

Abstract
Prolongation of the cardiac action potential (AP) and early after depolarizations (EADs) are electrical anomalies of cardiomyocytes that can lead to lethal arrhythmias and are potential liabilities for existing drugs and drug candidates in development. For example, long QT syndrome‐3 (LQTS3) is caused by mutations in the Nav1.5 sodium channel that debilitate channel inactivation and cause arrhythmias. We tested the hypothesis that a useful drug (i.e., mexiletine) with potential liabilities (i.e., potassium channel inhibition and adverse reactions) could be re‐engineered by dynamic medicinal chemistry to afford a new drug candidate with greater efficacy and less toxicity. Human cardiomyocytes were generated from LQTS3 patient‐derived induced pluripotent stem cells (hIPSCs) and normal hIPSCs to determine beneficial (on‐target) and detrimental effects (off‐target) of mexiletine and synthetic analogs, respectively. The approach combined “drug discovery” and "hit to lead” refinement and showed that iterations of medicinal chemistry and physiological testing afforded optimized compound 22. Compared to mexiletine, compound 22 showed a 1.85‐fold greater AUC and no detectable CNS toxicity at 100 mg/kg. In vitro hepatic metabolism studies showed that 22 was metabolized via cytochrome P‐450, as previously shown, and by the flavin‐containing monooxygenase (FMO). Deuterated‐22 showed decreased metabolism and showed acceptable cardiovascular and physicochemical properties.
Keywords: arrhythmias, deuterated phenyl mexiletines, human‐induced pluripotent stem cells‐derived cardiomyocytes, metabolism, mexiletine, phenyl mexiletine, QT prolongation
Scheme for identification of Lead compounds.
![]()
Abbreviations
- APD
action potential duration
- CM
cardiomyocytes
- CNS
central nervous system
- CYP
cytochrome P‐450
- DADs
delayed after depolarizations
- DETAPAC
diethylenetriaminepentaacetic acid
- DPT
dimethylaminoalkyl phenothiazine
- EAD
early after depolarizations
- FMO
flavin‐containing monooxygenase
- hERG
Ether‐à‐go‐go‐R related gene
- hIPSCs
human‐induced pluripotent stem cells
- I NaP
peak sodium current
- LQTS3, I NaL
, late sodium current, Long QT Syndrome‐3
- MAO
monoamine oxidase
- MMI
N‐methyl‐mercaptoimidazole
- PK
pharmacokinetics
- PVT
polymorphic ventricular tachycardia
- TAO
troleandomycin
- TdP
Torsades de Pointes
- VT
ventricular tachycardia
1. INTRODUCTION
Human‐induced pluripotent stem cell (hiPSC)‐derived cardiomyocytes provide a robust cell‐based way to study disease in a dish or drug development in a dish. 1 Drug candidates examined in LQTS3 patient‐derived hiPSC cardiomyocytes (pathogenic on‐target effects) can be compared side by side with normal hiPSC‐derived cardiomyocytes (off‐target effects) 2 to identify more viable drug candidates. LQTS3 patient‐derived hiPSC cardiomyocytes represent a pathological condition and can illustrate the effect of compounds for on‐target effects. Normal hiPSC‐derived cardiomyocytes can show the effects of compounds on off‐target endpoints. Thus, patient hiPSC‐derived cardiomyocytes can be used in dynamic medicinal chemistry studies to improve drug candidates by examining patient hiPSC‐derived and normal cardiomyocytes. Reengineering existing drugs that harbor some liability to improve a drug could lead to promising new drug candidates. Mexiletine (Figure 1) is one such example of a drug with liabilities that could be reengineered and that is the subject of this work.
FIGURE 1.

Chemical structure of mexiletine hydrochloride and phenyl mexiletine hydrochloride
Numerous drugs and drug candidates have been withdrawn from the market or development because of QT prolongation (Figure 2) and ventricular tachycardia (VT) and torsades de pointes (TdP). 3 To determine potency to cause QT prolongation, drug or drug candidates can be tested to inhibit the human cardiac potassium channel (hERG). 4 This does not completely predict QT prolongation because drugs can inhibit hERG without causing VT or TdP. 5 , 6 Use of human cardiomyocytes represents a non‐clinical approach to antiarrhythmic drug development.
FIGURE 2.

Representative shows sodium current for peak, early, and late sodium channel action (top). Representative ECG trace shows the Q, R, S, and T waves for a normal individual (middle trace in black); red trace shows a T wave for a LQTS3 patient with a delayed action potential duration. In response to mexiletine, the T wave for the LQTS3 patient reverts to a normal action potential duration (black trace). Representative potential of the membrane shows normal late sodium channel (black) and abnormal LQTS3 late sodium channel (red, lower trace). Note, the axis for membrane voltage is an order of magnitude greater than that depicted by the ECG
Mexiletine is an antiarrhythmic drug that inhibits muscle and neuronal sodium channels. 7 Cardiac ventricular action potential (AP) is started by opening the Nav1.5 sodium channel. This causes an initial large increase in inward Na+ current (i.e., peak Na+ current, I NaP) (Figure 2). Blockade of voltage‐gated sodium channels that inhibit generation and propagation of APs can be antiarrhythmic and prevent pathological firing in cardiac tissue. 8 In normal individuals, sodium channels rapidly inactivate with depolarization. In depolarization of cardiomyocytes, sodium channels open briefly and allow influx of sodium. This generates a large inward sodium current (I NaP). Next, upstroke of the AP and rapid deactivation of the late portion of the late sodium current (I NaL) occur. 9 Normally, I NaL is only a small part (i.e., ~0.1%) of the AP, but the amplitude of I NaL can be much greater in cardiomyocytes from LQTS3 patients with mutations in genes encoding a pathogenic cardiac sodium channel SCN5A. In individuals with cardiac disease, a small percentage of sodium channels do not inactivate and cause an I NaL current that can protract the ventricular AP. This is manifested by a lengthening of the QT interval on the ECG (Figure 2). Long QT syndrome (LQTS3) is caused by mutations in the Nav1.5 sodium channel (encoded by SCN5A) that debilitate channel inactivation and accelerate recovery from the inactivated state. 10 , 11 , 12 The increase in I NaL (Figure 2) opposes repolarization and prolongs the AP. 10 , 13 Thus, effects of SCN5A sodium channel mutations prolong the QT interval of the electrocardiogram. LQTS3 manifests in events of polymorphic ventricular tachycardia (PVT). In extreme cases, this can result in sudden cardiac death.
Mexiletine (Figure 1) has been shown to suppress excessive late I Na and is used to shorten the QTc interval and decrease arrhythmias in LQTS3 patients 14 and decrease risk of VT and ventricular fibrillation (VF). 15 Mexiletine is useful for treatment of LQTS3 16 , 17 but mexiletine also prolongs the cardiac AP mediated in part by inhibition of hERG and modulation of other undefined targets. Thus, concern about proarrhythmia has restricted its use, although cardiologists predetermine a safe and efficacious dose. However, mexiletine has additional liabilities. The FDA Approved Label states that instances of severe liver injury and blood dyscrasias (i.e., leukopenia or agranulocytosis) and other adverse reactions including reversible gastrointestinal and nervous system problems have been reported after mexiletine treatment. Mexiletine also has a relatively short half‐life (i.e., t 1/2 α‐phase 3–12 min and β‐phase 6–12 h 18 ) that necessitates multiple doses per day. Greater doses of mexiletine produce side effects in the central nervous system. 19
Mexiletine is metabolized by hydroxylation, deamination, and glucuronidation, although the molecular details are not completely clear 20 (Figure 3). Only about 10% of a dose is recovered as unchanged mexiletine. Mexiletine possesses a center of chirality and is subject to stereoselective binding to sodium channels 21 , 22 and stereoselective metabolism. 20 Sodium channel binding and metabolism favor the (R)‐enantiomer over the S‐enantiomer. (R)‐Mexiletine is about twofold more potent than (S)‐mexiletine to bind to cardiac sodium channels. 23 , 24 (R)‐Mexiletine is metabolized more rapidly than the (S)‐enantiomer. 25 Generally, metabolites of mexiletine are less potent than the parent drug with the possible exception of the meta‐hydroxylation metabolite. 20 In the presence of hepatic microsomes, (R)‐mexiletine largely undergoes aliphatic hydroxylation and (S)‐mexiletine is largely para‐hydroxylated. 26
FIGURE 3.

Representation of the metabolism of mexiletine
The purpose of this work was to show re‐engineering mexiletine to phenyl mexiletine analogs and then deuterated phenyl mexiletine analogs (Figure 4) and testing them against LQTS3 patient iPSC‐derived cardiomyocytes could lead to removal of a deleterious in vitro arrhythmia phenotype. Another purpose was to examine whether deuterated phenyl mexiletine analogs improved pharmaceutical properties. We synthesized and tested deuterated analogs because we observed a previously unreported metabolism: mexiletine and phenyl mexiletine analogs were metabolized by human flavin‐containing monooxygenase. Accordingly, we synthesized deuterated phenyl mexiletine analogs at the alpha carbon to decrease metabolism and improve pharmaceutical properties. To confirm in vitro observations of decreased metabolism for deuterated phenyl mexiletine analogs, selected compounds were administered to rats and pharmacokinetic studies were conducted. Compared to mexiletine, deuterated phenyl mexiletine analog (R)‐22 showed a 2.7‐fold increase in bioavailability. Administration of deuterated phenyl mexiletine analogs to rats showed significant improvement in pharmaceutical properties and confirmed that dynamic medicinal chemistry 27 and drug development can be used even with complex biological matrices such as human cardiomyocytes to elaborate promising drug candidates.
FIGURE 4.
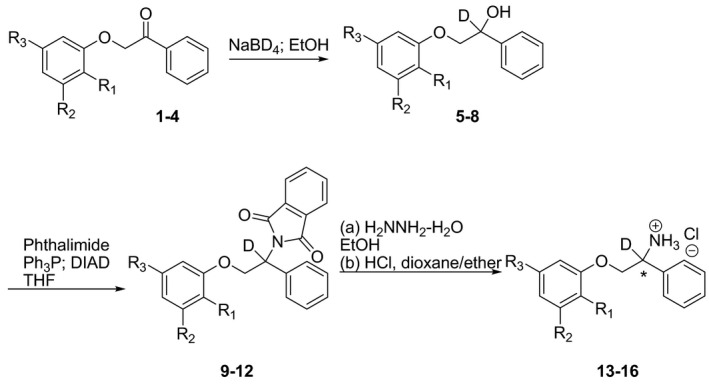
Synthetic scheme for the synthesis of deuterated phenyl mexiletines. The center of chirality is alpha to the amine
2. MATERIALS AND METHODS
2.1. General
Starting materials, reagents and solvents were purchased in the highest purity available from commercial suppliers and used as received. Mexiletine and (R)‐ and (S)‐mexiletine were purchased from Toronto Research. Mexiletine and synthetic phenyl mexiletine analogs were prepared and tested as hydrochloride salts unless otherwise noted. Hydrochloride salts were prepared by dissolution of the appropriate compound in a minimum amount of dichloromethane and addition of excess 2 M HCl in dioxane/ether. Phosphate buffered saline (PBS) was purchased from Life Technologies. Fluorescence was determined using a Tecan SPECTRAFluor Plus plate reader (Tecan). Luminescence was recorded on a Wallac Victor plate reader (PerkinElmer Inc.).
2.2. Biology experimental section
2.2.1. Cell culture of differentiated hiPSC‐CMs
MyCell (one differentiation batch from an LQTS3 patient) and iCell (multiple differentiation batches from a healthy individual) cardiomyocytes (Cellular Dynamics International) were thawed and placed in pre‐coated 384 plate wells (Greiner Bio‐One) with 0.1% (w/v) gelatin (Stem Cell Technologies) as described previously 28 and placed in a 37℃ 5% CO2 incubator. After 24 h, plating media were diluted by adding 80 µl of iCell Cardiomyocyte Maintenance Media (iCCMM), supplemented with 5 mM D‐glucose, for a final volume of 100 µl/well. The plates were incubated at 37℃ 5% CO2 for 48 h. Media were exchanged every other day by removing 50 µl of media and adding 50 µl of fresh iCCMM for 14 days prior to imaging. hiPSCs were dissociated using 0.5 mM EDTA (ThermoFisher Scientific) in PBS without CaCl2 or MgCl2 (Corning) for 7 min at room temperature.
2.2.2. Preparation of VF2.1.Cl loading solution and automated image acquisition
VF2.1.Cl dye used was synthesized as described previously 29 (Fluovolt, ThermoFisher). One µl of 2 mM VF2.1.Cl in DMSO was mixed with 1 µl of 10% pluronic F127 (diluted in 1.7 ml water) by agitating and centrifuging three times. Separately, Hoechst 33258 was diluted into Tyrode's solution (136 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, pH 7.4) to a concentration of 4 µg/ml. One ml of Hoechst/Tyrode's solution was added to a 1.7 ml VF2.1.Cl/Pluronic F127 mixture and vortexed for 10 s. Each test compound was diluted in Tyrode's solution to a 2x concentrated stock and warmed to 37℃ using a dry heat block prior to addition to cells. After rinsing to remove Tyrode/dye solution, the dissociated cells were placed back in a 37℃ 5% CO2 incubator for 10 min to recover. After recovery, 50 µl of solution was removed and 50 µl of 2x test compound stock was added to a well and incubated at 37℃ and 5% CO2 for 5 min before image acquisition. Time‐series images were acquired automatically using an IC200 KIC instrument (Vala Sciences) at an acquisition frequency of 100 Hz for a duration of 6.5 s or 33 Hz for 20 s, with excitation wavelength of 485/20 nm and emission filter 525/30 nm using a 0.75 NA 20x Nikon Apo VC objective. A single image of the Hoechst/Tyrode solution was acquired after the time series. Optimized dye loading and imaging conditions were replicated using both a different high content imager, the ImageXpress Micro XLS platform (Molecular Devices) and hiPSC‐CMs produced by a novel differentiation protocol. 30
2.2.3. Image analysis, physiological parameter calculations, and data analysis
Image analysis and physiological parameter calculations were conducted using Cyteseer (Vala Sciences) as previously described. 31 , 32 The output images from the IC200 KIC were loaded into Cyteseer and a whole‐well cardiac time‐series algorithm was executed on the image files. Physiological parameters (i.e., beat rate, normalized area under the peak trace [normalized peak integral], and APD25, APD50, APD75, and APD90) were automatically calculated for each time series. EADs were quantified automatically by identifying peaks following a local minimum above a user‐defined threshold above the diastolic interval minimum. Data tables were analyzed using Microsoft Excel 2013 and dose–response curves were calculated using GraphPad Prism 7 software (Prism).
Chemical stability of mexiletine and analogs at various temperatures and pH
A typical incubation for chemical stability contained 50–100 µM of test compound prepared in 50 mM PBS buffer (pH 3.0 or 7.4) with 1% ethanol. Test compounds examined for chemical stability were incubated at 37℃. An aliquot from each incubation was taken at various times and injected into an RP‐HPLC. Samples were run on a Hitachi D‐7000 HPLC system (Hitachi High Tech) using a L‐7100 analytical pump, L‐7400 UV‐Visible variable wavelength detector, and L‐7600 automatic sample injector. A Gemini C18 column (250 × 4.6 mm, 5 µm particle size; Phenomenex) with a C18 guard column (Phenomenex) was used for chromatographic separation of mexiletine and analogs. The mobile phase used was an isocratic system using 75% water (0.05% TFA) and 25% acetonitrile (0.05% TFA) with a flow rate of 1.25 ml/min and monitored at 275 nM. Disappearance of the analyte was monitored over time. A plot of the area under the curve for the normalized analyte versus time‐afforded half‐life values.
Metabolic stability studies in the presence of human liver S‐9
Human and mouse liver S‐9, highly purified cytochrome P‐450 (CYP)‐3A4, ‐3A5, or ‐2D6, or highly purified human FMO1 and FMO3 were obtained from BD Biosciences. NADP+ and other components of the NADPH‐generating system were from Sigma Aldrich. A typical incubation contained human liver S‐9 (0.5 mg of protein), or FMO (15 µg protein) or CYP (3 pmol protein), 6 mM potassium phosphate buffer (pH 7.4 or pH 8.4 for FMO), 50 µM test compound, an NADPH‐generating system consisting of 0.5 mM NADP+, 0.5 mM glucose‐6‐phosphate, 5 U/ml glucose‐6‐phosphate dehydrogenase, 1 mg/ml diethylenetriaminepentaacetic acid (DETAPAC), and 3 mM MgCl2 in a final incubation volume of 0.25 ml. Incubations were run for 0, 7, 15, 30, and 60 min with constant shaking at 37℃ in a water bath and were terminated by the addition of 0.75 ml of cold acetonitrile. After centrifugation at 2500 g for 5 min, the organic fraction was collected and solvent was removed with a stream of argon. The residue was reconstituted in 125 µl of MeOH and 125 µl H2O, mixed thoroughly, centrifuged at 13 000 rpm for 5 min and analyzed by high‐performance liquid chromatography as above. Selected samples were run on LCMS and confirmed observations made with HPLC. Disappearance of analyte was monitored over time. A plot of the area under the curve for the normalized analyte versus time‐afforded rates of disappearance.
Pharmacokinetics
Animal work was conducted in accordance with the Guide for Care and Use of Laboratory Animals as adopted by the National Institutes of Health. Formal approval was obtained from the IACUC of HBRI. For pharmacokinetic studies, cannulated male Sprague–Dawley rats (Charles River) weighing 250–290 g at the time of the experiment were housed individually. Except during testing, animals were given free access to food and water. Animals administered compounds via the oral route were deprived of food 10 h before the experiment. For determination of pharmacokinetic parameters, 2–4 jugular catheterized male Sprague–Dawley rats were administered test compound by oral (25 mg/kg) or by intravenous (I.V.) (5 mg/kg) routes of administration. At the appropriate time, blood was obtained, serum was separated by centrifugation, and an aliquot of serum was combined with an internal standard and sodium bicarbonate (20 mg) and extracted with 0.5 ml of acetonitrile. Samples were analyzed by an Agilent LCMS (Agilent) by the method described above. The area under the curve for the analyte was compared to a standard curve and expressed as ng of analyte/ml of serum. Pharmacokinetic parameters were determined with a WinNonlin‐Pro program (Pharsight, Inc.).
Safety studies
Formal approval was obtained from the IACUC of HBRI. Balb‐c mice (Charles River) weighing 22–24 g at the time of the experiment were housed in groups of four. Except during testing, animals were given free access to food and water. Animals were administered compounds as hydrochloride salts via the i.p. route (PEG water) and monitored for signs of central nervous system toxicity or behavioral pharmacology for 24 h.
2.3. Statistical analysis
Statistical analyses and graphical plots were done using GraphPad Prism. Concentration‐dependent IC50 curves were fitted using a log (inhibitor) versus response—variable slope (four parameters). Other values were calculated using non‐linear regression analysis of the mean and standard deviation (SD) of at least triplicate samples for each biological assay. Student t tests were used to calculate statistical significance for comparison between two groups. p‐values <.05 were considered significant. Spearman's rank‐correlation test was used to analyze the correlation between two ranked variables. The strength and direction of association between two ranked variables were defined by Spearman's rank correlation coefficient (ρ). p‐value <.05 was considered a significant correlation.
For the data of the toxicity studies of mexiletine and analogs, the Student t test was used for statistical evaluation of the difference between two means. Multiple means were analyzed by a randomized one‐way analysis of variance. When the analysis indicated that a significant difference existed, the means of each group were compared by the Student–Newman–Keuls test. The level of significance chosen was p < .05. For the study of mexiletine or analogs, Fisher's exact probability test was used in the case of quantal data.
2.4. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, 33 , 34 , 35 and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 36
3. RESULTS
3.1. Chemistry
Initially, structure–activity studies of compounds 19–22 were done to examine replacement of metabolically labile groups (i.e., ortho‐methyl substituents of mexiletine) with metabolically inert groups (i.e., ortho‐CF3 substituents). The results showed that replacement of ortho‐methyl groups with CF3 groups decreased metabolism and increased bioavailability. 37 We next examined the effect of alpha‐amino deuteration of mexiletine and phenyl mexiletine on pharmacological properties. Deuterated phenyl mexiletine analogs were prepared using the synthetic routes shown in Figure 4. A three‐step process was used starting from the requisite ketone. The procedure to synthesize the ketones was adapted from the literature. 38 In a typical synthesis, ketones 1–4 were treated with sodium borodeuteride (>99% deuterium) in ethanol at room temperature to generate the deutero alcohols 5–8. Deuterated alcohols were treated with phthalimide, triphenylphosphine, and diisopropyl azodicarboxylate in THF to afford the phthalimide derivatives 9–12. Treatment of the deuterated phthalimide derivatives with hydrazine hydrate in refluxing ethanol yielded the amines in free base form that were converted into their corresponding hydrochloride salts 13–16 by treatment with hydrogen chloride in dioxane/ether (Table 1). The compounds were fully characterized by spectral means. Compounds 13–16 were tested in vitro and in vivo. See Supplemental Information for characterization of compounds.
TABLE 1.
Chemical structures of deutero‐ and protio‐phenyl mexiletine analogs 13–16 and 19–22
| Compd | Substitution at the alpha carbon | R1 | R2 | R3 |
|---|---|---|---|---|
| 13 | Deutero‐ | H | CF3 | CF3 |
| 14 | Deutero‐ | H | CH3 | CH3 |
| 15 | Deutero‐ | CH3 | H | H |
| 16 | Deutero‐ | CF3 | H | H |
| 19 a | Protio‐ | H | CF3 | CF3 |
| 20 a | Protio‐ | H | CH3 | CH3 |
| 21 a | Protio‐ | CH3 | H | H |
| 22 a | Protio‐ | CF3 | H | H |
Compounds were synthesized as described previously. 39
3.2. Biological results
3.2.1. Effect of deutero‐ and protio‐phenyl mexiletine analogs on human cardiomyocytes
Phenyl mexiletine analogs were re‐engineered by chemical synthesis to afford deuterated analogs with increased on‐target potency (i.e., mutated sodium channel) and decreased off‐target (i.e., normal potassium channel) effects. The effect of deuterated phenyl mexiletine analogs was examined in the presence of normal and LQTS3 patient‐derived hiPSC cardiomyocytes (Table 2). Patient‐derived cardiomyocytes were derived from a pediatric LQTS3 patient with a mutation in the gene encoding the cardiac sodium channel SCN5A (F1473C). Thus, the effects of phenyl mexiletine analogs on cardiomyocytes with a channelopathy were compared to the effects on normal cardiomyocytes. Cardiomyocytes from LQTS3 patients show a small percentage of sodium channels that do not inactivate and cause a INaL current that can protract the ventricular AP. This is manifested by a lengthening of the QT interval on the ECG (Figure 2). LQTS3 cardiomyocytes have on average an increase in I NaL of 1.2% ±0.6. Cardiomyocytes from normal individuals have no detectable increase in I NaL. Any APD difference between normal and LQTS3 cardiomyocytes is discernible only above 37 beats/min based on Bonferroni correction (p value <.05). The increase in I NaL opposes repolarization and prolongs the AP in cardiomyocytes from LQTS3 patients but not normal individuals. Thus, a comparison of the effects of test compounds on cardiomyocytes from LQTS3 patients showed the effects of SCN5A sodium channel mutations ability to prolong the QT interval of the electrocardiogram and affords a cell model for drug development. The effect of test compounds on normal cardiomyocytes provides information about cellular toxicity.
TABLE 2.
Effect of mexiletine and substituted phenyl mexiletine and deuterated phenyl mexiletine analogs on cardiovascular properties in human iPSC‐derived cardiomyocytes
| Number | R= | LQT‐3 cells | Normal cells | |||||
|---|---|---|---|---|---|---|---|---|
| Cessation dose (µM) | EC50 AP shortening a (µM) | AP fold shortening | AP shortening dose (µM) | Cessation dose (µM) | EAD dose (µM) | EC50 AP prolongation a (µM) | ||
| Mexiletine | – | 1.8 | 1.3 | 22 | ‐ b | 200 c | 20.4 | |
| Phenyl Mexiletine |

|
66 | <0.8 | 1.2 | 2.5 | 133 | – | 4 |
| Bis‐CF3 19 |

|
66 | 23.1 | 1.3 | 22 | 133 | – | – |
| Bis‐CF3‐D 13 |

|
133 | – | 1.3 | 22 | 66 | – | – |
| Bis‐CH3 20 |

|
66 | 4.1 | 1.5 | 22 | 66 | – | – |
| Bis‐CH3‐D 14 |

|
22 | 0.9 | 1.3 | 2.5 | 22 | – | – |
| o‐Me 21 |

|
66 | <0.8 | 1.6 | 22 | 133 | – | – |
| o‐Me‐D 15 |

|
22 | – | – | – | 66 | – | – |
| o‐CF3 22 |

|
66 | <0.8 | 1.8 | 22 | 66 | – | – |
| o‐CF3‐D 16 |

|
66 | – | 1.4 | 22 | 22 | – | – |
Determined with kinetic imaging cytometer assay. Dose series of optical voltage traces (6 s, 100 fps) showing action potential (AP) shortening of LQTS3 patient hiPSC‐CMs (SCN5A F1473) or dose series for prolongation in normal hiPSC‐CMs. Dose–response (n = 4) showed effects of treatment on APD75 and indicates effect on AP duration at the point of 75% decay from peak height (APD75). The dose responsiveness is highly reproducible across experiments. Values have a range 5%–7%.
The symbol “‐” denotes that the indicated effect was not observed.
EAD dose indicates the concentration at which the compound induced early after depolarizations.
To quantify the effect of chemical structural modification on cardiomyocyte response, each test compound was tested in a dose–response study (i.e., 0–200 µM, nine concentrations, in triplicate) in LQTS3 human cardiomyocytes to determine the beneficial effects, quantified as the concentration of cessation of cell beating, an EC50 for shortening the APD, a fold shortening of the APD, and a concentration that caused shortening of the action potential and EADs. These results were compared to the concentration of a test compound required for cessation of cell beating, an EC50 for shortening the APD, and fold shortening of the APD in normal cardiomyocytes. Therefore, the detrimental effect on action potential prolongation and early after depolarizations (EADs) were most apparent in healthy normal donor hiPSC cardiomyocytes. Thus, the results from studies with normal cardiomyocytes served as a control and quantified the concentration of cessation of cell beating, a concentration at which EADs occurred and an EC50 for prolonging the APD (i.e., markers for cardiomyocyte toxicity).
Normal cardiomyocytes are not prone to arrhythmias. For this study, normal cardiomyocytes were synchronously contracting monolayers. LQTS3 cardiomyocytes, on the other hand, showed a predictable prolongation of the APD. Mexiletine reversed the APD in LQTS3 cardiomyocytes but did not in normal cardiomyocytes because cardiomyocytes from normal individuals lack the disease phenotype (i.e., cardiac sodium channel SCN5A (F1473C) mutation) (Table 2). Normal cardiomyocytes were, thus, used as control cells for general toxicity assessment. The important measurement is the effect of a new chemical entity on reversing prolongation of the APD in LQTS3 cells and showing no apparent toxicity observed in normal cardiomyocytes.
For deuterated analogs (except for 14 and 15), the cessation dose was the same or greater than non‐deuterated analogs. Compound 14 showed a lower EC50 value for shortening the APD. Compared to phenyl mexiletine, fold shortening for non‐deuterated phenyl mexiletines 19–22 was 8%–50% greater. Compared to phenyl mexiletine, fold shortening for deuterated phenyl mexiletines 13–16 was 8%–17% greater. In contrast to mexiletine, EADs were not observed for any of the phenyl mexiletine analogs tested in either LQTS3 or normal human cardiomyocytes (Table 2). The results of these studies showed that deuteration of the alpha‐aryl moiety of phenyl mexiletines afforded compounds that did not cause prolongation of the APD and caused fold shortening to occur at a lower dose than for non‐deuterated compounds (Table 2).
3.3. Chemical and metabolic stability
Initially, we examined the chemical stability of phenyl mexiletines and mexiletine at pH 3 and pH 7.4 and they were stable. 37 Mexiletine and compounds 13–22 have half‐lives of >15 days and >30 days at pH 3 and pH 7.4, respectively. The hepatic metabolism of mexiletine and compound 22 by highly purified CYPs as representative candidates of the two chemotypes was examined. In agreement with the literature, 40 mexiletine was metabolized by CYP3A4, CYP3A5, and CYP2D6 (i.e., 46.2 ± 13.6, 48.8 ± 21.2, and 6 ± 2.2 nmol/min/mg of protein, respectively). Compound 22 was metabolized by CYP3A4, CYP3A5, and CYP2D6 (i.e., 15.4 ± 6.7, 42.6 ± 4.4, and 43.6 ± 8.2 nmol/min/mg of protein, respectively).
The in vitro metabolism of (R)‐ and (S)‐21 and (R)‐ and (S)‐22 by human liver microsomes + NADPH was examined by HPLC. The T 1/2 of (R)‐ and (S)‐21 was 365 and 136 min, respectively. In the presence of human liver microsomes + NADPH, the metabolism of (R)‐ and (S)‐22 was minimal and was >95% stable after 1 h. The conclusion was that 22 was more metabolically stable than 21 and there was modest stereoselective metabolism of 21 (i.e., (S)‐enantiomer being metabolized to a greater extent than (R)‐21).
Based on these data, we elected to examine the in vitro metabolism of mexiletine, substituted phenyl mexiletines, and deuterated analogs with mouse liver S‐9, FMO, and CYP3A4. S‐9 was used because it contained the widest array of mexiletine drug‐metabolizing enzymes including CYPs, FMOs, and monoamine oxidase.
As a prelude to in vivo studies, the metabolism of mexiletine was compared to deuterated mexiletine and metabolism of phenyl mexiletine was compared with deuterated phenyl mexiletine. As shown in Table 3, phenyl mexiletines with alpha‐amino deuterium showed large kinetic isotope effects of the deuterium atom on metabolism as judged by compound disappearance analyzed by HPLC. For example, compared to mexiletine, alpha deuteration of mexiletine caused a 51% and 31% decrease in metabolism by mouse and human liver S‐9, respectively. In the presence of human FMO1, compared to mexiletine, alpha deuteration of mexiletine caused a 42% decrease in metabolism. Similarly, compared to phenyl mexiletine, alpha deuteration of phenyl mexiletine caused a 44% decrease in metabolism by mouse liver S‐9. In the presence of human FMO1, compared to phenyl mexiletine, alpha deuteration of phenyl mexiletine caused an 82% decrease in metabolism. In the presence of human CYP3A4, compared to phenyl mexiletine, alpha deuteration of phenyl mexiletine caused a 34% decrease in metabolism. Based on these results, we examined the effect of deuteration of phenyl mexiletine analogs on metabolic stability in vitro.
TABLE 3.
Effect of metabolism on deuterated mexiletine and phenyl mexiletine
| Condition | Mexiletine | D c ‐mexiletine, 17 | Phenyl mexiletine | D c ‐Phenyl mexiletine, 18 |
|---|---|---|---|---|
| Rate metabolized | Rate metabolized | Rate metabolized | Rate metabolized | |
| Mouse S‐9 a | 0.67 ± 0.04 | 0.33 ± 0.01 d | 0.21 ±.01 | 0.12 ± 0.01 e |
| Human S‐9 a | 0.33 ± 0.1 | 0.23 ± 0.01 d | 0.1 ±.01 | 0.12 ± 0.02 |
| Human FMO1 a | 5.33 ± 0.08 | 3.11 ± 0.05 d | 0.73 ± 0.01 | 0.13 ± 0.01 e |
| Human CYP3A4 b | 10.2 ± 0.24 | 12.9 ± 0.6 | 5.7 ± 0.21 | 3.7 ± 0.15 e |
Quantity expressed as nmol/min/mg of protein, 0.4 mg pooled male and female S‐9 hepatic protein/incubation; n = 3. 15 µg FMO1 enzyme/incubation; incubations (88 µM) were run for 30 min with shaking at 37℃ (n = 3).
Results are in nmol of metabolism/min/mg of CYP3A4. 13.4 µg CYP3A4 enzyme/incubation; incubations were run for 30 min with shaking at 37℃ (n = 3).
D stands for deuterium.
Statistically different than mexiletine, p < .05.
Statistically different from phenyl mexiletine, p < .05.
Based on the literature, 20 we surmised deuterated compounds (i.e., deuterated compounds 13–16) possessed the required metabolic stability (i.e., in liver S‐9 possessed a T1/2 > 60 min) and chemical stability (i.e., possess a T 1/2 > 30 days, pH 7.4, 37℃) for the deuterated test compounds to be taken forward for further in vitro and in vivo testing. We tested the hepatic metabolism of deuterated compounds 13–16 in vitro.
Metabolic stability studies of mexiletine and deuterated phenyl mexiletines were conducted with mouse and human liver S‐9, FMO1, FMO3, and CYP3A4. FMO1 and FMO3 were used because they are the major functionally active FMOs present in human kidney and liver, respectively. In good agreement with the literature, 41 mexiletine was metabolized under standard conditions (Tables 3 and 4). With the exception of compound 14 in mouse S‐9, compared to mexiletine, deuterated compounds 13, 15, and 16 were metabolized to a less degree as judged by HPLC (Table 4).
TABLE 4.
Metabolic stability of unlabeled mexiletine and deuterated analogs of phenyl mexiletine
| Compound | Mouse liver S‐9 (rate metabolized) a | Human FMO1 (rate metabolized) a | Human FMO3 (rate metabolized) a | Human CYP3A4 (rate metabolized) b |
|---|---|---|---|---|
| Mexiletine | 0.43 ± 0.01 | 14.88 ± 0.15 | ND c | 7.4 ± 0.12 |
| 13 | ND c | 1.77 ± 0.12 | 1.33 ± 0.11 | 2.73 ± 0.26 |
| 14 | 1.1 ± 0.09 | 3.11 ± 0.05 | 2.88 ± 0.09 | 2.48 ± 0.12 |
| 15 | ND c | 2.22 ± 0.04 | 0.11 ± 0.01 | 0.75 ± 0.01 |
| 16 | ND c | 0.67 ± 0.02 | 1.55 ± 0.04 | 0.75 ± 0.05 |
Quantity expressed as nmol/min/mg of protein, 0.4 mg pooled male and female S‐9 protein/incubation; 15 µg FMO1 enzyme/incubation; incubations were run for 30 min with shaking at 37℃.
Results are in nmol of metabolism/min/mg of CYP3A4. 13.4 µg CYP3A4 enzyme/incubation incubations were run for 30 min with shaking at 37℃.
ND stands for not detectable.
Mexiletine and compounds 13, 14, 15, and 16 were present at 88, 45, 65, 69, and 56 µM, respectively.
As discussed above, deuterated phenyl mexiletine analogs were synthesized and tested for metabolic stability in hepatic preparations or highly purified enzymes to determine if deuteration would decrease metabolism compared to mexiletine. Compared to mexiletine, data of Table 4, below, showed that deuterated analogs 13, 14, 15, and 16 were more metabolically stable. For example, compounds 13, 14, 15, and 16 were 8.5‐, 4.8‐, 6.7‐, and 22‐fold, respectively, less metabolized by human FMO1 compared to mexiletine. Compounds 13, 14, 15, and 16 were 2.7‐, 3‐, 9.9‐, and 9.9‐fold, respectively, less metabolized by human CYP3A4 compared to mexiletine. In general, compared to mexiletine, the effect of the deuterium isotope was quite apparent for deuterated compounds 13–16 compared to non‐deuterated mexiletine.
The effect of such a pronounced effect of deuterium on metabolic stability could translate to a large effect on pharmacological response, in vivo metabolism, a decrease in clearance, greater bioavailability, and greater efficacy. This was examined for selected compounds in safety and pharmacokinetic studies below.
3.4. In vivo studies
3.4.1. Behavioral studies
Mexiletine caused seizures at elevated doses in mice (Table 5). In our hands, after administration of 30 mg/kg (R)‐mexiletine, 0/4 mice had seizures or tremors. In contrast, after a dose of 30 mg/kg (S)‐mexiletine, 4/4 mice had seizures. After administration of 100 mg/kg (R)‐mexiletine, ¾ mice had seizures but after administration of 100 mg of (S)‐mexiletine, 7/7 mice had seizures. After administration of 200 mg/kg (R)‐mexiletine to mice, 1/4 mice had seizures. In contrast, mice administered (S)‐mexiletine at 200 mg/kg showed 3/4 have seizures and one animal died. Accordingly, central nervous system (CNS) toxicity appeared to be stereoselective. (R)‐Mexiletine was judged to produce less CNS toxicity than (S)‐mexiletine in mice at the doses examined. The effect was dose dependent (i.e., CNS toxicity and death for (S)‐mexiletine at 200 mg/kg was greater than 100 mg/kg and greater than 30 mg/kg compared to (R)‐mexiletine). Thus, this small dose‐escalation study showed (R)‐mexiletine was less toxic than (S)‐mexiletine. Based on these results, the potential for CNS toxicity was examined for phenyl mexiletine analogs 19–22. No apparent toxicity or seizures were observed for racemic 19–22 administered at 30 or 100 mg/kg in mice. All mice examined (4 animals) administered 19–22 at 30 or 100 mg/kg did not show any toxicity (i.e., seizures or deaths). Both enantiomers of 22 showed no CNS toxicity, whereas 21 showed detectable toxicity (100 mg/kg) (Table 5). While not a full dose‐escalation study, nevertheless phenyl mexiletine analogs 19–22 showed negligible CNS toxicity compared to mexiletine.
TABLE 5.
Effect of (R)‐ or (S)‐Mexiletine or Mexiletine analog treatment on behavior in mice
| Compound | Dose a (mg/kg) | Seizures immobilization b /dosed |
|---|---|---|
| (R)‐Mexiletine | 30 | 0/4 |
| (S)‐Mexiletine | 30 | 4/4 c |
| (R)‐Mexiletine | 100 | 3/4 |
| (S)‐Mexiletine | 100 | 7/7 |
| (R)‐Mexiletine | 200 | 1/4 |
| (S)‐Mexiletine | 200 | 3/4 d |
| (R)‐21 | 100 | 4/4 e |
| (S)‐21 | 100 | 3/4 e |
| (R)‐22 | 100 | 0/4 |
| (S)‐22 | 100 | 0/4 |
Mexiletines were administered in saline by i.p. injection.
Cumulative behavior during the first 20 min after dosing. After 2 h, surviving animals were largely recovered.
Significantly different from (R)‐mexiletine, p = .05, Fishers exact probability test.
One animal died in the first 20 min after dosing.
No seizures, only immobilization.
As described above, we showed that phenyl mexiletine compounds were sufficiently chemically and metabolically stable for in vivo studies and selected phenyl mexiletine compounds (i.e., 22 enantiomers) that showed no apparent in vivo CNS toxicity at the doses examined. On that basis, and in view of the lack of toxicity and in vitro potency of 22 (Table 2), we selected compound 22 for bioavailability studies.
3.5. Pharmacokinetic studies
Based on the data shown above, compound 22 was selected for pharmacokinetic (PK) studies. Representative examples are shown in Figure 5. Mexiletine and (S)‐ and (R)‐ and deutero‐22 were evaluated in male rats (N = 2–4 animals/dose) (Table 6). Animals were administered compounds by intravenous (5 mg/kg) or oral (25 mg/kg) routes of administration and plasma was collected over about 24 h. Doses were selected on the basis of pediatric dose of mexiletine (i.e., 15–25 mg/kg/day). PK parameters of mexiletine were compared to racemic 22, (R)‐22, (S)‐22, and alpha‐deuterated 22 (i.e., compound 16). For i.v. administration, as expected because the compounds are so similar to mexiletine, no significant difference was observed for T max, C max, and AUC, for 22 or certain analogs of 22 (Table 6). In contrast, oral administration of phenyl mexiletine analogs showed differences. For analogs of 22 tested, T max was markedly greater (i.e., 33%) than mexiletine. In addition, compared to mexiletine, AUC for racemic 22, and (S)‐22 and 16 were 18%, 18%, and 47% greater, respectively. Racemic 22, (S)‐22, (R)‐22, and alpha‐deuterated 22 (i.e., compound 16) showed a 20%, 20%, 48%, and 8% decrease in clearance compared to mexiletine. For 22 and each analog tested, the bioavailability (F) was greater than for mexiletine (Table 6). (R)‐22 showed 100% bioavailability that was threefold greater than mexiletine. In summary, compared to mexiletine, the data showed that 22 and analogs possessed improved PK properties. In agreement with metabolic stability studies, compared to the non‐deuterated compound 22, deuterated analog 16 showed 27% increased bioavailability compared to mexiletine.
FIGURE 5.

(A). Concentration of mexiletine (ng/ml) in mouse serum after an intravenous dose (5 mg/kg dose, black diamonds) or after an oral dose (25 mg/kg, red circles) as a function of time (hours). (B) Concentration of compound 16 (ng/ml) in mouse serum after an intravenous dose (5 mg/kg dosing, black diamonds) or after an oral dose (25 mg/kg, red circles) as a function of time (hours)
TABLE 6.
Effect of mexiletine and phenyl mexiletines on pharmacokinetic parameters in rats
| Compound | Route of administration | T max (h) | N a | C max b (ng/ml) | AUCc (h*ng/ml) | V dss d (L/kg) | CL e (L/h/kg) | T 1/2 (hr) |
|---|---|---|---|---|---|---|---|---|
| Mexiletine | i.v. | 0.17 | 3 | 289 ± 39 | 447 ± 47 | 197.3 ± 27 | 12.9 ± 1.2 | 10.6 |
| Oral | 1.0 | 3 | 167 ± 41 | 839 ± 456 | 473 ± 220 | 29.8 ± 14.0 | 11 | |
| F = 37% | ||||||||
| o‐CF3, 22 | i.v. | 0.17 | 3 | 250 ± 16 | 544 ± 27 | 42.5 ± 2.1 | 9.2 ± 0.5 | 3.2 |
| Oral | 1.5 | 4 | 226 ± 15 | 1008 ± 227 | 224 ± 46.8 | 23.9 ± 4.9 | 6.5 | |
| F = 38% | ||||||||
| o‐CF3‐D, 16 | i.v. | 0.17 | 3 | 224 ± 13 | 345 ± 25 | 86.8 ± 6.4 | 14.0 ± 1.0 | 4.3 |
| Oral | 1.5 | 4 | 238 ± 12 | 807 ± 11 | 435 ± 40 | 27.7 ± 0.4 | 10.9 | |
| F = 51% | ||||||||
| o‐CF3, (S)‐22 | i.v. | 0.17 | 2 | 219 ± 15 g | 407 ± 34 | 54.6 ± 3 | 10.0 ± 0.7 | 3.8 |
| Oral | 1.5 | 2 | 226 ± 15 g | 1008 ± 227 | 223.2 ± 49.1 | 23.8 ± 4.8 | 6.5 | |
| F = 42% | ||||||||
| o‐CF3, (R)‐22 | i.v. | 0.18 | 3 | 249 ± 2 | 280 ± 59 | 30.3 ± 6.3 | 17.5 ± 3.7 | 1.2 |
| Oral | 1.5 | 2 | 258 ± 2 g | 1556 ± 19 f | 118.5 ± 18.6 | 15.5 ± 0.2 f | 5.3 | |
| F = 100% |
The number of male rats for each route of administration, i.v. route (5 mg/kg) and oral route (2 mg/kg).
The maximum concentration in the serum.
AUC, area under the curve.
V dss is volume of distribution at steady state.
CL is clearance of test compound.
Statistically different from mexiletine, p = .05.
Represents a range of values indicated.
4. DISCUSSION
In prior studies, we developed moderate‐throughput physiological screens of LQTS3 patient‐derived hiPSC cardiomyocytes to characterize compounds that modulate cardiac electrophysiology and contractility. 28 The results were compared to normal cardiomyocytes that were used to ascertain toxicity of compounds because normal cardiomyocytes do not possess mutant sodium channels and pathophysiology. Difference between normal cardiomyocytes and LQTS3 cardiomyocytes likely reflect differences due to genetic variation. Normal and LQTS3 cardiomyocytes used in this study revealed similar maturity in different channels. The genetic variation is almost certainly due to genetic polymorphisms in voltage‐gated ion channels that cause drug‐induced arrhythmias in cardiomyocytes from a diseased individual (i.e., LQTS3 patient) not found in healthy (i.e., normal) cardiomyocytes.
Herein, mexiletine analogs were synthesized and tested and afforded compounds that reverted arrhythmia phenotypes in a hiPSC cardiomyocyte model of LQTS3. This provided the ability to directly detect AP anomalies and to compare compounds in normal and patient‐derived hiPSC cardiomyocytes. The results highlighted the use of hiPSC cardiomyocytes to characterize physiological effects of small molecules and showed this approach successfully led to new drug candidates to treat an inherited channel disorder. LQTS3 patient‐derived hiPSC cardiomyocytes were resistant to the adverse effects of mexiletine, reminiscent of the patient who tolerated a high dose. 42
Compared to mexiletine, substituted phenyl mexiletine analogs decreased prolongation of the AP. In addition, compared to non‐deuterated compounds, incorporation of alpha‐amino deuterium into phenyl mexiletine analogs did not significantly alter the cardiovascular properties of the molecule (i.e., AP shortening potency, Table 2). In general, the effect of phenyl mexiletines on cessation of cardiomyocyte beating in LQTS3 and normal cardiomyocytes was similar and combined with the observation none of the deuterated phenyl mexiletines examined caused EADs in normal cardiomyocytes, compared to mexiletine, 13–16 were judged to be relatively non‐toxic. Thus, alpha‐amino deuteration of phenyl mexiletine and analogs maintained the cardiopharmacological properties of protic precursor phenyl mexiletines examined.
4.1. Metabolism of mexiletine and phenyl mexiletine
The drug development program undertaken herein was directed at improving both pharmacological and pharmaceutical properties of mexiletine. Mexiletine is relatively efficiently metabolized in mammals. 40 The metabolism of phenyl mexiletine is less well characterized but it likely has similarities to mexiletine. We compared metabolism of mexiletine and phenyl mexiletine as well as deuterated analogs. This was facilitated by an efficient synthesis of deuterated phenyl mexiletine analogs. As described herein, the hepatic metabolism of deuterated mexiletine was less than protio‐mexiletine (Table 3). In the presence of mouse liver S‐9, human liver S‐9, and human FMO1, deuterated mexiletine was metabolized to a lesser extent than protio‐mexiletine. The effect of alpha‐amino deuteration on human CYP3A4‐mediated metabolism of mexiletine was not demonstrative. In the presence of mouse liver S‐9, human FMO1, and human CYP3A4, the metabolism of deuterated phenyl mexiletine was decreased. Compared to phenyl mexiletine, human liver S‐9 did not have a prominent effect on metabolism of deuterated phenyl mexiletine (Table 3). Similarly, compared to mexiletine, deutero‐phenyl mexiletine analogs (i.e., compounds 13–16) were generally less efficiently metabolized. Thus, in the presence of mouse liver S‐9, human FMO1, human FMO3, or CYP3A4, 13–16 were metabolized less efficiently than mexiletine (Table 4).
Compared to mexiletine, a large decrease in hepatic metabolism of 16 was observed. This could arise from the lack of metabolism of the o‐methyl aryl group because of replacement of methyl hydrogen atoms with fluorine atoms. Oxidative metabolism of a C‐F bond is known to be less efficient than oxidative metabolism of C‐H bonds. In addition, the phenyl group coupled with the alpha‐amino deuterium substituent apparently decreased metabolism proximal to the amine moiety. Decrease in metabolism largely due to a kinetic isotope effect upon introduction of a deuterium at the metabolic site of a molecule is possible because other effects (e.g., steric effects) are unlikely because of similarities of van der Waals volume of C‐H versus C‐D bonds. 43 It is possible that cleavage of C‐H (or C‐D) bonds at the alpha carbon is a, heretofore, significant unreported metabolic pathway for mexiletine (Figure 6). Although the N‐hydroxy metabolite of mexiletine has been described, 44 the enzymatic basis for N‐oxygenation has not been reported. Mexiletine N‐oxidation could arise via CYP metabolism. Alternatively, N‐oxygenation and oxidative deamination involving cleavage of the alpha‐amino C‐H bond could occur in principle via two routes: FMO‐dependent and/or monoamine oxidase (MAO)‐dependent pathways, 45 respectively. In the case of FMO, N‐oxygenation occurs twice to afford a di‐N‐hydroxy species that leads to the loss of the elements of water to produce an oxime. Ultimately, in the presence of metabolic or aqueous systems, oximes are hydrolyzed to ketones. In the case of monoamine oxidase‐catalyzed imine formation, direct C‐H bond cleavage occurs in the first step. Imines are likewise hydrolyzed to ketones. The magnitude of FMO‐mediated kinetic isotope effect is likely manifested in the first step of oxygenation. A related compound to mexiletine (i.e., amphetamine) did not show a significant C‐H (C‐D) metabolic kinetic isotope effect 46 but FMO is a thermally unstable enzyme and functional activity is highly dependent on quality of its source and the nature of how metabolism is conducted. 45 Amphetamine hydroxylamine showed a modest inverse deuterium kinetic isotope effect on further oxygenation. 47 Accordingly, the true intrinsic isotope effect (i.e., the full effect originating from the single isotopically sensitive step in catalysis) for alpha‐deutero mexiletine or phenyl mexiletine is likely due to terminal C‐H (C‐D) bond cleavage.
FIGURE 6.

Representation of metabolism of deuterated phenyl mexiletine to the corresponding ketone
As described above, less has been reported about the metabolism of phenyl mexiletines. As shown herein (Figure 6), phenyl mexiletines could be metabolized by FMO or MAO. 48 Both enzymatic pathways lead to the same ultimate ketone product. In either case, cleavage of the C‐D bond of alpha‐deutero phenyl mexiletines is required for ketone formation and this could result in a significant deuterium isotope effect on the deuterated compound and a decrease in metabolism. However, based on the metabolism data employing FMO, we favor a significant contribution from FMO (Tables 3 and 4).
4.2. Toxicity of mexiletine and phenyl mexiletine enantiomers
In small animals, it has been reported that (S)‐mexiletine is more toxic than (R)‐mexiletine. 49 In results shown herein, we also show that (S)‐mexiletine possesses greater CNS toxicity (i.e., seizures, death) than that observed for (R)‐mexiletine (Table 5). The closely related phenyl mexiletine homolog (i.e., compound 21) also showed significant CNS toxicity (Table 5). However, both ortho‐CF3 phenyl mexiletine homolog enantiomers (i.e., (R)‐ and (S)‐22) did not show CNS toxicity at 100 mg/kg. We elected to examine 22 in greater detail because of its lack of toxicity and decreased metabolism (based on compound 16, Table 4). Generally, a decrease in dose of mexiletine decreases the overall CNS toxicity in patients. The idea to examine compounds with decreased metabolism and decreased toxicity to decrease the amount and or number of doses to animals to improve bioavailability‐motivated study of 22 and deuterated analogs of 22. Pharmacokinetics were used to attempt to support this postulate.
Based on the metabolism data in Tables 3 and 4, compared to mexiletine, the pharmacokinetic properties of phenyl mexiletine analogs were predicted to be improved. Compared to mexiletine, the (R)‐enantiomer of 22 showed a 2.7‐fold increase in bioavailability and a 2‐fold decrease in CL (Table 6). As shown in Figure 5B, alpha‐amino deutero 16 showed a significant increase in AUC (Table 6). It is possible that the development of drug analogs of mexiletine with greater AUC and C max may allow fewer doses to patients. Frequent dosing and compliance with dosing is a major detriment to use of mexiletine, especially in children. Also, compared to mexiletine, the side effects of seizures may be decreased in analogs of the phenyl mexiletine class of compound examined due to the nature of the chemical structure. In addition, improved pharmacokinetic properties of phenyl mexiletines (e.g., deuterated phenyl mexiletines) may afford the use of lower doses and less adverse drug interactions. Phenyl mexiletines tested were observed to be considerably less CNS toxic compared to mexiletine. However, more work is needed to understand the full safety profile of novel mexiletine analogs.
In summary, use of relatively high‐throughput kinetic imaging and dynamic medicinal chemistry with LQTS3 patient hiPSC‐derived cardiomyocytes afforded mexiletine analogs with superior cardiovascular, metabolic and pharmacokinetic properties, and improved safety profiles.
DISCLOSURE
The authors reported no potential competing interest.
AUTHOR CONTRIBUTIONS
Participated in research design: JC, JGG, KO, MM. Conducted experiments: JC, JGG, KO, MJ, WM. Contributed new reagents or analytical tools: JC, JGG, KO, MJ. Performed data analysis: JC, JGG, WM, MM. Wrote or contributed to the writing of the manuscript: JC, JGG, WM, MM.
ETHICS STATEMENT
(1) This material is the authors’ own original work, which has not been previously published elsewhere. (2) The study is not currently being considered for publication elsewhere. (3) The study reflects the authors’ own research and analysis in a truthful and complete manner.
Supporting information
Supplementary Material
Gomez‐Galeno J, Okolotowicz K, Johnson M, McKeithan WL, Mercola M, Cashman JR. Human‐induced pluripotent stem cell‐derived cardiomyocytes: Cardiovascular properties and metabolism and pharmacokinetics of deuterated mexiletine analogs. Pharmacol Res Perspect. 2021;9:e00828. 10.1002/prp2.828
Funding information
We are grateful for the financial support of the California Institute for Regenerative Medicine (CIRM) (Grant Number TR4‐06857 to JRC and MM). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of CIRM or any other agency of the State of California. Flow cytometry and high‐throughput screening services were supported by NIH P30CA030199 at the Sanford‐Burnham‐Prebys Medical Discovery Institute
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the Human BioMolecular Research Institute but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are, however, available from the authors upon reasonable request and with permission of the Human BioMolecular Research Institute.
REFERENCES
- 1. Ma J, Guo L, Fiene SJ, et al. High purity human‐induced pluripotent stem cell‐derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol. 2011;301:H2006‐H2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mercola M, Colas A, Willems E. Induced pluripotent stem cells in cardiovascular drug discovery. Circ Res. 2013;112:534‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart. 2003;89:1363‐1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Waring MJ, Arrowsmith J, Leach AR, et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov. 2015;14:475‐486. [DOI] [PubMed] [Google Scholar]
- 5. Kramer J, Obejero‐Paz CA, Myatt G, et al. MICE models: superior to the HERG model in predicting Torsade de Pointes. Sci Rep. 2013;3:2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Redfern WS, Carlsson L, Davis AS, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32‐45. [DOI] [PubMed] [Google Scholar]
- 7. Singh S, Kerndt C, Zeltser R. Mexiletine. Treasure Island, FL: StatPearls; 2020. [Google Scholar]
- 8. De Luca A, Natuzzi F, Lentini G, Franchini C, Tortorella V, Conte Camerino D. Stereoselective effects of mexiletine enantiomers on sodium currents and excitability characteristics of adult skeletal muscle fibers. Naunyn Schmiedebergs Arch Pharmacol. 1995;352:653‐661. [DOI] [PubMed] [Google Scholar]
- 9. Catterall WA. Common modes of drug action on Na+ channels: local anesthetics, antiarrhythmics and anticonvulsants. Trends Pharmacol Sci. 1987;8:57‐65. [Google Scholar]
- 10. Bennett PB, Yazawa K, Makita N, George AL Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683‐685. [DOI] [PubMed] [Google Scholar]
- 11. Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805‐811. [DOI] [PubMed] [Google Scholar]
- 12. Wang Q, Shen J, Li Z, et al. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet. 1995;4:1603‐1607. [DOI] [PubMed] [Google Scholar]
- 13. Moss AJ, Kass RS. Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest. 2005;115:2018‐2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene‐specific therapy. Circulation. 1995;92:3381‐3386. [DOI] [PubMed] [Google Scholar]
- 15. Mazzanti A, Maragna R, Faragli A, et al. Gene‐specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3. J Am Coll Cardiol. 2016;67:1053‐1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Al‐Khatib SM, Stevenson WG, Ackerman MJ, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the american college of cardiology/american heart association task force on clinical practice guidelines and the heart rhythm society. J Am Coll Cardiol. 2018;72(14):e91–e220. 10.1016/j.jacc.2017.10.054 [DOI] [PubMed] [Google Scholar]
- 17. Priori SG, Blomstrom‐Lundqvist C, Mazzanti A, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2793‐2867. [DOI] [PubMed] [Google Scholar]
- 18. Campbell NP, Kelly JG, Adgey AA, Shanks RG. The clinical pharmacology of mexiletine. Br J Clin Pharmacol. 1978;6:103‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rudel R, Lehamann Horn F, Ricker K. The nondystrophic myotonias. In: Engel AG, Franzini‐Armstrong C, eds. Myology, Chapt 49. New York: McGraw‐Hill; 1994:1291‐1302. [Google Scholar]
- 20. Catalano A, Carocci A, Sinicropi MS. Mexiletine metabolites: a review. Curr Med Chem. 2015;22:1400‐1413. [DOI] [PubMed] [Google Scholar]
- 21. Turgeon J, Uprichard AC, Belanger PM, Harron DW, Grech‐Belanger O. Resolution and electrophysiological effects of mexiletine enantiomers. J Pharm Pharmacol. 1991;43:630‐635. [DOI] [PubMed] [Google Scholar]
- 22. Uprichard AC, Allen JD, Harron DW. Effects of tocainide enantiomers on experimental arrhythmias produced by programmed electrical stimulation. J Cardiovasc Pharmacol. 1988;11:235‐241. [PubMed] [Google Scholar]
- 23. Hill RJ, Duff HJ, Sheldon RS. Determinants of stereospecific binding of type I antiarrhythmic drugs to cardiac sodium channels. Mol Pharmacol. 1988;34:659‐663. [PubMed] [Google Scholar]
- 24. Igwemezie L, Kerr CR, McErlane KM. The pharmacokinetics of the enantiomers of mexiletine in humans. Xenobiotica. 1989;19:677‐682. [DOI] [PubMed] [Google Scholar]
- 25. Turgeon J, Fiset C, Giguere R, et al. Influence of debrisoquine phenotype and of quinidine on mexiletine disposition in man. J Pharmacol Exp Ther. 1991;259:789‐798. [PubMed] [Google Scholar]
- 26. Lanchote VL, Cesarino EJ, Santos VJ, Mere Junior Y, Santos SR. Enantioselectivity in the metabolism of mexiletine by conjugation in female patients with the arrhythmic form of chronic Chagas’ heart disease. Chirality. 1999;11:29‐32. [DOI] [PubMed] [Google Scholar]
- 27. Cashman JR, MacDougall JM. Dynamic medicinal chemistry in the elaboration of morphine‐6‐glucuronide analogs. Curr Top Med Chem. 2005;5(6):585‐594. [DOI] [PubMed] [Google Scholar]
- 28. McKeithan WL, Savchenko A, Yu MS, et al. An automated platform for assessment of congenital and drug‐induced arrhythmia with hiPSC‐derived cardiomyocytes. Front Physiol. 2017;8:766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miller EW. Small molecule fluorescent voltage indicators for studying membrane potential. Curr Opin Chem Biol. 2016;33:74‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cunningham TJ, Yu MS, McKeithan WL, et al. Id genes are essential for early heart formation. Genes Dev. 2017;31:1325‐1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cerignoli F, Charlot D, Whittaker R, et al. High throughput measurement of Ca(2)(+) dynamics for drug risk assessment in human stem cell‐derived cardiomyocytes by kinetic image cytometry. J Pharmacol Toxicol Methods. 2012;66:246‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu HR, Whittaker R, Price JH, et al. High throughput measurement of Ca++ dynamics in human stem cell‐derived cardiomyocytes by kinetic image cytometery: a cardiac risk assessment characterization using a large panel of cardioactive and inactive compounds. Toxicol Sci. 2015;148:503‐516. [DOI] [PubMed] [Google Scholar]
- 33. Harding SD, Sharman JL, Faccenda E, et al. NC‐IUPHAR . The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46(D1):D1091‐D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Attali B, Chandy KG, Giese MH, et al. Voltage‐gated potassium channels (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide Pharmacol CITE. 2019;2019(4). 10.2218/gtopdb/F81/2019.4 [DOI] [Google Scholar]
- 35. Catterall WA, Goldin AL, Waxman SG. Voltage‐gated sodium channels (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide Pharmacol CITE. 2019;2019(4). 10.2218/gtopdb/F82/2019.4 [DOI] [Google Scholar]
- 36. Alexander SPH, Mathie A, Peters JA, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: ion channels. Br J Pharmacol. 2019;176:S142‐S228. 10.1111/bph.14749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cashman JR, Ryan D, Mckeithan W, et al. Anti‐arrhythmic hit to lead refinement in a dish using patient‐derived iPSC cardiomyocytes. J Med Chem. 2021;64(9):5384‐5403. [DOI] [PubMed] [Google Scholar]
- 38. Huang K, Ortiz‐Marciales M, Stepanenko V, De Jesus M, Correa W. A practical and efficient route for the highly enantioselective synthesis of mexiletine analogues and novel beta‐thiophenoxy and pyridyl ethers. J Org Chem. 2008;73:6928‐6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ryan DA, Okolotowicz KJ, Mercola M, Cashman JR. Stereoselective synthesis of mexiletine and structural analogs with chiral tert‐butanesulfinamide. Tetrahedron Lett. 2015;56:4195‐4199. [Google Scholar]
- 40. Otani M, Fukuda T, Naohara M, et al. Impact of CYP2D6*10 on mexiletine pharmacokinetics in healthy adult volunteers. Eur J Clin Pharmacol. 2003;59:395‐399. [DOI] [PubMed] [Google Scholar]
- 41. Labbe L, Turgeon J. Clinical pharmacokinetics of mexiletine. Clin Pharmacokinet. 1999;37:361‐384. [DOI] [PubMed] [Google Scholar]
- 42. Terrenoire C, Wang K, Tung KW, et al. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol. 2013;141:61‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grant TG. Using deuterium in drug discovery: leaving the label in the drug. J Med Chem. 2014;57:3595‐3612. [DOI] [PubMed] [Google Scholar]
- 44. Labbe L, Abolfathi Z, Robitaille NM, St‐Maurice F, Gilbert M, Turgeon J. Stereoselective disposition of the antiarrhythmic agent mexiletine during the concomitant administration of caffeine. Ther Drug Monit. 1999;21:191‐199. [DOI] [PubMed] [Google Scholar]
- 45. Cashman JR. Monoamine oxidases and flavin‐containing monooxygenases. In: McQueen CA, ed. Comprehensive Toxicology. Vol 10. 3rd ed. Elsevier Science; 2018;87‐125. [Google Scholar]
- 46. Parli CJ, McMahon RE. The mechanism of microsomal deamination: heavy isotope studies. Drug Metab Dispos. 1973;1:337‐341. [PubMed] [Google Scholar]
- 47. Cashman JR, Xiong YN, Xu L, Janowsky A. N‐oxygenation of amphetamine and methamphetamine by the human flavin‐containing monooxygenase (Form 3): role in bioactivation and detoxication. J Pharmacol Exper Ther. 1999;288:1251‐1260. [PubMed] [Google Scholar]
- 48. Clarke DE, Lyles GA, Callingham BA. A comparison of cardiac and vascular clorgyline‐resistant amine oxidase and monoamine oxidase: inhibition by amphetamine, mexiletine and other drugs. Biochem Pharmacol. 1982;31:27‐35. [DOI] [PubMed] [Google Scholar]
- 49. Kamei J. Antinociceptive effects of the enantiomorphs of mexiletine and its main metabolite in streptozotocine‐induced diabetic mice. Nippon Shinkei Seishin Yakurigaku Zasshi. 2000;20:11‐16. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available from the Human BioMolecular Research Institute but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are, however, available from the authors upon reasonable request and with permission of the Human BioMolecular Research Institute.