

Keywords: aging, lipogenesis, liver, obesity, triglycerides
Abstract
Nonalcoholic fatty liver disease (NAFLD) is a spectrum of disorders ranging from hepatic steatosis [excessive accumulation of triglycerides (TG)] to nonalcoholic steatohepatitis, which can progress to cirrhosis and hepatocellular carcinoma. The molecular pathogenesis of steatosis and progression to more severe NAFLD remains unclear. Obesity and aging, two principal risk factors for NAFLD, are associated with a hyperadrenergic state. β-Adrenergic responsiveness in liver increases in animal models of obesity and aging, and in both is linked to increased hepatic expression of β2-adrenergic receptors (β2-ARs). We previously showed that in aging rodents intracellular signaling from elevated hepatic levels of β2-ARs may contribute to liver steatosis. In this study we demonstrate that injection of formoterol, a highly selective β2-AR agonist, to mice acutely results in hepatic TG accumulation. Further, we have sought to define the intrahepatic mechanisms underlying β2-AR mediated steatosis by investigating changes in hepatic expression and cellular localization of enzymes, transcription factors, and coactivators involved in processes of lipid accrual and disposition—and also functional aspects thereof—in livers of formoterol-treated animals. Our results suggest that β2-AR activation by formoterol leads to increased hepatic TG synthesis and de novo lipogenesis, increased but incomplete β-oxidation of fatty acids with accumulation of potentially toxic long-chain acylcarnitine intermediates, and reduced TG secretion—all previously invoked as contributors to fatty liver disease. Experiments are ongoing to determine whether sustained activation of hepatic β2-AR signaling by formoterol might be utilized to model fatty liver changes occurring in hyperadrenergic states of obesity and aging, and thereby identify novel molecular targets for the prevention or treatment of NAFLD.
NEW & NOTEWORTHY Results of our study suggest that β2-adrenergic receptor (β2-AR) activation by agonist formoterol leads to increased hepatic TG synthesis and de novo lipogenesis, incomplete β-oxidation of fatty acids with accumulation of long-chain acylcarnitine intermediates, and reduced TG secretion. These findings may, for the first time, implicate a role for β2-AR responsive dysregulation of hepatic lipid metabolism in the pathogenetic processes underlying NAFLD in hyperadrenergic states such as obesity and aging.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD), the most common chronic liver disease in individuals without alcohol abuse, encompasses a spectrum of pathological changes ranging from steatosis to more progressive injury (termed nonalcoholic steatohepatitis, or NASH), ultimately with potential development of cirrhosis and/or hepatocellular carcinoma (1–4). The prevalence of NAFLD has increased markedly, with ∼20%–30% of adults currently affected in the United States (1, 2, 5). End-stage hepatic failure and liver cancer resulting from advanced NAFLD are leading indications for liver transplantation (5, 6). Notably, NAFLD has been recognized to be the hepatic component of the metabolic syndrome, which consists of a cluster of disorders including obesity, insulin resistance, hyperglycemia and type 2 diabetes mellitus, dyslipidemia, and hypertension (1, 3). Whereas each of these associated disorders is well known to contribute to the development of cardiovascular disease, NAFLD itself appears to be a cardiovascular risk factor independent of other components of the metabolic syndrome (3). As a clinically significant metabolic risk factor, NAFLD has emerged as a focus of research into its causes and consequences, with an emphasis on identifying effective therapeutic options.
Hepatic steatosis, the earliest stage of NAFLD, is characterized by excessive accumulation of triglycerides (TG) in lipid droplets within hepatocytes (1). Under physiological conditions hepatic TG synthesis buffers against toxicity from free fatty acids accumulating within cells (7, 8). In the case of NAFLD, however, excess TG levels in liver predisposing to steatosis and progressive cellular injury appear to result from an imbalance between TG accumulation and removal. Hepatic TG formation is dependent on fatty acid uptake, de novo lipogenesis (fatty acid synthesis), and fatty acid esterification to TG, whereas TG levels in liver are reduced by processes of fatty acid β-oxidation as well as assembly and secretion of very low density lipoprotein (VLDL)-TG. Relative increases in processes controlling TG formation and/or decreases in mechanisms of TG removal have all been implicated in the development of hepatic steatosis clinically and in animal models of NAFLD (1, 9, 10).
Obesity, as a principal component of the metabolic syndrome, is the most common clinical risk factor for NAFLD. Prevalence and severity of the disorder also increase during aging (2). Both obesity and aging are associated with a hyperadrenergic state characterized by increased circulating levels of catecholamines and activation of the sympathetic nervous system (11, 12). Many critical adrenergic responses in liver and other target tissues are mediated by the β-adrenergic (β-AR) receptor-coupled adenylyl cyclase (AC)/cyclic AMP (cAMP)/cAMP-dependent protein kinase (protein kinase A) signaling cascade. Beta-ARs coupled to AC comprise a family of three receptor subtypes, i.e., β1-, β2- and β3-ARs; in human liver β-ARs are predominantly of the β2-AR subtype (13). Importantly, β-AR responsive AC activity in liver increases in animal models of both obesity and aging (14, 15). In earlier work with genetically obese mice, heightened β-adrenergic responsiveness in liver was linked to an increase in hepatic β2-AR levels (14). An age-related increase in β-AR linked AC activity appears to be unique to liver, perhaps reflecting a tissue-specific impairment in homeostatic processes operative in other tissues exhibiting reduced β-adrenergic responsiveness to increased sympathetic tone with age (16). We have demonstrated that increased β-AR stimulated AC activity correlates with fat accumulation in livers from aging rodents, and that in vitro overexpression of β2-ARs in hepatocytes from young rodents mimics the increased hepatocellular lipid content seen in old animals (17). In other experiments age-related increases in hepatic steatosis were found to be attenuated in β2-AR knockout mice (18). Recent work from our laboratory also shows that, while hepatic expression of both β2- and β1-AR subtypes is greater in senescent rodents than in younger animals, the β2-AR appears to be the principal β-AR subtype in liver regulated by agonist in hyperadrenergic states such as aging (16). Available evidence, then, implicates a role for β2-AR stimulated AC signaling in the development of hepatic steatosis, and by extension NAFLD, occurring with obesity and aging.
In the present study we have established an animal model to extend evidence for β2-AR linked lipid dysregulation in liver as a potential contributor to hepatic steatosis. We previously demonstrated that acute in vivo administration of the nonsubtype-selective β-AR agonist isoproterenol to rodents increases hepatic lipid content (17). We now report that treatment of mice with the long-acting, highly selective β2-AR agonist formoterol results in excessive hepatic lipid accumulation, and we delineate potential mechanisms underlying the development of steatosis in these animals. Clearly, systemic sympathetic activation stimulates β-AR mediated lipolysis in adipose tissue and increases the circulating pool of non-esterified fatty acids (NEFAs) entering the liver (4, 19). In this regard formoterol, used clinically as a bronchodilator in the treatment of asthma and chronic obstructive pulmonary disease, has been reported to increase plasma levels of NEFAs when administered to men (20). Notwithstanding the known effects of β-AR agonists on lipolysis, however, in the current study we have focused our efforts to identify processes involved in formoterol induced steatosis on intrinsic hepatic pathways of TG acquisition and removal previously implicated in the pathogenesis of NAFLD. Thus findings from formoterol treated mice may point to β2-AR mediated regulation of intrahepatic lipid metabolism as a modifiable determinant of steatosis, and in turn suggest novel therapeutic strategies against NAFLD as a chronic liver disease and metabolic risk factor for cardiovascular disease.
MATERIALS AND METHODS
Animals
Male C57Bl/6 mice (3.6–5.4 mo old) were purchased from Jackson Laboratory (Bar Harbor, ME) and housed in the Veterinary Medical Unit at the Audie L. Murphy Division, South Texas Veterans Health Care System (ALMD, STVHCS). Mice were fed a commercial chow (Teklad Diet LN-485; Envigo, Madison, WI) ad libitum. Animals were allowed to acclimatize in the animal care facility for at least two weeks before experiments were performed. All animals were treated in accordance with the guidelines approved by the Institutional Animal Care and Use Committee at the ALMD, STVHCS.
Formoterol Injection
Formoterol hemifumarate was purchased from R & D Systems (Minneapolis, MN). Formoterol was administered by intraperitoneal (i.p.) injection (20 µg/g body weight) twice over a 24-h period. Control animals were injected with saline vehicle twice over 24 h. Mice were euthanized 24 h after the first injection, and liver tissues and blood samples were collected. The dose and time course of formoterol injection were selected to coincide with the protocol we used previously for injection of mice with isoproterenol (17); we did not observe any mortality in mice injected with either agent (compared to saline treated animals) over the 24-h injection period.
Whole Body Measurements
Food consumption.
Food consumption by singly housed mice was measured essentially as described previously (21). The chow in the cage hopper and the spillage (the chow at the bottom of the cage) were weighed before and after two successive one-week control periods, and again following a 24-h experimental period of saline or formoterol administration; food consumption (g/24 h) was calculated as the difference between the amount of chow removed from the hopper and the spillage.
Quantitative magnetic resonance imaging.
Whole body composition analysis was performed on mice before and after 24-h saline or formoterol treatment by quantitative magnetic resonance (QMR) using an EchoMRI-900 (Echo Medical Systems, Houston, TX). Mice were placed in a thin walled plastic cylinder and subjected to a low intensity (0.05 Tesla) electromagnetic field for 70 s to measure body fat and lean mass.
Plasma Measurements
Blood samples from euthanized animals were collected in EDTA-coated tubes. The tubes were centrifuged at 1,000–2,000 g for 10 min in a refrigerated centrifuge to obtain plasma. Plasma levels of NEFAs were measured using an assay kit from Wako Diagnostics (Component numbers 999-34691, 995-34791, 991-34891, 993-34191, 276-76491; Mountain View, CA). Alanine transaminase (ALT) and aspartate transaminase (AST) activities and β-hydroxybutyrate levels were measured using assay kits (Cat. Nos. MAK052-1KT, MAK055-1KT, and MAK041-1KT, respectively) from Sigma-Aldrich (St. Louis, MO). Plasma insulin levels were determined using the Crystal Chem Ultra Sensitive Mouse Insulin ELISA Kit (Cat. No. 90080; Downers Grove, IL).
Hepatic Triglycerides
Hepatic triglycerides (TG) levels were determined using a Triglyceride Quantification Kit (Cat. No. K622-100) from BioVision Inc. (Milpitas, CA) following the manufacturer’s instructions. Briefly, 100 mg of frozen liver tissue were homogenized in 1 mL of 5% NP-40 solution, and the samples were then boiled twice before centrifugation to remove insoluble materials. Supernatant was diluted 50- to 100-fold for analysis. Liver TG was calculated from a standard curve and expressed as nmoL/mg of tissue.
Liver Tissues
Lipid content of liver tissues was visualized and quantified by staining with Oil Red O (ORO), as described previously (17). Briefly, frozen liver tissues were stained with ORO together with hematoxylin as a counterstain. At least three random fields of each stained specimen were photographed with an Olympus AX70 microscope equipped with a ×20 objective and ×1.25 multiplier (Olympus America Inc., Center Valley, PA). Red staining in each field was digitally extracted and quantified as a percentage of total area in each field.
Quantitative Real Time PCR
Total RNA was isolated from frozen liver tissues using TRI Reagent (Molecular Research Center, Cincinnati, OH) according to the manufacturer’s instructions. The RNA samples were treated with DNase I (Roche Diagnostics, Indianapolis, IN) and then reverse transcribed using a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific Inc., Grand Island, NY) in a thermocycler. Real time PCR assay was then performed with mouse TaqMan Gene Expression Assays (Thermo Fisher Scientific) for each gene of interest using an Applied Biosystems 7900 Sequence Detection System. In each experiment mRNA levels were normalized to β2-microglobulin which did not change under experimental conditions (data not shown). Individual normalized mRNA values were expressed as fold change relative to the normalized value from a single control animal (calibrator); in Fig. 1 only, for clarity of presentation of mRNA levels across a variety of lipid droplet-associated protein species, each mRNA value normalized to calibrator was expressed as fold change relative to the mean of normalized values from all control samples tested. The catalog number for all the mouse TaqMan Gene Expression Assays obtained from Thermo Fisher Scientific is 4331182 and the individual Assay IDs are as follows: β2-microglobulin (Mm00437762_m1); cell death inducing DNA fragmentation factor subunit alpha like effector (Cide)a (Mm00432554_m1); Cideb (Mm00438213_m1); Cidec (Mm00617672_m1); perilipin (Plin)2 (Mm00475794_m1); Plin3 (Mm04208646_g1); Plin5 (Mm00508852_m1); hypoxia-inducible lipid droplet-associated (Hilpda) protein (Mm00727638_s1); glycerol-3-phosphate acyl transferase (Gpat) (Mm00833328_m1); diacylglycerol acyltransferase (Dgat1) (Mm00515643_m1); lipin-1 (Mm00550511_m1); sterol regulatory element binding protein-1 (SREBP-1) (Mm00550338_m1); acetyl-CoA carboxylase (ACC) (Mm01304257_m1); fatty acid synthase (FAS) (Mm00662319_m1); peroxisome proliferator activated receptor alpha (PPARα) (Mm00440939_m1) and PPARγ coactivator 1 alpha (PGC1α) (Mm01208835_m1).
Figure 1.
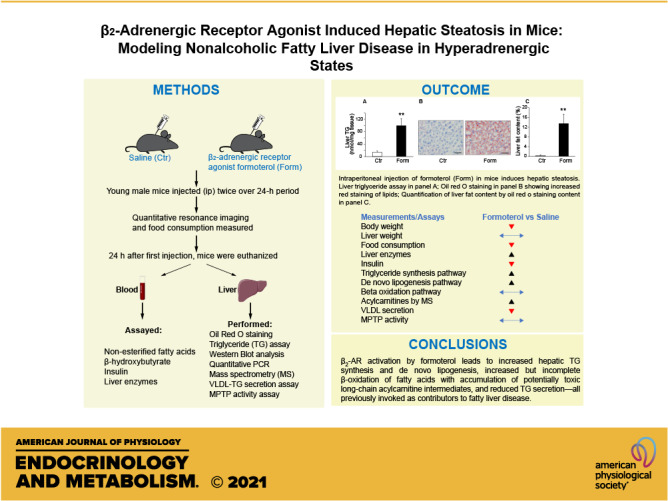
Intraperitoneal injection of formoterol (Form) induces hepatic steatosis and increases gene expression of lipid droplet-associated proteins in mice. A: liver triglyceride (TG) content was measured and normalized to tissue weight in young (3.6–5.4 mo old) male mice injected with Form (20 µg/g) or saline (Control, Ctr). n = 7 mice/group. B: representative Oil Red O (ORO)-stained liver sections of individual saline (Ctr) and Form-treated mice. Scale bar, 50 µm. C: quantitation of lipid levels in mouse liver determined by ORO staining. n = 6 mice/group. D: mRNA levels of lipid droplet-associated proteins in livers of mice injected with saline (Ctr) or Form measured by quantitative real time PCR. n = 5–7 mice/group. E: hepatic glycogen levels were measured using a glycogen assay kit (Cat. No. MAK016-1KT; Sigma-Aldrich) after Form treatment of mice and compared to levels from saline treated (Ctr) animals. n = 7 mice/group. Data are presented as means ± SE in all studies; *P < 0.05 vs. Ctr; **P < 0.01; ***P < 0.001.
Subcellular Fractionation
Nuclear, endoplasmic reticulum (ER) and cytosolic subcellular fractions were obtained using differential centrifugation. Briefly, liver tissue was homogenized in isotonic buffer using a teflon pestle and a glass homogenizer. After sequential centrifugations to collect the nuclear (1000 g pellet) and mitochondrial (15,000 g pellet) fractions, the post mitochondrial supernatant was centrifuged at 100,000 g for 1 h at 4°C. The resultant pellet yielded the crude ER fraction, while the remaining supernatant was collected as the cytosolic fraction. Crude ER proteins were homogenized in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors.
Western Blotting
Frozen liver pieces were homogenized in lysis buffer (50 mM NaCl, 1% NP-40, 50 mM Tris-HCl, pH 7.4) containing protease and phosphatase inhibitors (Thermo Fisher Scientific). Homogenates were rocked at 4°C for 60 min, followed by centrifugation at 14,000 g for 15 min at 4°C. The supernatant was collected and protein concentration was estimated by bicinchoninic acid (BCA) assay (Thermo Fisher Scientific). Protein samples (40–70 µg) from whole homogenates or ER, nuclear or cytosolic fractions were added to 2X Laemmli sample buffer (Bio-Rad, Hercules, CA) containing β-mercaptoethanol and then boiled. Samples were size-fractionated on SDS-PAGE gels and electroblotted onto PVDF membranes using iBlot Gel Transfer Device (Thermo Fisher Scientific). For most proteins of interest membranes were immunoblotted overnight at 4°C with a primary antibody followed by a secondary infrared fluorescent dye (IRDye) conjugated antibody (Cat. No. 926-32211 or Cat. No. 926-32212; LI-COR Biotechnology, Lincoln, NE) at 1:20,000 for 1 h at room temperature. Specific proteins were visualized using ODYSSEY CLx imager and blot intensity was analyzed using Image Studio software (LI-COR Biotechnology). Primary antibodies used against lipin-1 (sc-98450), SREBP-1 (sc-8984), nuclear PPARα (sc-9000), PGC1α (sc-13067), calnexin (sc-6465), and histone (sc-10809) were from Santa Cruz Biotechnology (Dallas, TX); MTTP (Cat. No. 612022) was from BD Biosciences (San Jose, CA); and tubulin (PA5-27108) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (MA5-15738) were from Thermo Fisher Scientific. For detection of FAS (sc-20140; Santa Cruz Biotechnology), PGC1α, PPARα (ab24509; Abcam, Cambridge, MA), and loading control actin (A2066; Sigma-Aldrich) in liver homogenates, following overnight incubation with the primary antibodies blots were washed and incubated with horseradish peroxidase-conjugated secondary antibody (Cat. No. 7074; Cell Signaling, Danvers, MA or Cat. No. 111-035-144; Jackson ImmunoResearch Laboratories, Inc, West Grove, PA). Membranes were then developed using ECL (enhanced chemiluminescent) reagent (Thermo Fisher Scientific), proteins were visualized by exposure to X-ray films (22), and band intensities were quantified with ImageJ software. Intensity of blotted proteins in each tissue sample was normalized against loading controls, followed by expression as fold change relative to the mean of normalized values from all control samples tested.
Extraction of Lipids from Liver and Quantitation of Acylcarnitines
Lipids were extracted from liver tissues in ice-cold chloroform/methanol (2:1) containing [13C4]palmitoylcarnitine, using a bead homogenizer (Biospec Products, Bartlesville, OK). After centrifugation, the chloroform layer was removed, dried in vacuo and reconstituted in 40% isopropanol. Lipid extracts were analyzed on a Thermo Fisher Q Exactive mass spectrometer (MS) with online separation using a Thermo Fisher/Dionex HPLC: column, Acclaim C30 (3 µm, 2.1 × 150 mm); mobile phase A, acetonitrile/water (40:60), 10 mM ammonium acetate; mobile phase B, acetonitrile/isopropanol (10:90), 10 mM ammonium acetate; flow rate, 250 µL/min; gradient, 40% B to 99% B in 15 min. Data-dependent analyses were conducted using one full MS scan followed by 10 tandem-MS scans (positive ion). TraceFinder and LipidSearch (Thermo Fisher Scientific) were employed for data processing. Acylcarnitine identification was based on accurate mass, fragment ions, library searching and comparison with authentic standards. Levels of acylcarnitines were expressed relative to the intensity of the internal standard, normalized to tissue weight.
Hepatic Very Low Density Lipoprotein-TG Secretion
Mice were injected with Triton WR-1339 (1,000 µg/g ip; Sigma-Aldrich) in saline, and VLDL-TG secretion from the liver was assessed by determining serial plasma TG levels over a 2-h period. In these experiments Triton WR-1339 was administered 22 h after the first injection of formoterol or saline.
Microsomal Triglyceride Transfer Protein Activity
Activity of microsomal triglyceride transfer protein (MTTP) in liver homogenates was measured using a fluorometric assay kit (Cat. No. MAK110-1KT) from Sigma-Aldrich following the manufacturer’s instructions. Briefly, liver tissues were homogenized in ice-cold lysis buffer (10 mM Tris, pH 7.4, 150 mM NaCl and 2 mM EDTA) containing protease inhibitors, sonicated, and centrifuged at 100 g for 10 min at 4°C to remove debris. Protein concentration was measured using the BCA method. Liver homogenate (50 µg in 10 µL assay buffer) was added to each well with 4 µL donor particle and 4 µL acceptor particle and 182 µL assay buffer. After 3-h incubation at 37°C, fluorescence was measured at excitation 465 nm and emission 535 nm. Standard curves were generated and activity was expressed as µM/h/mg protein.
Data Analysis
Data from multiple experiments were expressed as means ± standard error (SE). Statistical significance was determined by Student’s t test.
RESULTS
Formoterol Treatment Increases Hepatic Lipid Accumulation in Mice
The long-acting β2-AR agonist formoterol was utilized to determine if activation of β2-ARs promotes hepatic steatosis. Compared with control mice after saline injection, administration of formoterol (20 µg/g i.p.) markedly elevated TG content (P < 0.01) and lipid droplet accumulation (Fig. 1, A–C) in liver over 24 h. Quantitation of lipid droplets by ORO staining showed that hepatic fat content in formoterol-injected mice was distinctly higher than that measured in control animals injected with saline (P < 0.01; Fig. 1C). A marked increase in gene expression of lipid droplet-associated proteins such as cell death inducing DNA fragmentation factor subunit alpha like effector (Cide)a, Cideb and Cidec, perilipins (Plin2, Plin3, Plin5) and hypoxia-inducible lipid droplet-associated (Hilpda) protein was also observed upon formoterol treatment (Fig. 1D). Consistent with the known glycogenolytic function of cAMP/protein kinase A signaling in the liver, we found that hepatic glycogen content was reduced by 68% with formoterol treatment, compared to saline administration (Fig. 1E).
Whole Body Measurements and Biochemical Analysis of Plasma following Formoterol Treatment
Treatment of mice with formoterol for 24 h resulted in significant reduction of body weight and food consumption (both P < 0.001 vs. pretreatment measurements) whereas no changes in these parameters were noted in saline treated animals (Table 1). Quantitative magnetic resonance imaging revealed decreased percent fat content (expressed relative to body weight) and increased percent lean body weight after formoterol (P < 0.001) but not saline treatment. Liver weights were comparable in animals treated with formoterol or saline.
Table 1.
Whole body measurements before and after 24 h treatment with formoterol (Form) or saline
| Pretreatment | Posttreatment | |
|---|---|---|
| Body weight, g | ||
| Form | 27.5 ± 0.6 | 25.3 ± 0.6*** |
| Saline | 28.1 ± 0.5 | 27.9 ± 0.4 |
| Food consumption, g/24 h | ||
| Form | 4.1 ± 0.3 | 1.3 ± 0.1*** |
| Saline | 3.9 ± 0.2 | 4.2 ± 0.8 |
| Fat, % | ||
| Form | 5.5 ± 0.7 | 3.9 ± 0.8*** |
| Saline | 7.8 ± 0.7 | 8.6 ± 0.9* |
| Lean, % | ||
| Form | 94.1 ± 0.7 | 95.5 ± 0.8*** |
| Saline | 92.0 ± 0.7 | 91.0 ± 0.8** |
| Liver weight, g | ||
| Form | - | 1.34 ± 0.06 |
| Saline | - | 1.38 ± 0.05 |
Intraperitoneal injections of mice with Form (20 µg/g) or saline vehicle over 24 h, measurement of food consumption, and assessments of body fat and lean mass (%) by quantitative magnetic resonance imaging were carried out as described in materials and methods. Data are presented as means ± SE; n = 7–8 mice/group; *P < 0.05 vs. Pretreatment; **P < 0.01; ***P < 0.001.
Plasma biochemical measurements indicated that activity levels of the liver enzymes alanine transaminase (ALT) and aspartate transaminase (AST) were significantly elevated by formoterol compared to saline (control) treatment (Table 2). Further, plasma insulin levels were reduced in formoterol treated mice compared to control animals (P < 0.05). In contrast, plasma non-esterified, or free, fatty acids were unaffected by formoterol treatment. A trend for an increase in plasma β-hydroxybutyrate levels after formoterol treatment was observed when compared with values from control animals (0.05 < P < 0.1).
Table 2.
Biochemical parameters measured in plasma of mice treated with saline (Control, Ctr) or formoterol (Form)
| Ctr | Form | |
|---|---|---|
| ALT activity, nmoL/min/mL | 6.3 ± 0.3 | 8.6 ± 0.5** |
| AST activity, nmoL/min/mL | 22.6 ± 3.9 | 44.9 ± 6.1* |
| Insulin, ng/mL | 1.2 ± 0.2 | 0.6 ± 0.1* |
| Nonesterified fatty acids, µmoL/L | 439.5 ± 40.6 | 523.1 ± 121.2 |
| β-hydroxybutyrate, ng/µL | 42.3 ± 9.2 | 115.0 ± 39.2# |
ALT, alanine transaminase; AST, aspartate aminotransferase. Mice were treated with saline or Form for 24 h, as described in materials and methods. Data are presented as means ± SE; n = 7 mice/group; *P < 0.05 vs. Ctr; **P < 0.01; #0.05 < P < 0.1.
Formoterol Treatment Increases Expression of Hepatic Enzymes Involved in TG Synthesis
As we observed an increase in liver TG levels following formoterol treatment, we investigated whether formoterol increased expression of hepatic enzymes involved in the synthesis of TG from free fatty acids via the glycerol-3-phosphate pathway (9). Enzymes of this pathway, including glycerol-3-phosphate acyl transferase (Gpat) and diacylglycerol acyltransferase (Dgat), are constitutive residents of the endoplasmic reticulum (ER); in contrast, the lipin family of proteins, which possess phosphohydrolase (phosphatidic acid phosphatase) activity, are sequestered in the cytosol and participate enzymatically in the glycerol phosphate pathway only upon translocation to the ER. Quantitative real-time PCR revealed a significant increase (P < 0.001) in mRNA levels of Dgat1 in livers of mice treated with formoterol compared with saline treated controls. In addition, a trend for an increase in mRNA levels of Gpat and lipin-1 (0.05 < P < 0.1) was noted upon formoterol treatment (Fig. 2A). We also examined whether formoterol treatment increased protein levels of lipin-1 and its translocation to the ER. As shown in Fig. 2B, formoterol elevated lipin-1 protein levels (P < 0.05) in both the cytosolic and ER fractions.
Figure 2.

Expression of enzymes involved in hepatic triglyceride synthesis is increased upon formoterol (Form) treatment. A: hepatic mRNA expression of glycerol-3-phosphate acyltransferase (Gpat), lipin-1, and diacylglycerol acyltransferase 1 (Dgat1) was measured by quantitative real-time PCR in saline- (Control, Ctr) and Form-treated animals. n = 5 mice/group. B: protein levels of lipin-1 in endoplasmic reticulum (ER) and cytosolic (Cyto) compartments of liver were measured by western blot analysis. n = 7 mice/group. Inset: representative blots of lipin-1 protein levels in ER and Cyto fractions. Calnexin and tubulin were used as loading controls for ER and Cyto, respectively. Numbers represent molecular weight markers, kD. Data are presented as means ± SE; *P < 0.05 vs. Ctr; ***P < 0.001; #0.05<P < 0.1.
Expression of Key Regulators Controlling De Novo Lipogenesis Is Upregulated in Liver of Formoterol-Treated Mice
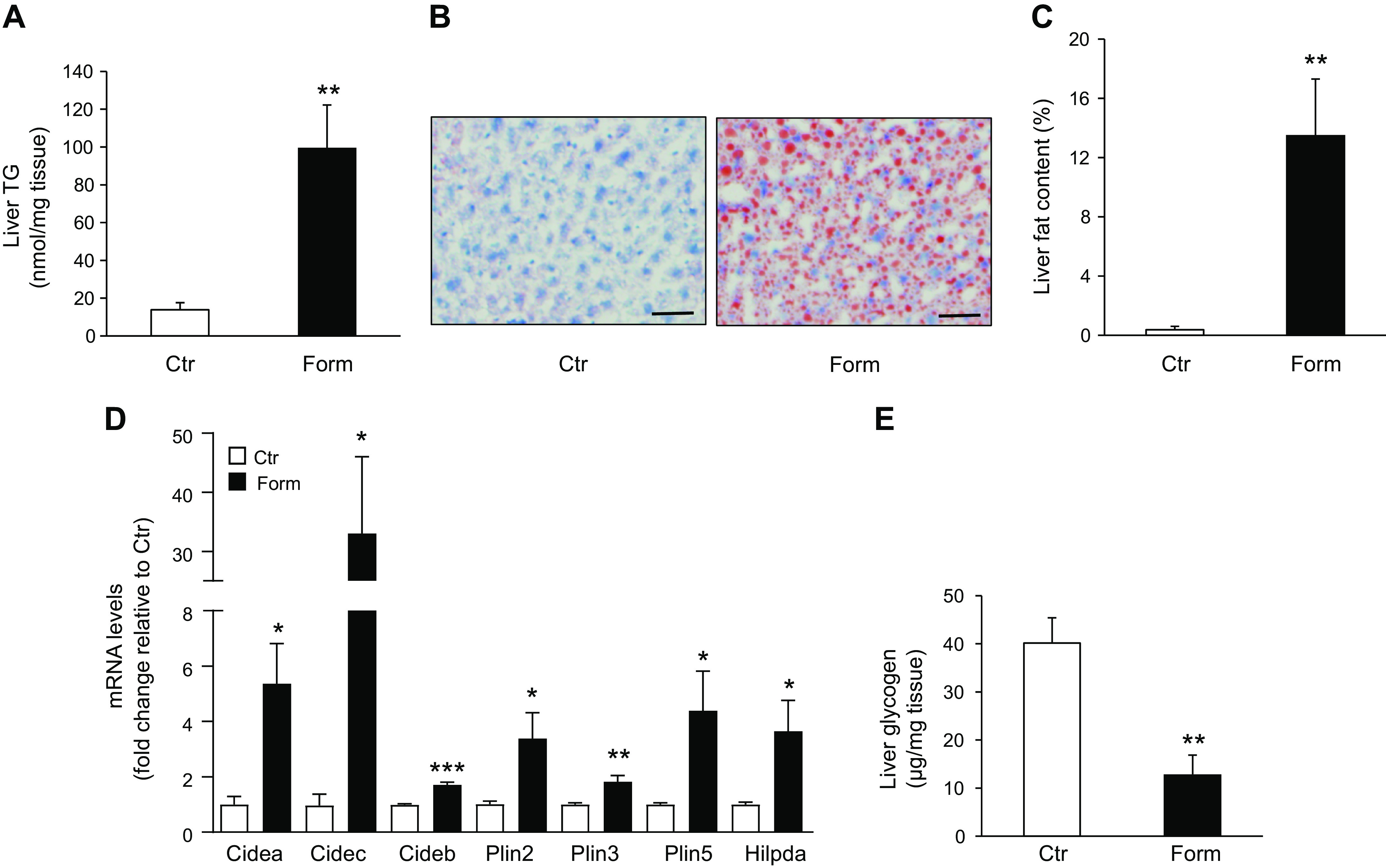
Fatty acids serving as the source for liver TG synthesis are derived either by uptake from the circulation or by de novo lipogenesis. The lipogenic transcription factor sterol regulatory element binding protein-1c (SREBP-1c), a critical regulator of de novo lipogenesis, is a member of the SREBP family of proteins including three isoforms SREBP-1a, SREBP-1c, and SREBP-2, encoded by two genes SREBF1 and SREBF2 (23). Although SREBP-2 regulates cholesterol synthesis, SREBP-1c is the predominant isoform controlling the expression of acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS), enzymes involved in hepatic fatty acid synthesis. SREBPs are synthesized as inactive precursors bound to the ER and upon activation are cleaved into their mature forms that enter the nucleus to activate lipogenic genes. Our results showed that in vivo treatment of mice with formoterol increased hepatic SREBP-1c mRNA levels (P < 0.01; Fig. 3A). Moreover, levels of the mature SREBP-1c protein were increased by formoterol (P < 0.05), without change in levels of the precursor isoform (Fig. 3B), perhaps suggesting that SREBP-1c induced by formoterol localized principally in the nuclear pool. A significant increase in hepatic ACC mRNA levels was also observed in formoterol treated animals (P < 0.05; Fig. 3A). Although we were unable to detect a significant effect of formoterol on FAS mRNA levels, FAS protein levels increased in liver following formoterol treatment (P < 0.05; Fig. 3C).
Figure 3.
Formoterol (Form) treatment upregulates mRNA and protein levels of factors involved in hepatic de novo lipogenesis. A: quantitative real-time PCR of liver samples from mice treated with Form or saline (Control, Ctr) was used to measure mRNA levels of sterol regulatory element binding protein-1c (SREBP-1c), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FAS) involved in de novo lipogenesis. n = 5 mice/group. B: Western blot analysis was used to measure protein levels of SREBP-1c upon Form or saline (Ctr) treatment of mice. Precursor (p)-SREBP-1c; mature (m)-SREBP-1c. n = 7 mice/group. Inset: representative blots of p-SREBP-1c and m-SREBP-1c protein levels; glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as loading control. Numbers represent molecular weight markers, kD. C: protein levels of FAS were measured by western blot analysis. n = 4 mice/group. Inset: Immunoblot of FAS levels; actin was used as loading control. Numbers represent molecular weight markers, kD. Data are presented as means ± SE; * P <0.05 vs. Ctr; **P < 0.01.
Nuclear Protein Levels of Lipin-1 and Expression of Transcriptional Activators Promoting β-Oxidation of Fatty Acids Are Increased in Liver of Formoterol-Treated Mice
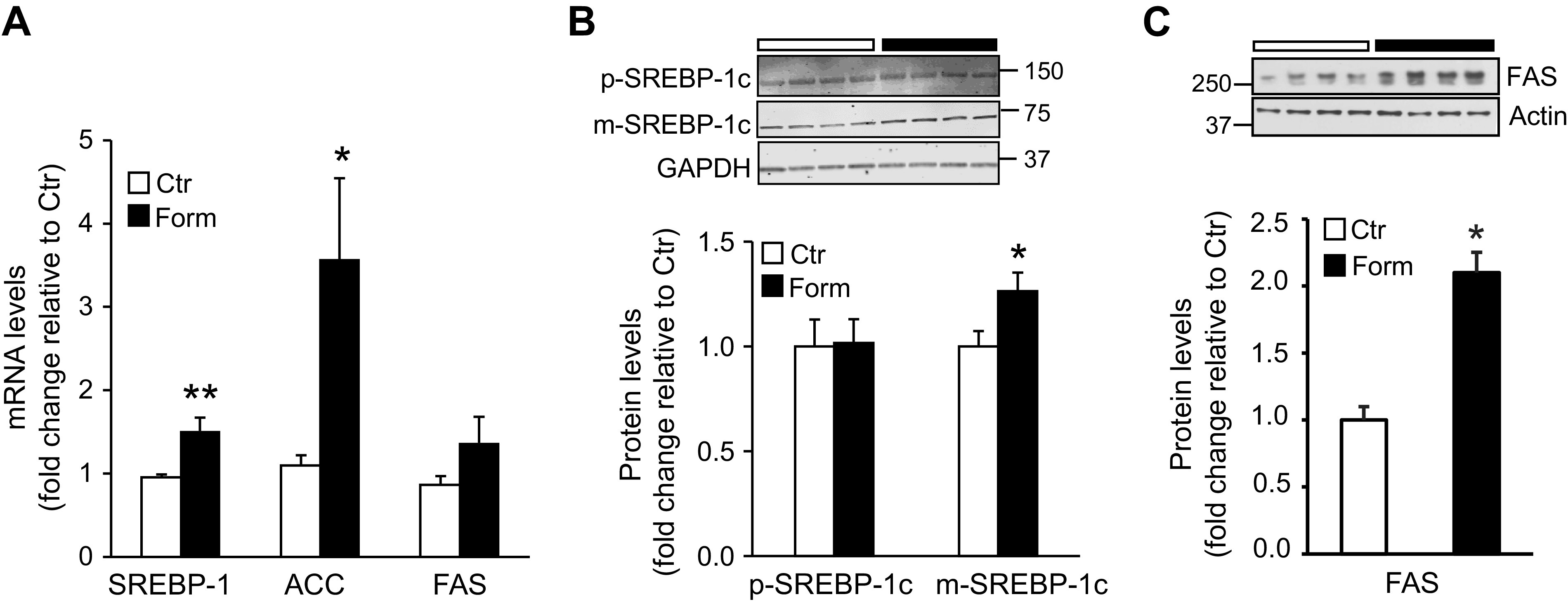
In other studies we investigated whether formoterol treatment might modify regulators of fatty acid β-oxidation. Whereas lipin-1 in the ER functions enzymatically in TG synthesis (see above), lipin-1 also localizes to the nucleus where it acts as a transcriptional co-regulator promoting β-oxidation of fatty acids by interacting with the transcription factor peroxisome proliferator activated receptor alpha (PPARα) and the PPARγ coactivator PGC1α (24, 25). As we observed an increase in lipin-1 protein levels in the cytosol and ER following formoterol treatment of mice (Fig. 2B), we determined whether formoterol would alter nuclear lipin-1 levels as well. In the same experiment PPARα and PGC1α expression levels were compared in livers of animals injected with formoterol or saline. Formoterol treatment resulted in a marked increase of nuclear lipin-1 protein levels (P < 0.05; Fig. 4A). In addition, the β2-AR agonist increased hepatic PPARα and PGC1α mRNA levels (P < 0.05; Fig. 4B) and also protein levels of the two transcriptional activators in whole liver homogenates (P < 0.01; Fig. 4C). However, formoterol treatment had no significant effect on nuclear PPARα and PGC1α protein levels (Fig. 4D). The apparent discrepancy between whole liver and nuclear protein levels of PPARα and PGC1α could reflect altered nuclear turnover of the two transcriptional regulators during heightened β-oxidation of fatty acids accumulating in liver following formoterol treatment (see also discussion).
Figure 4.
Formoterol (Form) treatment of mice upregulates lipin-1 nuclear protein levels and peroxisome proliferator activated receptor α (PPARα) and PPARγ coactivator 1α (PGC1α) mRNA and protein levels in liver. A: Western blot analysis was performed to measure lipin-1 protein levels in hepatic nuclear fractions from mice injected with saline (Ctr) or Form. n = 7 mice/group. Inset: representative Western blot depicting lipin-1 protein levels; histone was used as loading control. Numbers represent molecular weight markers, kD. B: quantitative RT-PCR was used to measure mRNA levels of PPARα and PGC1α in livers from Ctr and Form treated mice. n = 5 mice/group. PPARα and PGC1α protein levels in liver homogenates (C) and hepatic nuclear fractions (D) were measured by Western blot analysis. n = 4–7 mice/group. Insets to (C) and (D): representative blots of PPARα and PGC1α, with actin and histone as respective loading controls. Numbers represent molecular weight markers, kD. Data are expressed as means ± SE; *P < 0.05 vs. Ctr; **P < 0.01.
Levels of Long-Chain Acylcarnitines Are Increased in Liver of Mice following Formoterol Treatment
Acylcarnitines are intermediate metabolites of β-oxidation that accumulate when there is inefficient or incomplete β-oxidation of fatty acids (26). We determined the influence of formoterol administration on hepatic acylcarnitine levels as a surrogate for completion of fatty acid β-oxidation. Long-chain (14–20 carbon atoms) fatty acids are preferentially oxidized in the mitochondria. Of 12 long-chain acylcarnitines identified and quantified by HPLC-electrospray ionization-mass spectrometry in liver, the levels of all but one were significantly higher (P < 0.05) in samples from formoterol treated animals compared to saline treated controls (Fig. 5). Formoterol-stimulated increases in levels of long-chain acylcarnitines ranged from 1.5 [C20:4 carnitine] to 4.8 [C18:3 carnitine] times control levels; only C20:3 carnitine exhibited no change in levels upon formoterol treatment. Accumulation of long-chain acylcarnitines in livers from formoterol treated mice suggests incomplete mitochondrial oxidation of long-chain fatty acids in these animals. Of note, very long-chain (>20 carbon atoms) fatty acids are oxidized predominantly by peroxisomes rather than mitochondria (27). Interestingly, contrasting to the results for long-chain acylcarnitines, hepatic levels of three very long-chain acylcarnitines identified in our study were decreased by formoterol treatment relative to saline controls (Fig. 5).
Figure 5.
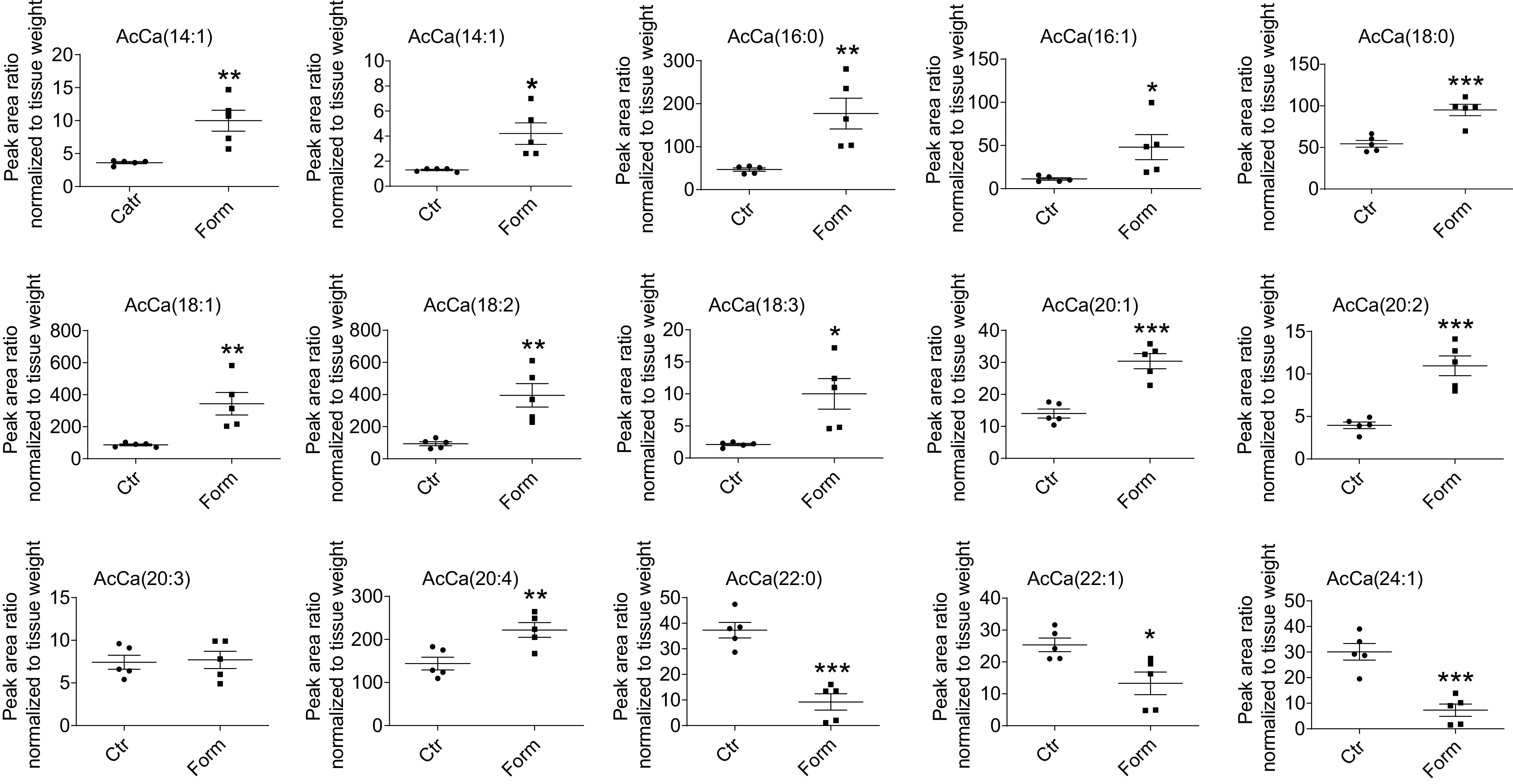
Hepatic levels of long-chain (14–20 carbon atoms) acylcarnitines (AcCa) are increased in response to formoterol (Form) treatment of mice. AcCa levels in lipid extracts of livers from Form- and saline (Control, Ctr)-treated animals were measured by HPLC-electrospray ionization-mass spectrometry as described in materials and methods. Numbers in parentheses designate the number of carbon atoms:number of double bonds. Data are expressed as means ± SE; n = 5 mice/group; *P < 0.05 vs. Ctr; **P < 0.01; ***P < 0.001.
Formoterol Treatment Leads to a Reduction of VLDL-TG Secretion from Liver
Triglyceride export from the liver occurs primarily via secretion into the circulation as VLDL-TG. In addition to increased hepatic lipogenesis, impaired secretion of VLDL-TG, or levels of VLDL-TG secretion insufficient to compensate for accumulation of TG in the liver, have been invoked as contributing to the development and progression of hepatic steatosis (1, 28). We therefore performed experiments to determine whether formoterol treatment decreases VLDL-TG secretion from the liver. Twenty-two hours after the initial i.p. injection of formoterol or saline control (see materials and methods), mice were injected with Triton WR-1339, a lipoprotein lipase inhibitor that prevents plasma VLDL-TG breakdown; and over the ensuing 2 h plasma TG levels were monitored as an index of VLDL-TG secretion from the liver. Triglyceride levels measured 0, 60, and 120 min after Triton injection increased progressively in plasma from saline treated mice, whereas in plasma from formoterol treated animals TG levels were significantly (P < 0.05) reduced compared to controls (Fig. 6A). To investigate whether a decline in VLDL-TG synthesis might account for the reduced VLDL-TG secretion observed in mice treated with formoterol, we examined hepatic levels and activity of the lipid transfer protein MTTP, a critical participant in lipidation of nascent VLDL particles in the ER (1). In these experiments, however, formoterol had no effect on control levels of MTTP protein in the ER fraction or MTTP lipid transfer activity in liver homogenates (Fig. 6, B and C).
Figure 6.
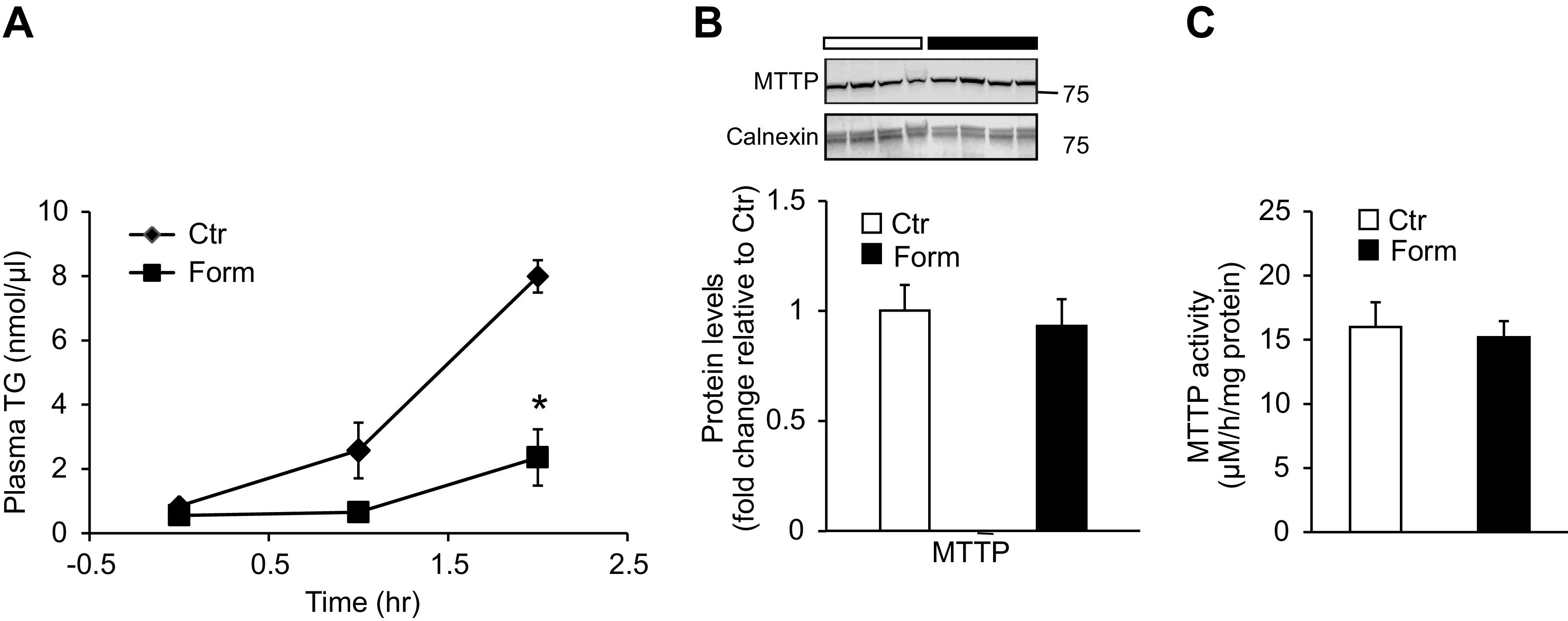
Formoterol (Form) treatment reduces VLDL-TG secretion from liver. A: hepatic VLDL-TG secretion was determined by measuring plasma TG concentrations at indicated times after Triton WR-1339 injection (time 0); Triton was administered 22 h after initial injection of saline (Control, Ctr) or Form. n = 4 mice/group. B: protein levels of microsomal triglyceride transfer protein (MTTP) were measured in the liver ER fraction by western blot analysis. n = 7 mice/group. Inset: representative western blot image depicting MTTP protein levels; calnexin was used as loading control. Numbers represent molecular weight markers, kD. C: lipid transfer activity of MTTP was measured in liver homogenates by fluorescence assay as described in materials and methods. n = 4 mice/group. Data are presented as means ± SE; *P < 0.05 vs Ctr. TG, triglyceride; VLDL, very low density lipoprotein.
DISCUSSION
In this study we demonstrated that in vivo treatment of mice with the β2-AR selective agonist formoterol increases hepatic TG levels and promotes lipid droplet accumulation and gene expression of lipid droplet-associated proteins (Fig. 1). Concomitantly, formoterol treatment was found to increase hepatic expression of enzymes involved in TG synthesis (Fig. 2) together with transcriptional and enzymatic regulators of de novo lipogenesis (Fig. 3). In other experiments formoterol augmented expression levels of nuclear factors activating fatty acid β-oxidation in liver (Fig. 4), yet interestingly increased accumulation of long-chain acylcarnitines, by-products of incomplete fatty acid oxidation (Fig. 5). A decrease in secretion of TG from the liver as VLDL-TG was also observed following formoterol treatment (Fig. 6). Taken together, our results suggest that systemic β2-AR activation in mice induces hepatic steatosis by modifying intrahepatic processes of lipid deposition, metabolism and secretion that may be involved in the development of NAFLD.
Lipid droplets are organelles that store TG and other neutral lipids within cells (23, 29–33). Accumulation of lipid droplets within hepatocytes induces perturbations of cellular structure leading to destructive processes (apoptosis, ballooning, and inflammation) characteristic of NAFLD. Accordingly, lipid droplets are thought to play a critical role in the pathology and progression of NAFLD (30, 31). Numerous proteins associated with lipid droplets, including the Cide family of proteins (Cidea, Cideb, Cidec), perilipins (Plins 1–5) and Hilpda, have emerged as important regulators of lipid droplet formation and growth (23, 29, 32, 33). Expression of Cidea and Cidec is highly correlated with the severity of hepatic steatosis in humans and not detected in normal liver specimens (33). Moreover, perilipin family members are variably upregulated in hepatocytes both in humans with NAFLD and in animal models of hepatosteatosis (29, 32, 33), whereas mice with high-fat diet-induced liver steatosis exhibit augmented hepatic expression of Hilpda (23). In the present study treatment of mice with the β2-AR agonist formoterol increased liver mRNA levels of Cidea, Cideb, Cidec, Plin2, Plin3, Plin5, and Hilpda (Fig. 1D). We previously reported that whole body β2-AR ablation in mice blunted increases in both liver TG levels and hepatic Cidea expression otherwise observed during senescent aging of wild type animals (16). Our current and earlier findings therefore implicate β2-AR signaling in lipid droplet protein expression patterns associated with liver steatosis and accompanying hepatocyte damage and dysfunction. This conclusion is supported by our observation that circulating levels of liver enzyme activities, which are widely used clinically to assess liver cell injury and are commonly elevated in NAFLD, were increased in formoterol treated mice compared to control animals (Table 2).
Hepatic steatosis reflects an imbalance between pathways leading to TG accumulation and processes controlling lipid metabolism and removal (1). Numerous lines of evidence suggest that our findings implicating a role for β2-AR signaling in multiple aspects of hepatic lipid acquisition may be applicable to the pathogenetic mechanisms underlying NAFLD. In the present study formoterol treatment of mice augmented hepatic mRNA levels of Dgat1, the final enzyme in the glycerol-3-phosphate pathway of TG synthesis, as well as ACC and FAS, enzymes involved in de novo synthesis of fatty acids; mRNA levels of Gpat, the first enzyme in the TG synthesis pathway, also tended to increase with formoterol treatment (Figs. 2 and 3). Moreover, mRNA levels encoding the lipogenic transcription factor SREBP-1c, which activates expression of ACC, FAS and other enzymes controlling de novo lipogenesis, were elevated in liver of formoterol treated animals (Fig. 3). These results resemble those in clinical studies of liver biopsies from patients with NAFLD, in which hepatic expression levels of Dgat1, ACC, FAS, and SREBP-1c were upregulated in comparison with levels in normal liver specimens (34). Further, investigations combining multiple stable isotope methodology with liver biopsy to determine sources of liver TG in patients with NAFLD revealed rates of de novo lipogenesis 4–5 times those measured in healthy subjects (4). In our own experiments formoterol also augmented mature but not precursor protein levels of SREBP-1c in liver (Fig. 3), but whether SREBP-1c cleavage and transport to the nucleus might represent processes by which β2-AR linked signaling could contribute to excessive hepatic TG accumulation in NAFLD is yet to be determined.
Other results from the current study raise the possibility that enhanced β2-AR signaling in liver may contribute to modifications in processes of hepatic lipid metabolism associated with NAFLD. In our experiments formoterol treatment of mice increased liver mRNA and protein levels of PPARα and PGC1α, transcriptional activators of genes controlling metabolism of fatty acids via mitochondrial β-oxidation (Fig. 4) (24, 25). We are not aware of any previous investigations reporting an effect of β2-AR signaling on expression levels of β-oxidation gene activators in liver; however, in skeletal muscle, where PGC1α expression exhibits an isoform profile differing from that in liver, β2-AR activation produces isoform-specific increases in PGC1α mRNA levels (35). Acetyl-CoA produced during β-oxidation is a substrate for energy production, either by oxidation in the tricarboxylic acid (TCA) cycle leading to ATP synthesis via mitochondrial respiration/electron transport or, when generated in excess, by conversion to ketone bodies such as β-hydroxybutyrate (1, 9). Plasma β-hydroxybutyrate levels measured in our study tended to be higher in formoterol treated mice compared to control animals (Table 2), further suggesting an increase in β-oxidation following formoterol administration. Similarly, preclinical and human studies have in many cases reported findings consistent with increased fatty acid β-oxidation in NAFLD, including elevations in liver PGC1α expression, lipid oxidation measured by indirect calorimetry, and circulating levels of β-hydroxybutyrate (36–38). In some studies of humans with NAFLD, however, contrasting findings such as reduced hepatic expression levels of PGC1α are not clearly consistent with upregulated β-oxidation (39).
Notwithstanding considerable evidence for increased β-oxidation associated with NAFLD, fatty acid metabolism in liver appears to be inadequate to accommodate the lipid overload occurring in states of hepatosteatosis, including under conditions of systemic β2-AR stimulation. Our results demonstrated that formoterol treatment of mice substantially increased hepatic levels of an array of long-chain acylcarnitines (Fig. 5), fatty acid β-oxidation intermediates thought to accumulate during incomplete, or impaired, β-oxidation and to participate in mitochondrial dysfunction (26, 40). Analogous to this finding, numerous studies have described accumulation of long-chain acylcarnitines in livers from patients with NAFLD and animals with dietary induced fatty liver; generally in these investigations the extent of acylcarnitine accumulation increased with progression of fatty liver changes from simple steatosis to NASH (40–43). Importantly, despite increases in TCA activity observed in fatty liver and persistent through progression to NASH, an apparently adaptive upregulation of mitochondrial respiration occurs in early NAFLD but is lost in steatohepatitis (37, 39, 41, 44). Although the precise functional relationships among mitochondrial β-oxidation, TCA activity, and respiration rates during progression of NAFLD remain incompletely understood, the notion of increased yet incomplete β-oxidation in NAFLD, with resultant accumulation of acylcarnitine intermediates toxic to mitochondrial function, appears to reflect a level of fatty acid oxidation in excess of that which can be processed for energy production through TCA and/or respiratory activities (26). Additional investigations will be required to determine whether β2-AR stimulation leads to changes in mitochondrial TCA and respiratory function approximating those seen clinically and in dietary animal models of NAFLD. Quantitation of acylcarnitines in our study was also noteworthy in revealing significantly reduced levels of very long-chain acylcarnitines in livers of formoterol treated mice compared to controls (Fig. 6). This finding may be related to the fact that, whereas long-chain fatty acids are oxidized mainly in mitochondria, very long-chain fatty acids are oxidized principally in peroxisomes, where they are shortened to long-chain fatty acids for further oxidation in mitochondria (27).
Another means by which heightened β2-AR signaling might promote hepatic steatosis is by inhibition of TG export from the liver as VLDL-TG. The main structural protein of VLDL is apolipoprotein B-100 (apoB), and assembly and secretion of VLDL particles require, among other steps, transfer of neutral lipids such as TG to nascent apoB by the lipid transfer protein MTTP (45). In the present work we demonstrated that treatment of mice with formoterol reduced secretion of VLDL-TG from the liver into the circulation (Fig. 6A). This observation extends earlier in vitro studies in which the β-AR agonist isoproterenol and cAMP analog were found to suppress secretion of TG from rat hepatocytes (46, 47). Evidence from a variety of clinical investigations points to a complex role for dysfunctional VLDL-TG secretion in the development of NAFLD and its progression to NASH. On the one hand, inactivating mutations in apoB and MTTP lead to defective VLDL export and fatty liver in humans (1, 9). In apparent contrast, VLDL-TG secretion rates measured in individuals with NAFLD are considerably greater than in control subjects without evident hepatosteatosis (28, 48); yet, as with the increase in fatty acid β-oxidation associated with steatosis (see above), the extent to which VLDL-TG secretion is elevated in NAFLD appears insufficient to offset excessive intrahepatic lipid accumulation. The failure of VLDL-TG secretion to accommodate to TG overload in NAFLD has been attributed to inadequate rates of apoB (and possibly MTTP) synthesis that may even fall to subnormal levels, especially with disease progression to NASH (28, 45, 48, 49). Thus have recent studies provided support for the idea that methods of increasing VLDL export may be valid therapies for NASH (50). In our experiments hepatic MTTP protein levels and lipid transfer activities were unaffected by formoterol treatment (Fig. 6B). However, in the context of evidence for limitations in apoB synthesis and VLDL secretion in NASH, further experiments are called for to determine the influence of β2-AR signaling on apoB expression and progression of fatty liver damage.
In our experiments we observed that formoterol treatment of mice increased lipin-1 protein levels in liver ER, nuclear and cytosolic fractions, and also tended to increase lipin-1 mRNA levels in whole liver preparations (Figs. 2 and 4). Effects of β2-AR agonist on lipin-1 expression in liver, as seen in the present study, are of particular interest in that this multifunctional protein influences several pathways underlying hepatic lipid accrual and disposal. Dual functions of lipin-1—in the ER as the phosphatidate phosphatase catalyzing the penultimate step in TG synthesis, and in the nucleus as a transcriptional coactivator participating with PPARα and PGC1α in promoting transcription of fatty acid β-oxidation genes–have been well described (24, 25). Elegant studies conducted in vitro and in vivo have also identified nuclear lipin-1 as a key factor mediating suppression of the SREBP-1 pathway of de novo lipogenesis by the nutrient sensitive kinase mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) (51). Other investigations point to a role for lipin-1 in the control of VLDL-TG assembly and secretion, although whether lipin-1 increases or decreases TG export by VLDL is controversial and apparently dependent on the experimental systems and conditions employed (52, 53). Intracellular trafficking of lipin-1 is directed in part by its phosphorylation status, such that phosphorylated lipin-1 is retained in the cytosol whereas dephosphorylation targets lipin-1 to its sites of action in ER and nuclear compartments (24, 25). Of note, cAMP and β2-AR agonists have previously been reported to increase lipin-1 expression in liver and skeletal muscle, respectively (54–56); and while the literature also suggests that catecholamines, β-AR agonist and/or cAMP may promote lipin-1 dephosphorylation and its localization to the ER in adipocytes and hepatocytes (57–59), our own findings do not address whether formoterol might influence lipin-1 translocation to any specific cellular compartment(s). In regulating numerous aspects of hepatic lipid metabolism also responsive to formoterol, however, lipin-1 could play a central role in at least some of the cellular processes by which β2-AR signaling in liver leads to hepatosteatosis.
A hallmark of NAFLD is its strong association with insulin resistance in liver and other insulin responsive targets such as adipose tissue and skeletal muscle. This association remains incompletely defined, however, as NAFLD may exist without insulin resistance under some clinical and experimental circumstances; and whether NAFLD is a result or cause of insulin resistance has been a subject of debate (1, 10, 60, 61). Notably obesity and aging, two prominent risk factors for NAFLD, are also associated with insulin resistance (10, 62, 63). In characterizing fatty liver in mice treated with formoterol, we did not formally assess hepatic insulin sensitivity. Nonetheless, livers of these animals might be expected to be insulin resistant, in that in pancreatic clamp studies we previously demonstrated acute induction of hepatic insulin resistance in rodents infused with the β-AR agonist isoproterenol (64). Earlier results from insulin clamp experiments in humans also showed that the mixed α/β-AR agonist epinephrine acutely impaired hepatic insulin sensitivity via a β-AR-mediated mechanism (65). Whereas insulin resistance is generally accompanied by increased circulating insulin levels, in the current study plasma insulin levels in mice treated for 24 h with formoterol were significantly reduced compared to levels in saline treated control animals (Table 1). This finding may be explained by the observation that formoterol treatment led to substantial reduction in food intake, with associated decrease in body weight (Table 2), at a time likely preceding equilibration of metabolic responsiveness to acutely induced insulin resistance. Comparable, transitory weight loss, thought to be related to short-term appetite suppression, has previously been reported in rodents treated with formoterol and other β2-AR agonists (66). Additional experiments will be required to document insulin resistance, and presumably elevated circulating insulin levels, in mice treated for longer periods with formoterol. Questions of cause-and-effect relationships between NAFLD and insulin resistance after formoterol treatment also arise from our current results and warrant more detailed study of β2-AR signaling mechanisms contributing to steatosis. For example, in the context of earlier suggestions that accumulation of fatty acid β-oxidation intermediates like acylcarnitines may impact negatively on insulin signaling (26), does the increase in hepatic long-chain acylcarnitines we have seen upon formoterol treatment (Fig. 5) play a role in β-AR mediated hepatic insulin resistance? Of further interest, insulin is a primary activator of SREBP-1c expression and activation; and hepatic insulin resistance associated with NAFLD is thought to be “selective” in that elevated circulating insulin levels accompanying insulin resistance continue to stimulate the SREBP-1c pathway of de novo lipogenesis even while failing to suppress liver gluconeogenesis (67). In our experiments the increases in SREBP-1c mRNA expression and mature SREBP-1c protein levels observed after acute administration of formoterol (Fig. 3) were dissociated from hyperinsulinemia, i.e., occurred in the presence of insulin levels that were lower than in saline treated controls. It remains to be clarified whether SREBP-1c expression and activation of the lipogenic pathway would be further modified in association with increasing plasma insulin levels likely to occur with recovery of appetite and food consumption upon more prolonged treatment with β2-AR agonist.
That food intake and body weight declined in formoterol treated animals (Table 1) raises the question of whether short term fasting, which is known to induce hepatic TG accumulation and steatosis (68), might have contributed to changes in liver fat metabolism implicated in the present study. The possibility of fasting as a confounder to interpretation of our results is especially pertinent in that fasting results in stimulation of sympathetic nervous system activity, which in turn may mediate some of the metabolic consequences of the food deprived state (68). However, a number of whole body and hepatic changes observed after formoterol treatment do not resemble those occurring with fasting. In our study male C57Bl/6 mice treated with formoterol over 24 h were not completely deprived of food, and consumed about one-third the amount of food measured before treatment with the β2-AR agonist (Table 1). Further, weight loss of ∼8% after formoterol treatment in these animals was half that previously observed in male C57Bl/6 mice after short term fasting; also, whereas liver weight was found to decline dramatically in fasted mice, we observed no difference in liver weights between formoterol- and saline-treated mice (Ref. 68; see also Table 1). Circulating liver enzyme activity levels, which were elevated after formoterol treatment (Table 1), are unchanged by fasting of mice (68). Although fasting and formoterol both result in reduced plasma insulin levels over the short term, hepatic insulin sensitivity is impaired by β-AR agonist but not during fasting (see previous paragraph, Table 2 and Ref. 69). Of particular note, C57Bl/6 mice treated with formoterol or fasted short term exhibit distinct changes in expression patterns of hepatic lipid regulatory molecules: whereas our results point to formoterol induced increases in expression of SREBP-1c, ACC, FAS, and PPARα (Figs. 2–4), previous work (69, 70; Kamat A., unpublished) demonstrated decreased, or unchanged, expression levels of these same enzymes and transcription factors in mice fasted short term. It should be noted that formoterol and fasting both result in increased expression levels of hepatic lipin-1, Dgat1, and PGC1α in C57Bl/6 mice (Figs. 2 and 4) (69, 71); also plasma levels of β-hydroxybutyrate appear to increase after formoterol treatment (Table 2) as they do during fasting (68). In aggregate, however, the differences observed between the effects of formoterol and fasting suggest to us that formoterol promotes liver steatosis by mechanisms to a large extent distinct from those exerted by fasting.
Adipose tissue lipolysis, with attendant release of NEFAs into the circulation, is subject to classical regulatory mechanisms principally involving inhibition by insulin and stimulation by adrenergic agonists acting through the β-AR/AC/cAMP signaling pathway. In NAFLD and other conditions associated with insulin resistance, the inhibitory influence of insulin on lipolysis is reduced; and increased circulating levels of NEFAs, especially from visceral adipose tissues with efferent blood flow into the portal vein, are presented to the liver for uptake and processing (72). Notably, elevated circulating NEFAs account for the majority (59%) of fatty acids incorporated into liver TG in subjects with NAFLD (4). In our current study of intrahepatic pathways underlying formoterol induced liver steatosis, we did not specifically evaluate the effect of the β2-AR agonist on adipose tissue lipolysis. Percent fat mass declined in formoterol-treated mice compared with saline-injected controls (Table 1). Whereas this finding could be compatible with β2-AR stimulated lipolysis and depletion of adipose tissue fat stores, we observed no change in plasma NEFA levels after formoterol administration (Table 2), possibly related to increased fatty acid uptake by the liver. In previous work from this laboratory the antilipolytic drug acipimox was found to have no effect on isoproterenol induced accumulation of hepatic lipid in mice, and we interpreted this result to suggest that steatosis in response to β-AR stimulation might occur primarily via mechanisms intrinsic to liver rather than by altered NEFA flux from adipose tissue (17). It should be emphasized here that a number of investigations in the literature bear on the question of whether β-AR mediated signaling pathways might play a role in excessive adipose tissue lipolysis and generation of circulating NEFAs in hyperadrenergic states—obesity and aging—linked to insulin resistance and NAFLD. The findings of these studies, however, have been variable and inconclusive. In human obesity, for example, β-adrenergic responsive lipolysis is reduced in subcutaneous adipose tissue yet appears to be increased in visceral adipose albeit by mechanisms that remain in question (72–74). Lipolysis as a function of age has been studied mainly in rodents, where adrenergic responsiveness variably decreases or shows no change with age depending on the fat depot studied and the experimental conditions employed (75); we are not aware of any published studies examining β-AR responsive lipolysis in visceral fat during aging.
In conclusion, we have shown that acute administration of the β2-AR agonist formoterol to mice induces liver steatosis; and our findings further suggest that hepatic lipid accumulation upon formoterol treatment reflects disruptions of intrahepatic lipid accumulation and disposal mirroring those underlying NAFLD. Fatty liver changes responsive to heightened hepatic β2-AR signaling are likely to be of particular relevance to NAFLD occurring in association with the hyperadrenergic states of obesity and aging. In this regard experiments are required to determine whether sustained activation of hepatic β2-AR signaling, by chronic treatment of mice with varying doses of formoterol, leads to the development and progression of fatty liver changes modeling NAFLD. We are currently investigating if the accumulation of hepatic long-chain acylcarnitines observed after acute formoterol exposure (Fig. 5) might over longer periods of time play a role in mitochondrial dysfunction, progression of liver damage to NASH, and/or development of hepatic insulin resistance (see also discussion above). Additional investigations into both time- and dose-dependent effects of formoterol may help to clarify apparent differences in our current findings from those of a recently published study showing reduced hepatic steatosis and improved whole-body insulin sensitivity in diet induced obese mice under conditions of chronic stimulation with low doses of the β2-AR agonist clenbuterol (76). Interestingly, augmented β2-AR signaling in liver may also influence progression of NAFLD to end-stage disease, e.g., hepatocellular carcinoma, which in previous studies has been linked to upregulated expression and function of hepatic β2-ARs (77, 78). Moreover, a number of preclinical observations implicate hepatic β2-AR signaling as a potential therapeutic target in the management of NAFLD: for example, mice have been shown to be protected from diet induced hepatosteatosis by ablation of functional protein kinase A, the downstream effector of classical β-AR signaling, in liver (79, 80); and in a recent study liver steatosis in mice fed a high-fat diet was associated with hepatic sympathetic overactivity and was reduced by liver sympathetic denervation (81). Thus ongoing studies into mechanisms underlying formoterol induced fatty liver changes may ultimately inform novel strategies to prevent or treat NAFLD and its progression as a chronic liver disease and metabolic risk factor.
GRANTS
This work was partially supported by Department of Veterans Affairs Biomedical Laboratory Research and Development Service Merit Review Awards (1I01BX001744 to A.K.; 2I01 BX000926 to G.G-C.), Translation Technology Resources pilot award to A.K. (Award Number UL1TR002645 from the National Center for Advancing Translational Sciences), and by the National Institutes of Health Grants R21 AA026922, R01 DK100603, and R01 DK121527 to M.Z. G.G-C. is also a recipient of Research Career Scientist Award IK6BX00361 from the Department of Veterans Affairs Biomedical Laboratory Research and Development Service. Mass spectrometry analyses were conducted in the University of Texas Health Science Center at San Antonio (UTHSCSA) Institutional Mass Spectrometry Laboratory, supported in part by UTHSCSA and by NIH grant 1S10RR031586-01 (to S.T.W.) for purchase of the mass spectrometer. The technical assistance of Xiaoli Gao is gratefully acknowledged.
DISCLOSURES
Y.S., J.P., H.W., F.D., P.A.A., G.G-C., M.Z., S.T.W., C-K.Y., M.S.K., and A.K. have nothing to disclose. J.L.B. is Chief Science Officer and has equity interests in ProbeTex, Inc.
AUTHOR CONTRIBUTIONS
Y.S., M.S.K., and A.K. conceived and designed research; Y.S., A.K., J.P., H.W., F.D., and P.A.A. performed experiments; Y.S., A.K., J.P., H.W., F.D., J.L.B., and S.T.W. analyzed data; Y.S., C.Y., M.S.K., A.K., G.G-C., J.L.B., M.Z., and S.T.W. interpreted results of experiments; Y.S., A.K., H.W., F.D., and P.A.A. prepared figures; Y.S. and A.K. drafted manuscript; Y.S., C.Y., M.S.K., A.K., G.G-C., J.L.B., M.Z., and S.T.W. edited and revised manuscript; M.S.K. and A.K. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of J. Pizzini: Department of Immunology and Microbiology, Baylor College of Medicine, Houston, Texas.
Present address of Y. Shi: Department of Family and Community Medicine, University of Texas Health Science Center at San Antonio, San Antonio, Texas.
REFERENCES
- 1.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science 332: 1519–1523, 2011. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, Harrison SA, Brunt EM, Sanyal AJ. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67: 328–357, 2018. doi: 10.1002/hep.29367. [DOI] [PubMed] [Google Scholar]
- 3.Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med 363: 1341–1350, 2010. doi: 10.1056/NEJMra0912063. [DOI] [PubMed] [Google Scholar]
- 4.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115: 1343–1351, 2005. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woo Baidal JA, Lavine JE. The intersection of nonalcoholic fatty liver disease and obesity. Sci Transl Med 8: 323rv1, 2016. doi: 10.1126/scitranslmed.aad8390. [DOI] [PubMed] [Google Scholar]
- 6.Ahmed M. Non-alcoholic fatty liver disease in 2015. World J Hepatol 7: 1450–1459, 2015. doi: 10.4254/wjh.v7.i11.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alves-Bezerra M, Cohen DE. Triglyceride metabolism in the liver. Compr Physiol 8: 1–8, 2017. doi: 10.1002/cphy.c170012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, Bhanot S, Monia BP, Li Y-X, Diehl AM. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 45: 1366–1374, 2007. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 9.Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol 48: 434–441, 2013. doi: 10.1007/s00535-013-0758-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stefan N, Kantartzis K, Haring HU. Causes and metabolic consequences of fatty liver. Endocr Rev 29: 939–960, 2008. doi: 10.1210/er.2008-0009. [DOI] [PubMed] [Google Scholar]
- 11.Thorp AA, Schlaich MP. Relevance of sympathetic nervous system activation in obesity and metabolic syndrome. J Diabetes Res 2015: 341583, 2015. doi: 10.1155/2015/341583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seals DR, Bell C. Chronic sympathetic activation: consequence and cause of age-associated obesity? Diabetes 53: 276–284, 2004. doi: 10.2337/diabetes.53.2.276. [DOI] [PubMed] [Google Scholar]
- 13.Kawai Y, Powell A, Arinze IJ. Adrenergic receptors in human liver plasma membranes: predominance of beta 2- and alpha 1-receptor subtypes. J Clin Endocrinol Metab 62: 827–832, 1986. doi: 10.1210/jcem-62-5-827. [DOI] [PubMed] [Google Scholar]
- 14.Begin-Heick N. Liver beta-adrenergic receptors, G proteins, and adenylyl cyclase activity in obesity-diabetes syndromes. Am J Physiol Cell Physiol 266: C1664–C1672, 1994. doi: 10.1152/ajpcell.1994.266.6.C1664. [DOI] [PubMed] [Google Scholar]
- 15.Katz MS, Dax EM, Gregerman RI. Beta adrenergic regulation of rat liver glycogenolysis during aging. Exp Gerontol 28: 329–340, 1993. doi: 10.1016/0531-5565(93)90060-Q. [DOI] [PubMed] [Google Scholar]
- 16.Shi Y, Shu ZJ, Wang H, Barnes JL, Yeh CK, Ghosh PM, Katz MS, Kamat A. Altered expression of hepatic beta-adrenergic receptors in aging rats: implications for age-related metabolic dysfunction in liver. Am J Physiol Regul Integr Comp Physiol 314: R574–R583, 2018. doi: 10.1152/ajpregu.00372.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh PM, Shu ZJ, Zhu B, Lu Z, Ikeno Y, Barnes JL, Yeh CK, Zhang BX, Katz MS, Kamat A. Role of beta-adrenergic receptors in regulation of hepatic fat accumulation during aging. J Endocrinol 213: 251–261, 2012. doi: 10.1530/JOE-11-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi Y, Shu ZJ, Xue X, Yeh CK, Katz MS, Kamat A. beta2-adrenergic receptor ablation modulates hepatic lipid accumulation and glucose tolerance in aging mice. Exp Gerontol 78: 32–38, 2016. doi: 10.1016/j.exger.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Lonnqvist F, Nyberg B, Wahrenberg H, Arner P. Catecholamine-induced lipolysis in adipose tissue of the elderly. J Clin Invest 85: 1614–1621, 1990. doi: 10.1172/JCI114612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee P, Day RO, Greenfield JR, Ho KK. Formoterol, a highly beta2-selective agonist, increases energy expenditure and fat utilisation in men. Int J Obes Relat Metab Disord 37: 593–597, 2013. doi: 10.1038/ijo.2012.90. [DOI] [PubMed] [Google Scholar]
- 21.Ikeno Y, Hubbard GB, Lee S, Richardson A, Strong R, Diaz V, Nelson JF. Housing density does not influence the longevity effect of calorie restriction. J Gerontol A Biol Sci Med Sci 60: 1510–1517, 2005. doi: 10.1093/gerona/60.12.1510. [DOI] [PubMed] [Google Scholar]
- 22.Das F, Maity S, Ghosh-Choudhury N, Kasinath BS, Ghosh Choudhury G. Deacetylation of S6 kinase promotes high glucose-induced glomerular mesangial cell hypertrophy and matrix protein accumulation. J Biol Chem 294: 9440–9460, 2019. doi: 10.1074/jbc.RA118.007023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DiStefano MT, Danai LV, Roth Flach RJ, Chawla A, Pedersen DJ, Guilherme A, Czech MP. The lipid droplet protein hypoxia-inducible gene 2 promotes hepatic triglyceride deposition by inhibiting lipolysis. J Biol Chem 290: 15175–15184, 2015. doi: 10.1074/jbc.M115.650184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris TE, Finck BN. Dual function lipin proteins and glycerolipid metabolism. Trends Endocrinol Metab 22: 226–233, 2011. doi: 10.1016/j.tem.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reue K, Brindley DN. Thematic review series: glycerolipids. Multiple roles for lipins/phosphatidate phosphatase enzymes in lipid metabolism. J Lipid Res 49: 2493–2503, 2008. doi: 10.1194/jlr.R800019-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schooneman MG, Vaz FM, Houten SM, Soeters MR. Acylcarnitines: reflecting or inflicting insulin resistance? Diabetes 62: 1–8, 2013. doi: 10.2337/db12-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reddy JK, Mannaerts GP. Peroxisomal lipid metabolism. Annu Rev Nutr 14: 343–370, 1994. doi: 10.1146/annurev.nu.14.070194.002015. [DOI] [PubMed] [Google Scholar]
- 28.Fujita K, Nozaki Y, Wada K, Yoneda M, Fujimoto Y, Fujitake M, Endo H, Takahashi H, Inamori M, Kobayashi N, Kirikoshi H, Kubota K, Saito S, Nakajima A. Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology 50: 772–780, 2009. doi: 10.1002/hep.23094. [DOI] [PubMed] [Google Scholar]
- 29.Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, Puri V, Yan QW, Miyoshi H, Mashek DG. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest 121: 2102–2110, 2011. doi: 10.1172/JCI46069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hall AM, Brunt EM, Chen Z, Viswakarma N, Reddy JK, Wolins NE, Finck BN. Dynamic and differential regulation of proteins that coat lipid droplets in fatty liver dystrophic mice. J Lipid Res 51: 554–563, 2010. doi: 10.1194/jlr.M000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mashek DG, Khan SA, Sathyanarayan A, Ploeger JM, Franklin MP. Hepatic lipid droplet biology: getting to the root of fatty liver. Hepatology 62: 964–967, 2015. doi: 10.1002/hep.27839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gross DA, Silver DL. Cytosolic lipid droplets: from mechanisms of fat storage to disease. Crit Rev Biochem Mol Biol 49: 304–326, 2014. doi: 10.3109/10409238.2014.931337. [DOI] [PubMed] [Google Scholar]
- 33.Xu L, Zhou L, Li P. CIDE proteins and lipid metabolism. Arterioscler Thromb Vasc Biol 32: 1094–1098, 2012. doi: 10.1161/ATVBAHA.111.241489. [DOI] [PubMed] [Google Scholar]
- 34.Kohjima M, Enjoji M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, Yada M, Yada R, Harada N, Takayanagi R, Nakamuta M. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med 20: 351–358, 2007. doi: 10.3892/ijmm.20.3.351. [DOI] [PubMed] [Google Scholar]
- 35.Miura S, Kai Y, Kamei Y, Ezaki O. Isoform-specific increases in murine skeletal muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) mRNA in response to beta2-adrenergic receptor activation and exercise. Endocrinology 149: 4527–4533, 2008. doi: 10.1210/en.2008-0466. [DOI] [PubMed] [Google Scholar]
- 36.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120: 1183–1192, 2001. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 37.Satapati S, Sunny NE, Kucejova B, Fu X, He TT, Mendez-Lucas A, Shelton JM, Perales JC, Browning JD, Burgess SC. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J Lipid Res 53: 1080–1092, 2012. doi: 10.1194/jlr.M023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, Baldi S, Ponti V, Pagano G, Ferrannini E, Rizzetto M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia 48: 634–642, 2005. doi: 10.1007/s00125-005-1682-x. [DOI] [PubMed] [Google Scholar]
- 39.Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, Schlensak M, Roden M. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab 21: 739–746, 2015. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 40.Peng KY, Watt MJ, Rensen S, Greve JW, Huynh K, Jayawardana KS, Meikle PJ, Meex RCR. Mitochondrial dysfunction-related lipid changes occur in non-alcoholic fatty liver disease progression. J Lipid Res 59: 1977–1986, 2018. doi: 10.1194/jlr.M085613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez-Carreras M, Del Hoyo P, Martin MA, Rubio JC, Martin A, Castellano G, Colina F, Arenas J, Solis-Herruzo JA. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 38: 999–1007, 2003. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 42.Lake AD, Novak P, Shipkova P, Aranibar N, Robertson DG, Reily MD, Lehman-McKeeman LD, Vaillancourt RR, Cherrington NJ. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino Acids 47: 603–615, 2015. doi: 10.1007/s00726-014-1894-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patterson RE, Kalavalapalli S, Williams CM, Nautiyal M, Mathew JT, Martinez J, Reinhard MK, McDougall DJ, Rocca JR, Yost RA, Cusi K, Garrett TJ, Sunny NE. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am J Physiol Endocrinol Metab 310: E484–E494, 2016. doi: 10.1152/ajpendo.00492.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab 14: 804–810, 2011. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab 22: 353–363, 2011. doi: 10.1016/j.tem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rasouli M, Zahraie M. Suppression of VLDL associated triacylglycerol secretion by both alpha- and beta-adrenoceptor agonists in isolated rat hepatocytes. Eur J Pharmacol 545: 109–114, 2006. doi: 10.1016/j.ejphar.2006.06.066. [DOI] [PubMed] [Google Scholar]
- 47.Bjornsson OG, Sparks JD, Sparks CE, Gibbons GF. Regulation of VLDL secretion in primary culture of rat hepatocytes: involvement of cAMP and cAMP-dependent protein kinases. Eur J Clin Invest 24: 137–148, 1994. doi: 10.1111/j.1365-2362.1994.tb00979.x. [DOI] [PubMed] [Google Scholar]
- 48.Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 134: 424–431, 2008. doi: 10.1053/j.gastro.2007.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Charlton M, Sreekumar R, Rasmussen D, Lindor K, Nair KS. Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology 35: 898–904, 2002. doi: 10.1053/jhep.2002.32527. [DOI] [PubMed] [Google Scholar]
- 50.Simon J, Nunez-Garcia M, Fernandez-Tussy P, Barbier-Torres L, Fernandez-Ramos D, Gomez-Santos B, Buque X, Lopitz-Otsoa F, Goikoetxea-Usandizaga N, Serrano-Macia M, Rodriguez-Agudo R, Bizkarguenaga M, Zubiete-Franco I, Gutierrez-de Juan V, Cabrera D, Alonso C, Iruzubieta P, Romero-Gomez M, van Liempd S, Castro A, Nogueiras R, Varela-Rey M, Falcon-Perez JM, Villa E, Crespo J, Lu SC, Mato JM, Aspichueta P, Delgado TC, Martinez-Chantar ML. Targeting hepatic glutaminase 1 ameliorates non-alcoholic steatohepatitis by restoring very-low-density lipoprotein triglyceride assembly. Cell Metab 31: 605–622.e10, 2020. doi: 10.1016/j.cmet.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146: 408–420, 2011. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Z, Gropler MC, Norris J, Lawrence JC, Jr., Harris TE, Finck BN. Alterations in hepatic metabolism in fld mice reveal a role for lipin 1 in regulating VLDL-triacylglyceride secretion. Arterioscler Thromb Vasc Biol 28: 1738–1744, 2008. doi: 10.1161/ATVBAHA.108.171538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bou Khalil M, Sundaram M, Zhang HY, Links PH, Raven JF, Manmontri B, Sariahmetoglu M, Tran K, Reue K, Brindley DN, Yao Z. The level and compartmentalization of phosphatidate phosphatase-1 (lipin-1) control the assembly and secretion of hepatic VLDL. J Lipid Res 50: 47–58, 2009. doi: 10.1194/jlr.M800204-JLR200. [DOI] [PubMed] [Google Scholar]
- 54.Manmontri B, Sariahmetoglu M, Donkor J, Bou Khalil M, Sundaram M, Yao Z, Reue K, Lehner R, Brindley DN. Glucocorticoids and cyclic AMP selectively increase hepatic lipin-1 expression, and insulin acts antagonistically. J Lipid Res 49: 1056–1067, 2008. doi: 10.1194/jlr.M800013-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pearen MA, Myers SA, Raichur S, Ryall JG, Lynch GS, Muscat GE. The orphan nuclear receptor, NOR-1, a target of beta-adrenergic signaling, regulates gene expression that controls oxidative metabolism in skeletal muscle. Endocrinology 149: 2853–2865, 2008. doi: 10.1210/en.2007-1202. [DOI] [PubMed] [Google Scholar]
- 56.Higashida K, Higuchi M, Terada S. Potential role of lipin-1 in exercise-induced mitochondrial biogenesis. Biochem Biophys Res Commun 374: 587–591, 2008. doi: 10.1016/j.bbrc.2008.07.079. [DOI] [PubMed] [Google Scholar]
- 57.Harris TE, Huffman TA, Chi A, Shabanowitz J, Hunt DF, Kumar A, Lawrence JC, Jr.. Insulin controls subcellular localization and multisite phosphorylation of the phosphatidic acid phosphatase, lipin 1. J Biol Chem 282: 277–286, 2007. doi: 10.1074/jbc.M609537200. [DOI] [PubMed] [Google Scholar]
- 58.Taylor SJ, Saggerson ED. Adipose-tissue Mg2+-dependent phosphatidate phosphohydrolase. Control of activity and subcellular distribution in vitro and in vivo. Biochem J 239: 275–284, 1986. doi: 10.1042/bj2390275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Butterwith SC, Martin A, Brindley DN. Can phosphorylation of phosphatidate phosphohydrolase by a cyclic AMP-dependent mechanism regulate its activity and subcellular distribution and control hepatic glycerolipid synthesis? Biochem J 222: 487–493, 1984. doi: 10.1042/bj2220487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grefhorst A, Hoekstra J, Derks TG, Ouwens DM, Baller JF, Havinga R, Havekes LM, Romijn JA, Kuipers F. Acute hepatic steatosis in mice by blocking beta-oxidation does not reduce insulin sensitivity of very-low-density lipoprotein production. Am J Physiol Gastrointest Liver Physiol 289: G592–G598, 2005. doi: 10.1152/ajpgi.00063.2005. [DOI] [PubMed] [Google Scholar]
- 61.Utzschneider KM, Kahn SE. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab 91: 4753–4761, 2006. doi: 10.1210/jc.2006-0587. [DOI] [PubMed] [Google Scholar]
- 62.Fink RI, Kolterman OG, Griffin J, Olefsky JM. Mechanisms of insulin resistance in aging. J Clin Invest 71: 1523–1535, 1983. doi: 10.1172/JCI110908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rowe JW, Minaker KL, Pallotta JA, Flier JS. Characterization of the insulin resistance of aging. J Clin Invest 71: 1581–1587, 1983. doi: 10.1172/jci110914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muscogiuri G, Kamat A, Balas B, Giaccari A, Defronzo RA, Musi N, Katz MS. beta-Adrenergic responsive induction of insulin resistance in liver of aging rats. Endocr Res 36: 74–82, 2011. doi: 10.3109/07435800.2010.539993. [DOI] [PubMed] [Google Scholar]
- 65.Deibert DC, DeFronzo RA. Epinephrine-induced insulin resistance in man. J Clin Invest 65: 717–721, 1980. doi: 10.1172/JCI109718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ryall JG, Sillence MN, Lynch GS. Systemic administration of beta2-adrenoceptor agonists, formoterol and salmeterol, elicit skeletal muscle hypertrophy in rats at micromolar doses. Br J Pharmacol 147: 587–595, 2006. doi: 10.1038/sj.bjp.0706669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 7: 95–96, 2008. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 68.Jensen TL, Kiersgaard MK, Sorensen DB, Mikkelsen LF. Fasting of mice: a review. Lab Anim 47: 225–240, 2013. doi: 10.1177/0023677213501659. [DOI] [PubMed] [Google Scholar]
- 69.Heijboer AC, Donga E, Voshol PJ, Dang ZC, Havekes LM, Romijn JA, Corssmit EPM. Sixteen hours of fasting differentially affects hepatic and muscle insulin sensitivity in mice. J Lipid Res 46: 582–588, 2005. doi: 10.1194/jlr.M400440-JLR200. [DOI] [PubMed] [Google Scholar]
- 70.Hashimoto T, Cook WS, Qi C, Yeldandi AV, Reddy JK, Rao MS. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem 275: 28918–28928, 2000. doi: 10.1074/jbc.M910350199. [DOI] [PubMed] [Google Scholar]
- 71.Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Kelly DP, Jr.. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab 4: 199–210, 2006. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 72.Fruhbeck G, Mendez-Gimenez L, Fernandez-Formoso JA, Fernandez S, Rodriguez A. Regulation of adipocyte lipolysis. Nutr Res Rev 27: 63–93, 2014. doi: 10.1017/S095442241400002X. [DOI] [PubMed] [Google Scholar]
- 73.Arner P. Catecholamine-induced lipolysis in obesity. Int J Obes Relat Metab Disord 23 Suppl 1: 10–13, 1999. doi: 10.1038/sj.ijo.0800789. [DOI] [PubMed] [Google Scholar]
- 74.Lafontan M, Langin D. Lipolysis and lipid mobilization in human adipose tissue. Prog Lipid Res 48: 275–297, 2009. doi: 10.1016/j.plipres.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 75.Gregerman RI. Aging and hormone-sensitive lipolysis: reconciling the literature. J Gerontol 49: B135–B139, 1994. doi: 10.1093/geronj/49.4.b135. [DOI] [PubMed] [Google Scholar]
- 76.Kalinovich A, Dehvari N, Aslund A, van Beek S, Halleskog C, Olsen J, Forsberg E, Zacharewicz E, Schaart G, Rinde M, Sandstrom A, Berlin R, Ostenson C-G, Hoeks J, Bengtsson T. Treatment with a beta-2-adrenoceptor agonist stimulates glucose uptake in skeletal muscle and improves glucose homeostasis, insulin resistance and hepatic steatosis in mice with diet-induced obesity. Diabetologia 63: 1603–1615, 2020. doi: 10.1007/s00125-020-05171-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen D, Xing W, Hong J, Wang M, Huang Y, Zhu C, Yuan Y, Zeng W. The beta2-adrenergic receptor is a potential prognostic biomarker for human hepatocellular carcinoma after curative resection. Ann Surg Oncol 19: 3556–3565, 2012. doi: 10.1245/s10434-012-2396-1. [DOI] [PubMed] [Google Scholar]
- 78.Wu FQ, Fang T, Yu LX, Lv GS, Lv HW, Liang D, Li T, Wang C-Z, Tan Y-X, Ding J, Chen Y, Tang L, Guo L-N, Tang S-H, Yang W, Wang H-Y. ADRB2 signaling promotes HCC progression and sorafenib resistance by inhibiting autophagic degradation of HIF1alpha. J Hepatol 65: 314–324, 2016. doi: 10.1016/j.jhep.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 79.Enns LC, Morton JF, Mangalindan RS, McKnight GS, Schwartz MW, Kaeberlein MR, Kennedy BK, Rabinovitch PS, Ladiges WC. Attenuation of age-related metabolic dysfunction in mice with a targeted disruption of the Cbeta subunit of protein kinase A. J Gerontol A Biol Sci Med Sci 64: 1221–1231, 2009. doi: 10.1093/gerona/glp133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.London E, Nesterova M, Sinaii N, Szarek E, Chanturiya T, Mastroyannis SA, Gavrilova O, Stratakis CA. Differentially regulated protein kinase A (PKA) activity in adipose tissue and liver is associated with resistance to diet-induced obesity and glucose intolerance in mice that lack PKA regulatory subunit type IIalpha. Endocrinology 155: 3397–3408, 2014. doi: 10.1210/en.2014-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hurr C, Simonyan H, Morgan DA, Rahmouni K, Young CN. Liver sympathetic denervation reverses obesity-induced hepatic steatosis. J Physiol 597: 4565–4580, 2019. doi: 10.1113/JP277994. [DOI] [PMC free article] [PubMed] [Google Scholar]