

Keywords: fibroblast, hepatocyte growth factor, MET, papillary renal cell carcinoma, spheroid
Abstract
Papillary renal cell carcinoma (pRCC) represents the second most common kidney cancer and can be distinguished from other types based on its unique histological architecture and specific pattern of genomic alterations. Sporadic type 1 pRCC is almost universally driven by focal or chromosomal amplification of the receptor tyrosine kinase MET, although the specific mode of its activation is unclear. Although the MET receptors found in human tumor specimens appear highly active, those found on the surface of in vitro-cultured tumor cells are only weakly activated in the absence of exogenous hepatocyte growth factor ligand. Furthermore, pRCC cells cultured in standard two-dimensional conditions with serum fail to respond functionally to MET knockdown or the selective MET inhibitor capmatinib despite clear evidence of kinase inhibition at the molecular level. To better model pRCC in vitro, we developed a three-dimensional coculture system in which renal tumor cells are layered on top of primary fibroblasts in a fashion that mimics the papillary architecture of human tumors. In this three-dimensional spheroid model, the tumor cells survive and proliferate in the absence of serum due to trophic support of hepatocyte growth factor-producing fibroblasts. Unlike tumor cells grown in monoculture, the proliferation of cocultured tumor cells is sensitive to capmatinib and parallels inhibition of MET kinase activity. These findings demonstrate the importance of stromal fibroblasts in pRCC and indicate that accurate in vitro representation of this disease requires the presence of both tumor and fibroblast cells in a structured coculture model.
NEW & NOTEWORTHY Two-dimensional monoculture of papillary renal cancer cells fails to replicate several features of the disease found in humans. We hypothesized that this discordance results from lack of trophic support from renal fibroblasts, which are involved in the architecture of human papillary renal tumors. We found that three-dimensional layering of renal cancer cells on top of a fibroblast core using magnetic bioprinting produces a structured spheroid that more faithfully mimics the behavior of human tumors.
INTRODUCTION
Renal cell carcinoma (RCC) represents one of the 10 most common solid tumor types with a lifetime risk of ∼1.6% in the United States (1). Like many other solid tumors, RCC can be subclassified into several different entities based on histological presentation and immunohistochemical staining (2–4). Of the seven subtypes of kidney cancer, papillary RCC (pRCC) represents ∼10–15% of all cases and can be identified based on its unique tissue architecture, which has the appearance of finger-like projections (papilla) composed of a single layer of cancer cells coating a protrusion of connective tissue (2, 3). The pathophysiological explanation for this prototypic appearance has not been clearly elucidated, although it suggests an intimate tumor-stroma relationship that is characteristic of many solid tumor types (5).
In addition to histological approaches, pRCC can also be distinguished from other subtypes of renal cancer by molecular subclassification schemes involving analysis of genomic alterations, RNA transcriptome profiles, or protein expression patterns (6–10). Recent studies have used these methods to demonstrate a relatively clear separation between two subtypes of pRCC (6, 11). Within these subtypes, type 1 patients are particularly characterized by genetic abnormalities in the MET oncogene, which is also associated with a hereditary form of pediatric pRCC (12). Although this latter cohort of pediatric patients is typically affected by activating point mutations within the tyrosine kinase domain of MET, the majority of sporadic cases (∼83%) are instead driven by chromosomal amplification of the wild-type gene located at 7q31. Both mechanisms appear to result in constitutive activation of the enzyme based on measurements of tyrosine phosphorylated MET in tumors (6, 11, 13). Because MET normally functions as a receptor for hepatocyte growth factor (HGF), it seems likely that its overexpression is sufficient for activation due to the presence of endogenous HGF ligand in the renal microenvironment.
Prior work has shown that HGF levels increase in the serum of patients with renal damage and that its production is critical for repair of damaged kidney tubules (14, 15). Although multiple renal cell types may produce HGF, interstitial fibroblasts are the most likely source for the bulk of this ligand (16, 17). As structural support cells for the kidney, interstitial fibroblasts represent an important source of trophic signals both during development and repair of the organ after damage (18). Previous studies have demonstrated that renal fibroblasts produce abundant levels of HGF, suggesting that the fibroblasts associated with the pRCC microenvironment may be an important driver of tumor cell growth in vivo (19).
In this study, we identify the shortcomings of monoculture for replicating the in vivo behavior of pRCC by demonstrating that standard two-dimensional (2-D) cell culture models involving serum stimulation fail to respond to MET inhibition, despite clear evidence of target engagement. We further demonstrate that a three-dimensional (3-D) system involving serum-free coculture of pRCC cells with human primary fibroblasts produces a more faithful model of this disease. These data support the notion that an exogenous source of HGF is required for stimulation of pRCC tumor cells and that interruption of this paracrine circuit underlies the efficacy of pharmacological MET inhibitors for the treatment of pRCC in vivo.
MATERIALS AND METHODS
Antibodies and Reagents
All primary antibodies, isotype controls, and DyLight-labeled secondary antibodies used for immunoblot analysis were purchased from Cell Signaling Technology (Danvers, MA). Anti-HGF and anti-IL6 neutralizing antibodies were obtained from R&D Systems (Minneapolis, MN). Further information for the antibody reagents is provided in Supplemental Table S1 (all Supplemental material is available at https://doi.org/10.6084/m9.figshare.14556078.v1). All chemical reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. The MET inhibitor capmatinib (INCB28060) was purchased from Selleck Chemicals (Houston, TX). Recombinant human HGF was obtained from Peprotech (Rocky Hill, NJ).
Cell Culture
The Caki2 and SKRC39 pRCC cell lines were provided as a gift by Dr. Bin Tean Teh as previously described (20). Normal immortalized human kidney (HK2) cells and Wi-38 fibroblasts were purchased from American Type Culture Collection (Manassas, VA). Renal lines were maintained in RPMI-1640 media supplemented with 2 mM GlutaMAX (Thermo Fisher Scientific, Grand Island, NY) and 5% FBS (Atlanta Biologicals, Flowery Branch, GA). Wi-38 fibroblasts were maintained in high-glucose (4.5 g/L) DMEM (Thermo Fisher) with 10% FBS. Cells were incubated at 37°C in a humidified tissue culture incubator with 5% CO2 and were passaged every 3–4 days or when they achieved a confluence of ∼95%.
For assays involving serum-free growth conditions, RPMI-1640 media containing 2 mM GlutaMAX were supplemented with selenium-insulin-transferrin supplement (1× SIT, Thermo Fisher) and with 1 mg/mL heparin (Sigma-Aldrich). Where indicated, plates were coated for 1 h at room temperature with either MaxGel or Engelbreth-Holm-Swarm mouse sarcoma matrix (Sigma-Aldrich) diluted in PBS or RPMI-1640 media, respectively. Excess liquid was aspirated, and plates were rinsed once with sterile PBS before being dried overnight in a tissue culture hood.
Quantification of 2-D Proliferation by Electrical Impedance Assay
The xCELLigence real-time cell analysis system (Acea Biosciences, San Diego, CA) for measuring 2-D cell proliferation of renal lines was used as previously described (20). Cells were plated at a density of 2,000 cells/well in a 96-well E-plate and allowed to adhere for 30 min at room temperature. The plates were then placed into the electrical impedance apparatus in a tissue culture incubator as previously indicated for the duration of the assay. Measurements were recorded on an hourly basis for 110 h. All conditions were performed in quadruplicate, and values were normalized to the initial impedance after 8 h of initial plating.
Quantification of 2-D Proliferation by Luminescence Assay
Cells were seeded to black-wall, 96-well culture plates (Greiner, Kremsmünster, Austria) in the indicated culture media. At the specified intervals after seeding, relative cellular abundance was measured using a Cell Titer Glo assay (Promega, Madison, WI), which produces an ATP-dependent luminescent signal proportional to cell number. Luminescent values were captured with a Synergy H1 plate reader (BioTek, Winooski, WT). Average luminescent values for each triplicate sample were obtained and plotted using Prism software (Mac version 6, GraphPad Software) to obtain a relative proliferation rate in arbitrary luminescent units per hour.
Quantification of Cell Cycle Progression by 5-Ethynyl-Deoxyuridine Incorporation
Identification of proliferating cells in the S-phase of the cell cycle was performed using copper(I)-catalyzed click chemistry after metabolic labeling of cells with the thymidine analog 5-ethynyl-deoxyuridine (EdU) (21). Cells were pulsed with 10 μM EdU for 2 h under normal cell culture conditions, detached by trypsinization, rinsed with PBS, and then fixed and permeabilized with 4% formaldehyde and 0.2% Triton X-100 in cold PBS for 30 min on ice. Cells were washed twice with PBS and then incubated for 20 min at room temperature with labeling solution [100 mM Tris (pH 8.5), 1 mM CuSO4, 10 µM Cy6-azide, and 100 mM ascorbate]. Cells were then washed twice with cold PBS and stored in the dark until analysis by flow cytometry. Unlabeled cells were used as a negative control to establish proper gating for flow cytometry. All experiments were performed in triplicate and averaged to obtain the percent EdU labeling efficiency.
Protein Extraction and Immunoblot Analysis
Cells were rinsed with cold PBS and harvested into 100 µL of lysis buffer [20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM sodium glycerophosphate, 1 mM sodium orthovanadate, 0.5% Nonidet P-40, 0.1% Brij35, and 0.1% sodium deoxycholate] supplemented with mammalian cell protease inhibitor cocktail (Sigma-Aldrich). Lysates were homogenized by brief sonication at 30% power on ice and cleared by centrifugation at 10,000 relative centrifugal forces for 5 min at 4°C. The concentration of each lysate was determined by a Bradford assay. Equal amounts of protein lysate (20–50 µg) were separated by reducing PAGE and transferred overnight to nitrocellulose membrane using a wet transfer tank (TE62 model, Hoefer, Holliston, MA). Membranes were blocked with 3% nonfat dry milk in Tris-buffered saline containing 0.05% Tween 20 and then probed overnight at 4°C with primary antibodies diluted 1:1,000 in Tris-buffered saline and Tween 20 with 3% BSA. After unbound primary antibody was washed off, the membranes were incubated for 1 h with goat anti-rabbit-DyLight-800 and goat anti-mouse-DyLight-680 secondary antibodies. Membranes were imaged with an Odyssey scanner (Li-Cor, Lincoln, NE) to produce images that were processed with Odyssey Infrared Imagining software (version 3.0.25) to ensure that signal was in the linear range before export as TIFF files. Full panel images of immunoblots with independent molecular weight markers are provided in Supplemental Fig. S1.
MET Knockdown by RNA Interference
Two MET-targeted siRNAs (s8700 and s8701, Silencer Select) and two control siRNAs [nontargeting (NT) and activated cell death control (ACDC)] were purchased from Ambion (Austin, TX). Lyophilized siRNAs were resuspended in nuclease-free resuspension buffer to a concentration of 20 µM and frozen in 50-µL aliquots. Transfection complexes composed of 50 nM siRNA and 2.5 µL/mL siLentFect lipid transfection reagent in RPMI-1640 basal media were incubated 30 min before exposure to cells (Bio-Rad, Hercules, CA). Culture media were aspirated from cells and replaced with the transfection complexes under normal tissue culture conditions to facilitate siRNA uptake. An equal volume of RPMI-1640 media containing 10% FBS was added 4 h later. After an overnight incubation, the transfection media were aspirated and replaced with complete culture media.
Quantification of MET Surface Expression by Flow Cytometry
Cells were detached from culture plates with enzyme-free detachment buffer (Thermo Fisher) at 48 h after siRNA transfection and filtered into polystyrene cytometry tubes (BD Biosciences, San Jose, CA). Pelleted cells were resuspended in 0.5 mL of blocking solution (3% BSA in PBS) and incubated for 1 h at 4°C with shaking. Cells were then pelleted and resuspended in 100 µL of primary antibody solution containing a 1:100 dilution of anti-MET(L6E7) mouse monoclonal antibody targeted to the extracellular domain of MET (Cell Signaling Technologies). The negative control for staining was performed with a 1:100 dilution of isotype mouse IgG obtained from the same commercial source. After incubation at 4°C with shaking for 1 h, cells were diluted with 1 mL of cold PBS, pelleted, and resuspended in 100 µL of secondary antibody solution containing a 1:1,000 dilution of Alexa Fluor 647-coupled goat anti-mouse antibody. Cells were incubated for an additional hour with secondary antibody, washed, resuspended in 300 µL of cold PBS containing 1 µM propidium iodide (PI), and then stored in the dark on ice until cytometry analysis.
Analysis of stained cells was performed with a FACScalibur flow cytometer (Becton Dickinson, San Jose, CA). Each sample was gated using the FL2 channel to exclude PI-positive cells. Live cells were assessed on the FL4 channel to quantify the intensity of Alexa Fluor 647 staining. Laser power for the FL4 channel was set to center the negative control cell population distribution at a mean fluorescent intensity (MFI) of 10 units on the log scale. No fewer than 200,000 cells were captured for each sample, and each condition was measured in duplicate to obtain an averaged MFI for each cell population.
Quantification of HGF Production by ELISA
Triplicate samples of conditioned media were harvested at 24-h intervals from renal or Wi-38 cells. Samples were stored at 4°C over the course of the assay and then diluted 1:5 with basal RPMI-1640 media before human HGF abundance was measured with a commercial ELISA assay (Thermo Fisher). The assay was performed according to the manufacturer’s instructions using positive control standards provided with the kit and serum-free media as a negative control.
2-D Coculture and Proliferation Analysis
Wi-38 cells were seeded at a 1:30 ratio from 10-cm dishes to 24-well dishes in complete media containing 10% serum. When the fibroblasts reached full confluency (3–5 days postplating), the media were removed and cells were rinsed twice with sterile Dulbecco’s PBS to remove residual serum. Renal cells growing in standard complete media were then detached by trypsinization, washed with Dulbecco’s PBS, and seeded separate on top of fibroblasts at a density of 50,000 cells/well in serum-free media containing heparin. Cell lysates from these cocultures were collected at various times to assay MET receptor activation and cell viability as indicated. Proliferation analysis done by measurement of EdU incorporation at 24 h after plating was performed as indicated above in Quantification of Cell Cycle Progression by 5-Ethynyl-Deoxyuridine Incorporation in MATERIALS AND METHODS. Time-course proliferation assays were performed in 96-well format in which 5,000 renal cells were seeded to confluent Wi-38 feeder layers. At 24-h increments after seeding, triplicate wells for each condition were rinsed with PBS and incubated for 10 min with 100 μL per well of Cell Titer Glo reagent as described above in Quantification of 2-D Proliferation by Luminesence Assay of MATERIALS AND METHODS. Wells containing only Wi-38 cells were used to control for the expected signal from fibroblasts alone.
3-D Spheroid Coculture and Proliferation Analysis
Wi-38 fibroblasts and renal cells grown on standard culture plates were incubated overnight with complete media containing 30 μL/mL of NanoShuttle magnetic nanoparticles coated with poly-l-lysine (Nano3D/Greiner, Frickenhausen, Germany). After overnight incubation, cells were detached with trypsin, resuspended in serum-free media, counted, and then seeded to a Corning ultralow-adherence 96-well U-bottom plate (Sigma-Aldrich). The 96-well plate was immediately inserted into a magnetic bioprinting rack containing small magnets under each well to aggregate the cells. After 3 h, the plate was removed from the rack and left to incubate at 37°C under normal tissue culture conditions. Successful spheroid production was evaluated qualitatively the following day based on microscopic visualization of cellular compaction (22).
3-D coculture experiments were performed by seeding pRCC renal lines on top of Wi-38 spheroids that had been freshly established 3 h before renal cell addition. Two distinct coculture models were used for immunoblot and proliferation analyses, respectively. For immunoblot analysis, an initial sphere of 10,000 fibroblasts was overlaid with 50,000 renal cells and allowed to aggregate on the magnetic rack for an additional 3 h. For proliferation analysis, an initial sphere of 2,000 fibroblasts was overlaid with 10,000 renal cells. This reduction in size allows for longer incubation without media changes, which is necessary for longitudinal assays. Time-course evaluation of proliferation was identical to that described for 2-D cocultures with the exception that the Cell Titer Glo 3-D reagent was added as a 2× concentrate to avoid removing culture media (Promega).
Statistical Analysis
Each experiment with triplicate samples was repeated a minimum of three times to ensure that results could be replicated. Data reporting proliferation rates were analyzed with GraphPad Prism 6 software for line fitting, with statistical significance determined by a two-tailed Student’s t test. Statistical significance is reported at P < 0.05 as indicated in the figures.
RESULTS
Cellular Models of pRCC Depend on Exogenous Stimulation to Drive Proliferation
Relatively few cellular models of pRCC are currently available for research purposes. We chose to work with Caki2 and SKRC39 cell lines, which have been previously validated as models of this disease (23–25). Caki2 cells were originally derived from a renal tumor of mixed histology with primarily clear cell features (26); however, when transplanted into nude mice, they produce tumors with clear papillary histology (27). SKRC39 cells were derived from a soft tissue metastasis of a patient with renal cancer rather than from a primary tumor itself (24). When transplanted into nude mice, they also produce tumors with papillary renal histology. Whether these cell lines represent type 1 or type 2 pRCC is somewhat unclear from prior studies, which prompted us to examine their growth and cell signaling characteristics compared with normal proximal tubule cells.
Both pRCC cell lines proliferate more rapidly in vitro than HK2 cells, which are immortalized normal renal proximal tubule cells obtained from a human kidney biopsy (Fig. 1A) (28). In standard 2-D culture on treated plastic, all three cell lines were dependent on external mitogenic stimulation by FBS for cell cycle entry and proliferation, although the maximal growth response was higher in pRCC lines than for normal HK2 cells (Fig. 1B and Supplemental Fig. S2). Relative to both HK2 and SKRC39 cells, the Caki2 cell line expressed about twofold higher levels of surface MET receptor as analyzed by flow cytometry (Fig. 1C). Next-generation sequencing analysis of this line has demonstrated that the MET gene has a wild-type sequence, which together with its overexpression of MET suggest that Caki2 cells may represent an appropriate model for type 1 pRCC (29).
Figure 1.

Characterization of MET activation in standard renal cell monocultures. The renal cell models used in this project included normal immortalized proximal tubule epithelia (HK2) and two papillary renal cell carcinoma lines (Caki2 and SKRC39). A: the proliferation rate of each cell line was measured using a Cell Titer Glo luminescence assay in serum-free media (SFM) or in media with 10% FBS. Luminescent values in arbitrary units (a.u.) were captured from quadruplicate wells at regular intervals for 72 h and fit to exponential growth equations to obtain growth rates. B: the proliferation rate of each cell line in four different media conditions (basal, 1% FBS, 5% FBS, and 10% FBS) was measured using the assay described in A. The significance of the growth rate in FBS was determined for each cell line relative to its proliferation rate in basal media without serum (*P < 0.05). C: cell surface expression of MET was measured by flow cytometry for each of the three cell lines and normalized to HK2 cell expression. Data represent averages of four separate assays with error bars representing the standard deviation of these experimental replicates. D: cell cycle entry of renal lines under subconfluent and confluent conditions as measured by 5-ethynyl-deoxyuridine (EdU) incorporation. Data represent the average of three biological replicates with error bars representing the standard deviation (*P < 0.05). E: representative immunoblot analysis of cells treated with 5% FBS or 100 ng/mL recombinant human hepatocyte growth factor ligand for 15 min. Cells were cultured under subconfluent (S) or fully confluent (C) conditions to evaluate contact inhibition of signal transduction. Activation of MET is indicated by phosphorylation at tyrosine 1234 (pMETY1234). Transmission of signals from activated receptors to their downstream pathways was determined by evaluating the phosphorylation of STAT3, AKT, ERK, and ribosomal S6 protein at the indicated sites. Detection of β-actin was included as a loading control for each sample.
The responsiveness of normal and pRCC cell lines to contact inhibition and ligand stimulation provides further information on the functional identity of each cell line. As demonstrated by EdU incorporation, HK2 cells almost completely exited the cell cycle after forming confluent, polarized monolayers (Fig. 1D). In contrast, both of the transformed pRCC cell lines continued to progress through the cell cycle even after becoming fully confluent in 2-D culture, although SKRC39 cells did so at a significantly reduced rate compared with subconfluent conditions. For Caki2 cells, the lack of functional contact inhibition was paralleled by MET receptor activation in response to both serum (FBS) and recombinant human HGF (Fig. 1E and Supplemental Fig. S1). Treatment of Caki2 cells with either stimulus led to MET phosphorylation that remained elevated even under high-density culture conditions. Furthermore, Caki2 cells also displayed increased autoactivation of MET under confluent conditions, which may be related to the mutational loss of protein tyrosine phosphatase receptor type J, which was previously identified in these cells by exome sequencing (29). Together, these data argue strongly that Caki2 cells represent an accurate model of type 1 pRCC in which the MET receptor remains active and responsive to ligand regardless of cell density.
In contrast to Caki2 cells, both HK2 and SKRC39 cells displayed robust contact inhibition of MET phosphorylation under high-density culture (Fig. 1E). Although both cell lines activated MET in response to FBS and HGF stimulation under subconfluent conditions, they displayed blunted MET activation in response to mitogenic stimulation after reaching confluency. The contact-mediated inhibition of MET phosphorylation in HK2 cells mirrored their functional exit from the cell cycle, as expected for normal epithelial cells (Fig. 1, D and E). The discordance between MET phosphorylation and cell cycle entry in confluent SKRC39 cells, however, indicates that their transformed phenotype is independent of the HGF/MET axis, which is more consistent with a type 2 pRCC identity.
The downstream pathways associated with receptor tyrosine kinase signaling in renal cells largely followed the expected response to treatment with FBS or HGF (Fig. 1E). All three renal cell lines showed robust activation of growth factor signaling pathways, including the RAS/MAPK pathway (phospho-ERKT202/Y204), the phosphatidylinositol 3-kinase/AKT pathway (phospho-AKTS473), and the mammalian target of rapamycin pathway (phospho-S6S235/S236), when treated with either stimulus. In contrast, the JAK/STAT pathway (phospho-STAT3Y705) was not engaged by HGF or FBS stimulation in these cells but instead was driven by autocrine signaling that increased with cell density. Of particular concern for these cellular models was the lack of concordance between MET phosphorylation and downstream pathway activation during FBS stimulation. The RAS/MAPK and phosphatidylinositol 3-kinase/AKT pathways in particular remained active under FBS stimulation even when MET phosphorylation was decreased due to contact inhibition. This is perhaps to be expected given the complexity of mitogenic factors found in serum, which likely activate other growth factor receptors that are redundant to MET. Our previous work has identified the epidermal growth factor receptor and insulin-like growth factor 1 receptor as potential mediators of this effect, which brings into question the utility of using FBS as a mitogenic stimulant for studies involving the HGF/MET signaling pathway in pRCC (20).
Acute Inhibition of MET Activity Fails to Attenuate pRCC Cell Proliferation
The increased expression of MET in type 1 pRCC is thought to be a driving force behind disease growth and progression. Model systems failing to recapitulate this reliance on MET signaling, therefore, would have limited utility for further therapeutic discovery. Thus, we sought to confirm the tumor suppressive effects of MET signaling inhibition with our cell lines in vitro. Surprisingly, we were unable to substantiate a significant role for MET signaling in any 2-D culture model despite approaching pathway inhibition by three distinct means. We first demonstrated that acute knockdown of MET with two distinct siRNAs had virtually no impact on the proliferation of either Caki2 or SKRC39 cells, despite the clear efficacy of knockdown based on flow cytometric validation of MET protein levels (Fig. 2, A–D). This effect was not a function of the method being used to measure proliferation (electrical impedance assay), as transfection of cells with a positive control siRNA that universally kills human cells (ACDC siRNA) induced significant toxicity within 36 h of the initial transfection (Fig. 2, A and C).
Figure 2.
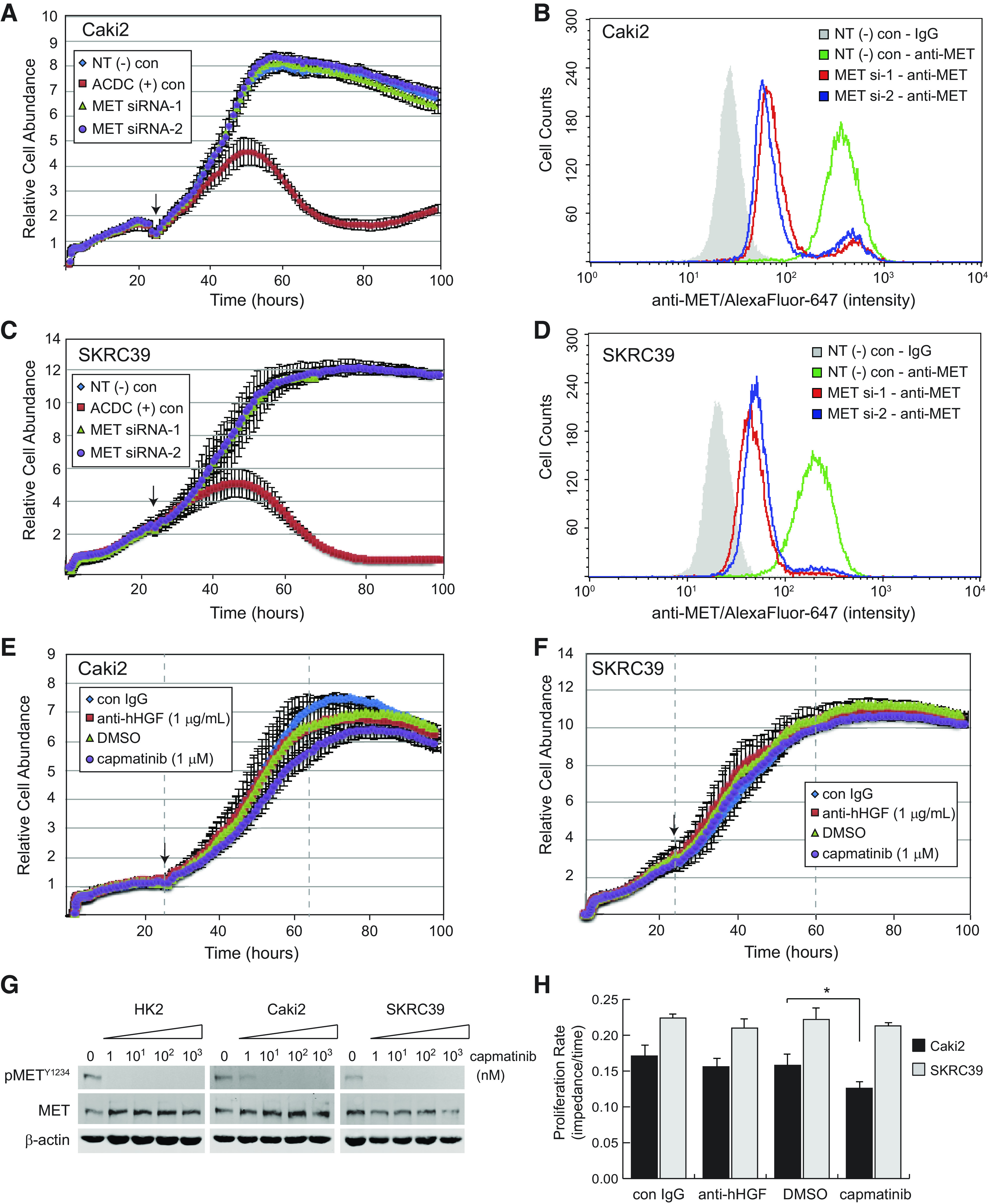
Inhibition of human hepatocyte growth factor (hHGF)/MET signaling in standard two-dimensional monoculture fails to impact papillary renal cell carcinoma cell proliferation. A, C, E, and F: Caki2 or SKRC39 cell proliferation was continuously monitored using the xCELLigence real-time cell analysis system for 110 h. The arrow on each figure indicates the 24-h time point at which siRNA transfection (A and C) or addition of the indicated drug (E and F) was performed. B and D: flow cytometry histograms of MET expression in control or MET knockdown lines to validate effective siRNA silencing of MET. Negative control cells were stained with mouse isotype IgG to establish the background signal for the assay (gray shaded histogram). G: immunoblot analysis of cells treated with the indicated nanomolar concentrations of capmatinib for 24 h in complete media containing 5% FBS. Activation of MET is indicated by autophosphorylation at tyrosine 1234 (pMETY1234); actin was included as a loading control for each sample. H: proliferation rates measured as the linear slope (impedance/time) of curves between the gray dashed lines in E and F. The significance of growth rate change was determined for each cell line relative to its matching negative control condition (*P < 0.05). ACDC, cell death positive control; con, control; NT(−), nontargeting negative control.
Because acute knockdown of MET protein levels by RNA interference only leads to an 85−90% reduction of the receptor, we next measured the proliferation of Caki2 and SKRC39 cells in the presence of capmatinib, a potent and selective inhibitor of the MET tyrosine kinase domain (Fig. 2, E and F) (30). This drug is effective in blocking MET activation at levels as low as 10 nM, demonstrating its potency against MET (Fig. 2G). Despite using capmatinib at 1 µM concentration, which is nearly 100 times the concentration required for target engagement, very little impact was observed on proliferation or survival of either cell line (Fig. 2H). Growth of Caki2 cells was modestly attenuated by the drug (∼20%), whereas SKRC39 cells showed no difference in their growth rate compared with vehicle (DMSO)-treated cells.
In addition to approaching the role of MET signaling by inhibiting receptor activity, we also perturbed the pathway at the level of its ligand, HGF. Consistent with the receptor-directed methods involving the drug capmatinib, we found that treatment of both pRCC cell lines with high doses of an anti-HGF neutralizing antibody had no impact on their in vitro proliferation (Fig. 2, E, F, and H). The results from these three distinct approaches collectively suggest that activation of the MET receptor under standard 2-D conditions is dispensable for growth of pRCC cells. This observation stands in stark contrast to findings from in vivo xenograft models of pRCC, indicating that a more robust model is required for in vitro studies of this form of renal cancer (31).
Primary Human Fibroblasts Produce HGF in Response to Serum or Heparin
As previously noted, the tissue architecture of pRCC suggests that interactions between tumor and stromal cells are important to the etiology of this disease. Because renal fibroblasts are known to secrete HGF in the context of acute kidney injury (19), we surmised that coculture of our tumor cell lines with fibroblasts might serve as a more effective and representative model system in vitro. Unfortunately, primary human renal interstitial fibroblasts are not readily available, so we instead turned to a more generic source of primary human fibroblasts to test this hypothesis.
Among the various cell lines that have been described, Wi-38 cells are perhaps the best-characterized commercial line of primary fibroblasts (32). To validate these cells as a support layer for growth of pRCC cells, we first determined whether they produce HGF at levels that could produce a mitogenic stimulus for tumor cells. Culture of Wi-38 cells in media containing various levels of FBS demonstrated a time-dependent increase in HGF concentration in response to increasing amounts of FBS (Fig. 3A). Similar experiments with all three of the renal lines failed to produce any measurable HGF in conditioned media out to 96 h, indicating that these tumor cells are incapable of autocrine MET activation.
Figure 3.

Primary human fibroblasts produce hepatocyte growth factor (HGF) in response to serum or heparin. A: ELISA measurement of human HGF production by Wi-38 primary fibroblasts as a function of time and FBS concentration. Measurements were taken at 24-h intervals from triplicate wells for each sample. B: 72-h ELISA measurements of human HGF secreted by Wi-38 fibroblasts under the indicated culture conditions. Cells were grown on standard tissue culture plastic (uncoated) or on culture plates that were precoated with either a synthetic (MaxGel) or biological [Engelbreth-Holm-Swarm (EHS)] extracellular matrix (ECM). Heparin was used at 1.0 mg/mL. Basal media, RPMI-1640. C: ELISA measurements of human HGF produced by Wi-38 primary fibroblasts as a function of time and serially diluted capmatinib concentration. D: slope of the linear plots in C providing the production rate for HGF by Wi-38 fibroblasts. For all A−D, error bars represent the standard deviation of triplicate measurements. Significance was determined for each set of samples relative to the SFM or vehicle (DMSO) condition at the same time point (*P < 0.05). SFM, serum-free media; SIT, selenium-insulin-transferrin supplement.
Because we sought to remove the confounding effects of exogenous serum in our coculture model, we were particularly interested in developing a serum-free means of HGF production by Wi-38 fibroblasts. Prior work with fibroblasts indicated that growth on particular substrates or in the presence of heparin could significantly increase HGF secretion by these cells (33, 34). To this end, we tested the effects of culturing Wi-38 cells on plates coated with synthetic (MaxGel) or biologically derived (Engelbreth-Holm-Swarm tumor) extracellular matrix proteins in the presence of 1 mg/mL heparin or 5% FBS. Measurement of HGF concentrations in conditioned media after 72 h demonstrated a roughly threefold increase in the production of HGF in response to heparin independent of the surface substrate being used (Fig. 3B). Notably, the effect of heparin was similar to that of 5% FBS, indicating that use of this molecule could be an effective replacement for serum when used in conjunction with a defined culture supplement (1× SIT) to promote cell survival. The combination of heparin and SIT without additional extracellular matrix additives was therefore used in subsequent coculture experiments of fibroblasts with renal cells.
Primary Human Fibroblasts Lack Responsiveness to MET Inhibition
An important caveat to the proposed coculture model concerned the potential for Wi-38 cells to engage in both autocrine and paracrine signaling with HGF, which would confound our ability to interpret the cell type-specific impact of MET inhibition. To control for this possibility, we asked whether production of HGF by Wi-38 fibroblasts would be sensitive to MET inhibition by capmatinib. We found that doses of capmatinib up to 1 µM caused a <15% decrease in HGF production by Wi-38 cells, suggesting that use of this inhibitor in a coculture model would not interrupt the paracrine signaling of fibroblasts to pRCC cells (Fig. 3, C and D).
An explanation for this lack of effect is found by the observation that Wi-38 cells fail to express the MET receptor. Although flow cytometry and immunoblotting clearly demonstrate that all three of the renal cell lines express MET and can be inhibited by capmatinib, neither method is able to detect MET protein in Wi-38 cells (Fig. 4, A and B). This finding suggests that any effect of capmatinib in the context of coculture is a result of a loss of MET signaling in tumor cells only and not due to secondary effects on fibroblasts.
Figure 4.

Fibroblast-derived hepatocyte growth factor (HGF) activates epithelial MET in papillary renal cell carcinoma (pRCC) cell tumor cells. A: flow cytometry histogram of MET receptor surface expression in Wi-38 fibroblasts, HK2 normal proximal tubule cells, and the two pRCC cell lines (Caki2 and SKRC39). HK2 cells stained with mouse isotype IgG were used to establish the background signal for the assay (gray shaded histogram). B: immunoblots of whole cell lysates for activated and total MET expression in all four cell lines indicated above. Actin was included as a control for protein loading. C: immunoblots of whole cell lysates after 30 min of treatment with the indicated conditions. Conditioned media (CM) from Wi-38 fibroblasts were harvested after 72 h of culture and then pretreated overnight with the indicated controls or inhibitors of HGF/MET signaling prior to treatment of renal cells. con, RPMI-1640 + 5% FBS; veh, 0.1% DMSO; CAP, 100 nM capmatinib; IgG, 1 µg/mL nonspecific mouse IgG; αHGF, 1 µg/mL human HGF neutralizing antibody; αIL6, 1 µg/mL interleukin-6 neutralizing antibody.
Conditioned Media From Human Fibroblasts Stimulates MET Activation in pRCC Cells
Before proceeding with the coculture experiments, we first wanted to demonstrate that the soluble form of HGF produced by Wi-38 cells is capable of activating MET on the renal lines. To this end, we harvested media from Wi-38 fibroblasts after 72 h of culture and then incubated the conditioned media with various inhibitors of HGF or MET before stimulating renal cells. The conditioned media were then used to stimulate each of the three renal lines for 30 min, after which protein lysates were extracted and analyzed by immunoblot analysis (Fig. 4C). In addition to inhibitors of MET and HGF, we also included IL-6 neutralizing antibodies as a positive control since fibroblasts are known to produce this cytokine, which signals through the JAK/STAT3 pathway (35).
Consistent with our prediction, we observed that HGF produced by Wi-38 cells was able to activate MET phosphorylation (phospho-Y1234) above the basal levels promoted by serum in Caki2 and SKRC39 cell lines (Fig. 4C). Both of these cell lines appear to be more sensitive than normal HK2 cells to the relatively low level of HGF produced by fibroblasts, which showed very little activation of MET above the basal level found in serum. Importantly, both capmatinib and HGF neutralizing antibodies clearly decreased the ability of Wi-38 conditioned media to activate MET in the renal lines. This effect was specific to MET activation and had no impact on STAT3 phosphorylation (phospho-Y705) by the conditioned media. In contrast, IL6 neutralizing antibodies decreased activation of STAT3 without impacting the activation of MET. Together, these results confirm that Wi-38 cells are capable of producing HGF at levels that can stimulate MET signaling in pRCC cells, indicating that they should be able to serve as a suitable support layer for growth of renal cells in serum-free conditions.
A 2-D Coculture Model Demonstrates HGF-MET Engagement but Lacks Biological Functionality
Our initial attempt at coculture of fibroblasts with the three renal lines involved growing Wi-38 cells to full confluency on 2-D plastic in complete media containing serum for 3–5 days. When the fibroblasts reached full confluency, the media were removed and each of the renal cell lines was then separately seeded on top of this feeder layer in serum-free media containing heparin to stimulate HGF production. Unlike direct culture on plastic, seeding of the three renal lines on top of fibroblasts produced a more rounded morphology due to the absence of cell surface contacts normally seen on plastic (Fig. 5A).
Figure 5.
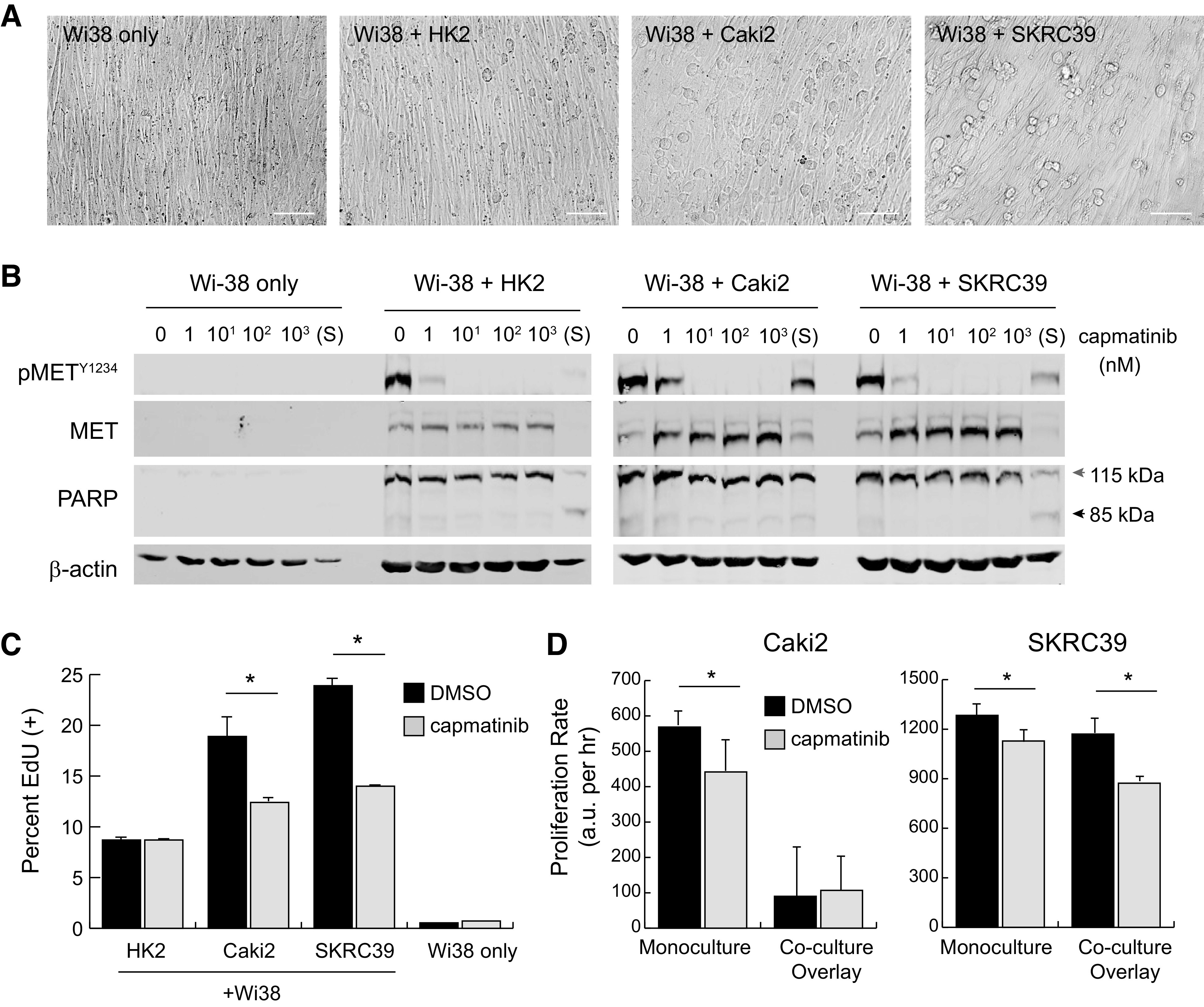
Two-dimensional overlay coculture induces paracrine hepatocyte growth factor (HGF)/MET signaling but has limited impact on tumor cell proliferation. Wi-38 fibroblasts were grown to confluency in 24-well plates in serum-free media and then overlaid with 50,000 renal cells to create two-dimensional cocultures. A: images of different mono- or cocultures at 24 h postplating. Scale bar = 125 µm. B: immunoblots from whole cell lysates of two-dimensional cultures at 24 h postplating with the indicated nanomolar concentrations of capmatinib or after 1 h of treatment with 1 µM staurosporine (S). Poly(ADP-ribose) polymerase (PARP) cleavage from 115 to 85 kDa was included as an indicator of cell death; actin was included as a control for protein loading. C: active S-phase entry of coculture cells treated with capmatinib (100 nM) or vehicle (DMSO) at 72 h, as quantified by 5-ethynyl-deoxyuridine (EdU) incorporation. Values represent the average of three replicates with error bars indicating the standard deviation. Significance was determined by pairwise comparison for each set of samples (*P < 0.05). D: the proliferation rate of cocultures treated with capmatinib (100 nM, gray) or DMSO (black) was measured using a Cell Titer Glo luminescence assay. Luminescent values were captured from triplicate wells at regular intervals for 72−96 h and fit to linear equations to obtain growth rates with values of arbitrary luminescent units (a.u.) per hour. The significance of growth rate difference between vehicle and drug conditions is shown for each cell line (*P < 0.05).
To evaluate whether MET receptors were activated by fibroblast-derived HGF, cocultures were established in a 24-well format along with various concentrations of capmatinib added to the serum-free media. After 24 h of culture, the combined contents of each well were harvested with lysis buffer and analyzed by immunoblot analysis. Monocultures of confluent Wi-38 cells were used as controls to demonstrate that the MET protein observed in cocultures was exclusively derived from renal cells. As shown in Fig. 5B, MET receptors appear to be strongly activated on all three renal lines in this coculture paradigm. Furthermore, capmatinib was fully effective at engaging its target at 10 nM concentration.
At 24 h after seeding, even complete inhibition of MET appeared not to impact survival of renal cells based both on visual inspection of the cultures and absence of poly(ADP-ribose) polymerase (PARP) cleavage in lysates (Fig. 5B). In contrast, even a short pulse of the cells for 1 h with a 1 µM concentration of the pan-kinase inhibitor staurosporine was sufficient to induce rapid cell death indicated by cleavage of PARP (Fig. 5B). These results indicate that inhibition of HGF-MET signaling is not acutely cytotoxic to renal cells in this 2-D coculture model.
In addition to testing for cytotoxic effects of MET inhibition, we also sought to understand whether renal cells seeded on top of the Wi-38 monolayer were able to proliferate and, if so, whether this aspect of the model depended on HGF-MET signaling. To test this feature of the assay, we performed two separate measurements of proliferation. We initially tested proliferative capacity after only 24 h of culture using a pulse-chase assay with EdU. This analog of thymidine is effectively incorporated into DNA during the S-phase of the cell cycle and can be detected using fluorescent copper-catalyzed click chemistry followed by flow cytometry (Fig. 5C and Supplemental Fig. S2, B–D) (23). Treatment with capmatinib resulted in a 30–40% decrease in EdU labeling for both pRCC cell lines, but had no impact on HK2 cells. As expected, the highly confluent Wi-38 cells themselves showed little incorporation of EdU due to their contact-inhibited state at the time the assay was performed.
We next performed a longer-term experiment that involved quantifying relative cell numbers over the course of 3–4 days in culture. In this assay, we compared growth of the renal lines in monoculture versus coculture and in the presence of 100 nM capmatinib or vehicle (0.1% DMSO) as a control. Interestingly, we observed that HK2 cells progressively detached from the Wi-38 feeder layer and lost viability when cultured for more than 24–48 h, suggesting that they require a more stable attachment surface to enable progression through the cell cycle. In contrast, both Caki2 and SKRC39 cell lines remained adherent and viable throughout the course of the experiment; however, proliferation of Caki2 cells in coculture was extremely low compared with monoculture and showed no difference in the presence or absence of MET inhibition (Fig. 5D and Supplemental Fig. S3). SKRC39 cells showed similar proliferation rates under both monoculture and coculture conditions and displayed a more pronounced reduction in proliferation rate when treated with capmatinib. Although these experiments demonstrated that 2-D coculture promotes more biologically faithful activation of the HGF-MET signaling pathway than monoculture with serum, the relatively weak impact of MET inhibition on Caki2 cells limits the utility of this coculture paradigm.
A 3-D Coculture Model More Faithfully Mimics Pathway Engagement and Biological Function
One of the more significant means to functionally distinguish between transformed pRCC cells and immortalized HK2 is by growing these cells in nonadherent spheroid conditions. In this context, HK2 cells fail to adhere to one another and rapidly lose viability, similar to what we observed after 24 h in the fibroblast coculture model. In contrast, both Caki2 and SKRC39 cells are able to survive and proliferate (22). This observation prompted us to ask whether a 3-D spheroid growth model could be adapted to serum-free coculture conditions between Wi-38 fibroblasts and the renal lines.
To address this question, we turned to a commercially available magnetic bioprinting system as a means to produce structured, 3-D spheroid cocultures (24). In this system, both Wi-38 fibroblasts and renal cells growing in standard 2-D culture were magnetized by endocytic uptake of magnetic nanoparticles, which allow the detached cells to be aggregated in a controlled fashion using a magnetic rack (Fig. 6A). Due to the relative paucity of known methods for coculture of renal cells, we tried several different approaches and cell ratios to generate spheroids with this method. We found that simply mixing the cells together in serum-free media and then plating them led to poor adhesion, which resulted in the spheroids disaggregating after removal from the magnetic rack. In contrast, when an initial spheroid of Wi-38 cells was made and then coated with a second layer of renal cells, stable spheroids could be formed that would remain intact for up to a week depending on the number of initial cells seed to each spheroid (Fig. 6B). The limiting factor in this case is nutrient and oxygen diffusion, as larger spheroids tended to lose viability more rapidly than small spheroids.
Figure 6.
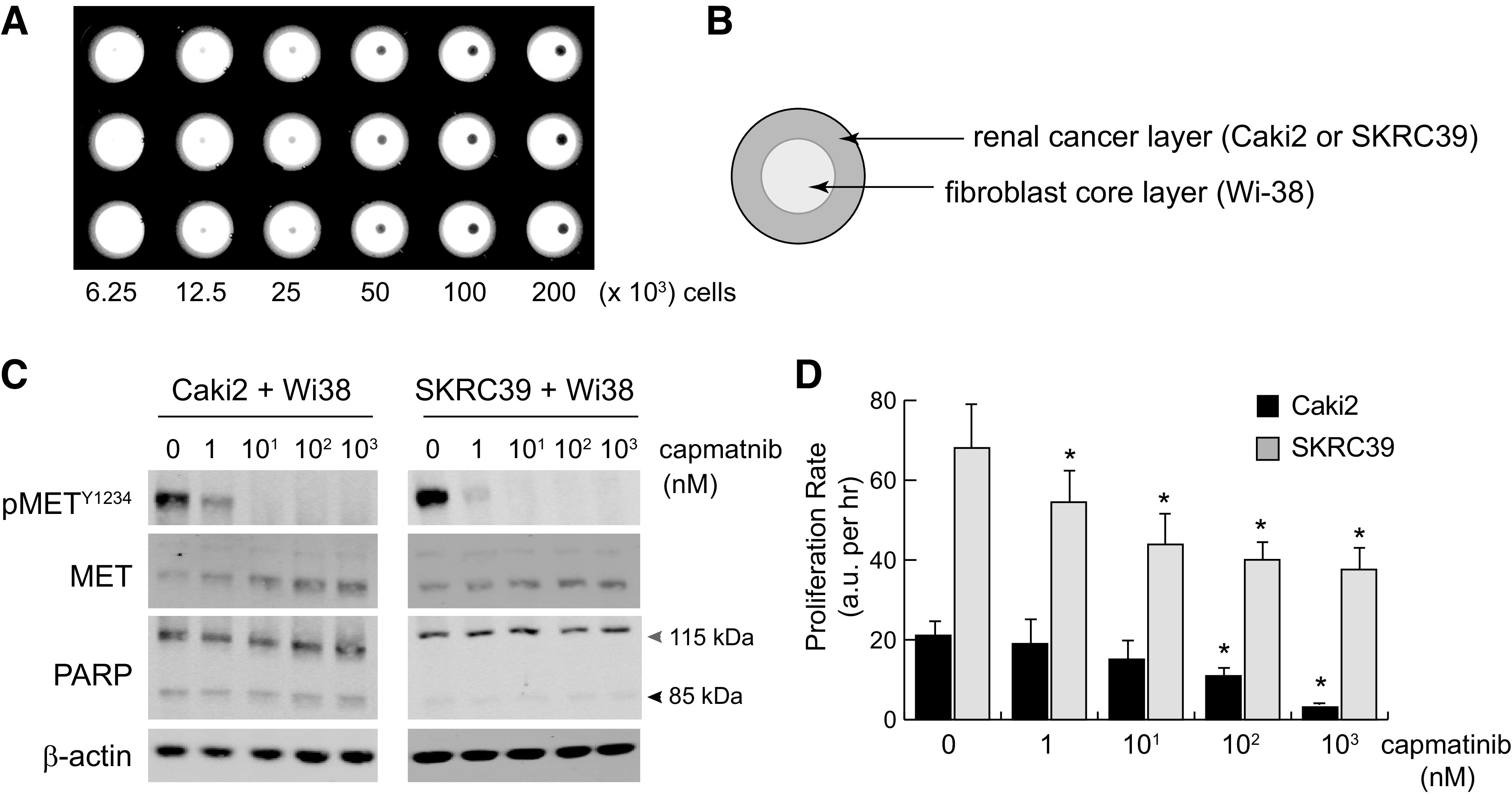
Three-dimensional tumor spheroid coculture reproduces paracrine hepatocyte growth factor (HGF)/MET signaling and is sensitive to pharmacological MET inhibition. Three-dimensional spheroids were created by incubating magnetized cells in nonadherent 96-well plates placed above a magnet. Plates were removed from the magnet after 3 h to allow for normal spheroid compaction resulting from cell-cell adhesion. A: low-magnification image of spheroids plated at a twofold serial density of 200,000–6,250 cells/well. B: to create coculture spheroids, Wi-38 fibroblasts were used to form the inner core layer, which was then coated with an outer layer of cancer cells 3 h later. Cells were cultured in serum-free media with heparin to promote HGF production and paracrine signaling. C: immunoblots from single spheroid lysates at 24 h postplating with the indicated nanomolar concentrations of capmatinib. Poly(ADP-ribose) polymerase (PARP) cleavage from 115 to 85 kDa was included as an indicator of cell death; actin was included as a control for protein loading. D: the proliferation rate of cocultured spheroids treated with the indicated concentration of capmatinib was measured using a Cell Titer Glo three-dimensional luminescence assay. Luminescent values were captured from three individual spheroids for each condition at regular intervals for 72–96 h and fit to linear equations to obtain growth rates with values of arbitrary luminescent units (a.u.) per hour. The significance of growth rate difference for each condition was determined relative to the same line grown in vehicle only (0 nM) (*P < 0.05).
To evaluate HGF-MET signaling and responsiveness to inhibition, we performed immunoblots on entire spheroids composed of 50,000 pRCC cells seeded around 10,000 Wi-38 cells after 72 h of culture with various concentrations of capmatinib (Fig. 6C). As with the 2-D coculture model, we observed strong basal activation of MET that appeared to be specific to renal cells and was absent from spheroids composed of Wi-38 fibroblasts only. MET phosphorylation was effectively inhibited by drug concentrations as low as 10 nM, indicating that capmatinib can effectively diffuse into the pRCC layer of spheroids. Also consistent with the previous model, we saw little evidence for cytotoxicity in response to MET inhibition in either cell line. Although Caki2 cells did show modest levels of PARP cleavage indicative of cell death, there was no increase in this effect even at the highest concentration of drug (Fig. 6C).
As a final validation of this model, we again evaluated the proliferation of cells in spheroids using a luminescent readout of relative cell numbers. To minimize issues of nutrient diffusion that could limit the duration of this assay, we seeded smaller spheroids composed of 10,000 pRCC cells seeded around 2,000 Wi-38 cells. Measurements were taken from lysed spheroids at 24-h increments after seeding and used to plot their growth rate over 96 h (Fig. 6D and Supplemental Fig. S4). As before, Caki2 cells proliferated at a significantly lower rate than SKRC39 cells, although both lines maintained a positive slope over the duration of the experiment. Importantly, we observed that both lines responded to capmatinib in a dose-responsive fashion that better parallels target engagement at the molecular level and recapitulates the published in vivo data regarding MET inhibition in pRCC (Fig. 6, C and D). The efficacy of capmatinib was notably higher for Caki2 cells in this model, which is consistent with their larger dependence on the HGF/MET signaling axis compared with SKRC39 cells. These data suggest that a 3-D, serum-free coculture model using fibroblasts with renal cells produces the most faithful in vitro model of pRCC to date.
DISCUSSION
It has been realized for some time that most cancers, and in particular solid tumors, cannot be accurately understood in isolation from the microenvironment from which they emerge (36). Each microenvironment in the body contains a multiplicity of cell types, a unique extracellular matrix, and a distinctive cohort of signaling ligands that collectively determine how cancer develops in that specific location. During the course of cancer progression, the microenvironment undergoes continuous change in which cancer cells shape local conditions to their favor and respond to both metabolic and immunologic challenges from the broader systemic environment (37). This remarkable complexity is incompletely understood and, as such, is often neglected in standard in vitro studies of cancer. This fact plays at least some part in the translational failure of many therapies that showed significant promise in the early stages of discovery.
In this study, we identified two key limitations to the standard model of cellular monoculture in the context of pRCC. The first of these limitations is the fact that growth of tumor cells with exogenous serum as a source of growth factors fails to replicate the typical reliance of type 1 pRCC on the HGF-MET axis. None of the three inhibitory methods we used to disrupt HGF-MET signaling had a significant impact on tumor cell survival or proliferation when FBS was present, which indicates that this rather general mitogenic stimulus diverges significantly from the renal microenvironment. We have previously observed that other receptor tyrosine kinases, most notably the epithelial growth factor receptor and insulin-like growth factor 1 receptor, display higher levels of phosphorylation when MET is downregulated in pRCC cells, suggesting that serum provides other forms of trophic stimuli in addition to HGF (20). Although it could be argued that simply replacing FBS with recombinant human HGF would itself improve the in vitro model, this approach neglects the value of continuous production and presentation of the ligand at physiological levels when fibroblasts are used as a feeder layer for pRCC cells. Furthermore, it also assumes that HGF production is the only reason for the presence of fibroblasts within tumors.
The second limitation of standard 2-D monoculture models is the nonphysiological attachment surface on which adherent cells grow. Given the specific architecture of papillary tumors, it is likely that much more than a simple ligand-receptor interaction is involved in the communication between tumor cells and fibroblasts. This notion is reinforced by the fact that while type 2 pRCC tumors continue to display papillary histology, they do not depend on the HGF/MET axis for initiation or growth (6, 11). This limitation is also addressed by 3-D coculture of tumor cells with fibroblasts. The model we used here requires physical cell-cell contacts to maintain continuous viability and functional cross talk between pRCC cells and fibroblasts. The precise nature of these interactions is unclear at this point, although differential expression of cadherins has been described in the etiology of pRCC and in heterotypic interactions between cancer cells and fibroblasts (38–40). Our model should prove useful for further studies of these interactions in both type 1 and type 2 pRCC.
Another particular strength of the model described in this study is its more faithful topological arrangement of tumor cells relative to fibroblasts. In contrast to prior work with pancreatic cancer cells, we failed to produce cohesive spheroids by simply mixing fibroblasts with pRCC cells and magnetically printing them simultaneously (41). Instead, we found that consecutive printing of fibroblasts and tumor cells in concentric layers produced spheroids that compacted appropriately over the first 24 h and then remained metabolically viable for out to 96 h (22). In retrospect, this finding is perhaps unsurprising because coordinated layering of pRCC cells on top of fibroblasts more closely mimics the papillary architecture of human tumors than a random mosaic of both cell types. With further optimization, it may be possible to extend the duration of time over which these spheroids remain viable, which would allow for in vitro experimentation with a wider array of patient-derived pRCC cells and renal fibroblasts that better reflect the cell types that compose human tumors. Our 3-D spheroid model also awaits genetic confirmation using stable knockdown of MET, which cannot be achieved with the siRNA approach we used here.
A final point of interest regarding this model involves our observation that normal human proximal tubule cells (HK2 cell line) are unable to survive in either type of coculture model when adherence to Wi-38 fibroblasts is required. The capacity of coculture to distinguish between normal and cancerous renal cells makes this a powerful approach for defining the repertoire of mutations required to transform a normal renal epithelial cell into the various types of cancer found in the kidney. Although it is clear that MET amplification is part of this process in type 1 pRCC, for instance, simple overexpression of MET in renal cells is insufficient to mediate transformation. Recent publication of large-scale genomic studies of human pRCC provide the raw data required to generate hypotheses regarding the combinations of genes involved in cancer genesis and progression (6, 11). Use of 3-D coculture in conjunction with genomic alteration tools such as CRISPR/cas9 provides an unbiased approach to rapidly screen and identify mutational combinations that cooperate with known oncogenes such as MET to fully transform kidney cells.
SUPPLEMENTAL DATA
Supplemental Table S1 and Supplemental Figs. S1–S4: https://doi.org/10.6084/m9.figshare.14556078.v1.
GRANTS
This work was supported by National Cancer Institute Grant 1R15CA192094 and by Calvin University with support from the Calvin Research Fellowship program.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.V. and B.D.L. conceived and designed research; K.A.R., S.M.L., C.V., and B.D.L. performed experiments; K.A.R., S.M.L., C.V., and B.D.L. analyzed data; K.A.R., S.M.L., C.V., and B.D.L. interpreted results of experiments; B.D.L. prepared figures; B.D.L. drafted manuscript; B.D.L. edited and revised manuscript; K.A.R., C.V., and B.D.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Bin Tean Teh (Cancer Science Institute, National University of Singapore) for the provision of the Caki2 and SKRC39 cell lines used in this study.
Present addresses: K. A. Rosette, Pacific University, Physician Assistant Studies, 190 SE 8th Avenue, Hillsboro, OR 97123; S. M. Lander, Feinberg School of Medicine, Northwestern University, 420 E Superior St, Chicago, IL 60611; C. VanOpstall, Covance CLS, 8211 Scicor Dr, Indianapolis, IN 46214.
REFERENCES
- 1.American Cancer Society. Key Statistics About Kidney Cancer? [Online]. https://www.cancer.org/cancer/kidney-cancer/about/key-statistics.html [7 June 2021]. [Google Scholar]
- 2.Allory Y, Bazille C, Vieillefond A, Molinie V, Cochand-Priollet B, Cussenot O, Sibony M. Profiling and classification tree applied to renal epithelial tumours. Histopathology 52: 158–166, 2008. doi: 10.1111/j.1365-2559.2007.02900.x. [DOI] [PubMed] [Google Scholar]
- 3.Shuch B, Amin A, Armstrong AJ, Eble JN, Ficarra V, Lopez-Beltran A, Martignoni G, Rini BI, Kutikov A. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. Eur Urol 67: 85–97, 2015. doi: 10.1016/j.eururo.2014.04.029. [DOI] [PubMed] [Google Scholar]
- 4.Truong LD, Shen SS. Immunohistochemical diagnosis of renal neoplasms. Arch Pathol Lab Med 135: 92–109, 2011. doi: 10.5858/2010-0478-RAR.1. [DOI] [PubMed] [Google Scholar]
- 5.Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol 15: 366–381, 2018. doi: 10.1038/s41571-018-0007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, Davis C, et al.; Cancer Genome Atlas Research Network. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med 374: 135–145, 2016. doi: 10.1056/NEJMoa1505917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furge KA, Dykema K, Petillo D, Westphal M, Zhang Z, Kort EJ, Teh BT. Combining differential expression, chromosomal and pathway analyses for the molecular characterization of renal cell carcinoma. Can Urol Assoc J 1: S21–S27, 2007. doi: 10.5489/cuaj.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang XJ, Sugimura J, Schafernak KT, Tretiakova MS, Han M, Vogelzang NJ, Furge K, Teh BT. Classification of renal neoplasms based on molecular signatures. J Urol 175: 2302–2306, 2006. doi: 10.1016/S0022-5347(06)00255-2. [DOI] [PubMed] [Google Scholar]
- 9.Yang XJ, Tan M-H, Kim HL, Ditlev JA, Betten MW, Png CE, Kort EJ, Futami K, Furge KA, Takahashi M, Kanayama H-O, Tan PH, Teh BS, Luan C, Wang K, Pins M, Tretiakova M, Anema J, Kahnoski R, Nicol T, Stadler W, Vogelzang NG, Amato R, Seligson D, Figlin R, Belldegrun A, Rogers CG, Teh BT. A molecular classification of papillary renal cell carcinoma. Cancer Res 65: 5628–5637, 2005. doi: 10.1158/0008-5472.CAN-05-0533. [DOI] [PubMed] [Google Scholar]
- 10.Furge KA, Lucas KA, Takahashi M, Sugimura J, Kort EJ, Kanayama H, Kagawa S, Hoekstra P, Curry J, Yang XJ, Teh BT. Robust classification of renal cell carcinoma based on gene expression data and predicted cytogenetic profiles. Cancer Res 64: 4117–4121, 2004. doi: 10.1158/0008-5472.CAN-04-0534. [DOI] [PubMed] [Google Scholar]
- 11.Durinck S, Stawiski EW, Pavía-Jiménez A, Modrusan Z, Kapur P, Jaiswal BS, et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet 47: 13–21, 2015. doi: 10.1038/ng.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet 16: 68–73, 1997. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura T, Mizuno S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc Jpn Acad Ser B Phys Biol Sci 86: 588–610, 2010. doi: 10.2183/pjab.86.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagaike M, Hirao S, Tajima H, Noji S, Taniguchi S, Matsumoto K, Nakamura T. Renotropic functions of hepatocyte growth factor in renal regeneration after unilateral nephrectomy. J Biol Chem 266: 22781–22784, 1991. [PubMed] [Google Scholar]
- 15.Zhou D, Tan RJ, Lin L, Zhou L, Liu Y. Activation of hepatocyte growth factor receptor, c-met, in renal tubules is required for renoprotection after acute kidney injury. Kidney Int 84: 509–520, 2013. doi: 10.1038/ki.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boor P, Floege J. The renal (myo-)fibroblast: a heterogeneous group of cells. Nephrol Dial Transplant 27: 3027–3036, 2012. doi: 10.1093/ndt/gfs296. [DOI] [PubMed] [Google Scholar]
- 17.Farris AB, Colvin RB. Renal interstitial fibrosis: mechanisms and evaluation. Curr Opin Nephrol Hypertens 21: 289–300, 2012. doi: 10.1097/MNH.0b013e3283521cfa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan RJ, Zhou D, Liu Y. Signaling crosstalk between tubular epithelial cells and interstitial fibroblasts after kidney injury. Kidney Dis (Basel) 2: 136–144, 2016. doi: 10.1159/000446336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gui Y, Lu Q, Gu M, Wang M, Liang Y, Zhu X, Xue X, Sun X, He W, Yang J, Zhao AZ, Xiao B, Dai C. Fibroblast mTOR/PPARγ/HGF axis protects against tubular cell death and acute kidney injury. Cell Death Differ 26: 2774–2789, 2019. doi: 10.1038/s41418-019-0336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Looyenga BD, Furge KA, Dykema KJ, Koeman J, Swiatek PJ, Giordano TJ, West AB, Resau JH, Teh BT, MacKeigan JP. Chromosomal amplification of leucine-rich repeat kinase-2 (LRRK2) is required for oncogenic MET signaling in papillary renal and thyroid carcinomas. Proc Natl Acad Sci USA 108: 1439–1444, 2011. doi: 10.1073/pnas.1012500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci USA 105: 2415–2420, 2008. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tseng H, Gage JA, Shen T, Haisler WL, Neeley SK, Shiao S, Chen J, Desai PK, Liao A, Hebel C, Raphael RM, Becker JL, Souza GR. A spheroid toxicity assay using magnetic 3D bioprinting and real-time mobile device-based imaging. Sci Rep 5: 13987, 2015. doi: 10.1038/srep13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brodaczewska KK, Szczylik C, Fiedorowicz M, Porta C, Czarnecka AM. Choosing the right cell line for renal cell cancer research. Mol Cancer 15: 83, 2016. doi: 10.1186/s12943-016-0565-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebert T, Bander NH, Finstad CL, Ramsawak RD, Old LJ. Establishment and characterization of human renal cancer and normal kidney cell lines. Cancer Res 50: 5531–5536, 1990. [PubMed] [Google Scholar]
- 25.Furge KA, Chen J, Koeman J, Swiatek P, Dykema K, Lucin K, Kahnoski R, Yang XJ, Teh BT. Detection of DNA copy number changes and oncogenic signaling abnormalities from gene expression data reveals MYC activation in high-grade papillary renal cell carcinoma. Cancer Res 67: 3171–3176, 2007. doi: 10.1158/0008-5472.CAN-06-4571. [DOI] [PubMed] [Google Scholar]
- 26.Fogh J, Fogh JM, Orfeo T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst 59: 221–226, 1977. doi: 10.1093/jnci/59.1.221. [DOI] [PubMed] [Google Scholar]
- 27.Pulkkanen KJ, Parkkinen JJ, Kettunen MI, Kauppinen RA, Lappalainen M, Ala-Opas MY, Yla-Herttuala S. Characterization of a new animal model for human renal cell carcinoma. In Vivo 14: 393–400, 2000. [PubMed] [Google Scholar]
- 28.Ryan MJ, Johnson G, Kirk J, Fuerstenberg SM, Zager RA, Torok-Storb B. HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int 45: 48–57, 1994. doi: 10.1038/ki.1994.6. [DOI] [PubMed] [Google Scholar]
- 29.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, et al. Next-generation characterization of the cancer cell line encyclopedia. Nature 569: 503–508, 2019. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X, Wang Q, Yang G, Marando C, Koblish HK, Hall LM, Fridman JS, Behshad E, Wynn R, Li Y, Boer J, Diamond S, He C, Xu M, Zhuo J, Yao W, Newton RC, Scherle PA. A novel kinase inhibitor, INCB28060, blocks c-MET-dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin Cancer Res 17: 7127–7138, 2011. doi: 10.1158/1078-0432.CCR-11-1157. [DOI] [PubMed] [Google Scholar]
- 31.Schuller AG, Barry ER, Jones RDO, Henry RE, Frigault MM, Beran G, Linsenmayer D, Hattersley M, Smith A, Wilson J, Cairo S, Déas O, Nicolle D, Adam A, Zinda M, Reimer C, Fawell SE, Clark EA, D’Cruz CM. The MET inhibitor AZD6094 (savolitinib, HMPL-504) induces regression in papillary renal cell carcinoma patient-derived xenograft models. Clin Cancer Res 21: 2811–2819, 2015. doi: 10.1158/1078-0432.CCR-14-2685. [DOI] [PubMed] [Google Scholar]
- 32.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 25: 585–621, 1961. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 33.Matsumoto K, Tajima H, Okazaki H, Nakamura T. Heparin as an inducer of hepatocyte growth factor. J Biochem 114: 820–826, 1993. doi: 10.1093/oxfordjournals.jbchem.a124262. [DOI] [PubMed] [Google Scholar]
- 34.Sakiyama R, Fukuta K, Matsumoto K, Furukawa M, Takahashi Y, Nakamura T. Stimulation of hepatocyte growth factor production by heparin-derived oligosaccharides. J Biochem 141: 653–660, 2007. doi: 10.1093/jb/mvm067. [DOI] [PubMed] [Google Scholar]
- 35.Peterfi L, Yusenko MV, Kovacs G. IL6 shapes an inflammatory microenvironment and triggers the development of unique types of cancer in end-stage kidney. Anticancer Res 39: 1869–1874, 2019. doi: 10.21873/anticanres.13294. [DOI] [PubMed] [Google Scholar]
- 36.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 144: 646–674, 2011. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 37.McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship. J Clin Oncol 28: 4022–4028, 2010. doi: 10.1200/JCO.2010.28.4257. [DOI] [PubMed] [Google Scholar]
- 38.Behnes CL, Hemmerlein B, Strauss A, Radzun H-J, Bremmer F. N-cadherin is differentially expressed in histological subtypes of papillary renal cell carcinoma. Diagn Pathol 7: 95, 2012. doi: 10.1186/1746-1596-7-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li G, Satyamoorthy K, Herlyn M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res 61: 3819–3825, 2001. [PubMed] [Google Scholar]
- 40.Omelchenko T, Fetisova E, Ivanova O, Bonder EM, Feder H, Vasiliev JM, Gelfand IM. Contact interactions between epitheliocytes and fibroblasts: formation of heterotypic cadherin-containing adhesion sites is accompanied by local cytoskeletal reorganization. Proc Natl Acad Sci USA 98: 8632–8637, 2001. doi: 10.1073/pnas.151247698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noel P, Muñoz R, Rogers GW, Neilson A, Von Hoff DD, Han H. Preparation and metabolic assay of 3-dimensional spheroid co-cultures of pancreatic cancer cells and fibroblasts. J Vis Exp 2017: 56081, 2017. doi: 10.3791/56081. [DOI] [PMC free article] [PubMed] [Google Scholar]