi: 10.1096/fj.201902397R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laudes IJ, Chu JC, Huber-Lang M, Guo RF, Riedemann NC, Sarma JV, Mahdi F, Murphy HS, Speyer CL, Lu KT, Lambris JD, Zetoune FS, Ward PA. Expression and function of C5a receptor in mouse microvascular endothelial cells. J Immunol 169: 5962–5970, 2002. doi: 10.4049/jimmunol.169.10.5962. [DOI] [PubMed] [Google Scholar]
- 41.Ding BS, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, Mittal V, Kobayashi H, Shido K, Lyden D, Sato TN, Rabbany SY, Rafii S. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 468: 310–315, 2010. doi: 10.1038/nature09493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sato T, El-Assal ON, Ono T, Yamanoi A, Dhar DK, Nagasue N. Sinusoidal endothelial cell proliferation and expression of angiopoietin/Tie family in regenerating rat liver. J Hepatol 34: 690–698, 2001. doi: 10.1016/s0168-8278(00)00109-4. [DOI] [PubMed] [Google Scholar]
- 43.Coulon S, Legry V, Heindryckx F, Van Steenkiste C, Casteleyn C, Olievier K, Libbrecht L, Carmeliet P, Jonckx B, Stassen J-M, Van Vlierberghe H, Leclercq I, Colle I, Geerts A. Role of vascular endothelial growth factor in the pathophysiology of nonalcoholic steatohepatitis in two rodent models. Hepatology 57: 1793–1805, 2013. doi: 10.1002/hep.26219. [DOI] [PubMed] [Google Scholar]
- 44.Coulon S, Francque S, Colle I, Verrijken A, Blomme B, Heindryckx F, De Munter S, Prawitt J, Caron S, Staels B, Van Vlierberghe H, Van Gaal L, Geerts A. Evaluation of inflammatory and angiogenic factors in patients with non-alcoholic fatty liver disease. Cytokine 59: 442–449, 2012. doi: 10.1016/j.cyto.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 45.Yang L, Kwon J, Popov Y, Gajdos GB, Ordog T, Brekken RA, Mukhopadhyay D, Schuppan D, Bi Y, Simonetto D, Shah VH. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology 146: 1339–1350.e1, 2014. doi: 10.1053/j.gastro.2014.01.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lefere S, Van de Velde F, Hoorens A, Raevens S, Van Campenhout S, Vandierendonck A, Neyt S, Vandeghinste B, Vanhove C, Debbaut C, Verhelst X, Van Dorpe J, Van Steenkiste C, Casteleyn C, Lapauw B, Van Vlierberghe H, Geerts A, Devisscher L. Angiopoietin-2 promotes pathological angiogenesis and is a therapeutic target in murine nonalcoholic fatty liver disease. Hepatology 69: 1087–1104, 2019. doi: 10.1002/hep.30294. [DOI] [PubMed] [Google Scholar]
- 47.Winkler M, Staniczek T, Kurschner SW, Schmid CD, Schonhaber H, Cordero J, Kessler L, Mathes A, Sticht C, Neßling M, Uvarovskii A, Anders S, Zhang X-J, von Figura G, Hartmann D, Mogler C, Dobreva G, Schledzewski K, Géraud C, Koch P-S, Goerdt S. Endothelial GATA4 controls liver fibrosis and regeneration by preventing a pathogenic switch in angiocrine signaling. J Hepatol 74: 380–393, 2021. doi: 10.1016/j.jhep.2020.08.033. [DOI] [PubMed] [Google Scholar]
- 48.Verweij FJ, Revenu C, Arras G, Dingli F, Loew D, Pegtel DM, Follain G, Allio G, Goetz JG, Zimmermann P, Herbomel P, Del Bene F, Raposo G, van Niel G. Live tracking of inter-organ communication by endogenous exosomes in vivo. Dev Cell 48: 573–589.e4, 2019. doi: 10.1016/j.devcel.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 49.Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 67: 963–972, 2018. doi: 10.1136/gutjnl-2017-315691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hirsova P, Ibrahim SH, Verma VK, Morton LA, Shah VH, LaRusso NF, Gores GJ, Malhi H. Extracellular vesicles in liver pathobiology: small particles with big impact. Hepatology 64: 2219–2233, 2016. doi: 10.1002/hep.28814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Povero D, Eguchi A, Niesman IR, Andronikou N, de Mollerat Du Jeu X, Mulya A, Berk M, Lazic M, Thapaliya S, Parola M, Patel HH, Feldstein AE. Lipid-induced toxicity stimulates hepatocytes to release angiogenic microparticles that require Vanin-1 for uptake by endothelial cells. Sci Signal 6: ra88, 2013. doi: 10.1126/scisignal.2004512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tomita K, Freeman BL, Bronk SF, LeBrasseur NK, White TA, Hirsova P, Ibrahim SH. CXCL10-mediates macrophage, but not other innate immune cells-associated inflammation in murine nonalcoholic steatohepatitis. Sci Rep 6: 28786, 2016. doi: 10.1038/srep28786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nourshargh S, Marelli-Berg FM. Transmigration through venular walls: a key regulator of leukocyte phenotype and function. Trends Immunol 26: 157–165, 2005. doi: 10.1016/j.it.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 54.Coito AJ. Leukocyte transmigration across endothelial and extracellular matrix protein barriers in liver ischemia/reperfusion injury. Curr Opin Organ Transplant 16: 34–40, 2011. doi: 10.1097/MOT.0b013e328342542e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wei A, Gu Z, Li J, Liu X, Wu X, Han Y, Pu J. Clinical adverse effects of endothelin receptor antagonists: insights from the meta-analysis of 4894 patients from 24 randomized double-blind placebo-controlled clinical trials. J Am Heart Assoc 5: e003896, 2016. doi: 10.1161/JAHA.116.003896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carr RM. VCAM-1: closing the gap between lipotoxicity and endothelial dysfunction in nonalcoholic steatohepatitis. J Clin Invest 131: e147556, 2021. doi: 10.1172/JCI147556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, Charlton MR, Shah VH, Malhi H, Gores GJ. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology 150: 956–967, 2016. doi: 10.1053/j.gastro.2015.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]