

Keywords: heart, insulin, metabolism, signal transduction
Abstract
Insulin receptors are highly expressed in the heart and vasculature. Insulin signaling regulates cardiac growth, survival, substrate uptake, utilization, and mitochondrial metabolism. Insulin signaling modulates the cardiac responses to physiological and pathological stressors. Altered insulin signaling in the heart may contribute to the pathophysiology of ventricular remodeling and heart failure progression. Myocardial insulin signaling adapts rapidly to changes in the systemic metabolic milieu. What may initially represent an adaptation to protect the heart from carbotoxicity may contribute to amplifying the risk of heart failure in obesity and diabetes. This review article presents the multiple roles of insulin signaling in cardiac physiology and pathology and discusses the potential therapeutic consequences of modulating myocardial insulin signaling.
INTRODUCTION
Activation of insulin signaling has important effects on cellular metabolic, growth, and survival pathways. Insulin receptors (IRs) are ubiquitously expressed. Although the majority of studies of insulin signaling have been performed in cells and organs that regulate systemic metabolic homeostasis such as adipose tissue, skeletal muscle, liver, and brain, insulin signaling also plays an important role in other organs such as the heart. As a muscular pump that is uniquely adapted to contract millions of time during a lifetime, without fatigue, the heart is uniquely adapted to generate large quantities of ATP to fuel its contractile apparatus and ionic pumps. Myocardial metabolism is largely governed by substrate availability. Therefore, diurnal and feeding-related changes in circulating insulin influences cardiac metabolism both by its modulation of the periphery to regulate myocardial substrate supply and by direct effects on the myocardium. Although insulin signaling in the cardiomyocyte is not required for maintaining cardiac metabolism in nonstressed hearts (1), studies by many groups have revealed unique roles for insulin signaling in cardiomyocytes that transcend its metabolic effects. This review, written as part of a series celebrating the 100th anniversary of the discovery of insulin, provides an overview of the pleiotropic aspects of myocardial insulin action in health and disease and highlights the aspects of insulin signal transduction and its consequences that might be unique to the heart.
INSULIN SIGNALING AND METABOLIC REGULATION IN THE HEART
Cardiomyocytes exhibit robust expression of insulin receptors (IRs) and nearly equivalent expression of the closely related insulin-like-growth-factor 1 receptor (IGF1R). Significant redundancy and overlap in pathways engaged by these two receptors exists in the myocardium (2). As described in multiple other cell types, both receptors interact with insulin receptor substrate proteins (IRS1 and IRS2) that serve as signaling hubs to transduce insulin signals to downstream pathways via phosphoinositide-3-kinase (PI3K) and protein kinase B (PKB or Akt) or via members of the ERK signaling pathway (Fig. 1). Akt activation phosphorylates many downstream signaling intermediates. These include tuberous sclerosis complex isoform 2 (TSC2) and proline-rich Akt substrate of 40 kDa (Pras40) to increase mechanistic target of rapamycin (mTOR) activity, glycogen synthase kinase 3 (GSK3) that regulates glycogen metabolism and cell survival pathways, nitric oxide synthase isoform 3 (NOS3) (eNOS) that regulates nitric oxide and cyclic guanosine monophosphate (cGMP) generation, antiapoptotic mediators such as BCL2-associated agonist of cell death promoter (BAD) and members of the forkhead (forkhead box O, FOXO) family of transcriptional regulators (3, 4) (Fig. 1). Together, activation of insulin signaling modulates diverse cellular processes within cardiomyocytes ranging from metabolism, to cell growth, cell survival, and suppression of apoptosis and autophagy (5). Novel mechanisms involved in the activation of insulin signaling in cardiomyocytes include a requirement for NOX2/4 generated ROS (6). There are targets of insulin action that might be unique to cardiomyocytes, such as the regulation of ion channel function (7).
Figure 1.

Schematic representations of signaling intermediates described in cardiomyocytes that are activated downstream of the insulin or insulin-like-growth-factor 1 (IGF-1) receptors. Activation of insulin signaling intermediates mediate pleiotropic effects that are summarized. 4E-BP1, eukaryotic translation initiation factor 4E-binding protein 1; Akt1/2, protein kinase B isoforms 1 and 2; BAD, BCL2 associated agonist of cell death promoter; CD36, cluster of differentiation 36, a.k.a. platelet glycoprotein 4, fatty acid translocase (FAT), scavenger receptor class B member 3 (SCARB3), and glycoproteins 88 (GP88), IIIb (GPIIIB), or IV (GPIV); CRK, proto-oncogene c-Crk; eIF4E, eukaryotic translation initiation factor; eNOS (NOS3), nitric oxide synthase isoform 3; ERK1/2, extracellular signal-regulated kinase; FOXO, forkhead box O; FYN, proto-oncogene tyrosine-protein kinase Fyn; GLUT4, solute carrier family 2, facilitated glucose transporter member 4; GMP, guanosine monophosphate; GRB2, growth factor receptor-binding protein 2; GS, glycogen synthase; GSK3, glycogen synthase kinase 3; GTP, guanosine-5'-triphosphate; IRS1/2, insulin receptor substrate isoforms 1 and 2; MEK, MAPK (mitogen-activated protein kinases/ERK (extracellular signal-regulated) kinase; mTOR, mechanistic target of rapamycin; NCK, non-catalytic region of tyrosine kinase adaptor protein 1; NO, nitric oxide; NOX2, NADPH oxidase isoform 2; p70S6, ribosomal protein S6 kinase beta-1; PDPK1, 3-phosphoinositide dependent protein kinase 1; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PKCζ, protein kinase C zeta isoform; PKG, protein kinase G; PTP, protein tyrosine phosphatase; RAF, rapidly accelerated fibrosarcoma; RAS, RAS guanosine triphosphatase; SOS, Son of Sevenless guanine nucleotide exchange factors; TSC1/2, tuberous sclerosis complex isoforms 1 and 2; ULK1, Unc-51 like autophagy activating kinase.
An important downstream effector of insulin action that has been well-studied in skeletal muscle and adipocytes is the glucose transporter protein solute carrier family 2, facilitated glucose transporter member 4 (GLUT4) (8). Similar to the case in these cells, activation of insulin signaling promotes GLUT4 translocation to increase cardiomyocyte glucose uptake via mechanisms similar to those described in other cell types and also by mechanisms involving the Golgi-dependent compartment (Fig. 1) (9, 10). Rates of insulin-mediated glucose uptake in cardiomyocytes from mice with cardiomyocyte-restricted knockout (KO) of IRS1 or IRS2 are similar to those observed in wild-type mice, indicating functional and signaling redundancy (11). Thus, activation of insulin receptor substrate isoforms IRS1 or IRS2 is sufficient to fully activate insulin-mediated glucose uptake and GLUT4 translocation in the heart. In contrast, Akt isoforms appear to differentially regulate myocardial metabolism. Germline deletion of Akt2 but not Akt1 is associated with reduced insulin-stimulated glucose uptake in the heart and reduced ischemia tolerance (12). Although this model is confounded by systemic hyperglycemia and insulin resistance, a specific role for Akt2 in regulating cardiomyocyte glucose uptake was also demonstrated following siRNA-mediated gene silencing in vitro (13). Moreover, in streptozotocin (STZ)-induced diabetic FVB mice, impaired myocardial glucose utilization correlated with impaired phosphorylation of Akt2, whereas Akt1 phosphorylation was unchanged (13).
It is important to note that GLUT4 translocation is induced by cardiac muscle contraction, thus the fold increase in glucose uptake in insulin-stimulated contracting hearts is less than changes observed in cultured cardiomyocytes or in skeletal muscle or adipose tissue (1). Moreover, genetic deletion of insulin receptors in the heart is associated with increased GLUT4 content in the heart and increased rates of basal glycolysis that are as high as glycolysis rates in insulin-perfused wild-type hearts (1). Thus, insulin likely plays a minor role in the direct physiological regulation of myocardial glucose uptake in vivo. GLUT4-mediated glucose uptake in the heart may serve a major role in the response of the heart to ischemia and acute hemodynamic stress, as revealed in studies of mice with cardiomyocyte GLUT4 deficiency (14–16). Acute insulin stimulation has been reported to promote cardioprotection and although some of this might be due to GLUT4 translocation, the beneficial impact of insulin during reperfusion following myocardial ischemia includes additional pathways mediated by activation of PI3K, Akt, and protein kinase C (PKC) (17–19).The relationship between insulin signaling and ischemic preconditioning (IPC) is more complex. Although, insulin protects against ischemia/reperfusion (I/R) injury, pretreatment with insulin abrogates IPC, which is mediated by Akt activation, as this inhibition is also phenocopied by transgenic Akt overexpression in cardiomyocytes (20).
In the fasting state in vivo, the heart relies almost exclusively on fatty acids (FAs) as its main metabolic substrate, reflecting increased availability of FAs, arising from increased lipolysis when circulating insulin concentrations are low (21). Under conditions of euglycemic hyperinsulinemia, myocardial glucose uptake is increased relative to that of fatty acids, whose circulating levels are suppressed (1, 22, 23). However, in response to feeding the heart will utilize absorbed fatty acids despite the associated increase in circulating insulin (24, 25). Classic studies decades ago, confirmed that insulin directly regulates myocardial glucose metabolism (26, 27). In isolated hearts, although GLUT4 translocation is increased (28, 29) and phosphofructokinase isoform 2 (PFK2) is activated by insulin (30) the fold increase in glycolysis is not as large as the increase in glucose oxidation (31, 32). This reflects in part, the already elevated levels of myocardial glycolysis secondary to insulin-independent mechanisms of GLUT4 translocation. Additional mechanisms for this difference include direct insulin-mediated activation of glucose-oxidation by mitochondrial targeting of Akt (31). The activation of glucose oxidation by insulin leads to suppression of mitochondrial fatty acid oxidation in the heart, via reversal of the Randle’s cycle. In addition, insulin might also directly suppress fatty acid oxidation (FAO) in part by inhibiting adenosine monophosphate-activated protein kinase (AMPK) activity (33), which decreases the phosphorylation of acetyl CoA carboxylase (ACC), leading to increased malonyl CoA generation and suppression of FAO (34). Despite the net effect of insulin to suppress FAO, insulin signaling stimulates the translocation of the fatty acid transporter CD36 from an intracellular compartment to the sarcolemma to increase FA uptake via a mechanism that requires Akt2 and PKCzeta isoform (PKCζ Fig. 1). These signals also regulate GLUT4 trafficking (35, 36). Thus, short-term insulin stimulation might direct FA into synthetic pathways such as triglycerides (TG), which represent an important source of FA that is oxidized by the beating heart (37).
Although this review will not focus on insulin signaling in the vasculature, the large endothelial surfaces within the heart (endocardium and coronary microcirculation) means that insulin signaling in the heart likely reflects signaling phenomena that take place within endothelial cells. The most widely studied downstream mediator of insulin action in endothelial cells is the activation of NOS3 (eNOS) phosphorylation by insulin and Akt, leading to increased nitric oxide (NO) generation to promote vasodilatation. However, NOS3 is also phosphorylated by other kinases including AMPK and also by shear stress (38). Thus, mice with genetic deletion of insulin receptors in endothelium are in fact not hypertensive on the basis of NO deficiency, but were paradoxically hypotensive (39). This may reflect increased sensitivity of NOS3 to shear stress or activity of other kinases. Impaired myocardial insulin-mediated NO generation has been described in insulin resistant states. Although insulin-stimulated Akt phosphorylation might be reduced, NOS3 activity or abundance is also likely perturbed via other mechanisms such as NOS3 uncoupling mediated in part by oxidative stress or lipid intermediates such as ceramide (40–43).
INSULIN SIGNALING AND CARDIAC HYPERTROPHY
Insulin Signaling and Physiological Cardiac Hypertrophy
Based on initial observations that absence of insulin signaling in the heart during development reduces heart size (Fig. 2A) (1, 3), our group performed a series of studies that sought to understand the contribution of insulin and growth factor signaling to physiological hypertrophy and the accompanying metabolic adaptations. The initial study focused on PI3K and Akt (44). Using a swim training model of exercise, this study showed that PI3K signaling coordinates the physiological hypertrophic response and the mitochondrial adaptation characterized by increased mitochondrial oxidative capacity. Importantly, whereby 3-phosphoinositide dependent protein kinase 1 (Pdpk1) that transduces PI3K activation to Akt was required for the hypertrophic response to exercise, it was dispensable for the mitochondrial adaptation to exercise, suggesting that other non-Akt targets of PI3K may mediate this effect (45). Indeed, persistent Akt activation promotes pathological hypertrophy and represses mitochondrial oxidative capacity (Fig. 2C) (46). Interestingly, in mice with cardiomyocyte-restricted deletion of IGF1R (CIGFKO), no differences in basal cardiac size was observed, which indicates that IGF1 signaling does not regulate prenatal cardiac size. However, the hypertrophic and mitochondrial adaptations of these mice to exercise swim training was prevented (47). Mechanistically, the absence of IGF1R signaling induced mitochondrial stress and AMPK activation, which antagonized the hypertrophic response (Fig. 2A). Cross talk between insulin and IGF1 signaling likely also contributes to physiological cardiac hypertrophy. This is supported by evidence that defects in exercise-induced hypertrophy were exacerbated by additional allelic loss of IR in CIGFKO mice (2). Insulin receptor substrates (IRS) activity is also required for physiological cardiac hypertrophy. IRS1 expression regulates developmental myocardial size, but IRS2 does not. However, both isoforms play nonredundant roles in the hypertrophic and bioenergetic response to exercise training, with loss of either IRS1 or IRS2 in cardiomyocytes both preventing physiological hypertrophy in response to exercise training (Fig. 2C) (11).
Figure 2.

Summary of lessons learned from mutant mice with inactivation of insulin receptors (IRs) and insulin-like-growth-factor 1 receptors (IGF1R), or other insulin signaling intermediates selectively in cardiomyocytes. A: consequences of cardiomyocyte-restricted deletion of IGF1R or IRs in nonstressed hearts. B: consequences of cardiomyocyte-restricted deletion of the IRs in the cardiac response to metabolic or hemodynamic stress. C: consequence of inactivation of insulin receptor substrate isoform 1 (IRS1), IRS2, phosphoinositide 3-kinase (PI3K), 3-phosphoinositide dependent protein kinase 1 (PDPK1) or protein kinase B (Akt)1 or 2 respectively on cardiac structure, function, and mitochondrial metabolism. D: mechanisms and adaptations following combined inactivation of the IR and IGF1R or IRS1 and IRS2, respectively.
Insulin Signaling and Pathological Hypertrophy
The relationship between myocardial insulin signaling and pathological cardiac hypertrophy is complex with evidence indicating that either insufficient or excessive insulin signaling can both promote pathological ventricular remodeling (Figs. 2B and 3). Mice lacking insulin receptors in cardiomyocytes (CIRKO) develop accelerated left ventricular (LV) remodeling following pressure overload (48). Similar responses were reported following isoproterenol infusions and mechanisms included increased apoptosis, increased cell death, and decreased angiogenesis driven in part by repressed hypoxia inducible factor 1 alpha isoform (HIF1α) (49). Similar mechanistic findings were confirmed by others (50). Treatment of these mice with an insulin sensitizing peroxisome proliferator-activated receptor gamma isoform (PPARγ) ligand, also induced pathologic hypertrophy on the basis of volume overload (51). Although CIRKO mice and CIGFKO mice reveal no obvious pathology when unstressed, genetic and combined deletion of the IR and IGF1R in cardiomyocytes led to catastrophic heart failure, indicating a critical role for IR and IGF1R signaling in maintaining cardiomyocyte viability (Fig. 2D) (52). Similar observations were reported in mice with constitutive deletion of IRS1 and IRS2 in cardiomyocytes (Fig. 2D) (53). In this model, unrestrained autophagy appeared to be the important mechanism. This study also defined the essential role of insulin signaling in perinatal nutrient sensing that suppresses the increase in myocardial autophagy that develops in the peripartum period before feeding begins (53). Studies in an independent model of combined IRS1 and IRS2 deletion in the heart also identified an important role for dephosphorylated and constitutively nuclear FOXO 1, as genetic deletion of FOXO1 ameliorated these phenotypes (54, 55). The transcriptional targets of FOXO1 that promote this maladaptive response remain to be elucidated. Constitutive knockout of Pdpk1 in cardiomyocytes also mirrors the consequence of loss of IRS1 and IRS2 on cardiac structure and function (56). Mice with inducible deletion of Akt1 and Akt2 also develop a lethal cardiomyopathy. These studies identified additional mechanisms such as loss of the gap junction proteins Cx43, which would impair intercellular communication (57). Therefore, it will be of interest to determine if inducible reduction of IR and IGF1R or IRS1/IRS2 signaling in adult cardiomyocytes would induce heart failure by similar or distinct mechanisms. These KO models identify an essential role for intact insulin and IGF1 signaling for maintaining myocardial structure and function under basal conditions and in response to acute stressors (Fig. 2).
Figure 3.

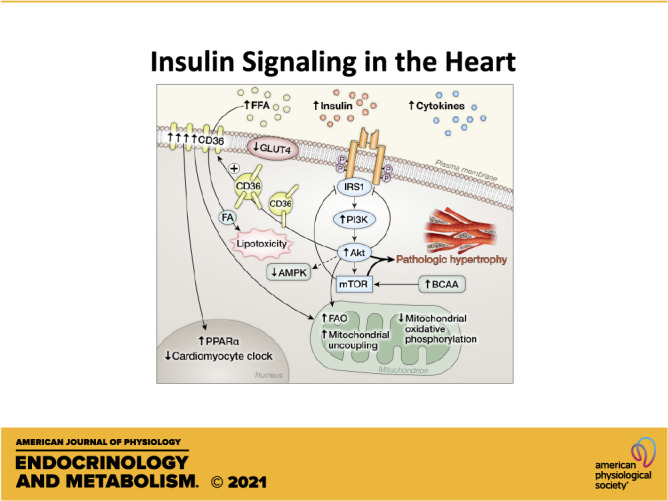
Schematic representation of adaptations in insulin signaling pathways in the heart under conditions of systemic insulin resistance characterized by metabolic disturbances such as hyperinsulinemia and increased circulating fatty acids. Hyperinsulinemia activates phosphoinositide 3-kinase (PI3K), protein kinase B (Akt), and mechanistic target of rapamycin (mTOR) signaling in the heart that drives cardiac hypertrophy and remodeling. Activation of this signaling module promotes serine phosphorylation that limits insulin receptor substrate isoform 1 (IRS1) activation. Akt activation promotes translocation of CD36 that increases the uptake of fatty acids that promotes lipotoxicity, increases mitochondrial fatty acid (FA) utilization, and mediates transcriptional changes in the nucleus that alter myocardial gene expression. Solute carrier family 2, facilitated glucose transporter member 4 (GLUT4) content and translocation is impaired despite increased activation of Akt.
In contrast, pathological cardiac hypertrophy and heart failure under clinically relevant circumstances, such as pressure overload or postischemic remodeling might be mediated in part by excessive activation of insulin signaling pathways in cardiomyocytes. Augmented or excessive insulin signaling may drive pathological LV remodeling (58), and is mediated by IRS1 but not IRS2 (Fig. 3) (59). Moreover, there is cross talk between insulin receptors, IRS proteins, and components of the β adrenergic system particularly β2AR. Activation of IR by insulin induces protein kinase A (PKA) and G-protein receptor kinase 2 (GRK2)-mediated phosphorylation of the β2AR, which promotes β2AR coupling to the inhibitory G-protein, Gi. This pathway is exacerbated by diet-induced obesity (DIO) and leads to induction of phosphodiesterase PDE4D that degrades cGMP, which dampens myocardial contractile function (Fig. 4). Inhibition of this GRK-mediated pathway reverses ventricular dysfunction in this model, raising the possibility that modulation of myocardial insulin signaling could represent an important strategy for managing obesity-associated heart failure (60). IRS2 is required for forming the IR-GRK2-β2AR complex and is required for GRK-mediated β2AR internalization, which in turn globally attenuates βAR signaling (61). Induction of GRK2 in heart failure impairs IRS1 signaling by directly phosphorylating IRS1 and reducing its activity (62). Activation of the renin-aldosterone system is implicated in the pathogenesis of ventricular remodeling in heart failure. Aldosterone impairs insulin and β-AR signaling in the heart via GRK2 (63). Mechanistically, there is increased phosphorylation of IRS1 at Ser307P that correlates with reduced β1AR signaling. These molecular changes were reversed following pharmacological mineralocorticoid receptor inhibition and correlated with LV recovery. Altered branch chain amino metabolism has been described in failing hearts (64–66). Accumulation of branch chain amino acids (BCAAs) have been postulated to contribute to insulin resistance by activating mTOR signaling which phosphorylates IRS1 on ser636/639, leading to impaired insulin signal transduction (67). mTOR-independent mechanisms by which leucine impairs insulin-mediated GLUT4 translocation have also been described (68).
Figure 4.
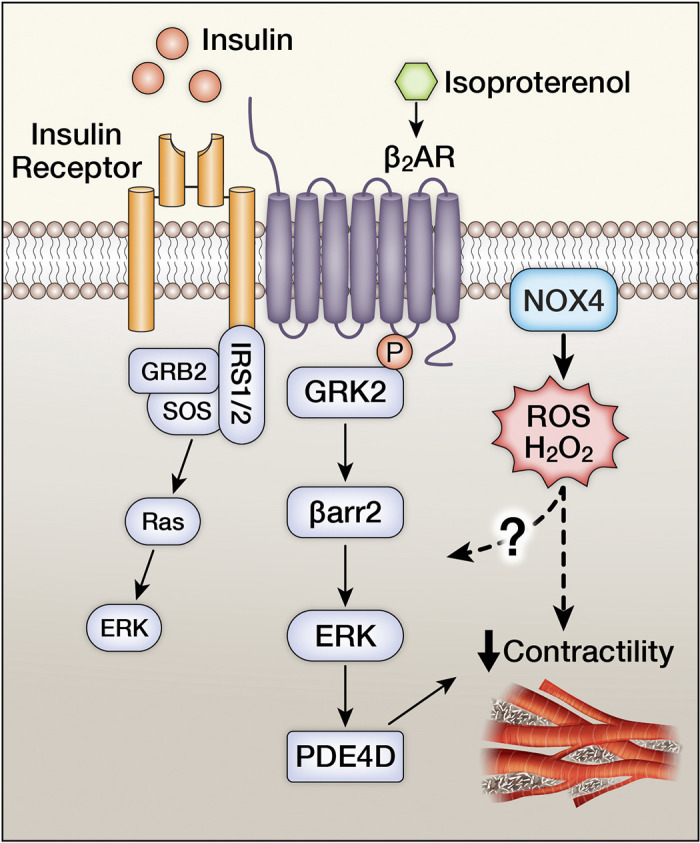
Schematic representation of insulin receptor (IR) and beta adrenergic receptor (β2AR) cross talk that impact myocardial contractility. IR/β2AR physically interact to promote a G-protein receptor kinase (GRK2)-mediated phosphorylation of the β2AR leading to β-arrestin (βarr2)-mediated activation of ERK that leads to the induction of the phosphodiesterase isoform PDE4D that catalyze the hydrolysis of 3′ cyclic phosphate bond of cyclic nucleotides such as cAMP and cGMP leading to impaired myocardial contractility. NADPH oxidase (NOX4)-derived H2O2 has been recently shown to contribute to the repression of βAR-mediated myocardial contractility, when insulin receptors are concomitantly activated.
Metabolic mechanisms exist by which persistent activation of insulin signaling in the myocardium could accelerate LV remodeling. Increased glucose uptake increases glucose-6-phosphate (G6P), which directly activates mTOR to promote pathological remodeling (69, 70). In heart failure, some of the mechanisms driving this increase in glucose availability are insulin independent. In pressure overload induced hypertrophy modeled by transverse aortic constriction (TAC), basal glucose uptake might be increased (71–73) although insulin-mediated GLUT4 translocation and insulin-augmented glycolysis and glucose oxidation are decreased. Basal Akt phosphorylation is increased but is not further increased by exogenous insulin (46, 74). Angiotensin 2-induced cardiac hypertrophy reduced insulin-mediated glucose uptake via pyruvate dehydrogenase kinase (PDK4) and sirtuin 3 (Sirt3)-dependent mechanisms that increased the phosphorylation and acetylation of pyruvate dehydrogenase (PDH), to limit glucose oxidation (75). Both loading and unloading transiently reduces insulin responsiveness (76). The chronically infarcted rat heart has reduced GLUT4, which could contribute to the reduction in insulin-mediated glucose uptake (77). Some studies have not observed a reduction in insulin-mediated glucose uptake in the failing heart. However, the ability of insulin to increase glucose oxidation and suppress FA oxidation was impaired (78). The disparate responses of glucose uptake versus substrate oxidation would be predicted to amplify the mismatch between glucose uptake and glycolysis with glucose oxidation. Glycolysis-glucose oxidation mismatch is increasingly recognized as an important metabolic driver of altered ventricular remodeling. This concept has been confirmed in studies in humans (79), and in mouse models in which mitochondrial pyruvate utilization is genetically impaired (80–83).
Reduced autophagy has been proposed to contribute to the pathophysiology of adverse ventricular remodeling. Signaling via IR and IGF1R suppresses autophagy (53, 84, 85). Thus, intrinsic persistent activation of insulin signaling pathways, that characterize the failing heart or secondary to hyperinsulinemia and insulin resistance that accompanies heart failure, could contribute to autophagy suppression. Insulin signals through both IRs and IGF1Rs to mediate this effect as was demonstrated in hyperinsulinemic mice lacking insulin receptors in the heart and may involve additional signaling mechanisms beyond Akt, such as ERK-mediated pathways that activate mTOR (84, 86). Aging is associated with ventricular remodeling. Interestingly, knockout of IGF-1 in cardiomyocytes attenuates myocardial aging in mice (87) and reducing insulin signaling in Drosophila heart attenuates cardiac aging (88). Inhibition of PI3K attenuates cardiac aging (89). However, it is unclear if these salutary effects on cardiac aging are secondary to constitutive activation of autophagy.
INSULIN RESISTANCE IN THE HEART
Genetic Models of Myocardial Insulin Resistance
Studies in mice with genetic deletion of insulin receptors have shed important insight into physiological roles of myocardial insulin signaling and consequences of myocardial insulin resistance. Constitutive deletion of insulin receptors in cardiomyocytes (CIRKO) identified an important role for insulin signaling in the developmental regulation of cardiac size, underscoring its role as a growth factor that mediates this effect via Akt (1, 3). These studies also provided the first direct evidence that insulin signaling constitutively regulates mitochondrial oxidative capacity, as evidenced by reduced FAO and glucose oxidation, an adaptive increase in glycolysis and induction of GLUT4. Direct evidence for mitochondrial impairment was confirmed by Boudina et al. (90), demonstrating that mitochondria isolated from hearts of CIRKO mice exhibited impaired mitochondrial respiratory capacity and ATP production for multiple substrates due to reduced oxidative phosphorylation (OXPHOS) capacity, that was further exacerbated by mitochondrial uncoupling induced by reactive oxygen species (ROS) overproduction (Fig. 2A). Proteomics analysis revealed reduced levels of mitochondrial proteins that regulate tricarboxylic acid cycle (TCA) activity and FAO. We subsequently discovered that insulin induces mitochondrial fusion via Akt-mTOR-mediated optic atrophy 1 (OPA1) induction (91). The baseline defect in CIRKO mitochondria led to accelerated mitochondrial dysfunction in response to postischemic cardiac remodeling (Fig. 2B) (92). Although mitochondria may be targets of insulin action in a cell autonomous manner, cardiac mitochondria are also modified by metabolic consequences of systemic insulin resistance or insulin deficiency (93). For example, in insulin-deficient diabetes, mitochondrial dysfunction, i.e., decreased oxidative capacity, that was associated with remodeling of the mitochondrial proteome was evident, despite preserved myocardial insulin sensitivity (94, 95). However, when genetic insulin resistance was superimposed, phenotypes were exacerbated and associated with increased ROS generation and exacerbated mitochondrial uncoupling (Fig. 2B) (96). Proteomics analysis revealed distinct changes that may be due to changes in the metabolic milieu versus consequences of altered myocardial insulin signaling. Specifically, diabetes was associated with reduced expression of TCA enzymes but induction of FAO proteins, whereas genetic reduction of insulin signaling reduced FAO proteins in addition to TCA proteins (96). Similarly, whereas most isoforms of acyl CoA synthase (Acsl) were regulated by insulin-deficient diabetes, only the Acsl6 isoform was directly regulated by insulin signaling (97). These and other observations underscore the importance of distinguishing between myocardial changes that are secondary to systemic consequences of insulin resistance or insulin deficiency versus direct cell-autonomous consequences of reduced insulin signaling in cardiomyocytes.
Myocardial Adaptations to Generalized Insulin Resistance
Multiple cardiac adaptations develop within the abnormal systemic milieu that characterizes insulin-resistant states such as obesity, type 2 diabetes, and heart failure (Fig. 3) (5, 98). Generally speaking, obesity is characterized by decreased myocardial glucose oxidation, reduced insulin stimulation of glucose transport and glycolysis, increased myocardial FA utilization, increased myocardial oxygen consumption () and decreased cardiac efficiency that precedes the development of hyperglycemia (32, 99, 100). An important mechanism for reduced cardiac efficiency is mitochondrial uncoupling, mediated in part by a combination of impaired mitochondrial oxidative phosphorylation leading to ROS overproduction, which activates uncoupling proteins (101, 102). The ability of insulin to suppress FAO is reduced in the face of increased myocardial FA utilization. This could be due in part to attenuation by FA of insulin’s ability to suppress AMPK, which has been described in the context of ischemia/reperfusion (103).
Many studies have attempted to elucidate the time course of alterations in insulin signaling pathways and myocardial metabolism that develop in the evolution of insulin-resistant conditions. In mice, a reduction in insulin-mediated glucose uptake is one of the earliest responses to be detected, despite preserved insulin signaling to Akt/PKB (28). This was accounted for by a reduction in GLUT4 protein levels. Importantly, persistent Akt activation also impairs GLUT4 translocation in the heart (29). Molecular mechanisms that impair GLUT4 translocation in the heart in this context are incompletely understood, but reduced levels of the vesicle-associated membrane protein (VAMP3) have been implicated (104). Similar findings were reported in a study examining surgical samples from human hearts obtained from individuals with diabetes that revealed reduced myocardial glucose uptake, reduced GLUT4 translocation, and reduced GLUT4 content despite increased insulin signaling to PI3K and Akt. In addition, these authors reported that similar molecular changes were present in individuals with pathologic cardiac hypertrophy and heart failure in the absence of diabetes indicating shared pathophysiology in failing hearts, characterized by hyperactivation of proximal insulin signaling, but with defective glucose uptake (105). Although most studies in obese rodent and large animal models have revealed decreased insulin-mediated glucose uptake, differences in the extent of changes in insulin signaling have been reported, which likely reflect the duration or the severity of obesity and diabetes at the time that these studies were performed. For example in the GK rat, GLUT4 proteins were reduced but insulin-stimulated Akt phosphorylation was normal, despite reduced IR, IRS1-phosphorylation, and reduced insulin-mediated IRS1-PI3K association (106, 107). Likewise, porcine models of obesity and ob/ob mice manifested decreased proximal insulin signaling and insulin-mediated glucose uptake (32, 108).
Increased myocardial FAO, characteristic of obesity is mediated in part by transcriptional induction of regulators of FA metabolism that occur in response to increased FA supply. In the studies described earlier in this section, describing an early defect in GLUT4 protein and translocation that was evident as early as 2 wk following high-fat feeding, an increase in FAO was evident only after 5 wk, occurring in concert with induction of peroxisome proliferator-activated receptor alpha isoform (PPARα) targets (28). However, translocation of the FA transporter CD36 to the sarcolemma on the basis of activation of insulin signaling occurs very early in the cardiac adaptation to DIO, and may precede alterations in glucose transport (Fig. 3) (109). It has been postulated that this initial influx of fatty acids may increase the susceptibility of the heart to lipotoxicity that contributes to insulin resistance and ultimately to contractile dysfunction (110). The molecular mechanisms governing the differences in CD36 and GLUT4 trafficking in the adaptation of the heart to DIO are incompletely understood. For example, mammalian uncoordinated-18 (MUNC 18) was identified in skeletal muscle as a regulator of GLUT4 but not CD36 trafficking (111). However in cardiac muscle, MUNC18 does not regulate GLUT4 or CD36 trafficking (112). Other proteins that regulate vesicle transporter trafficking include isoforms of vesicle-associated membrane proteins (VAMPS). Various VAMP isoforms share regulatory roles in the trafficking of both GLUT4 and CD36 in response to diverse proximal signaling inputs, such as Vamp2 and Vamp5 that are regulated by insulin and Vamp3 that is regulated by AMPK. However, Vamp7 may selectively regulate GLUT4 trafficking and Vamp4 may selectively regulate CD36 trafficking (113). Thus, under conditions of chronic lipid overload, CD36 and GLUT4 recycling are differentially regulated, with CD36 going constitutively to the sarcolemma whereas GLUT4 is sequestered in the intracellular compartment. It is noteworthy that CD36 translocation can be specifically reduced by activating the vacuolar ATPase (114) opening the possibility that limiting lipid overload could prevent the subsequent defects in insulin signaling and glucose transport that develops in the cardiac adaptation to lipid overload.
Altered myocardial insulin signaling in insulin-deficient diabetes should be interpreted in the context of accompanying metabolic changes such as increased lipolysis, and increased FA utilization by the heart, which could independently impair insulin signaling. Following 3 mo of STZ-induced diabetes, basal levels of Akt phosphorylation is reduced (13), and in some studies the activation of Akt (total) or Akt2 phosphorylation by insulin is impaired (13, 115), and was related to induction of the Akt2 inhibitor Tribbles3 (13). However, in studies of diabetes of shorter duration in STZ-treated rat heart or in the Akita model of type 1 diabetes, insulin-stimulated Akt activation was preserved (94, 116, 117). It should be noted that type 1 diabetes reduces myocardial GLUT4 levels, thus in models in which insulin-stimulated activation of Akt might be preserved, insulin-stimulated glucose uptake or glycogen synthesis is reduced (116, 117). In type 1 diabetic humans with suboptimal glycemic control (A1C 7.8–8.8) studied under euglycemic (basal) conditions, myocardial glucose utilization normalized to ambient circulating insulin concentrations were reduced, suggesting insulin resistance. However, under conditions of hyperinsulinemic euglycemia, these differences were abolished and myocardial glucose utilization was then equivalent to that of nondiabetic controls (23). Taken together, these data indicate that abnormalities in insulin signaling in the myocardium in type 1 diabetes are variable and dependent on the duration and severity of hyperglycemia.
The relationship between myocardial insulin resistance or insulin deficiency and autophagy is complex. This reflects consequences of reduced insulin signaling, which would be predicted to increase autophagy, versus consequences of lipotoxicity, which reduces autophagic flux and hyperinsulinemic activation of IGF1R, which could also suppress autophagy (84, 118, 119). Although the functional consequences directly attributable to acquired insulin resistance in the resting heart are incompletely understood and likely multifactorial, there is consensus that impaired myocardial insulin action might contribute to maladaptive responses of the heart to acute ischemic or hemodynamic stress (103, 120).
Molecular Mechanisms Associated with Myocardial Insulin Resistance
Multiple mechanisms have been described that may contribute to impaired myocardial insulin action in the context of obesity, diabetes, or heart failure. Some, but not all of these mechanisms have been identified as contributing to insulin resistance in other cell types (5). Mechanisms include changes at the level of insulin receptors and IRS proteins, activation of other kinases and phosphatases, lipotoxicity, branch chain amino acid (BCAA) accumulation, and inflammation (Fig. 3). Serine phosphorylation of IRS1 on residues such as Ser307 reduces insulin signaling. Evidence for this has been described in insulin resistant hearts, potentially as a consequence of increased mTOR signaling (67, 121) but also by increased Rho Kinase signaling (122). Mechanisms associated with increased degradation of IRS proteins have also been described. These include constitutive activation of PI3K by persistent activation of insulin signaling that may reduce IRS2 expression in the heart (123), and induction of the microRNA mir128-3p (124). There are few studies that have examined IRS1 in human cardiac tissues. However, in failing human hearts, IRS1 tyrosine phosphorylation is increased, consistent with hyperactivation of insulin signaling (59). Interestingly, the Gly1057Asp polymorphism of the IRS2 gene in humans is associated with increased epicardial fat (125). Constitutive activation of PI3K has been associated with induction of phosphatases such as phosphatase and tensin homolog (PTEN) (123), which would limit insulin signal transduction downstream of PI3K. Impaired myocardial insulin signaling is associated with activation of FOXO1 transcriptional signaling following its dephosphorylation (126). Activation of FOXO1 was shown to further modulate insulin signaling by suppressing PP2A leading to hyperactivation of Akt signaling, which in turn inhibited proximal insulin signaling (127). Increased insulin sensitivity and protection from endoplasmic reticulum (ER) stress and obesity-related cardiac dysfunction in protein tyrosine phosphatase 1B (PTP1B)-deficient hearts supports the concept that increased phosphatase activity could impair myocardial insulin signaling and potentially contribute to cardiac dysfunction in insulin-resistant states (128, 129).
Lipotoxicity has been implicated as a contributor to altered myocardial insulin signaling in the context of obesity. Mouse models harboring mutations or transgenes that alter triglyceride metabolism to prevent lipotoxicity, also increase myocardial insulin sensitivity. For example, overexpression of adipose triglyceride lipase (ATGL) or knockout of stearoyl CoA desaturase isoform 1 (SCD1) for reasons that are incompletely understood increases rates of glucose oxidation and reduces FAO. In both of these models, myocardial insulin signaling is preserved or increased even in the face of caloric excess (130–133). In other lipotoxic models, reducing accumulated lipid intermediates such as ceramide also lowered FAO and increased glucose utilization concurrently with improved ventricular function (134). The impact of reversing lipotoxicity on insulin signaling has not been systematically determined in all such models, but would be anticipated to increase insulin sensitivity. In contrast, transgenic mice overexpressing PPARα develop lipotoxicity, characterized by increased FAO and decreased glucose utilization. As predicted, myocardial insulin signaling is impaired in these hearts (135). A mismatch between FA uptake and mitochondrial oxidative capacity to completely oxidize these lipids would be expected to promote accumulation of lipid intermediates that could impair insulin signaling in the heart (136). Incomplete or impaired myocardial mitochondrial FA metabolism in obesity correlates with accumulation of diacylglycerol (DAG), which has been implicated in reducing insulin signaling in other cell types (137). Additional mechanisms that could limit FA oxidative capacity include chronic hyperinsulinemia, which suppresses sterol regulatory-element binding protein (SREBP) and reduces the expression of uncoupling protein isoform 3 (UCP3) (138). Similarly, insulin resistance in sucrose fed rats is associated with decreased UCP3, which decreases post-IR mitochondrial FA uptake (139). In both of these instances, these changes correlate with reduced myocardial recovery following ischemia/reperfusion. High-fat diet (HFD) decreases BCAA oxidation leading to their accumulation that activates mTOR signaling to promote insulin resistance (140). HFD increases lysine acetylation of β-oxidation enzymes, increasing their activity to increase FAO, via mechanisms that are partially SIRT1 dependent. Increased acetylation of Akt was also described in this study, which antagonized insulin-stimulated Akt phosphorylation (74).
Inflammatory cytokines and secreted molecules from adipose tissue (adipokines) or liver (hepatokines) have been implicated in the pathophysiology of myocardial insulin resistance in the context of obesity or heart failure (141). Leptin-deficient models have striking abnormalities in cardiac metabolism and insulin signaling. Interestingly, some of these changes are independent of obesity, but are mediated by central actions of leptin (142). Exogenous resistin impairs glucose transport in cardiomyocytes without altering insulin signaling (143). However, resistin overexpression in cultured cells and in hearts impair myocardial insulin signaling via the AMPK/mTOR/p70(S6K) and apoptosis signal-regulating kinase 1/JNK/IRS1 pathways (144). The hepatokine fetuinB binds to the IR in the STZ-HFD diabetes model and exacerbates myocardial injury post-MI. Repression of fetuin B promotes myocardial insulin signaling and this correlates with improved recovery of contractile function (145). HFD increases myocardial inflammation, promoting suppressor of cytokine signaling isoform 2 (SOCS2)-mediated serine phosphorylation of IRS1 (146). Fructose feeding increases transcriptional markers of inflammation and matrix remodeling in concert with decreased insulin signaling, changes in which are reversed by exercise training (147). Cardiac-released myokines (cardiokines) such as MG53 (aka TRIM 72) may impair systemic insulin action (148). Also, its induction in cardiomyocytes impairs insulin signaling and promotes lipotoxicity (149). TRIM 72 TG mice also exhibit IRS1 degradation and reduced physiological/IGF1 signaling to Akt and mTOR (150).
Generalized insulin resistance has been associated with disruptions in the circadian clock (151). Studies in cardiomyocytes have illustrated intact and functional circadian clock machinery (152), and their activity was impaired by STZ-induced diabetes (153). Mice with genetic mutations in core circadian clock machinery in the heart develop many of the characteristics that are associated with the adaptation of the heart to obesity. Cardiomyocyte-specific Clock mutant (CCM) mice, exhibit increased FAO and reduced cardiac efficiency which parallels adaptations that develop in the heart in response to obesity and insulin resistance (154). CCM mice develop altered expression of multiple insulin signaling components in the heart, including p85α and Akt. Both baseline and insulin-mediated Akt activation was augmented in cardiomyocyte-specific Bmal1 knockout (CBK) and CCM hearts (relative to littermate controls). However, insulin-mediated glucose utilization (both oxidative and nonoxidative) and AS160 phosphorylation were attenuated in CBK hearts, potentially secondary to decreased inhibitor-1. Consistent with increased Akt activation in CBK hearts, mTOR signaling was persistently increased, which was associated with attenuation of autophagy, augmented rates of protein synthesis, and pathological cardiac hypertrophy (155).
Modulation of Myocardial Insulin Resistance
Myocardial insulin resistance, characterized by impaired insulin-mediated glucose uptake, repression of GLUT4, and reduced mitochondrial oxidative capacity could represent an adaptation that protects the heart from the consequences of substrate overload, which has been termed “carbo-toxicity” describing the combined deleterious consequences of lipotoxicity and glucotoxicity (98, 156). An increase in glucose availability could be cardioprotective in the context of ischemia, pressure overload, or aging (15, 16, 157–159). However, in the presence of substrate excess from multiple sources such as increased myocardial fatty acid delivery as occurs in insulin-resistant states, failure to downregulate glucose uptake is deleterious (160, 161). The myocardial adaptations to obesity are reversed by weight loss induced by caloric restriction (162) or following ileal transposition (bariatric surgery) (163). In obese mice subjected to pressure overload, a low-fat diet or caloric restriction increases insulin-mediated glucose oxidation that correlates with reduced phosphorylation of PDH, decreased phosphorylation of AMPK and ACC, and reduced acetylation of fatty acid β-oxidation enzymes (164). In general, therapies such as PPARγ ligands thiazolidinediones that improve systemic insulin sensitivity will improve myocardial insulin action (165). Altering dietary lipid composition could also reverse myocardial insulin resistance, as ω3 FAs supplementation was shown to prevent myocardial insulin resistance in vitro under hyperinsulinemic conditions (166). Exercise activates AMPK in the heart (167) and reverses some of the defects in myocardial insulin signaling observed in the fructose-fed model of obesity and insulin resistance (147, 168). Moreover, exercise overtraining-induced inflammation that reduces skeletal muscle insulin sensitivity continues to increase insulin signaling in heart (169). Direct AMPK activation restores insulin responsiveness in hearts with prior insulin resistance (170), by mechanisms that might be independent of cross talk with mTOR signaling (171). AMPK activation may also increase myocardial insulin sensitivity without reversing markers of lipotoxicity (172).
Additional therapeutic strategies seeking to modulate insulin signaling defects in the heart in the context of obesity and insulin resistance have been described. For example, IGF-1 treatment antagonizes many of the potentially adverse changes in insulin signaling associated with DIO, underscoring the important cross talk between both signaling pathways (173). Activation of the renin-angiotensin system and oxidative stress has been implicated in obesity-associated insulin resistance in the heart and in other tissues (174). Captopril restored insulin-responsive Akt signaling and the ability of insulin to modulate metabolism, namely to suppress FAO and to increase glucose oxidation in ob/ob mice (175). Also, antioxidant treatment restored insulin sensitivity in the hearts of DIO mice (176). In the context of heart failure, antagonizing glucagon signaling in hearts postmyocardial infarction [permanent left anterior descending artery (LAD) occlusion] attenuated left ventricular hypertrophy, improved LV function, activated the IRS-1/Akt/AS160/GSK-3β pathway, increased GLUT4 expression, and reduced pyruvate dehydrogenase phosphorylation. These changes which correlated with reduced accumulation of BCAAS and reduced mTOR signaling led to increased insulin-mediated glucose oxidation (120). The multiple signaling abnormalities with nonoverlapping targets that develop in the heart in obesity and diabetes raise challenges when a single pathway is therapeutically targeted. For example, activation of protein kinase D in the context of diet-induced obesity increased myocardial glucose utilization while antagonizing CD36 translocation. However under normal diet conditions, these changes induced cardiac hypertrophy and a cardiomyopathy (177). In cultured cardiomyocytes, where insulin resistance was induced by exposure to palmitate or to increased insulin concentrations in the media, AMPK overexpression prevented insulin resistance but not lipotoxicity whereas PKD overexpression had the opposite effect preventing lipid overload, but not insulin resistance (172).
SUMMARY AND CONCLUSIONS
Insulin signaling plays pleiotropic roles in the heart ranging from metabolism to hypertrophy to autophagy and cell survival. There is significant redundancy in signaling via IR and IGF1R. However, signaling through both receptors is unequivocally required for cardiomyocyte survival. Short-term insulin treatment may provide cardioprotection following ischemia. Insulin signaling in the heart adapts to pathophysiological stressors such as substrate excess that occurs in insulin resistant states and to hyperinsulinemia or insulin-deficiency. Moreover, hemodynamic stressors such as pressure overload and ischemia may lead to significant perturbations in insulin signaling, which could contribute to heart failure progression. It remains to be definitively proven if modulating myocardial insulin signaling in disease states will alter heart failure progression or limit ventricular remodeling. Much remains to be done to further elucidate the many nuances of myocardial insulin signaling and to determine therapeutic applications of this knowledge.
GRANTS
Work from the Abel laboratory, cited in this review, has been supported by grants from the National Institutes of Health RO1-HL127764, RO1-HL112413, RO1-HL12357, R01-HL108379, R01-DK092065, U01-HL087947, RO1-HL73167, RO1-HL074259, R21-DK073590, U01-HL70525, R03-DK058073, R21-HL62886, the American Heart Association EIA, 16SFRN31810000, 20SFRN3512012, and research awards from the American Diabetes Association.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author.
AUTHOR CONTRIBUTIONS
E.D.A. prepared figures; drafted manuscript; and approved final version of manuscript.
ACKNOWLEDGMENTS
I thank Teresa Ruggle for assistance with the figures.
REFERENCES
- 1.Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, Zhang D, Cooksey RC, McClain DA, Litwin SE, Taegtmeyer H, Severson D, Kahn CR, Abel ED. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest 109: 629–639, 2002. doi: 10.1172/JCI13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ikeda H, Shiojima I, Ozasa Y, Yoshida M, Holzenberger M, Kahn CR, Walsh K, Igarashi T, Abel ED, Komuro I. Interaction of myocardial insulin receptor and IGF receptor signaling in exercise-induced cardiac hypertrophy. J Mol Cell Cardiol 47: 664–675, 2009. doi: 10.1016/j.yjmcc.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shiojima I, Yefremashvili M, Luo Z, Kureishi Y, Takahashi A, Tao J, Rosenzweig A, Kahn CR, Abel ED, Walsh K. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J Biol Chem 277: 37670–37677, 2002. doi: 10.1074/jbc.M204572200. [DOI] [PubMed] [Google Scholar]
- 4.Skurk C, Izumiya Y, Maatz H, Razeghi P, Shiojima I, Sandri M, Sato K, Zeng L, Schiekofer S, Pimentel D, Lecker S, Taegtmeyer H, Goldberg AL, Walsh K. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J Biol Chem 280: 20814–20823, 2005. doi: 10.1074/jbc.M500528200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riehle C, Abel ED. Insulin signaling and heart failure. Circ Res 118: 1151–1169, 2016. doi: 10.1161/CIRCRESAHA.116.306206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinhorn B, Sartoretto JL, Sorrentino A, Romero N, Kalwa H, Abel ED, Michel T. Insulin-dependent metabolic and inotropic responses in the heart are modulated by hydrogen peroxide from NADPH-oxidase isoforms NOX2 and NOX4. Free Radic Biol Med 113: 16–25, 2017. doi: 10.1016/j.freeradbiomed.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Izquierdo A, Pereira RO, Wende AR, Punske BB, Abel ED, Tristani-Firouzi M. The absence of insulin signaling in the heart induces changes in potassium channel expression and ventricular repolarization. Am J Physiol Heart Circ Physiol 306: H747–H754, 2014. doi: 10.1152/ajpheart.00849.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klip A, McGraw TE, James DE. Thirty sweet years of GLUT4. J Biol Chem 294: 11369–11381, 2019. doi: 10.1074/jbc.REV119.008351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abel ED. Glucose transport in the heart. Front Biosci 9: 201–215, 2004. doi: 10.2741/1216. [DOI] [PubMed] [Google Scholar]
- 10.Doenst T, Cedars AM, Taegtmeyer H. Insulin-stimulated glucose transport is dependent on Golgi function in isolated working rat heart. J Mol Cell Cardiol 32: 1481–1488, 2000. doi: 10.1006/jmcc.2000.1181. [DOI] [PubMed] [Google Scholar]
- 11.Riehle C, Wende AR, Zhu Y, Oliveira KJ, Pereira RO, Jaishy BP, Bevins J, Valdez S, Noh J, Kim BJ, Moreira AB, Weatherford ET, Manivel R, Rawlings TA, Rech M, White MF, Abel ED. Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol Cell Biol 34: 3450–3460, 2014. doi: 10.1128/MCB.00426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeBosch B, Sambandam N, Weinheimer C, Courtois M, Muslin AJ. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J Biol Chem 281: 32841–32851, 2006. doi: 10.1074/jbc.M513087200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu J, Yan X, Dai X, Wang Y, Lin Q, Xiao J, Zhou S, Zhang J, Wang K, Zeng J, Xin Y, Barati MT, Zhang C, Bai Y, Li Y, Epstein PN, Wintergerst KA, Li X, Tan Y, Cai L. Metallothionein preserves Akt2 activity and cardiac function via inhibiting TRB3 in diabetic hearts. Diabetes 67: 507–517, 2018. [Erratum in Diabetes 69: 267, 2020]. doi: 10.2337/db17-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abel ED, Kaulbach HC, Tian R, Hopkins JC, Duffy J, Doetschman T, Minnemann T, Boers ME, Hadro E, Oberste-Berghaus C, Quist W, Lowell BB, Ingwall JS, Kahn BB. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest 104: 1703–1714, 1999. doi: 10.1172/JCI7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian R, Abel ED. Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation 103: 2961–2966, 2001. doi: 10.1161/01.cir.103.24.2961. [DOI] [PubMed] [Google Scholar]
- 16.Wende AR, Kim J, Holland WL, Wayment BE, O'Neill BT, Tuinei J, Brahma MK, Pepin ME, McCrory MA, Luptak I, Halade GV, Litwin SE, Abel ED. Glucose transporter 4-deficient hearts develop maladaptive hypertrophy in response to physiological or pathological stresses. Am J Physiol Heart Circ Physiol 313: H1098–H1108, 2017. doi: 10.1152/ajpheart.00101.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer-Rasokat U, Beyersdorf F, Doenst T. Insulin addition after ischemia improves recovery of function equal to ischemic preconditioning in rat heart. Basic Res Cardiol 98: 329–336, 2003. doi: 10.1007/s00395-003-0414-y. [DOI] [PubMed] [Google Scholar]
- 18.Fischer-Rasokat U, Doenst T. Insulin-induced improvement of postischemic recovery is abolished by inhibition of protein kinase C in rat heart. J Thorac Cardiovasc Surg 126: 1806–1812, 2003. doi: 10.1016/S0022-5223(03)01229-7. [DOI] [PubMed] [Google Scholar]
- 19.Zaha V, Francischetti I, Doenst T. Insulin improves postischemic recovery of function through PI3K in isolated working rat heart. Mol Cell Biochem 247: 229–232, 2003. doi: 10.1023/A:1024183527668. [DOI] [PubMed] [Google Scholar]
- 20.Fullmer TM, Pei S, Zhu Y, Sloan C, Manzanares R, Henrie B, Pires KM, Cox JE, Abel ED, Boudina S. Insulin suppresses ischemic preconditioning-mediated cardioprotection through Akt-dependent mechanisms. J Mol Cell Cardiol 64: 20–29, 2013. doi: 10.1016/j.yjmcc.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murashige D, Jang C, Neinast M, Edwards JJ, Cowan A, Hyman MC, Rabinowitz JD, Frankel DS, Arany Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 370: 364–368, 2020. doi: 10.1126/science.abc8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrett EJ, Schwartz RG, Francis CK, Zaret BL. Regulation by insulin of myocardial glucose and fatty acid metabolism in the conscious dog. J Clin Invest 74: 1073–1079, 1984. doi: 10.1172/JCI111474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peterson LR, Herrero P, McGill J, Schechtman KB, Kisrieva-Ware Z, Lesniak D, Gropler RJ. Fatty acids and insulin modulate myocardial substrate metabolism in humans with type 1 diabetes. Diabetes 57: 32–40, 2008. doi: 10.2337/db07-1199. [DOI] [PubMed] [Google Scholar]
- 24.Labbe SM, Grenier-Larouche T, Croteau E, Normand-Lauziere F, Frisch F, Ouellet R, Guerin B, Turcotte EE, Carpentier AC. Organ-specific dietary fatty acid uptake in humans using positron emission tomography coupled to computed tomography. Am J Physiol Endocrinol Metab 300: E445–E453, 2011. doi: 10.1152/ajpendo.00579.2010. [DOI] [PubMed] [Google Scholar]
- 25.Labbe SM, Grenier-Larouche T, Noll C, Phoenix S, Guerin B, Turcotte EE, Carpentier AC. Increased myocardial uptake of dietary fatty acids linked to cardiac dysfunction in glucose-intolerant humans. Diabetes 61: 2701–2710, 2012. doi: 10.2337/db11-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen TM, Goodwin GW, Guthrie PH, Taegtmeyer H. Effects of insulin on glucose uptake by rat hearts during and after coronary flow reduction. Am J Physiol Heart Circ Physiol 273: H2170–H2177, 1997. doi: 10.1152/ajpheart.1997.273.5.H2170. [DOI] [PubMed] [Google Scholar]
- 27.Russell RR III, Cline GW, Guthrie PH, Goodwin GW, Shulman GI, Taegtmeyer H. Regulation of exogenous and endogenous glucose metabolism by insulin and acetoacetate in the isolated working rat heart. A three tracer study of glycolysis, glycogen metabolism, and glucose oxidation. J Clin Invest 100: 2892–2899, 1997. doi: 10.1172/JCI119838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wright JJ, Kim J, Buchanan J, Boudina S, Sena S, Bakirtzi K, Ilkun O, Theobald HA, Cooksey RC, Kandror KV, Abel ED. Mechanisms for increased myocardial fatty acid utilization following short-term high-fat feeding. Cardiovasc Res 82: 351–360, 2009. doi: 10.1093/cvr/cvp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Y, Pereira RO, O'Neill BT, Riehle C, Ilkun O, Wende AR, Rawlings TA, Zhang YC, Zhang Q, Klip A, Shiojima I, Walsh K, Abel ED. Cardiac PI3K-Akt impairs insulin-stimulated glucose uptake independent of mTORC1 and GLUT4 translocation. Mol Endocrinol 27: 172–184, 2013. doi: 10.1210/me.2012-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mouton V, Toussaint L, Vertommen D, Gueuning MA, Maisin L, Havaux X, Sanchez-Canedo C, Bertrand L, Dequiedt F, Hemmings BA, Hue L, Rider MH. Heart 6-phosphofructo-2-kinase activation by insulin requires PKB (protein kinase B), but not SGK3 (serum- and glucocorticoid-induced protein kinase 3). Biochem J 431: 267–275, 2010. doi: 10.1042/BJ20101089. [DOI] [PubMed] [Google Scholar]
- 31.Karwi QG, Wagg CS, Altamimi TR, Uddin GM, Ho KL, Darwesh AM, Seubert JM, Lopaschuk GD. Insulin directly stimulates mitochondrial glucose oxidation in the heart. Cardiovasc Diabetol 19: 207, 2020. doi: 10.1186/s12933-020-01177-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mazumder PK, O'Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 53: 2366–2374, 2004. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 33.Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem 278: 39422–39427, 2003. [Erratum in J Biol Chem 279: 2332, 2004]. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 34.Gamble J, Lopaschuk GD. Insulin inhibition of 5' adenosine monophosphate-activated protein kinase in the heart results in activation of acetyl coenzyme A carboxylase and inhibition of fatty acid oxidation. Metabolism 46: 1270–1274, 1997. doi: 10.1016/S0026-0495(97)90229-8. [DOI] [PubMed] [Google Scholar]
- 35.Jain SS, Luiken JJ, Snook LA, Han XX, Holloway GP, Glatz JF, Bonen A. Fatty acid transport and transporters in muscle are critically regulated by Akt2. FEBS Lett 589: 2769–2775, 2015. doi: 10.1016/j.febslet.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 36.Luiken JJ, Ouwens DM, Habets DD, van der Zon GC, Coumans WA, Schwenk RW, Bonen A, Glatz JF. Permissive action of protein kinase C-zeta in insulin-induced CD36- and GLUT4 translocation in cardiac myocytes. J Endocrinol 201: 199–209, 2009. doi: 10.1677/JOE-09-0046. [DOI] [PubMed] [Google Scholar]
- 37.Banke NH, Wende AR, Leone TC, O'Donnell JM, Abel ED, Kelly DP, Lewandowski ED. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circ Res 107: 233–241, 2010. doi: 10.1161/CIRCRESAHA.110.221713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang QJ, McMillin SL, Tanner JM, Palionyte M, Abel ED, Symons JD. Endothelial nitric oxide synthase phosphorylation in treadmill-running mice: role of vascular signalling kinases. J Physiol 587: 3911–3920, 2009. doi: 10.1113/jphysiol.2009.172916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vicent D, Ilany J, Kondo T, Naruse K, Fisher SJ, Kisanuki YY, Bursell S, Yanagisawa M, King GL, Kahn CR. The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J Clin Invest 111: 1373–1380, 2003. doi: 10.1172/JCI15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bharath LP, Ruan T, Li Y, Ravindran A, Wan X, Nhan JK, Walker ML, Deeter L, Goodrich R, Johnson E, Munday D, Mueller R, Kunz D, Jones D, Reese V, Summers SA, Babu PV, Holland WL, Zhang QJ, Abel ED, Symons JD. Ceramide-initiated protein phosphatase 2A activation contributes to arterial dysfunction in vivo. Diabetes 64: 3914–3926, 2015. doi: 10.2337/db15-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Symons JD, Hu P, Yang Y, Wang X, Zhang QJ, Wende AR, Sloan CL, Sena S, Abel ED, Litwin SE. Knockout of insulin receptors in cardiomyocytes attenuates coronary arterial dysfunction induced by pressure overload. Am J Physiol Heart Circ Physiol 300: H374–H381, 2011. doi: 10.1152/ajpheart.01200.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Symons JD, McMillin SL, Riehle C, Tanner J, Palionyte M, Hillas E, Jones D, Cooksey RC, Birnbaum MJ, McClain DA, Zhang QJ, Gale D, Wilson LJ, Abel ED. Contribution of insulin and Akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circ Res 104: 1085–1094, 2009. doi: 10.1161/CIRCRESAHA.108.189316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang QJ, Holland WL, Wilson L, Tanner JM, Kearns D, Cahoon JM, Pettey D, Losee J, Duncan B, Gale D, Kowalski CA, Deeter N, Nichols A, Deesing M, Arrant C, Ruan T, Boehme C, McCamey DR, Rou J, Ambal K, Narra KK, Summers SA, Abel ED, Symons JD. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes 61: 1848–1859, 2012. doi: 10.2337/db11-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Neill BT, Kim J, Wende AR, Theobald HA, Tuinei J, Buchanan J, Guo A, Zaha VG, Davis DK, Schell JC, Boudina S, Wayment B, Litwin SE, Shioi T, Izumo S, Birnbaum MJ, Abel ED. A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab 6: 294–306, 2007. doi: 10.1016/j.cmet.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Noh J, Wende AR, Olsen CD, Kim B, Bevins J, Zhu Y, Zhang QJ, Riehle C, Abel ED. Phosphoinositide dependent protein kinase 1 is required for exercise-induced cardiac hypertrophy but not the associated mitochondrial adaptations. J Mol Cell Cardiol 89: 297–305, 2015. doi: 10.1016/j.yjmcc.2015.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wende AR, O'Neill BT, Bugger H, Riehle C, Tuinei J, Buchanan J, Tsushima K, Wang L, Caro P, Guo A, Sloan C, Kim BJ, Wang X, Pereira RO, McCrory MA, Nye BG, Benavides GA, Darley-Usmar VM, Shioi T, Weimer BC, Abel ED. Enhanced cardiac Akt/protein kinase B signaling contributes to pathological cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion-targeted nuclear genes. Mol Cell Biol 35: 831–846, 2015. doi: 10.1128/MCB.01109-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, Wayment BE, Litwin SE, Holzenberger M, LeRoith D, Abel ED. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol 22: 2531–2543, 2008. doi: 10.1210/me.2008-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol 285: H1261–H1269, 2003. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 49.McQueen AP, Zhang D, Hu P, Swenson L, Yang Y, Zaha VG, Hoffman JL, Yun UJ, Chakrabarti G, Wang Z, Albertine KH, Abel ED, Litwin SE. Contractile dysfunction in hypertrophied hearts with deficient insulin receptor signaling: possible role of reduced capillary density. J Mol Cell Cardiol 39: 882–892, 2005. doi: 10.1016/j.yjmcc.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 50.He Z, Opland DM, Way KJ, Ueki K, Bodyak N, Kang PM, Izumo S, Kulkarni RN, Wang B, Liao R, Kahn CR, King GL. Regulation of vascular endothelial growth factor expression and vascularization in the myocardium by insulin receptor and PI3K/Akt pathways in insulin resistance and ischemia. Arterioscler Thromb Vasc Biol 26: 787–793, 2006. doi: 10.1161/01.ATV.0000209500.15801.4e. [DOI] [PubMed] [Google Scholar]
- 51.Sena S, Rasmussen IR, Wende AR, McQueen AP, Theobald HA, Wilde N, Pereira RO, Litwin SE, Berger JP, Abel ED. Cardiac hypertrophy caused by peroxisome proliferator- activated receptor-γ agonist treatment occurs independently of changes in myocardial insulin signaling. Endocrinology 148: 6047–6053, 2007. doi: 10.1210/en.2006-1559. [DOI] [PubMed] [Google Scholar]
- 52.Laustsen PG, Russell SJ, Cui L, Entingh-Pearsall A, Holzenberger M, Liao R, Kahn CR. Essential role of insulin and insulin-like growth factor 1 receptor signaling in cardiac development and function. Mol Cell Biol 27: 1649–1664, 2007. doi: 10.1128/MCB.01110-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riehle C, Wende AR, Sena S, Pires KM, Pereira RO, Zhu Y, Bugger H, Frank D, Bevins J, Chen D, Perry CN, Dong XC, Valdez S, Rech M, Sheng X, Weimer BC, Gottlieb RA, White MF, Abel ED. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J Clin Invest 123: 5319–5333, 2013. doi: 10.1172/JCI71171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qi Y, Xu Z, Zhu Q, Thomas C, Kumar R, Feng H, Dostal DE, White MF, Baker KM, Guo S. Myocardial loss of IRS1 and IRS2 causes heart failure and is controlled by p38alpha MAPK during insulin resistance. Diabetes 62: 3887–3900, 2013. doi: 10.2337/db13-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qi Y, Zhu Q, Zhang K, Thomas C, Wu Y, Kumar R, Baker KM, Xu Z, Chen S, Guo S. Activation of Foxo1 by insulin resistance promotes cardiac dysfunction and beta-myosin heavy chain gene expression. Circ Heart Fail 8: 198–208, 2015. doi: 10.1161/CIRCHEARTFAILURE.114.001457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mora A, Davies AM, Bertrand L, Sharif I, Budas GR, Jovanovic S, Mouton V, Kahn CR, Lucocq JM, Gray GA, Jovanovic A, Alessi DR. Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity to hypoxia. EMBO J 22: 4666–4676, 2003. doi: 10.1093/emboj/cdg469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ock S, Lee WS, Kim HM, Park KS, Kim YK, Kook H, Park WJ, Lee TJ, Abel ED, Kim J. Connexin43 and zonula occludens-1 are targets of Akt in cardiomyocytes that correlate with cardiac contractile dysfunction in Akt deficient hearts. Biochim Biophys Acta Mol Basis Dis 1864: 1183–1191, 2018. doi: 10.1016/j.bbadis.2018.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shimizu I, Minamino T, Toko H, Okada S, Ikeda H, Yasuda N, Tateno K, Moriya J, Yokoyama M, Nojima A, Koh GY, Akazawa H, Shiojima I, Kahn CR, Abel ED, Komuro I. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J Clin Invest 120: 1506–1514, 2010. doi: 10.1172/JCI40096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Riehle C, Weatherford ET, Wende AR, Jaishy BP, Seei AW, McCarty NS, Rech M, Shi Q, Reddy GR, Kutschke WJ, Oliveira K, Pires KM, Anderson JC, Diakos NA, Weiss RM, White MF, Drakos SG, Xiang YK, Abel ED. Insulin receptor substrates differentially exacerbate insulin-mediated left ventricular remodeling. JCI Insight 5: e134920, 2020. doi: 10.1172/jci.insight.134920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Q, Liu Y, Fu Q, Xu B, Zhang Y, Kim S, Tan R, Barbagallo F, West T, Anderson E, Wei W, Abel ED, Xiang YK. Inhibiting insulin-mediated beta2-adrenergic receptor activation prevents diabetes-associated cardiac dysfunction. Circulation 135: 73–88, 2017. doi: 10.1161/CIRCULATIONAHA.116.022281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu Q, Xu B, Parikh D, Cervantes D, Xiang YK. Insulin induces IRS2-dependent and GRK2-mediated beta2AR internalization to attenuate betaAR signaling in cardiomyocytes. Cell Signal 27: 707–715, 2015. doi: 10.1016/j.cellsig.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ciccarelli M, Chuprun JK, Rengo G, Gao E, Wei Z, Peroutka RJ, Gold JI, Gumpert A, Chen M, Otis NJ, Dorn GW II, Trimarco B, Iaccarino G, Koch WJ. G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation 123: 1953–1962, 2011. doi: 10.1161/CIRCULATIONAHA.110.988642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cannavo A, Marzano F, Elia A, Liccardo D, Bencivenga L, Gambino G, Perna C, Rapacciuolo A, Cittadini A, Ferrara N, Paolocci N, Koch WJ, Rengo G. Aldosterone jeopardizes myocardial insulin and beta-adrenergic receptor signaling via G protein-coupled receptor kinase 2. Front Pharmacol 10: 888, 2019. doi: 10.3389/fphar.2019.00888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen M, Gao C, Yu J, Ren S, Wang M, Wynn RM, Chuang DT, Wang Y, Sun H. Therapeutic effect of targeting branched-chain amino acid catabolic flux in pressure-overload induced heart failure. J Am Heart Assoc 8: e011625, 2019. doi: 10.1161/JAHA.118.011625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang Y, Zhou M, Sun H, Wang Y. Branched-chain amino acid metabolism in heart disease: an epiphenomenon or a real culprit? Cardiovasc Res 90: 220–223, 2011. doi: 10.1093/cvr/cvr070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, Rau CD, Shah S, Ilkayeva O, Gui WJ, William NS, Wynn RM, Newgard CB, Cai H, Xiao X, Chuang DT, Schulze PC, Lynch C, Jain MK, Wang Y. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation 133: 2038–2049, 2016. doi: 10.1161/CIRCULATIONAHA.115.020226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Uddin GM, Zhang L, Shah S, Fukushima A, Wagg CS, Gopal K, Al Batran R, Pherwani S, Ho KL, Boisvenue J, Karwi QG, Altamimi T, Wishart DS, Dyck JRB, Ussher JR, Oudit GY, Lopaschuk GD. Impaired branched chain amino acid oxidation contributes to cardiac insulin resistance in heart failure. Cardiovasc Diabetol 18: 86, 2019. doi: 10.1186/s12933-019-0892-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Renguet E, Ginion A, Gelinas R, Bultot L, Auquier J, Robillard Frayne I, Daneault C, Vanoverschelde JL, Des Rosiers C, Hue L, Horman S, Beauloye C, Bertrand L. Metabolism and acetylation contribute to leucine-mediated inhibition of cardiac glucose uptake. Am J Physiol Heart Circ Physiol 313: H432–H445, 2017. doi: 10.1152/ajpheart.00738.2016. [DOI] [PubMed] [Google Scholar]
- 69.Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, Roy RJ, Matherne GP, Berr SS, Terwelp M, Scott B, Carranza S, Frazier OH, Glover DK, Dillmann WH, Gambello MJ, Entman ML, Taegtmeyer H. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J Am Heart Assoc 2: e004796, 2013. doi: 10.1161/JAHA.113.004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sharma S, Guthrie PH, Chan SS, Haq S, Taegtmeyer H. Glucose phosphorylation is required for insulin-dependent mTOR signalling in the heart. Cardiovasc Res 76: 71–80, 2007. doi: 10.1016/j.cardiores.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kolwicz SC Jr, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res 90: 194–201, 2011. doi: 10.1093/cvr/cvr071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shao D, Tian R. Glucose transporters in cardiac metabolism and hypertrophy. Compr Physiol 6: 331–351, 2015. doi: 10.1002/cphy.c150016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang L, Jaswal JS, Ussher JR, Sankaralingam S, Wagg C, Zaugg M, Lopaschuk GD. Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ Heart Fail 6: 1039–1048, 2013. doi: 10.1161/CIRCHEARTFAILURE.112.000228. [DOI] [PubMed] [Google Scholar]
- 74.Alrob OA, Sankaralingam S, Ma C, Wagg CS, Fillmore N, Jaswal JS, Sack MN, Lehner R, Gupta MP, Michelakis ED, Padwal RS, Johnstone DE, Sharma AM, Lopaschuk GD. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc Res 103: 485–497, 2014. doi: 10.1093/cvr/cvu156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mori J, Alrob OA, Wagg CS, Harris RA, Lopaschuk GD, Oudit GY. ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: a critical role of PDK4. Am J Physiol Heart Circ Physiol 304: H1103–H1113, 2013. doi: 10.1152/ajpheart.00636.2012. [DOI] [PubMed] [Google Scholar]
- 76.Doenst T, Goodwin GW, Cedars AM, Wang M, Stepkowski S, Taegtmeyer H. Load-induced changes in vivo alter substrate fluxes and insulin responsiveness of rat heart in vitro. Metabolism 50: 1083–1090, 2001. doi: 10.1053/meta.2001.25605. [DOI] [PubMed] [Google Scholar]
- 77.Murray AJ, Lygate CA, Cole MA, Carr CA, Radda GK, Neubauer S, Clarke K. Insulin resistance, abnormal energy metabolism and increased ischemic damage in the chronically infarcted rat heart. Cardiovasc Res 71: 149–157, 2006. doi: 10.1016/j.cardiores.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 78.Amorim PA, Nguyen TD, Shingu Y, Schwarzer M, Mohr FW, Schrepper A, Doenst T. Myocardial infarction in rats causes partial impairment in insulin response associated with reduced fatty acid oxidation and mitochondrial gene expression. J Thorac Cardiovasc Surg 140: 1160–1167, 2010. doi: 10.1016/j.jtcvs.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 79.Diakos NA, Navankasattusas S, Abel ED, Rutter J, McCreath L, Ferrin P, McKellar SH, Miller DV, Park SY, Richardson RS, Deberardinis R, Cox JE, Kfoury AG, Selzman CH, Stehlik J, Fang JC, Li DY, Sg D. Evidence of glycolysis up-regulation and pyruvate mitochondrial oxidation mismatch during mechanical unloading of the failing human heart: implications for cardiac reloading and conditioning. JACC Basic Transl Sci 1: 432–444, 2016. doi: 10.1016/j.jacbts.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cluntun AA, Badolia R, Lettlova S, Parnell KM, Shankar TS, Diakos NA, Olson KA, Taleb I, Tatum SM, Berg JA, Cunningham CN, Van Ry T, Bott AJ, Krokidi AT, Fogarty S, Skedros S, Swiatek WI, Yu X, Luo B, Merx S, Navankasattusas S, Cox JE, Ducker GS, Holland WL, McKellar SH, Rutter J, Drakos SG. The pyruvate-lactate axis modulates cardiac hypertrophy and heart failure. Cell Metab 33: 629–648.e10, 2021. doi: 10.1016/j.cmet.2020.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fernandez-Caggiano M, Kamynina A, Francois AA, Prysyazhna O, Eykyn TR, Krasemann S, Crespo-Leiro MG, Vieites MG, Bianchi K, Morales V, Domenech N, Eaton P. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat Metab 2: 1223–1231, 2020. doi: 10.1038/s42255-020-00276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McCommis KS, Kovacs A, Weinheimer CJ, Shew TM, Koves TR, Ilkayeva OR, Kamm DR, Pyles KD, King MT, Veech RL, DeBosch BJ, Muoio DM, Gross RW, Finck BN. Nutritional modulation of heart failure in mitochondrial pyruvate carrier-deficient mice. Nat Metab 2: 1232–1247, 2020. doi: 10.1038/s42255-020-00296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Y, Taufalele PV, Cochran JD, Robillard-Frayne I, Marx JM, Soto J, Rauckhorst AJ, Tayyari F, Pewa AD, Gray LR, Teesch LM, Puchalska P, Funari TR, McGlauflin R, Zimmerman K, Kutschke WJ, Cassier T, Hitchcock S, Lin K, Kato KM, Stueve JL, Haff L, Weiss RM, Cox JE, Rutter J, Taylor EB, Crawford PA, Lewandowski ED, Des Rosiers C, Abel ED. Mitochondrial pyruvate carriers are required for myocardial stress adaptation. Nat Metab 2: 1248–1264, 2020. [Erratum in Nat Metab 2: 1498, 2020]. doi: 10.1038/s42255-020-00288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pires KM, Torres NS, Buffolo M, Gunville R, Schaaf C, Davis K, Selzman CH, Gottlieb RA, Boudina S. Suppression of cardiac autophagy by hyperinsulinemia in insulin receptor-deficient hearts is mediated by insulin-like growth factor receptor signaling. Antioxid Redox Signal 31: 444–457, 2019. doi: 10.1089/ars.2018.7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, Del Campo A, Toro B, Battiprolu PK, Aranguiz P, Chiong M, Yakar S, Gillette TG, Hill JA, Abel ED, Leroith D, Lavandero S. Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res 93: 320–329, 2012. doi: 10.1093/cvr/cvr321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pires KM, Buffolo M, Schaaf C, David Symons J, Cox J, Abel ED, Selzman CH, Boudina S. Activation of IGF-1 receptors and Akt signaling by systemic hyperinsulinemia contributes to cardiac hypertrophy but does not regulate cardiac autophagy in obese diabetic mice. J Mol Cell Cardiol 113: 39–50, 2017. doi: 10.1016/j.yjmcc.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ock S, Lee WS, Ahn J, Kim HM, Kang H, Kim HS, Jo D, Abel ED, Lee TJ, Kim J. Deletion of IGF-1 receptors in cardiomyocytes attenuates cardiac aging in male mice. Endocrinology 157: 336–345, 2016. doi: 10.1210/en.2015-1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wessells RJ, Fitzgerald E, Cypser JR, Tatar M, Bodmer R. Insulin regulation of heart function in aging fruit flies. Nat Genet 36: 1275–1281, 2004. doi: 10.1038/ng1476. [DOI] [PubMed] [Google Scholar]
- 89.Inuzuka Y, Okuda J, Kawashima T, Kato T, Niizuma S, Tamaki Y, Iwanaga Y, Yoshida Y, Kosugi R, Watanabe-Maeda K, Machida Y, Tsuji S, Aburatani H, Izumi T, Kita T, Shioi T. Suppression of phosphoinositide 3-kinase prevents cardiac aging in mice. Circulation 120: 1695–1703, 2009. doi: 10.1161/CIRCULATIONAHA.109.871137. [DOI] [PubMed] [Google Scholar]
- 90.Boudina S, Bugger H, Sena S, O'Neill BT, Zaha VG, Ilkun O, Wright JJ, Mazumder PK, Palfreyman E, Tidwell TJ, Theobald H, Khalimonchuk O, Wayment B, Sheng X, Rodnick KJ, Centini R, Chen D, Litwin SE, Weimer BE, Abel ED. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 119: 1272–1283, 2009. doi: 10.1161/CIRCULATIONAHA.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D, Zhu Y, Kuzmicic J, Pennanen C, Lopez-Crisosto C, Jana F, Ferreira J, Noguera E, Chiong M, Bernlohr DA, Klip A, Hill JA, Rothermel BA, Abel ED, Zorzano A, Lavandero S. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFkappaB-Opa-1 signaling pathway. Diabetes 63: 75–88, 2014. doi: 10.2337/db13-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sena S, Hu P, Zhang D, Wang X, Wayment B, Olsen C, Avelar E, Abel ED, Litwin SE. Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. J Mol Cell Cardiol 46: 910–918, 2009. doi: 10.1016/j.yjmcc.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bugger H, Abel ED. Mitochondria in the diabetic heart. Cardiovasc Res 88: 229–240, 2010. doi: 10.1093/cvr/cvq239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bugger H, Boudina S, Hu XX, Tuinei J, Zaha VG, Theobald HA, Yun UJ, McQueen AP, Wayment B, Litwin SE, Abel ED. Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes 57: 2924–2932, 2008. doi: 10.2337/db08-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bugger H, Chen D, Riehle C, Soto J, Theobald HA, Hu XX, Ganesan B, Weimer BC, Abel ED. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic akita mice. Diabetes 58: 1986–1997, 2009. doi: 10.2337/db09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bugger H, Riehle C, Jaishy B, Wende AR, Tuinei J, Chen D, Soto J, Pires KM, Boudina S, Theobald HA, Luptak I, Wayment B, Wang X, Litwin SE, Weimer BC, Abel ED. Genetic loss of insulin receptors worsens cardiac efficiency in diabetes. J Mol Cell Cardiol 52: 1019–1026, 2012. doi: 10.1016/j.yjmcc.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Durgan DJ, Smith JK, Hotze MA, Egbejimi O, Cuthbert KD, Zaha VG, Dyck JR, Abel ED, Young ME. Distinct transcriptional regulation of long-chain acyl-CoA synthetase isoforms and cytosolic thioesterase 1 in the rodent heart by fatty acids and insulin. Am J Physiol Heart Circ Physiol 290: H2480–H2497, 2006. doi: 10.1152/ajpheart.01344.2005. [DOI] [PubMed] [Google Scholar]
- 98.Nakamura M, Sadoshima J. Cardiomyopathy in obesity, insulin resistance and diabetes. J Physiol 598: 2977–2993, 2020. doi: 10.1113/JP276747. [DOI] [PubMed] [Google Scholar]
- 99.Boardman N, Hafstad AD, Larsen TS, Severson DL, Aasum E. Increased O2 cost of basal metabolism and excitation-contraction coupling in hearts from type 2 diabetic mice. Am J Physiol Heart Circ Physiol 296: H1373–H1379, 2009. doi: 10.1152/ajpheart.01264.2008. [DOI] [PubMed] [Google Scholar]
- 100.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146: 5341–5349, 2005. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 101.Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 112: 2686–2695, 2005. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 102.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56: 2457–2466, 2007. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 103.Folmes CD, Clanachan AS, Lopaschuk GD. Fatty acids attenuate insulin regulation of 5'-AMP-activated protein kinase and insulin cardioprotection after ischemia. Circ Res 99: 61–68, 2006. doi: 10.1161/01.RES.0000229656.05244.11. [DOI] [PubMed] [Google Scholar]
- 104.Schwenk RW, Angin Y, Steinbusch LK, Dirkx E, Hoebers N, Coumans WA, Bonen A, Broers JL, van Eys GJ, Glatz JF, Luiken JJ. Overexpression of vesicle-associated membrane protein (VAMP) 3, but not VAMP2, protects glucose transporter (GLUT) 4 protein translocation in an in vitro model of cardiac insulin resistance. J Biol Chem 287: 37530–37539, 2012. doi: 10.1074/jbc.M112.363630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cook SA, Varela-Carver A, Mongillo M, Kleinert C, Khan MT, Leccisotti L, Strickland N, Matsui T, Das S, Rosenzweig A, Punjabi P, Camici PG. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur Heart J 31: 100–111, 2010. doi: 10.1093/eurheartj/ehp396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Desrois M, Sidell RJ, Gauguier D, Davey CL, Radda GK, Clarke K. Gender differences in hypertrophy, insulin resistance and ischemic injury in the aging type 2 diabetic rat heart. J Mol Cell Cardiol 37: 547–555, 2004. doi: 10.1016/j.yjmcc.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 107.Desrois M, Sidell RJ, Gauguier D, King LM, Radda GK, Clarke K. Initial steps of insulin signaling and glucose transport are defective in the type 2 diabetic rat heart. Cardiovasc Res 61: 288–296, 2004. doi: 10.1016/j.cardiores.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 108.Lee J, Xu Y, Lu L, Bergman B, Leitner JW, Greyson C, Draznin B, Schwartz GG. Multiple abnormalities of myocardial insulin signaling in a porcine model of diet-induced obesity. Am J Physiol Heart Circ Physiol 298: H310–H319, 2010. doi: 10.1152/ajpheart.00359.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu Y, Steinbusch LKM, Nabben M, Kapsokalyvas D, van Zandvoort M, Schonleitner P, Antoons G, Simons PJ, Coumans WA, Geomini A, Chanda D, Glatz JFC, Neumann D, Luiken J. Palmitate-induced vacuolar-type H(+)-ATPase inhibition feeds forward into insulin resistance and contractile dysfunction. Diabetes 66: 1521–1534, 2017. doi: 10.2337/db16-0727. [DOI] [PubMed] [Google Scholar]
- 110.Luiken J, Nabben M, Neumann D, Glatz JFC. Understanding the distinct subcellular trafficking of CD36 and GLUT4 during the development of myocardial insulin resistance. Biochim Biophys Acta Mol Basis Dis 1866: 165775, 2020. doi: 10.1016/j.bbadis.2020.165775. [DOI] [PubMed] [Google Scholar]
- 111.Jain SS, Snook LA, Glatz JF, Luiken JJ, Holloway GP, Thurmond DC, Bonen A. Munc18c provides stimulus-selective regulation of GLUT4 but not fatty acid transporter trafficking in skeletal muscle. FEBS Lett 586: 2428–2435, 2012. doi: 10.1016/j.febslet.2012.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Habets DD, Thurmond DC, Coumans WA, Bonen A, Glatz JF, Luiken JJ. Munc18c is not rate-limiting for glucose and long-chain fatty acid uptake in the heart. Mol Cell Biochem 322: 81–86, 2009. doi: 10.1007/s11010-008-9942-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schwenk RW, Dirkx E, Coumans WA, Bonen A, Klip A, Glatz JF, Luiken JJ. Requirement for distinct vesicle-associated membrane proteins in insulin- and AMP-activated protein kinase (AMPK)-induced translocation of GLUT4 and CD36 in cultured cardiomyocytes. Diabetologia 53: 2209–2219, 2010. doi: 10.1007/s00125-010-1832-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang S, Wong LY, Neumann D, Liu Y, Sun A, Antoons G, Strzelecka A, Glatz JFC, Nabben M, Luiken J. Augmenting vacuolar H+-ATPase function prevents cardiomyocytes from lipid-overload induced dysfunction. Int J Mol Sci 21: 1520, 2020. doi: 10.3390/ijms21041520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huisamen B. Protein kinase B in the diabetic heart. Mol Cell Biochem 249: 31–38, 2003. [PubMed] [Google Scholar]
- 116.Maria Z, Campolo AR, Lacombe VA. Diabetes alters the expression and translocation of the insulin-sensitive glucose transporters 4 and 8 in the atria. PLoS One 10: e0146033, 2015. doi: 10.1371/journal.pone.0146033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang PH, Almahfouz A, Giorgino F, McCowen KC, Smith RJ. In vivo insulin signaling in the myocardium of streptozotocin-diabetic rats: opposite effects of diabetes on insulin stimulation of glycogen synthase and c-Fos. Endocrinology 140: 1141–1150, 1999. doi: 10.1210/endo.140.3.6595. [DOI] [PubMed] [Google Scholar]
- 118.Jaishy B, Zhang Q, Chung HS, Riehle C, Soto J, Jenkins S, Abel P, Cowart LA, Van Eyk JE, Abel ED. Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity. J Lipid Res 56: 546–561, 2015. doi: 10.1194/jlr.M055152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Riehle C, Abel ED. Insulin regulation of myocardial autophagy. Circ J 78: 2569–2576, 2014. doi: 10.1253/circj.cj-14-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Karwi QG, Zhang L, Wagg CS, Wang W, Ghandi M, Thai D, Yan H, Ussher JR, Oudit GY, Lopaschuk GD. Targeting the glucagon receptor improves cardiac function and enhances insulin sensitivity following a myocardial infarction. Cardiovasc Diabetol 18: 1, 2019. doi: 10.1186/s12933-019-0806-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Turdi S, Ge W, Hu N, Bradley KM, Wang X, Ren J. Interaction between maternal and postnatal high fat diet leads to a greater risk of myocardial dysfunction in offspring via enhanced lipotoxicity, IRS-1 serine phosphorylation and mitochondrial defects. J Mol Cell Cardiol 55: 117–129, 2013. doi: 10.1016/j.yjmcc.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 122.Soliman H, Nyamandi V, Garcia-Patino M, Varela JN, Bankar G, Lin G, Jia Z, MacLeod KM. Partial deletion of ROCK2 protects mice from high-fat diet-induced cardiac insulin resistance and contractile dysfunction. Am J Physiol Heart Circ Physiol 309: H70–H81, 2015. doi: 10.1152/ajpheart.00664.2014. [DOI] [PubMed] [Google Scholar]
- 123.Niizuma S, Inuzuka Y, Okuda J, Kato T, Kawashima T, Tamaki Y, Iwanaga Y, Yoshida Y, Kosugi R, Watanabe-Maeda K, Machida Y, Tsuji S, Aburatani H, Izumi T, Kita T, Kimura T, Shioi T. Effect of persistent activation of phosphoinositide 3-kinase on heart. Life Sci 90: 619–628, 2012. doi: 10.1016/j.lfs.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 124.Ruiz-Velasco A, Zi M, Hille SS, Azam T, Kaur N, Jiang J, Nguyen B, Sekeres K, Binder P, Collins L, Pu F, Xiao H, Guan K, Frey N, Cartwright EJ, Muller OJ, Wang X, Liu W. Targeting mir128-3p alleviates myocardial insulin resistance and prevents ischemia-induced heart failure. eLife 9: e54298, 2020. doi: 10.7554/eLife.54298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mazzoccoli G, Dagostino MP, Fontana A, Grandone E, Favuzzi G, Tiscia G, Margaglione M, de Matthaeis A, Greco A, Vendemiale G. Influence of the Gly1057Asp variant of the insulin receptor substrate 2 (IRS2) on insulin resistance and relationship with epicardial fat thickness in the elderly. Exp Gerontol 47: 988–993, 2012. doi: 10.1016/j.exger.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 126.Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, Shelton JM, Gerard RD, Rothermel BA, Gillette TG, Lavandero S, Hill JA. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest 122: 1109–1118, 2012. doi: 10.1172/JCI60329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ni YG, Wang N, Cao DJ, Sachan N, Morris DJ, Gerard RD, Kuro OM, Rothermel BA, Hill JA. FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc Natl Acad Sci USA 104: 20517–20522, 2007. doi: 10.1073/pnas.0610290104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kandadi MR, Panzhinskiy E, Roe ND, Nair S, Hu D, Sun A. Deletion of protein tyrosine phosphatase 1B rescues against myocardial anomalies in high fat diet-induced obesity: Role of AMPK-dependent autophagy. Biochim Biophys Acta 1852: 299–309, 2015. doi: 10.1016/j.bbadis.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 129.Wang S, Chen X, Nair S, Sun D, Wang X, Ren J. Deletion of protein tyrosine phosphatase 1B obliterates endoplasmic reticulum stress-induced myocardial dysfunction through regulation of autophagy. Biochim Biophys Acta Mol Basis Dis 1863: 3060–3074, 2017. doi: 10.1016/j.bbadis.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 130.Dobrzyn P, Sampath H, Dobrzyn A, Miyazaki M, Ntambi JM. Loss of stearoyl-CoA desaturase 1 inhibits fatty acid oxidation and increases glucose utilization in the heart. Am J Physiol Endocrinol Metab 294: E357–E364, 2008. doi: 10.1152/ajpendo.00471.2007. [DOI] [PubMed] [Google Scholar]
- 131.Kienesberger PC, Pulinilkunnil T, Sung MM, Nagendran J, Haemmerle G, Kershaw EE, Young ME, Light PE, Oudit GY, Zechner R, Dyck JR. Myocardial ATGL overexpression decreases the reliance on fatty acid oxidation and protects against pressure overload-induced cardiac dysfunction. Mol Cell Biol 32: 740–750, 2012. doi: 10.1128/MCB.06470-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pulinilkunnil T, Kienesberger PC, Nagendran J, Sharma N, Young ME, Dyck JR. Cardiac-specific adipose triglyceride lipase overexpression protects from cardiac steatosis and dilated cardiomyopathy following diet-induced obesity. Int J Obes (Lond) 38: 205–215, 2014. doi: 10.1038/ijo.2013.103. [DOI] [PubMed] [Google Scholar]
- 133.Pulinilkunnil T, Kienesberger PC, Nagendran J, Waller TJ, Young ME, Kershaw EE, Korbutt G, Haemmerle G, Zechner R, Dyck JR. Myocardial adipose triglyceride lipase overexpression protects diabetic mice from the development of lipotoxic cardiomyopathy. Diabetes 62: 1464–1477, 2013. doi: 10.2337/db12-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res 49: 2101–2112, 2008. doi: 10.1194/jlr.M800147-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Park SY, Cho YR, Finck BN, Kim HJ, Higashimori T, Hong EG, Lee MK, Danton C, Deshmukh S, Cline GW, Wu JJ, Bennett AM, Rothermel B, Kalinowski A, Russell KS, Kim YB, Kelly DP, Kim JK. Cardiac-specific overexpression of peroxisome proliferator-activated receptor-alpha causes insulin resistance in heart and liver. Diabetes 54: 2514–2524, 2005. doi: 10.2337/diabetes.54.9.2514. [DOI] [PubMed] [Google Scholar]
- 136.Atkinson LL, Kozak R, Kelly SE, Onay Besikci A, Russell JC, Lopaschuk GD. Potential mechanisms and consequences of cardiac triacylglycerol accumulation in insulin-resistant rats. Am J Physiol Endocrinol Metab 284: E923–E930, 2003. doi: 10.1152/ajpendo.00360.2002. [DOI] [PubMed] [Google Scholar]
- 137.Zhang L, Ussher JR, Oka T, Cadete VJ, Wagg C, Lopaschuk GD. Cardiac diacylglycerol accumulation in high fat-fed mice is associated with impaired insulin-stimulated glucose oxidation. Cardiovasc Res 89: 148–156, 2011. doi: 10.1093/cvr/cvq266. [DOI] [PubMed] [Google Scholar]
- 138.Harmancey R, Haight DL, Watts KA, Taegtmeyer H. Chronic hyperinsulinemia causes selective insulin resistance and down-regulates uncoupling protein 3 (UCP3) through the activation of sterol regulatory element-binding protein (SREBP)-1 transcription factor in the mouse heart. J Biol Chem 290: 30947–30961, 2015. doi: 10.1074/jbc.M115.673988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Harmancey R, Vasquez HG, Guthrie PH, Taegtmeyer H. Decreased long-chain fatty acid oxidation impairs postischemic recovery of the insulin-resistant rat heart. FASEB J 27: 3966–3978, 2013. doi: 10.1096/fj.13-234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fillmore N, Wagg CS, Zhang L, Fukushima A, Lopaschuk GD. Cardiac branched-chain amino acid oxidation is reduced during insulin resistance in the heart. Am J Physiol Endocrinol Metab 315: E1046–E1052, 2018. doi: 10.1152/ajpendo.00097.2018. [DOI] [PubMed] [Google Scholar]
- 141.Chanda D, Oligschlaeger Y, Geraets I, Liu Y, Zhu X, Li J, Nabben M, Coumans W, Luiken J, Glatz JFC, Neumann D. 2-Arachidonoylglycerol ameliorates inflammatory stress-induced insulin resistance in cardiomyocytes. J Biol Chem 292: 7105–7114, 2017. doi: 10.1074/jbc.M116.767384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sloan C, Tuinei J, Nemetz K, Frandsen J, Soto J, Wride N, Sempokuya T, Alegria L, Bugger H, Abel ED. Central leptin signaling is required to normalize myocardial fatty acid oxidation rates in caloric-restricted ob/ob mice. Diabetes 60: 1424–1434, 2011. doi: 10.2337/db10-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Graveleau C, Zaha VG, Mohajer A, Banerjee RR, Dudley-Rucker N, Steppan CM, Rajala MW, Scherer PE, Ahima RS, Lazar MA, Abel ED. Mouse and human resistins impair glucose transport in primary mouse cardiomyocytes, and oligomerization is required for this biological action. J Biol Chem 280: 31679–31685, 2005. doi: 10.1074/jbc.M504008200. [DOI] [PubMed] [Google Scholar]
- 144.Kang S, Chemaly ER, Hajjar RJ, Lebeche D. Resistin promotes cardiac hypertrophy via the AMP-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways. J Biol Chem 286: 18465–18473, 2011. doi: 10.1074/jbc.M110.200022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Xing W, Tan Y, Li K, Tian P, Tian F, Zhang H. Upregulated hepatokine fetuin B aggravates myocardial ischemia/reperfusion injury through inhibiting insulin signaling in diabetic mice. J Mol Cell Cardiol 151: 163–172, 2021. doi: 10.1016/j.yjmcc.2020.03.002. [DOI] [PubMed] [Google Scholar]
- 146.Ko HJ, Zhang Z, Jung DY, Jun JY, Ma Z, Jones KE, Chan SY, Kim JK. Nutrient stress activates inflammation and reduces glucose metabolism by suppressing AMP-activated protein kinase in the heart. Diabetes 58: 2536–2546, 2009. doi: 10.2337/db08-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Stanisic J, Koricanac G, Kostic M, Stojiljkovic M, Culafic T, Romic S, Tepavcevic S. Low-intensity exercise in the prevention of cardiac insulin resistance-related inflammation and disturbances in NOS and MMP-9 regulation in fructose-fed ovariectomized rats. Appl Physiol Nutr Metab 44: 1219–1229, 2019. doi: 10.1139/apnm-2018-0785. [DOI] [PubMed] [Google Scholar]
- 148.Wu HK, Zhang Y, Cao CM, Hu X, Fang M, Yao Y, Jin L, Chen G, Jiang P, Zhang S, Song R, Peng W, Liu F, Guo J, Tang L, He Y, Shan D, Huang J, Zhou Z, Wang DW, Lv F, Xiao RP. Glucose-sensitive myokine/cardiokine MG53 regulates systemic insulin response and metabolic homeostasis. Circulation 139: 901–914, 2019. doi: 10.1161/CIRCULATIONAHA.118.037216. [DOI] [PubMed] [Google Scholar]
- 149.Liu F, Song R, Feng Y, Guo J, Chen Y, Zhang Y, Chen T, Wang Y, Huang Y, Li CY, Cao C, Zhang Y, Hu X, Xiao RP. Upregulation of MG53 induces diabetic cardiomyopathy through transcriptional activation of peroxisome proliferation-activated receptor alpha. Circulation 131: 795–804, 2015. doi: 10.1161/CIRCULATIONAHA.114.012285. [DOI] [PubMed] [Google Scholar]
- 150.Ham YM, Mahoney SJ. Compensation of the AKT signaling by ERK signaling in transgenic mice hearts overexpressing TRIM72. Exp Cell Res 319: 1451–1462, 2013. doi: 10.1016/j.yexcr.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 151.Oosterman JE, Wopereis S, Kalsbeek A. The circadian clock, shift work, and tissue-specific insulin resistance. Endocrinology 161: bqaa180, 2020. doi: 10.1210/endocr/bqaa180. [DOI] [PubMed] [Google Scholar]
- 152.Durgan DJ, Young ME. The cardiomyocyte circadian clock: emerging roles in health and disease. Circ Res 106: 647–658, 2010. doi: 10.1161/CIRCRESAHA.109.209957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Young ME, Wilson CR, Razeghi P, Guthrie PH, Taegtmeyer H. Alterations of the circadian clock in the heart by streptozotocin-induced diabetes. J Mol Cell Cardiol 34: 223–231, 2002. doi: 10.1006/jmcc.2001.1504. [DOI] [PubMed] [Google Scholar]
- 154.Bray MS, Shaw CA, Moore MW, Garcia RA, Zanquetta MM, Durgan DJ, Jeong WJ, Tsai JY, Bugger H, Zhang D, Rohrwasser A, Rennison JH, Dyck JR, Litwin SE, Hardin PE, Chow CW, Chandler MP, Abel ED, Young ME. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function, metabolism, and gene expression. Am J Physiol Heart Circ Physiol 294: H1036–H1047, 2008. doi: 10.1152/ajpheart.01291.2007. [DOI] [PubMed] [Google Scholar]
- 155.McGinnis GR, Tang Y, Brewer RA, Brahma MK, Stanley HL, Shanmugam G, Rajasekaran NS, Rowe GC, Frank SJ, Wende AR, Abel ED, Taegtmeyer H, Litovsky S, Darley-Usmar V, Zhang J, Chatham JC, Young ME. Genetic disruption of the cardiomyocyte circadian clock differentially influences insulin-mediated processes in the heart. J Mol Cell Cardiol 110: 80–95, 2017. doi: 10.1016/j.yjmcc.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Taegtmeyer H, Beauloye C, Harmancey R, Hue L. Insulin resistance protects the heart from fuel overload in dysregulated metabolic states. Am J Physiol Heart Circ Physiol 305: H1693–H1697, 2013. doi: 10.1152/ajpheart.00854.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Liao R, Jain M, Cui L, D’Agostino J, Aiello F, Luptak I, Ngoy S, Mortensen RM, Tian R. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 106: 2125–2131, 2002. doi: 10.1161/01.CIR.0000034049.61181.F3. [DOI] [PubMed] [Google Scholar]
- 158.Luptak I, Yan J, Cui L, Jain M, Liao R, Tian R. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation 116: 901–909, 2007. doi: 10.1161/CIRCULATIONAHA.107.691253. [DOI] [PubMed] [Google Scholar]
- 159.Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, Anderson SM, Abel ED. Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J Am Heart Assoc 2: e000301, 2013. doi: 10.1161/JAHA.113.000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wende AR, Schell JC, Ha CM, Pepin ME, Khalimonchuk O, Schwertz H, Pereira RO, Brahma MK, Tuinei J, Contreras-Ferrat A, Wang L, Andrizzi CA, Olsen CD, Bradley WE, Dell'Italia LJ, Dillmann WH, Litwin SE, Abel ED. Maintaining myocardial glucose utilization in diabetic cardiomyopathy accelerates mitochondrial dysfunction. Diabetes 69: 2094–2111, 2020. doi: 10.2337/db19-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation 119: 2818–2828, 2009. doi: 10.1161/CIRCULATIONAHA.108.832915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sankaralingam S, Abo Alrob O, Zhang L, Jaswal JS, Wagg CS, Fukushima A, Padwal RS, Johnstone DE, Sharma AM, Lopaschuk GD. Lowering body weight in obese mice with diastolic heart failure improves cardiac insulin sensitivity and function: implications for the obesity paradox. Diabetes 64: 1643–1657, 2015. doi: 10.2337/db14-1050. [DOI] [PubMed] [Google Scholar]
- 163.Yan Z, Chen W, Liu S, Zhang G, Sun D, Hu S. Myocardial insulin signaling and glucose transport are up-regulated in Goto-Kakizaki type 2 diabetic rats after ileal transposition. Obes Surg 22: 493–501, 2012. doi: 10.1007/s11695-012-0604-5. [DOI] [PubMed] [Google Scholar]
- 164.Karwi QG, Zhang L, Altamimi TR, Wagg CS, Patel V, Uddin GM, Joerg AR, Padwal RS, Johnstone DE, Sharma A, Oudit GY, Lopaschuk GD. Weight loss enhances cardiac energy metabolism and function in heart failure associated with obesity. Diabetes Obes Metab 21: 1944–1955, 2019. doi: 10.1111/dom.13762. [DOI] [PubMed] [Google Scholar]
- 165.Sidell RJ, Cole MA, Draper NJ, Desrois M, Buckingham RE, Clarke K. Thiazolidinedione treatment normalizes insulin resistance and ischemic injury in the zucker fatty rat heart. Diabetes 51: 1110–1117, 2002. doi: 10.2337/diabetes.51.4.1110. [DOI] [PubMed] [Google Scholar]
- 166.Franekova V, Angin Y, Hoebers NT, Coumans WA, Simons PJ, Glatz JF, Luiken JJ, Larsen TS. Marine omega-3 fatty acids prevent myocardial insulin resistance and metabolic remodeling as induced experimentally by high insulin exposure. Am J Physiol Cell Physiol 308: C297–C307, 2015. doi: 10.1152/ajpcell.00073.2014. [DOI] [PubMed] [Google Scholar]
- 167.Coven DL, Hu X, Cong L, Bergeron R, Shulman GI, Hardie DG, Lh Y. Physiological role of AMP-activated protein kinase in the heart: graded activation during exercise. Am J Physiol Endocrinol Metab 285: E629–E636, 2003. doi: 10.1152/ajpendo.00171.2003. [DOI] [PubMed] [Google Scholar]
- 168.Stanisic J, Koricanac G, Culafic T, Romic S, Stojiljkovic M, Kostic M, Pantelic M, Tepavcevic S. Low intensity exercise prevents disturbances in rat cardiac insulin signaling and endothelial nitric oxide synthase induced by high fructose diet. Mol Cell Endocrinol 420: 97–104, 2016. doi: 10.1016/j.mce.2015.11.032. [DOI] [PubMed] [Google Scholar]
- 169.Oliveira LDC, de Morais GP, da Rocha AL, Teixeira GR, Pinto AP, de Vicente LG, Pauli JR, de Moura LP, Mekary RA, Ropelle ER, Cintra DE, da Silva ASR. Excessive treadmill training enhances the insulin signaling pathway and glycogen deposition in mice hearts. J Cell Biochem 120: 1304–1317, 2019. doi: 10.1002/jcb.27092. [DOI] [PubMed] [Google Scholar]
- 170.Bertrand L, Ginion A, Beauloye C, Hebert AD, Guigas B, Hue L, Vanoverschelde JL. AMPK activation restores the stimulation of glucose uptake in an in vitro model of insulin-resistant cardiomyocytes via the activation of protein kinase B. Am J Physiol Heart Circ Physiol 291: H239–H250, 2006. doi: 10.1152/ajpheart.01269.2005. [DOI] [PubMed] [Google Scholar]
- 171.Ginion A, Auquier J, Benton CR, Mouton C, Vanoverschelde JL, Hue L, Horman S, Beauloye C, Bertrand L. Inhibition of the mTOR/p70S6K pathway is not involved in the insulin-sensitizing effect of AMPK on cardiac glucose uptake. Am J Physiol Heart Circ Physiol 301: H469–H477, 2011. doi: 10.1152/ajpheart.00986.2010. [DOI] [PubMed] [Google Scholar]
- 172.Steinbusch LK, Dirkx E, Hoebers NT, Roelants V, Foretz M, Viollet B, Diamant M, van Eys G, Ouwens DM, Bertrand L, Glatz JF, Luiken JJ. Overexpression of AMP-activated protein kinase or protein kinase D prevents lipid-induced insulin resistance in cardiomyocytes. J Mol Cell Cardiol 55: 165–173, 2013. doi: 10.1016/j.yjmcc.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 173.Zhang Y, Yuan M, Bradley KM, Dong F, Anversa P, Ren J. Insulin-like growth factor 1 alleviates high-fat diet-induced myocardial contractile dysfunction: role of insulin signaling and mitochondrial function. Hypertension 59: 680–693, 2012. [Erratum in Hypertension 63: e92, 2014]. doi: 10.1161/HYPERTENSIONAHA.111.181867. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 174.Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 61: 21–28, 2018. doi: 10.1007/s00125-017-4390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tabbi-Anneni I, Buchanan J, Cooksey RC, Abel ED. Captopril normalizes insulin signaling and insulin-regulated substrate metabolism in obese (ob/ob) mouse hearts. Endocrinology 149: 4043–4050, 2008. doi: 10.1210/en.2007-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ilkun O, Wilde N, Tuinei J, Pires KM, Zhu Y, Bugger H, Soto J, Wayment B, Olsen C, Litwin SE, Abel ED. Antioxidant treatment normalizes mitochondrial energetics and myocardial insulin sensitivity independently of changes in systemic metabolic homeostasis in a mouse model of the metabolic syndrome. J Mol Cell Cardiol 85: 104–116, 2015. doi: 10.1016/j.yjmcc.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Dirkx E, van Eys GJ, Schwenk RW, Steinbusch LK, Hoebers N, Coumans WA, Peters T, Janssen BJ, Brans B, Vogg AT, Neumann D, Glatz JF, Luiken JJ. Protein kinase-D1 overexpression prevents lipid-induced cardiac insulin resistance. J Mol Cell Cardiol 76: 208–217, 2014. doi: 10.1016/j.yjmcc.2014.08.017. [DOI] [PubMed] [Google Scholar]