

PD-L1 plays an important role in inhibiting T-cell activity and driving tumor cell escape from immune surveillance by binding its ligand, PD-1, on T cells.1 Several intracellular and extracellular factors, such as interferon-γ (IFN-γ), MYC, transforming growth factor β, and miR-200, may modulate PD-L1 expression by transcriptional and posttranscriptional mechanisms.2–5 Here we discovered that ENO1, a novel protein, is involved in the regulation of PD-L1. ENO1 could destabilize PD-L1 and decrease its expression by promoting PD-L1 ubiquitination and subsequent proteasomal degradation in lung cancer cells. A lung cancer tissue microarray also showed that the expression of ENO1 was negatively correlated with PD-L1. Furthermore, overexpression of ENO1 sensitized tumor cells to specific T-cell killing and increased T-cell-mediated antitumor immunity in syngeneic mouse models.
In this study, we first used immunoprecipitation coupled with mass spectrometry (IP-MS) to seek new regulators of PD-L1 expression. Coomassie blue staining of SDS-polyacrylamide gel electrophoresis revealed an ~50 kDa band in V5-bound immunoprecipitates (Supplementary Fig. S1) and MS identified a sequence consisting of ten consecutive amino acids from ENO1 in the band (Supplementary Fig. S2). A co-IP experiment proved that ENO1 interacted with PD-L1 in H1299 and SPC-A-1 cells with increased PD-L1 and ENO1 expression (Fig. 1A and Supplementary Fig. S3). Then, we detected whether ENO1 protein accumulation contributes to modulating the expression of PD-L1. As shown in Fig. 1B, ectopic expression of ENO1 greatly decreased the PD-L1 protein level in a dose-dependent manner. Knockdown of ENO1 induced an increase in PD-L1 expression (Fig. 1C). Ectopic expression or knockdown of ENO1 also significantly affected the cell-surface PD-L1 protein level, as shown by fluorescence-activated cell sorting (Fig. 1D, E). To recapitulate the above results in human cancer patients for future clinical application, we performed immunohistochemical staining of ENO1 and PD-L1 in 208 human lung cancer specimens. ENO1 and PD-L1 expression presented a strong negative correlation in the clinical samples (P < 0.001, r = −0.603) (Supplementary Fig. S4).
Fig. 1. ENO1 destabilized PD-L1 to sensitize lung cancer cells to T-cell killing and promote antitumor immunity.
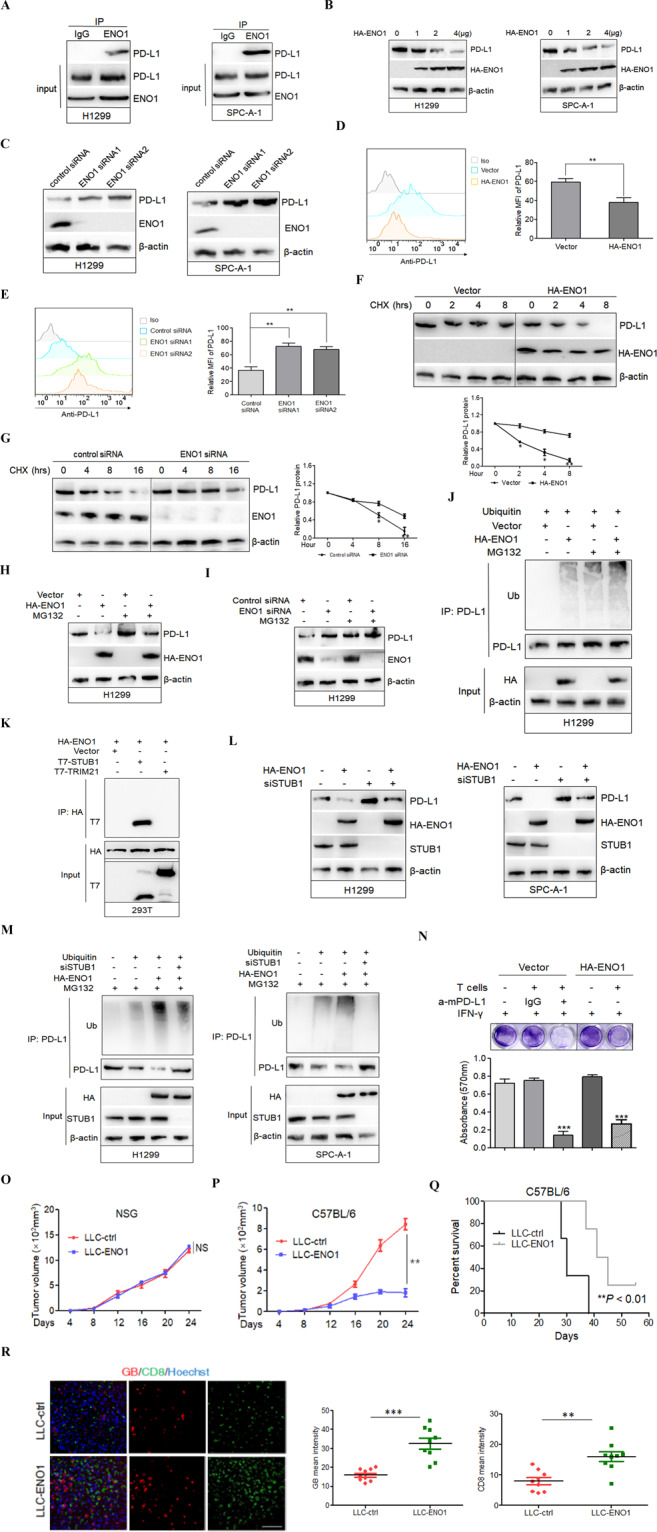
A Co-IP detected the interaction between ENO1 and PD-L1 in the lysates of natural H1299 or SPC-A-1 cells. B, C Western blot analysis showed the expression of PD-L1 in H1299 and SPC-A-1 cells transiently transfected with HA-ENO1 or ENO1 siRNA. D, E The expression of cell-surface PD-L1 in H1299 and SPC-A-1 cells transiently transfected with HA-ENO1 or ENO1 siRNA was determined by FACS (left panel). Iso, isotype control. Panels on the right show relative protein amounts in different groups. Error bars represent ±SD of triplicate experiments. The two-tailed Student’s t-test was used. **P < 0.01. F, G A CHX chase experiment was used to detect the protein stability of PD-L1 in H1299 and SPC-A-1 cells transiently transfected with HA-ENO1 (upper panel) or ENO1 siRNA (left panel). PD-L1 expression levels were quantified by densitometric analysis (TotalLab 2.01) and statistically analyzed from three independent experiments and are presented in the lower panel or right panel. *P < 0.05; **P < 0.01. H, I H1299 cells were transfected with HA-ENO1 vector or ENO1 siRNA and treated with MG132 or dimethyl sulfoxide (DMSO) for another 5 h. Western blot analysis of PD-L1 expression. J H1299 cells were transiently transfected with the indicated constructs. PD-L1 was immunoprecipitated and subjected to a ubiquitination assay. Ub indicates ubiquitin modification. K Co-IP detected the interaction between ENO1 and STUB1 or TRIM21 in 293T cells transiently transfected with the HA-ENO1 plasmid and the T7-STUB1 or T7-TRIM21 plasmid. L Western blot analysis of PD-L1 expression in STUB1-knockdown H1299 or SPC-A-1 cells with or without HA-ENO1 transfection. M H1299 or SPC-A-1 cells were transiently transfected with the indicated constructs. PD-L1 was immunoprecipitated and subjected to a ubiquitination assay. N Top, T-cell-mediated tumor cell-killing assay in LLC cells stably transfected with empty vector or HA-mENO1 vector as indicated. Bottom, quantification of cell viability. Data represent the mean ± SD of three independent experiments, ***P < 0.001. O, P Tumor growth of LLC cells stably transfected with empty or HA-mENO1 vector (as LLC-ctrl and LLC-ENO1) in NSG mice and C57BL/6 mice. Tumor volumes were measured at the indicated time points (n = 6 mice per group). Data represent mean ± SD. NS denotes no significance. **P < 0.01. Q Survival of C57BL/6 mice bearing LLC-ctrl or LLC-ENO1 tumors (n = 6 mice per group). **P < 0.01 by log-rank test. R Left, representative immunostaining images of CD8 (a CTL marker) and granzyme B (GB, showing T-cell activity) in LLC-ctrl or LLC-ENO1 tumor masses. Scale bar, 200 μm. Hoechst was used for nuclear counterstaining. Right, quantification of CD8 and GB signals by ImageJ. Data represent the mean ± SD n = 9. Three tissue slides per tumor, three mice per group. **P < 0.01. ***P < 0.001.
To determine whether downregulation of the PD-L1 protein by ENO1 occurs at the transcriptional level, real-time PCR was performed. The results showed that ENO1 transfection had no effect on PD-L1 mRNA levels in H1299 cells (Supplementary Fig. S5), indicating that ENO1 regulates PD-L1 expression mainly at the posttranscriptional level. Therefore, we detected the impact of ENO1 on PD-L1 protein stability. As shown in Fig. 1F, G, the half-life of PD-L1 was remarkably shorter in HA-ENO1-transfected H1299 cells but largely extended in ENO1 small interfering RNA-transfected H1299 cells, as shown by a chase experiment with the protein synthesis inhibitor cycloheximide. These results imply that ENO1 could promote the degradation of PD-L1 in vitro. Then, we treated H1299 cells with the proteasome inhibitor MG132 and found that altered PD-L1 levels in cells with ENO1 overexpression or depletion could be restored by incubation with MG132 (Fig. 1H, I), suggesting that ENO1 mediates the proteasomal degradation of PD-L1. Ubiquitination is a key process that leads to the proteasomal degradation of cellular proteins.6 A signal for polyubiquitinated PD-L1 was detected upon ENO1 transfection by ubiquitination ladder assay; moreover, the ubiquitination of PD-L1 was further enhanced in the presence of MG132 (Fig. 1J). These results demonstrate that ENO1 downregulates PD-L1 expression by promoting its ubiquitination and subsequent proteasome-mediated degradation.
Three types of enzymes are required for the ubiquitination process: ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), and ubiquitin ligases (E3s), among which only E3s define the target specificity of the ubiquitination reaction.7 To further investigate the mechanism of ENO1-induced PD-L1 proteasomal degradation, we identified STUB1 and TRIM21 as putative E3 ligases for PD-L1 via MS (Supplementary Fig. S6A, B). We readily detected PD-L1 associated with STUB1 and TRIM21 in lysates by co-IP experiments (Supplementary Fig. S7). However, half-life analysis indicated that the overexpression of STUB1 but not TRIM21 destabilized PD-L1 expression (Supplementary Fig. S8), suggesting that STUB1 enzyme activity is involved in PD-L1 destabilization. Consistently, downregulation of STUB1 increased PD-L1 expression and the PD-L1 protein half-life (Supplementary Fig. S9). Moreover, STUB1 could ubiquitinate PD-L1 in a dose-dependent manner (Supplementary Fig. S10). The interaction between STUB1 and ENO1 was also observed in transfected 293T cells (Fig. 1K). The blockade of STUB1 expression inhibited PD-L1 degradation induced by ENO1 in H1299 and SPC-A-1 cells (Fig. 1L). In addition, a signal for polyubiquitinated PD-L1 was detected upon ENO1 induction, while ubiquitin could no longer be detected or was detected at a lower level after STUB1 was knocked down (Fig. 1M). Collectively, these results verify STUB1 as an E3 ligase that participates in the ENO1-mediated proteasomal degradation of PD-L1.
Next, we isolated CD8+ T cells from the mouse spleen and performed a T-cell killing assay. As shown in Fig. 1N, LLC cells stably transfected with the HA-mENO1 plasmid, which exhibited reduced mPD-L1 cell surface expression upon IFN-γ treatment compared with that in the control (Supplementary Fig. S11), were more sensitive to T-cell killing, results that were similar to those following mPD-L1 antibody blockade. To test the possibility that ENO1 inhibits tumor growth by activating CD8+ cytotoxic T lymphocytes (CTLs), a major effector of antitumor immunity that eliminates cancer cells by secreting granzyme B (GB),8 we injected immunodeficient NSG (NOD-Prkdcscid Il2rgnull) and immunocompetent C57BL/6 mice with control cells or cells stably transfected with HA-mENO1 showing reduced mPD-L1 expression (Supplementary Fig. S12). The results revealed that ENO1 significantly reduced tumor growth (Fig. 1P) and increased survival (Fig. 1Q) of the immunocompetent C57BL/6 mice but had no effects on tumor growth in the NSG mice (Fig. 1O). We also analyzed the CD8+ CTL population and CTL activity using GB release as an indicator by staining tumor sections. ENO1 in tumors derived from immunocompetent C57BL/6 mice markedly increased the CD8+ CTL population and the amount of GB release compared with those in the controls (Fig. 1R). These data suggested that increased levels of ENO1 resulted in decreased levels of PD-L1, which enhanced CD8+ CTL activation, promoting killing ability and delaying tumor growth; thus, the LLC-ENO1 mice lived longer. In line with the results of survival analysis of the C57BL/6 mice, low ENO1 expression in 208 human lung cancer tissues appeared to correlate with poor cancer patient survival (Supplementary Fig. S13, left panel). Receiver operating characteristic analysis revealed that ENO1 expression showed good prognostic value [area under curve 0.747, 95% confidence interval 0.674–0.820] (Supplementary Fig. S13, right panel).
In summary, our data reveal the role of ENO1 in decreasing PD-L1 stability and efficiently boosting antitumor immunity, suggesting a new immune checkpoint-blockade strategy for lung cancer.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Fund (81972617 and 81772948).
Author contributions
C.Y.Z. and D.G. designed the experiments and wrote the paper. C.Y.Z., K.P.Z., and J.G. performed the experiments. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Chunyi Zhang, Kunpeng Zhang.
Contributor Information
Chunyi Zhang, Email: cyzhang@fudan.edu.cn.
Di Ge, Email: ge.di@zs-hospital.sh.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41423-021-00710-y.
References
- 1.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016;8:328rv324. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casey SC, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–231. doi: 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee SJ, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274) FEBS Lett. 2006;580:755–762. doi: 10.1016/j.febslet.2005.12.093. [DOI] [PubMed] [Google Scholar]
- 4.Mariathasan S, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noman MZ, et al. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology. 2017;6:e1263412. doi: 10.1080/2162402X.2016.1263412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hochstrasser M. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 7.Zheng N, Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu. Rev. Biochem. 2017;86:129–157. doi: 10.1146/annurev-biochem-060815-014922. [DOI] [PubMed] [Google Scholar]
- 8.Lim SO, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–939. doi: 10.1016/j.ccell.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.