

Keywords: cell therapy, first-in-human, intestine, organoids, translation
Abstract
Recent advances in intestinal organoid research, along with encouraging preclinical proof-of-concept studies, have revealed significant therapeutic potential for induced pluripotent stem cell (iPSC)-derived organoids in the healing and replacement of severely injured or diseased bowel (Finkbeiner et al. Biol Open 4: 1462–1472, 2015; Kitano et al. Nat Commun 8: 765, 2017; Cruz-Acuna et al. Nat Cell Biol 19: 1326–1335, 2017). To fully realize the tremendous promise of stem cell organoid-based therapies, careful planning aligned with significant resources and efforts must be devoted demonstrating their safety and efficacy to meet critical regulatory requirements. Early recognition of the inherent preclinical and clinical obstacles that occur with the novel use of pluripotent stem cell-derived products will accelerate their bench-to-bedside translation (Neofytou et al. J Clin Invest 125: 2551–2557, 2015; O’Brien et al. Stem Cell Res Ther 6: 146, 2015; Ouseph et al. Cytotherapy 17: 339–343, 2015). To overcome many of these hurdles, a close and effective collaboration is needed between experts from various disciplines, including basic and clinical research, product development and manufacturing, quality assurance and control, and regulatory affairs. Therefore, the purpose of this article is to outline the critical areas and challenges that must be addressed when transitioning laboratory-based discovery, through an investigational new drug (IND) application to first-in-human clinical trial, and to encourage investigators to consider the required regulatory steps from the earliest stage of the translational process. The ultimate goal is to provide readers with a draft roadmap that they could use while navigating this exciting cell therapy space.
INTRODUCTION
It is increasingly clear that stem cell technology has an enormous potential to advance medical therapy by personalizing regenerative medicine and providing new solutions to conditions with a current unmet medical need. With the establishment of new methods to grow stem cells derived from human biopsy or reprogrammed somatic cells into three-dimensional (3-D) structures termed “organoids,” it became possible to develop functional and complex human living tissues in laboratory conditions, which could then be used to replace damaged or diseased organs (1–3). The feasibility of such experimental approach has been established for enteroids and colonoids, which are generated from small intestinal and colonic tissues, respectively, and that contain cells of epithelial lineage only (1, 3). Both enteroids and colonoids can be expanded in vitro and engrafted onto damaged mouse intestine and contribute to regeneration of functional epithelium in vivo (4–8). Although this highlights the translational potential of primary-tissue-derived intestinal stem cells for the treatment of disorders in which the mucosal layer of intestinal tissue is affected, the clinical utility of this cell source will be limited in conditions in which reconstruction of a full thickness of intestinal wall (including mucosa, submucosa, and muscularis layers) is required. For instance, in patients with transmural damage to the entire bowel that may be due to inflammatory bowel disease, necrotizing enterocolitis, ischemia, or bowel radiation. It will also be required in conditions where bowel length or regional loss (ileum) is compromised resulting in short bowel syndrome (9, 10). For such severely diseased patients, the bench-to-bedside translation approach may be challenging due to the limited ability to obtain starting material for the establishment of enteroid cultures. Human intestinal organoids (HIOs), which are established from blood-derived iPSCs and which when transplanted give rise to epithelium and all supporting layers of smooth muscle cells found in human intestine, may represent a more viable treatment strategy (11). Moreover, HIOs have been previously shown to functionally integrate enteric nervous system derived from iPSCs and to develop intestinal vasculature through contribution from the host (11, 12). Thus, when combined with synthetic or biological (decellularized) scaffolds they can be used to engineer intestinal grafts in vitro using an autologous and easily accessed patient material (blood) (13–15).
As our area of expertise lies in the generation of iPSC-derived human intestinal tissues, in this review, we will focus on the translation of iPSC-derived HIO therapy to the clinic. It is important to note that although some considerations are common to all cell therapies (e.g., origin of ancillary materials, and aseptic cell processing, etc.), others are more unique to certain types of products (autologous cell lots vs. large allogeneic cell banks). Therefore, our goal is to outline some of the main points of emphasis that we have identified while laying down a groundwork for transition of our organoid-based therapy to the clinic rather than attempting to create a “one-size-fits-all” roadmap. In line with that, many of the considerations presented here are unique to iPSC-derived tissues and not biopsy-derived enteroids, which represent a very distinct approach that was recently reviewed elsewhere (16).
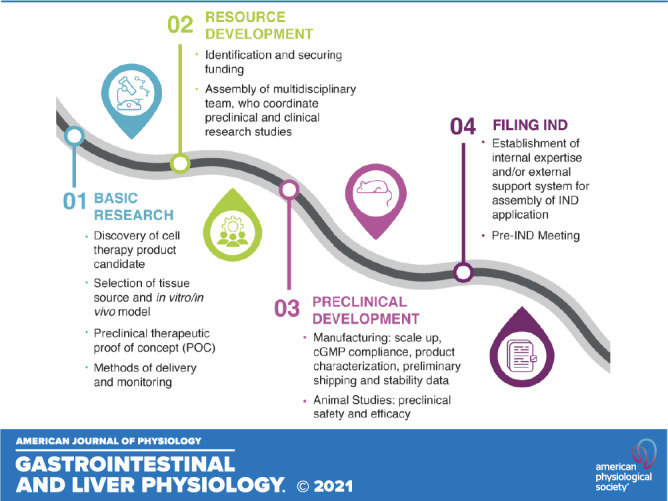
Given the remarkable ability of the intestinal organoid-derived cells to spontaneously self-renew and self-organize into properly differentiated functional cell types in a dish as well as to reach appropriate cellular composition, engraftment, and vascularization upon transplantation into an animal host, they represent an excellent source of tissue for regenerative medicine applications (2, 11, 17). Despite their vast potential, positioning organoid-based therapy for success is not as clear cut, given several key challenges that need to be addressed on their path to clinical translation. These include relatively low reproducibility or insufficient scalability of organoid manufacturing, use of bioprocesses that are not compliant with quality and regulatory guidelines, and potential tumorigenicity and immunogenicity issues (18–20). Understanding and mitigating risks associated with these challenges are the first and foremost responsibilities of the investigator during the translation phase of the project. The inherent paradox in new stem cell therapy development is the assessment of new product safety in the context in which it has never been used previously. This makes the risk assessment process unclear and challenging. Thus, it is critical to embrace a patient-centric approach when evaluating the strength of the supporting data in relationship to the known or anticipated risks of potential harm to the patient. In addition, researchers or clinicians who are engaged in the translation of intestinal organoid research to the clinic should be cognizant of the scientific, technical, and regulatory requirements that need to be addressed at various phases of the process. Therefore, the goal of this review is to outline the draft roadmap for organoid-based therapy to assist stem cell therapy developers in planning of research and allocation of appropriate resources to enable a clear demonstration of safety and efficacy of these new cellular products (Fig. 1).
Figure 1.

Roadmap to organoid-based therapy. The roadmap identifies the four critical areas that need to be considered when developing a cellular therapy intended for evaluation in human clinical studies under an investigational new drug (IND) application: basic research, resource and preclinical development, and IND assembly and submission. POC, proof-of-concept; CGMP, current good manufacturing practice.
Early-Phase Considerations
Identification of the specific indication of use and appropriate patient population.
Addressing unmet medical needs is a key criterion for stem cell therapy development. It is widely accepted that identification of the disease state or other clinical problem that lacks tangible therapeutic solutions or results in poor clinical outcome might justify the use of iPSC-derived cell products in the clinical context. Nevertheless, the importance of early identification of the appropriate patient population often remains underappreciated in determining the type of preclinical data necessary for investigational new drug (IND) submission (57). A uniform patient population with poor but predictable prognosis, few alternative treatment options, and as few comorbidities as possible would be ideal. However, a balance should be maintained between the severity of the underlying illness and the ability of the investigator to adequately monitor and attribute adverse events associated with the study product. Although it might be justifiable to allow a greater risk to patients with no alternative treatment options and poor prognosis, trials performed in these populations characterized by an extremely advanced disease state might fail to provide firm evidence on the safety of the studied cell product. First, because the life expectancy of these patients might be lower than the time required for the accurate evaluation of the disease progression, and second, because the occurrence of the disease-mediated comorbidities might complicate data interpretation. Thus, well-defined indication of use and characteristics of target population should be considered in early phases of stem cell therapy development. This process requires dedicated and open discussions with clinicians and experts who are specialized in the treatment of the specific condition, to better comprehend the technical and logistical obstacles that will need to be addressed. Then working backward, key decisions during the cell therapy development can be taken based on the patient-centered benefit-risk assessment.
One of the potential early applications of HIO-based therapy may be the use of iPSC-derived tissues to regenerate patient’s bowel following intestinal transplant rejection. Patients affected by this condition can be identified well in advance, providing enough time to generate quality-controlled autologous cell banks established from patient’s own cells. Importantly, rejection is a clinical condition that may result in complete loss of the transplanted bowel often leading to removal of the graft followed by relisting and retransplantation and carries a high risk of patient’s death, thus justifying the risk of the new therapy. Finally, the presence of stoma in those individuals provides an easy access to the intestinal lumen facilitating both delivery of cell therapy product as well as posttransplant monitoring of tissue engraftment and healing. We have identified this clinical scenario as our first stepping-stone toward the creation of the roadmap for translating the HIO-based therapy to the clinic. With the experience gained from focusing on these rare conditions, we will be poised to further establish organoid-based therapeutic approaches that could then be applied into the treatment of more common conditions. One potential condition will likely include nonhealing intestinal ulcers, luminal areas of bowel devoid of mucosal epithelial cells with exposed mucosal mesenchyme, as they provide a potential target site for HIO’s engraftment. These nonhealing ulcers may develop as a common postoperative complication after Roux-en-Y gastric bypass (RYGB) or after complex esophageal reconstruction. New methodologies to generate and transplant organoids representing the gastric antrum, fundus, and esophagus are currently being developed, laying the foundation for the development of autologous organoid-based therapies targeting the upper gastrointestinal track. Moreover, the use of genome engineering techniques such as clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 to selectively correct the disease-causing variant within the stem cell may expand the use of organoid transplant to the treatment of monogenic congenital disorders such as congenital tufting enteropathy and microvillus inclusion disease. However, a number of techniques still need to be further optimized, including more robust large-scale production of organoids, ablation of native epithelium, and engraftment protocols to ensure feasibility of this approach.
Advanced planning and maintenance of high level of scientific rigor.
Despite the recent progress in cell therapy in general and iPSC-based approaches, which have demonstrated their potential benefits to patients, translation of these findings into humans is still inefficient. This is often because of an underestimation of how important it is to adhere to good laboratory practice (GLP) principles and to maintain the highest level of scientific rigor while generating the data to support an IND application. Therefore, we believe that advanced planning is critical during the early phases of stem cell therapy development along with paying close attention to detail, including appropriate documentation and monitoring of all of the studies, to avoid any unnecessary repetition of the experiments. We strongly encourage investigators who are new to the stem cell therapy field to define early and carefully what is the clinical expectation for their cell therapy product and then work backward to establish specifications ensuring its quality, safety, and efficacy.
Considerations for tissue sourcing.
Investing efficient time and effort toward the development of stem cell organoid-based therapies at this point requires a clear understanding of the cell source that is most likely to provide the safest first-in-human approach. An important consideration should be the choice of tissue source for the development of the stem cell therapy product, choosing among the options of autologous, allogeneic HLA-matched, or allogeneic HLA-modified iPSCs (Fig. 2). Ideally, the same cell source should be used in all initial experiments that will support the IND application. Autologous cells are harvested from and readministered to the same patient, without the need for immunosuppression and with a low likelihood of immunological rejection. Yet these benefits of using an autologous source need to be weighed against several negative considerations. The first drawback is the high cost and extensive time investment involved in autologous cell banking, which usually precludes acute treatment modalities and may be impractical to clinical application for a large group of patients. Moreover, a recent study revealed the appearance of de novo mutations in mitochondrial DNA (mtDNA) of autologous iPSCs, which can arise during the cell reprogramming stage, long-term culture, or differentiation into a final cell therapy product, leading to the formation of neoantigens capable of eliciting immune recognition and rejection in the transplant recipient (21). This mutational process highlights the need for implementation of screening for mtDNA mutations of all potential iPSC-derived autologous cell products. Another consideration is the possible presence of patient-specific germline mutations that contribute to the disease, which would need to be genetically modified (e.g., by CRISPR/Cas9 or other techniques) before being utilized as a source of iPSCs.
Figure 2.

Potential approaches for organoid-based therapy dependent on the source of induced pluripotent stem cells (iPSCs). A: autologous organoid-based therapy uses a patient’s own peripheral blood cells (PMBCs) to derive clinical cell therapy product. PBMCs are reprogrammed into iPSCs followed by in vitro expansion of iPSCs, differentiation into organoids and transplantation into the patient. B: allogeneic human leukocyte antigen (HLA)-matched organoid-based therapy generates organoids from PBMCs of a healthy HLA homozygous donor in a process similar to that in A; iPSCs are expanded in vitro, differentiated into organoids, and transplanted into an HLA-matched patient. C: allogeneic HLA-modified organoid-based therapy can use iPSCs derived from various healthy donors. iPSCs are then genetically modified to remove expression of major HLA molecules and to prevent immunological rejection. Created in such way universal iPSCs can be expanded in vitro, differentiated into organoid, and transplanted into patient independently of haplotypes of HLA. Created with BioRender.com.
As an alternative, HLA-matched allogeneic iPSCs can be obtained from selected healthy donors who are homozygous for a given HLA locus and manufactured at scale in the form of “off-the-shelf” cell therapy product. Although perfect matching of allogeneic cellular therapeutics to all recipients is not possible, access to HLA-matched cell lines might reduce the risk of rejection of cellular therapy and decrease required dosage of immunosuppression in HLA-matched recipients, providing benefit to a greater number of individuals globally. Unfortunately, even in the case of HLA-matched allogeneic transplantation, there is a risk for rejection driven by the presence of minor histocompatibility antigens. Thus, significant efforts are currently underway to develop hypoimmunogenic iPSCs that are able to avoid immune rejection even in mismatched allogeneic recipients and in the absence of immunosuppression. Recently, promising results using gene engineering have been obtained, demonstrating that inactivation of major histocompatibility complex (MHC) class I and II genes along with increased CD47 expression prevents a vigorous immune response against histoincompatible cells (22). However, the long-term effects and safety profile of this approach are still under investigation. In addition, the capability of these cells to differentiate toward intestinal cell lineage and to elicit critical biological responses including regeneration, proliferation, and cell migration is unknown and, therefore, would need to be evaluated before considering their use for first-in-human trials.
Early implementation of current good manufacturing practice-compliant cell production.
It is strongly encouraged that all supporting data for a future IND application are generated using a product that is identical or highly comparable with the one intended for eventual clinical use. Early implementation of a well-defined manufacturing process and comprehensive product characterization can help to fast-track cell therapy development. Over the past few years, significant efforts have been devoted to establishing common global standards for the generation of clinical-grade human iPSC products (23–25). Use of chemically defined and preferably animal-free (xeno) and serum-free cell culture systems, rigorous quality testing, document control, and standard operating procedures carried out according to current good manufacturing practices (CGMPs) enforced by the Food and Drug Administration (FDA) will facilitate product consistency, traceability, and reproducibility. The current good manufacturing practice (CGMP) regulations incorporate flexibility to support the use of modern technologies and innovative approaches such as iPSCs while allowing investigators to decide how best to implement scientifically sound design, processing methods, and testing procedures (26). Each manufactured batch of cellular products should satisfy specific release criteria; however, these may vary between different types of cell therapies, yet should always include the product’s purity, identity, potency, and sterility. Initial work using CGMP-compliant processes can be developed in a basic science laboratory; however, transferring the work to a GMP facility needs to be considered early in the process. Active collaborations with a GMP facility are usually needed to understand the documentation and compliance processes needed to translate the work to this setting. It is important to note that CGMPs represent the minimal safety requirements required in therapeutic development. Implementation of even more comprehensive, modern quality systems and risk management approaches that exceed these minimum standards is highly recommended to ensure a successful IND.
Reliable method to deliver cell therapy product.
Another critical factor to consider is the expected method of delivery of the stem cell therapy product to the anatomical site of interest. Often it may require surgical intervention or the use of novel delivery devices that could potentially pose additional safety risks to the patient. As each implantation site has a different vulnerability, the cell delivery method may result in variable levels of toxicity to both the cells and the endogenous tissue at the relevant site of administration. Thus, it is critical for cell therapy developers to ensure that the cell delivery method and/or device does not induce significant damage to the cellular product or to the delivery system itself, that it ensures maintenance of cell product identity, and that it does not elicit any adverse biological reactions in the patient. For the initiation of our first-in-human study, clinical-grade culture methods and endoscopic delivery approaches are both under development.
Moving from Silos to Synergy in Regenerative Medicine
Multiple disciplines are invariably involved in translating a basic science discovery into a therapeutic product, making knowledge sharing and effective collaboration across the disciplines foundational to the success of the newly developed cell therapy in the clinic. Once proof-of-concept data has demonstrated the potential therapeutic value of the candidate, it is time to engage experts representing diverse but complementary areas of expertise, including basic and clinical research, project management, biostatistics, regulatory affairs, product development and manufacturing, and quality assurance and control. Moving away from academic research silos to work in synergy within a multidisciplinary study team allows for the establishment of the essential toolset to accelerate the development of novel cellular therapies. Table 1 provides a potential list of core members of a translational study team and summarizes their main roles and responsibilities (27).
Table 1.
Translational study team members, roles, and responsibilities
| Team Member(s) | Roles and Responsibilities |
|---|---|
| Principal investigator | Ultimate responsibility for the project |
| Study design for evaluation of the safety and efficacy of therapeutic candidate (preclinical phase of the project) | |
| Design and execution of the clinical trial protocol (clinical phase of the project) | |
| Analysis and interpretation of data | |
| Basic research team | Assessment of the product’s safety and efficacy in in vitro and in vivo disease models (preclinical phase of the project) |
| Clinical research team | Subjects’ enrollment, treatment, and follow-up care (clinical phase of the project) |
| Project manager | Responsibility for meeting project timeline and sticking to budget |
| Resource allocation and management | |
| Development and implementation of the strategy for mitigation of risks | |
| Facilitation of technology transfer | |
| Product development and manufacturing experts | Responsibility for translating research protocols into current good manufacturing procedure (CGMP)-compliant manufacturing procedures for the large-scale production of clinical grade material |
| Regulatory experts | Regulatory oversight |
| Involvement in the preparation of documents for review by regulatory authorities, such as US Food and Drug Administration (FDA), Institutional Review Board (IRB), and Data and Safety Monitoring Board (DSMB) | |
| Quality assurance experts | Responsibility for the compliance of the product and manufacturing processes with the CGMP regulations and meeting predefined product release criteria |
| Support in addressing product manufacturing issues | |
| Compliance audits | |
| Quality control experts | Release testing of the clinical product |
| Generation of data in support of CGMP compliance | |
| Specific area experts | Provide an expert knowledge and support in the missing areas of expertise specific for the product of indication of use, i.e., support of transplant specialist might be needed during the development of cell replacement therapy product |
Currently, stem cell research leading to the development of new cell therapies is primarily conducted by academic researchers who are supported by traditional external funding sources (i.e., National Institutes of Health-sponsored research grants). Thus, the creation of a translational study team faces a number of challenges, which include 1) limited access to the areas of expertise and professionals that are typically not available in an academic setting (i.e., specialist in quality control and assurance), 2) limited funding opportunities that could support the costs of product transition from the early proof-of-concept stage into clinical evaluation, and 3) tendency of academic researchers to work in discipline-specific silos that often contain robust international research communities but that rarely interact with one another.
Moving from silos to synergy in regenerative medicine will require a paradigm shift and the establishment of effective codevelopment partnerships between academia and private entities such as biopharmaceutical companies or nonprofit organizations. These types of alliances can be grouped around the access to four vital components: 1) technological knowledge and expertise in stem cell biology and biomaterials (for combinational products); 2) manufacture of the clinical product and/or technological expertise; 3) commercialization, regulatory, and/or clinical trial expertise; and 4) financing. Although technological knowledge in the field of regenerative medicine is largely concentrated in academia, expertise in commercialization, regulation, and clinical trials may often be missing in this setting. Therefore, input from both industry experts and regulatory bodies in these critical areas of cell therapy development will be required to better navigate the complex route from bench to bedside. Moreover, codevelopment partnerships provide an alternative funding stream to academia benefiting from the financial resources of the pharmaceutical industry and to industries gaining the access to governmental grants or support from disease foundations (28).
Nevertheless, it is important to acknowledge the uniqueness of stem cell-based therapies in terms of their regulatory, commercialization, and infrastructure requirements. The regulatory environment for cell therapies differs significantly from that of conventional products. In addition, autologous cell therapies do not align with current pharmaceutical business models, highlighting the necessity to develop new approaches to commercialization. Furthermore, effective fostering of stem cell clinical trials and successful implementation of FDA-approved cell therapies in the clinic will require an enhancement of the existing clinical infrastructures with the requisite scientific, technical, and/or medical expertise and operational efficiencies in the area of stem cell-based regenerative medicine. These unique considerations have led to the creation of “translational centers” with a goal to accelerate the transition of stem cell-based treatments into sustainable patient care programs, including Canadian Centre for Regenerative Medicine (CCRM), UK Cell Therapy Catapult, Center for Stem Cell and Organoid Medicine (CuSTOM) at Cincinnati Children’s Hospital Medical Center (CCHMC), and the City of Hope Alpha Stem Cell Clinic (ASCC) (29, 30). Both US-based initiatives, CuSTOM and ASCC support realization of stem cell-based medicine through 1) identification of the most promising cellular therapies and acceleration of their path toward human trials, 2) establishment of an infrastructure and expertise necessary to support conduction of stem cell clinical trials, and 3) coordination of the patient care and new protocol implementation to ensure the highest safety and effectiveness of the novel treatments. It is important to highlight the multidisciplinary character of these centers/networks that breaks the domain-specific silos and brings together experts in stem cell research, regulatory affairs, and CGMP-compliant cell manufacturing and clinical staff. This highly synergistic structure and unique models of translation along with emerging educational programs (i.e., Master of Science in Translational Medicine, which offers an in-depth understanding of how to translate basic research into medical products) will provide the springboard for translating cell therapies to the clinic.
Development of Organoids into Cell Therapy Products
The biological complexity of human intestinal organoids is a double-edged sword, in that the greater complexity enhances its efficacy and potential clinical success, yet at the same time challenges its translation and regulatory approval. Our preclinical therapeutic proof-of-concept (POC) studies have demonstrated that the mesenchymal cells represent a critical component of intestinal organoids ensuring successful engraftment of intestinal epithelium in the animal rat injury model (Poling H. M., Sundaram N., Fisher G. W., Shiley J. R., Singh A., Cortez A. R., Wells J. M., Mayhew C. N., Takebe T., Mahe M. M, and Helmrath M. A., unpublished observation). In addition, in contrast to enteroids, which can repopulate only the epithelial lining of diseased or damaged bowel, iPSC-derived organoids have a high potential to regenerate all the required cell sources needed for full-thickness intestinal reconstruction. However, these findings also bring additional considerations at the level of risk assessment, as well as challenges in the reproducibility and consistency across various lots of organoid-based therapy products. Therefore, in addition to considerations, which are common for all cell therapies (e.g., compliance of manufacturing processes with CGMP standards, biobanking requirements) and that have been described thoroughly by others (31–33), organoids face their own unique challenges to clinical translation. In the next paragraph, we will discuss some of the major hurdles associated with the technical development of organoids into the cell therapy products.
Novel solutions to improve reproducibility and quality control of organoids production.
Because organoid formation requires the synergetic differentiation and morphogenesis of multiple cell lineages closely interacting with each other, this process is inherently difficult to control, resulting in heterogeneous outcomes with highly variable yields and quality of organoids (34–41). Such inconsistency represents a major concern for the transition of protocols from small-scale research laboratories to large-scale CGMP-compliant product manufacturing methods that are required for the clinical use. A bottleneck in intestinal organoid production has been identified at the stage of spontaneous generation of spheroids from the hindgut cultures and their subsequent differentiation into mature organoids. The efficacy of spheroid budding is strongly affected by the density and induction efficacy of endodermal cultures, resulting in unsuccessful or very low-yield production of organoids from culture with an inadequate level of confluency or differentiation (target: 100% confluent monolayer consisted of 85%–90% SOX17/FOXA2-double positive cells) (42). In addition, only a small fraction (∼13%) of hindgut spheroids that bud out from the interconnected layers of posterior endoderm and mesoderm are able to mature into intestinal organoids (43).
These inefficiencies, in combination with the lack of quantitative and unbiased methods for process monitoring or quality control, may lead to great inconsistency and poor quality of the generated product. Therefore, the development of new process engineering approaches enabling consistent and high-yield production of organoids is critical to make these platforms tractable for clinical applications. In addition, identification of the parameters (i.e., specific cell markers) predictive of product safety and efficacy will be essential to allow for an appropriate control and real-time monitoring of the organoid manufacture process. Only when we are able to move away from subjective assessments or “judgment calls” from well-trained cell culture experts as to the level of organoid differentiation and quality, inevitably prone to error, we will approach the realization of organoids medicine. To address this challenge, the establishment of automated and quantitative methods, supported by machine learning and advanced robotics, could assist cell therapy developers tremendously. Some of these advances recently have been reported for the derivation and characterization of iPSCs; however, no major progress has been made in the field of organoids (44–46). Further advances can only come from a highly collaborative team of biologists, bioengineers, data scientists, and/or experts in process automation and control, again highlighting the importance of breaking down silos and enabling cross-disciplinary collaborations.
New methods of delivery and monitoring of the transplant recipient.
The success of the therapeutic approaches based on intestinal organoids will require the development of new endoscopic delivery methods, or else the adaptation of the existing endoscopic tools, to allow organoid transplantation into human host. Considerable effort should be devoted to the identification of the appropriate delivery vehicle, i.e., extracellular matrix (ECM) that is able to support the survival, engraftment, and retention of intestinal organoids at the site of administration. A recent study showed that synthetic PEG-4MAL hydrogel can be delivered via endoscopic techniques, promoting localized organoid engraftment and enhanced healing of murine colonic mucosal wounds in vivo (47). This finding should form the basis for the development of HIO-based therapies to treat human gastrointestinal diseases involving intestinal epithelial wounds (i.e., inflammatory bowel disease). However, it is still not clear whether this strategy could be used in conditions where larger portions of human intestine are damaged or missing, as seen in patients with irreversible intestinal failure or short bowel syndrome. This consideration is particularly important, given that this patient population is characterized by poor clinical outcomes, frequent need of retransplantation, and limited treatment options, thereby representing an appropriate target for the first-in-human studies (48–51). An alternative approach to treat these patients described recently is the transplantation of cell sheets that can be fabricated from a patient’s own cells and delivered via a 3-D printed endoscopic device (52). However, additional research is required to assess whether the grafts formed using this method are fully functional and are able to persist for extended periods without causing adverse effects.
In addition to new surgical procedures and techniques, there is a need for the development of new visualization technologies that could greatly benefit organoids applications in the clinic. To date, noninvasive monitoring techniques to evaluate the engraftment and persistence of cell therapy product, as well as the presence of any off-target effects, primarily have been applied in the setting of small animal studies (rodents). Some of these methods and equipment (i.e., micro-PET imaging system) likely could be adapted and further optimized for use in human trials. However, large animal models (i.e., pigs) that have organs comparable in size and physiology with those of humans will need to be established to evaluate and perfect these new techniques. Current progress on the development of these models and available imaging technologies have been reviewed by others (53–55).
Potential Regulatory Pathways for Organoid-Based Therapies
Once preclinical studies have confirmed the potential value of a “lead” therapeutic candidate and all of the procedures related to product manufacturing, storage, and shipment have been developed and successfully validated, the investigator and/or cell therapy developer is ready to file an IND application with the FDA. The FDA in the United States and the European Medicines Agency (EMA) in Europe have jurisdiction over the commercialization of stem cell therapies in their respective territories. As discussed earlier, organoids (and other stem cell products) are considered as highly complex and significantly different from traditional medicines, in the way they are made, how they are administered, and in the type of benefits they may provide. From the regulatory perspective, they can be designated as regenerative medicine therapies (cell therapies), therapeutic tissue engineering products, human cell and tissue products, or combination products. Since the regulations for these types of biologics were established relatively recently, there is no one-size-fits-all formula as to how best they should be regulated within the existing regulatory framework. Instead, the approach is being tailored to the needs of each individual stem cell/organoids-based therapy. Therefore, it is critical that investigators develop skills and relationships that allow them to successfully navigate this complex regulatory environment. We highly recommend that the investigator becomes familiar with country-specific processes, reads and follows all the guidance documents, engages with the regulatory agencies early in the development process, and seeks external regulatory support if in-house resources are limited or unavailable. It is important to highlight that both parties—regulatory agencies and investigators—share the same goal of bringing safe and efficacious novel therapy to patients. Thus, the specific criteria or required deliverables for any given cell therapy product can be established through close interactions and discussions with the FDA, ensuring the safety, efficacy, and timely delivery of the new product to the market.
The Office of Tissues and Advanced Therapies [OTAT; formerly the Office of Cellular, Tissue and Gene Therapies (OCTGT)] in the Center for Biologics Evaluation and Research, FDA (OTAT/CBER/FDA) is responsible for the regulation of cellular and gene therapy products. The FDA offers several expedited programs to speed up development of exceptionally promising therapies for serious or life-threatening conditions. Table 2 (56) provides a summary of the criteria and benefits of these expedited programs, including the Fast Track and Breakthrough Therapy Designation (FTD and BTD) and the Regenerative Medicine Advanced Therapy (RMAT) designation. Requests for the specific designation for the cell therapy products are reviewed by OTAT in the context of an IND submitted by the investigator.
Table 2.
Key features of the expedited programs (Fast Track Designation, Breakthrough Therapy Designation, and Regenerative Medicine Advanced Therapies)
| Qualifying Criteria | Benefits |
|---|---|
| Fast Track Designation (FTD) | |
| A drug (including biological product) is intended to | • More frequent meetings with FDA |
| treat a serious condition, and nonclinical or clinical data demonstrate potential to address an unmet medical need | • Rolling review*• Possible eligibility for Accelerated Approval (AA), Priority Review (PR) |
| Breakthrough Therapy Designation (BTD) | |
| A drug (including biological product) is intended to treat a serious condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on a clinically significant endpoint(s) over available therapies | • All the benefits of FTD• Intensive guidance from FDA on efficient drug development, beginning as early as phase 1•Organizational commitment involving FDA senior managers |
| Regenerative Medicine Advanced Therapies (RMAT) | |
| A drug is a regenerative medicine therapy intended to treat, modify, reverse, or cure a serious condition, and preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such disease or condition | • All the benefits of BTD• Early interactions to discuss any potential surrogate or intermediate endpoints• Methods to support accelerated approval and satisfy postapproval requirements |
*Rolling review means that an applicant (e.g., investigator) can submit completed section(s) of its marketing application, such as a Biologics License Application (BLA) for review by FDA, rather than waiting until every section of the BLA is completed before submitting the entire application for review.
Conclusion/Outlook
Translation of iPSC methods to generate organ-specific tissue for clinical application is complex and requires a multidisciplinary approach, which is well beyond the expertise of a traditional academic research laboratory. Identification of the specific clinical problem that the therapy will approach and the choice of the source of stem cells are the key delineators on the way of intestinal organoids to the clinic. Appreciation of the need for early implementation of good manufacturing and traceability practices may significantly accelerate the transition from bench to the bedside. The draft roadmap presented here was an attempt to provide a detailed process that we will use to translate our extensive basic research experience into clinical application. We hope that information included here will be valuable to those of you who are also preparing to move their new cellular therapy into first-in-human clinical trials.
GRANTS
This work was supported by the National Institute of Diabetes and Digestive and Kidney Disorders Grant U01DK103117, by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health Grant R01GM012345, and by the Pursuing Our Potential Together effort of Cincinnati Children’s Hospital.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.K. drafted manuscript; M.K., M.T., T.B., W.C.B., T.C.W., J.C.N., and M.A.H. edited and revised manuscript; M.K. and M.A.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Rachael Kiniyalocts for the creation of figures.
REFERENCES
- 1.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459: 262–265, 2009. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 2.Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn AM, Shroyer NF, Wells JM. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470: 105–109, 2011. doi: 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.VanDussen KL, Marinshaw JM, Shaikh N, Miyoshi H, Moon C, Tarr PI, Ciorba MA, Stappenbeck TS. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 64: 911–920, 2015. doi: 10.1136/gutjnl-2013-306651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yui S, Nakamura T, Sato T, Nemoto Y, Mizutani T, Zheng X, Ichinose S, Nagaishi T, Okamoto R, Tsuchiya K, Clevers H, Watanabe M. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5(+) stem cell. Nat Med 18: 618–623, 2012. doi: 10.1038/nm.2695. [DOI] [PubMed] [Google Scholar]
- 5.Sugimoto S, Ohta Y, Fujii M, Matano M, Shimokawa M, Nanki K, Date S, Nishikori S, Nakazato Y, Nakamura T, Kanai T, Sato T. Reconstruction of the human colon epithelium in vivo. Cell Stem Cell 22: 171–176.e5, 2018. doi: 10.1016/j.stem.2017.11.012. [DOI] [PubMed] [Google Scholar]
- 6.Khalil HA, Hong SN, Rouch JD, Scott A, Cho Y, Wang J, Lewis MS, Martin MG, Dunn JCY, Stelzner MG. Intestinal epithelial replacement by transplantation of cultured murine and human cells into the small intestine. PLoS One 14: e0216326, 2019. doi: 10.1371/journal.pone.0216326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergenheim F, Seidelin JB, Pedersen MT, Mead BE, Jensen KB, Karp JM, Nielsen OH. Fluorescence-based tracing of transplanted intestinal epithelial cells using confocal laser endomicroscopy. Stem Cell Res Ther 10: 148, 2019. doi: 10.1186/s13287-019-1246-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukuda M, Mizutani T, Mochizuki W, Matsumoto T, Nozaki K, Sakamaki Y, Ichinose S, Okada Y, Tanaka T, Watanabe M, Nakamura T. Small intestinal stem cell identity is maintained with functional Paneth cells in heterotopically grafted epithelium onto the colon. Genes Dev 28: 1752–1757, 2014. doi: 10.1101/gad.245233.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanderhoof JA, Langnas AN. Short-bowel syndrome in children and adults. Gastroenterology 113: 1767–1778, 1997. doi: 10.1053/gast.1997.v113.pm9352883. [DOI] [PubMed] [Google Scholar]
- 10.Stollman NH, Neustater BR, Rogers AI. Short-bowel syndrome. Gastroenterologist 4: 118–128, 1996. [PubMed] [Google Scholar]
- 11.Watson CL, Mahe MM, Múnera J, Howell JC, Sundaram N, Poling HM, Schweitzer JI, Vallance JE, Mayhew CN, Sun Y, Grabowski G, Finkbeiner SR, Spence JR, Shroyer NF, Wells JM, Helmrath MA. An in vivo model of human small intestine using pluripotent stem cells. Nat Med 20: 1310–1314, 2014. doi: 10.1038/nm.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Workman MJ, Mahe MM, Trisno S, Poling HM, Watson CL, Sundaram N, Chang C-F, Schiesser J, Aubert P, Stanley EG, Elefanty AG, Miyaoka Y, Mandegar MA, Conklin BR, Neunlist M, Brugmann SA, Helmrath MA, Wells JM. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat Med 23: 49–59, 2017. doi: 10.1038/nm.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finkbeiner SR, Freeman JF, Wieck MM, El-Nachef W, Altheim CH, Tsai Y-H, Huang S, Dyal R, White ES, Grikscheit TC, Teitelbaum DH, Spence JR. Generation of tissue-engineered small intestine using embryonic stem cell-derived human intestinal organoids. Biol Open 4: 1462–1472, 2015. doi: 10.1242/bio.013235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitano K, Schwartz DM, Zhou H, Gilpin SE, Wojtkiewicz GR, Ren X, Sommer CA, Capilla AV, Mathisen DJ, Goldstein AM, Mostoslavsky G, Ott HC. Bioengineering of functional human induced pluripotent stem cell-derived intestinal grafts. Nat Commun 8: 765, 2017. doi: 10.1038/s41467-017-00779-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schweinlin M, Wilhelm S, Schwedhelm I, Hansmann J, Reitscher R, Jurowich C, Wales H, Metzger M. Development of an advanced primary human in vitro model of the small intestine. Tissue Eng Part C Methods 22: 873–883, 2016. doi: 10.1089/ten.TEC.2016.0101. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura T, Sato T. Advancing intestinal organoid technology toward regenerative medicine. Cell Mol Gastroenterol Hepatol 5: 51–60, 2018. doi: 10.1016/j.jcmgh.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cortez AR, Poling HM, Brown NE, Singh A, Mahe MM, Helmrath MA. Transplantation of human intestinal organoids into the mouse mesentery: a more physiologic and anatomic engraftment site. Surgery 164: 643–650, 2018. doi: 10.1016/j.surg.2018.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vives J, Batlle-Morera L. The challenge of developing human 3D organoids into medicines. Stem Cell Res Ther 11: 72, 2020. doi: 10.1186/s13287-020-1586-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng W, Datta P, Wu Y, Dey M, Ayan B, Dababneh A, Ozbolat IT. Challenges in bio-fabrication of organoid cultures. Adv Exp Med Biol 1107: 53–71, 2018. doi: 10.1007/5584_2018_216. [DOI] [PubMed] [Google Scholar]
- 20.Huch M, Knoblich JA, Lutolf MP, Martinez-Arias A. The hope and the hype of organoid research. Development 144: 938–941, 2017. doi: 10.1242/dev.150201. [DOI] [PubMed] [Google Scholar]
- 21.Deuse T, Hu X, Agbor-Enoh S, Koch M, Spitzer MH, Gravina A, Malik A, Marishta A, Peters B, Kosaloglu-Yalcin Z, Yang Y, Rajalingam R, Wang D, Nashan B, Kiefmann R, Reichenspurner H, Valanthreine H, Weissman IL, Schrepfer S. De novo mutations in mitochondrial DNA of iPSCs produce immunogenic neoepitopes in mice and humans. Nat Biotechnol 37: 1137–1144, 2019. doi: 10.1038/s41587-019-0227-7. [DOI] [PubMed] [Google Scholar]
- 22.Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, Thaye r WO, Wahl A, Garcia JV, Reichenspurner H, Davis MM, Lanier LL, Schrepfer S. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat Biotechnol 37: 252–258, 2019. doi: 10.1038/s41587-019-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan S, Stacey GN, Akazawa C, Aoyama N, Baptista R, Bedford P, Griscelli AB, Chandra A, Elwood N, Girard M, Kawamata S, Hanatani T, Latsis T, Lin S, Ludwig TE, Malygina T, Mack A, Mountford JC, Noggle S, Pereira LV, Price J, Sheldon M, Srivastava A, Stachelscheid H, Velayudhan SR, Ward NJ, Turner ML, Barry J, Song J. Quality control guidelines for clinical-grade human induced pluripotent stem cell lines. Regen Med 13: 859–866, 2018. doi: 10.2217/rme-2018-0095. [DOI] [PubMed] [Google Scholar]
- 24.Yaffe MP, Noggle SA, Solomon SL. Raising the standards of stem cell line quality. Nat Cell Biol 18: 236–237, 2016. doi: 10.1038/ncb3313. [DOI] [PubMed] [Google Scholar]
- 25.Huang CY, Liu C-L, Ting C-Y, Chiu Y-T, Cheng Y-C, Nicholson MW, Hsieh PCH. Human iPSC banking: barriers and opportunities. J Biomed Sci 26: 87, 2019. doi: 10.1186/s12929-019-0578-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Food and Drug Administration. Facts About the Current Good Manufacturing Practices (CGMPs). https://www.fda.gov/drugs/pharmaceutical-quality-resources/facts-about-current-good-manufacturing-practices-cgmps. [June 25, 2018].
- 27.Ouseph S, Tappitake D, Armant M, Wesselschmidt R, Derecho I, Draxler R, Wood D, Centanni JM. Cellular Therapies Clinical Research Roadmap: lessons learned on how to move a cellular therapy into a clinical trial. Cytotherapy 17: 339–343, 2015. doi: 10.1016/j.jcyt.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.French A, Suh JY, Suh CY, Rubin L, Barker R, Bure K, Reeve B, Brindley DA. Global strategic partnerships in regenerative medicine. Trends Biotechnol 32: 436–440, 2014. doi: 10.1016/j.tibtech.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 29.Takebe T, Wells JM, Helmrath MA, Zorn AM. Organoid center strategies for accelerating clinical translation. Cell Stem Cell 22: 806–809, 2018. doi: 10.1016/j.stem.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perrin M, Kim T, Stan R, Giesie P, Tabor J, Le Verche V, Johnson S, Lomax GP, Zaia JA. Role of nursing competencies for accelerating clinical trials in stem cell clinics. Stem Cells Transl Med 7: 6–10, 2018. doi: 10.1002/sctm.17-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neofytou E, O’Brien CG, Couture LA, Wu JC. Hurdles to clinical translation of human induced pluripotent stem cells. J Clin Invest 125: 2551–2557, 2015. doi: 10.1172/JCI80575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abranches E, Spyrou S, Ludwig T. GMP banking of human pluripotent stem cells: a US and UK perspective. Stem Cell Res 45: 101805, 2020. doi: 10.1016/j.scr.2020.101805. [DOI] [PubMed] [Google Scholar]
- 33.Doss MX, Sachinidis A. Current challenges of iPSC-based disease modeling and therapeutic implications. Cells 8: 403, 2019. doi: 10.3390/cells8050403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 51: 297–305, 2010. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swierczek B, Ciemerych MA, Archacka K. From pluripotency to myogenesis: a multistep process in the dish. J Muscle Res Cell Motil 36: 363–375, 2015. doi: 10.1007/s10974-015-9436-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian L, Deshmukh A, Ye Z, Jang Y-Y. Efficient and controlled generation of 2D and 3D bile duct tissue from human pluripotent stem cell-derived spheroids. Stem Cell Rev Rep 12: 500–508, 2016. doi: 10.1007/s12015-016-9657-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang L, Geng Z, Nickel T, Johnson C, Gao L, Dutton J, Hou C, Zhang J. Differentiation of human induced-pluripotent stem cells into smooth-muscle cells: two novel protocols. PLoS One 11: e0147155, 2016. doi: 10.1371/journal.pone.0147155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zahabi A, Shahbazi E, Ahmadieh H, Hassani S-N, Totonchi M, Taei A, Masoudi N, Ebrahimi M, Aghdami N, Seifinejad A, Mehrnejad F, Daftarian N, Salakdeh GH, Baharvand H. A new efficient protocol for directed differentiation of retinal pigmented epithelial cells from normal and retinal disease induced pluripotent stem cells. Stem Cells Dev 21: 2262–2272, 2012. doi: 10.1089/scd.2011.0599. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Klos M, Wilson GF, Herman AM, Lian X, Raval KK, Barron MR, Hou L, Soerens AG, Yu J, Palecek SP, Lyons GE, Thomson JA, Herron TJ, Jalife J, Kamp TJ. Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: the matrix sandwich method. Circ Res 111: 1125–1136, 2012. doi: 10.1161/CIRCRESAHA.112.273144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matthys OB, Hookway TA, McDevitt TC. Design principles for engineering of tissues from human pluripotent stem cells. Curr Stem Cell Rep 2: 43–51, 2016. doi: 10.1007/s40778-016-0030-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purpura KA, Bratt-Leal AM, Hammersmith KA, McDevitt TC, Zandstra PW. Systematic engineering of 3D pluripotent stem cell niches to guide blood development. Biomaterials 33: 1271–1280, 2012. doi: 10.1016/j.biomaterials.2011.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCracken KW, Howell JC, Wells JM, Spence JR. Generating human intestinal tissue from pluripotent stem cells in vitro. Nat Protoc 6: 1920–1928, 2011. doi: 10.1038/nprot.2011.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arora N, Alsous JI, Guggenheim JW, Mak M, Munera J, Wells JM, Kamm RD, Asada HH, Shvartsman SY, Griffith LG. A process engineering approach to increase organoid yield. Development 144: 1128–1136, 2017. doi: 10.1242/dev.142919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan K, Zhang S, Zhang Y, Lu J, Holcombe M, Zhang X. A machine learning assisted, label-free, non-invasive approach for somatic reprogramming in induced pluripotent stem cell colony formation detection and prediction. Sci Rep 7: 13496, 2017. doi: 10.1038/s41598-017-13680-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paull D, Sevilla A, Zhou H, Hahn AK, Kim H, Napolitano C, et al. Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods 12: 885–892, 2015. doi: 10.1038/nmeth.3507. [DOI] [PubMed] [Google Scholar]
- 46.Tristan CA, Ormanoglu P, Slamecka J, Malley C, Chu P-H, Jovanovic VM, Gedik Y, Bonney C, Barnaeva E, Braisted J, Mallanna SK, Dorjsuren D, Iannotti MJ, Voss TC, Michael S, Simeonov A, Singeç I. Robotic high-throughput biomanufacturing and functional differentiation of human pluripotent stem cells. bioRxiv 2020.08.03.235242. doi: 10.1101/2020.08.03.235242. [DOI] [PMC free article] [PubMed]
- 47.Cruz-Acuna R, Quiros M, Farkas AE, Dedhia PH, Huang S, Siuda D, Garcia-Hernandez V, Miller AJ, Spence JR, Nusrat A, Garcia AJ. Synthetic hydrogels for human intestinal organoid generation and colonic wound repair. Nat Cell Biol 19: 1326–1335, 2017. doi: 10.1038/ncb3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ekser B, Kubal CA, Fridell JA, Mangus RS. Comparable outcomes in intestinal retransplantation: single-center cohort study. Clin Transplant 32: e13290, 2018. doi: 10.1111/ctr.13290. [DOI] [PubMed] [Google Scholar]
- 49.Kubal CA, Pennington C, Fridell J, Ekser B, Muhaylov P, Mangus R. Challenges with intestine and multivisceral re-transplantation: importance of timing of re-transplantation and optimal immunosuppression. Ann Transplant 23: 98–104, 2018. doi: 10.12659/AOT.908052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Selvaggi G, Khan FA. Overview of intestinal and multivisceral transplantation. UpToDate. (Online). https://www.uptodate.com/contents/overview-of-intestinal-and-multivisceral-transplantation. [2020 Sep 16].
- 51.Trevizol AP, David AI, Yamashita ET, Pecora RA, D’Albuquerque LA. Intestinal and multivisceral retransplantation results: literature review. Transplant Proc 45: 1133–1136, 2013. doi: 10.1016/j.transproceed.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 52.Maeda M, Kanai N, Kobayashi S, Hosoi T, Takagi R, Ohki T, Muragaki Y, Yamato M, Eguchi S, Fukai F, Okano T. Endoscopic cell sheet transplantation device developed by using a 3-dimensional printer and its feasibility evaluation in a porcine model. Gastrointest Endosc 82: 147–152, 2015. doi: 10.1016/j.gie.2015.01.062. [DOI] [PubMed] [Google Scholar]
- 53.Harding J, Roberts RM, Mirochnitchenko O. Large animal models for stem cell therapy. Stem Cell Res Ther 4: 23, 2013. doi: 10.1186/scrt171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alstrup A, Winterdahl M. Imaging techniques in large animals. Scand J Lab Anim Sci 36: 55–66, 2009. [Google Scholar]
- 55.Yang P. Is reliable in vivo detection of stem cell viability possible in a large animal model of myocardial injury? Circulation 126: 388–390, 2012. doi: 10.1161/CIRCULATIONAHA.112.119305. [DOI] [PubMed] [Google Scholar]
- 56.Food and Drug Administration. Expedited Programs for Regenerative Medicine Therapies for Serious Conditions. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/expedited-programs-regenerative-medicine-therapies-serious-conditions. FDA-2017-D-6159. [May 16, 2019].
- 57.O'Brien T, Creane M, Windebank AJ, Terzic A, Dietz AB. Translating stem cell research to the clinic: a primer on translational considerations for your first stem cell protocol. Stem Cell Res Ther 6: 146, 2015. doi: 10.1186/s13287-015-0145-7. [26296990] [DOI] [PMC free article] [PubMed] [Google Scholar]