

Abstract
Obesity-related asthma often presents with more severe symptoms than non-obesity-related asthma and responds poorly to current treatments. Both insulin resistance and hyperinsulinemia are common in obesity. We have shown that increased insulin mediates airway hyperreactivity in diet-induced obese rats by causing neuronal M2 muscarinic receptor dysfunction, which normally inhibits acetylcholine release from parasympathetic nerves. Decreasing insulin with streptozotocin prevented airway hyperreactivity and M2 receptor dysfunction. The objective of the present study was to investigate whether pioglitazone, a hypoglycemic drug, prevents airway hyperreactivity and M2 receptor dysfunction in obese rats. Male rats fed a low- or high-fat diet were treated with pioglitazone or PBS by daily gavage. Body weight, body fat, fasting insulin, and bronchoconstriction and bradycardia in response to electrical stimulation of vagus nerves and to aerosolized methacholine were recorded. Pilocarpine, a muscarinic receptor agonist, was used to measure M2 receptor function. Rats on a high-fat diet had potentiated airway responsiveness to vagal stimulation and dysfunctional neuronal M2 receptors, whereas airway responsiveness to methacholine was unaffected. Pioglitazone reduced fasting insulin and prevented airway hyperresponsiveness and M2 receptor dysfunction but did not change inflammatory cytokine mRNA expression in alveolar macrophages. High-fat diet, with and without pioglitazone, had tissue-specific effects on insulin receptor mRNA expression. In conclusion, pioglitazone prevents vagally mediated airway hyperreactivity and protects neuronal M2 muscarinic receptor function in obese rats.
Keywords: asthma, insulin, M2 muscarinic receptors, obesity, pioglitazone
INTRODUCTION
Eight percent of American adults have asthma (1). In individuals with obesity, this is increased to 12% (1). Patients with asthma and obesity have more frequent exacerbations and hospitalizations, leading to higher healthcare costs and reduced quality of life. Additionally, patients with asthma and obesity respond poorly to existing asthma therapeutics (2–5). In the United States, 42.4% of adults and 18.5% of children are obese (6), so there is an urgent need for effective treatments for obesity-related asthma symptoms.
Airway hyperreactivity is a key pathophysiological feature of asthma and can result from loss of neuronal M2 muscarinic receptor function (7–10). Parasympathetic nerves control airway tone and mediate bronchoconstriction via release of acetylcholine onto M3 muscarinic receptors on airway smooth muscle, resulting in smooth muscle contraction and bronchoconstriction. M2 muscarinic receptors on parasympathetic nerves normally inhibit release of acetylcholine and limit bronchoconstriction (11). Dysfunction of neuronal M2 receptors leads to increased acetylcholine release and airway hyperreactivity as has been described in humans with asthma (9, 10) and animal models of asthma (12–15). However, the mechanisms of M2 dysfunction differs with different models of airway hyperreactivity (12–15).
Insulin resistance and hyperinsulinemia are common in individuals with obesity and have been associated with increased asthma risk (16, 17). We have shown that M2 muscarinic receptor function is decreased by insulin (7). Conversely, when insulin is reduced by streptozotocin in naïve rats, M2 receptor function is increased on nerves innervating the airway and ileum, suppressing nerve-mediated contraction (18, 19). Obese rats that have high levels of insulin have reduced neuronal M2 function and increased bronchoconstriction in response to parasympathetic nerve stimulation (7). Depleting insulin in these animals with streptozotocin reversed airway hyperresponsiveness and protected neuronal M2 receptor function (7). Supplemental insulin administered to streptozotocin-treated rats restored airway hyperreactivity and M2 receptor dysfunction. Together, these data argue strongly for a central role for hyperinsulinemia in obesity-related M2 receptor dysfunction, leading to airway hyperreactivity.
Insulin receptors are expressed as two isoforms from alternative splicing: insulin receptor isoform A (IR-A) and insulin receptor isoform B (IR-B) (20). Although they have a similar affinity for insulin (21), IR-A internalizes more rapidly than IR-B (22) and is associated with a decrease in metabolic signaling of insulin, whereas IR-B is responsible for most of the metabolic actions of insulin (21). IR-A and IR-B are differentially expressed in tissues (20). In adipose tissue, dysregulation of the IR-A/IR-B ratio is associated with obesity-related insulin resistance (20). Although it has been reported that IR-A expression is increased by hyperinsulinemia (23), a direct role for insulin in the regulation of insulin receptor splicing is controversial.
Pioglitazone, an agonist for nuclear receptor peroxisome proliferator-activated receptor-γ (PPAR-γ), decreases hepatic glucose output and increases insulin-dependent glucose uptake in skeletal muscle and, possibly, in the liver and adipose tissues (24). Thus, pioglitazone lowers the plasma insulin level by increasing removal of glucose in peripheral tissues. In this study, we tested whether decreasing insulin with pioglitazone protects M2 receptor function and reduces airway hyperreactivity in diet-induced obese rats.
METHODS
Animals
Obese-prone male rats were purchased from Charles River Laboratory (Wilmington, MA), shipped in filtered crates and kept in high-efficiency particulate-filtered air. Obese-prone rats are outbred and have a heterogenetic background, similar to humans (25), because they were originally developed from wild-type Sprague-Dawley rats by selectively breeding of rats that gained the most weight. As a consequence of this selective breeding, their offspring are prone to become obese when fed a high-fat diet (26). Animals were handled in accordance with the standards established by the US Animal Welfare Acts set forth in National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committee at Oregon Health & Science University.
Rats at 5–6 wk of age were all similar weight (140 ± 10 g) before being switched to either a high-fat diet (60% of calories from fat, Test Diet 58126, St. Louis, MO) or a low-fat diet (13% of calories from fat, Test Diet 58124) for 5 wk (Fig. 1A) while their food intake and body weight were monitored. Rats on a high-fat diet were gavaged daily with pioglitazone (3 mg/kg, E6910-10MG, Sigma-Aldrich, St. Louis, MO) or PBS (300 µL). At the end of 5 wk of diet treatment, rats were fasted overnight (16 h) before body fat was analyzed by quantitative magnetic resonance (EchoMRI-5000, EchoMRI, Houston, TX). Briefly, nonanesthetized and unrestrained rats were placed into a holding tube and placed into a calibrated EchoMRI system. This system uses nuclear magnetic resonance relaxometry to detect different spin relaxation rates in variable tissues, such as free water, fat, and lean tissue, and uses a preinstalled and precalibrated software to calculate grams of body water, fat, and lean mass. Body fat was presented as a percentage of body weight. Animals were then anesthetized for physiological measurements of airway and cardiac function. Blood and tissues were collected. Blood glucose was measured using a glucose meter (The OneTouch Ultra2, LifeScan, Milpitas, CA). Plasma insulin was measured by ELISA (no. 10–1250-01, Mercodia, Winston Salem, NC).
Figure 1.
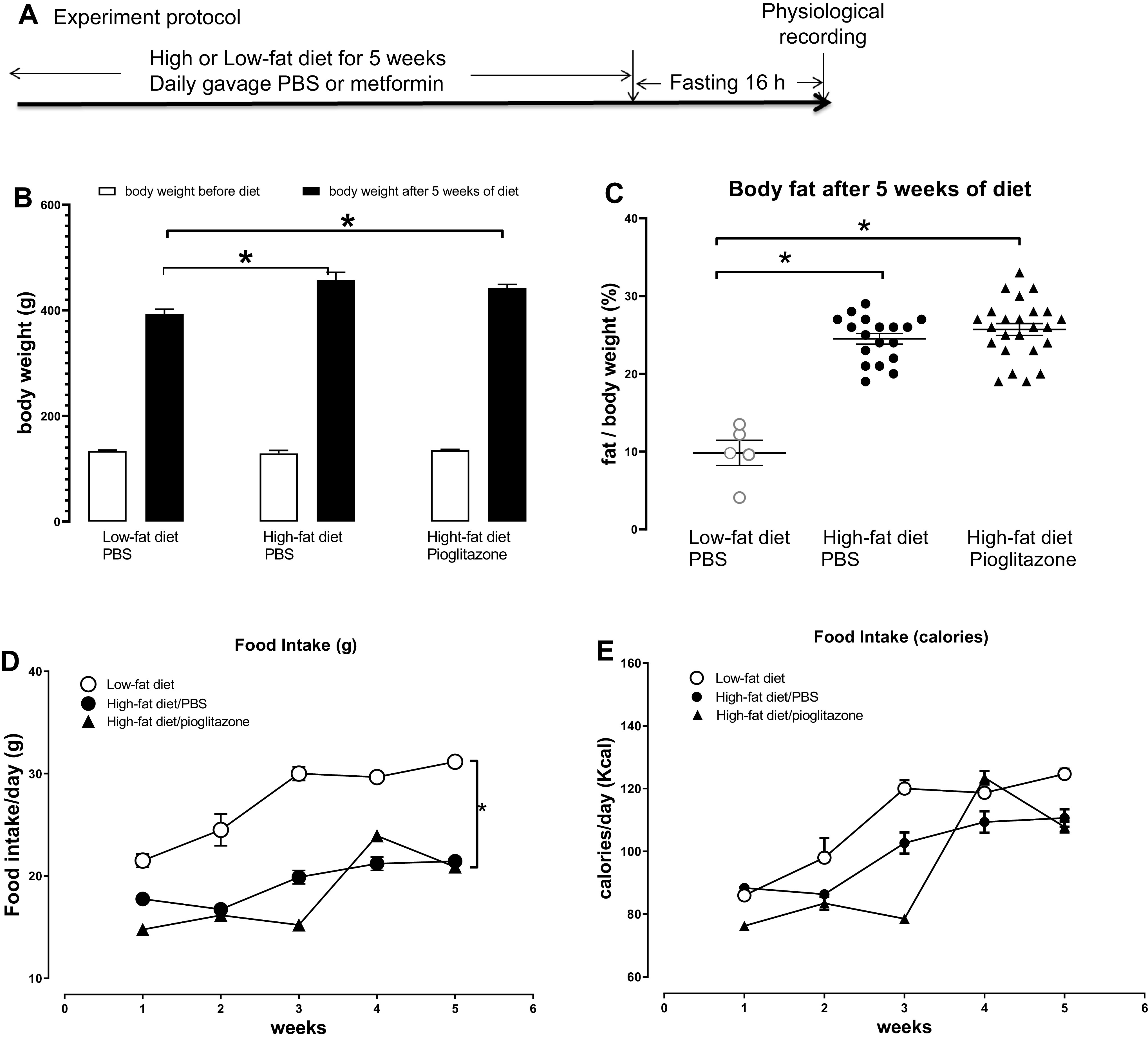
Experiment protocol, animal body weight, and food intake. A: obese-prone rats were fed a high-fat diet or low-fat diet for 5 wk. Animals on a high-fat diet were treated with either pioglitazone (3 mg/kg) or PBS (control) by daily gavage. After 5 wk, all rats were fasted overnight (16 h) and airway physiology was tested the next day. B: rats were weighed before and every week during the diet treatment. High-fat diet-fed rats gained more body weight than that in low-fat diet-fed rats, but pioglitazone did not prevent the increase of body weight gain induced by high-fat diet. C: the percentage of body fat was significantly higher in rats fed a high-fat diet than that in rats fed a low-fat diet, and pioglitazone did not prevent the increase of body fat induced by a high-fat diet. Although the food intake every week was higher in rats on a low-fat diet (D), calories consumed every week (E) was not significantly different. n = 5–16. *P < 0.05.
Anesthesia and Measurement of Pulmonary Inflation Pressure and Heart Rate
Bronchoconstriction in response to electrical stimulation of both vagus nerves was measured, as previously described (27). Rats were anesthetized with urethane (1.6 g/kg ip, Sigma-Aldrich), chemically sympathectomized with guanethidine (5 mg/kg iv, 30 min before recording, Sigma-Aldrich), and paralyzed with succinylcholine (10 mg/mL, infused into a jugular vein at a rate of 0.155 mL/min, Sigma-Aldrich). Animals were ventilated (tidal volume: 2.5 mL, 100 breaths/min, Harvard Apparatus, South Natick, MA) via a tracheal cannula. The sidearm of tracheal cannula was linked to a pressure transducer to measure pulmonary inflation pressure (Ppi, in mmH2O), and bronchoconstriction was measured as the increase in Ppi above baseline inflation pressure produced by the ventilator. Heart rate (beats/min) and blood pressure (mmHg) were measured from a pressure transducer attached to a cannula inserted into the carotid artery. Baseline heart rate, blood pressure, and pulmonary inflation pressure (Table 1) were recorded using Lab Chart software (AD Instruments, Colorado Springs, CO). Body temperature was maintained at 37°C using a heating blanket.
Table 1.
Pioglitazone did not change pulmonary inflation pressure or cardiovascular parameters at baseline
| Diet and Treatment Group | Ppi mmH2O | Heart Rate, beats/min | Systolic BP, mmHg | Diastolic BP, mmHg | Number of Animals |
|---|---|---|---|---|---|
| Low-fat diet + PBS | 77.0 ± 2.8 | 403 ± 14 | 85 ± 3 | 53 ± 3 | 11 |
| High-fat diet+ PBS | 80.4 ± 1.8 | 376 ± 15 | 89 ± 5 | 49 ± 3 | 22 |
| High-fat diet + Pioglitazone | 83.26 ± 3.4 | 386 ± 16 | 95 ± 6 | 50 ± 2 | 19 |
Ppi, pulmonary inflation pressure.
Vagally Induced Bronchoconstriction and Bradycardia
Rats were vagotomized by cutting both vagus nerves to remove input from the central nervous system. The distal ends of both vagus nerves were placed on platinum electrodes and immersed in mineral oil. Electrical stimulation of both vagus nerves (2–30 Hz, 20 V, 0.4-ms pulse duration, for 6 s at 40-s intervals) produced frequency-dependent bronchoconstriction and bradycardia that recovered upon cessation of electrical stimulation. Atropine (1 mg/kg) was administrated by intravenous injection to verify whether the vagally induced bronchoconstriction and bradycardia were mediated by release of acetylcholine, a muscarinic receptor agonist, onto muscarinic receptors.
Neuronal M2 Muscarinic Receptor Function
Electrical stimulation of both vagus nerves (10 Hz, 5 V, 0.4-ms pulse duration, for 6 s at 40-s intervals) resulted in reproducible bronchoconstriction. Pilocarpine is a muscarinic agonist (28) with functional selectivity for M2 over M3 muscarinic receptors at a low dose (<100 µg/kg) (13). In vivo, pilocarpine (0.001–100 µg/kg) inhibits vagally induced bronchoconstriction by stimulating neuronal M2 muscarinic receptors to limit acetylcholine release (13), whereas higher doses (≥100 µg/kg) induce bronchoconstriction via stimulating M3 receptors on airway smooth muscle (13). Vagally induced bronchoconstriction was measured in the absence and presence of increasing doses of pilocarpine (0.001–100 µg/kg iv), and data are expressed as the percentage of vagally induced bronchoconstriction in the absence of pilocarpine.
Methacholine-Induced Bronchoconstriction and Bradycardia
Muscarinic receptor function on airway smooth muscle and cardiac muscle was tested by measuring bronchoconstriction and bradycardia in response to aerosolized methacholine (1–30 mM, 20 µL for 20 s, Sigma-Aldrich) via nebulizer (AG-AL1100, Kent Scientific) in sedated, ventilated, and vagotomized rats.
Bronchoalveolar Lavage and Alveolar Macrophage Culture
Upon completion of physiological measurements, 5 mL of cold, sterile PBS containing 100 U/mL penicillin and 100 µg/mL streptomycin (Hyclone, GE Healthcare, Chicago, IL) were instilled in the tracheal cannula, withdrawn, and collected. This was repeated four more times. Bronchoalveolar lavage (BAL) fluid was centrifuged at 300 g for 10 min, and the supernatant was aspirated. Cells were resuspended in 10 mL of media (at 37°C) consisting of RPMI (Cellgro, Corning, NY), 10% heat-inactivated FBS (Hyclone), 2 µM l-glutamine (Sigma-Aldrich), and penicillin-streptomycin and incubated in a 100-mm2 tissue culture dish for 2 h at 37°C and 5% CO2 and 95% O2 to allow macrophages to adhere before media with nonattached cells were removed. Attached macrophages were rinsed three times with sterile PBS (at 37°C) and collected for RNA isolation.
RNA Isolation and PCR
Rat lung, liver, and BAL macrophage mRNA were isolated using an RNeasy Mini Kit (Qiagen, Valencia, CA), whereas brain and adipose mRNA were isolated using an RNeasy Lipid Tissue Mini Kit (Qiagen). RNA was reverse transcribed to cDNA using random hexamers and Superscript III (Invitrogen, Carlsbad, CA). Cytokine and total insulin receptor mRNA expression was quantified using QuantiTect SYBR Green (Qiagen) and real-time RT-PCR (7500 Fast Real-Time PCR System, Applied Biosystems, Foster City, CA). Primer sequences are shown in Table 2. Cytokine and total insulin receptor mRNA data were analyzed using the ΔΔCT method (where CT is threshold cycle) and normalized to 18S rRNA (29). Data were then graphed as a fold changes from the average RNA expression in animals on a low-fat diet.
Table 2.
Primer sequences
| Gene | Forward | Reverse |
|---|---|---|
| 18S | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
| Rat IR A | GTTTTTGTTCCCAGGCCATC | GATGGTGGAGGAGATGTTGG |
| Rat IR B | GTCCCCACCTTTTGAGTCTG | CACCATTGCCTGAAGAGGTT |
| Rat TNFα | CCCAGACCCTCACACTCAGAT | TTGTCCCTTGAAGAGAACCTG |
| Rat IL-1β | CACCTTCTTTTCCTTCATCTT T | GTCGTTGCTTGTCTCTCCTTG |
| Rat TGFβ | GAGAGCCCTGGATACCAACTA | GTGTGTCCAGGCTCCAAATGT |
| Rat IL-6 | ACTTCACAAGTCGGAGGCTT | TTCTGACAGTGCATCATCGCT |
To generate a series of standards for the quantification of copy numbers of IR-A and IR-B mRNA in tissue, mRNA from the rat brain (where IR-A expression is abundant) and mRNA from the rat liver (where IR-B expression is abundant) were isolated and reverse transcribed to cDNA, as described above. Conventional RT-PCR (7500 Fast Real-Time PCR System, Applied Biosystems) was used to detect IR-A in brain tissue and IR-B in liver tissue. The PCR products were electrophoresed on an agarose gel, extracted (QIAquick Gel extraction, Qiagen), and then quantified using a spectrometer (Spectronic BioMate 3, Thermo Fisher Scientific, Waltham, MA). The number of copies in these samples were calculated as follows: [cDNA amount (ng) × (6.022 × 1023)]/[length (base pairs) × (1 × 109) × 650], where 650 is the average weight of a base pair. Using this value, the highest standard (1010 copies) was prepared and then serial diluted to obtain a series of standards (1010-101) for both IR-A and IR-B.
To determine the number of copies of IR-A and IR-B in lung, adipose, and brain tissues, isolated mRNA was reverse transcribed to cDNA and measured by real-time RT-PCR. Using the CT values generated by the standard curve for IR-A and IR-B, as described above, the number IR-A and IR-B mRNA copies in each tissue sample was calculated.
Statistical Analysis
Bronchoconstriction and bradycardia in response to electrical stimulation of the vagus nerves and to aerosolized methacholine as well as bronchoconstriction and bradycardia in the presence of increasing doses of pilocarpine were analyzed using repeated-measures ANOVA. Daily food intake was analyzed using two-way ANOVA with Bonferroni correction. All other data were analyzed by one-way ANOVA with Bonferroni correction. All data were analyzed with Prism 8.0 software (GraphPad, La Jolla, CA). P values of <0.05 were considered statistically significant.
RESULTS
Pioglitazone Did Not Prevent High-Fat Diet-Induced Gain in Body Weight and Body Fat
Rat average body weight among groups was similar before the start of the low- or high-fat diet (Fig. 1B). After 5 wk, rats on a high-fat diet had significantly more body weight (Fig. 1B) and body fat (Fig. 1C) than rats on a low-fat diet. Pioglitazone did not prevent diet-induced increases in either body weight or fat (Fig. 1, B and C). Food consumption increased with age (Fig. 1, D and E). Although rats on a low-fat diet consumed more grams of food than those on a high-fat diet (Fig. 1D), caloric intake was not affected by diet or by pioglitazone (Fig. 1E).
Pioglitazone Reduced High-Fat Diet-Induced Hyperinsulinemia
Levels of fasting glucose were similar across all groups of animals, regardless of diet or pioglitazone treatment (Fig. 2A). In contrast, rats on a high-fat diet had elevated fasting insulin compared with rats on a low-fat diet, and this increase was prevented by pioglitazone (Fig. 2B).
Figure 2.
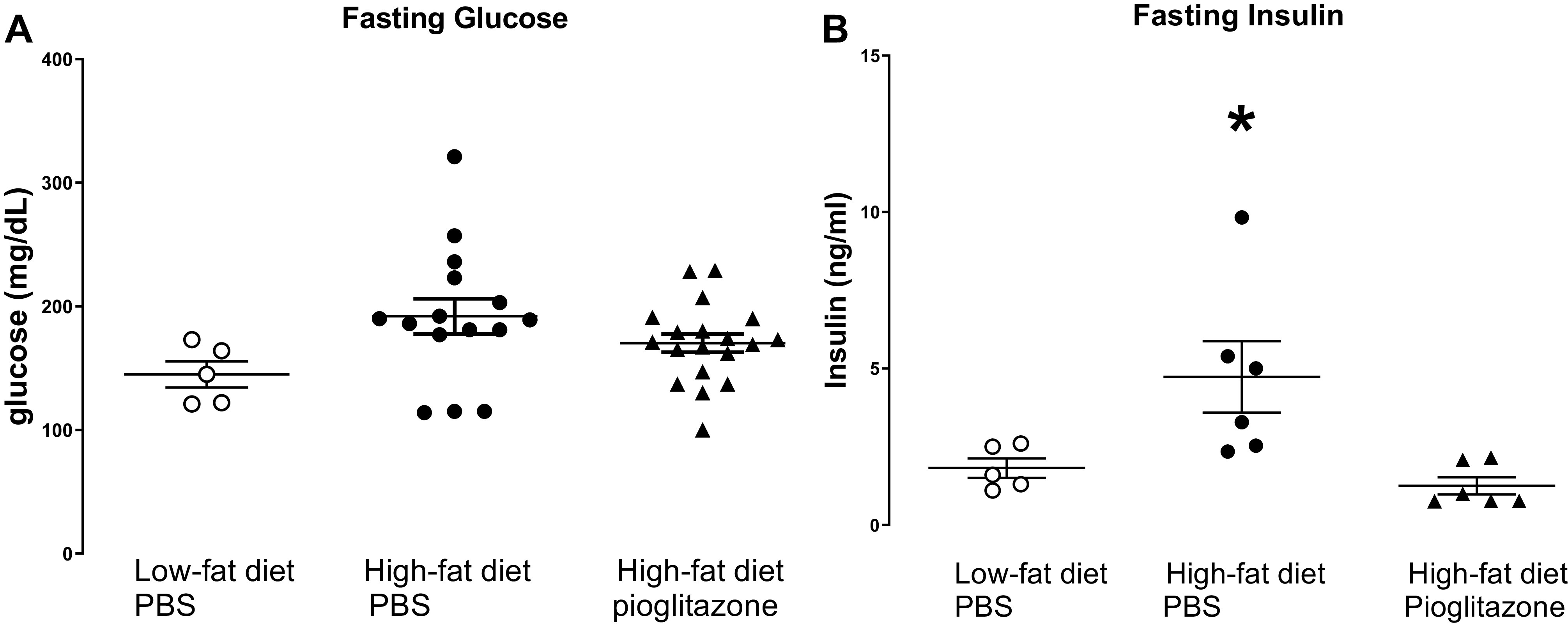
Fasting glucose and insulin. After 5 wk of a low-fat diet or high-fat diet and treated with pioglitazone (3 mg/kg) or PBS by daily gavage, rats were fasted overnight and blood glucose (A) and plasma insulin (B) were measured the following day. n = 5–18. *P < 0.05 compared with all other groups.
Pioglitazone Prevented High-Fat Diet-Induced Airway Hyperreactivity
To assess nerve-mediated airway hyperreactivity, bronchoconstriction in response to electrical stimulation of the vagus nerve was measured. In anesthetized rats, baseline Ppi, heart rate, and blood pressure were similar among the groups (Table 1). Vagally induced bronchoconstriction was significantly potentiated in rats on a high-fat diet compared with rats eating a low-fat diet (Fig. 3A). Pioglitazone treatment significantly reduced vagally mediated bronchoconstriction in rats fed a high-fat diet, such that bronchoconstrictions were similar to those rats fed a low-fat diet (Fig. 3A). Bronchoconstriction in response to aerosolized methacholine (1–30 mM) was not different among groups of rats, regardless of diet or pioglitazone treatment (Fig. 3B).
Figure 3.
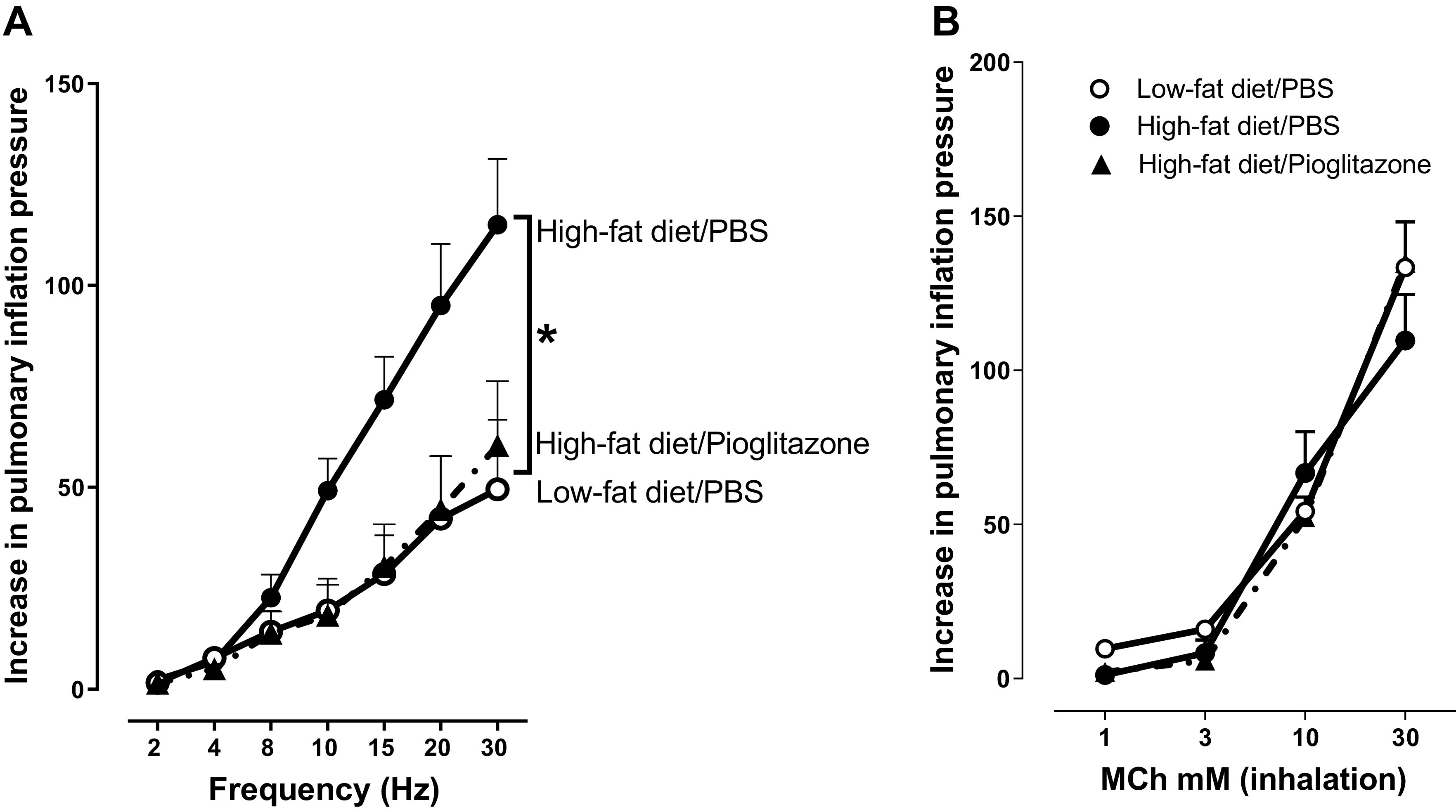
Airway physiology in vagotomized rats. A airway responsiveness was measured by electrically stimulating the vagus nerves with increasing frequency to cause frequency-dependent bronchoconstriction (measured as an increase in pulmonary inflation pressure) in vagotomized, anesthetized rats. B airway smooth muscle function was determined by measuring increased pulmonary inflation pressure in response to increasing concentrations (1–30 mM) of aerosolized methacholine (MCh) delivered in 20 µL saline in vagotomized, anesthetized rats. n = 4–6. *P < 0.05.
Vagally induced bradycardia in rats fed a high-fat diet was greater than bradycardia measured in rats fed a low-fat diet (Fig. 4A), although this did not reach statistical significance. Pioglitazone treatment tended to improve vagally induced bradycardia (Fig. 4A). Bradycardia induced by aerosolized methacholine was similar among rats regardless of diet or treatment with pioglitazone (Fig. 4B).
Figure 4.
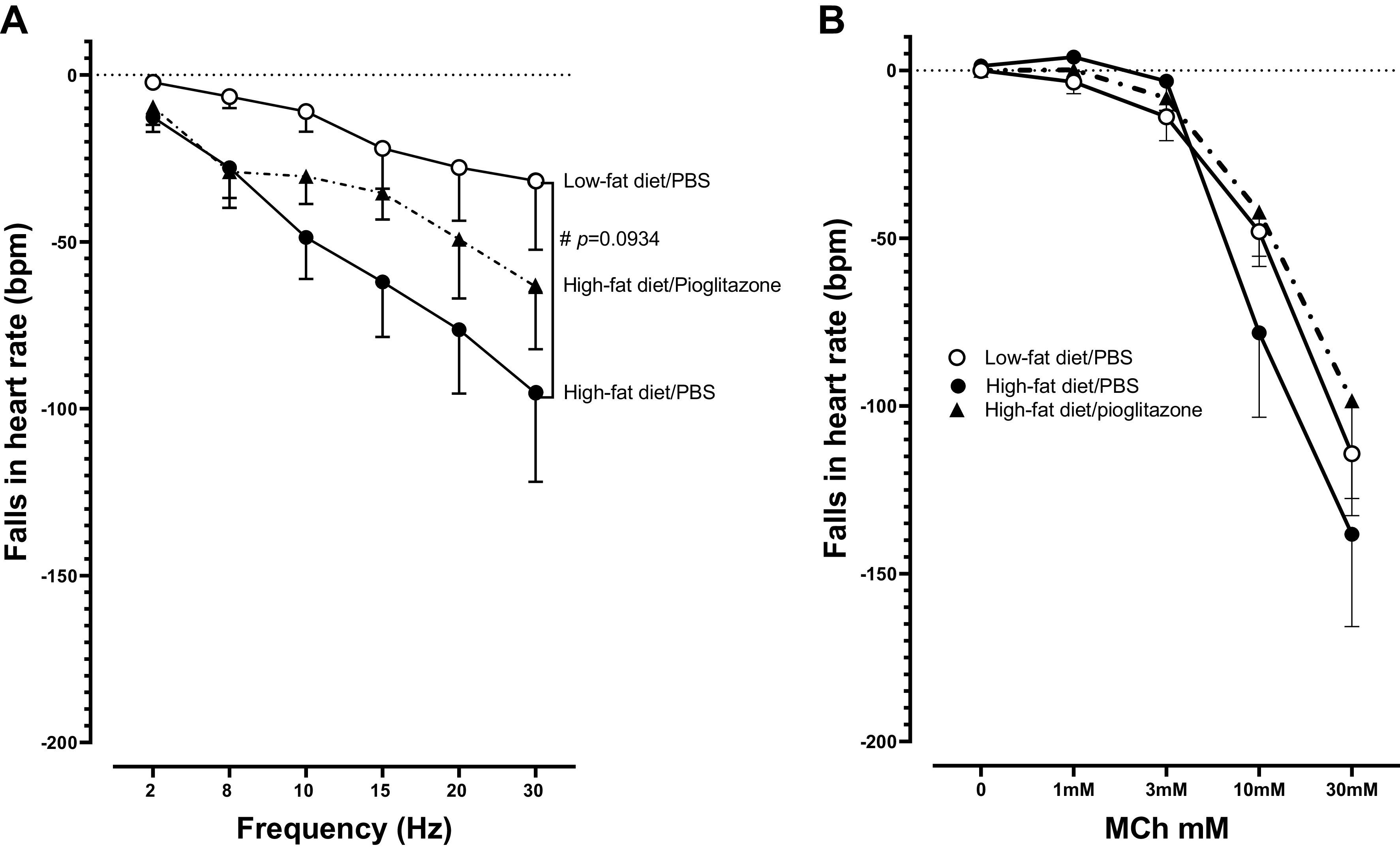
Bradycardia induced by vagus nerve stimulation (A) and by inhaled methacholine (B) in vagotomized, anesthetized rats. Rats were fed a low-fat or high-fat diet and either treated with pioglitazone (3 mg/kg) or PBS by daily gavage for 5 wk. There was a nonsignificant trend toward greater bradycardia in response to vagal stimulation in high-fat diet-fed rats compare with low-fat diet-fed rats, regardless of pioglitazone treatment (A). #P = 0.0934. Inhaled MCh-induced bradycardia was not different among groups of rats (B). n = 4–6.
Bronchoconstriction and bradycardia responses to aerosolized methacholine were only graphed up to 30 mM (Figs. 3B and 4B), as concentrations higher than 30 mM appeared to induce sudden cardiac death in most of the rats on a high-fat diet.
Pioglitazone Prevented High-Fat Diet-Induced Neuronal M2 Receptor Dysfunction in the Lungs
To test neuronal M2 receptor function on parasympathetic nerves, bronchoconstriction before and after the administration of pilocarpine was measured. Before administration of pilocarpine to rats, bronchoconstriction in response to electrical stimulation of the vagus nerve at 5 V and 10 Hz was repeatable and similar between rats fed a low-fat diet and rats fed a high-fat diet, treated with either PBS or pioglitazone (Fig. 5A). In rats fed a low-fat diet, pilocarpine decreased bronchoconstrictions induced by electrical stimulation of vagus nerve (5 V, 10 Hz) in a dose-dependent manner (Fig. 5B) by activating neuronal M2 receptors to decrease acetylcholine release from parasympathetic nerves, indicating functional M2 receptors. In these rats, the highest dose of pilocarpine (100 µg/kg iv) reduced bronchoconstriction by 87% ± 2% (Fig. 5B). In rats fed a high-fat diet and treated with PBS, pilocarpine still reduced bronchoconstriction, but this response was significantly attenuated compared with the response in low-fat diet-fed rats. At the highest dose, pilocarpine only reduced bronchoconstriction by 50% ± 7% (Fig. 5B), indicating neuronal M2 receptor dysfunction. Pioglitazone significantly improved M2 muscarinic receptor function in high-fat diet-fed rats, with the highest dose of pilocarpine inducing a 76% ± 8% reduction in bronchoconstriction (Fig. 5B).
Figure 5.

Neuronal M2 muscarinic receptor function was measured in vagotomized, anesthetized rats. Rats were fed a low-fat or high-fat diet and either treated with pioglitazone (3 mg/kg) or PBS by daily gavage for 5 wk. M2 muscarinic receptor function on parasympathetic nerves was measured using pilocarpine, a muscarinic receptor agonist that is physiologically selective for M2 over M3 receptors. Bronchoconstriction was measured in response to electrical stimulation of both vagus nerves (5 V, 10 Hz, for 6 s at 40-s intervals). There was no difference in vagally induced bronchoconstriction among groups before administration of pilocarpine (A). Pilocarpine dose dependently inhibited vagally induced bronchoconstriction. Pilocarpine-induced inhibition was significantly reduced in rats on a high-fat diet (filled circles); it was not reduced in high-fat diet rats treated with pioglitazone (filled triangles; B). n = 4–6. #P = 0.0029; *P = 0.0161.
Effects of High-Fat Diet and Pioglitazone on Cytokine and Insulin Receptor mRNA Expression in Alveolar Macrophages
Macrophages were the dominant leukocyte in the lung irrespective of diet or treatment (data not shown). Airway macrophages are a major source of inflammatory cytokines, including TNF-α, IL-1β, and IL-6, which have all been shown to modulate the function and expression of M2 receptors (30, 31). Thus, we investigated whether alveolar macrophage cytokine expression was affected by diet or pioglitazone treatment. A high-fat diet significantly reduced TNF-α (Fig. 6A) and transforming growth factor (TGF)-β (Fig. 6B) mRNA expression in alveolar macrophages compared with a low-fat diet, and pioglitazone did not prevent this decrease. IL-1β was slightly decreased as well in those rats fed a high-fat diet, regardless of treatment, but this did not reach significance (Fig. 6C). IL-6 was not affected by a high-fat diet (Fig. 6D) regardless of treatment. High-fat diet with or without pioglitazone treatment had no effect on insulin receptor mRNA expression in alveolar macrophages (Fig. 6E).
Figure 6.
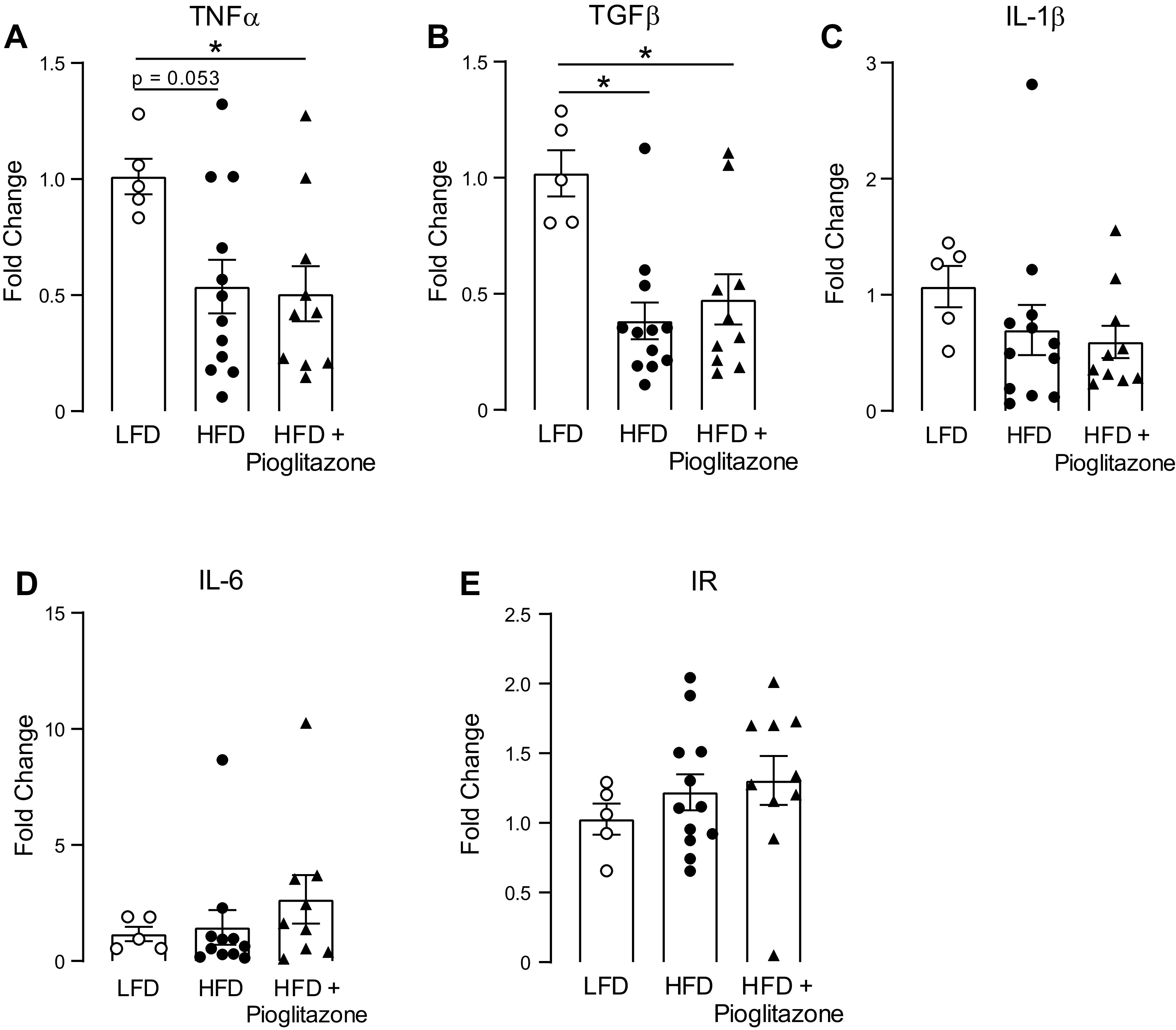
Cytokine and insulin receptor mRNA expression in alveolar macrophages. Alveolar macrophages were isolated from bronchoalveolar lavage fluid collected from rats fed a low-fat diet (LFD) or a high-fat diet (HFD) and treated with pioglitazone (3 mg/kg) or PBS by daily gavage for 5 wk. Tumor necrosis factor-α (TNF-α; A), transforming growth factor-β (TGF-β; B), interleukin (IL)-1β (C), IL-6 (D), and insulin receptor (IR; E) mRNA were quantified by real-time RT-PCR. n = 5–8. *P < 0.05.
Effects of Pioglitazone and High-Fat Diet on Insulin Receptor mRNA Expression
Because insulin plays a key role in obesity-related airway hyperreactivity and neuronal M2 receptor dysfunction and the expression of insulin receptor modulates the effect of insulin, we investigated whether insulin receptor expression in different tissues was changed by diet or pioglitazone treatment. High-fat diet slightly increased insulin receptor mRNA expression in adipose tissue; however, this did not reach statistical significance (Fig. 7A). High-fat diet had no effect on insulin receptor expression in the brain or lung (Fig. 7, B and C). Pioglitazone significantly reduced insulin receptor expression in adipose tissue (Fig. 7A), had no effect on insulin receptor expression in the brain (Fig. 7B), but significantly increased insulin receptor expression in the lung (Fig. 7C).
Figure 7.
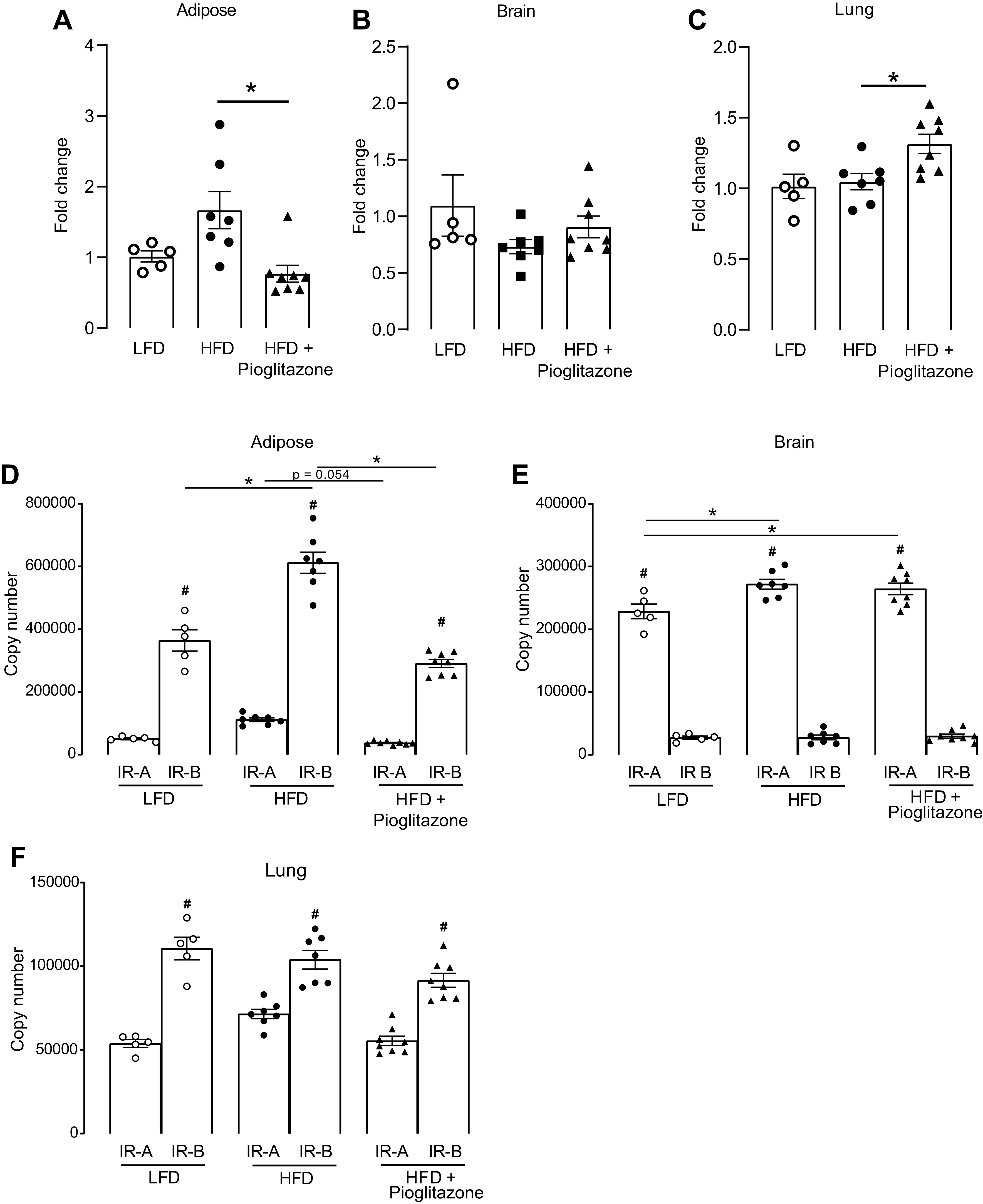
Quantification of total insulin receptor and insulin receptor isoform mRNA expression. Adipose, brain, and lung tissues were collected from rats that were fed either a low-fat diet (LFD) or a high-fat diet (HFD) and treated with pioglitazone (3 mg/kg) or PBS by daily gavage for 5 wk. mRNA was isolated from adipose (A and D), brain (B and E), and lung (C and F) tissue. Total insulin receptor expression (A–C), insulin receptor isoform A (IR-A), and insulin receptor isoform B (IR-B) mRNA expression (D–F) were quantified using real-time RT-PCR. n = 5–8. *P < 0.05 between treatment groups; #P < 0.05 between IR-A and IR-B within the same treatment group.
Both IR-A and IR-B mRNA were expressed in adipose tissue and in the brain and lung; however, their expression patterns differed (Fig. 7, D–F). IR-B was the dominant isoform expressed in the adipose tissue (Fig. 7D) and the lung (Fig. 7F), whereas IR-A was the dominant isoform expressed in the brain (Fig. 7E). Neither high-fat diet nor pioglitazone changed the dominate isoform among these tissues (Fig. 7, D–F). In adipose tissue (Fig. 7D), dominant IR-B was significantly increased in rats on a high-fat diet and this effect was prevented by pioglitazone (Fig. 7D). In addition, pioglitazone also greatly reduced the expression of IR-A in adipose tissue in rats on a high-fat diet (Fig. 7D). In the brain (Fig. 7E), dominant IR-A was significantly increased in rats on a high-fat diet but pioglitazone did not prevent this increase. In the lung (Fig. 7F), neither high-fat diet nor pioglitazone had any effect on the expression of IR-A or IR-B.
DISCUSSION
Obesity is a prominent risk factor for the development and severity of asthma, and obese asthmatics respond poorly to common asthma therapeutics (2–5). Understanding the pathophysiology of obesity-related asthma is crucial for developing new therapeutic targets. Obesity frequently leads to insulin resistance and compensatory hyperinsulinemia. High levels of insulin are associated with moderate persistent asthma in adolescents with obesity (32) and with decreased lung function (33). Furthermore, inhaled insulin significantly reduces lung function in both nonasthmatics and asthmatics (34). We have previously reported that hyperinsulinemia mediates vagus nerve-induced airway hyperreactivity by reducing neuronal M2 muscarinic receptor function on parasympathetic nerves (7). In the current study, we show that pioglitazone, a drug used to treat diabetes, reduced fasting insulin. In pioglitazone-treated obese rats, airway hyperresponsiveness to vagal stimulation was prevented and neuronal M2 receptor function was protected. This suggests that pioglitazone indirectly protected neuronal M2 receptor function by reducing circulating insulin, indicating that pharmacotherapy with pioglitazone may be beneficial for the treatment of asthma in hyperinsulinemic patients with obesity. This is consistent with our previous study that showed that decreasing insulin with streptozotocin prevented airway hyperreactivity in obese rats by protecting neuronal M2 receptor function. In both the previous study and our current study, airway response to aerosolized methacholine was similar among groups of rats, suggesting that the changes of airway hyperresponsiveness in these obese animals were mediated by airway nerves, not smooth muscle.
Airway neuronal M2 receptor function was tested with the use of the muscarinic agonist pilocarpine. Although pilocarpine can act on all the subtypes of muscarinic receptors, the effect of pilocarpine on bronchoconstriction is dose dependent. Pilocarpine stimulates M2 muscarinic receptors and inhibits vagally induced bronchoconstriction at doses lower than those doses that will induce sustained bronchoconstriction via stimulating M3 muscarinic receptors in vivo (35). The low doses of pilocarpine used in our experiments have been reported to produce minimal bronchoconstriction (13), mediated by M3 muscarinic receptors on the smooth muscle, and therefore unlikely interfered with the assessment of pilocarpine on neuronal M2 muscarinic receptors. In high-fat diet-induced obese rats, the inhibitory effect of pilocarpine on vagally induced bronchoconstriction was significantly reduced compared with low-fat diet-fed rats, indicating dysfunctional neuronal M2 muscarinic receptors in diet-induced obese rats. In pioglitazone-treated rats, the effect of pilocarpine on vagally induced bronchoconstriction was restored and similar to that in low-fat diet-fed rats, suggesting that pioglitazone protected neuronal M2 muscarinic receptor function in obese rats. Pioglitazone may protect neuronal M2 muscarinic receptor function through an indirect mechanism via inhibiting high-fat diet-induced hyperinsulinemia. We have shown that increased circulating insulin in diet-induced obese rats reduces neuronal M2 receptor function and causes airway hyperreactivity. Reducing insulin by streptozotocin can restore neuronal M2 receptor function on parasympathetic nerves in those diet-induced obese rats (7). Here, pioglitazone treatment reduced fasting insulin in obese rats (Fig. 2), and thus similarly may protect neuronal M2 muscarinic receptor function in obese rats via reducing circulating insulin.
M2 muscarinic receptors are also expressed on cardiac muscle to slow heart rate. In this study, hyperinsulinemic obese rats had normal baseline heart rate (Table 1), and high-fat diet did not affect vagus nerve stimulation or methacholine-induced bradycardia. Thus, although pioglitazone prevented neuronal M2 muscarinic receptor dysfunction in the lungs, cardiac M2 receptors remained unaffected by obesity or pioglitazone.
Airway smooth muscle M3 receptor function was tested by measuring bronchoconstriction induced by inhaled methacholine in vagotomized rats. Animals with intact vagus nerves are often used to test obesity-related airway hyperreactivity (36–38). However, we have to choose vagotomized rats because we (39) and others (40) have previously shown that there is an underappreciated, but substantial, vagally mediated reflex component to inhaled and intravenous methacholine. If the vagus nerve is intact, sensory nerves in the lungs are stimulated by inhaled methacholine, sending signal to the brain and back to the lungs via the parasympathetic nerves, leading to acetylcholine release and bronchoconstriction. Thus, in animals with intact vagus nerves, bronchoconstriction in response to inhaled methacholine is a combination of bronchoconstriction induced by the reflex response and by the direct action of methacholine on M3 muscarinic receptor on airway smooth muscle. Therefore, to measure bronchoconstriction induced only by methacholine on M3 muscarinic receptors on airway smooth muscle, we used vagotomized rats. The lack of a potentiated response to inhaled methacholine in vagotomized obese rats clearly demonstrated that airway hyperresponsiveness was not due to an increase in M3 muscarinic receptor numbers or function on airway smooth muscle.
Macrophages are the dominant leukocyte in the lung and are important for maintaining tissue homeostasis in both lung and adipose tissue. Some proinflammatory cytokines released from adipose macrophages, including TNF-α, IL-1β, IL-6, and TGF-β, are also elevated in patients with obesity (41, 42). Also, importantly, TNFα, IL-1β, and TGF-β have all been shown to affect M2 receptor function or expression (30, 31). In this study, we found that high-fat diet did not increase cytokine expression in alveolar macrophages, and actually decreased the expression of TNF-α and TGF-β. Thus, it is likely that hyperinsulinemia, not cytokines, is the mediator of obesity-induced airway hyperreactivity. Moreover, although pioglitazone suppresses airway inflammation in antigen-challenged mice (43, 44), studies in patients with asthma have not found improvement in airway inflammation. This is consistent with our finding that pioglitazone did not affect levels of TNF-α, TGF-β, IL-1, or IL-6 in obese rats (Fig. 6).
In type 2 diabetes, skeletal muscle, liver, and adipose tissue become insulin resistant, whereas other tissues do not become insulin resistant (45, 46). In a study of diet-induced obesity in mice, the liver became insulin resistant at 6 wk and white adipose tissue at 12 wk, whereas skeletal muscle remained insulin sensitive at 12 wk (45). In another study, insulin administration still activated inflammatory cells in insulin-resistant patients (46). Insulin binds to insulin receptors to activate the cascade of insulin receptor cell signaling pathway. Thus, insulin sensitivity in tissues may be regulated on many levels, including by differential expression of two insulin receptor isoforms (20, 47). IR-A is primarily expressed in fetal tissue, binds to insulin-like growth factor, and has a role in growth, whereas IR-B is more common in adult tissue and regulates most of insulin’s metabolic actions. IR-B is predominantly expressed in insulin-sensitive tissues (48). An increase in IR-B has been found in adipocytes and skeletal muscle isolated from patients with diabetes (49, 50). Other studies have found no difference in the expression of the isoforms with obesity or diabetes (51).
In our study, total insulin receptor expression in adipose tissue or in the brain or lung was not affected by high-fat diet and subsequent hyperinsulinemia. However, in adipose tissue, IR-B was increased in rats on a high-fat diet and this increase was prevented with pioglitazone. Pioglitazone also decreased expression of IR-A in adipose tissue. High-fat diet significantly increased IR-A in the brain, and pioglitazone did not prevent this increase. Neither obesity nor pioglitazone affected insulin receptor expression or the ratio of subtypes. Further investigation will be required to understand the role of changes in insulin receptor subtypes in obesity and insulin resistance.
Although pioglitazone is primarily used for the treatment of diabetes, several studies have tested the effects of pioglitazone in airway disease. In an early case report on two patients with diabetes and asthma, pioglitazone ameliorated their asthma symptoms (52). A large cohort study of diabetics with asthma also found that those taking pioglitazone had fewer exacerbations and had less need for oral steroids (53). Since then, pioglitazone has not been found effective in clinical trials involving two trials in obese asthmatic patients (54, 55) and in one trial in nonobese mild asthma patients (56). Notably, diabetes was an exclusion factor for two of these trials (56) and circulating insulin levels were not measured in any of these studies. In this context, our finding that hyperinsulinemia is responsible for obesity-related airway hyperreactivity and that pioglitazone reverses this hyperreactivity by decreasing insulin may underscore the need to identify obese asthmatics with hyperinsulinemia for clinical trials of pioglitazone.
In summary, pioglitazone prevents obesity-induced airway hyperreactivity and M2 muscarinic receptor dysfunction in obese rats with hyperinsulinemia. These data corroborate our previous data and other studies suggesting a central role for insulin in obesity-related asthma. Therapeutic strategies that reduce insulin, including the use of pioglitazone, may be beneficial specifically in patients with obesity, asthma, as well as hyperinsulinemia.
GRANTS
This work was supported by National Institutes of Health Grants R01HL131525, R01HL113023, R01AR061567, R01HL124165, and R01ES017592.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.J.P., A.D.F., D.B.J., and Z.N. conceived and designed research; B.J.P. and Z.N. performed experiments; B.J.P. and Z.N. analyzed data; B.J.P., A.D.F., D.B.J., and Z.N. interpreted results of experiments; B.J.P. and Z.N. prepared figures; B.J.P. and Z.N. drafted manuscript; B.J.P., A.D.F., D.B.J., and Z.N. edited and revised manuscript; B.J.P., A.D.F., D.B.J., and Z.N. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Lauren Hales Beck, Jessica Maung, and Gina Calco for technical assistance.
REFERENCES
- 1.Centers for Disease Control and Prevention. Current Asthma Prevalence by Weight Status Among Adults: United States, 2001–2014. NCHS Data Brief No. 239, March 2016. https://www.cdc.gov/nchs/products/databriefs/db239.htm. [PubMed]
- 2.Lessard A, Turcotte H, Cormier Y, Boulet LP. Obesity and asthma: a specific phenotype? Chest 134: 317–323, 2008. doi: 10.1378/chest.07-2959. [DOI] [PubMed] [Google Scholar]
- 3.Sutherland TJ, Cowan JO, Young S, Goulding A, Grant AM, Williamson A, Brassett K, Herbison GP, Taylor DR. The association between obesity and asthma: interactions between systemic and airway inflammation. Am J Respir Crit Care Med 178: 469–475, 2008. doi: 10.1164/rccm.200802-301OC. [DOI] [PubMed] [Google Scholar]
- 4.Boulet LP, Franssen E. Influence of obesity on response to fluticasone with or without salmeterol in moderate asthma. Respir Med 101: 2240–2247, 2007. doi: 10.1016/j.rmed.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 5.Schatz M, Hsu JW, Zeiger RS, Chen W, Dorenbaum A, Chipps BE, Haselkorn T. Phenotypes determined by cluster analysis in severe or difficult-to-treat asthma. J Allergy Clin Immunol 133: 1549–1556, 2014. doi: 10.1016/j.jaci.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 6.Centers for Disease Control and Prevention. U.S. Overweight & Obesity Data & Statistics. https://www.cdc.gov/obesity/data/.
- 7.Nie Z, Jacoby DB, Fryer AD. Hyperinsulinemia potentiates airway responsiveness to parasympathetic nerve stimulation in obese rats. Am J Respir Cell Mol Biol 51: 251–261, 2014. doi: 10.1165/rcmb.2013-0452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costello RW, Jacoby DB, Fryer AD. Pulmonary neuronal M2 muscarinic receptor function in asthma and animal models of hyperreactivity. Thorax 53: 613–618, 1998. doi: 10.1136/thx.53.7.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minette PA, Barnes PJ. Prejunctional inhibitory muscarinic receptors on cholinergic nerves in human and guinea pig airways. J Appl Physiol (1985) 64: 2532–2537, 1988. doi: 10.1152/jappl.1988.64.6.2532. [DOI] [PubMed] [Google Scholar]
- 10.Minette PA, Lammers JW, Dixon CM, McCusker MT, Barnes PJ. A muscarinic agonist inhibits reflex bronchoconstriction in normal but not in asthmatic subjects. J Appl Physiol (1985) 67: 2461–2465, 1989. doi: 10.1152/jappl.1989.67.6.2461. [DOI] [PubMed] [Google Scholar]
- 11.Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev 50: 279–290, 1998. [PubMed] [Google Scholar]
- 12.Fryer AD, Jacoby DB. Parainfluenza virus infection damages inhibitory M2 muscarinic receptors on pulmonary parasympathetic nerves in the guinea-pig. Br J Pharmacol 102: 267–271, 1991. doi: 10.1111/j.1476-5381.1991.tb12164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fryer AD, Wills-Karp M. Dysfunction of M2-muscarinic receptors in pulmonary parasympathetic nerves after antigen challenge. J Appl Physiol (1985) 71: 2255–2261, 1991. doi: 10.1152/jappl.1991.71.6.2255. [DOI] [PubMed] [Google Scholar]
- 14.Schultheis AH, Bassett DJ, Fryer AD. Ozone-induced airway hyperresponsiveness and loss of neuronal M2 muscarinic receptor function. J Appl Physiol (1985) 76: 1088–1097, 1994. doi: 10.1152/jappl.1994.76.3.1088. [DOI] [PubMed] [Google Scholar]
- 15.Proskocil BJ, Bruun DA, Thompson CM, Fryer AD, Lein PJ. Organophosphorus pesticides decrease M2 muscarinic receptor function in guinea pig airway nerves via indirect mechanisms. PLoS One 5: e10562, 2010. doi: 10.1371/journal.pone.0010562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Husemoen LL, Glumer C, Lau C, Pisinger C, Morch LS, Linneberg A. Association of obesity and insulin resistance with asthma and aeroallergen sensitization. Allergy 63: 575–582, 2008. doi: 10.1111/j.1398-9995.2007.01613.x. [DOI] [PubMed] [Google Scholar]
- 17.Cottrell L, Neal WA, Ice C, Perez MK, Piedimonte G. Metabolic abnormalities in children with asthma. Am J Respir Crit Care Med 183: 441–448, 2011. doi: 10.1164/rccm.201004-0603OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belmonte KE, Jacoby DB, Fryer AD. Increased function of inhibitory neuronal M2 muscarinic receptors in diabetic rat lungs. Br J Pharmacol 121: 1287–1294, 1997. doi: 10.1038/sj.bjp.0701274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coulson FR, Jacoby DB, Fryer AD. Increased function of inhibitory neuronal M2 muscarinic receptors in trachea and ileum of diabetic rats. Br J Pharmacol 135: 1355–1362, 2002. doi: 10.1038/sj.bjp.0704602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moller DE, Yokota A, Caro JF, Flier JS. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol Endocrinol 3: 1263–1269, 1989. doi: 10.1210/mend-3-8-1263. [DOI] [PubMed] [Google Scholar]
- 21.Escribano O, Beneit N, Rubio- LC, Lopez-Pastor AR, Gomez- HA. The role of insulin receptor isoforms in diabetes and its metabolic and vascular complications. J Diabetes Res 2017: 1403206, 2017. doi: 10.1155/2017/1403206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogt B, Carrascosa JM, Ermel B, Ullrich A, Haring HU. The two isotypes of the human insulin receptor (HIR-A and HIR-B) follow different internalization kinetics. Biochem Biophys Res Commun 177: 1013–1018, 1991. doi: 10.1016/0006-291x(91)90639-o. [DOI] [PubMed] [Google Scholar]
- 23.Huang Z, Bodkin NL, Ortmeyer HK, Zenilman ME, Webster NJ, Hansen BC, et al. Altered insulin receptor messenger ribonucleic acid splicing in liver is associated with deterioration of glucose tolerance in the spontaneously obese and diabetic rhesus monkey: analysis of controversy between monkey and human studies. J Clin Endocrinol Metab 81: 1552–1556, 1996. doi: 10.1210/jcem.81.4.8636366. [DOI] [PubMed] [Google Scholar]
- 24.Kemnitz JW, Elson DF, Roecker EB, Baum ST, Bergman RN, Meglasson MD. Pioglitazone increases insulin sensitivity, reduces blood glucose, insulin, and lipid levels, and lowers blood pressure, in obese, insulin-resistant rhesus monkeys. Diabetes 43: 204–211, 1994. doi: 10.2337/diab.43.2.204. [DOI] [PubMed] [Google Scholar]
- 25.Giles ED, Jackman MR, MacLean PS. Modeling diet-induced obesity with obesity-prone rats: implications for studies in females. Front Nutr 3: 50, 2016. doi: 10.3389/fnut.2016.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levin BE, Dunn-Meynell AA, Balkan B, Keesey RE. Selective breeding for diet-induced obesity and resistance in Sprague-Dawley rats. Am J Physiol Regul Integr Comp Physiol 273: R725–R730, 1997. doi: 10.1152/ajpregu.1997.273.2.R725. [DOI] [PubMed] [Google Scholar]
- 27.Nie Z, Jacoby DB, Fryer AD. Etanercept prevents airway hyperresponsiveness by protecting neuronal M2 muscarinic receptors in antigen-challenged guinea pigs. Br J Pharmacol 156: 201–210, 2009. doi: 10.1111/j.1476-5381.2008.00045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang SZ, el-Fakahany EE. Application of transfected cell lines in studies of functional receptor subtype selectivity of muscarinic agonists. J Pharmacol Exp Ther 266: 237–243, 1993. [PubMed] [Google Scholar]
- 29.Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol, 2006. Chapter 15: Unit 15.8. doi: 10.1002/0471142727.mb1508s73. [DOI] [PubMed] [Google Scholar]
- 30.Haddad EB, Rousell J, Lindsay MA, Barnes PJ. Synergy between tumor necrosis factor alpha and interleukin 1beta in inducing transcriptional down-regulation of muscarinic M2 receptor gene expression. Involvement of protein kinase A and ceramide pathways. J Biol Chem 271: 32586–32592, 1996. doi: 10.1074/jbc.271.51.32586. [DOI] [PubMed] [Google Scholar]
- 31.Haddad EB, Rousell J, Mak JC, Barnes PJ. Transforming growth factor-beta 1 induces transcriptional down-regulation of m2 muscarinic receptor gene expression. Mol Pharmacol 49: 781–787, 1996. [PubMed] [Google Scholar]
- 32.Morishita R, Franco MD, Suano-Souza FI, Sole D, Puccini RF, Strufaldi MW. Body mass index, adipokines and insulin resistance in asthmatic children and adolescents. J Asthma 53: 478–484, 2016. doi: 10.3109/02770903.2015.1113544. [DOI] [PubMed] [Google Scholar]
- 33.Kim KM, Kim SS, Lee SH, Song WJ, Chang YS, Min KU, Cho SH. Association of insulin resistance with bronchial hyperreactivity. Asia Pac Allergy 4: 99–105, 2014. doi: 10.5415/apallergy.2014.4.2.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mudaliar S, Henry RR. Inhaled insulin in patients with asthma and chronic obstructive pulmonary disease. Diabetes Technol Ther 9: S83–S92, 2007. doi: 10.1089/dia.2007.0217. [DOI] [PubMed] [Google Scholar]
- 35.Fryer AD, Maclagan J. Muscarinic inhibitory receptors in pulmonary parasympathetic nerves in the guinea-pig. Br J Pharmacol 83: 973–978, 1984. doi: 10.1111/j.1476-5381.1984.tb16539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fricke K, Vieira M, Younas H, Shin MK, Bevans-Fonti S, Berger S, Lee R, D'Alessio FR, Zhong Q, Nelson AM, Loube J, Sanchez I, Hansel NN, Mitzner W, Polotsky VY. High fat diet induces airway hyperresponsiveness in mice. Sci Rep 8: 6404, 2018. doi: 10.1038/s41598-018-24759-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnston RA, Zhu M, Rivera-Sanchez YM, Lu FL, Theman TA, Flynt L, Shore SA. Allergic airway responses in obese mice. Am J Respir Crit Care Med 176: 650–658, 2007. doi: 10.1164/rccm.200702-323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chong L, Zhang W, Yu G, Zhang H, Zhu L, Li H, Shao Y, Li C. High-fat-diet induces airway hyperresponsiveness partly through activating CD38 signaling pathway. Int Immunopharmacol 56: 197–204, 2018. doi: 10.1016/j.intimp.2018.01.033. [DOI] [PubMed] [Google Scholar]
- 39.Wagner EM, Jacoby DB. Methacholine causes reflex bronchoconstriction. J Appl Physiol (1985) 86: 294–297, 1999. doi: 10.1152/jappl.1999.86.1.294. [DOI] [PubMed] [Google Scholar]
- 40.McAlexander MA, Gavett SH, Kollarik M, Undem BJ. Vagotomy reverses established allergen-induced airway hyperreactivity to methacholine in the mouse. Respir Physiol Neurobiol 212–214: 20–24, 2015. doi: 10.1016/j.resp.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gosset P, Tsicopoulos A, Wallaert B, Vannimenus C, Joseph M, Tonnel AB, Capron A. Increased secretion of tumor necrosis factor alpha and interleukin-6 by alveolar macrophages consecutive to the development of the late asthmatic reaction. J Allergy Clin Immunol 88: 561–571, 1991. doi: 10.1016/0091-6749(91)90149-i. [DOI] [PubMed] [Google Scholar]
- 42.Fain JN, Tichansky DS, Madan AK. Transforming growth factor beta1 release by human adipose tissue is enhanced in obesity. Metabolism 54: 1546–1551, 2005. doi: 10.1016/j.metabol.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 43.Narala VR, Ranga R, Smith MR, Berlin AA, Standiford TJ, Lukacs NW, Reddy RC. Pioglitazone is as effective as dexamethasone in a cockroach allergen-induced murine model of asthma. Respir Res 8: 90, 2007. doi: 10.1186/1465-9921-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meng X, Sun X, Zhang Y, Shi H, Deng W, Liu Y, Wang G, Fang P, Shuanying Y. PPARγ agonist PGZ attenuates OVA-induced airway inflammation and airway remodeling via RGS4 signaling in mouse model. Inflammation 41: 2079–2089, 2018. doi: 10.1007/s10753-018-0851-2. [DOI] [PubMed] [Google Scholar]
- 45.Kleemann R, van Erk M, Verschuren L, van den Hoek AM, Koek M, Wielinga PY, Jie A, Pellis L, Bobeldijk-Pastorova I, Kelder T, Toet K, Wopereis S, Cnubben N, Evelo C, van Ommen B, Kooistra T. Time-resolved and tissue-specific systems analysis of the pathogenesis of insulin resistance. PLoS One 5: e8817, 2010. doi: 10.1371/journal.pone.0008817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghanim H, Green K, Abuaysheh S, Batra M, Kuhadiya ND, Patel R, Makdissi A, Dhindsa S, Chaudhuri A, Dandona P. Suppressive effect of insulin on the gene expression and plasma concentrations of mediators of asthmatic inflammation. J Diabetes Res 2015: 1–7, 2015. doi: 10.1155/2015/202406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Konner AC, Bruning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab 16: 144–152, 2012. doi: 10.1016/j.cmet.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 48.Benecke H, Flier JS, Moller DE. Alternatively spliced variants of the insulin receptor protein. Expression in normal and diabetic human tissues. J Clin Invest 89: 2066–2070, 1992. doi: 10.1172/JCI115819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sesti G, Marini MA, Tullio AN, Montemurro A, Borboni P, Fusco A, Accili D, Lauro R. Altered expression of the two naturally occurring human insulin receptor variants in isolated adipocytes of non-insulin-dependent diabetes mellitus patients. Biochem Biophys Res Commun 181: 1419–1424, 1991. doi: 10.1016/0006-291x(91)92097-4. [DOI] [PubMed] [Google Scholar]
- 50.Kellerer M, Sesti G, Seffer E, Obermaier-Kusser B, Pongratz DE, Mosthaf L, Häring HU. Altered pattern of insulin receptor isotypes in skeletal muscle membranes of type 2 (non-insulin-dependent) diabetic subjects. Diabetologia 36: 628–632, 1993. doi: 10.1007/BF00404072. [DOI] [PubMed] [Google Scholar]
- 51.Anderson CM, Henry RR, Knudson PE, Olefsky JM, Webster NJ. Relative expression of insulin receptor isoforms does not differ in lean, obese, and noninsulin-dependent diabetes mellitus subjects. J Clin Endocrinol Metab 76: 1380–1382, 1993. doi: 10.1210/jcem.76.5.7684396. [DOI] [PubMed] [Google Scholar]
- 52.Hashimoto Y, Nakahara K. Improvement of asthma after administration of pioglitazone. Diabetes Care 25: 401, 2002. doi: 10.2337/diacare.25.2.401. [DOI] [PubMed] [Google Scholar]
- 53.Rinne ST, Feemster LC, Collins BF, Au DH, Perkins M, Bryson CL, O'Riordan TG, Liu C-F. Thiazolidinediones and the risk of asthma exacerbation among patients with diabetes: a cohort study. Allergy Asthma and Clinical Immunology 10: 34, 2014. doi: 10.1186/1710-1492-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dixon AE, Subramanian M, DeSarno M, Black K, Lane L, Holguin F. A pilot randomized controlled trial of pioglitazone for the treatment of poorly controlled asthma in obesity. Respir Res 16: 143, 2015. doi: 10.1186/s12931-015-0303-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaler M, Barochia AV, Weir NA, Cuento RA, Stylianou M, Roth MJ, Filie AC, Vaughey EC, Nathan SD, Levine SJ. A randomized, placebo-controlled, double-blinded, crossover trial of pioglitazone for severe asthma. J Allergy Clin Immunol 140: 1716–1718, 2017. doi: 10.1016/j.jaci.2017.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anderson JR, Mortimer K, Pang L, Smith KM, Bailey H, Hodgson DB, Shaw DE, Knox AJ, Harrison TW. Evaluation of the PPAR-gamma agonist pioglitazone in mild asthma: a double-blind randomized controlled trial. PLoS One 11: e0160257, 2016. doi: 10.1371/journal.pone.0160257. [DOI] [PMC free article] [PubMed] [Google Scholar]