

Abstract
Furin is a proprotein convertase that regulates the activation and the inactivation of multiple proteins including matrix metalloproteinases, integrins, and cytokines. It is a serine endoprotease that localizes to the plasma membrane and can be secreted into the extracellular space. The role of furin in regulating inflammation in isolated canine airway smooth muscle tissues was investigated. The treatment of airway tissues with recombinant furin (rFurin) inhibited the activation of Akt and eotaxin secretion induced by IL-13, and it prevented the IL-13-induced suppression of smooth muscle myosin heavy chain expression. rFurin promoted a differentiated phenotype by activating β1-integrin proteins and stimulating the activation of the adhesome proteins vinculin and paxillin by talin. Activated paxillin induced the binding of Akt to β-parvin IPP [integrin-linked kinase (ILK), PINCH, parvin] complexes, which inhibits Akt activation. Treatment of tissues with a furin inhibitor or the depletion of endogenous furin using shRNA resulted in Akt activation and inflammatory responses similar to those induced by IL-13. Furin inactivation or IL-13 caused talin cleavage and integrin inactivation, resulting in the inactivation of vinculin and paxillin. Paxillin inactivation resulted in the coupling of Akt to α-parvin IPP complexes, which catalyze Akt activation and an inflammatory response. The results demonstrate that furin inhibits inflammation in airway smooth muscle induced by IL-13 and that the anti-inflammatory effects of furin are mediated by activating integrin proteins and integrin-associated signaling complexes that regulate Akt-mediated pathways to the nucleus. Furin may have therapeutic potential for the treatment of inflammatory conditions of the lungs and airways.
Keywords: airway inflammation, airway smooth muscle, IL-13, integrin complexes, signal transduction
INTRODUCTION
Airway inflammation causes changes in the function and properties of the smooth muscle tissues within the airways as well as in other resident lung tissues. Inflammatory cytokines induce airway smooth muscle tissues to transition from a differentiated smooth muscle phenotype to a synthetic inflammatory phenotype, characterized by the upregulation of Akt-mediated pathways that promote the synthesis and secretion of cytokines (1–8). Changes in phenotypic expression in airway smooth muscle tissues can also be induced by modulations in the extracellular environment such as alterations in tissue structure, extracellular matrix composition, or external mechanical forces (1, 3, 7, 9–12). Treatment with the protease elastase, which degrades extracellular matrix proteins, induces an inflammatory phenotype in airway smooth muscle tissues in vitro (4). Chronic mechanical loading can inhibit the responses of airway smooth muscle tissues to an inflammatory stimulus (1, 3, 7).
Transmembrane integrin proteins act as sensors for alterations in the extracellular matrix, mechanical tension, and other extracellular conditions and transduce signals from the extracellular environment to intracellular signaling pathways that regulate the airway smooth muscle phenotype. The extracellular domains of integrin proteins bind to extracellular matrix proteins and link individual smooth muscle cells to each other, whereas the cytoplasmic domains of integrin proteins link to multiprotein signaling complexes (adhesomes) that regulate nuclear signaling pathways that can modulate phenotypic expression (13–16). Integrin-mediated signaling pathways regulate transitions in the airway smooth muscle phenotype between an inflammatory and a differentiated state (1, 3, 7). These transitions can be induced by alterations in the extracellular matrix or in the mechanical tension imposed on the muscle tissues.
Furin is a member of the subtilisin-and-kexin-type protein convertase superfamily of serine endoproteases. It regulates the activation and the inactivation of a number of extracellular and transmembrane proteins, including matrix metalloproteinases, integrins, and cytokines, through targeted proteolytic activity that cleaves substrates at a specific sequence of basic residues (17–19). Furin is widely expressed in almost all mammalian cell and tissue types (19). It is trafficked to the plasma membrane, where it resides on the cell surface and anchors to the membrane via its C-terminal transmembrane domain. A soluble form of catalytically active furin can be shed into the extracellular space by cleavage of its transmembrane C-terminal domain (17, 19, 20). There is evidence that the activity of furin is upregulated in a number of pathological conditions, such as atherosclerosis, cancer, and pathogen infection, and in several inflammatory disorders, including asthma (19, 21–23). Furin has been proposed to promote the metastatic spreading of cancer cells and tissue invasion by remodeling the extracellular matrix to promote cell motility (21, 24).
Several studies have demonstrated an anti-inflammatory immunoprotective effect of furin (25–28). In an experimental murine model of arthritis, treatment with furin suppressed inflammation and prevented increases in the arthritis score, joint destruction, and bone loss, whereas the systemic administration of a furin inhibitor to these animals enhanced these parameters (26). An anti-inflammatory function of furin has also been shown in myeloid cells in mice in vivo (25). However, the role for furin in regulating inflammation in tissues within the lungs and airways has not been determined. We hypothesized that furin might regulate the expression of the inflammatory phenotype in airway smooth muscle.
We evaluated the effects of furin on inflammatory responses of airway smooth muscle tissues induced by the cytokine, interleukin-13 (IL-13)—a key cytokine associated with airway inflammation in asthma (29, 30). In airway smooth muscle, IL-13 induces the activation of STAT6 and the synthetic kinase Akt, which stimulates the synthesis and secretion of eotaxin (1, 2, 7, 31–34). Akt activation also suppresses the differentiated smooth muscle phenotype by inhibiting the localization of the transcription factor serum response factor (SRF) to the nucleus, thus suppressing the transcription of smooth muscle phenotype-specific proteins (35, 36).
Our results demonstrate that furin has an anti-inflammatory effect on airway smooth muscle tissues and that it suppresses the responses of airway smooth muscle to IL-13. We found that the anti-inflammatory effects of furin are mediated by integrin proteins and components of their associated signaling complexes that regulate pathways to the nucleus that determine phenotypic expression.
MATERIALS AND METHODS
Reagents and Antibodies
Sources of primary antibodies are as follows: rabbit anti-human polyclonal furin (Cat. No. PA1-062, Invitrogen) and rabbit anti-human polyclonal Akt phospho-Ser473 (Cat. No. 44-621G, Invitrogen); mouse anti-human monoclonal Akt (Cat. No. 9271, Cell Signaling); mouse anti-human monoclonal smooth muscle myosin heavy chain (SmMHC) and mouse anti-human α-actinin (Cat. No. M7786 and Cat. No. A5044, Sigma); mouse anti-human monoclonal paxillin (Cat. No. 610569, BD Transduction); rabbit anti-human polyclonal paxillin phospho-tyrosine 118 (Cat. No. 44-722G, Invitrogen); mouse anti-human monoclonal vinculin (clone hVIN-1, v9131 Sigma); rabbit anti-human polyclonal vinculin phospho-tyrosine 1065 (Cat. No. 44-1078G, Invitrogen); rabbit anti-canine cardiac polyclonal vinculin (custom made by BABCO, Richmond, CA); mouse anti-human integrin β1-activated clone HUTS-4 (Cat. No. MAB2079z, Sigma-Aldrich); rabbit anti-human monoclonal integrin β1 (Cat. No. 14-0299-82, Invitrogen); mouse anti-human monoclonal NH2-terminal talin (Cat. No. MAB1676, Millipore); and rabbit anti-human polyclonal α-parvin (Cat. No. ab154654) and rabbit anti-human monoclonal β-parvin (Cat. No. ab154840, Abcam). Secondary antibodies were horseradish peroxidase-conjugated anti-mouse IgG and anti-rabbit IgG (Amersham Biosciences) and IR Dye 800CW goat anti-rabbit IgG and IR Dye 680LT goat anti-mouse IgG (Li-Cor). Validation of the specificity of antibodies against activated β1-integrin and furin is shown in Figs. 3, 4, and 5. All other antibodies have been validated in previous studies (1, 7, 37–39).
Figure 3.
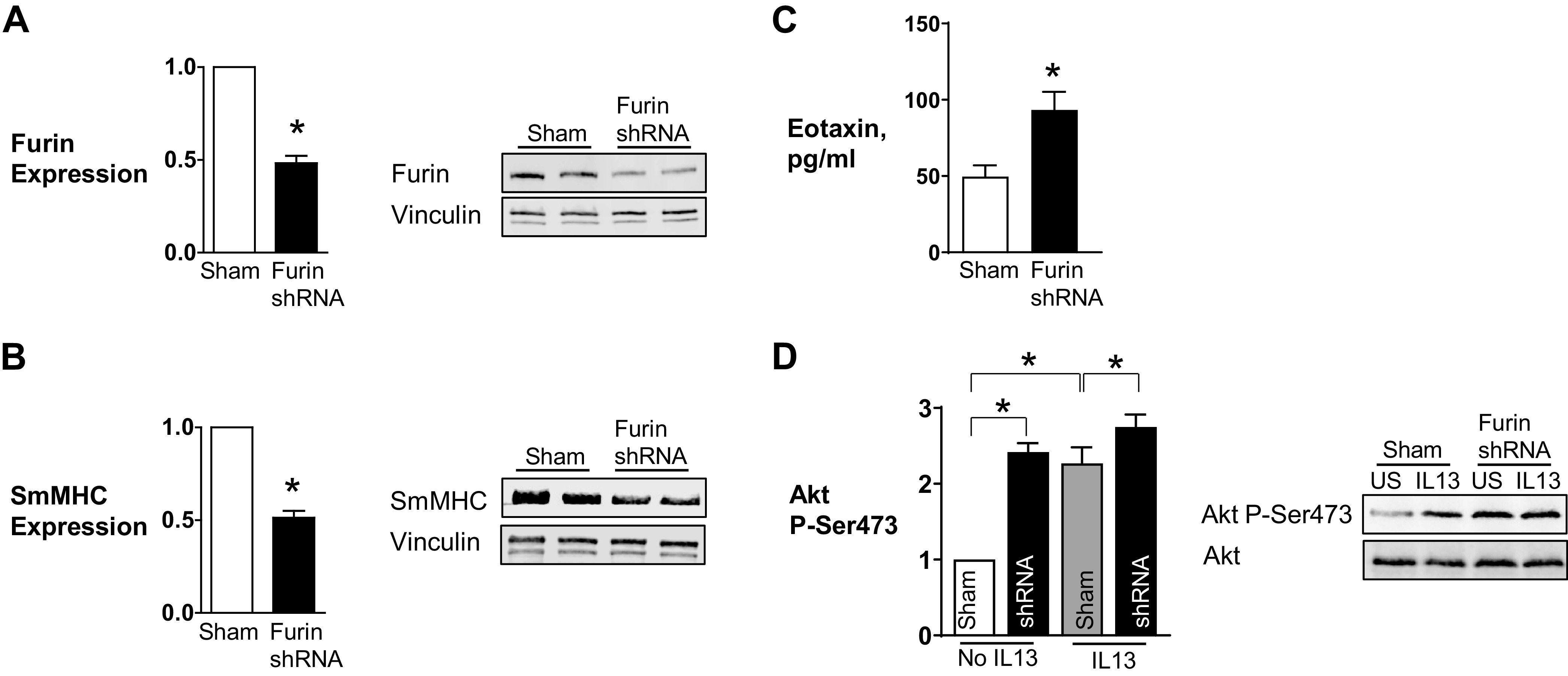
Depletion of furin from airway smooth muscle tissues increases Akt phosphorylation and decreases smooth muscle myosin heavy chain (SmMHC) expression. A, B: representative immunoblots and mean expression levels of endogenous furin protein in extracts from tissues treated with furin shRNA or sham treated. ShRNA significantly reduced expression of furin (n = 10, P = 0.0001) (A) and SmMHC (n = 9, P = 0.0001) (B). C: the depletion of furin caused a significant increase in eotaxin secretion (n = 4, P = 0.0272). D: representative immunoblots of Akt from extracts of sham-treated or furin-depleted muscle tissues stimulated with IL-13 or unstimulated (US). Furin depletion resulted in a significant increase in Akt phosphorylation at Ser473 (n = 7, P = 0.0001). Furin inhibition also significantly potentiated IL-13-induced Akt phosphorylation (n = 7, P = 0.019). Data were analyzed by using a paired Student’s t test (A–C) or one-way ANOVA (D). Values are means ± SE. *Significant difference between groups.
Figure 4.

Furin regulates the activation of β1-integrins and integrin-associated adhesion proteins in airway smooth muscle tissues. A: the effects of furin on the activation on β1-integrins were evaluated using an antibody (HUTS-4) that selectively reacts with activated β1-integrin. Representative immunoblots from extracts of recombinant furin (rFurin)-treated or -untreated muscle tissues stimulated with IL-13 or unstimulated (US). The incubation of tissues with rFurin caused the activation of β1-integrins (n = 9, P = 0.0001). rFurin also increased integrin activation in the presence of IL-13 (n = 9, P = 0.0001). B: representative immunoblots from extracts of rFurin-treated or -untreated muscle tissues stimulated with IL-13 or unstimulated (US). rFurin significantly increased vinculin and paxillin phosphorylation in airway smooth muscle tissues (vinculin: n = 5, P = 0.0004; paxillin: n = 6, P = 0.0001). rFurin also increased vinculin and paxillin phosphorylation in the presence of IL-13 (vinculin: n = 5, P = 0.0025; paxillin: n = 6, P = 0.0002). Data were analyzed by using one-way ANOVA. All values are means ± SE. *Significant difference between groups. rF, rFurin.
Figure 5.

Furin Inhibitor II inhibits the activation of β1-integrins and integrin-associated adhesion proteins by acetylcholine (Ach) in airway smooth muscle tissues. A: representative immunoblots from extracts of inhibitor treated or untreated muscle tissues stimulated with ACh or unstimulated (US). ACh stimulation resulted in a marked increase in the activation of β1-integrin (n = 8, P = 0.0001) and incubation with Furin Inhibitor II inhibited the increase of activated β1-integrin induced by ACh (n = 8, P = 0.0001). B: representative immunoblots from extracts of untreated or inhibitor-treated muscle tissues stimulated with ACh or unstimulated (US). The increases in vinculin Tyr1065 phosphorylation and paxillin Tyr118 phosphorylation in response to 10−5 M ACh were significantly inhibited in tissues treated with Furin Inhibitor II (vinculin: n = 7, P = 0.0001; paxillin: n = 8, P = 0.0001). Data were analyzed by using one-way ANOVA. All values are means ± SE. *Significant difference between groups. Inh, Furin Inhibitor II.
Other materials and reagents used were as follows: recombinant canine IL-13, recombinant human furin (rhFurin), and human eotaxin-1 ELISA kit (R&D Systems) and Furin inhibitor II (Sigma). Furin shRNA plasmids (Cat. No. SC-40595) were purchased from Santa Cruz Biotechnology, (Dallas, TX).
Preparation of Smooth Muscle Tissues
All procedures were in accordance with the procedures approved by the Institutional Animal Care and Use Committee (IUCAC) of Indiana University School of Medicine under the National Research Council’s Guide for the Care and Use of Laboratory Animals. The Indiana University Laboratory Animal Resource Center (LARC) at Indiana University School of Medicine procured mongrel dogs (20–30 kg, either sex) from LBL Kennels, Reelsville, Indiana. LARC personnel euthanized animals by an intravenous injection of Fatal-Plus (Vortech Pharmaceuticals, Ltd, Dearborn, MI) [pentobarbital sodium, 390 mg/mL; propylene glycol, 0.01 mg/mL; ethyl alcohol, 0.29 mg/mL; benzyl alcohol (preservative), 0.2 mg/mL] at a dose of ∼0.3 mL/kg. A tracheal segment was immediately removed and immersed in physiological saline solution (composition in mM: 110 NaCl, 3.4 KCl, 2.4 CaCl2, 0.8 MgSO4, 25.8 NaHCO3, 1.2 KH2PO4, and 5.6 glucose). Strips of tracheal smooth muscle (in mm: 1.0 wide × 0.2–0.5 thick × 15 long) were dissected free of connective and epithelial tissues and maintained within a tissue bath at 37°C. Muscle tissues were equilibrated and attached to metal mounts to maintain them at a constant length.
Tissues were treated overnight with 100 µM Furin Inhibitor II or 0.5 µg/mL recombinant furin (rFurin). Furin Inhibitor II (Hexa-d-arginine) is a polyarginine peptide that acts as a competitive inhibitor of furin (40–42). Tissues were treated with furin shRNA plasmids to deplete endogenous furin. Tissues were stimulated for 30 min or incubated overnight with 50 ng/mL IL-13 to elicit inflammatory responses (1, 7). Tissues were treated for 5 min with 10−5 M acetylcholine (ACh) to elicit contractile responses.
Immunoblots
Muscle tissues were pulverized in liquid N2 using a mortar and pestle before the extraction of proteins. Proteins were extracted from pulverized muscle tissues for electrophoresis and Western blotting using a buffer containing 20 mM Tris-HCl at pH 7.4, 2% Triton X-100, 0.3%–0.6% SDS, 2 mM EDTA, phosphatase inhibitors (2 mM sodium orthovanadate, 2 mM molybdate, and 2 mM sodium pyrophosphate), and protease inhibitors (2 mM benzamidine, 0.5 mM aprotinin, and 1 mM phenylmethylsulfonyl fluoride). Each sample was centrifuged, and the supernatant was then boiled for 5 min in sample buffer (1.5% dithiothreitol, 2% SDS, 80 mM Tris-HCl at pH 6.8, 10% glycerol, and 0.01% bromophenol blue). Proteins were separated by 6%–8% SDS-PAGE and transferred to nitrocellulose membranes. The nitrocellulose membrane was blocked with 2%–5% milk or Li-Cor Odyssey blocking buffer (TBS) for 1 h and probed with primary antibodies against proteins of interest overnight followed by secondary antibodies for 1 h. Proteins were visualized by enhanced chemiluminescence or by infrared fluorescence using an Li-Cor Odyssey imager.
For immunoprecipitation, pulverized muscle strips were mixed with extraction buffer containing 50 mM Tris-HCl at pH 7.6, 1% Nonidet P-40 (NP-40), 2 mM EDTA, 10% glycerol, phosphatase inhibitors (in mM: 2 sodium orthovanadate, 2 molybdate, and 2 sodium pyrophosphate), and protease inhibitors (in mM: 2 benzamidine, 0.5 aprotinin, and 1 phenylmethylsulfonyl fluoride). Muscle extracts were precleared with protein A/G UltraLink Resin and incubated overnight with antibodies against α-parvin, β-parvin, or N-terminal talin at 4°C followed by 2-h incubation with protein A/G UltraLink beads. Immunocomplexes were washed three times using washing buffer containing 50 mM Tris-HCl (pH 7.6), 150 mM NaCl, and 0.1% Triton X-100. Proteins in immunoprecipitates were separated by 8% SDS-PAGE, transferred to nitrocellulose membranes, and quantified by immunoblot.
Depletion of Endogenous Furin Protein
Furin protein was depleted from muscle tissues by introducing furin shRNA plasmids into the tissues by the method of reversible permeabilization as described previously (39, 43). We have previously demonstrated that this technique results in the successful transfection of 80%–90% of the cells within the tissue strip (39, 44–46). The strips were incubated successively in each of the following solutions: solution 1 at 4°C for 120 min containing (in mM) 10 EGTA, 5 Na2ATP, 120 KCl, 2 MgCl2, and 20 N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES); solution 2 at 4°C overnight containing (in mM) 0.1 EGTA, 5 Na2ATP, 120 KCl, 2 MgCl2, 20 TES, and 20 Furin shRNA plasmids; solution 3 at 4°C for 30 min containing (in mM) 0.1 EGTA, 5 Na2ATP, 120 KCl, 10 MgCl2, 20 TES; and solution 4 at 22°C for 90 min containing (in mM) 110 NaCl, 3.4 KCl, 0.8 MgSO4, 25.8 NaHCO3, 1.2 KH2PO4, and 5.6 dextrose. Solutions 1–3 were maintained at pH 7.1 and aerated with 100% O2. Solution 4 was maintained at pH 7.4 and was aerated with 95% O2 and 5% CO2. After 30 min in solution 4, CaCl2 was added gradually to reach a final concentration of 2.4 mM. The strips were then incubated in a CO2 incubator at 37°C for 2 days in serum-free DMEM containing 5 mM Na2ATP, 100 U/mL penicillin, 100 µg/mL streptomycin, 50 µg/mL kanamycin, 2.5 µg/mL antifungal, and 20 µg/mL furin siRNA to allow for the expression of the recombinant proteins. The depletion of furin in transfected muscle tissues was confirmed by immunoblot. Sham-treated tissues were subjected to the same procedures except that no plasmids were added to solution 2.
Measurement of Eotaxin Secretion from Muscle Tissues
Eotaxin secreted from muscle tissues was measured in the incubation medium as described previously (7). Tracheal muscle tissue strips were incubated with 0.5 µg/mL rFurin or 100 µM Furin Inhibitor II overnight or transfected with furin shRNA. Some tissues were also stimulated with 50 ng/mL IL-13. The incubation medium from each muscle tissue was collected and concentrated to 100 µL using centrifugal filters. The concentration of eotaxin-1 in each sample was determined by ELISA using the human CCL11/eotaxin antibody and quantified using a standard curve. All samples and standards were measured in duplicate.
Statistical Analysis
Comparisons among multiple treatment groups of tissues were performed by ANOVA with repeated measures using Tukey’s test for post hoc analysis. Comparisons between two groups were performed using paired two-tailed Student’s t tests. Values of n refer to the number of tissues used to obtain mean values. P < 0.05 was considered statistically significant.
RESULTS
Furin Inhibits the Inflammatory Responses of Airway Smooth Muscle Tissues to IL-13
We evaluated the effect of furin on the responses of airway smooth muscle to stimulation with IL-13. Isolated tracheal smooth muscle tissues were incubated overnight with or without rFurin and then stimulated with IL-13 for 30 min before measuring the activation of Akt, or they were stimulated with IL-13 overnight to evaluate SmMHC expression and eotaxin secretion. The activation of Akt was assessed by measuring the phosphorylation of Akt at Ser473, which correlates with Akt kinase activity (47, 48).
IL-13 induced a significant increase in Akt activation and eotaxin secretion and suppressed the expression of SmMHC, as observed in our previous studies (3, 4, 7, 8) (Fig. 1). The treatment of tissues with rFurin inhibited Akt activation and eotaxin secretion in response to stimulation with IL-13 and prevented the IL-13-induced suppression of SmMHC expression. The treatment of tissues with rFurin alone did not significantly affect Akt activation or SmMHC expression compared with untreated tissues (Fig. 1, B and C). These results demonstrate that the treatment of airway smooth muscle tissues with rFurin suppresses inflammatory responses to IL-13.
Figure 1.

Recombinant furin (rFurin) prevents inflammatory signaling pathways in airway smooth muscle tissues. Airway smooth muscle tissues were treated with 0.5 µg/mL furin overnight and then incubated with or without 50 ng/mL IL-13 overnight or for 30 min. A: eotaxin secretion was significantly increased by stimulation with IL-13 (n = 6, P = 0.0001). rFurin significantly inhibited IL-13-induced eotaxin secretion (n = 6, P = 0.0133). B: IL-13 significantly increases Akt phosphorylation (n = 9, P = 0.0001). Furin significantly inhibits Akt phosphorylation induced by IL-13 (n = 9, P = 0.0001). C: expression of smooth muscle myosin heavy chain (SmMHC) was significantly inhibited in tissues stimulated with IL-13 (n = 7, P = 0.0001). Furin prevents inhibition of SmMHC expression induced by IL-13 (n = 7, P = 0.2526). All statistical analyses were performed by using one-way ANOVA with repeated measures. Values are means ± SE. *Significant difference between groups. rFu, rFurin; US, unstimulated.
The Inhibition of Furin Activity Results in Airway Smooth Muscle Inflammation
The role of endogenous furin in airway smooth muscle inflammation was evaluated by inhibiting furin activity using Furin Inhibitor II. Isolated tracheal smooth muscle tissues were incubated overnight with or without Furin Inhibitor II and then stimulated with IL-13 for 30 min before measuring the activation of Akt, or they were incubated overnight concurrently with Furin Inhibitor II and IL-13 for the measurement of eotaxin secretion or SmMHC expression.
The treatment of tissues with Furin Inhibitor II alone caused significant increases in eotaxin secretion and Akt phosphorylation and suppressed SmMHC expression (Fig. 2). The inflammatory effects of Furin Inhibitor II on airway smooth muscle tissues were analogous to those caused by the stimulation with IL-13. Thus, the inhibition of endogenous furin activity promotes an inflammatory response in airway smooth muscle tissues.
Figure 2.

Inhibition of furin activity promotes inflammatory signaling pathways in airway smooth muscle tissues. Airway smooth muscle tissues were treated with 100 µM Furin Inhibitor II in the presence or absence of 50 ng/mL IL-13 for measurement of Akt phosphorylation, smooth muscle myosin heavy chain (SmMHC), or eotaxin secretion. A: eotaxin secretion was significantly increased by Furin Inhibitor II (P = 0.0487) or IL-13 (P = 0.0001) in airway smooth muscle (ASM) tissues (n = 6). Inhibition of furin activity did not significantly increase eotaxin secretion induced by IL-13 (n = 6, P = 0.116). B: treatment of tissues with Furin Inhibitor II or IL-13 results in a significant increase in Akt phosphorylation (n = 8, P = 0.0001). C: expression of SmMHC was significantly inhibited by Furin Inhibitor II or IL-13 (n = 6, P = 0.0001). Furin inhibition did not significantly depress the expression of SmMHC of ASM tissues after IL-13 stimulation (n = 6, P = 0.058). Statistical analysis was performed by using one-way ANOVA with repeated measures. Values are means ± SE. *Significant difference between groups. Inh, Furin Inhibitor II; US, unstimulated.
We also evaluated the role of endogenous furin in airway smooth muscle inflammation by depleting tissues of endogenous furin using furin shRNA (Fig. 3). Treatment of the tissues with furin shRNA caused the depletion of endogenous furin by ∼50% compared with sham-treated tissues and led to a significant decrease in the expression of SmMHC (Fig. 3, A and B). Furin depletion also resulted in a significant increase in eotaxin secretion and Akt phosphorylation at Ser473 (Fig. 3, C and D). The stimulation of furin-depleted tissues with IL-13 slightly increased Akt activation relative to the stimulation of sham-treated tissues with IL-13 alone. These results demonstrate that endogenous furin promotes the expression of a differentiated phenotype in the airway smooth muscle by suppressing the activation of Akt and the synthesis of inflammatory mediators.
Furin Regulates the Activation of β1-Integrins and Integrin-Associated Adhesome Proteins in Airway Smooth Muscle Tissues
Cellular stimulation can induce the activation of transmembrane integrin proteins, which increases their adhesion to extracellular matrix ligands and triggers the activation of proteins within integrin-associated adhesome complexes (14, 49–52). In airway smooth muscle, vinculin and paxillin form a complex in the cytoplasm that can be recruited to integrin adhesomes, where both proteins undergo activation (37, 38, 53, 54). Vinculin activation involves a conformational change that requires its phosphorylation on Tyr1065 and facilitates its binding to the integrin-cross-linking protein talin and to actin filaments at the adhesome (37, 38). Paxillin remains in a complex with vinculin but undergoes tyrosine phosphorylation by focal adhesion kinase (FAK) within the adhesome complex (38, 54, 55).
We investigated the role of furin in regulating integrin activation and the phosphorylation and activation of vinculin and paxillin (Fig. 4). Tissues were incubated overnight with or without rFurin and then stimulated with IL-13 for 30 min or not stimulated. Integrin activation was evaluated by immunoblot using an antibody that selectively reacts with activated β1-integrin (HUTS-4) (56, 57). The activation of vinculin and paxillin was evaluated by determining the phosphorylation of vinculin at Tyr1065 and phosphorylation of paxillin at Tyr118 (37, 38, 44).
The treatment of tissues with rFurin induced the activation of β1-integrins and significantly increased the phosphorylation of both vinculin and paxillin (Fig. 4). The stimulation of tissues with IL-13 by itself for 30 min did not activate integrins or induce vinculin or paxillin phosphorylation, but β1-integrin activation and the activation of vinculin and paxillin that was induced by treatment with rFurin were not inhibited by IL-13.
The effect of inhibiting furin activity on integrin activation and on paxillin and vinculin phosphorylation was evaluated in tissues incubated overnight with or without Furin Inhibitor II and then stimulated with 10−5 M acetylcholine (ACh) for 5 min to induce integrin activation and the phosphorylation of vinculin and paxillin (Fig. 5). The stimulation of tissues with 10−5M ACh significantly increased integrin activation and the phosphorylation of vinculin and paxillin in untreated tissues. When tissues were pretreated with Furin Inhibitor II before stimulation with ACh, ACh did not induce the activation of integrins, vinculin, or paxillin. The treatment of unstimulated muscles with Furin Inhibitor II did not have a significant effect on integrin activation or on the phosphorylation of vinculin or paxillin; however, the low basal levels of activation of these proteins would make any decreases in protein phosphorylation caused by furin inhibition difficult to detect. These results demonstrate that furin catalyzes β1-integrin activation and activation of the adhesome proteins vinculin and paxillin and that furin activity is necessary for integrin activation induced by ACh.
Furin Prevents IL-13-Induced Talin Cleavage in Airway Smooth Muscle Tissues
The adhesion molecule talin binds to β-integrin proteins, actin filaments, and vinculin and forms a foundation for the assembly of integrin-associated adhesome complexes. In airway smooth muscle, vinculin and paxillin are recruited to integrin adhesion sites in response to contractile stimulation, where vinculin binds to talin catalyzing vinculin activation. This is a necessary step in tension generation in airway smooth muscle during active contraction (37, 38, 53).
Talin (270 kD) consists of an N-terminal 50-kD head domain and an elongated helical rod of ∼220 kD connected by a linker region (58, 59). Talin can undergo cleavage in the linker region mediated by calpain 2 in response to external stimuli that results in the separation of the head and rod domains (60, 61). We investigated the effect of furin on talin cleavage and the ability of talin to bind to and activate vinculin.
Airway smooth muscle tissues were depleted of endogenous furin using shRNA, or furin activity was inhibited by incubating tissues with Furin Inhibitor II (Fig. 6A). Talin cleavage was analyzed by immunoblot using an antibody against the head domain of talin. The depletion of endogenous furin resulted in an increase in talin cleavage, as indicated by an increase in the head domain of talin and a decrease in full-length talin (Fig. 6A). The treatment of tissues with Furin Inhibitor II also caused talin cleavage.
Figure 6.
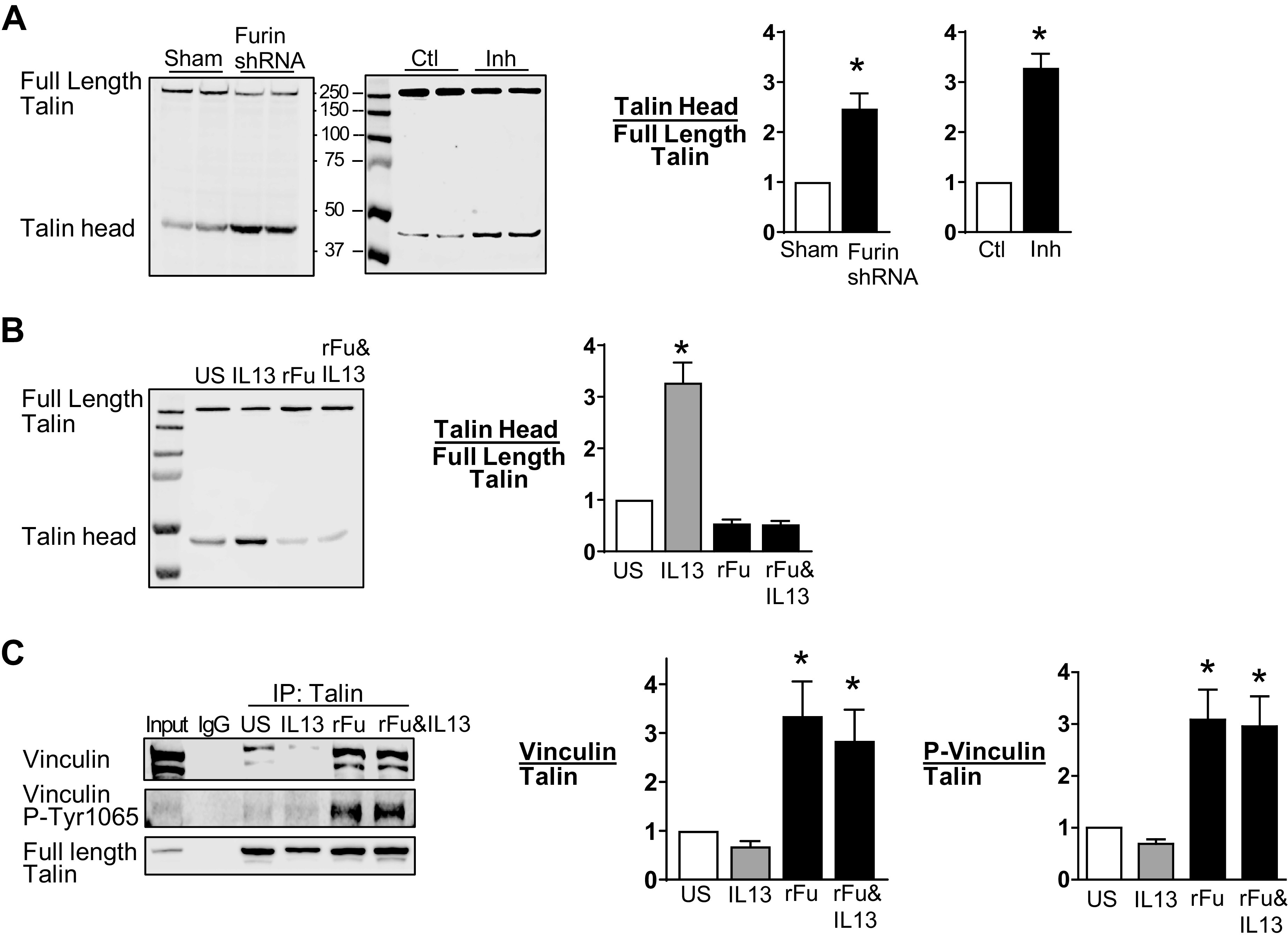
Furin prevents IL-13-induced talin cleavage in airway smooth muscle tissues. A: representative immunoblots from extracts of tissues treated with furin shRNA or sham treated and treated with or without Furin Inhibitor II (Inh). Full-length talin and the talin head domain were analyzed by immunoblot with an N-terminal antibody to talin. Furin depletion significantly increased the ratio of the talin head domain to full-length talin (n = 6, P = 0.005). Furin inhibition also significantly increased the ratio of the talin head domain to full-length talin (n = 15, P = 0.0001). B: representative immunoblot of extracts from tissues treated with or without IL-13 in the presence or absence of recombinant furin (rFurin). IL-13 significantly increased the ratio of the talin head to full-length talin (n = 12, P = 0.0001). rFurin prevented the increase in talin cleavage induced by IL-13 (n = 12, P = 0.2972). C: talin was immunoprecipitated from tissue extracts, and immunocomplexes were blotted for vinculin and Tyr1065 phosphorylated vinculin. Vinculin and phosphorylated vinculin were significantly higher in tissues treated with rFurin or rFurin and IL-13 compared with untreated tissues (vinculin: P = 0.0026 for rFurin, P = 0.0184 for rFurin and IL-13; phosphorylated vinculin: P = 0.0014 for rFurin, P = 0.0026 for rFurin and IL-13) (n = 7). Data were analyzed by using a paired Student’s t test (A) or one-way ANOVA (B and C). All values are means ± SE. *Significant difference between groups. ctl, control; rFu, rFurin; US, unstimulated.
We then investigated the effect of IL-13 and rFurin on talin cleavage by incubating tissues overnight with rFurin, IL-13, or with rFurin and IL-13 concurrently (Fig. 6B). Treatment of the tissues with IL-13 caused an increase in talin cleavage relative to untreated tissues, whereas the treatment of tissues with rFurin alone did not cause talin cleavage. The incubation of tissues concurrently with rFurin and IL-13 prevented IL-13-induced talin cleavage (Fig. 6B).
The effect of furin on the interaction of talin with vinculin was determined by evaluating the coprecipitation of vinculin with talin in extracts of untreated tissues or from tissues treated overnight with IL-13, rFurin, or IL-13 and rFurin (Fig. 6C). In unstimulated tissues and in tissues stimulated with IL-13, little vinculin coimmunoprecipitated with talin and little phosphorylated vinculin was detected, indicating that most of the vinculin was in an inactive conformation and was not bound to talin. The treatment of tissues with rFurin resulted in a significant increase in the coprecipitation of phosphorylated vinculin with talin in tissues irrespective of whether they were treated with IL-13 (Fig. 6C).
These results show that furin promotes integrin activation and the assembly of adhesome complexes and that it inhibits talin cleavage induced by IL-13, thus preventing the inactivation of vinculin and paxillin.
Furin Suppresses Akt Activation and Airway Smooth Muscle Inflammation by Activating β-Parvin IPP Complexes
The heterotrimeric IPP complex, which consists of integrin-linked kinase (ILK), particularly interesting new cysteine-histidine-rich protein (PINCH), and α-parvin or β-parvin, is maintained as a stable complex in the airway smooth muscle that acts as a critical regulator of integrin-mediated signaling pathways involved in phenotypic modulation (3, 62). The differential coupling of Akt to α-parvin versus β-parvin IPP complexes regulates phenotypic transitions between a differentiated state and an inflammatory synthetic state in airway smooth muscle tissues (3). The binding of α-parvin IPP complexes to Akt promotes Akt activation and an inflammatory phenotype, whereas the binding of β-parvin IPP complexes to Akt suppresses Akt activation and promotes a differentiated phenotype. Paxillin plays a critical role in switching the coupling of Akt between α-parvin and β-parvin IPP complexes: α-parvin IPP complexes bind to unphosphorylated paxillin, whereas β-parvin IPP complexes bind to phosphorylated paxillin (3). The binding of Akt to β-parvin prevents its binding to ILK, which prevents Akt activation by ILK and promotes a differentiated phenotype (3, 63).
We evaluated the effect of furin on the interactions of α-parvin and β-parvin IPP complexes with Akt and paxillin. Smooth muscle tissues were treated overnight with IL-13, furin, or with IL-13 and furin. α-Parvin and β-parvin IPP complexes were immunoprecipitated using antibodies to α-parvin or β-parvin, as PINCH and ILK both coprecipitate in parvin immunocomplexes (3). We analyzed the association of each parvin complex with phosphorylated and unphosphorylated Akt and with phosphorylated and unphosphorylated paxillin under each condition (Fig. 7).
Figure 7.

Furin promotes the association of Akt with β-parvin IPP [integrin-linked kinase (ILK), PINCH, parvin] complexes, which prevents IL-13-induced Akt activation. α-Parvin or β-parvin was immunoprecipitated from extracts of tissues treated with or without IL-13, recombinant furin (rFurin), or IL-13 and rFurin. A: α-parvin and β-parvin immunocomplexes were immunoblotted for Akt, pS473-Akt, paxillin, pY118-paxillin, and α-parvin or β-parvin. B: rFurin stimulated a significant increase in the association of phosphorylated paxillin with β-parvin IPP complexes (n = 4, P = 0.0001) and a significant decrease in the association of unphosphorylated paxillin with α-parvin complexes (n = 5, P = 0.0251). IL-13 stimulates the association of unphosphorylated paxillin with α-parvin IPP complexes (n = 5, P = 0.0018) and not with β-parvin complexes (n = 4, P = 0.6808). Furin prevents the interaction of unphosphorylated paxillin with α-parvin complexes in response to IL-13 (n = 5, P = 0.7880) and stimulates the interaction of phosphorylated paxillin with β-parvin complexes (n = 4, P = 0.0001). C: IL-13 stimulates the activation of Akt by α-parvin IPP complexes (n = 5, P = 0.0001). Furin alone does not stimulate the activation of Akt by α-parvin IPP complexes (n = 5, P = 0.0423), but furin prevents the activation of Akt by α-parvin IPP complexes caused by stimulation with IL-13 (n = 4, P = 0.7941). Statistical analysis was performed by using one-way ANOVA with repeated measures. Values are means ± SE. *Significant difference between groups. ns, no significant difference; rFu, rFurin; US, unstimulated.
The treatment of tissues with rFurin stimulated the interaction of β-parvin IPP complexes with phosphorylated paxillin and unphosphorylated Akt. In contrast, the treatment of tissues with IL-13 stimulated the interaction of α-parvin IPP complexes with unphosphorylated paxillin and phosphorylated Akt (Fig. 7, B and C). In tissues treated with both rFurin and IL-13, phosphorylated paxillin and unphosphorylated Akt associated with β-parvin IPP complexes; thus, Akt activation and the inflammatory responses to IL-13 were inhibited.
The effect of Furin inhibitor II on the interactions of α-parvin and β-parvin IPP complexes with paxillin and Akt was also evaluated by coimmunoprecipitation analysis (Fig. 8). The treatment of tissues with Furin Inhibitor II promoted the association of α-parvin IPP complexes with unphosphorylated paxillin and phosphorylated Akt. As furin inhibition does not stimulate paxillin phosphorylation, it preferentially promotes the association of Akt with α-parvin IPP complexes, which results in Akt activation and leads to an inflammatory phenotype similar to that caused by IL-13.
Figure 8.

Furin inhibition promotes the activation of Akt by α-parvin IPP [integrin-linked kinase (ILK), PINCH, parvin] complexes in airway smooth muscle tissues. α-Parvin or β-parvin was immunoprecipitated from extracts of tissues treated with or without IL-13, Furin Inhibitor II, or IL-13 and Furin Inhibitor II. A: α-parvin and β-parvin immunocomplexes were immunoblotted for Akt, pS473-Akt, paxillin, pY118-paxillin, and α-parvin or β-parvin. B: IL-13 and Furin Inhibitor II each stimulates a significant increase in the association of unphosphorylated paxillin with α-parvin IPP complexes (IL-13: n = 5, P = 0.0001; Furin Inhibitor II: n = 5, P = 0.0017). Furin Inhibitor II stimulates a significant decrease in the association of paxillin with β-parvin IPP complexes (n = 4, P = 0.0001). C: IL-13 and Furin Inhibitor II each induces Akt activation by stimulating the association of Akt with α-parvin IPP complexes (IL-13: n = 5, P = 0.0009; Furin Inhibitor II: n = 5, P = 0.0006). Furin Inhibitor II stimulates a significant decrease in the association of Akt with β-parvin IPP complexes (n = 4, P = 0.0001). Statistical analysis was performed by using one-way ANOVA with repeated measures. Values are means ± SE. *Significant difference between groups. Inh, Furin Inhibitor II; ns, no significant difference; US, unstimulated.
The results of these experiments demonstrate that furin regulates integrin-associated signaling pathways mediated by IPP complexes to induce phenotypic transitions in airway smooth muscle tissues. Furin causes the activation of pathways that suppress inflammation and promote a differentiated phenotype, whereas the inhibition of furin leads to the activation of Akt and promotes inflammatory responses in airway smooth muscle tissues.
DISCUSSION
Our results demonstrate that furin is a potent anti-inflammatory agent that suppresses the responses of airway smooth muscle to stimulation with the inflammatory cytokine IL-13. The treatment of airway smooth muscle tissues with exogenous rFurin inhibits the activation of Akt and eotaxin secretion in muscle tissues in response to IL-13 and promotes the expression of a differentiated smooth muscle phenotype. Conversely, a reduction in furin activity in smooth muscle tissues caused by furin inhibition or by the depletion of endogenous furin protein results in the activation of Akt and stimulates eotaxin secretion, while suppressing the expression of a differentiated smooth muscle phenotype. This indicates that endogenous furin is constitutively active and that it promotes the maintenance of a differentiated phenotype in airway smooth muscle.
We found that furin inhibits airway smooth muscle inflammation by inducing the activation of transmembrane integrins and integrin-associated adhesome signaling complex proteins that regulate signaling pathways to the nucleus (Fig. 9). Furin stimulates the binding of vinculin to talin causing the activation of vinculin and of the adaptor protein paxillin. The depletion of furin or its inhibition resulted in integrin inactivation and talin cleavage and prevented the binding of vinculin to talin, thereby inhibiting the activation of both vinculin and paxillin. In airway smooth muscle, the regulation of paxillin tyrosine phosphorylation plays a critical role in the differential activation of α-parvin versus β-parvin IPP complexes that control Akt-mediated nuclear signaling pathways that lead to the expression of an inflammatory phenotype versus a differentiated phenotype (3).
Figure 9.

Proposed mechanism for the regulation of inflammation by furin in airway smooth muscle. A: furin activates integrin-mediated signaling pathways regulating Akt activity and phenotype expression in airway smooth muscle. Furin promotes the cross linking of β integrin proteins by talin, resulting in integrin activation and the high-affinity binding of integrin proteins to the extracellular matrix. Vinculin is activated by its binding to full-length talin, facilitating phosphorylation of its binding partner paxillin. Phosphorylated paxillin binds to β-parvin IPP (ILK, PINCH, parvin) complexes, which promotes the preferential binding of Akt to β-parvin instead of integrin-linked kinase (ILK). This prevents the activation of Akt by ILK and suppresses Akt-mediated synthetic pathways that induce an inflammatory phenotype. This promotes the localization of serum response factor (SRF) to the nucleus and the expression of a differentiated phenotype. α-Parvin IPP complexes are inactive because they are not recruited by phosphorylated paxillin. B: effect of furin inhibition or IL-13 stimulation on integrin-mediated signaling pathways regulating Akt activity and phenotype expression in airway smooth muscle. Stimulation with IL-13 or treatment with furin inhibitor leads to talin cleavage, which prevents the cross linking of β integrins by talin, resulting in integrin inactivation. This prevents the high-affinity binding of integrins to the extracellular matrix. Vinculin remains in an inactive complex with paxillin because vinculin cannot be activated by cleaved talin, and paxillin phosphorylation is therefore prevented. Unphosphorylated paxillin binds to α-parvin IPP complexes, which promotes the binding of Akt directly to ILK. ILK induces Akt activation resulting in an inflammatory phenotype. SRF localization to the nucleus is inhibited by activated Akt, which suppresses the expression of a differentiated smooth muscle phenotype. β-Parvin IPP complexes remain inactive, as they are not recruited by unphosphorylated paxillin. smMHC, smooth muscle myosin heavy chain.
Talin binds to β-integrin proteins, actin filaments, and vinculin and forms a foundation for the assembly of integrin-associated adhesome complexes (58, 59). The N-terminal domain of talin forms the head and includes binding sites for the β1-integrin cytoplasmic tail. The talin rod domain contains multiple binding sites for vinculin, and the C-terminus of the rod domain is responsible for the formation of talin homodimers that enable talin to cross link actin filaments via a C-terminal F-actin binding site (59). By binding to the cytoplasmic tails of integrin proteins, dimeric talin molecules elicit integrin clustering and activation, resulting in their high-affinity binding to extracellular matrix proteins. They also link integrins to the actin cytoskeleton by cross linking actin filaments and form a scaffold for the assembly of adhesome signaling modules (16, 49, 59, 64). Vinculin undergoes a conformational shift when it binds to the talin rod domain that promotes its binding to actin filaments, further strengthening adhesion linkages and providing a foundation for the assembly of signaling modules within integrin adhesome complexes (37, 38, 65–68). The shift in vinculin conformation also promotes the activation of paxillin, an adaptor protein that regulates the assembly of signaling modules that mediate nuclear signaling pathways (37, 38).
Our results showed that the inhibition of endogenous furin activity or stimulation of the tissues with IL-13 led to the cleavage of talin, which prevented the binding of vinculin to talin and inhibited the activation of vinculin and paxillin. Cleavage of talin in the linker region is mediated by calpain 2 and separates the talin head and rod domains (60, 61). Although the head domain of talin can bind to the cytoplasmic tail of β integrins, the interaction of integrins with full-length talin dimers may be necessary to convert the integrin to a high-affinity active state that binds to extracellular matrix ligands (59).
Furin might regulate integrin activation indirectly through its effects on components of the extracellular matrix or by directly regulating the activation of integrin proteins (17, 19, 59). Furin can be anchored to the plasma membrane via its C-terminal transmembrane domain with its catalytically active N-terminal domain extending into the extracellular space. In addition, the proteolytic cleavage of furin at its C-terminus can separate the transmembrane domain from the catalytic domain, resulting in the shedding of catalytically active furin into the extracellular space (17). Furin is thus positioned to regulate the activation or the inactivation of both extracellular and membrane proteins, and its substrates include growth factors and cytokines, collagens, matrix metalloproteinases, and integrins. In isolated cultured vascular smooth muscle cells, furin activity is critical for endoproteolytic processing of αv integrin precursors, and it is necessary for the activation of integrin-receptor mediated signaling by FAK and paxillin during vascular smooth muscle cell adhesion and motility (69).
In airway smooth muscle, paxillin plays a critical role in the differential recruitment of α-parvin and β-parvin IPP complexes. α-Parvin and β-parvin heterotrimeric IPP complexes are present in distinct cytoplasmic pools that are maintained in an inactive conformation and are separately recruited to adhesome complexes in response to exogenous stimuli, where they catalyze phenotype transitions in airway smooth muscle tissues by regulating the activation of Akt (3, 36). The differential recruitment and activation of α-parvin versus β-parvin IPP complexes is regulated by paxillin: tyrosine phosphorylated paxillin binds selectively to β-parvin IPP complexes, whereas unphosphorylated paxillin selectively binds to α-parvin IPP complexes (3). The interaction of α-parvin IPP complexes with paxillin catalyzes the binding of Akt to ILK, which induces its activation. The interaction of β-parvin IPP complexes with paxillin catalyzes the binding of Akt to β-parvin, which prevents Akt activation by hindering its binding to ILK (3, 63). Akt mediates pathways to the nucleus that promote cytokine synthesis and inhibit the translocation of the transcription factor SRF to the nucleus, thus inhibiting the expression of smooth muscle-specific proteins (36). We found that the treatment of airway smooth muscle tissues with furin catalyzed integrin activation and paxillin phosphorylation, which promoted the binding of β-parvin IPP complexes to paxillin and suppressed Akt activation. Conversely, furin inhibitors and IL-13 did not stimulate paxillin phosphorylation, which facilitated the interaction of paxillin with α-parvin IPP complexes and promoted Akt activation. These results document a molecular process in airway smooth muscle by which furin regulates transitions between differentiated and inflammatory phenotypes by regulating integrin activation and talin cleavage.
The changes in phenotypic expression induced by furin are similar to those that are induced by other conditions that alter the extracellular milieu of airway smooth muscle (1, 3, 4, 7). The treatment of airway smooth muscle tissues with the serine proteinase elastase, which is secreted by neutrophils and degrades extracellular matrix proteins, modulates airway smooth muscle phenotype by modulating signaling through integrin complexes (4). The proteolytic activity of elastase leads to talin cleavage and the inactivation of vinculin, paxillin, and FAK, resulting in Akt activation and an inflammatory phenotype in airway smooth muscle (4). The protease activity of elastase has an opposite effect to the protease activity of furin, which activates integrins and suppresses inflammation.
Airway smooth muscle tissues maintained under low mechanical tension exhibit an inflammatory phenotype characterized by Akt activation, eotaxin secretion, and a decrease in the expression of smooth muscle phenotype-specific proteins, whereas maintaining them under high tension promotes the expression of a differentiated phenotype and suppresses responses to inflammatory mediators (1, 3, 7). Mechanically induced changes in phenotypic expression are induced by mechanosensitive changes in the activation of FAK and paxillin that regulate the ILK-dependent activation of Akt by α-parvin and β-parvin IPP complexes (3, 7). Integrin activation can be elicited by the binding of integrins to extracellular matrix proteins or by mechanical tension imposed on integrin proteins, both of which may be affected by protease treatment or by imposing mechanical tension on the airway tissues. Thus, the alterations in phenotypic expression and cytoskeletal signaling that these conditions induce may result from outside-in signaling by integrin proteins.
Our results demonstrate that furin has a potent anti-inflammatory effect on airway smooth muscle; thus, furin may have therapeutic potential for the treatment of inflammatory conditions of the lungs and airways. Our findings are consistent with previous studies demonstrating anti-inflammatory protective effects of furin treatment (25–27). The systemic administration of furin to an experimental mouse model of arthritis reduced the expression of metalloproteinases in the joints and prevented increases in the arthritis score, joint destruction, and bone loss, whereas the systemic administration of a furin inhibitor, α1-PDX, to these animals enhanced these parameters (26). An anti-inflammatory function of furin has also been shown in myeloid cells in mice in vivo (25), where the loss of furin was found to reduce peripheral tolerance in mice in vivo (28). These studies suggest that the therapeutic use of furin or its derivatives may be beneficial in some inflammatory conditions.
Furin activity has also been extensively linked to cancer progression and metastasis, where it has been proposed to facilitate cell motility by remodeling the extracellular matrix to promote tissue invasion and the metastatic spreading of cancer cells (19, 21, 24). The enzymatic activity of furin has also been exploited by a variety of viral and bacterial pathogens to enhance their virulence and spread (17). The potential of furin inhibition as a viable cancer therapy has led to the development of a variety of reagents that inhibit furin activity or furin expression, some of which are now being tested in clinical trials for cancer therapy (22, 70, 71). Thus, while our evidence suggests that furin has beneficial anti-inflammatory effects, there is also evidence for pathophysiological effects. If the substrates of furin activity that regulate its beneficial anti-inflammatory effects could be determined, this might provide a basis for the design of focused therapeutic agents that selectively harness the anti-inflammatory potential of furin’s enzymatic activity.
GRANTS
These studies were supported by National Heart, Lung, and Blood Institute (NHLBI) Grant HL29289.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.W., Y.H., W.Z., and S.J.G. conceived and designed research; Y.W., Y.H., and W.Z. performed experiments; Y.W., Y.H., W.Z., and S.J.G. analyzed data; Y.W., Y.H., W.Z., and S.J.G. interpreted results of experiments; Y.W., Y.H., W.Z., and S.J.G. prepared figures; Y.W., Y.H., W.Z., and S.J.G. drafted manuscript; Y.W., Y.H., W.Z., and S.J.G. edited and revised manuscript; Y.W., Y.H., W.Z., and S.J.G. approved final version of manuscript.
REFERENCES
- 1.Desai LP, Wu Y, Tepper RS, Gunst SJ. Mechanical stimuli and IL-13 interact at integrin adhesion complexes to regulate expression of smooth muscle myosin heavy chain in airway smooth muscle tissue. Am J Physiol Lung Cell Mol Physiol 301: L275–L284, 2011. doi: 10.1152/ajplung.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirst SJ, Hallsworth MP, Peng Q, Lee TH. Selective induction of eotaxin release by interleukin-13 or interleukin-4 in human airway smooth muscle cells is synergistic with interleukin-1beta and is mediated by the interleukin-4 receptor alpha-chain. Am J Respir Crit Care Med 165: 1161–1171, 2002. doi: 10.1164/ajrccm.165.8.2107158. [DOI] [PubMed] [Google Scholar]
- 3.Huang Y, Gunst SJ. Phenotype transitions induced by mechanical stimuli in airway smooth muscle are regulated by differential interactions of parvin isoforms with paxillin and Akt. Am J Physiol Lung Cell Mol Physiol 318: L1036–L1055, 2020. doi: 10.1152/ajplung.00506.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lockett AD, Wu Y, Gunst SJ. Elastase alters contractility and promotes an inflammatory synthetic phenotype in airway smooth muscle tissues. Am J Physiol Lung Cell Mol Physiol 314: L626–L634, 2018. doi: 10.1152/ajplung.00334.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore PE, Church TL, Chism DD, Panettieri RA Jr, Shore SA. IL-13 and IL-4 cause eotaxin release in human airway smooth muscle cells: a role for ERK. Am J Physiol Lung Cell Mol Physiol 282: L847–L853, 2002. doi: 10.1152/ajplung.00245.2001. [DOI] [PubMed] [Google Scholar]
- 6.Tliba O, Panettieri RA Jr.. Noncontractile functions of airway smooth muscle cells in asthma. Annu Rev Physiol 71: 509–535, 2009. doi: 10.1146/annurev.physiol.010908.163227. [DOI] [PubMed] [Google Scholar]
- 7.Wu Y, Huang Y, Gunst SJ. Focal adhesion kinase (FAK) and mechanical stimulation negatively regulate the transition of airway smooth muscle tissues to a synthetic phenotype. Am J Physiol Lung Cell Mol Physiol 311: L893–L902, 2016. doi: 10.1152/ajplung.00299.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Y, Zhang W, Gunst SJ. S100A4 is secreted by airway smooth muscle tissues and activates inflammatory signaling pathways via receptors for advanced glycation end products. Am J Physiol Lung Cell Mol Physiol 319: L185–L195, 2020. doi: 10.1152/ajplung.00347.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dekkers BG, Spanjer AI, van der Schuyt RD, Kuik WJ, Zaagsma J, Meurs H. Focal adhesion kinase regulates collagen I-induced airway smooth muscle phenotype switching. J Pharmacol Exp Ther 346: 86–95, 2013. doi: 10.1124/jpet.113.203042. [DOI] [PubMed] [Google Scholar]
- 10.Shkumatov A, Thompson M, Choi KM, Sicard D, Baek K, Kim DH, Tschumperlin DJ, Prakash YS, Kong H. Matrix stiffness-modulated proliferation and secretory function of the airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 308: L1125–L1135, 2015. doi: 10.1152/ajplung.00154.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tschumperlin DJ, Drazen JM. Chronic effects of mechanical force on airways. Annu Rev Physiol 68: 563–583, 2006. doi: 10.1146/annurev.physiol.68.072304.113102. [DOI] [PubMed] [Google Scholar]
- 12.Wright DB, Trian T, Siddiqui S, Pascoe CD, Johnson JR, Dekkers BG, Dakshinamurti S, Bagchi R, Burgess JK, Kanabar V, Ojo OO. Phenotype modulation of airway smooth muscle in asthma. Pulm Pharmacol Ther 26: 42–49, 2013. doi: 10.1016/j.pupt.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 13.Arnaout MA, Mahalingam B, Xiong JP. Integrin structure, allostery, and bidirectional signaling. Annu Rev Cell Dev Biol 21: 381–410, 2005. doi: 10.1146/annurev.cellbio.21.090704.151217. [DOI] [PubMed] [Google Scholar]
- 14.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 110: 673–687, 2002. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 15.Winograd-Katz SE, Fassler R, Geiger B, Legate KR. The integrin adhesome: from genes and proteins to human disease. Nat Rev Mol Cell Biol 15: 273–288, 2014. doi: 10.1038/nrm3769. [DOI] [PubMed] [Google Scholar]
- 16.Zaidel-Bar R, Geiger B. The switchable integrin adhesome. J Cell Sci 123: 1385–1388, 2010. doi: 10.1242/jcs.066183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braun E, Sauter D. Furin-mediated protein processing in infectious diseases and cancer. Clin Transl Immunol 8: e1073, 2019. doi: 10.1002/cti2.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein-Szanto AJ, Bassi DE. Proprotein convertase inhibition: paralyzing the cell’s master switches. Biochem Pharmacol 140: 8–15, 2017. doi: 10.1016/j.bcp.2017.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol 3: 753–766, 2002. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vey M, Schafer W, Berghofer S, Klenk HD, Garten W. Maturation of the trans-Golgi network protease furin: compartmentalization of propeptide removal, substrate cleavage, and COOH-terminal truncation. J Cell Biol 127: 1829–1842, 1994. doi: 10.1083/jcb.127.6.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bassi DE, Fu J, Lopez de Cicco R, Klein-Szanto AJ. Proprotein convertases: “master switches” in the regulation of tumor growth and progression. Mol Carcinog 44: 151–161, 2005. doi: 10.1002/mc.20134. [DOI] [PubMed] [Google Scholar]
- 22.Couture F, Kwiatkowska A, Dory YL, Day R. Therapeutic uses of furin and its inhibitors: a patent review. Expert Opin Ther Pat 25: 379–396, 2015. doi: 10.1517/13543776.2014.1000303. [DOI] [PubMed] [Google Scholar]
- 23.Kermani NZ, Song WJ, Badi Y, Versi A, Guo Y, Sun K, Bhavsar P, Howarth P, Dahlen SE, Sterk PJ, Djukanovic R, Adcock IM, Chung KF; U-BIOPRED Consortium. Sputum ACE2, TMPRSS2 and FURIN gene expression in severe neutrophilic asthma. Respir Res 22: 10, 2021. doi: 10.1186/s12931-020-01605-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khatib AM, Siegfried G, Chretien M, Metrakos P, Seidah NG. Proprotein convertases in tumor progression and malignancy: novel targets in cancer therapy. Am J Pathol 160: 1921–1935, 2002. doi: 10.1016/S0002-9440(10)61140-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cordova ZM, Gronholm A, Kytola V, Taverniti V, Hamalainen S, Aittomaki S, Niininen W, Junttila I, Ylipaa A, Nykter M, Pesu M. Myeloid cell expressed proprotein convertase FURIN attenuates inflammation. Oncotarget 7: 54392–54404, 2016. doi: 10.18632/oncotarget.11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin H, Ah Kioon MD, Lalou C, Larghero J, Launay JM, Khatib AM, Cohen-Solal M. Protective role of systemic furin in immune response-induced arthritis. Arthritis Rheum 64: 2878–2886, 2012. doi: 10.1002/art.34523. [DOI] [PubMed] [Google Scholar]
- 27.Oksanen A, Aittomaki S, Jankovic D, Ortutay Z, Pulkkinen K, Hamalainen S, Rokka A, Corthals GL, Watford WT, Junttila I, O’Shea JJ, Pesu M. Proprotein convertase FURIN constrains Th2 differentiation and is critical for host resistance against Toxoplasma gondii. J Immunol 193: 5470–5479, 2014. doi: 10.4049/jimmunol.1401629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pesu M, Watford WT, Wei L, Xu L, Fuss I, Strober W, Andersson J, Shevach EM, Quezado M, Bouladoux N, Roebroek A, Belkaid Y, Creemers J, O’Shea JJ. T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature 455: 246–250, 2008. doi: 10.1038/nature07210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venkayya R, Lam M, Willkom M, Grunig G, Corry DB, Erle DJ. The Th2 lymphocyte products IL-4 and IL-13 rapidly induce airway hyperresponsiveness through direct effects on resident airway cells. Am J Respir Cell Mol Biol 26: 202–208, 2002. doi: 10.1165/ajrcmb.26.2.4600. [DOI] [PubMed] [Google Scholar]
- 30.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science 282: 2258–2261, 1998. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 31.Grunstein MM, Hakonarson H, Leiter J, Chen M, Whelan R, Grunstein JS, Chuang S. IL-13-dependent autocrine signaling mediates altered responsiveness of IgE-sensitized airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 282: L520–L528, 2002. doi: 10.1152/ajplung.00343.2001. [DOI] [PubMed] [Google Scholar]
- 32.Hakonarson H, Maskeri N, Carter C, Grunstein MM. Regulation of TH1- and TH2-type cytokine expression and action in atopic asthmatic sensitized airway smooth muscle. J Clin Invest 103: 1077–1087, 1999. doi: 10.1172/JCI5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laporte JC, Moore PE, Baraldo S, Jouvin MH, Church TL, Schwartzman IN, Panettieri RA , Jr, Kinet JP, Shore SA. Direct effects of interleukin-13 on signaling pathways for physiological responses in cultured human airway smooth muscle cells. Am J Respir Crit Care Med 164: 141–148, 2001. doi: 10.1164/ajrccm.164.1.2008060. [DOI] [PubMed] [Google Scholar]
- 34.Peng Q, Matsuda T, Hirst SJ. Signaling pathways regulating interleukin-13-stimulated chemokine release from airway smooth muscle. Am J Respir Crit Care Med 169: 596–603, 2004. doi: 10.1164/rccm.200307-888OC. [DOI] [PubMed] [Google Scholar]
- 35.Kaplan-Albuquerque N, Garat C, Desseva C, Jones PL, Nemenoff RA. Platelet-derived growth factor-BB-mediated activation of Akt suppresses smooth muscle-specific gene expression through inhibition of mitogen-activated protein kinase and redistribution of serum response factor. J Biol Chem 278: 39830–39838, 2003. doi: 10.1074/jbc.M305991200. [DOI] [PubMed] [Google Scholar]
- 36.Wu Y, Huang Y, Herring BP, Gunst SJ. Integrin-linked kinase regulates smooth muscle differentiation marker gene expression in airway tissue. Am J Physiol Lung Cell Mol Physiol 295: L988–L997, 2008. doi: 10.1152/ajplung.90202.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Y, Day RN, Gunst SJ. Vinculin phosphorylation at Tyr1065 regulates vinculin conformation and tension development in airway smooth muscle tissues. J Biol Chem 289: 3677–3688, 2014. doi: 10.1074/jbc.M113.508077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang Y, Zhang W, Gunst SJ. Activation of vinculin induced by cholinergic stimulation regulates contraction of tracheal smooth muscle tissue. J Biol Chem 286: 3630–3644, 2011. doi: 10.1074/jbc.M110.139923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang W, Gunst SJ. Non-muscle (NM) myosin heavy chain phosphorylation regulates the formation of NM myosin filaments, adhesome assembly and smooth muscle contraction. J Physiol 595: 4279–4300, 2017. doi: 10.1113/JP273906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernot D, Stalin J, Stocker P, Bonardo B, Scroyen I, Alessi MC, Peiretti F. Plasminogen activator inhibitor 1 is an intracellular inhibitor of furin proprotein convertase. J Cell Sci 124: 1224–1230, 2011. doi: 10.1242/jcs.079889. [DOI] [PubMed] [Google Scholar]
- 41.Cameron A, Appel J, Houghten RA, Lindberg I. Polyarginines are potent furin inhibitors. J Biol Chem 275: 36741–36749, 2000. doi: 10.1074/jbc.M003848200. [DOI] [PubMed] [Google Scholar]
- 42.Kacprzak MM, Peinado JR, Than ME, Appel J, Henrich S, Lipkind G, Houghten RA, Bode W, Lindberg I. Inhibition of furin by polyarginine-containing peptides: nanomolar inhibition by nona-D-arginine. J Biol Chem 279: 36788–36794, 2004. doi: 10.1074/jbc.M400484200. [DOI] [PubMed] [Google Scholar]
- 43.Zhang W, Gunst SJ. S100A4 is activated by RhoA and catalyses the polymerization of non-muscle myosin, adhesion complex assembly and contraction in airway smooth muscle. J Physiol 598: 4573–4590, 2020. doi: 10.1113/JP280111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang DD, Turner CE, Gunst SJ. Expression of non-phosphorylatable paxillin mutants in canine tracheal smooth muscle inhibits tension development. J Physiol 553: 21–35, 2003. doi: 10.1113/jphysiol.2003.045047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang W, Gunst SJ. Dynamic association between alpha-actinin and beta-integrin regulates contraction of canine tracheal smooth muscle. J Physiol 572: 659–676, 2006. doi: 10.1113/jphysiol.2006.106518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang W, Wu Y, Du L, Tang DD, Gunst SJ. Activation of the Arp2/3 complex by N-WASp is required for actin polymerization and contraction in smooth muscle. Am J Physiol Cell Physiol 288: C1145–C1160, 2005. doi: 10.1152/ajpcell.00387.2004. [DOI] [PubMed] [Google Scholar]
- 47.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541–6551, 1996. doi: 10.1002/j.1460-2075.1996.tb01045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Persad S, Attwell S, Gray V, Mawji N, Deng JT, Leung D, Yan J, Sanghera J, Walsh MP, Dedhar S. Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: critical roles for kinase activity and amino acids arginine 211 and serine 343. J Biol Chem 276: 27462–27469, 2001. doi: 10.1074/jbc.M102940200. [DOI] [PubMed] [Google Scholar]
- 49.Kim C, Ye F, Ginsberg MH. Regulation of integrin activation. Annu Rev Cell Dev Biol 27: 321–345, 2011. doi: 10.1146/annurev-cellbio-100109-104104. [DOI] [PubMed] [Google Scholar]
- 50.Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA. Talin binding to integrin beta tails: a final common step in integrin activation. Science 302: 103–106, 2003. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 51.Tanentzapf G, Martin-Bermudo MD, Hicks MS, Brown NH. Multiple factors contribute to integrin-talin interactions in vivo. J Cell Sci 119: 1632–1644, 2006. doi: 10.1242/jcs.02859. [DOI] [PubMed] [Google Scholar]
- 52.Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, Campbell ID. Structural basis of integrin activation by talin. Cell 128: 171–182, 2007. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 53.Opazo Saez A, Zhang W, Wu Y, Turner CE, Tang DD, Gunst SJ. Tension development during contractile stimulation of smooth muscle requires recruitment of paxillin and vinculin to the membrane. Am J Physiol Cell Physiol 286: C433–C447, 2004. doi: 10.1152/ajpcell.00030.2003. [DOI] [PubMed] [Google Scholar]
- 54.Zhang W, Huang Y, Gunst SJ. The small GTPase RhoA regulates the contraction of smooth muscle tissues by catalyzing the assembly of cytoskeletal signaling complexes at membrane adhesion sites. J Biol Chem 287: 33996–34008, 2012. doi: 10.1074/jbc.M112.369603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang DD, Gunst SJ. Depletion of focal adhesion kinase by antisense depresses contractile activation of smooth muscle. Am J Physiol Cell Physiol 280: C874–C883, 2001. doi: 10.1152/ajpcell.2001.280.4.C874. [DOI] [PubMed] [Google Scholar]
- 56.Luque A, Gomez M, Puzon W, Takada Y, Sanchez-Madrid F, Cabanas C. Activated conformations of very late activation integrins detected by a group of antibodies (HUTS) specific for a novel regulatory region (355-425) of the common beta 1 chain. J Biol Chem 271: 11067–11075, 1996. doi: 10.1074/jbc.271.19.11067. [DOI] [PubMed] [Google Scholar]
- 57.Mould AP, Barton SJ, Askari JA, McEwan PA, Buckley PA, Craig SE, Humphries MJ. Conformational changes in the integrin beta A domain provide a mechanism for signal transduction via hybrid domain movement. J Biol Chem 278: 17028–17035, 2003. doi: 10.1074/jbc.M213139200. [DOI] [PubMed] [Google Scholar]
- 58.Critchley DR. Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Annu Rev Biophys 38: 235–254, 2009. doi: 10.1146/annurev.biophys.050708.133744. [DOI] [PubMed] [Google Scholar]
- 59.Klapholz B, Brown NH. Talin - the master of integrin adhesions. J Cell Sci 130: 2435–2446, 2017. doi: 10.1242/jcs.190991. [DOI] [PubMed] [Google Scholar]
- 60.Bate N, Gingras AR, Bachir A, Horwitz R, Ye F, Patel B, Goult BT, Critchley DR. Talin contains a C-terminal calpain2 cleavage site important in focal adhesion dynamics. PLoS One 7: e34461, 2012. doi: 10.1371/journal.pone.0034461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, Huttenlocher A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat Cell Biol 6: 977–983, 2004. doi: 10.1038/ncb1175. [DOI] [PubMed] [Google Scholar]
- 62.Legate KR, Montañez E, Kudlacek O, Füssler R. ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol 7: 20–31, 2006. doi: 10.1038/nrm1789. [DOI] [PubMed] [Google Scholar]
- 63.Stiegler AL, Draheim KM, Li X, Chayen NE, Calderwood DA, Boggon TJ. Structural basis for paxillin binding and focal adhesion targeting of β-parvin. J Biol Chem 287: 32566–32577, 2012. doi: 10.1074/jbc.M112.367342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Calderwood DA, Campbell ID, Critchley DR. Talins and kindlins: partners in integrin-mediated adhesion. Nat Rev Mol Cell Biol 14: 503–517, 2013. doi: 10.1038/nrm3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bakolitsa C, Cohen DM, Bankston LA, Bobkov AA, Cadwell GW, Jennings L, Critchley DR, Craig SW, Liddington RC. Structural basis for vinculin activation at sites of cell adhesion. Nature 430: 583–586, 2004. doi: 10.1038/nature02610. [DOI] [PubMed] [Google Scholar]
- 66.Chen H, Choudhury DM, Craig SW. Coincidence of actin filaments and talin is required to activate vinculin. J Biol Chem 281: 40389–40398, 2006. doi: 10.1074/jbc.M607324200. [DOI] [PubMed] [Google Scholar]
- 67.Cohen DM, Kutscher B, Chen H, Murphy DB, Craig SW. A conformational switch in vinculin drives formation and dynamics of a talin-vinculin complex at focal adhesions. J Biol Chem 281: 16006–16015, 2006. doi: 10.1074/jbc.M600738200. [DOI] [PubMed] [Google Scholar]
- 68.Johnson RP, Craig SW. An intramolecular association between the head and tail domains of vinculin modulates talin binding. J Biol Chem 269: 12611–12619, 1994. doi: 10.1016/S0021-9258(18)99920-5. [DOI] [PubMed] [Google Scholar]
- 69.Kappert K, Furundzija V, Fritzsche J, Margeta C, Kruger J, Meyborg H, Fleck E, Stawowy P. Integrin cleavage regulates bidirectional signalling in vascular smooth muscle cells. Thromb Haemost 103: 556–563, 2010. doi: 10.1160/TH09-07-0478. [DOI] [PubMed] [Google Scholar]
- 70.Oh J, Barve M, Matthews CM, Koon EC, Heffernan TP, Fine B, Grosen E, Bergman MK, Fleming EL, DeMars LR, West L, Spitz DL, Goodman H, Hancock KC, Wallraven G, Kumar P, Bognar E, Manning L, Pappen BO, Adams N, Senzer N, Nemunaitis J. Phase II study of Vigil(R) DNA engineered immunotherapy as maintenance in advanced stage ovarian cancer. Gynecol Oncol 143: 504–510, 2016. doi: 10.1016/j.ygyno.2016.09.018. [DOI] [PubMed] [Google Scholar]
- 71.Rocconi RP, Grosen EA, Ghamande SA, Chan JK, Barve MA, Oh J, Tewari D, Morris PC, Stevens EE, Bottsford-Miller JN, Tang M, Aaron P, Stanbery L, Horvath S, Wallraven G, Bognar E, Manning L, Nemunaitis J, Shanahan D, Slomovitz BM, Herzog TJ, Monk BJ, Coleman RL. Gemogenovatucel-T (Vigil) immunotherapy as maintenance in frontline stage III/IV ovarian cancer (VITAL): a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Oncol 21: 1661–1672, 2020. doi: 10.1016/S1470-2045(20)30533-7. [DOI] [PubMed] [Google Scholar]