

Abstract
Adipose tissue homeostasis plays a central role in cardiovascular physiology, and the presence of thermogenically active brown adipose tissue (BAT) has recently been associated with cardiometabolic health. We have previously shown that adipose tissue-specific deletion of HuR (Adipo-HuR−/−) reduces BAT-mediated adaptive thermogenesis, and the goal of this work was to identify the cardiovascular impacts of Adipo-HuR−/−. We found that Adipo-HuR−/− mice exhibit a hypercontractile phenotype that is accompanied by increased left ventricle wall thickness and hypertrophic gene expression. Furthermore, hearts from Adipo-HuR−/− mice display increased fibrosis via picrosirius red staining and periostin expression. To identify underlying mechanisms, we applied both RNA-seq and weighted gene coexpression network analysis (WGCNA) across both cardiac and adipose tissue to define HuR-dependent changes in gene expression as well as significant relationships between adipose tissue gene expression and cardiac fibrosis. RNA-seq results demonstrated a significant increase in proinflammatory gene expression in both cardiac and subcutaneous white adipose tissue (scWAT) from Adipo-HuR−/− mice that is accompanied by an increase in serum levels of both TNF-α and IL-6. In addition to inflammation-related genes, WGCNA identified a significant enrichment in extracellular vesicle-mediated transport and exosome-associated genes in scWAT, whose expression most significantly associated with the degree of cardiac fibrosis observed in Adipo-HuR−/− mice, implicating these processes as a likely adipose-to-cardiac paracrine mechanism. These results are significant in that they demonstrate the spontaneous onset of cardiovascular pathology in an adipose tissue-specific gene deletion model and contribute to our understanding of how disruptions in adipose tissue homeostasis may mediate cardiovascular disease.
NEW & NOTEWORTHY The presence of functional brown adipose tissue in humans is known to be associated with cardiovascular health. Here, we show that adipocyte-specific deletion of the RNA binding protein HuR, which we have previously shown to reduce BAT-mediated thermogenesis, is sufficient to mediate a spontaneous development of cardiac hypertrophy and fibrosis. These results may have implications on the mechanisms by which BAT function and adipose tissue homeostasis directly mediate cardiovascular disease.
Keywords: adipose tissue, cardiovascular disease, cardiac hypertrophy, fibrosis, HuR
INTRODUCTION
Adipose tissue has long been linked to cardiovascular disease (CVD) through its central role in obesity, but work over the past two decades has since established a clear endocrine role for adipose tissue that is dependent on both adipocyte composition (e.g., white vs. brown fat) as well as anatomical location (e.g., visceral vs. subcutaneous) (1). When energy intake exceeds expenditure, excess caloric energy is stored in white adipose tissue (WAT) as triglycerides. In contrast, beige and brown adipose tissue (BAT) drive energy expenditure through uncoupled thermogenic metabolism in their conserved evolutionary role in thermoregulation. Specifically, accumulation of WAT tends to be more proinflammatory in nature, especially under conditions of chronic energy surplus, and exerts deleterious effects on cardiovascular health (2–4).
On the other hand, the presence of metabolically active BAT has been demonstrated to positively correlate with myocardial health (1, 5–7). Functional BAT, previously only believed to be in infants and children, was recently discovered in adult humans (8, 9) and is considered a potential therapeutic target in obesity due to its high metabolic potential, which is capable of accounting for up to 20% of the total whole body energy expenditure (10–12). A recent retrospective analysis of over 50,000 patients found that the presence of functional BAT independently correlated with decreased hypertension, coronary artery disease, and congestive heart failure (13), demonstrating a clear and direct role for BAT in cardiovascular health.
Human antigen R (HuR) is a widely-expressed RNA binding protein that interacts with specific AU-rich domains in target mRNAs and directly regulates expression by modulating mRNA stability and/or translation (14). We have previously demonstrated a direct role for HuR expression in cardiomyocytes during the progression of pathological cardiac hypertophy (15, 16). Additional recently published work was among the first to show the in vivo role of HuR on adipocyte physiology, specifically showing that adipocyte-specific deletion of HuR in mice (Adipo-HuR−/− mice) resulted in a reduction in both WAT and BAT mass that was accompanied by a profound deficit in BAT-mediated thermogenesis when subjected to acute cold challenge (17). Li et al. (18) recently showed an inverse correlation between HuR expression in subcutaneous WAT (scWAT) and obesity in human patients, suggesting a clinical link for HuR expression in metabolic syndrome and raising the question as to whether loss of HuR expression in adipose tissue may play a functional role in obesity-associated comorbidities such as CVD. In light of the growing interest in the adipose tissue-to-myocardium signaling axis, the goal of this work was to explore HuR-mediated cross talk between adipose tissue and the myocardium.
Here, we demonstrate that adipocyte-specific deletion of HuR in mice mediates the spontaneous development of left ventricular cardiac hypertrophy and fibrosis that appears to be mechanistically driven by an increase in inflammatory gene expression in subcutaneous white adipose tissue.
MATERIALS AND METHODS
Animal Models
All mouse studies were approved by the University of Cincinnati Institutional Animal Care and Use Committee (IACUC). HuR-floxed mice were described by Ghosh et al. (19) and obtained from Jackson Labs (Stock No. 021431). To generate an adipose-specific HuR deletion model (Adipo-HuR−/−), HuR-floxed mice were crossed with mice harboring an adiponectin-specific Cre recombinase transgene (AdipoQ-Cre) (Jackson Labs, Stock No. 010803). Nonfloxed Adipo-Cre+ littermates were used as controls. Mice were housed in the University of Cincinnati vivarium at 23°C. All data presented are from 15- to 25-wk-old mice.
Echocardiography
All echocardiographic studies were performed as previously described (15). Briefly, mice were sedated with isoflurane and body temperature was maintained at 37°C during imaging. Using a Vevo 2100, parasternal images were obtained in long axis in two-dimensional mode and motion (M)-mode for quantification. These were then analyzed using VevoStrain software (Vevo 2100, v1.1.1 B1455, Visualsonic, Toronto, ON, Canada).
LV Pressure Catheterization
As a terminal procedure, mice were weighed and anesthetized using inhaled isoflurane. Terminal development of LV pressure was determined using a 1.2-French catheter pressure transducer (Scisense FTH-1211B-0018) advanced through the carotid into the LV as previously described (2, 20).
Tissue and Plasma Processing
Mice were sedated with isoflurane and subsequently euthanized via thoracotomy. Blood was collected into syringes containing 3.2% sodium citrate, and plasma was isolated via centrifugation at 4,000 g at room temperature and flash frozen in liquid nitrogen. Tissues were removed, weighed, and flash frozen in liquid nitrogen for further analysis. Plasma catecholamine (epinephrine, norepinephrine; Abnova KA1877), IL-6 (Bioss BSKM1004), and TNF-α (Bioss BSKM1002) levels were assessed using ELISA kits according to the manufacturers’ instructions.
Histological Analysis
Frozen hearts were embedded in OCT, cut in 10-µM thick sections, and stained using Picrosirius red (Polysciences, Inc.) as previously described (21). To determine the presence of neutral lipids, tissues were stained with oil red O (Sigma Aldrich, 0.3% solution) and counterstained with hematoxylin and eosin.
RNA Isolation and qPCR
RNA was isolated using a Macherey-Nagel NucleoSpin RNA kit and cDNA was synthesized using iScript RT Supermix (Bio-Rad). Samples were run on a Bio-Rad CFX96 Touch to assess mRNA levels of BNP, RCAN, and 18S. Primers are as listed: BNP, F: 5′- AAGTCCTAGCCAGTCTCCAGA-3′, R: 5′- GAGCTGTCTCTGGGCCATTTC-3′; RCAN, F: 5′- GGGCCAAATTTGAATCCCTCTTC-3′, R: 5′- GGAGCCAGGTGTGAACTTCC-3′; 18S, F: 5′- AGTCCCTGCCCTTTGTACACA-3′, and R: 5′- CCGAGGGCCTCACTAAACC-3′.
Protein Isolation and Western Blotting
Total protein was isolated from crushed tissue in RIPA buffer with 0.5 mM DTT, 0.2 mM sodium-orthovanadate, and protease inhibitor (Halt Protease and Phosphatase Inhibitor Cocktail; ThermoFisher). Then, 25 µg of protein extract per lane was separated on a 10% polyacrylamide gel and transferred to a nitrocellulose membrane. Blocking was performed for 1 h at room temperature using 5% BSA milk in 0.1% Tween 20, tris-buffered saline (T-TBS). Primary antibodies for Periostin (Novus Biologicals NBP1-30042), PERK (Cell Signaling, 3192), and GAPDH (Santa Cruz Biotechnology sc25778) were incubated overnight at 4°C, and secondary antibodies were incubated for 1–2 h at room temperature in T-TBS. Images were captured and analyzed using a ChemiDoc Imaging System and ImageLab software (BioRad).
RNA Sequencing and Weighted Gene Coexpression Network Analysis
RNA was isolated from cardiac and adipose tissue as previously described (16, 17) and RNA sequencing was done by the Cincinnati Children’s Hospital Medical Center (CCHMC) DNA Sequencing and Genotyping Core. Sequence read mapping, principal component analysis, and differential expression analysis were done using CLC Genomics Workbench (v20.0.2, Qiagen) as previously described (15, 17). Gene ontology analysis was done using the NIH DAVID Bioinformatics Functional Annotation Tool with an EASE score/P value threshold of 0.05 as previously described (22–24). Significant differential expression between groups was defined as an absolute fold change of greater than or equal to 1.5 and a false discovery rate adjusted (FDR) P value of less than or equal to 0.05. For WGCNA, coexpression network construction, module detection, association of expression modules with phenotype data, and identification of hub genes within expression modules were done as previously described (25, 26). Hub genes for each module were defined as the top 10 genes with the highest module membership. All RNA-seq expression data are available in the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) (GSE174193).
Statistical Analysis
All data (other than RNA-seq and WGCNA) were analyzed in a blinded fashion and are presented as means ± SE, unless otherwise noted. Statistical comparisons between two population means were done using Student’s t test in GraphPad Prism (v. 9.1.0).
RESULTS
Cardiac Function in Adipo-HuR−/− Mice
Examination of cardiac function in Adipo-HuR−/− and wild-type littermate control mice by echocardiography showed an increase in left ventricular (LV) ejection fraction and fractional shortening (Fig. 1, A and B) in hearts from Adipo-HuR−/− mice compared with littermate controls. Despite no increase in LV mass relative to body mass in Adipo-HuR−/− mice (Supplemental Fig. S1; see https://doi.org/10.6084/m9.figshare.14567733), our results showed decreased LV chamber volume (Fig. 1, C and D), and increased LV septal and posterior wall thickness (Fig. 1, E and F) relative to total LV mass. Lack of a significant change in LV mass indexed to body weight is unsurprising in this model as we have previously shown Adipo-HuR−/− mice to have decreased body weight (17).
Figure 1.

Functional assessment of cardiac function in hearts from mice with an adipocyte-specific deletion of HuR. Echocardiographic analysis shows that hearts from Adipo-HuR−/− mice have increased % ejection fraction (A) and fractional shortening (B), with decreased LV chamber volume (C and D), and increased LV septal and posterior wall thickness (E and F). LV pressure catheterization shows an exacerbation in the rates of positive and negative developed pressure (G and H). n = 21 control and 18 mice/group for control and Adipo-HuR−/− mice, respectively, in (A–F). n = 4 control and 5 mice/group for control and Adipo-HuR−/− mice, respectively for (G and H). *P ≤ 0.05. HuR, human antigen R.
Invasive hemodynamic assessment via LV pressure catheterization also showed an increased rate of positive and negative pressure generation in Adipo-HuR−/− hearts (Fig. 1, G and H). See Table 1 for all echocardiography and hemodynamic data. Together, these data are indicative of a state of hypercontractile cardiac hypertrophy in Adipo-HuR−/− mice.
Table 1.
All unnormalized echocardiography and LV pressure catheterization data
| Control | Adipo-HuR−/− | P Value | |
|---|---|---|---|
| Echocardiography result | |||
| Heart rate, beats/min | 492 ± 9 | 479 ± 8 | 0.2840 |
| LV end-systolic diameter, mm | 3.19 ± .076 | 2.77 ± .072 | 0.0003 |
| LV end-diastolic diameter, mm | 4.28 ± .077 | 3.87 ± .073 | 0.0004 |
| LV end-systolic volume, mm | 41.19 ± 2.14 | 29.35 ± 1.88 | 0.0002 |
| LV end-diastolic volume, mm | 82.95 ± 3.44 | 65.32 ± 2.94 | 0.0004 |
| Stroke volume, µL | 41.8 ± 1.94 | 35.9 ± 1.35 | 0.0210 |
| Ejection fraction, % | 50.5 ± 1.42 | 55.3 ± 1.37 | 0.0200 |
| Fractional shortening, % | 25.6 ± 0.89 | 28.4 ± 0.89 | 0.0315 |
| Cardiac output, ml/min | 17.2 ± 0.85 | 20.8 ± 1.09 | 0.0146 |
| Intraventricular septal wall thickness, mm | 0.907 ± 0.06 | 0.933 ± 0.06 | 0.7604 |
| LV posterior wall thickness, mm | 0.853 ± 0.04 | 0.806 ± 0.05 | 0.4771 |
| LV mass, mg | 118.9 ± 9.6 | 100.9 ± 6 | 0.2007 |
| LV catheterization hemodynamic result | |||
| Femoral blood pressure, mmHg | 74.4 ± 3.2 | 75.5 ± 2.9 | 0.8093 |
| Mean LV pressure, mmHg | 103.5 ± 4.1 | 105.5 ± 6 | 0.8013 |
| +dP/dt, mmHg/s | 9,314 ± 366 | 11,367 ± 722 | 0.0522 |
| −dP/dt, mmHg/s | −9,017 ± 469 | −11,343 ± 636 | 0.0264 |
HuR, human antigen R; LV, left ventricular.
Molecular Assessment of Cardiac Hypertrophy and Fibrosis
qPCR assessment of hypertrophic marker gene expression showed increased expression of both BNP and RCAN in hearts from Adipo-HuR−/− mice (Fig. 2, A and B). Assessment of cardiac fibrosis via picrosirius red collagen staining found increased interstitial fibrosis throughout the myocardium of Adipo-HuR−/− mice (Fig. 2, C and D). This was accompanied by a significant increase in protein expression of periostin, a marker for active myofibroblast ECM remodeling, and the ER-stress marker PERK (Fig. 2, E–G). Thus, molecular signaling within the myocardium was found to be consistent with pathological hypertrophy.
Figure 2.

Molecular assessment of cardiac hypertrophy and fibrosis. Gene expression of the hypertrophic markers BNP and RCAN show increased expression in Adipo-HuR−/− mice (A and B). Hearts from Adipo-HuR−/− mice also display increased fibrosis compared with controls as shown by representative picrosirius red staining (C and D). Western blotting shows a significant increase in PERK and periostin protein expression in Adipo-HuR−/− hearts (F and G). n ≥ 11 per group for (A–B), ≥6 per group for (C and D), and ≥4 per group for (E and F). *P ≤ 0.05. BNP, brain natriuretic peptide; HuR, human antigen R; PERK, protein kinase R-like endoplasmic reticulum kinase; RCAN, residual channel attention network.
Interestingly, we did not observe the development of cardiac hypertrophy or fibrosis in the hearts of female Adipo-HuR−/− mice. Echocardiography showed no difference in cardiac function between Adipo-HuR−/− and controls (Supplemental Fig. S2A; see https://doi.org/10.6084/m9.figshare.14573274). Similarly, we observed no difference in hypertrophic gene expression (Supplemental Fig. S2B) or fibrosis via picrosirius red (Supplemental Fig. S2C) or periostin expression (Supplemental Fig. S2D). This result is consistent with other observed sex differences in BAT-mediated cardiac effects (7, 13), as well as existing data in both humans and mice demonstrating protection from cardiovascular disease in premenopausal females (27–29).
Circulating Catecholamine Levels
Adrenergic signaling response to cold activates thermogenic pathways, but also mediates increased cardiac contractility upon acute exposure and pathological cardiac hypertrophy with prolonged exposure (30). Thus, to rule out adrenergic-induced cardiac pathology as a result of cold-intolerance in Adipo-HuR−/− mice, we show that plasma levels of epinephrine and norepinephrine were not increased (Fig. 3, A and B, respectively). This demonstrates that the cardiac hypertrophy and fibrosis observed in Adipo-HuR−/− mice is not driven by increased adrenergic signaling.
Figure 3.

Serum levels of epinephrine (A) and norepinephrine (B) as determined by ELISA. n ≥ 5 per group. **P ≤ 0.01.
Similarly, it should also be noted that we observed no difference in blood pressure (Table 1), mean LV pressure (Table 1), cardiac lipid deposition (Supplemental Fig. S3; see https://doi.org/10.6084/m9.figshare.14573286), or expression of glucose transporters (glut1 and glut4; Supplemental Fig. S4, B and C; see https://doi.org/10.6084/m9.figshare.14568483) or fatty acid binding protein (fabp4; Supplemental Fig. S4D) between control and Adipo-HuR−/− mice.
RNA-Seq Assessment of Cardiac Gene Expression
Transcriptome-wide RNA-sequencing was used to identify the full breadth of cardiac gene expression changes upon HuR deletion in adipose tissue. Principal component analysis showed a clear distinction between cardiac transcriptomes in Adipo-HuR−/− mice and littermate controls (Fig. 4A). We identified a total of 476 significantly dysregulated genes, as defined by a fold change of ≥1.5 and a false discovery rate (FDR)-corrected P value of ≤0.05 (Supplemental Table S1; see https://doi.org/10.6084/m9.figshare.13326524). Of these significant genes, 218 were significantly downregulated and 258 were significantly upregulated relative to expression in control hearts. Gene ontology (GO) analysis identified a significant enrichment in GO terms for inflammatory gene expression among significantly upregulated genes in hearts from Adipo-HuR−/− mice (Fig. 4B and Supplemental Table S2; see https://doi.org/10.6084/m9.figshare.13326659). In addition to an increase in many individual proinflammatory genes, RNA-seq analysis also confirmed a significant increase in profibrotic and hypertrophic genes (Fig. 4C).
Figure 4.
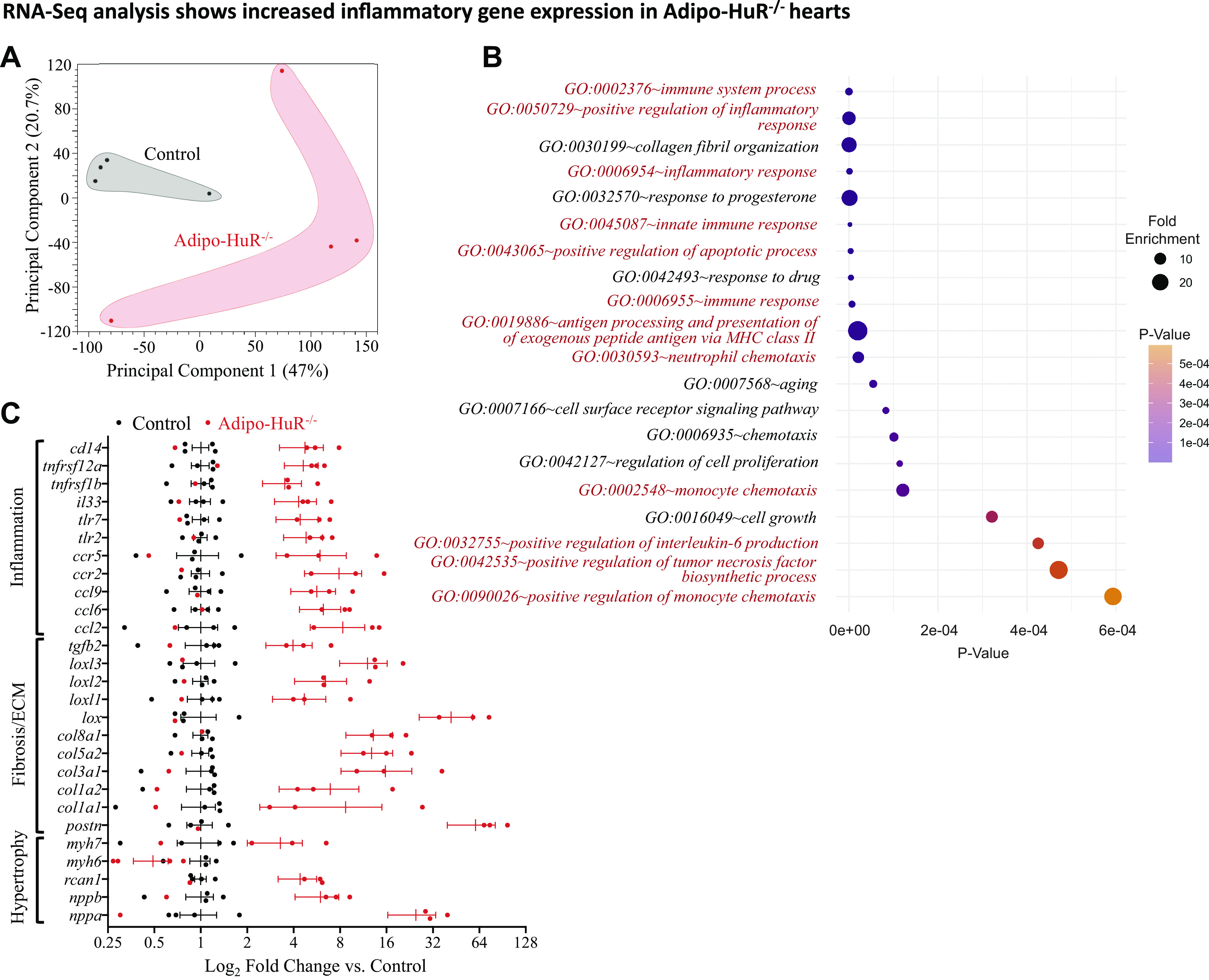
RNA-seq analysis of gene expression in control and Adipo-HuR−/− hearts. Principal component analysis (PCA) plot shows clustering of gene expression patterns from control and Adipo-HuR−/− hearts (A). Gene ontology (GO) analysis shows a significant enrichment in inflammation-associated genes, indicated in red (B). RNA-seq analysis shows a significant increase in hypertrophic, fibrotic, and inflammatory gene expression (C); all genes shown in C are significant as defined by fold change ≥ 1.5 and FDR P ≤ 0.05. n = 4 per group. FDR, false discovery rate; HuR, human antigen R.
RNA-Seq Assessment of Adipose Tissue Gene Expression
We previously reported RNA-seq analysis of HuR-dependent changes in BAT (17), but found nothing that would be obviously indicative of cardiovascular disease. Here, we again applied RNA-seq (with a statistical cutoff of a fold change of ≥1.5 and an FDR P value of ≤0.05) to identify transcriptome-wide HuR-dependent gene expression in scWAT. Results showed that although HuR mediates expression of 588 genes in BAT, 1117 HuR-dependent genes were identified in scWAT (Fig. 5A; see Supplemental Table S3; https://doi.org/10.6084/m9.figshare.13326680) for a list of all significant differentially expressed genes). Interestingly, many of the HuR-dependent gene expression changes in BAT were decreases in expression, but most HuR-dependent changes in scWAT were increases in expression, suggesting differing roles for HuR-mediated gene expression in the two tissues (Fig. 5B).
Figure 5.

RNA-seq analysis of gene expression in control and Adipo-HuR−/− scWAT. Venn diagram of HuR-dependent changes in gene expression changes in BAT and scWAT shows a unique set of HuR-dependent genes in the two adipose depots (A). The majority (381/588; 65%) of the significant HuR-dependent changes in gene expression in BAT are down-regulated, whereas the majority (774/1117; 69%) are up-regulated in scWAT (B). Expression of beige adipocyte marker genes in scWAT (C). Gene ontology analysis showing classification by percent of all enriched HuR-dependent GO terms in scWAT (D) and the top 20 enriched HuR-dependent GO terms among genes with significantly increased expression (E). RNA-seq determined expression change of proinflammatory cytokines in scWAT that have previously been suggested to mediate cardiac pathology through secreted/paracrine signaling (F). n = 4 per group. Serum levels of TNF-α (G) and IL-6 (H) as determined by ELISA. n ≥ 5 per group. *P ≤ 0.05. **P ≤ 0.01. BAT, brown adipose tissue; GO, gene ontology; HuR, human antigen R; scWAT, subcutaneous white adipose tissue.
Among the significant gene expression changes in scWAT, we observed an increase in many prothermogenic genes/beige adipocyte markers (Fig. 5C), which is not surprising given thermogenic deficiency previously reported in Adipo-HuR−/− mice (17). Gene ontology (GO) analysis of HuR-dependent gene expression changes in scWAT showed a significant enrichment in proinflammatory mediators, as ∼33% of all significant GO terms were associated with inflammation/immune processes (Fig. 5D). Many of the top 20 enriched GO terms among HuR-dependent up-regulated genes in scWAT are inflammation-associated GO terms (Fig. 5E; see Supplemental Table S4; https://doi.org/10.6084/m9.figshare.13326692) for a list of all significant GO terms). Among these increased inflammatory gene products are several proinflammatory cytokines which play a detrimental role in cardiac physiology (Fig. 5F) (31–34). Accordingly, we also showed an increase in plasma levels of TNF-α and IL-6 (Fig. 5, G and H, respectively), suggesting that this large increase in inflammatory gene expression in scWAT results in an increase in systemic inflammation.
Association of Adipose Tissue Expression Networks with Cardiac Phenotype
To more specifically determine how changes in gene expression within adipose tissue may underlie specific pathological changes in the heart, we applied weighted gene coexpression network analysis (WGCNA) (Fig. 6A) (25, 26). First, the adipose tissue transcriptomes of both BAT and scWAT were independently clustered into coexpressed gene modules (Supplemental Fig. S5; see https://doi.org/10.6084/m9.figshare.14568621) followed by correlation of each module with cardiac phenotype through a parallel analysis of module-phenotype correlation (Fig. 6B) and average significance to phenotype of all genes within each module (Fig. 6C). The degree of cardiac fibrosis, as quantitatively determined by the percent of total picrosirius red stain (as shown in Fig. 2D), was used as a phenotype marker of CVD for correlation analysis, and significantly associated gene modules were defined as those with significant module-phenotype correlation (R2 ≥ 0.7 and P-value ≤ 0.05; Fig. 6B) as well as high gene significance to fibrosis within the module (average gene significance score ≥ 0.7 and P-value ≤ 0.05; Fig. 6C).
Figure 6.

WGCNA association of scWAT gene expression with cardiac hypertrophy phenotype. Workflow used to identify and associate coexpressed gene modules with phenotype independent of genotype (A). Correlation of each gene expression module with percent cardiac fibrosis was used as a first filter to identify significant modules (B). Average gene signficance relative to percent fibrosis was calculated for each module as a second filter for identification of significant modules (C). Enriched GO terms among genes from the significant fibrosis-associated coexpression modules midnightblue (D) and skyblue (E). GO, gene ontology; scWAT, subcutaneous white adipose tissue; WGCNA, weighted gene coexpression network analysis.
Only one coexpression module within the BAT transcriptome (Steelblue; Fig. 6B; top) was found to have a significant module-phenotype correlation, but failed to meet the gene significance criteria (Fig. 6C; top). However, we identified two significant scWAT coexpression modules (Midnightblue and Skyblue) that passed both criteria (Fig. 6, B and C; bottom). Overall significance of these modules was confirmed through linear correlation demonstrating a strong relationship between individual gene significance to phenotype and module membership (Fig. S5C).
Subsequent gene ontology analysis of individual genes within these two coexpression modules revealed a significant enrichment for gene-associated protein transport and vesicle-mediated transport (Fig. 6, D and E; related GO terms indicated by red font). Furthermore, vesicle-mediated transport (GO Term: 0016192) is the only GO Term that was significantly enriched in both modules, and analysis of enriched GO terms by cellular compartment revealed a significant enrichment in extracellular exosome (GO Term: 0070062) associated genes in both modules, along with additional vesicle transport and Golgi-associated terms (Fig. S5D).
To determine which genes have the highest likelihood of having a causal relationship to cardiac fibrosis within each module, individual hub genes were identified as those that have the highest statistical module membership as previously described (Table 2) (25). Congruent with the GO results from both WGCNA and RNA-seq, the majority of the hub genes for both modules have been previously shown to play a significant role in either exosomal/vesicular mediated transport or inflammation (denoted by bold italicized font in Table 2). The association of inflammation-related genes in scWAT with cardiac pathology is consistent with the inflammatory phenotype observed in both tissues, but these results also suggest paracrine signaling via extracellular vesicles as a likely mediator of adipose tissue driven cardiac pathology in Adipo-HuR−/− mice.
Table 2.
Hub gene identification within significant LVW/BW associated modules
| Gene Sig Fibrosis | P Value | Module Membership | P Value | Relevant Biological Function | |
|---|---|---|---|---|---|
| Midnightblue | |||||
| Symbol | |||||
| Mid2 | 0.76 | 0.0175 | 0.9879 | 6.29E-07 | E3 ubiquitin ligase ( 35 ); Identified in human urinary exosomes ( 36 ) |
| Slc36a4 | 0.79 | 0.0112 | 0.9846 | 1.48E-06 | Amino acid transporter; Regulates Golgi-associated mTORC1 ( 37 ) |
| Extl3 | 0.78 | 0.0126 | 0.9824 | 2.32E-06 | Mediates TN-α-induced NF-κB activity ( 38 ); identified in milk fat exosomes ( 39 ) |
| Tmem87b | 0.79 | 0.0109 | 0.9816 | 2.75E-06 | Endosome-to-Golgi retrograde transport ( 40 ); mutation linked with restrictive cardiomyopathy ( 41 ); identified in human tumor-derived exosomes ( 42 ) |
| Rab3gap2 | 0.85 | 0.0035 | 0.9778 | 5.25E-06 | Family of proteins that regulates exocytosis of neurotransmitters and hormones; identified in human neuroblastoma-derived exosomes ( 43 ) |
| Man2b2 | 0.88 | 0.0020 | 0.9776 | 5.43E-06 | lysosomal degradation of glycoproteins; identified in human urine exosomes ( 44 ) |
| Aph1b | 0.82 | 0.0062 | 0.9773 | 5.67E-06 | Male-specific association with atherosclerosis (45,46) |
| Gnas | 0.75 | 0.0203 | 0.9763 | 6.63E-06 | GPCR signaling via guanine nucleotide binding; associated with diastolic function in patients with heart failure (47); sex-dependent association with obesity (48); identified in exosomes from multiple sources (20, 49–55) |
| Tmem241 | 0.70 | 0.0340 | 0.9732 | 1.01E-05 | Contributes to increased triglyceride levels (56) |
| Dse | 0.83 | 0.0054 | 0.9727 | 1.08E-05 | Golgi associated protein linked with ECM-related Ehlers-Danlose syndrome ( 57 ) |
| Skyblue | |||||
| Rnf10 | 0.79 | 0.0108 | 0.9875 | 7.14E-07 | Diabetic vascular remodeling via vascular smooth muscle cell proliferation (58); Aging related inflammation (59) |
| Rnft2 | 0.70 | 0.0353 | 0.9829 | 2.13E-06 | Negative regulator of IL-3-mediated inflammation (60) |
| Mmp15 | 0.77 | 0.0159 | 0.9802 | 3.55E-06 | Adipose expression associated with obesity-mediated insulin resistance ( 61 ); Found in human tumor-derived exosomes ( 42 ) |
| Bicd2 | 0.76 | 0.0177 | 0.9764 | 6.48E-06 | Required for exosome transport in motor neurons ( 62 ) |
| Dnaaf9 | 0.72 | 0.0287 | 0.9761 | 6.74E-06 | Unknown |
| Ust | 0.84 | 0.0049 | 0.9760 | 6.90E-06 | Tumor cell adhesion and migration (63,64); regulates Wnt signaling (65) |
| Myo19 | 0.69 | 0.0413 | 0.9747 | 8.24E-06 | Intracellular transport of mitochondria (66,67) |
| Ckap4 | 0.74 | 0.0222 | 0.9730 | 1.04E-05 | Found in multiple human tumor-derived exosomes (42, 68–70); Identified in adipocyte-derived exosomes in rats (71) |
| Rhoc | 0.70 | 0.0352 | 0.9724 | 1.12E-05 | Found in multiple human tumor-derived exosomes (42,43, 68, 70, 72); Regulates exosome secretion (73) |
| Cyp4f16 | 0.77 | 0.0157 | 0.9707 | 1.38E-05 | Metabolism of proinflammatory compounds (74) |
ECM, extracellular matrix; GPCR, G-protein-coupled receptors.
DISCUSSION
In this work, we demonstrate that mice with adipocyte-specific deletion of HuR develop spontaneous cardiac pathology marked by a hypercontractile state and increases in both LV wall thickness and hypertrophic gene expression. These mice also showed an increase in cardiac fibrosis with increased expression of fibrotic (periostin) and ER stress (PERK) proteins. Increased PERK expression indicates activation of the unfolded protein response (UPR) and ER stress, which has been shown to be induced in multiple forms of CVD and is increased in failing human hearts (75, 76). Consistent with these results, RNA-seq analysis also shows a broad increase in hypertrophic, fibrotic, and inflammatory marker genes in these hearts confirming pathological hypertrophy and fibrosis. This result is significant and intriguing in that the cardiovascular pathology is driven by adipocyte-specific signaling in the absence of glucose intolerance, changes in circulating lipid levels, or obesity. All data presented are from lean mice maintained on control chow diet, and we have previously shown that Adipo-HuR−/− mice have no differences in glucose tolerance, plasma phospholipids, cholesterol, triglycerides, or nonesterified fatty acids (17). Thus, this model may be useful in identifying novel mechanisms by which adipose tissue homeostasis affects cardiac physiology in the absence of traditional adipose-associated cardiac comorbidities.
Both scWAT inflammation and BAT dysfunction have been previously associated with CVD through disparate independent mechanisms (1). In our prior work, we demonstrated the role of adipocyte-specific HuR deletion on the function of BAT, including a full RNA-seq analysis of HuR-dependent gene expression in BAT (17), but found nothing that would suggest a CVD-mediating effect emanating from direct HuR-dependent changes in BAT. Similarly, our WGCNA herein found no significant correlations between gene expression in BAT and cardiac fibrosis in Adipo-HuR−/− mice. However, data presented here showed a large degree of gene expression in scWAT that appears to be the result of direct compensation for the loss of BAT-mediated thermogenesis. For example, many gene markers of thermogenesis suggesting the beiging of scWAT, such as ucp1, elovl3, and cidea, were significantly increased in scWAT from Adipo-HuR−/− mice, suggestive of compensatory beiging of scWAT. Interestingly though, the majority of gene expression changes in scWAT, as categorized by gene ontology analysis, were associated with immune and inflammatory processes. This increased inflammatory gene expression in scWAT was accompanied by a systemic increase in circulating IL-6 and TNF-α, both of which have been shown to mediate CVD (33, 77, 78).
To expand upon the standard RNA-seq analysis and identify potentially unique gene expression changes within the scWAT that are likely to underlie specific pathological changes in the heart, we applied weighted gene coexpression network analysis (WGCNA) (25, 26). WGCNA has been shown to be a powerful tool to directly correlate gene expression changes with phenotype and is an excellent predictor of causal relationships (79–81). The results of this analysis identified two coexpressed gene modules in scWAT that showed a significant correlation with cardiac hypertrophy, and GO analysis revealed a strong enrichment of vesicle mediated transport-associated genes within these modules. We also identified an enrichment in apoptosis and inflammation-related GO terms that further support a role for the increase in inflammatory genes identified herein. However, hub gene analysis for these two modules showed that a large number of them have been previously found to be either contained within extracellular exosomes or play a mechanistic role in exosome secretion (8/10 for midnightblue and 4/10 for skyblue module hub genes). Although the specific role of each of these genes in adipose tissue paracrine signaling and their impact on cardiac physiology remains to be seen, these results strongly implicate vesicle-mediated transport as a likely mechanism for scWAT-mediated paracrine signaling to the myocardium. Future work will need to further delineate the degree to which increased inflammation and vesicle-mediated transport from scWAT contributes to CVD.
The potential translational impact of these results and to what extent these mechanisms play a broader role in the adipose-myocardial signaling axis remain to be fully elucidated. We observed a decrease in HuR expression in scWAT from obese wild-type mice (Supplemental Fig. S6; see https://doi.org/10.6084/m9.figshare.14568624), confirming a previously published report of decreased HuR expression in scWAT from obese humans (18). Here, we showed that deletion of HuR specifically in adipose tissue is sufficient to induce CVD in the absence of obesity, but a direct mechanistic role for decreased adipose tissue expression of HuR in promoting CVD in humans remains unexplored. For example, HuR has been previously shown to be a mediator of inflammatory signaling (82–86), but the specific mechanisms by which loss of HuR expression in scWAT drives inflammatory signaling remains unknown. Another important question is whether the loss of BAT-mediated thermogenesis directly contributes to the CVD observed in our model. An inverse clinical correlation between CVD and functional declines in BAT associated with aging, obesity, and other CVD comorbidities has been recently noted (13), but the loss of functional BAT has yet to be conclusively shown to play a direct causal role in CVD. This work is among the first to show a CVD phenotype in an adipocyte-specific deletion model and contributes to our knowledge base regarding the complex nature of adipose tissue and its role in cardiovascular health and disease.
SUPPLEMENTAL DATA
Fig. S1 https://doi.org/10.6084/m9.figshare.14567733, Fig. S2 https://doi.org/10.6084/m9.figshare.14573274, Fig. S3 https://doi.org/10.6084/m9.figshare.14573286, Fig. S4 https://doi.org/10.6084/m9.figshare.14568483, Fig. S5 https://doi.org/10.6084/m9.figshare.14568621, Fig. S6 https://doi.org/10.6084/m9.figshare.14568624.
Table S1 https://doi.org/10.6084/m9.figshare.13326524, Table S2 https://doi.org/10.6084/m9.figshare.13326659, Table S3 https://doi.org/10.6084/m9.figshare.13326680, Table S4 https://doi.org/10.6084/m9.figshare.13326692.
GRANTS
This work was partially supported by the American Heart Association Transformational Project Award (19TPA34910086; to M. Tranter) and National Institutes of Health (NIH) Grant R01 HL132111 (to M. Tranter). L. C. Green and S. Slone are supported by AHA Predoctoral Fellowships (20PRE35210795 and 20PRE35230020, respectively). O. Kanisicak is supported by the NIH Grant R01HL148598 and the American Heart Association Career Development Award (18CDA34110117). P. Alam is supported by the American Heart Association postdoctoral Grant (20POST35200267).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.R.G., S.R.A., A.G., A.P.O., and M.T. conceived and designed research; A.R.G., S.R.A., A.G., L.C.G., S.M.F., S.S., M.L.N., P.A., and M.T. performed experiments; A.R.G., S.R.A., A.G., L.C.G., S.S., M.L.N., P.A., and M.T. analyzed data; A.R.G., S.R.A., A.G., S.M.F., J.B.B., A.P.O., O.K., and M.T. interpreted results of experiments; M.T. prepared figures; A.R.G., S.R.A., and M.T. drafted manuscript; A.R.G., S.R.A., and M.T. edited and revised manuscript; A.R.G., S.R.A., S.S., M.L.N., P.A., J.B.B., O.K., and M.T. approved final version of manuscript.
REFERENCES
- 1.Anthony SR, Guarnieri AR, Gozdiff A, Helsley RN, Phillip Owens A, Tranter M. Mechanisms linking adipose tissue inflammation to cardiac hypertrophy and fibrosis. Clin Sci (Lond) 133: 2329–2344, 2019. doi: 10.1042/CS20190578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuster JJ, Ouchi N, Gokce N, Walsh K. Obesity-induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ Res 118: 1786–1807, 2016. doi: 10.1161/CIRCRESAHA.115.306885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McLaughlin T, Lamendola C, Liu A, Abbasi F. Preferential fat deposition in subcutaneous versus visceral depots is associated with insulin sensitivity. J Clin Endocrinol Metab 96: E1756–E1760, 2011. doi: 10.1210/jc.2011-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neeland IJ, Poirier P, Després J-P. Cardiovascular and metabolic heterogeneity of obesity: clinical challenges and implications for management. Circulation 137: 1391–1406, 2018. doi: 10.1161/CIRCULATIONAHA.117.029617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinckard KM, Shettigar VK, Wright KR, Abay E, Baer LA, Vidal P, Dewal RS, Das D, Duarte-Sanmiguel S, Hernández-Saavedra D, Arts PJ, Lehnig AC, Bussberg V, Narain NR, Kiebish MA, Yi F, Sparks LM, Goodpaster BH, Smith SR, Pratley RE, Lewandowski ED, Raman SV, Wold LE, Gallego-Perez D, Coen PM, Ziolo MT, Stanford KI. A novel endocrine role the BAT-released lipokine 12,13-diHOME to mediate cardiac function. Circulation 143: 145–159, 2021. doi: 10.1161/CIRCULATIONAHA.120.049813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stanford KI, Middelbeek RJW, Townsend KL, An D, Nygaard EB, Hitchcox KM, Markan KR, Nakano K, Hirshman MF, Tseng Y-H, Goodyear LJ. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest 123: 215–223, 2013. doi: 10.1172/JCI62308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thoonen R, Ernande L, Cheng J, Nagasaka Y, Yao V, Miranda-Bezerra A, Chen C, Chao W, Panagia M, Sosnovik DE, Puppala D, Armoundas AA, Hindle A, Bloch KD, Buys ES, Scherrer-Crosbie M. Functional brown adipose tissue limits cardiomyocyte injury and adverse remodeling in catecholamine-induced cardiomyopathy. J Mol Cell Cardiol 84: 202–211, 2015. doi: 10.1016/j.yjmcc.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng Y-H, Doria A, Kolodny GM, Kahn CR. Identification and importance of brown adipose tissue in adult humans. N Engl J Med 360: 1509–1517, 2009. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts J, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med 360: 1500–1508, 2009. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 10.Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 77: 731–758, 1997. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 11.Thyagarajan B, Foster MT. Beiging of white adipose tissue as a therapeutic strategy for weight loss in humans. Horm Mol Biol Clin Investig 31: 1500, 2017. doi: 10.1515/hmbci-2017-0016. [DOI] [PubMed] [Google Scholar]
- 12.Townsend K, Tseng Y-H. Brown adipose tissue: Recent insights into development, metabolic function and therapeutic potential. Adipocyte 1: 13–24, 2012. doi: 10.4161/adip.18951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Becher T, Palanisamy S, Kramer DJ, Eljalby M, Marx SJ, Wibmer AG, Butler SD, Jiang CS, Vaughan R, der HSX, Mark A, Cohen P. Brown adipose tissue is associated with cardiometabolic health. Nat Med 27: 58–65, 2021. doi: 10.1038/s41591-020-1126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doller A, Pfeilschifter J, Eberhardt W. Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell Signal 20: 2165–2173, 2008. doi: 10.1016/j.cellsig.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Green LC, Anthony SR, Slone S, Lanzillotta L, Nieman ML, Wu X, Robbins N, Jones SM, Roy S, Owens AP, Aubé J, Xu L, Lorenz JN, Blaxall BC, Rubinstein J, Benoit JB, Tranter M. Human antigen R as a therapeutic target in pathological cardiac hypertrophy. JCI Insight 4: e121541, 2019. doi: 10.1172/jci.insight.121541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slone S, Anthony SR, Wu X, Benoit JB, Aubé J, Xu L, Tranter M. Activation of HuR downstream of p38 MAPK promotes cardiomyocyte hypertrophy. Cell Signal 28: 1735–1741, 2016. doi: 10.1016/j.cellsig.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anthony SR, Guarnieri A, Lanzillotta L, Gozdiff A, Green LC, O'Grady K, Helsley RN, Owens Iii AP, Tranter M. HuR expression in adipose tissue mediates energy expenditure and acute thermogenesis independent of UCP1 expression. Adipocyte 9: 335–345, 2020. doi: 10.1080/21623945.2020.1782021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Gong L, Liu S, Zhang Y, Zhang C, Tian M, Lu H, Bu P, Yang J, Ouyang C, Jiang X, Wu J, Zhang Y, Min Q, Zhang C, Zhang W. Adipose HuR protects against diet-induced obesity and insulin resistance. Nat Commun 10: 2375, 2019. doi: 10.1038/s41467-019-10348-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh M, Aguila HL, Michaud J, Ai Y, Wu M-T, Hemmes A, Ristimaki A, Guo C, Furneaux H, Hla T. Essential role of the RNA-binding protein HuR in progenitor cell survival in mice. J Clin Invest 119: 3530–3543, 2009. doi: 10.1172/JCI38263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, García-Santos G, Ghajar C, Nitadori-Hoshino A, Hoffman C, Badal K, Garcia BA, Callahan MK, Yuan J, Martins VR, Skog J, Kaplan RN, Brady MS, Wolchok JD, Chapman PB, Kang Y, Bromberg J, Lyden D. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 18: 883–891, 2012. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hadi AM, Mouchaers KTB, Schalij I, Grunberg K, Meijer GA, Vonk-Noordegraaf A, van der Laarse WJ, Beliën JAM. Rapid quantification of myocardial fibrosis: a new macro-based automated analysis. Cell Oncol (Dordr) 34: 343–354, 2011. doi: 10.1007/s13402-011-0035-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57, 2009. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 23.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13, 2009. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tranter M, Ren X, Forde T, Wilhide ME, Chen J, Sartor MA, Medvedovic M, Jones WK. NF-kappaB driven cardioprotective gene programs; Hsp70.3 and cardioprotection after late ischemic preconditioning. J Mol Cell Cardiol 49: 664–672, 2010. doi: 10.1016/j.yjmcc.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9: 559,2008. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4: Article17, 2005. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 27.Mauvais-Jarvis F, Bairey Merz N, Barnes PJ, Brinton RD, Carrero J-J, DeMeo DL, De Vries GJ, Epperson CN, Govindan R, Klein SL, Lonardo A, Maki PM, McCullough LD, Regitz-Zagrosek V, Regensteiner JG, Rubin JB, Sandberg K, Suzuki A. Sex and gender: modifiers of health, disease, and medicine. Lancet 396: 565–582, 2020. [Erratum inLancet396: 668, 2020]. doi: 10.1016/S0140-6736(20)31561-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villar AV, Llano M, Cobo M, Expósito V, Merino R, Martín-Durán R, Hurlé MA, Nistal JF. Gender differences of echocardiographic and gene expression patterns in human pressure overload left ventricular hypertrophy. J Mol Cell Cardiol 46: 526–535, 2009. doi: 10.1016/j.yjmcc.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 29.Wu JC, Nasseri BA, Bloch KD, Picard MH, Scherrer-Crosbie M. Influence of sex on ventricular remodeling after myocardial infarction in mice. J Am Soc Echocardiogr 16: 1158–1162, 2003. doi: 10.1067/S0894-7317(03)00648-5. [DOI] [PubMed] [Google Scholar]
- 30.Fu Y, Xiao H, Zhang Y. Beta-adrenoceptor signaling pathways mediate cardiac pathological remodeling. Front Biosci (Elite Ed) 4: 1625–1637, 2012. doi: 10.2741/484. [DOI] [PubMed] [Google Scholar]
- 31.De Angelis E, Pecoraro M, Rusciano MR, Ciccarelli M, Popolo A. Cross-talk between neurohormonal pathways and the immune system in heart failure: a review of the literature. Int J Mol Sci 20: 1698, 2019.doi: 10.3390/ijms20071698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghali R, Habeichi NJ, Kaplan A, Tannous C, Abidi E, Bekdash A, Farhat R, Itani H, Jurjus A, Booz GW, Mallat Z, Zouein FA. IL-33 induces type-2-cytokine phenotype but exacerbates cardiac remodeling post-myocardial infarction with eosinophil recruitment, worsened systolic dysfunction, and ventricular wall rupture. Clin Sci (Lond) 134: 1191–1218, 2020. doi: 10.1042/CS20200402. [DOI] [PubMed] [Google Scholar]
- 33.Gullestad L, Ueland T, Vinge LE, Finsen A, Yndestad A, Aukrust P. Inflammatory cytokines in heart failure: mediators and markers. Cardiology 122: 23–35, 2012. doi: 10.1159/000338166. [DOI] [PubMed] [Google Scholar]
- 34.Tamaki S, Mano T, Sakata Y, Ohtani T, Takeda Y, Kamimura D, Omori Y, Tsukamoto Y, Ikeya Y, Kawai M, Kumanogoh A, Hagihara K, Ishii R, Higashimori M, Kaneko M, Hasuwa H, Miwa T, Yamamoto K, Komuro I. Interleukin-16 promotes cardiac fibrosis and myocardial stiffening in heart failure with preserved ejection fraction. PLoS One 8: e68893, 2013. doi: 10.1371/journal.pone.0068893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zanchetta ME, Meroni G. Emerging roles of the TRIM E3 ubiquitin ligases MID1 and MID2 in cytokinesis. Front Physiol 10: 274, 2019. doi: 10.3389/fphys.2019.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzales PA, Pisitkun T, Hoffert JD, Tchapyjnikov D, Star RA, Kleta R, Wang NS, Knepper MA. Large-scale proteomics and phosphoproteomics of urinary exosomes. J Am Soc Nephrol 20: 363–379, 2009. doi: 10.1681/ASN.2008040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan S-J, Snell C, Turley H, Li J-L, McCormick R, Perera SMW, Heublein S, Kazi S, Azad A, Wilson C, Harris AL, Goberdhan DCI. PAT4 levels control amino-acid sensitivity of rapamycin-resistant mTORC1 from the Golgi and affect clinical outcome in colorectal cancer. Oncogene 35: 3004–3015, 2016. doi: 10.1038/onc.2015.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mizuno K, Irie S, Sato TA. Overexpression of EXTL3/EXTR1 enhances NF-kappaB activity induced by TNF-alpha. Cell Signal 13: 125–130, 2001. doi: 10.1016/s0898-6568(00)00144-3. [DOI] [PubMed] [Google Scholar]
- 39.Reinhardt TA, Sacco RE, Nonnecke BJ, Lippolis JD. Bovine milk proteome: quantitative changes in normal milk exosomes, milk fat globule membranes and whey proteomes resulting from Staphylococcus aureus mastitis. J Proteomics 82: 141–154, 2013. doi: 10.1016/j.jprot.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Hirata T, Fujita M, Nakamura S, Gotoh K, Motooka D, Murakami Y, Maeda Y, Kinoshita T. Post-Golgi anterograde transport requires GARP-dependent endosome-to-TGN retrograde transport. Mol Biol Cell 26: 3071–3084, 2015. doi: 10.1091/mbc.E14-11-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu H-C, Coughlin CR, Geiger EA, Salvador BJ, Elias ER, Cavanaugh JL, Chatfield KC, Miyamoto SD, Shaikh TH. Discovery of a potentially deleterious variant in TMEM87B in a patient with a hemizygous 2q13 microdeletion suggests a recessive condition characterized by congenital heart disease and restrictive cardiomyopathy. Cold Spring Harb Mol Case Stud 2: a000844, 2016. doi: 10.1101/mcs.a000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang B, Peng P, Chen S, Li L, Zhang M, Cao D, Yang J, Li H, Gui T, Li X, Shen K. Characterization and proteomic analysis of ovarian cancer-derived exosomes. J Proteomics 80: 171–182, 2013. doi: 10.1016/j.jprot.2012.12.029. [DOI] [PubMed] [Google Scholar]
- 43.Keerthikumar S, Gangoda L, Liem M, Fonseka P, Atukorala I, Ozcitti C, Mechler A, Adda CG, Ang C-S, Mathivanan S. Proteogenomic analysis reveals exosomes are more oncogenic than ectosomes. Oncotarget 6: 15375–15396, 2015. doi: 10.18632/oncotarget.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Principe S, Jones EE, Kim Y, Sinha A, Nyalwidhe JO, Brooks J, Semmes OJ, Troyer DA, Lance RS, Kislinger T, Drake RR. In-depth proteomic analyses of exosomes isolated from expressed prostatic secretions in urine. Proteomics 13: 1667–1671, 2013. doi: 10.1002/pmic.201200561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu L, Liu Y, Liu C, Zhang Z, Du Y, Zhao H. Analysis of gene expression profile identifies potential biomarkers for atherosclerosis. Mol Med Rep 14: 3052–3058, 2016. doi: 10.3892/mmr.2016.5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Loo KMJ, Dejaegere T, van Zweeden M, van Schijndel JE, Wijmenga C, Trip MD, Martens GJM. Male-specific association between a gamma-secretase polymorphism and premature coronary atherosclerosis. PLoS One 3: e3662, 2008. doi: 10.1371/journal.pone.0003662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alves AJ, Goldhammer E, Ribeiro F, Eynon N, Ben-Zaken Cohen S, Duarte JA, Viana JL, Sagiv M, Oliveira J. GNAS A-1121G variant is associated with improved diastolic dysfunction in response to exercise training in heart failure patients. Int J Sports Med 34: 274–280, 2012. doi: 10.1055/s-0032-1316365. [DOI] [PubMed] [Google Scholar]
- 48.Weinstein LS, Xie T, Qasem A, Wang J, Chen M. The role of GNAS and other imprinted genes in the development of obesity. Int J Obes (Lond) 34: 6–17, 2010. doi: 10.1038/ijo.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buschow SI, van Balkom BWM, Aalberts M, Heck AJR, Wauben M, Stoorvogel W. MHC class II-associated proteins in B-cell exosomes and potential functional implications for exosome biogenesis. Immunol Cell Biol 88: 851–856, 2010. doi: 10.1038/icb.2010.64. [DOI] [PubMed] [Google Scholar]
- 50.Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G. Cells release prions in association with exosomes. Proc Natl Acad Sci USA 101: 9683–9688, 2004. doi: 10.1073/pnas.0308413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He M, Qin H, Poon TCW, Sze S-C, Ding X, Co NN, Ngai S-M, Chan T-F, Wong N. Hepatocellular carcinoma-derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and RNAs. Carcinogenesis 36: 1008–1018, 2015. doi: 10.1093/carcin/bgv081. [DOI] [PubMed] [Google Scholar]
- 52.Pisitkun T, Shen R-F, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci USA 101: 13368–13373, 2004. doi: 10.1073/pnas.0403453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Skogberg G, Gudmundsdottir J, van der Post S, Sandström K, Bruhn S, Benson M, Mincheva-Nilsson L, Baranov V, Telemo E, Ekwall O. Characterization of human thymic exosomes. PLoS One 8: e67554, 2013. doi: 10.1371/journal.pone.0067554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Welton JL, Khanna S, Giles PJ, Brennan P, Brewis IA, Staffurth J, Mason MD, Clayton A. Proteomics analysis of bladder cancer exosomes. Mol Cell Proteomics 9: 1324–1338, 2010. doi: 10.1074/mcp.M000063-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu R, Greening DW, Rai A, Ji H, Simpson RJ. Highly-purified exosomes and shed microvesicles isolated from the human colon cancer cell line LIM1863 by sequential centrifugal ultrafiltration are biochemically and functionally distinct. Methods 87: 11–25, 2015. doi: 10.1016/j.ymeth.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 56.Rodríguez A, Gonzalez L, Ko A, Alvarez M, Miao Z, Bhagat Y, Nikkola E, Cruz-Bautista I, Arellano-Campos O, Muñoz-Hernández LL, Ordóñez-Sánchez M-L, Rodriguez-Guillen R, Mohlke KL, Laakso M, Tusie-Luna T, Aguilar-Salinas CA, Pajukanta P. Molecular characterization of the lipid genome-wide association study signal on chromosome 18q11.2 implicates HNF4A-mediated regulation of the TMEM241 gene. Arterioscler Thromb Vasc Biol 36: 1350–1355, 2016. doi: 10.1161/ATVBAHA.116.307182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Müller T, Mizumoto S, Suresh I, Komatsu Y, Vodopiutz J, Dundar M, Straub V, Lingenhel A, Melmer A, Lechner S, Zschocke J, Sugahara K, Janecke AR. Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers-Danlos syndrome. Hum Mol Genet 22: 3761–3772, 2013. doi: 10.1093/hmg/ddt227. [DOI] [PubMed] [Google Scholar]
- 58.Li S, Yu G, Huang W, Wang R, Pu P, Chen M. RING finger protein 10 is a potential drug target for diabetic vascular complications. Mol Med Rep 20: 931–938, 2019. doi: 10.3892/mmr.2019.10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cao X, Liu L, Zhang Y, Yang Y. Reduced RING finger protein 10 expression in macrophages is associated with aging-related inflammation. FEBS Open Bio 11: 386–394, 2021. doi: 10.1002/2211-5463.13049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tong Y, Lear TB, Evankovich J, Chen Y, Londino JD, Myerburg MM, Zhang Y, Popescu ID, McDyer JF, McVerry BJ, Lockwood KC, Jurczak MJ, Liu Y, Chen BB. The RNFT2/IL-3Rα axis regulates IL-3 signaling and innate immunity. JCI Insight 5: e133652, 2020. doi: 10.1172/jci.insight.133652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tinahones FJ, Coín-Aragüez L, Mayas MD, Garcia-Fuentes E, Hurtado-Del-Pozo C, Vendrell J, Cardona F, Calvo R-M, Obregon M-J, Bekay El R. Obesity-associated insulin resistance is correlated to adipose tissue vascular endothelial growth factors and metalloproteinase levels. BMC Physiol 12: 4–8, 2012. doi: 10.1186/1472-6793-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rossor AM, Sleigh JN, Groves M, Muntoni F, Reilly MM, Hoogenraad CC, Schiavo G. Loss of BICD2 in muscle drives motor neuron loss in a developmental form of spinal muscular atrophy. Acta Neuropathol Commun 8: 34, 2020. doi: 10.1186/s40478-020-00909-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferguson BW, Datta S. Role of heparan sulfate 2-o-sulfotransferase in prostate cancer cell proliferation, invasion, and growth factor signaling. Prostate Cancer 2011: 893208, 2011. doi: 10.1155/2011/893208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nikolovska K, Spillmann D, Haier J, Ladányi A, Stock C, Seidler DG. Melanoma cell adhesion and migration is modulated by the uronyl 2-O sulfotransferase. PLoS One 12: e0170054, 2017. doi: 10.1371/journal.pone.0170054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cadwalader EL, Condic ML, Yost HJ. 2-O-sulfotransferase regulates Wnt signaling, cell adhesion and cell cycle during zebrafish epiboly. Development 139: 1296–1305, 2012. doi: 10.1242/dev.078238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adikes RC, Unrath WC, Yengo CM, Quintero OA. Biochemical and bioinformatic analysis of the myosin-XIX motor domain. Cytoskeleton (Hoboken) 70: 281–295, 2013. doi: 10.1002/cm.21110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quintero OA, DiVito MM, Adikes RC, Kortan MB, Case LB, Lier AJ, Panaretos NS, Slater SQ, Rengarajan M, Feliu M, Cheney RE. Human Myo19 is a novel myosin that associates with mitochondria. Curr Biol 19: 2008–2013, 2009. doi: 10.1016/j.cub.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Demory Beckler M, Higginbotham JN, Franklin JL, Ham A-J, Halvey PJ, Imasuen IE, Whitwell C, Li M, Liebler DC, Coffey RJ. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol Cell Proteomics 12: 343–355, 2013. doi: 10.1074/mcp.M112.022806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kimura H, Yamamoto H, Harada T, Fumoto K, Osugi Y, Sada R, Maehara N, Hikita H, Mori S, Eguchi H, Ikawa M, Takehara T, Kikuchi A. CKAP4, a DKK1 receptor, is a biomarker in exosomes derived from pancreatic cancer and a molecular target for therapy. Clin Cancer Res 25: 1936–1947, 2019. doi: 10.1158/1078-0432.CCR-18-2124. [DOI] [PubMed] [Google Scholar]
- 70.Park JE, Tan HS, Datta A, Lai RC, Zhang H, Meng W, Lim SK, Sze SK. Hypoxic tumor cell modulates its microenvironment to enhance angiogenic and metastatic potential by secretion of proteins and exosomes. Mol Cell Proteomics 9: 1085–1099, 2010. doi: 10.1074/mcp.M900381-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee J-E, Moon P-G, Lee I-K, Baek M-C. Proteomic analysis of extracellular vesicles released by adipocytes of Otsuka Long-Evans Tokushima fatty (OLETF) rats. Protein J 34: 220–235, 2015. doi: 10.1007/s10930-015-9616-z. [DOI] [PubMed] [Google Scholar]
- 72.Mathivanan S, Lim JWE, Tauro BJ, Ji H, Moritz RL, Simpson RJ. Proteomics analysis of A33 immunoaffinity-purified exosomes released from the human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Mol Cell Proteomics 9: 197–208, 2010. doi: 10.1074/mcp.M900152-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hu S-Q, Zhang Q-C, Meng Q-B, Hu A-N, Zou J-P, Li X-L. Autophagy regulates exosome secretion in rat nucleus pulposus cells via the RhoC/ROCK2 pathway. Exp Cell Res 395: 112239, 2020. doi: 10.1016/j.yexcr.2020.112239. [DOI] [PubMed] [Google Scholar]
- 74.Sehgal N, Agarwal V, Valli RK, Joshi SD, Antonovic L, Strobel HW, Ravindranath V. Cytochrome P4504f, a potential therapeutic target limiting neuroinflammation. Biochem Pharmacol 82: 53–64, 2011. doi: 10.1016/j.bcp.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 75.Jensen BC, Bultman SJ, Holley D, Tang W, de Ridder G, Pizzo S, Bowles D, Willis MS. Upregulation of autophagy genes and the unfolded protein response in human heart failure. Int J Clin Exp Med 10: 1051–1058, 2017. [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang G, Wang X, Gillette TG, Deng Y, Wang ZV. Unfolded protein response as a therapeutic target in cardiovascular disease. Curr Top Med Chem 19: 1902–1917, 2019. doi: 10.2174/1568026619666190521093049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meléndez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 56: 225–231, 2010. doi: 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhao L, Cheng G, Jin R, Afzal MR, Samanta A, Xuan Y-T, Girgis M, Elias HK, Zhu Y, Davani A, Yang Y, Chen X, Ye S, Wang O-L, Chen L, Hauptman J, Vincent RJ, Dawn B. Deletion of interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ Res 118: 1918–1929, 2016[Erratum inCirc Res126: e35, 2020]. doi: 10.1161/CIRCRESAHA.116.308688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haas BE, Horvath S, Pietiläinen KH, Cantor RM, Nikkola E, Weissglas-Volkov D, Rissanen A, Civelek M, Cruz-Bautista I, Riba L, Kuusisto J, Kaprio J, Tusie-Luna T, Laakso M, Aguilar-Salinas CA, Pajukanta P. Adipose co-expression networks across Finns and Mexicans identify novel triglyceride-associated genes. BMC Med Genomics 5: 61, 2012. doi: 10.1186/1755-8794-5-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang M, Wang J, Liu J, Zhu L, Ma H, Zou J, Wu W, Wang K. Systematic prediction of key genes for ovarian cancer by co-expression network analysis. J Cell Mol Med 24: 6298–6307, 2020. doi: 10.1111/jcmm.15271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang X, Duanmu J, Fu X, Li T, Jiang Q. Analyzing and validating the prognostic value and mechanism of colon cancer immune microenvironment. J Transl Med 18: 324–314, 2020. doi: 10.1186/s12967-020-02491-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gurgis FMS, Yeung YT, Tang MXM, Heng B, Buckland M, Ammit AJ, Haapasalo J, Haapasalo H, Guillemin GJ, Grewal T, Munoz L. The p38-MK2-HuR pathway potentiates EGFRvIII-IL-1β-driven IL-6 secretion in glioblastoma cells. Oncogene 34: 2934–2942, 2015. doi: 10.1038/onc.2014.225. [DOI] [PubMed] [Google Scholar]
- 83.Katsanou V, Papadaki O, Milatos S, Blackshear PJ, Anderson P, Kollias G, Kontoyiannis DL. HuR as a negative posttranscriptional modulator in inflammation. Mol Cell 19: 777–789, 2005. doi: 10.1016/j.molcel.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 84.Kim Y, Noren Hooten N, Dluzen DF, Martindale JL, Gorospe M, Evans MK. Post-transcriptional regulation of the inflammatory marker C-reactive protein by the RNA-binding protein HuR and miR-637. Mol Cell Biol 35: 4212–4221, 2015. doi: 10.1128/MCB.00645-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Srikantan S, Gorospe M. HuR function in disease. Front Biosci (Landmark Ed) 17: 189–205, 2012. doi: 10.2741/3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yiakouvaki A, Dimitriou M, Karakasiliotis I, Eftychi C, Theocharis S, Kontoyiannis DL. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J Clin Invest 122: 48–61, 2012. doi: 10.1172/JCI45021. [DOI] [PMC free article] [PubMed] [Google Scholar]