

Abstract
We tested the hypothesis that adiponectin deficiency attenuates cardiac and coronary microvascular function and prevents exercise training-induced adaptations of the myocardium and the coronary microvasculature in adult mice. Adult wild-type (WT) or adiponectin knockout (adiponectin KO) mice underwent treadmill exercise training or remained sedentary for 8–10 wk. Systolic and diastolic functions were assessed before and after exercise training or cage confinement. Vasoreactivity of coronary resistance arteries was assessed at the end of exercise training or cage confinement. Before exercise training, ejection fraction and fractional shortening were similar in adiponectin KO and WT mice, but isovolumic contraction time was significantly lengthened in adiponectin KO mice. Exercise training increased ejection fraction (12%) and fractional shortening (20%) with no change in isovolumic contraction time in WT mice. In adiponectin KO mice, both ejection fraction (−9%) and fractional shortening (−12%) were reduced after exercise training and these decreases were coupled to a further increase in isovolumic contraction time (20%). In sedentary mice, endothelium-dependent dilation to flow was higher in arterioles from adiponectin KO mice as compared with WT mice. Exercise training enhanced dilation to flow in WT mice but decreased flow-induced dilation in adiponectin KO mice. These data suggest that compensatory mechanisms contribute to the maintenance of cardiac and coronary microvascular function in sedentary mice lacking adiponectin; however, in the absence of adiponectin, cardiac and coronary microvascular adaptations to exercise training are compromised.
NEW & NOTEWORTHY We report that compensatory mechanisms contribute to the maintenance of cardiac and coronary microvascular function in sedentary mice in which adiponectin has been deleted; however, when mice lacking adiponectin are subjected to the physiological stress of exercise training, beneficial coronary microvascular and cardiac adaptations are compromised or absent.
Keywords: ejection fraction, endothelium, fractional shortening, heart, vasodilation
INTRODUCTION
Obesity, characterized by excessive adipose tissue, is a major risk factor for type II diabetes mellitus and is linked to hypertension, coronary artery disease, and heart failure (1–4). Adipose tissue is a highly reactive endocrine organ that produces adiponectin, an adipokine linked to the maintenance of healthy vascular function (5–8). Circulating adiponectin is derived primarily from healthy mature adipocytes, but is also produced locally in cardiomyocytes, microvascular endothelial cells, and other tissues (8–11). Adiponectin promotes the upregulation of endothelial nitric oxide synthase activity, helps maintain differentiation of vascular smooth muscle cells, suppresses vascular inflammation, and reduces cardiac cellular apoptosis after ischemia/reperfusion injury (6, 11–18). Circulating adiponectin decreases with obesity (19, 20) and has been linked to downregulated nitric oxide synthase activity, dedifferentiation of vascular smooth muscle, increased endothelial cell proliferation, and excessive superoxide formation (11, 14, 17, 21).
Adiponectin may be especially important to cardiac health; lower circulating adiponectin is associated with greater left ventricular mass index and increased coronary artery disease severity (2, 22). Shibata et al. (18) found that pressure overload induced exaggerated concentric hypertrophy and greater impairment of cardiac output in adiponectin-knockout mice as compared with wild-type mice. In addition, these authors reported that chronic supplementation with adiponectin attenuated these cardiac maladaptations to pressure overload. Acute infusion of monomeric adiponectin in pigs increases cardiac systolic contractility, coronary perfusion, and nitric oxide production (23). Adiponectin deficiency has also been shown to be predictive of future adverse cardiac events (24).
Although numerous reports indicate that lack of adiponectin contributes to the progression of cardiovascular disease (2, 18, 22, 25), little evidence is available with regard to the role of adiponectin in cardiac and coronary adaptations that occur with exercise training. Aerobic exercise training is a broadly used physiological stress that promotes eccentric left ventricular hypertrophy, coronary angiogenesis and is important in delayed progression or reversal of many obesity-related cardiac pathologies including diastolic dysfunction (11, 26–30); however, the role of adiponectin in exercise training-induced left ventricular remodeling, in either physiological or pathophysiological conditions, has not been explored.
Coronary blood flow is actively regulated by the coronary resistance vasculature to ensure that metabolic demand is met (31–34). We have previously shown that exercise training improves diastolic function and the functional responses of both the endothelium and smooth muscle of coronary arterioles in aged rats (30, 35). Improved contractile function of coronary arterioles from aged, exercise-trained rats was associated with an increase in circulating adiponectin and expression of its downstream effector, AMP-activated protein kinase (35). In a diabetic mouse model, exercise training improved endothelium-dependent vasodilation of the aorta through an adiponectin-dependent reduction in inflammation and oxidative stress (36, 37). Given that adiponectin regulates smooth muscle phenotype (5, 35), we chose to investigate the role of adiponectin in improvements of coronary resistance artery function and physiological remodeling of the left ventricle that occur in response to aerobic exercise training. In this study, we hypothesized that physiological adaptation of the myocardium and improvement of endothelium-dependent function of coronary resistance arteries induced by aerobic exercise training are impaired or absent in mice lacking adiponectin.
METHODS
Animals
Adult (12 wk) homozygous male adiponectin knockout (adiponectin KO; strain 008195) and wild-type (WT; C57BL/6) mice were obtained from The Jackson Laboratory (15). Mice were housed under a 12-h:12-h light-dark cycle in a temperature-controlled environment and given access to standard mouse chow and water ad libitum. Each genotype was randomized into either exercise training or sedentary groups. All animal procedures were approved by the Institutional Animal Care and Use Committee at Florida State University and conformed to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (8th ed., 2011).
Exercise Training
Mice were habituated to standard treadmill exercise by walking on the treadmill at 4 m/min (no incline), 5 min/day, for 3 days. Exercise training took place on a treadmill at 12 m/min (5° incline), 60 min/day, 5 days/wk for at least 8 wk. The duration of training was increased during the initial 4 wk until a 60-min duration was obtained. Exercise-trained mice were euthanized ≥48 h after the last exercise bout to exclude any acute effect of exercise (38).
Echocardiography
Cardiac function was assessed before and after 8 wk of exercise training. All imaging were performed using a Vevo 2100 high-resolution in vivo imaging system (Visual Sonics, Canada). All measurements were performed by an experienced investigator who was blinded to genotype and exercise condition. Data were analyzed post hoc using Visual Sonics Vevo Lab analysis software. Mice were anesthetized with 3% isoflurane, placed supine on a heating pad set to 37°C, and maintained at 2% isoflurane. Once hair was removed with Nair, transmission gel was used to obtain parasternal short axis B-mode images of the left ventricular systolic and diastolic dimensions, as described previously (39, 40). The mitral valve flow parameters were taken in the apical view with pulsed wave Doppler to index diastolic function. Fractional shortening was calculated as %FS = (LVd – LVS)/LVd × 100; ejection fraction was calculated as %EF = (LVEDV – LVESV)/LVEDV × 100 (41). Left ventricular wall-to-lumen ratio was calculated as the sum of the anterior and posterior left ventricular walls divided by the inner diameter during systole. All data were collected within 15 min of beginning anesthesia to limit its impact on cardiac function (42).
Microvessel Preparation
Mice were weighed and anesthetized with 3% isoflurane-O2 mix and euthanized by excision of the heart, which was immediately placed in cold physiological saline solution (PSS). PSS contained 145 mM NaCl, 4.7 mM KCl, 2.0 mM CaCl2, 1.17 mM MgSO4, 1.2 mM NaH2PO4, 5.0 mM glucose, 2.0 mM pyruvate, 0.02 mM EDTA, 3.0 mM MOPS buffer, and 1% bovine serum albumin (pH 7.4). Second- and third-order resistance arteries (<150 µm) (43) were isolated from the main coronary artery distribution in the left ventricular free wall. Resistance arteries were cannulated on pipettes of matched tip resistance (within ∼1%) and pressurized to 60 cmH2O in a Lucite chamber that contained warm (37°C), filtered PSS. Next, the chamber was placed on an inverted microscope equipped with a video camera and video caliper to measure intraluminal diameter in real time. Resistance arteries free of leaks were then equilibrated for ∼1 h. The warm PSS was replaced every 20 min during the course of each experiment. At the end of each experiment, vessels were placed in Ca2+-free PSS with 100 µM sodium nitroprusside to determine maximal diameter. Stock solutions of drugs were prepared in double-distilled water and frozen at −20°C. Fresh dilutions for experimental use were made daily.
Evaluation of Vasodilatory Responses
After development of a steady level of spontaneous tone, vasodilatory responses to intraluminal flow were obtained by exposing coronary resistance arteries to graded increases in intraluminal flow without changes to intraluminal pressure, as described previously (44). Briefly, this was accomplished by moving hydrostatic reservoirs in equal and opposite directions so that mean intraluminal pressure remained constant. Diameters were measured in response to incremental pressure differences of 2, 4, 10, 20, 40, and 60 cmH2O (45). Concentration-dependent dose-response curves for acetylcholine (ACh; 1 × 10−9 to 1 × 10−4M) and diethylamine-NONOate (Dea-NONOate; 1 × 10−9 to 1 × 10−4M) were determined by incremental additions of the drugs to the vessel bath. Resistance arteries were reequilibrated for at least 30 min to redevelop tone between successive determinations of vasodilatory responses.
Evaluation of Active Vasoconstriction
To determine the myogenic response, pressure was decreased to 0 cmH2O and then increased from 10 to 130 cmH2O in 20 cmH2O increments. Vasoconstriction to endothelin-1 was assessed by incremental addition of the drug to the vessel bath (1 × 10−11 to 3 × 10−7M).
Data Analysis
Development of spontaneous tone was determined as the percent constriction relative to maximal diameter and calculated as follows:
where Dmax is the maximal inner diameter recorded at a pressure of 60 cmH2O during Ca2+-free conditions and Db is the steady-state baseline diameter. The responses to intraluminal flow, ACh, and DEA-NONO-ate are expressed as percent relaxation and calculated by:
where Ds is the arteriolar diameter measured after the addition of each dose, DB is the diameter recorded immediately before initiation of the flow/concentration-diameter curves, and DM is the maximal diameter for the arteriole. Myogenic responses to pressure increments were normalized according to the maximal diameter formula:
where Ds is the steady-state diameter after each pressure change, and Dm is the maximal diameter measured at a pressure of 60 cmH2O. Responses to endothelin were expressed as percent constriction as calculated by:
where DB is the baseline diameter immediately before addition of the first dose of vasoconstrictor agonist, and Ds is the steady-state diameter measured after addition of each dose. Coronary wall-to-lumen ratio was calculated according to the following equation:
where WT1, WT2, and WT3 are wall thicknesses measured at three independent points of the vessel wall, and Dmax is the maximal inner diameter, with all these measurements recorded at a pressure of 60 cmH2O during Ca2+-free conditions.
Muscle Oxidative Capacity
To determine training efficacy, soleus muscles were removed and immediately stored at −80°C for later assessment of citrate synthase activity, a marker of oxidative capacity according to the method of Srere (46).
Adiponectin ELISA
Blood was collected from the thoracic cavity in serum separator tubes at the time of death. After centrifuging blood for 5 min, the serum was extracted and kept at −80°C for assessment of total adiponectin by ELISA (ELM-Adiponectin; Raybio).
Statistics
Data were analyzed with commercially available statistical software package (IBM SPSS version 25). Cardiac measurements made pre- and postexercise training were compared by three-way ANOVA (geno-type, exercise training, time) with one repeated measure (time). Group differences in animal and vessel characteristics were assessed by two-way ANOVA (genotype and exercise training). Coronary vessel responses were evaluated with a three-way ANOVA (genotype, exercise training, dose/time) with one repeated measure (dose/time). Post hoc analyses were performed using Bonferroni’s test for pairwise comparison if either significant main effects or significant interaction was found. Statistical significance was set as P ≤ 0.05. All data are presented as means ± SE; n, number of animals.
RESULTS
Body weight was higher in sedentary adiponectin KO mice as compared with sedentary WT mice after 8–10 wk of cage confinement. Exercise training decreased body mass in the adiponectin KO mice only (Table 1), resulting in an increase in HW/BW in this group. In sedentary mice, left ventricle weight/body weight (LV/BW, Table 1) was higher in WT as compared with adiponectin KO mice. In exercise-trained mice, absolute LV weight (mg, Table 1) was higher in WT as compared with adiponectin KO mice. Exercise training also increased soleus weight/body weight in the WT, but not the adiponectin KO mice (Table 1). Citrate synthase activity of the soleus muscle was higher in the soleus muscle of sedentary adiponectin KO mice when compared with soleus muscle of sedentary WT mice. Exercise training increased citrate synthase activity of soleus muscle in WT mice by 21.6%, confirming the efficacy of the exercise training regimen (Table 1). In contrast, exercise training did not increase citrate synthase activity in soleus muscle of adiponectin KO mice (Table 1), suggesting that a ceiling for citrate synthase activity was reached in adiponectin KO mice before exercise training, limiting any further increase in response to exercise training. WT mice had total adiponectin plasma levels in the range of 9.5 ± 0.3 µg/mL. In contrast, plasma adiponectin was not detectable in the adiponectin KO mice, confirming knockout of adiponectin (P < 0.001 vs. WT).
Table 1.
Wild-type and adiponectin knockout exercise-trained and sedentary mice characteristics
| WT |
KO |
|||
|---|---|---|---|---|
| Sed | Ex | Sed | Ex | |
| Animal characteristics | ||||
| n | 19 | 24 | 12 | 18 |
| Body weight, g | 28.0 ± 0.6 | 28.6 ± 0.4 | 29.9 ± 0.5* | 27.6 ± 0.4† |
| LV, mg | 97.7 ± 2.4 | 101.5 ± 2.8 | 96.2 ± 2.5 | 94.6 ± 2.7* |
| LV/BW, mg/g | 3.49 ± 0.07 | 3.54 ± 0.06 | 3.21 ± 0.07* | 3.42 ± 0.06 |
| HW, mg | 121 ± 2.9 | 126 ± 2.6 | 122 ± 2.5 | 122 ± 1.5 |
| HW/BW, mg/g | 4.3 ± 0.8 | 4.4 ± 0.8 | 4.0 ± 0.8 | 4.3 ± 0.5† |
| Soleus, mg | 13.8 ± 0.5 | 16.4 ± 0.7† | 14.7 ± 0.9 | 14.4 ± 0.7* |
| Soleus normalized to body weight, mg/g | 0.48 ± 0.01 | 0.57 ± 0.02† | 0.49 ± 0.01 | 0.52 ± 0.03 |
| Soleus, citrate synthase activity, µmol·min−1·g wet wt−1 | 16.47 ± 0.75 | 20.03 ± 0.72† | 18.97 ± 0.85* | 19.21 ± 0.75 |
| Serum adiponectin, µg/mL | 9.7 ± 0.39 | 9.5 ± 0.37 | 0 ± 0* | 0 ± 0* |
| Serum adiponectin/body wt, µg/mL/g | 0.34 ± 0.02 | 0.32 ± 0.02 | 0 ± 0* | 0 ± 0* |
| Coronary vessel characteristics | ||||
| Maximal diameter, µm | 118 ± 5 | 135 ± 6 | 115 ± 9 | 135 ± 9 |
| Spontaneous tone, % | ||||
| Myogenic | 20 ± 4 | 18 ± 2 | 16 ± 2 | 17 ± 1 |
| Flow | 21 ± 1 | 25 ± 2 | 22 ± 1 | 21 ± 2 |
| DEA-NONO-ate | 27 ± 5 | 34 ± 4 | 28 ± 4 | 25 ± 2 |
| Ach | 21 ± 1 | 29 ± 5 | 19 ± 2 | 23 ± 2 |
Values are means ± SE; n, number of mice. BW, body weight; EX, exercise trained; HW, heart weight; KNO, knockout; LV, left ventricle; SED, sedentary; WT, wild type. Significance set at P < 0.05, †effect of exercise training and *effect of genotype to matched WT.
Systolic Function
Baseline (i.e., preexercise training; Pre) measurements of ejection fraction and fractional shortening were not different across groups (Table 2, Pre). Baseline isovolumic contraction time (IVCT) was significantly higher in the adiponectin KO mice as compared with WT mice (Table 2, Pre). In WT mice, exercise training increased ejection fraction (Fig. 1, A and B) and fractional shortening (Fig. 2, A and B). In contrast, both ejection fraction and fractional shortening decreased in response to exercise training in adiponectin KO mice (Figs. 1B and 2B). Consistent with an exercise training-induced increase in systolic contractile function, left ventricular inner diameter during systole (LVIDs, Table 2) was decreased and left ventricular posterior wall during systole (LVPWs, Table 2) was increased by 8 wk of treadmill exercise training in WT mice. In adiponectin KO mice, treadmill exercise training increased left ventricular inner diameter during systole (LVIDs, Table 2) and tended to decrease left ventricular posterior wall during systole (LVPWs, Table 2, P = 0.06). In WT mice, ejection fraction and fractional shortening decreased during 8 wk of cage confinement; however, ejection fraction and fractional shortening did not change during cage confinement in adiponectin KO mice (Figs. 1A and 2A and Table 2, Post). In WT mice, isovolumic contraction time increased during 8 wk of cage confinement (Fig. 3B and Table 2), whereas isovolumic contraction time was maintained during 8 wk of exercise training in WT mice (Fig. 3B and Table 2). In contrast, isovolumic contraction time increased during exercise training, but not during cage confinement, in adiponectin KO mice (Fig. 3B and Table 2).
Table 2.
Cardiac systolic parameters
| WT SED | WT EX | KO SED | KO EX | |
|---|---|---|---|---|
| n | 8 | 6 | 6 | 11 |
| Pre | ||||
| Ejection fraction, % | 70.9 ± 1.7 | 68.5 ± 2.0 | 70.2 ± 2.0 | 73.2 ± 1.5 |
| Fractional shortening, % | 39.7 ± 1.4 | 37.4 ± 1.6 | 39.0 ± 1.6 | 41.7 ± 1.1 |
| IVCT, ms | 13.89 ± 1.08 | 13.02 ± 1.25 | 18.42 ± 1.25* | 19.21 ± 0.92* |
| Heart rate, beats/min | 568 ± 8 | 533 ± 9 | 513 ± 9 | 509 ± 7 |
| Wall:lumen | 0.57 ± 0.03 | 0.63 ± 0.04 | 0.53 ± 0.04 | 0.52 ± 0.03 |
| LVIDs, mm | 2.09 ± 0.12 | 2.09 ± 0.14 | 2.17 ± 0.14 | 2.22 ± 0.1 |
| LVPWs, mm | 1.52 ± 0.09 | 1.33 ± 0.11 | 1.57 ± 0.11 | 1.46 ± 0.08 |
| Post | ||||
| Ejection fraction, % | 66.1 ± 2.5§ | 77.1 ± 2.9†§ | 70.8 ± 2.9 | 66.4 ± 2.1§* |
| Fractional shortening, % | 35.7 ± 2§ | 44.9 ± 2.9†§ | 39.4 ± 2.4 | 36.3 ± 1.8§ |
| IVCT, ms | 22.53 ± 1.6§ | 11.65 ± 1.6 | 20.9 ± 1.8 | 24.2 ± 1.3§* |
| Heart rate, beats/min | 524 ± 22 | 556 ± 25 | 545 ± 25 | 504 ± 18 |
| Wall:lumen | 0.64 ± 0.05 | 0.65 ± 0.06 | 0.61 ± 0.06 | 0.57 ± 0.04 |
| LVIDs, mm | 2.29 ± 0.13 | 1.66 ± 0.18†§ | 2.13 ± 0.16 | 2.46 ± 0.11§* |
| LVPWs, mm | 1.30 ± 0.06§ | 1.61 ± 0.1†§ | 1.40 ± 0.07 | 1.29 ± 0.05* |
Values are means ± SE; n, number of mice. EX, exercise trained; IVCT, isovolumic contraction time; KO, knockout; LVIDs, left ventricular internal diameter at end-systole; LVPWs, left ventricular posterior wall systole; SED, sedentary; WT, wild type. Significance set at P < 0.05, †effect of exercise training, *effect of genotype to matched WT, and §vs. Pre.
Figure 1.
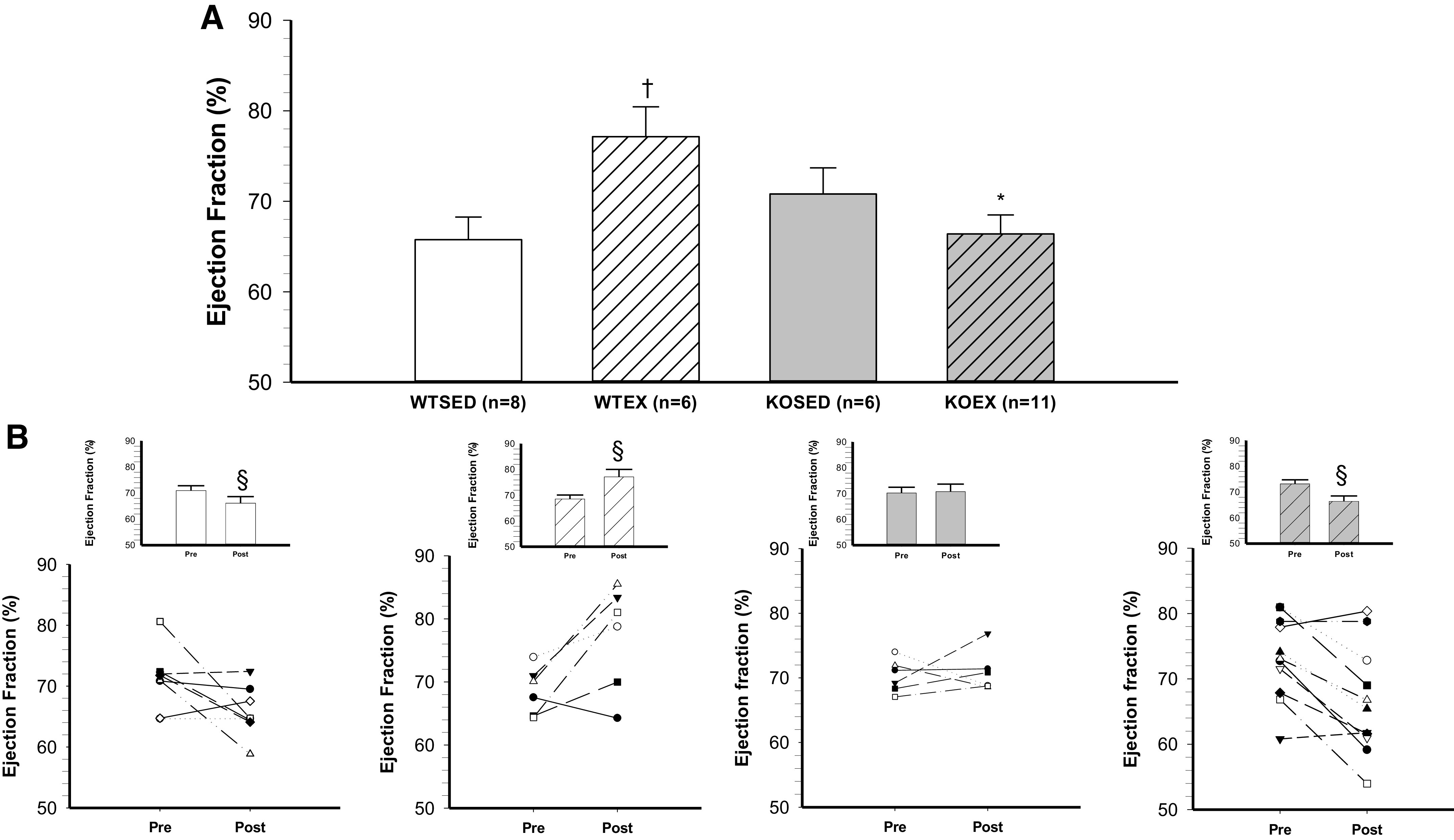
Effect of genotype and exercise training on ejection fraction. A: ejection fraction in wild-type sedentary (WTSED), wild-type trained (WTEX), adiponectin KO sedentary (adiponectin KOSED), and adiponectin KO trained (adiponectin KOEX). B: individual values before (pre) and after 8-wk exercise training or cage confinement. Values are means ± SE. Three-way ANOVA (genotype, exercise training, time) with one repeated measure (time). *Genotype effect; †exercise training effect; §significant pre/post difference, P ≤ 0.05. KO, knockout.
Figure 2.
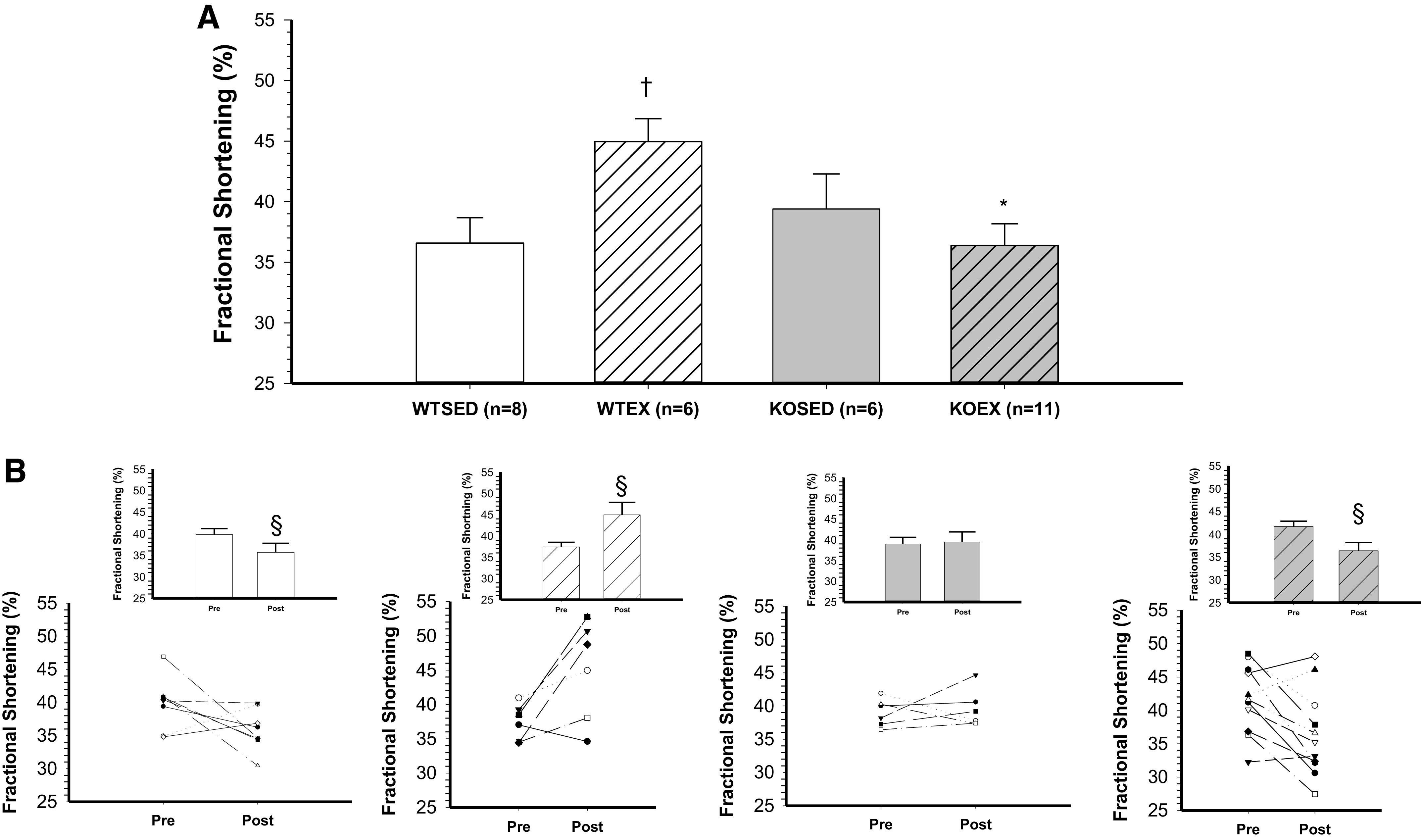
Effect of genotype and exercise training on fractional shortening. A: fractional shortening in wild-type sedentary (WTSED), wild-type trained (WTEX), adiponectin KO sedentary (adiponectin KOSED), and adiponectin KO trained (adiponectin KOEX). B: individual values before (pre) and after 8-wk exercise training or cage confinement. Values are means ± SE. Three-way ANOVA (genotype, exercise training, time) with one repeated measure (time). *Genotype effect; †exercise training effect; §significant pre/post difference, P ≤ 0.05. KO, knockout.
Figure 3.
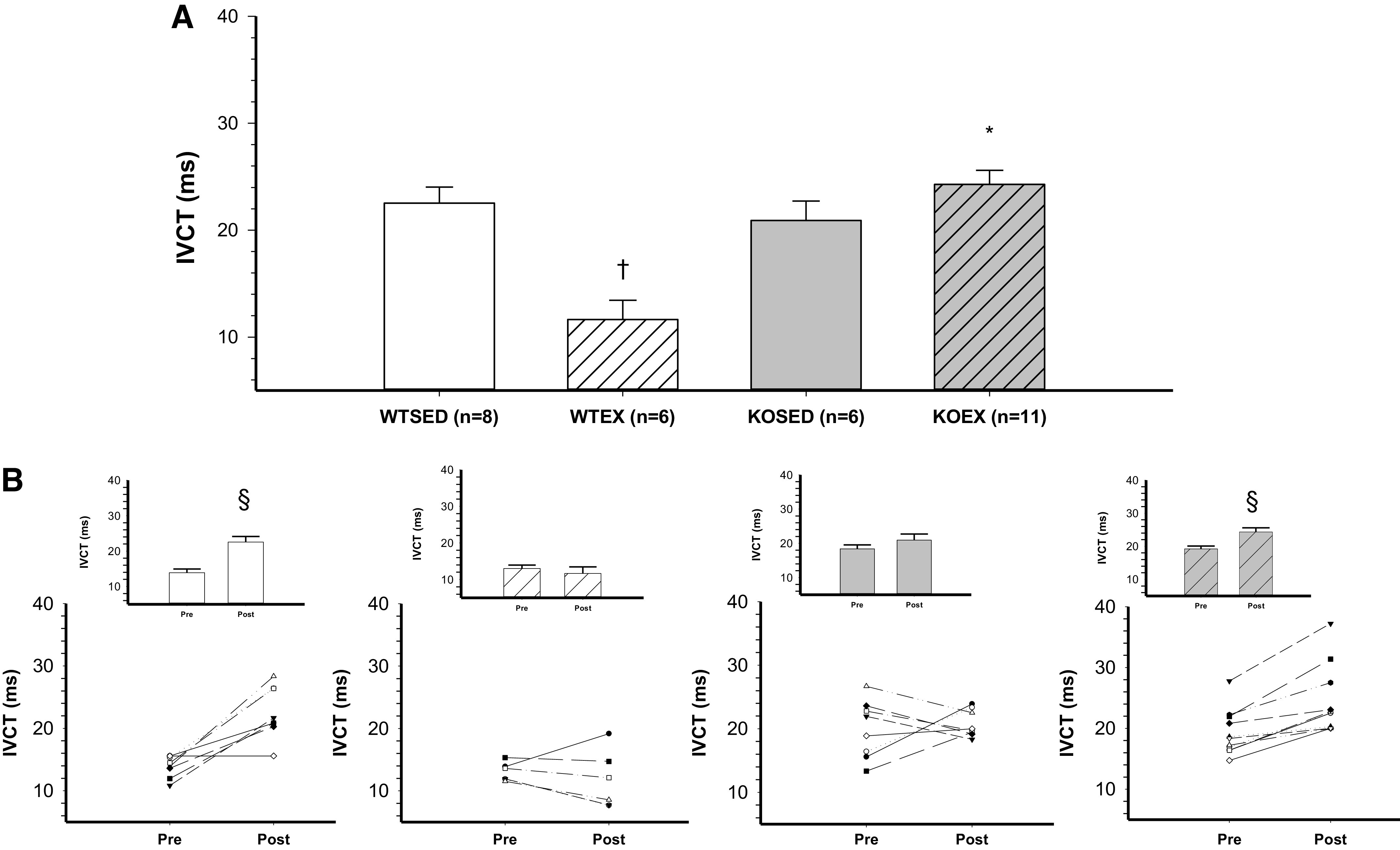
Effect of genotype and exercise training on isovolumic contraction time. A: isovolumic contraction time in wild-type sedentary (WTSED), wild-type trained (WTEX), adiponectin KO sedentary (adiponectin KOSED), and adiponectin KO trained (adiponectin KOEX). B: individual values before (pre) and after 8-wk exercise training or cage confinement. Values are means ± SE. Three-way ANOVA (genotype, exercise training, time) with one repeated measure (time). *Genotype effect; †exercise training effect; §significant pre/post difference, P ≤ 0.05. KO, knockout.
Diastolic Function
At baseline (i.e., pre-exercise training; Pre), diastolic parameters (isovolumic relaxation time, E/a ratio, and left ventricular diameter during diastole) were similar between WT and adiponectin KO mice (Table 3). In WT mice, isovolumic relaxation time increased during sedentary cage confinement; however, this prolongation of isovolumic relaxation time was mitigated by exercise training, so that at the end of 8 wk of training/cage confinement, isovolumic relaxation time was significantly shorter in exercise-trained WT mice as compared to sedentary WT mice (training effect). In contrast to WT mice, isovolumic relaxation time increased slightly in adiponectin KO mice in response to exercise training (Table 3, P = 0.06 post vs. pre), so that postexercise training isovolumic relaxation time was significantly lower in WT EX versus adiponectin KO EX mice (Table 3). Myocardial performance index was higher at baseline in the KO animals and remained higher after exercise training (Table 3). After cage confinement, sedentary WT mice showed a myocardial performance index that increased significantly from baseline values and was higher than the sedentary KO mice. E/a ratio was not altered by exercise training in either WT or adiponectin KO mice.
Table 3.
Cardiac diastolic parameters
| WT SED | WT EX | KO SED | KO EX | |
|---|---|---|---|---|
| n | 8 | 6 | 6 | 11 |
| Pre | ||||
| IVRT, ms | 14.5 ± 1.1 | 16.4 ± 1.3 | 15.7 ± 1.3 | 16.2 ± 0.99 |
| Mitral A velocity, mm/s | 680 ± 68 | 501 ± 79 | 547 ± 79 | 553 ± 58 |
| Mitral E velocity, mm/s | 823 ± 77 | 756 ± 89 | 896 ± 89 | 841 ± 66 |
| MPI | 0.79 ± 0.07 | 0.75 ± 0.08 | 1.14 ± 0.08* | 1.03 ± 0.06* |
| Mitral E/A ratio | 1.24 ± 0.09 | 1.54 ± 0.10 | 1.64 ± 0.10 | 1.56 ± 0.08 |
| LVIDd, mm | 3.48 ± 0.14 | 3.35 ± 0.16 | 3.69 ± 0.16 | 3.74 ± 0.12 |
| LVPWd, mm | 1.04 ± 0.1 | 0.84 ± 0.12 | 1.11 ± 0.12 | 1.08 ± 0.09 |
| Post | ||||
| IVRT, ms | 20.3 ± 1.3§ | 14.4 ± 1.5† | 16.2 ± 1.5* | 19.1 ± 1.1* |
| Mitral A velocity, mm/s | 645 ± 39 | 578 ± 45 | 638 ± 44 | 563 ± 32 |
| Mitral E velocity, mm/s | 867 ± 38 | 726 ± 44 | 911 ± 44 | 843 ± 32 |
| MPI | 1.37 ± 0.07§ | 0.70 ± 0.09† | 1.10 ± 0.09* | 1.19 ± 0.06* |
| Mitral E/A ratio | 1.36 ± 0.10 | 1.35 ± 0.09 | 1.44 ± 0.12 | 1.55 ± 0.09 |
| LVIDd, mm | 3.60 ± 0.16 | 3.11 ± 0.20† | 3.66 ± 0.19 | 3.89 ± 0.14* |
| LVPWd, mm | 0.92 ± 0.05 | 1.08 ± 0.12 | 0.99 ± 0.06 | 0.92 ± 0.04 |
Values are means ± SE; n, number of mice. EX, exercise trained; IVRT, isovolumic relaxation time; KO, knockout; LVIDd, left ventricular internal diameter at end-diastole; LVPWd, left ventricular posterior wall diastole; MPI, myocardial performance index; SED, sedentary; WT, wild type. Significance set at P < 0.05, †effect of exercise training, *effect of genotype to matched WT, and §vs. Pre.
Vasodilatory Responses to Flow
In the absence of exercise training (after 8 wk of sedentary cage confinement), vasodilatory responses to flow were greater in coronary resistance arteries of adiponectin KO mice as compared with those of WT mice (Fig. 4A). Exercise training increased vasodilation to flow in coronary resistance arteries of WT mice (Fig. 4C). In contrast, in adiponectin KO mice, exercise training reduced coronary vasodilation to flow (Fig. 4D). Thus, after exercise, flow-induced vasodilation was greater in coronary resistance arteries of WT mice as compared with those of adiponectin KO mice (Fig. 4B).
Figure 4.

Endothelium-dependent flow-induced vasodilation of coronary resistance arteries from sedentary wild-type and adiponectin KO (WTSED and adiponectin KOSED, respectively) and exercise trained (WTEX and adiponectin KOEX, respectively). A and B: effect of genotype on endothelium-dependent vasodilation. C and D: effect of exercise training on endothelium-independent vasodilation. Values are means ± SE. Three-way ANOVA (genotype, exercise training, time) with one repeated measure (time). *Genotype effect; †exercise training effect; P ≤ 0.05. KO, knockout.
Vasodilatory Responses to Acetylcholine
Receptor-mediated, endothelium-dependent vasodilation of coronary resistance arteries was assessed with acetylcholine. In sedentary mice, acetylcholine-induced vasodilation was decreased in coronary resistance arteries from adiponectin KO mice as compared with those from WT mice (Fig. 5A). Surprisingly, exercise training reduced responsiveness to acetylcholine in resistance arteries from WT mice (Fig. 5C); but had no impact on vasodilatory responses to acetylcholine in the adiponectin KO mice (Fig. 5D). Thus, after exercise training, no differences were detected in acetylcholine-induced vasodilation of coronary resistance arteries from WT versus adiponectin KO mice (Fig. 5B).
Figure 5.

Endothelium-dependent acetylcholine-induced vasodilation of coronary resistance arteries from sedentary wild-type and adiponectin KO (WTSED and adiponectin KOSED, respectively) and exercise trained (WTEX and adiponectin KOEX, respectively). A and B: effect of genotype on endothelium-dependent vasodilation. C and D: effect of exercise training on endothelium-dependent vasodilation. Values are means ± SE. Three-way ANOVA (genotype, exercise training, time) with one repeated measure (time). *Genotype effect; †exercise training effect; P ≤ 0.05. ‡indicates genotype effect; P < 0.10. KO, knockout.
Vasodilatory Responses to DEA-NONOate
Responsiveness to the nitric oxide donor DEA-NONOate was decreased in coronary resistance arteries from sedentary adiponectin KO mice as compared with those from sedentary WT mice (Fig. 6A). Exercise training reduced vasodilation to DEA-NONOate in coronary resistance arteries from WT mice (Fig. 6C), but did not affect vasodilation to DEA-NONOate in coronary resistance arteries from adiponectin KO mice (Fig. 6D). Because exercise altered responses to DEA-NONOate in coronary arteries from WT, but not adiponectin KO mice, differences in responsiveness to DEA-NONOate between WT and adiponectin KO mice were eliminated (Fig. 6B).
Figure 6.

Endothelium-independent DEA-NONOate-induced vasodilation of coronary resistance arteries from sedentary wild-type and adiponectin KO (WTSED and adiponectin KOSED, respectively) and exercise trained (WTEX and adiponectin KOEX, respectively). A and B: effect of genotype on endothelium-independent vasodilation. C and D: effect of exercise training on endothelium-independent vasodilation. Values are means ± SE. Three-way ANOVA (genotype, exercise training, time) with one repeated measure (time). *Genotype effect; †exercise training effect; P ≤ 0.05. ‡indicates genotype effect; P < 0.10. KO, knockout.
Vasoconstrictor Responses to Pressure and Endothelin-1
No differences in constrictor responses to pressure (the myogenic response) or endothelin-1 were detected between coronary resistance arteries from WT and adiponectin KO mice, and exercise training did not impact vasoconstrictor responses to pressure or endothelin-1 in coronary resistance arteries from either WT or adiponectin KO mice (Supplemental Figs. S1 and S2; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.14308022.
Resistance Artery Remodeling
Wall-to-lumen ratio was higher in coronary resistance arteries from sedentary WT mice as compared with coronary resistance arteries from sedentary KO mice and exercise trained-WT and adiponectin KO mice (Fig. 7). These results indicate that either deletion of adiponectin or exercise training-induced expansive remodeling of coronary resistance arteries, but that no greater remodeling was induced in arteries in which the stimulus of exercise training was combined with the effects of adiponectin deletion.
Figure 7.

Wall-to-lumen ratio (Wall:Lumen) in coronary resistance arteries from sedentary wild-type and adiponectin KO (WTSED and adiponectin KOSED, respectively) and exercise trained (WTEX and adiponectin KOEX, respectively). Three-way ANOVA (genotype, exercise training, time) with one repeated measure (time). *Genotype effect; †exercise training effect; P ≤ 0.05. KO, knockout.
DISCUSSION
The present study demonstrates that adaptations of ventricular function, ventricular relaxation, and coronary resistance artery responsiveness to long-term aerobic exercise training are impaired in adiponectin-deficient mice. In WT mice, ejection fraction and fractional shortening were increased by exercise training. In contrast, adiponectin KO mice had significant declines in left ventricular ejection fraction and fractional shortening in response to exercise training when compared with the WT mice (Figs. 1 and 2). Prior to exercise training, systolic parameters were similar in WT and adiponectin KO mice, with the exception of isovolumic contraction time, which was prolonged in adiponectin KO mice relative to their WT counterparts. IVCT was further prolonged by exercise training in the adiponectin KO mice after exercise training and was significantly higher than the WT exercise-trained mice. Diastolic filling (Mitral E and A velocity and left ventricular inner diameter during diastole) was not altered by exercise training; however, isovolumic relaxation time was reduced by exercise training in WT mice and increased in adiponectin KO mice. In sedentary mice, flow-induced vasodilation was actually higher in coronary resistance arteries from adiponectin KO mice as compared with WT mice; however, flow-induced vasodilation of coronary resistance arteries was increased by exercise training in WT mice and reduced by exercise training in adiponectin KO mice. Altogether, these data suggest that, before an exercise stimulus, congenital deletion of adiponectin results in a cardiac and coronary phenotype in which systolic function is preserved, in part, by prolonged contraction time and increased responsiveness to flow in the coronary resistance vasculature. In the absence of adiponectin, physiological adaptation to the stress of exercise training is impaired, and both ventricular function and flow-induced dilation of coronary resistance arteries are reduced by aerobic exercise training.
Exercise Training and Cardiac Systolic Function
Left ventricular hypertrophy is a well-demonstrated central adaptation to long-term aerobic exercise training, leading to increased cardiac output and oxygen delivery to working muscle (47, 48). As expected, 8 wk of aerobic exercise training led to a significant increase in ejection fraction (11%) and fractional shortening (9%) in WT mice (Figs. 2 and 3). This physiological adaptation of the left ventricle to aerobic exercise training was further confirmed by an increase in left ventricular posterior wall thickness and a decrease in left ventricular inner diameter at end-systole in WT mice (Table 2). Thus, 8 wk of aerobic exercise training enhanced the function of the left ventricle in WT mice. In contrast, aerobic exercise training reduced ejection fraction (−9%) and fractional shortening (−12%) (Figs. 2 and 3) in adiponectin KO mice. There was a trend (P = 0.06) for reduced left ventricular posterior wall thickness in adiponectin KO mice (Table 2). Our results in WT mice agree with reports of increased myocardial contractility induced by aerobic exercise training in healthy adult rats (28, 49, 50). Beyond left ventricular hypertrophy, improvements in contractility may stem from several mechanisms including increased Ca2+ sensitivity of the myofilaments (51), increased activity of the Na+/Ca2+ exchanger, and increased Ca2+ ATPase activity within the sarcolemma and sarcoplasmic reticulum (50, 52, 53). At baseline (before exercise training), isovolumic contraction time was increased in adiponectin KO mice relative to WT mice (Table 2); however, ejection fraction and fractional shortening were preserved. This suggests that cardiac function in adiponectin KO mice is maintained in the absence of a physiological stressor like exercise training. The increased baseline isovolumic contraction time suggests that Ca2+ flux or sensitivity and dynamic contraction of the left ventricle are impacted by deletion of adiponectin. Exercise training caused a further prolongation of isovolumic contraction time in adiponectin KO mice, suggesting that the stress of exercise training exacerbates potentially detrimental changes to dynamic contraction induced by deletion of adiponectin. Since adaptations of Ca2+ homeostasis in cardiac myocytes contribute to enhancement of myocardial contractility in endurance-trained animals (50, 51), disruption of Ca2+ homeostasis in adiponectin KO mice may contribute to the decline in myocardial performance that occurred in response to exercise training. Prior investigations have shown that adiponectin activates phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt), which preserves the homeostasis of sarcoplasmic reticulum Ca2+ and prevents endoplasmic reticulum stress after ischemia/reperfusion injury (54). Future investigations will need to focus on the interactive effects of adiponectin signaling and exercise training on Ca2+ flux and Ca2+ sensitivity of myofilaments in cardiac myocytes to determine whether alterations of Ca2+ homeostasis contribute to the reduced ventricular function in exercise-trained adiponectin KO mice.
Exercise Training and Cardiac Diastolic Function
Abnormal diastolic function is a recognized independent predictor of all-cause mortality (55, 56). Left ventricular diastolic function is determined by active/passive processes and effective chamber compliance (57, 58). Isovolumic relaxation was prolonged, and myocardial performance index was higher in the WT sedentary group after 8 wk of cage confinement. This finding in WT sedentary mice is in contrast to previous work showing relatively stable isovolumic relaxation periods over similar ages to the mice in the current investigation (59). Our results also show a higher baseline myocardial performance index in the knockout animals, indicating a potential genotype-specific diastolic dysfunction (Table 3) (60, 61). In WT mice, exercise training reduced isovolumic relaxation time, whereas isovolumic relaxation time was increase by exercise training in adiponectin KO mice. After exercise training, the adiponectin KO mice maintained an elevated myocardial performance index. We have previously shown that exercise training restores diastolic function in aged animals (30). Our current data suggest that abnormal cardiac function may begin to emerge in young adiponectin KO mice before being stressed by aerobic exercise training and that an exercise training stimulus does not improve the diastolic function of these adiponectin KO mice.
In mice with aldosterone-induced hypertension, lack of adiponectin exacerbates negative ventricular remodeling and diastolic dysfunction (62). Isovolumic relaxation requires adequate Ca2+ regulation (52, 57). Adiponectin has been shown to upregulate Ca2+-ATPase activity and to inhibit endoplasmic reticulum stress (54). In adiponectin knockout mice fed a high-fat diet, Ca2+ clearance is impaired in cardiomyocytes (63). These reports on the role of adiponectin and regulation of Ca2+ in cardiac myocytes suggest that reduced Ca2+-ATPase activity may underlie our findings of prolonged isovolumic relaxation time in exercise-trained adiponectin KO mice. In addition, systolic performance has been shown to impact diastolic relaxation (58). Less deformation of the cardiac tissue during systole, like that shown in the trained adiponectin-deficient mice, has also been shown to lead to slower relaxation rates of the left ventricle (58, 64). Further investigation is needed to determine the effect of adiponectin deletion on systolic dysfunction and Ca2+ reuptake insufficiency and related diastolic relaxation and filling, both under basal conditions and in response to exercise training.
Exercise Training and Coronary Resistance Artery Responsiveness
Adiponectin increases fatty acid uptake and insulin sensitivity in cardiac myocytes (65); however, the role of adiponectin in metabolic adaptations of the heart in response to exercise training has not been investigated. Metabolic adjustments and immune responses to acute exercise training are impaired in the skeletal muscle of adiponectin-deficient mice (66). Multiple mechanisms that include intravascular hemodynamic signals (flow and pressure), extravascular compressive forces (tissue pressure), local metabolites, and neural and hormonal influences contribute to regulation of coronary vascular resistance to blood flow, ensuring that coronary blood flow meets cardiac oxygen demand (67). Of these mechanisms, long-term metabolic influences are particularly important in adaptation of the coronary resistance vasculature, as indicated by the coronary microvascular dysfunction that accompanies diabetes, metabolic syndrome, and aging (35, 67, 68). We have reported that the expression of adiponectin receptors and AMPK are reduced in coronary arterioles with advancing age (35), but that late-life exercise training can reverse these age-related changes in adiponectin signaling. In the current study, we were surprised to find that flow-induced vasodilation was increased in coronary resistance arteries from sedentary adiponectin KO as compared with those from sedentary WT mice. It is possible that congenital deletion of adiponectin stimulates compensatory metabolic adaptation of the myocardium that also stimulates an increase in flow-induced vasodilation of coronary resistance arteries. In contrast, when WT mice undergo exercise training, this stimulates an increase in endothelium-dependent, flow-induced vasodilatory function, but the same exercise training induces a decrease in endothelium-dependent, flow-induced vasodilatory function in coronary resistance arteries of adiponectin KO mice. These findings suggest that compensatory adaptive mechanisms of coronary resistance arteries are triggered by congenital deletion of adiponectin, but further adaptation is not possible when the stress of exercise training is imposed on the phenotypically adapted coronary resistance vasculature. Similarly, morphological adaptation, a decrease in wall-to-lumen ratio (expansive remodeling) (69), was present in coronary resistance arteries of adiponectin KO mice, but further reduction of wall-to-lumen ratio, as seen in arteries from exercise-trained WT mice was absent in arteries from exercise-trained adiponectin KO mice. These data suggest that changes in cardiac function, and possibly cardiac metabolism that occur with germline deletion of adiponectin stimulate adaptation of the coronary resistance vasculature. Although these vascular adaptations may provide for adequate matching of blood flow to cardiac demand in the adiponectin KO mice, further stress created by exercise training bouts may lead to an overall decline in the vasodilatory capacity of coronary resistance arteries.
Although our finding that exercise training reduced acetylcholine-induced dilation in coronary resistance arteries may seem to be at odds with our finding that exercise training enhanced flow-mediated vasodilation, this finding is not completely surprising. Acetylcholine activates muscarinic receptors on the endothelium and the vascular smooth muscle, and the vasoactive responses of coronary arteries and arterioles to acetylcholine varies greatly between species (70–73), and even between strains of mice (74). The mediators of acetylcholine-induced vasodilation also differ between coronary arteries and resistance vessels in the mouse heart (73, 74). If flow-induced vasodilation of murine coronary resistance arteries is mediated predominantly by endothelial nitric oxide, as in rat coronary resistance arteries (75), whereas acetylcholine-induced vasodilation of murine coronary resistance arteries is mediated by prostacyclin or endothelium-derived hyperpolarizing factor (EDHF), these differences may account for the differential effects of exercise training on vasodilation to acetylcholine and flow. Park et al. studied acetylcholine-induced vasodilation in coronary arterioles of mice fed a high-fat diet housed in cages with running wheels. In the study by Park et al., vasodilation to acetylcholine was impaired in mice that were fed a high-fat diet, and this effect of a high-fat diet was reversed by wheel running; however, these investigators did not study the effects of wheel running in lean mice fed a low-fat diet. To our knowledge, the effects of exercise training on acetylcholine-induced vasodilation of coronary resistance arteries has not been reported previously in young, healthy mice. The reduction of the acetylcholine-induced vasodilation by exercise training may be an indicator that exercise training increases contractile responses linked to muscarinic receptor activation in the vascular smooth muscle (75, 76). It is also possible that the exercise training-induced reduction of acetylcholine-induced vasodilation is due to a reduction of a NO-independent pathway in the endothelium of these vessels. Finally, our data also indicate that responsiveness to Dea-NONOate was reduced in coronary resistance arteries from exercise-trained wild type mice and in arteries from sedentary AdipoKO mice. These results, along with the reduced vasodilatory responses to acetylcholine, suggest that changes in the smooth muscle may be occurring with exercise training and deletion of adiponectin, leading to reduction of both endothelium-dependent and endothelium-independent vasodilation.
Surprisingly, both exercise training and deletion of adiponectin led to increased flow-induced vasodilation of coronary resistance arteries (Fig. 4). In coronary resistance arteries of rats, flow-induced vasodilation is predominantly NO mediated (75, 76). Exercise training has been reported to increase flow-induced, NO-mediated vasodilation of resistance arteries and arterioles in a number of vascular beds (45, 77–79), including the coronary circulation, and exposure to flow increases expression of endothelial nitric oxide synthase in porcine coronary arterioles (80). In contrast, adiponectin receptors have been shown to possess inherent ceramidase activity (81), and in coronary arterioles from humans, manipulation of neutral ceramidase activity away from production of sphingosine-1-phosphate-favored H2O2-mediated flow-induced vasodilation (82). Inhibition of sphingosine kinase and sphingosine-1-phosphate production-enhanced EDHF-mediated vasodilation of mesenteric resistance arteries (83), but reduced NO-dependent vasodilation in the aorta. At baseline (before exercise training), it is possible that lack of adiponectin led to enhanced flow-induced dilation of coronary resistance arteries (Fig. 4A) through a compensatory increase in H2O2-mediated dilation. In contrast, exercise training in WT mice, most likely led to an increase in flow-induced dilation (Fig. 4C) through an increase in NO-mediated dilation. When exercise training was superimposed upon deletion of adiponectin, the enhancement of flow-induced vasodilation that had occurred with germline deletion of adiponectin was reversed, leading to impaired responsiveness to flow in coronary resistance arteries from KO EX mice (Fig. 4D). Further experiments are needed to elucidate the signaling mechanisms that lead to these changes in flow-induced vasodilation in coronary resistance arteries in response to exercise training in the presence and absence of adiponectin expression. These changes are likely due to local adiponectin signaling in the vasculature rather than due to changes in circulating adiponectin, as serum adiponectin did not increase with exercise training.
Limitations
Global germline knockout of adiponectin may stimulate compensatory vascular mechanisms that were not considered in the current study (84, 85). We did not assess blood pressure of the mice in the current study, and while there are no data to suggest that adiponectin-deficient mice have increased blood pressure and afterload, further work is needed to confirm this and should be taken into consideration when interpreting the current results. Acute deletion of adiponectin from adipose tissue has recently been shown to have greater negative effects on energy homeostasis when compared with whole body germline deletion in which compensatory mechanisms were activated (86). As such, our results may underestimate the contribution of adiponectin to exercise training-induced adaptations of cardiac and coronary function. To assess the immediate role of adiponectin in cardiovascular adaptation to exercise training, future experiments will need to be performed using acute deletion or blockade of adiponectin.
Our finding of an increase in isovolumic relaxation time in WT mice as age increased from 12 to 20 wk may be reflective of effects of anesthesia on heart rate during echocardiographic assessment. All pre- and post-exercise training measurements were made by the same individual, with roughly the same timeframe for anesthesia exposure, and no statistical differences in average heart rate during echocardiography were detected; however, in individual mice, differences in IVRT can be influenced by duration of anesthesia exposure and related changes in heart rate during the experimental period.
Conclusions
The present study demonstrates that physiological adaptations of the heart and coronary resistance vasculature to 8 wk of aerobic exercise training are impaired or absent in adiponectin KO mice. Our current results suggest that adiponectin, which is reduced in a number of cardiovascular disease states (11), may contribute to cardiovascular improvements that occur when exercise training is used therapeutically in patients with cardiovascular disease. If adiponectin contributes to the beneficial cardiovascular adaptations that occur with exercise training, then adiponectin signaling may present a therapeutic target that could be used as an adjuvant to exercise or as an exercise mimetic in patients with limited ability to exercise.
GRANTS
This work was funded by National Institutes of Health Grants R01-HL-128683 (to J. R. Pinto) and R56-AG-068156 (to J. M. Muller-Delp).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.M.M-D. conceived and designed research; J.T.C., K.M.D.J., H.P., P.G., E.C.R-F., B.U., and J.M.M-D. performed experiments; J.T.C., K.M.D.J., H.P., J.R.P., P.G., E.C.R-F., B.U., B.J.B., and J.M.M-D. analyzed data; J.T.C., K.M.D.J., J.R.P., P.G., M.D.D., B.J.B., and J.M.M-D. interpreted results of experiments; J.T.C. and J.M.M-D. prepared figures; J.T.C. drafted manuscript; J.T.C., J.R.P., P.G., M.D.D., B.J.B., and J.M.M-D. edited and revised manuscript; J.T.C., K.M.D.J., H.P., J.R.P., P.G., E.C.R., B.U., M.D.D., B.J.B., and J.M.M-D. approved final version of manuscript.
REFERENCES
- 1.Zarzour A, Kim HW, Weintraub NL. Understanding obesity-related cardiovascular disease: it's all about balance. Circulation 138: 64–66, 2018. doi: 10.1161/CIRCULATIONAHA.118.034454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel JV, Abraheem A, Dotsenko O, Creamer J, Gunning M, Hughes EA, Lip GY. Circulating serum adiponectin levels in patients with coronary artery disease: relationship to atherosclerotic burden and cardiac function. J Intern Med 264: 593–598, 2008. doi: 10.1111/j.1365-2796.2008.02007.x. [DOI] [PubMed] [Google Scholar]
- 3.Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan RS. Obesity and the risk of heart failure. N Engl J Med 347: 305–313, 2002. doi: 10.1056/NEJMoa020245. [DOI] [PubMed] [Google Scholar]
- 4.Hall JE. The kidney, hypertension, and obesity. Hypertension 41: 625–633, 2003. doi: 10.1161/01.HYP.0000052314.95497.78. [DOI] [PubMed] [Google Scholar]
- 5.Ding M, Carrao AC, Wagner RJ, Xie Y, Jin Y, Rzucidlo EM, Yu J, Li W, Tellides G, Hwa J, Aprahamian TR, Martin KA. Vascular smooth muscle cell-derived adiponectin: a paracrine regulator of contractile phenotype. J Mol Cell Cardiol 52: 474–484, 2012. doi: 10.1016/j.yjmcc.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldstein BJ, Scalia RG, Ma XL. Protective vascular and myocardial effects of adiponectin. Nat Clin Pract Cardiovasc Med 6: 27–35, 2009. doi: 10.1038/ncpcardio1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu W, Cheng KK, Vanhoutte PM, Lam KS, Xu A. Vascular effects of adiponectin: molecular mechanisms and potential therapeutic intervention. Clin Sci (Lond) 114: 361–374, 2008. doi: 10.1042/CS20070347. [DOI] [PubMed] [Google Scholar]
- 8.Yoo JK, Hwang MH, Luttrell MJ, Kim HK, Meade TH, English M, Segal MS, Christou DD. Higher levels of adiponectin in vascular endothelial cells are associated with greater brachial artery flow-mediated dilation in older adults. Exp Gerontol 63: 1–7, 2015. doi: 10.1016/j.exger.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pineiro R, Iglesias MJ, Gallego R, Raghay K, Eiras S, Rubio J, Dieguez C, Gualillo O, Gonzalez-Juanatey JR, Lago F. Adiponectin is synthesized and secreted by human and murine cardiomyocytes. FEBS Lett 579: 5163–5169, 2005. doi: 10.1016/j.febslet.2005.07.098. [DOI] [PubMed] [Google Scholar]
- 10.Natarajan R, Salloum FN, Fisher BJ, Kukreja RC, Fowler AA 3rd.. Hypoxia inducible factor-1 upregulates adiponectin in diabetic mouse hearts and attenuates post-ischemic injury. J Cardiovasc Pharmacol 51: 178–187, 2008. doi: 10.1097/FJC.0b013e31815f248d. [DOI] [PubMed] [Google Scholar]
- 11.Becic T, Studenik C, Hoffmann G. Exercise increases adiponectin and reduces leptin levels in prediabetic and diabetic individuals: systematic review and meta-analysis of randomized controlled trials. Med Sci (Basel) 6: 97, 2018. doi: 10.3390/medsci6040097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res 96: 939–949, 2005. doi: 10.1161/01.RES.0000163635.62927.34. [DOI] [PubMed] [Google Scholar]
- 13.Denzel MS, Scimia MC, Zumstein PM, Walsh K, Ruiz-Lozano P, Ranscht B. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J Clin Invest 120: 4342–4352, 2010. doi: 10.1172/JCI43464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang H, Judd RL. Adiponectin regulation and function. Compr Physiol 8: 1031–1063, 2018. doi: 10.1002/cphy.c170046. [DOI] [PubMed] [Google Scholar]
- 15.Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ, Ma XL. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation 115: 1408–1416, 2007. doi: 10.1161/CIRCULATIONAHA.106.666941. [DOI] [PubMed] [Google Scholar]
- 16.Hattori Y, Suzuki M, Hattori S, Kasai K. Globular adiponectin upregulates nitric oxide production in vascular endothelial cells. Diabetologia 46: 1543–1549, 2003. doi: 10.1007/s00125-003-1224-3. [DOI] [PubMed] [Google Scholar]
- 17.Mahadev K, Wu X, Donnelly S, Ouedraogo R, Eckhart AD, Goldstein BJ. Adiponectin inhibits vascular endothelial growth factor-induced migration of human coronary artery endothelial cells. Cardiovasc Res 78: 376–384, 2008. doi: 10.1093/cvr/cvn034. [DOI] [PubMed] [Google Scholar]
- 18.Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, Kumada M, Sato K, Schiekofer S, Ohashi K, Funahashi T, Colucci WS, Walsh K. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med 10: 1384–1389, 2004. doi: 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Achari AE, Jain SK. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int J Mol Sci 18: 1321, 2017. doi: 10.3390/ijms18061321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257: 79–83, 1999. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 21.Motoshima H, Wu X, Mahadev K, Goldstein BJ. Adiponectin suppresses proliferation and superoxide generation and enhances eNOS activity in endothelial cells treated with oxidized LDL. Biochem Biophys Res Commun 315: 264–271, 2004. doi: 10.1016/j.bbrc.2004.01.049. [DOI] [PubMed] [Google Scholar]
- 22.Sharma S, Colangelo LA, Lloyd-Jones D, Jacobs DR, Gross MD Jr, Gidding SS, Greenland P. Longitudinal associations between adiponectin and cardiac structure differ by hypertensive status: coronary artery risk development in young adults. Cardiovasc Endocrinol 5: 57–63, 2016. doi: 10.1097/xce.0000000000000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grossini E, Prodam F, Walker GE, Sigaudo L, Farruggio S, Bellofatto K, Marotta P, Molinari C, Mary D, Bona G, Vacca G. Effect of monomeric adiponectin on cardiac function and perfusion in anesthetized pig. J Endocrinol 222: 137–149, 2014. doi: 10.1530/JOE-14-0170. [DOI] [PubMed] [Google Scholar]
- 24.Kojima S, Funahashi T, Otsuka F, Maruyoshi H, Yamashita T, Kajiwara I, Shimomura H, Miyao Y, Fujimoto K, Sugiyama S, Sakamoto T, Yoshimura M, Ogawa H. Future adverse cardiac events can be predicted by persistently low plasma adiponectin concentrations in men and marked reductions of adiponectin in women after acute myocardial infarction. Atherosclerosis 194: 204–213, 2007. doi: 10.1016/j.atherosclerosis.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 25.Wang ZV, Scherer PE. Adiponectin, the past two decades. J Mol Cell Biol 8: 93–100, 2016. doi: 10.1093/jmcb/mjw011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Danaei G, Ding EL, Mozaffarian D, Taylor B, Rehm J, Murray CJ, Ezzati M. The preventable causes of death in the United States: comparative risk assessment of dietary, lifestyle, and metabolic risk factors. PLoS Med 6: e1000058, 2009[Erratumin PLoS Med8, 2011]. doi: 10.1371/journal.pmed.1000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piche ME, Poirier P, Lemieux I, Despres JP. Overview of epidemiology and contribution of obesity and body fat distribution to cardiovascular disease: an update. Prog Cardiovasc Dis 61: 103–113, 2018. doi: 10.1016/j.pcad.2018.06.004. [DOI] [PubMed] [Google Scholar]
- 28.Weiner RB, Baggish AL. Exercise-induced cardiac remodeling. Prog Cardiovasc Dis 54: 380–386, 2012. doi: 10.1016/j.pcad.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Pressler A, Schwarz S, Christle J, Heinrich C, Edelmann F, Halle M. The best treatment approach to pre-clinical diastolic dysfunction: think about exercise training! J Am Coll Cardiol 64: 529–530, 2014. doi: 10.1016/j.jacc.2014.03.054. [DOI] [PubMed] [Google Scholar]
- 30.Hotta K, Chen B, Behnke BJ, Ghosh P, Stabley JN, Bramy JA, Sepulveda JL, Delp MD, Muller-Delp JM. Exercise training reverses age-induced diastolic dysfunction and restores coronary microvascular function. J Physiol 595: 3703–3719, 2017. doi: 10.1113/JP274172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev 88: 1009–1086, 2008. doi: 10.1152/physrev.00045.2006. [DOI] [PubMed] [Google Scholar]
- 32.Duncker DJ, Bache RJ, Merkus D. Regulation of coronary resistance vessel tone in response to exercise. J Mol Cell Cardiol 52: 802–813, 2012. doi: 10.1016/j.yjmcc.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 33.Hanna MA, Taylor CR, Chen B, La HS, Maraj JJ, Kilar CR, Behnke BJ, Delp MD, Muller-Delp JM. Structural remodeling of coronary resistance arteries: effects of age and exercise training. J Appl Physiol (1985) 117: 616–623, 2014. doi: 10.1152/japplphysiol.01296.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shipley RD, Muller-Delp JM. Aging decreases vasoconstrictor responses of coronary resistance arterioles through endothelium-dependent mechanisms. Cardiovasc Res 66: 374–383, 2005. doi: 10.1016/j.cardiores.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 35.Muller-Delp JM, Hotta K, Chen B, Behnke BJ, Maraj JJ, Delp MD, Lucero TR, Bramy JA, Alarcon DB, Morgan HE, Cowan MR, Haynes AD. Effects of age and exercise training on coronary microvascular smooth muscle phenotype and function. J Appl Physiol (1985) 124: 140–149, 2018. doi: 10.1152/japplphysiol.00459.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S, Zhang H, Chen J, Dellsperger KC, Hill MA, Zhang C. Adiponectin abates diabetes-induced endothelial dysfunction by suppressing oxidative stress, adhesion molecules, and inflammation in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 303: H106–H115, 2012. doi: 10.1152/ajpheart.00110.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee S, Park Y, Dellsperger KC, Zhang C. Exercise training improves endothelial function via adiponectin-dependent and independent pathways in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 301: H306–H314, 2011. doi: 10.1152/ajpheart.01306.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spier SA, Delp MD, Meininger CJ, Donato AJ, Ramsey MW, Muller-Delp JM. Effects of ageing and exercise training on endothelium-dependent vasodilatation and structure of rat skeletal muscle arterioles. J Physiol 556: 947–958, 2004. doi: 10.1113/jphysiol.2003.060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yalcin F, Kucukler N, Cingolani O, Mbiyangandu B, Sorensen L, Pinherio A, Abraham MR, Abraham TP. Evolution of ventricular hypertrophy and myocardial mechanics in physiological and pathological hypertrophy. J Appl Physiol (1985) 126: 354–362, 2019. doi: 10.1152/japplphysiol.00199.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dieseldorff Jones KM, Vied C, Valera IC, Chase PB, Parvatiyar MS, Pinto JR. Sexual dimorphism in cardiac transcriptome associated with a troponin C murine model of hypertrophic cardiomyopathy. Physiol Rep 8: e14396, 2020. doi: 10.14814/phy2.14396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Broberg CS, Pantely GA, Barber BJ, Mack GK, Lee K, Thigpen T, Davis LE, Sahn D, Hohimer AR. Validation of the myocardial performance index by echocardiography in mice: a noninvasive measure of left ventricular function. J Am Soc Echocardiogr 16: 814–823, 2003. doi: 10.1067/S0894-7317(03)00399-7. [DOI] [PubMed] [Google Scholar]
- 42.Stypmann J. Doppler ultrasound in mice. Echocardiography 24: 97–112, 2007. doi: 10.1111/j.1540-8175.2006.00358.x. [DOI] [PubMed] [Google Scholar]
- 43.Chilian WM, Eastham CL, Marcus ML. Microvascular distribution of coronary vascular resistance in beating left ventricle. Am J Physiol Heart Circ Physiol 251: H779–788, 1986. doi: 10.1152/ajpheart.1986.251.4.H779. [DOI] [PubMed] [Google Scholar]
- 44.Muller-Delp J, Spier SA, Ramsey MW, Lesniewski LA, Papadopoulos A, Humphrey JD, Delp MD. Effects of aging on vasoconstrictor and mechanical properties of rat skeletal muscle arterioles. Am J Physiol Heart Circ Physiol 282: H1843–1854, 2002. doi: 10.1152/ajpheart.00666.2001. [DOI] [PubMed] [Google Scholar]
- 45.Sindler AL, Delp MD, Reyes R, Wu G, Muller-Delp JM. Effects of ageing and exercise training on eNOS uncoupling in skeletal muscle resistance arterioles. J Physiol 587: 3885–3897, 2009. doi: 10.1113/jphysiol.2009.172221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srere P. Citrate synthase. In: Methods in Enzymology. New York: Elsevier, 1969, p. 3–11. [Google Scholar]
- 47.Hawley JA, Hargreaves M, Joyner MJ, Zierath JR. Integrative biology of exercise. Cell 159: 738–749, 2014. doi: 10.1016/j.cell.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 48.Wagner PD. Determinants of maximal oxygen transport and utilization. Annu Rev Physiol 58: 21–50, 1996. doi: 10.1146/annurev.ph.58.030196.000321. [DOI] [PubMed] [Google Scholar]
- 49.Bocalini DS, Carvalho EV, de Sousa AF, Levy RF, Tucci PJ. Exercise training-induced enhancement in myocardial mechanics is lost after 2 weeks of detraining in rats. Eur J Appl Physiol 109: 909–914, 2010. doi: 10.1007/s00421-010-1406-x. [DOI] [PubMed] [Google Scholar]
- 50.Wisloff U, Loennechen JP, Falck G, Beisvag V, Currie S, Smith G, Ellingsen O. Increased contractility and calcium sensitivity in cardiac myocytes isolated from endurance trained rats. Cardiovasc Res 50: 495–508, 2001. doi: 10.1016/s0008-6363(01)00210-3. [DOI] [PubMed] [Google Scholar]
- 51.Diffee GM, Seversen EA, Titus MM. Exercise training increases the Ca2+ sensitivity of tension in rat cardiac myocytes. J Appl Physiol (1985) 91: 309–315, 2001. doi: 10.1152/jappl.2001.91.1.309. [DOI] [PubMed] [Google Scholar]
- 52.Pierce GN, Sekhon PS, Meng HP, Maddaford TG. Effects of chronic swimming training on cardiac sarcolemmal function and composition. J Appl Physiol (1985) 66: 1715–1721, 1989. doi: 10.1152/jappl.1989.66.4.1715. [DOI] [PubMed] [Google Scholar]
- 53.Tibbits GF, Kashihara H, O'Reilly K. O'Reilly K. Na+-Ca2+ exchange in cardiac sarcolemma: modulation of Ca2+ affinity by exercise. Am J Physiol Cell Physiol 256: C638–C643, 1989. doi: 10.1152/ajpcell.1989.256.3.C638. [DOI] [PubMed] [Google Scholar]
- 54.Guo J, Bian Y, Bai R, Li H, Fu M, Xiao C. Globular adiponectin attenuates myocardial ischemia/reperfusion injury by upregulating endoplasmic reticulum Ca2+-ATPase activity and inhibiting endoplasmic reticulum stress. J Cardiovasc Pharmacol 62: 143–153, 2013. doi: 10.1097/FJC.0b013e31829521af. [DOI] [PubMed] [Google Scholar]
- 55.Redfield MM, Jacobsen SJ, Burnett JC, Mahoney DW Jr, Bailey KR, Rodeheffer RJ. Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA 289: 194–202, 2003. doi: 10.1001/jama.289.2.194. [DOI] [PubMed] [Google Scholar]
- 56.Thomas L, Marwick TH, Popescu BA, Donal E, Badano LP. Left atrial structure and function, and left ventricular diastolic dysfunction: JACC state-of-the-art review. J Am Coll Cardiol 73: 1961–1977, 2019. doi: 10.1016/j.jacc.2019.01.059. [DOI] [PubMed] [Google Scholar]
- 57.Ogilvie LM, Edgett BA, Huber JS, Platt MJ, Eberl HJ, Lutchmedial S, Brunt KR, Simpson JA. Hemodynamic assessment of diastolic function for experimental models. Am J Physiol Heart Circ Physiol 318: H1139–H1158, 2020. doi: 10.1152/ajpheart.00705.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishimura RA, Tajik AJ. Evaluation of diastolic filling of left ventricle in health and disease: Doppler echocardiography is the clinician’s Rosetta Stone. J Am Coll Cardiol 30: 8–18, 1997. doi: 10.1016/s0735-1097(97)00144-7. [DOI] [PubMed] [Google Scholar]
- 59.Hinton RB Jr, Alfieri CM, Witt SA, Glascock BJ, Khoury PR, Benson DW, Yutzey KE. Mouse heart valve structure and function: echocardiographic and morphometric analyses from the fetus through the aged adult. Am J Physiol Heart Circ Physiol 294: H2480–H2488, 2008. doi: 10.1152/ajpheart.91431.2007. [DOI] [PubMed] [Google Scholar]
- 60.Gao S, Ho D, Vatner DE, Vatner SF. Echocardiography in mice. Curr Protoc Mouse Biol 1: 71–83, 2011. doi: 10.1002/9780470942390.mo100130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lakoumentas JA, Panou FK, Kotseroglou VK, Aggeli KI, Harbis PK. The Tei index of myocardial performance: applications in cardiology. Hellenic J Cardiol 46: 52–58, 2005. [PubMed] [Google Scholar]
- 62.Sam F, Duhaney TA, Sato K, Wilson RM, Ohashi K, Sono-Romanelli S, Higuchi A, De Silva DS, Qin F, Walsh K, Ouchi N. Adiponectin deficiency, diastolic dysfunction, and diastolic heart failure. Endocrinology 151: 322–331, 2010. doi: 10.1210/en.2009-0806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guo R, Zhang Y, Turdi S, Ren J. Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochim Biophys Acta 1832: 1136–1148, 2013. doi: 10.1016/j.bbadis.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brutsaert DL, Rademakers FE, Sys SU. Triple control of relaxation: implications in cardiac disease. Circulation 69: 190–196, 1984. doi: 10.1161/01.cir.69.1.190. [DOI] [PubMed] [Google Scholar]
- 65.Fang X, Palanivel R, Cresser J, Schram K, Ganguly R, Thong FS, Tuinei J, Xu A, Abel ED, Sweeney G. An APPL1-AMPK signaling axis mediates beneficial metabolic effects of adiponectin in the heart. Am J Physiol Endocrinol Metab 299: E721–E729, 2010. doi: 10.1152/ajpendo.00086.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Diniz TA, Aquino Junior JCJ, Mosele FC, Cabral-Santos C, Lima Junior EA, Teixeira AAS, Lira FS, Rosa Neto JC. Exercise-induced AMPK activation and IL-6 muscle production are disturbed in adiponectin knockout mice. Cytokine 119: 71–80, 2019. doi: 10.1016/j.cyto.2019.03.009. [DOI] [PubMed] [Google Scholar]
- 67.Goodwill AG, Dick GM, Kiel AM, Tune JD. Regulation of coronary blood flow. Compr Physiol 7: 321–382, 2017. doi: 10.1002/cphy.c160016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murthy VL, Naya M, Foster CR, Gaber M, Hainer J, Klein J, Dorbala S, Blankstein R, Di Carli MF. Association between coronary vascular dysfunction and cardiac mortality in patients with and without diabetes mellitus. Circulation 126: 1858–1868, 2012. doi: 10.1161/CIRCULATIONAHA.112.120402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prior BM, Lloyd PG, Yang HT, Terjung RL. Exercise-induced vascular remodeling. Exerc Sport Sci Rev 31: 26–33, 2003. doi: 10.1097/00003677-200301000-00006. [DOI] [PubMed] [Google Scholar]
- 70.Entzeroth M, Doods HN, Mayer N. Characterization of porcine coronary muscarinic receptors. Naunyn Schmiedebergs Arch Pharmacol 341: 432–438, 1990. doi: 10.1007/BF00176336. [DOI] [PubMed] [Google Scholar]
- 71.Skovsted GF, Tveden-Nyborg P, Lindblad MM, Hansen SN, Lykkesfeldt J. Vitamin C deficiency reduces muscarinic receptor coronary artery vasoconstriction and plasma tetrahydrobiopterin concentration in guinea pigs. Nutrients 9: 691, 2017. doi: 10.3390/nu9070691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park Y, Booth FW, Lee S, Laye MJ, Zhang C. Physical activity opposes coronary vascular dysfunction induced during high fat feeding in mice. J Physiol 590: 4255–4268, 2012. doi: 10.1113/jphysiol.2012.234856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Muller-Delp JM, Lubahn DB, Nichol KE, Philips BJ, Price EM, Curran EM, Laughlin MH. Regulation of nitric oxide-dependent vasodilation in coronary arteries of estrogen receptor-alpha-deficient mice. Am J Physiol Heart Circ Physiol 285: H2150–H2157, 2003. doi: 10.1152/ajpheart.00966.2002. [DOI] [PubMed] [Google Scholar]
- 74.Gwozdz P, Drelicharz L, Kozlovski VI, Chlopicki S. Prostacyclin but not nitric oxide, is the major mediator of acetylcholine-induced vasodilatation in the isolated mouse heart. Pharmacol Rep 59: 545–552, 2007. [PubMed] [Google Scholar]
- 75.LeBlanc AJ, Shipley RD, Kang LS, Muller-Delp JM. Age impairs Flk-1 signaling and NO-mediated vasodilation in coronary arterioles. Am J Physiol Heart Circ Physiol 295: H2280–H2288, 2008. doi: 10.1152/ajpheart.00541.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.LeBlanc AJ, Reyes R, Kang LS, Dailey RA, Stallone JN, Moningka NC, Muller-Delp JM. Estrogen replacement restores flow-induced vasodilation in coronary arterioles of aged and ovariectomized rats. Am J Physiol Regul Integr Comp Physiol 297: R1713–R1723, 2009[Erratum inAm J Physiol Regul Integr Comp Physiol298: R1446, 2010]. doi: 10.1152/ajpregu.00178.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spier SA, Delp MD, Stallone JN, DominguezJM , 2nd, Muller-Delp JM. Exercise training enhances flow-induced vasodilation in skeletal muscle resistance arteries of aged rats: role of PGI2 and nitric oxide. Am J Physiol Heart Circ Physiol 292: H3119–H3127, 2007. doi: 10.1152/ajpheart.00588.2006. [DOI] [PubMed] [Google Scholar]
- 78.Robinson AT, Fancher IS, Sudhahar V, Bian JT, Cook MD, Mahmoud AM, Ali MM, Ushio-Fukai M, Brown MD, Fukai T, Phillips SA. Short-term regular aerobic exercise reduces oxidative stress produced by acute in the adipose microvasculature. Am J Physiol Heart Circ Physiol 312: H896–H906, 2017. doi: 10.1152/ajpheart.00684.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun D, Huang A, Koller A, Kaley G. Adaptation of flow-induced dilation of arterioles to daily exercise. Microvasc Res 56: 54–61, 1998. doi: 10.1006/mvre.1998.2083. [DOI] [PubMed] [Google Scholar]
- 80.Woodman CR, Muller JM, Rush JW, Laughlin MH, Price EM. Flow regulation of ecNOS and Cu/Zn SOD mRNA expression in porcine coronary arterioles. Am J Physiol Heart Circ Physiol 276: H1058–H1063, 1999. doi: 10.1152/ajpheart.1999.276.3.H1058. [DOI] [PubMed] [Google Scholar]
- 81.Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, Wade MR, Tenorio VM, Kuo MS, Brozinick JT, Zhang BB, Birnbaum MJ, Summers SA, Scherer PE. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 17: 55–63, 2011. doi: 10.1038/nm.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schulz ME, Katunaric B, Hockenberry JC, Gutterman DD, Freed JK. Manipulation of the sphingolipid rheostat influences the mediator of flow-induced dilation in the human microvasculature. J Am Heart Assoc 8: e013153, 2019. doi: 10.1161/JAHA.119.013153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mulders AC, Mathy MJ, Meyer zu Heringdorf D, ter Braak M, Hajji N, Olthof DC, Michel MC, Alewijnse AE, Peters SL. Activation of sphingosine kinase by muscarinic receptors enhances NO-mediated and attenuates EDHF-mediated vasorelaxation. Basic Res Cardiol 104: 50–59, 2009. doi: 10.1007/s00395-008-0744-x. [DOI] [PubMed] [Google Scholar]
- 84.Levine HJ, Forwand SA, McIntyre KM, Schechter E. Effect of afterload on force-velocity relations and contractile element work in the intact dog heart. Circ Res 18: 729–744, 1966. doi: 10.1161/01.res.18.6.729. [DOI] [PubMed] [Google Scholar]
- 85.Cho EJ, Caracciolo G, Khandheria BK, Steidley DE, Scott R, Abhayaratna WP, Chandrasekaran K, Sengupta PP. Tissue Doppler image-derived measurements during isovolumic contraction predict exercise capacity in patients with reduced left ventricular ejection fraction. JACC Cardiovasc Imaging 3: 1–9, 2010. doi: 10.1016/j.jcmg.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 86.Xia JY, Sun K, Hepler C, Ghaben AL, Gupta RK, An YA, Holland WL, Morley TS, Adams AC, Gordillo R, Kusminski CM, Scherer PE. Acute loss of adipose tissue-derived adiponectin triggers immediate metabolic deterioration in mice. Diabetologia 61: 932–941, 2018. doi: 10.1007/s00125-017-4516-8. [DOI] [PMC free article] [PubMed] [Google Scholar]